

Abstract
Tumor‐targeting protein toxins are composed of a toxic enzyme coupled to a specific cell binding domain that targets cancer‐associated antigens. The anti‐tumor treatment by targeted toxins is accompanied by dose‐limiting side effects. The future prospects of targeted toxins for therapeutic use in humans will be determined by reduce side effects. Certain plant secondary metabolites (saponins) were shown to increase the efficacy of a particular epidermal growth factor receptor (EGFR)‐targeted toxin, paralleled by a tremendous decrease of side effects.
This study was conducted in order to investigate the effects of substituting different toxin moieties fused to an EGF ligand binding domain on the augmentative ability of saponins for each against therapeutic potential of the saponin‐mediated efficacy increase for different anti‐tumor toxins targeting the EGFR.
We designed several EGFR‐targeted toxins varying in the toxic moiety. Each targeted toxin was used in combination with a purified saponin (SA1641), isolated from the ornamental plant Gypsophila paniculata L. SA1641 was characterized and the SA1641‐mediated efficacy increase was investigated on EGFR‐transfected NIH‐3T3 cells.
We observed a high dependency of the SA1641‐mediated efficacy increase on the nature of toxin used for the construction of the targeted toxin, indicating high specificity.
Structural alignments revealed a high homology between saporin and dianthin‐30, the two toxic moieties that benefit most from the combination with SA1641.
We further demonstrate that SA1641 did not influence the plasma membrane permeability, indicating an intracellular interaction of SA1641 and the toxin components of targeted toxins. Surface plasmon resonance measurements point to a transient binding of SA1641 to the toxin components of targeted toxins.
Keywords: Toxin efficacy, Cancer therapy, Saponin, Targeted toxin
1. Introduction
The concept of targeted tumor therapy goes back to Paul Ehrlich who postulated that antibodies fused to toxic components might serve as magic bullets (Silverstein, 2002). For the most part, targeted toxins are chimeric fusion proteins that consist of two components: a toxic protein with enzymatic activity and a cell binding domain that targets tumor‐associated antigens. Cell binding domains can be either monoclonal antibodies, fragments of antibodies (e.g. single chain antibodies, scFv) or natural ligands such as growth factors or cytokines (Fuchs and Bachran, 2009). Several such targeted toxins are in clinical trials and DAB (389) IL‐2 (Ontak®), a fusion between diphtheria toxin (DT) lacking cell binding domain and interleukin‐2, has been approved by the Food and Drug Administration (Woo et al., 2010; Frankel et al., 2002).
Specific cell binding ligands may be chemically linked to or genetically fused to bacterial or plant toxins such as DT from Corynebacterium diphtheria, Pseudomonas exotoxin A (PE) from Pseudomonas aeruginosa or saporin from Saponaria officinalis L. DT and PE are ADP‐ribosyl transferases (EC 2.4.2.36) that transfer an ADP‐ribose to the eukaryotic elongation factor 2 (EF2), a process that halts protein synthesis. Saporin, dianthin‐30 and ricin‐A‐chain (RTA) are ribosomal RNA (rRNA) N‐glycosylases (EC 3.2.2.22) that remove an adenine residue at position 4324 from the 28S ribosomal RNA (Stirpe and Battelli, 2006). Usually these toxic enzymes have high turnover numbers (Fermani et al., 2009). In theory this should facilitate efficient cell killing by one internalized molecule. Targeted toxins have to escape from the endosome after internalization by endocytosis to reach the cytosol, where they inhibit protein synthesis. Alternatively they can be either exocytosed via recycling endosomes or degraded in lysosomes.
Beside lysosomal degradation an inefficient endosomal escape is one of the toxicity limiting steps after endocytosis of the targeted toxin (Varkouhi et al., 2010). The design of methods that would improve the efficiency of the endosomal escape process would therefore be likely to improve the cytotoxic efficacy of targeted toxins (Pirie et al., 2010). The inefficient endosomal escape of targeted toxins into the cytosol necessitates that relatively high doses of drug are used that are associated with serious adverse side effects such as vascular leak syndrome (Kreitman, 2006).
We have demonstrated in earlier publications that a composite of plant secondary metabolites (Saponinum album) enhanced the toxicity of a targeted toxin composed of human epidermal growth factor (EGF) and saporin (referred to as Sap‐EGF) by up to a million‐fold (Bachran et al., 2010). In spite of the substantial improvement by Saponinum album it must be considered that Saponinum album is an exceedingly complex mixture of various saponin molecules that consist of a triterpenoid backbone (aglycone) and one or two branched carbohydrate chains attached to the aglycone (Weng et al., 2010). Saponinum album is isolated by solvent extraction of the roots of Gypsophila paniculata L. (Baby's Breath), a common ornamental plant. The efficacy of the combination treatment of Saponinum album and Sap‐EGF was also shown in vivo in BALB/c mice (Bachran et al., 2009). The therapeutic benefit of the combination anti‐tumor therapy with Saponinum album is based on the reduction of the targeted toxin dosage and the concomitant increase in efficacy.
With respect to the application of targeted toxins in cancer therapy it would be a major step forward to improve the selective cytotoxic effect of arbitrary targeted toxins by the use of saponins. Such a combination treatment (saponin + targeted toxin) could then be progressed and developed as a new platform technology in targeted tumor therapies to increase the cytosolic delivery of targeted toxins to tumor cells.
To test this hypothesis we constructed different targeted toxins consisting of EGF as targeting moiety and either dianthin‐30, saporin, RTA (rRNA N‐glycosylases) or truncated forms of DT and PE (ADP‐ribosyl transferases) as toxic moieties. All targeted toxins were combined with a saponin (SA1641) that was isolated from Saponinum album. SA1641 was characterized and the SA1641‐mediated augmentation of efficacy was determined.
2. Materials and methods
2.1. Molecular cloning of targeted toxins
Native saporin and RTA were obtained from Sigma–Aldrich (Steinheim, Germany). Oligonucleotides were purchased from Metabion (Martinsried, Germany). Dianthin‐30 (EC 3.2.2.22) was subcloned by PCR from a plasmid containing the cDNA for dianthin‐30 (a gift from Marco Colombatti, University of Verona). The forward primer (5′‐AGC CGC GGC CAC AGC ATA CAC ATT AAA‐3′) contained a SacII and the reverse primer (5′‐GGC TAG CCT TCG GTC TAC CTA AAT ACC‐3′) a NheI cleavage site. The PCR product was subcloned into pJET1.2 (CloneJET™ PCR Cloning Kit, Fermentas, St. Leon‐Rot, Germany). The insert DNA was then ligated into the pET11d (Novabiochem, Schwalbach, Germany) expression vector via SacII/NheI, which already contained the cDNA for human EGF and the motif 5′‐CAT CAT CAT CAT CAT CAT‐3′ coding for a His‐tag (Heisler et al., 2003). The plasmid (Dia‐EGFpET11d) was propagated in DH5α cells (ZYMO RESEARCH, Irvine, USA). Sequencing was done using ABI PRISM 310 Genetic analyzer (Perkin–Elmer, Foster City, CA). Sap‐EGF and Hissaporin were cloned previously (Heisler et al., 2003).
RTA (EC 3.2.2.22, amino acids 1–268 of ricin) was released by NcoI/NheI from a plasmid containing the cDNA for RTA (provided by Marco Colombatti, University of Verona) and the sequence 5′‐CAT CAT CAT CAT CAT CAT‐3′ for the His‐tag. The insert was ligated via NcoI/NheI into a pET11d expression vector that already contained the cDNA for EGF. EGF was amplified from cDNA generated from dimethylsulfoxide‐differentiated HL‐60 cells (Heisler et al., 2003). The cDNAs (RTApET11d and EGFpET11d) used for the cloning of RTA‐EGFpET11d were sequenced and the plasmid RTA‐EGFpET11d was multiplied in DH5α cells as described before.
For the construction of EGF‐ETA′ (EC 2.4.2.36, domain II/III, amino acids 276–638 of Pseudomonas exotoxin A) EGF was amplified by PCR from Sap‐EGF using the forward primer 5′‐CTT GCA AAG CTT GCT AGC CCC GGG AAT AGT GAC‐3′ (HindIII cleavage site) and the reverse primer 5′‐CAG CTA GCG GCC GCG CGC AGT TCC CAC CAC TTC AG‐3′ (NotI cleavage site). The PCR product was ligated via HindIII/NotI into a pET27b (Novabiochem, Schwalbach, Germany) variant that already contained the cDNA for ETA′ and a 5′‐CAT CAT CAT CAT CAT CAT CAT CAT CAT CAC‐3′ motif (the cDNA for ETA′ was a generous gift from Stefan Barth, Fraunhofer IME, Aachen). The plasmid EGF‐ETA′pET27b was also propagated in DH5α cells and sequenced in an ABI PRISM 310 Genetic analyzer. DT390 (EC 2.4.2.36, amino acids 1−390) was amplified by RT‐PCR from C. diphtheria and fused to EGF as described elsewhere in detail (Bachran et al., 2005).
2.2. Expression and purification of fusion proteins
Plasmids (HisSappET11d, Sap‐EGFpET11d, Dia‐EGFpET11d, RTA‐EGFpET11d, DT390EGFpET11d, EGF‐ETA′pET27b) were transformed into Escherichia coli Rosetta DE pLysS (Novabiochem, Schwalbach, Germany). Cells were grown overnight at 30 °C in Lysogeny Broth (LB) supplemented with either 100 μg/mL ampicillin (pET11d constructs) or 30 μg/mL kanamycin (EGF‐ETA′pET27b). After centrifugation (5 min, 3000g) cells were resuspended in 2 L LB containing 50 μg/mL ampicillin or 30 μg/mL kanamycin and grown to a density of A 550 0.8–1. Isopropyl β‐d‐1‐thiogalactopyranoside (IPTG, 2 mL, 1 M) was added and cells were further grown for either 3 h at 37 °C (HisSappET11d, Sap‐EGFpET11d, Dia‐EGFpET11d, DT390EGFpET11d) or 3 h at 30 °C (RTA‐EGFpET11d, EGF‐ETA′pET27b). Cells were harvested by centrifugation (10 min, 5000g, 4 °C) and the cell pellets were either stored in 20 mL of phosphate‐buffered saline (PBS, pH 7.4) (HisSappET11d, Sap‐EGFpET11d, Dia‐EGFpET11d, EGF‐ETA′pET27b) or in 20 mL of triethanol amine buffer (20 mM, pH 8.3) (RTA‐EGFpET11d) at −20 °C. Inclusion bodies (DT390EGFpET11d) were solubilized in 8 M Urea and DT390EGF was purified and renatured as described elsewhere (Bachran et al., 2005). After thawing, fusion proteins (Hissaporin, Sap‐EGF, Dia‐EGF, EGF‐ETA′, RTA‐EGF) were released by FRENCH® Press at 1500 psi. The solution was centrifuged (30 min, 4 °C) and adjusted to 10 mM imidazole. All constructs contained a N‐terminal His‐tag and were applied to nickel‐nitrilotriacetate chromatography (Ni‐NTA Agarose, Qiagen, Hilden Germany). After elution with imidazole (20–250 mM) fractions were analyzed by SDS‐PAGE (12%). Fractions containing Dia‐EGF, Sap‐EGF and Hissaporin were concentrated in Amicon centrifugal filter devices (30 kDa or 5 kDa, Millipore, Eschborn, Germany) and imidazole was removed by buffer exchange (PBS) with PD10 columns (GE Healthcare, Munich, Germany). The protein concentration was determined by bicinchoninic assay (Pierce, Rockford, USA).
Fractions containing EGF‐ETA′ and RTA‐EGF were applied to anion exchange chromatography (Q Sepharose Fast Flow, GE Healthcare, Munich, Germany). EGF‐ETA′ was eluted by a sodium chloride (0.05–0.3 M) gradient in l‐histidine buffer (20 mM, pH 6.3). RTA‐EGF was eluted by a sodium chloride (0.05–0.5 M) gradient in a triethanol amine buffer (20 mM, pH 8.3). After SDS‐PAGE (12%) fractions were pooled and samples were processed as described before.
2.3. Determination of enzymatic activity
The rRNA N‐glycosylase activities of Hissaporin, Sap‐EGF, Dia‐EGF, RTA‐EGF, native saporin and RTA were determined by an adenine releasing assay that is based on the release of adenine molecules from herring sperm DNA (hsDNA) (Fermani et al., 2009). Briefly, 30 pmol targeted toxin or toxin were mixed with 10 μl (100 μg) hsDNA and volume made up to 50 μl by acetate buffer (pH 4, 100 mM KCl). The mixture was incubated for 1 h at 50 °C. Controls were either incubated with native saporin or RTA or only with acetate buffer and hsDNA. After centrifugation through filtration devices (molecular mass cut off 5 kDa), the absorbance of the filtrate was measured at 260 nm by a Nano Drop spectrometer (ND‐1000, Peqlab, Erlangen, Germany). Amount of the released adenine was determined against an adenine calibration curve.
To determine the enzymatic activity of EGF‐ETA′ and DT390EGF, an ADP‐ribosylation assay was performed (Collier and Kandel, 1971). His‐tagged EF2 was expressed in Saccharomyces cerevisiae (TKY675) and purified by metal chelate chromatography as described elsewhere (Bachran et al., 2007). For the ADP‐ribosylation assay 2 μL EF2 (0.5 μg/μL), 0.5 μL 6‐Biotin‐17‐NAD+ (2.5 × 10−4 M, Trevigen, Gaithersburg, USA) and either EGF‐ETA′ (1.6 μL with 1.2 μg/μL), DT390EGF (2.6 μL with 0.74 μg/μL) or 3 μL native diphtheria toxin (DT) (0.3 μg/μL) (Sigma–Aldrich, Steinheim, Germany) were mixed and volumes were made up to 24 μL with reaction buffer (0.05 M Tris, pH 7.6, 1 mM EDTA, 1 mM dithiothreitol). Samples were incubated for 1 h at 37 °C and biotinylated EF2 was detected by Western blotting with peroxidase‐conjugated streptavidin (Sigma–Aldrich, Steinheim, Germany).
2.4. Isolation of SA1641 from saponinum album
SA1641 was isolated by high performance liquid chromatography from Saponinum album (Merck, Darmstadt, Germany) and analyzed by electron spray ionization time‐of‐flight mass spectrometry (m/z 1641.7325) as described elsewhere (Weng et al., 2009a). Purity was determined by thin layer chromatography (data not shown). SA1641 (8 mg) was dissolved in 600 μL pyridine‐d5 and 1H/13C NMR analyses were performed using a Bruker DRX600 and a Bruker AV600 NMR spectrometer. The assignments were based on Double Quantum Filtered‐Correlated Spectroscopy (DQF‐COSY), Total Correlation Spectroscopy (TOCSY), Heteronuclear Single Quantum Coherence–Total Correlation Spectroscopy (HSQC‐TOCSY) and Heteronuclear Multiple Quantum Correlation (HMBC) experiments at 600/150 MHz. XWINNMR and topspin were used as the acquisition software.
2.5. Membrane integrity assay
To investigate the influence of SA1641 on the plasma membrane integrity HER14 cells were seeded in gelatin‐coated 24‐well plates and grown to approximately 80% confluency. Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum (BioChrom KG, Berlin, Germany), 100 U/mL penicillin, and 100 μg/mL streptomycin was used as cell culture medium. Medium was aspirated and 300 μL fresh medium supplemented with 4 μg/mL propidium iodide was added to each well. SA1641 with a final conc. of 5 μg/mL (3 μM), 10 μg/mL (6 μM), 20 μg/mL (12 μM) or 40 μg/mL (24 μM) was added. Digitonin, a saponin that is known to influence membrane permeability by association with membrane sterol (Hsuchen and Feingold, 1973) was used as positive control (final conc. 3, 6, 12 or 24 μM). Cells were incubated for further 30 min, medium was removed and 200 μL trypsin per well was added. Each 200 μL were mixed with 300 μL PBS supplemented with 10% fetal bovine serum. Cells were analyzed by a FACScalibur (Becton Dickinson, Heidelberg, Germany).
2.6. Cytotoxicity assay
HER14 cells (Swiss mouse embryo NIH‐3T3 cells transfected with human EGFR, obtained from E.J. Zoelen, Department of Cell Biology, University of Nijmegen, The Netherlands) were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum (BioChrom KG, Berlin, Germany), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were cultivated at 37 °C, 5% CO2, and 95% humidity. Cells were seeded in gelatin‐coated (0.1%) 96‐well plates (2000 cells/well) and grown for 24 h. Cells were washed (100 μL PBS/well) and covered with cell culture medium (180 μL/well) either supplemented with 5 μg/mL SA1641 or not. Thereafter 20 μL of either Sap‐EGF (final conc. 0.1 fM–100 nM), Dia‐EGF (final conc. 0.1 fM to 100 nM), RTA‐EGF (final conc. 0.1–1000 nM), EGF‐ETA′ (final conc. 0.02–200 nM) or DT390EGF (final conc. 0.01–100 nM) was added. Controls were added with only 20 μL phosphate‐buffered saline or 5 μg/mL SA1641. For the combination experiments with various concentrations of SA1641 and targeted toxins cells were incubated with 0–10 μg/mL SA1641 and 0.01 nM Dia‐EGF or Sap‐EGF. Cytotoxicity of SA1614 was determined by incubation HER14 cells with different concentrations (2–80 μg/mL) SA1641. After 48 h cytotoxicity was determined by MTT‐assay (Mosmann, 1983).
2.7. Binding of targeted toxins
HER14 Cells were seeded in gelatin‐coated 96‐well plates (2000 cells/well) and grown for 72 h. Medium was removed and 50 μL of pre‐cooled Dulbecco's Modified Eagle Medium supplemented with either Sap‐EGF, Dia‐EGF, RTA‐EGF, DT390EGF, EGF‐ETA′ or EGF (each 200 nM) was added. Cells were incubated at 4 °C for 2 h, washed twice with 200 μL phosphate‐buffered saline supplemented with Ca2+/Mg2+ (PBS +/+) and fixed with 150 μL paraformaldehyde (1%) for 15 min. Cells were washed twice with 200 μL PBS +/+ and blocked with 200 μL blocking solution (100 mL/L fetal bovine serum and 20 g/L bovine serum albumin, 30 min). Cells were then incubated with 100 μL of either a monoclonal mouse anti‐His‐tag antibody (#34660, Qiagen, Hilden Germany, 100 ng/mL in PBS +/+) solution (in case of Sap‐EGF, Dia‐EGF, RTA‐EGF, DT390EGF) or with 100 μL of a rabbit anti‐Pseudomonas exotoxin A antibody (#P2318, Sigma–Aldrich, Steinheim, Germany, 1:2000) solution (EGF‐ETA′). EGF treated cells were incubated with 100 μL of rabbit polyclonal anti‐human EGF antibody (#ab9695, Abcam, Cambridge, UK, 100 ng/mL in PBS +/+). After 30 min incubation, cells were washed 3× with 200 μL PBS +/+ supplemented with 0.05% Tween 20 (PBST). Bound antibodies were detected with 100 μL of peroxidase‐conjugated rabbit polyclonal antibody to mouse IgG (#P0260, 650 ng/mL in PBS+/+, Dako, Hamburg, Germany), or peroxidase‐conjugated goat polyclonal antibody to rabbit IgG (#P0448, 500 ng/mL, in PBS+/+, Dako, Hamburg, Germany). Cells were washed with 200 μL PBST (4×) and incubated with 100 μL of 40 mM citric acid, pH 3.95, supplemented with 3, 3', 5, 5'‐tetramethylbenzidine solution (0.2 mg/mL) and hydrogen peroxide (0.1 μg/mL). After 60 min 50 μL 2 M sulfuric acid was added to each well and absorbance was measured at 450 nm (reference 490 nm).
2.8. Surface plasmon resonance (SPR) measurements
SPR measurements were performed at 25 °C on a BIACORE X instrument (GE Healthcare, Freiburg, Germany). HisSaporin was immobilized on a CM5 (carboxymethylated) sensor chip (GE Healthcare, Freiburg, Germany) by amine coupling at ∼770 response units (RU) according to the manufacturer's instructions. For the coupling procedure protein was diluted in 10 mM sodium acetate, pH 5.0 and injected at 5 μL/min using HBS‐EP running buffer (GE Healthcare), consisting of 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant P20. Unreacted free binding sites were blocked with 1 M ethanol amine (GE Healthcare), pH 8.5. A control surface to discriminate the unspecific binding was obtained by ethanol amine deactivation of the activated dextran surface, according to the amine coupling protocol.
Binding analyses were performed with a running buffer, containing 1× Dulbecco's PBS buffer w/o Ca & Mg (PAA Laboratories GmbH, Coelbe, Germany) or 10 mM sodium acetate pH 5 at 25 °C. SA1641 was diluted with the running buffer to 5, 10 or 20 μg/mL concentration and 100 μl of each sample were injected at a flow rate of 30 μL/min. The association phase was set to 200 s, followed by a 180 s dissociation phase. Final sensorgrams were obtained by subtraction of reference lane data. To regenerate the chip after each run, a 60 s regeneration was performed with 4 M MgCl2. Running buffer was injected between each sample to avoid carryover effects. Binding curves were analyzed with BIA evaluation software 4.1.1.
2.9. Statistical analysis
Statistical analysis has been performed using GraphPad Prism 4.0 (GraphPad Software Inc.)
3. Results
3.1. Enzymatic activity
All constructs contained EGF as binding domain and different toxic enzymes with either N‐glycosylase such as saporin, dianthin‐30, RTA (amino acids 1 to 268 of ricin) or ADP‐ribosyl transferase (DT390, ETA') activity. DT390 comprises the first 390 amino acids of diphtheria toxin and ETA' comprises domain II and III (amino acids 276–638) of Pseudomonas exotoxin A. DT390, ETA' and RTA do not contain the toxin's natural binding domains. The enzymatic activity was determined for all targeted toxins since it is one of the main prerequisites for exerting cytotoxicity.
In an adenine releasing assay recombinant Hissaporin that was used for the construction of Sap‐EGF, native saporin and RTA released 312, 397 and 22 pmol adenine/pmol toxin/h (Table 1). Although the activity is reduced in the fusion proteins, Sap‐EGF (240 pmol adenine/pmol toxin/h) and also Dia‐EGF (218 pmol adenine/pmol toxin/h) released a high number of adenine molecules from hsDNA and RTA‐EGF is also still active, releasing 17 pmol adenine/pmol toxin/h.
Table 1.
Enzymatic activity of N‐glycosylases (N‐GA) and N‐GA based targeted toxins
| Targeted toxins | Adenine release (pmol adenine/pmol toxin/h) |
|---|---|
| Saporin‐EGF | 240 ± 4 |
| Dianthin‐30‐EGF | 218 ± 10 |
| RTA‐EGF | 17 ± 1 |
| N‐glycosylases | |
| HisSaporin‐3 | 312 ± 1 |
| nSaporin | 397 ± 9 |
| nRTA | 22 ± 2 |
In general it can be stated that the fusion of EGF to the N‐glycosylases causes a moderate decrease (20–23%) of the enzymatic activity compared to the single toxins.
The enzymatic activity of EGF‐ETA', DT390EGF and native DT was estimated by detecting ADP‐ribosylated elongation factor 2 (EF2) in a Western blot. As shown in Figure 1, EGF‐ETA' and DT390EGF showed comparable ADP‐ribosyl transferase activities. Native diphtheria toxin (DT) was used as positive control.
Figure 1.

Determination of ADP‐ribosyl transferase activity. Elongation factor 2 was incubated with biotinylated NAD+ and the corresponding enzyme. The enzymatic activities of the targeted toxins were determined in a Western blot that is based on the detection of biotinylated elongation factor 2 via streptavidin‐HRP. EGF‐ETA' and DT390EGF showed comparable ADP‐ribosyl transferase activities. Native diphtheria toxin (DT) was used as positive control.
3.2. Binding of targeted toxins
A further prerequisite to achieve toxicity of targeted toxins on target cells is the binding to their target receptors. In a cell based ELISA we determined the binding of Sap‐EGF, Dia‐EGF, RTA‐EGF, DT390EGF, EGF‐ETA′ and EGF (used as positive control) to NIH‐3T3 cells that have been stably transfected with human EGF receptor (EGFR). Transfected cells are referred to as HER14 cells. As shown in Figure 2 all targeted toxins bound to the cells. Since we had to use different antibodies (for Sap‐EGF, Dia‐EGF, RTA‐EGF and DT390EGF anti‐His‐tag, for EGF‐ETA′ anti‐PE and for EGF anti‐EGF) direct quantitative comparisons of the binding activities were not possible. However for this study it was adequate to show a principal binding to the cells.
Figure 2.
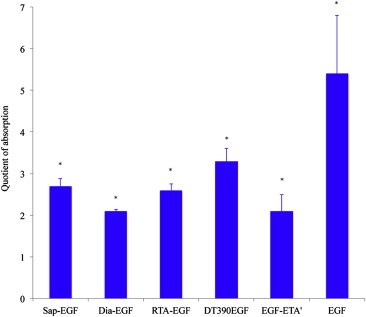
Binding of targeted toxins and EGF to HER14 cells. Binding activity was determined in a cell based ELISA. All targeted toxins and EGF bound to the cells. Sap‐EGF, Dia‐EGF, RTA‐EGF and DT390EGF were detected with an anti‐His‐, EGF‐ETA' with an anti‐PE‐ and EGF with an anti‐EGF antibody. Each column represents the mean ± SD of one experiment performed in quadruplicate, n = 4. Values (expressed as quotient of absorption) were normalized to control cells that were only treated with antibodies. ∗Significant to control (anti‐His, anti‐PE, anti‐EGF), Mann‐Whitney‐Test, p ≤ 0.04.
3.3. Isolation and cytotoxicity of SA1641
Saponinum album is a complex mixture of saponins. In a preliminary study we already isolated SA1641 from Saponinum album (Weng et al., 2009a). Up to now detailed structural data for SA1641 is missing and a hypothetical structure has been proposed. In this study we isolated SA1641 to homogeneity from Saponinum album and characterized it by intensive one and two dimensional NMR spectroscopy (for NMR data, see supplementary data). SA1641 (Figure 3A) was identified as: 3‐O‐β‐d‐xylopyranosyl‐(1 → 3)‐[β‐d‐galactopyranosyl‐(1→2)]‐β‐d‐glucuronopyranosyl gypsogenin 28‐o‐(6‐deoxy‐β‐d‐glucopyranosyl)‐(1 → 4)‐[β‐d‐xylopyranosyl‐(1 → 3)‐β‐d‐xylopyranosyl(1 → 4)]‐α‐l‐rhamnopyranosyl‐(1 → 2)‐β‐d‐fucopyranoside.
Figure 3.
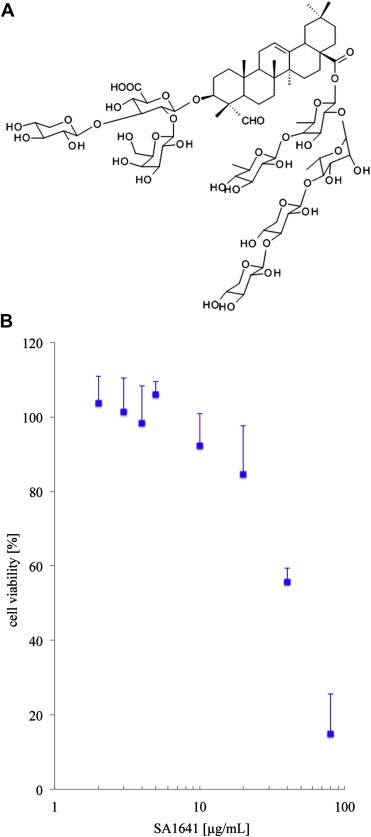
SA1641 isolated from Gypsophila paniculata L. (A) Structure of SA1641. It consists of a hydrophobic C30 backbone (gypsogenin) and two branched carbohydrate chains attached to gypsogenin. (B) Cytotoxicity of SA1641 on HER14 cells. SA1641 was non‐cytotoxic on HER14 cells over a wide concentration range. Cell viability was determined by MTT‐assay. Each point represents the mean ± SD of 3 independent experiments, n = 3.
As shown in Figure 3B SA1641 was well tolerated by HER14 cells. The lack of cytotoxicity was a prerequisite for further investigations on the SA1641‐mediated efficacy increase of targeted toxins. For the combination experiments with targeted toxins 5 μg/mL SA1641 was used. At this concentration SA1641 showed no significant cytotoxicity on HER14 cells. Due to the low cytotoxicity (Figure 3B) SA1641 is a potential candidate for a combinatorial anti‐tumor therapy with targeted toxins.
3.4. Efficacy of targeted toxins in combination with SA1641
Following characterization of targeted toxins and SA1641, we started to investigate the SA1641‐mediated efficacy increase of Sap‐EGF, Dia‐EGF, RTA‐EGF, DT390EGF and EGF‐ETA'.
Within a range of 1 to 100 nM DT390EGF, we observed differences in the efficacy in combination with SA1641 compared to the single application of DT390EGF (Figure 4A). Although this increase was statistically significant (p ≤ 0.02) the overall SA1641‐mediated toxicity enhancement was rather weak. The comparison of the SI50 (50% cell survival) values revealed a 12‐fold increase in cytotoxicity due to combination of DT390EGF with SA1641 (Table 2). The combination of SA1641 with EGF‐ETA' did not increase the cytotoxicity (Figure 4B) and no significant differences in the dose response curves with and without SA1641 were observed. The dose response curve of RTA‐EGF with SA1641 significantly differed from the curve obtained for single RTA‐EGF (Figure 4C) within the concentration range 10–1000 nM (p ≤ 0.02). The overall SA1641‐mediated efficacy increase was higher for RTA‐EGF compared to DT390EGF, but negligible in relation to Dia‐EGF and Sap‐EGF.
Figure 4.
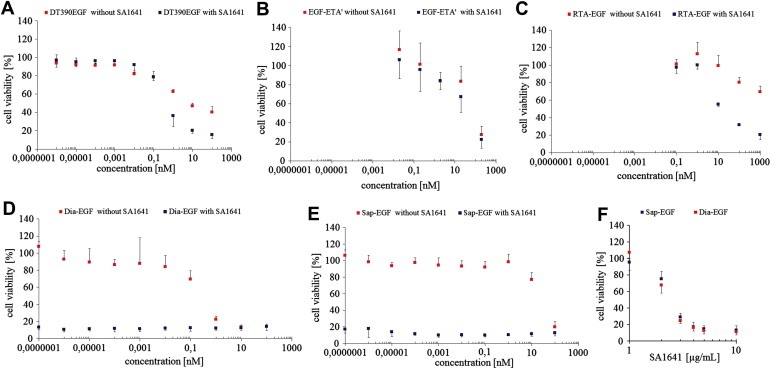
Efficacy of targeted toxins in combination with SA1641. HER14 cells were incubated with different targeted toxins (A–E) in the absence (red symbols) or presence (blue symbols) of 5 μg/mL SA1641 or with different concentrations of SA1641 in combination with Sap‐EGF (blue symbols) or Dia‐EGF (red symbols) (0.01 nM) (F). Cell viability was determined after 48 h by MTT‐assay. The slight differences between SA1641‐treated and untreated cells in (A–C) was significant for DT390EGF and RTA‐EGF but not for EGF‐ETA'. The tremendous difference for Dia‐EGF and Sap‐EGF (D–E) was significant. (F) Various concentrations of SA1641 were combined with Sap‐EGF or Dia‐EGF (both 0.01 nM). At 3 μg/mL SA1641 started to be effective in enhancing the efficacy of Sap‐EGF and Dia‐EGF. Each point represents the mean ± SD of at least 3 independent experiments performed in triplicates. t‐test, p ≤ 0.02.
Table 2.
SA1641‐mediated efficacy increase of different targeted toxins
| SI50 (nM)a | TEFb | ||
|---|---|---|---|
| Without SA1641 | With SA1641 | ||
| RTA‐EGF | >1000 | 61 | 16.3 |
| DT390EGF | 8.8 | 0.73 | 12 |
| EGF‐ETA' | 128 | 103 | 1.2 |
| Dianthin‐30‐EGF | 0.45 | <0.000 0001 | >4,000,000 |
| Saporin‐EGF | 57 | <0.000 0001 | >4,000,000 |
50% cell survival.
TEF, toxicity enhancement factor (SI50 without SA1641/SI50 with SA1641), represents the x‐fold SA1641‐mediated efficacy increase of targeted toxins.
By comparing the graphs of Dia‐EGF with and without SA1641 it is evident that SA1641 caused a tremendous efficacy increase of Dia‐EGF (Figure 4D). The combination of Dia‐EGF with SA1641 results in a dose response curve that is parallel and close to the abscissa indicating significant cytotoxicity down to ≤0.1 fM. This remarkable toxicity increase in combination with SA1641 was also observed for Sap‐EGF (Figure 4E). However, in the absence of SA1641 Sap‐EGF was less cytotoxic (SI50 value 57 nM) than Dia‐EGF (SI50 value 0.45 nM). These two targeted toxin's cytotoxicity was at least 4,000,000‐fold increased by SA1641 (Table 2). It is interesting to note that in combination with fixed concentrations (0.01 nM) of Dia‐EGF and Sap‐EGF, SA1641 started to be highly effective at 3 μg/mL, but not at 1 or 2 μg/mL (Figure 4F). This indicates a saponin threshold concentration above which SA1641 starts to be effective in augmenting the efficacy of Dia‐EGF and Sap‐EGF.
3.5. Membrane integrity assay
In general saponins are known to influence the membrane permeability of cellular membranes by association with cholesterol and subsequently pore formation.
This facilitates the entry of extracellular material into the cell. To determine if SA1641 does lead to pore formation and an influx of extracellular material we performed a membrane integrity assay, that is based on a saponin induced uptake of propidium iodide (PI) into the cell. Once inside the cell, PI intercalates into the DNA and exhibits strong fluorescence at 617 nm. SA1641 did not lead to an increased influx of PI (Figure 5). Only a high SA1641 concentration of 40 μg/mL (24 μM) caused a slight membrane permeabilization.
Figure 5.
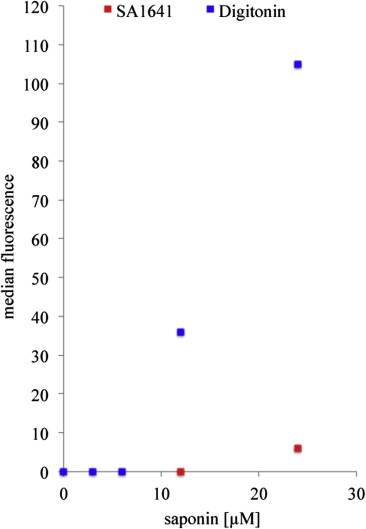
Membrane integrity assay. HER14 cells were incubated with propidium iodide and SA1641 (red symbols) or digitonin (blue symbols) (0–24 μM). The saponin induced influence on membrane permeability was determined by measuring the uptake of PI into the cells using flow cytometry. Y‐achsis represents medians of the fluorescence histograms. Compared to digitonin, SA1641 exhibited no membrane permeabilizing effects. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
This indicates that the SA1641‐mediated efficacy increase of Dia‐EGF and Sap‐EGF is not based on an unspecific uptake of Dia‐EGF and Sap‐EGF through SA1641 induced pores in the plasma membrane. Digitonin, a saponin that influences the membrane permeability (Hsuchen and Feingold, 1973) was used as positive control. Compared to SA1641 digitonin lead to a tremendous influx of PI into the cells.
3.6. Surface plasmon resonance (SPR) measurements
To investigate if SA1641 binds to isolated toxin, Hissaporin was immobilized to 770 resonance units (RU) on a carboxy methylated dextran gold surface. In SPR spectroscopy adsorption/desorption events are determined as a shift in the refractive index at a gold surface.
Via their EGF domain Sap‐EGF and Dia‐EGF are delivered into early and than late endosomes. Finally they reach the lysosomes for degradation. The pH in early endosomes was shown to be 6.1–6.8, in late endosomes 4.8–6 and in lysosomes 4.5 (Huotari and Helenius, 2011). We assume that SA1641 mediates the endosomal and/or lysosomal escape of Sap‐EGF and Dia‐EGF into the cytosol by an intra‐endosomal/intra‐lysosomal interaction with targeted toxin. If we take this hypothesis into consideration an acidic pH should influence the binding of SA1641 to Hissaporin. We therefore investigated the effect of a neutral (pH 7.4) and an acidic (pH 5) running buffer on the binding of SA1641 to immobilized Hissaporin.
At 5, 10 and 20 μg/mL SA1641 bound in neutral and in acidic buffer to Hissaporin (Figure 6). However compared to the SA1641 samples that were run in neutral buffer, a pH of 5 led to a strong increase of the binding of SA1641 to Hissaporin. This was observed for all concentrations of SA1641 (Figure 6).
Figure 6.
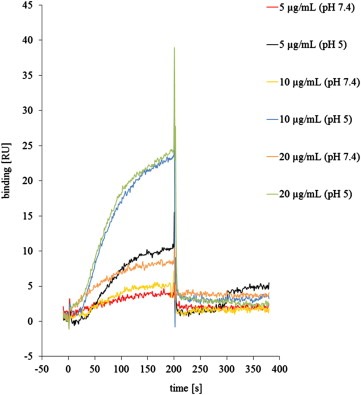
Binding of SA1641 to Hissaporin. Binding of SA1641 to Hissaporin was determined by surface plasmon resonance (SPR). Hissaporin was immobilized on a sensor chip with a shift of approximately 770 resonance units (RU) and SA1641 was injected at following concentrations: 5, 10 or 20, μg/mL with PBS (pH 7.4) or sodium acetate (pH 5) running buffer. Each concentration was applied in duplicate. At pH 5 the binding of SA1641 to Hissaporin was increased.
4. Discussion
The anti‐tumor therapy with targeted toxins can be accompanied by severe side effects such as vascular leak syndrome and hepatic or renal toxicities (Kreitman, 2006). These effects are probably the result of non‐specific uptake of targeted toxins by non‐target cells located in the endothelium, liver or kidney. Any technology minimizing the non‐specific toxicity whilst augmenting the anti‐tumoral efficacy of targeted toxins would be a valuable tool for improving the therapeutic index of these drugs.
This study was undertaken in order to investigate the potential therapeutic anti‐tumor effects of SA1641 used in combination with targeted toxins comprised of EGF as the binding ligand and different toxins. We aimed to show the challenges and chances of a combinatorial application of a purified saponin (SA1641) and targeted toxins.
Thus, we constructed a panel of five different targeted toxins against EGFR (comprising RTA, dianthin‐30, DT390, ETA' and saporin as toxin domains fused to EGF) and tested the cytotoxicity of each in the presence and absence of a pure saponin (SA1641) isolated from Saponinum album. EGF is endocytosed via clathrin‐mediated endocytosis followed by transport into early and subsequently late endosomes (Sigismund et al., 2008). By fusing the various toxins to EGF we were able to direct them into the endosomal transport system, whose permeability is probably modulated by SA1641. This allowed us to make direct comparisons between each targeted toxin and the augmentative effects of SA1641 on each.
While the toxicity of EGF‐ETA', DT390EGF and RTA‐EGF was increased only up to 16‐fold, the efficacy of Dia‐EGF and Sap‐EGF was augmented million‐fold (Table 2). This indicates that different toxins are affected differently by SA1641. We therefore aligned the sequences of DT390, ETA', RTA and dianthin‐30 with the sequence of saporin that served as template (alignments are shown in the supplementary data). The following sequence identities with saporin were determined: ETA' 9.5%, DT390 16.7%, RTA 23.9%, dianthin‐30 72.2%. Based on these results we suggest a relationship between the amino acid sequence of the toxin moiety and the ability of SA1641 to augment the efficacy of targeted toxins.
It is known from certain protein transduction domains such as human immunodeficiency virus‐1 TAT protein transduction domain that they are basic peptides rich in arginine and lysine residues with high isoelectric points (IP) (Caron et al., 2004). It is conspicuous that saporin (IP = 9.45) and dianthin‐30 (IP = 9.48) are also basic proteins each containing more than 30 arginine and lysine residues while DT390 (IP = 5.1), ETA' (IP = 4.8) and RTA (IP = 7.1) exhibit quite low calculated IPs. However the efficacy of a construct of a pancreatic RNase I (IP = 9.1) fused to EGF was not enhanced (see supplementary data) by SA1641. We therefore assume that the SA1641‐mediated efficacy increase is not dependent on high IPs.
It is conceivable that the efficacy of specific transduction domains responsible for dragging the toxins into the cytosol becomes more effective in the presence of SA1641. This may be facilitated by an intra‐endosomal/intra‐lysosomal interaction of SA1641 and the toxin component of targeted toxin. We could show a direct binding of SA1641 to Hissaporin by surface plasmon resonance spectroscopy (Figure 6). It is interesting to note that the binding of SA1641 to Hissaporin was increased at pH 5 compared to a pH of 7.4.
In previous studies with radiolabelled Saponinum album we have shown that saponins were internalized by ECV‐304 cells (Weng et al., 2009b). We assume that SA1641 is co‐internalized within vesicles that contain receptor bound targeted toxins. These vesicles mature to early/late endosomes and lysosomes. The pH is concomitantly decreasing from 7.4 (extracellular) to 6.1–6.8 (early endosomes), 4.8–6 (late endosomes) and 4.5 (lysosomes). During the proton‐pump driven generation of an acidic milieu within the vesicles, SA1641 starts to interact with the toxin component of targeted toxin in a pH dependent manner. This facilitates the endosomal/lysosomal escape of targeted toxin into the cytosol.
This study supports the thesis on specific SA1641‐sensitive transduction domains where we speculate that these domains become more effective by a transient association with SA1641 in an acidic milieu.
Herein lays also the relevance of the present study: (i) sequence alignments can be employed to predict a potential SA1641‐mediated efficacy increase of a certain targeted toxin and (ii) after identifying the relevant SA1641‐sensitive transduction domains within the saporin molecule, scientists will be able to fuse these domains to arbitrary targeted toxins. This would result in a new class of targeted toxins that can be defined as Saponin‐Sensitized Targeted Toxins.
5. Conclusions
Certain saponins were shown to dramatically improve the tolerance of a saporin‐based targeted toxin. This study showed the potential impact of saponins on targeted tumor therapies in general.
For this purpose several targeted toxins containing the toxic enzymes saporin, dianthin‐30, truncated forms of ricin, of diphtheria toxin and Pseudomonas exotoxin A fused to human epidermal growth factor as cell binding domain, were constructed. Each targeted toxin was co‐incubated with a saponin (SA1641), isolated from G. paniculata L., on epidermal growth factor receptor‐expressing NIH‐3T3 cells. We observed high efficacy increases for the targeted toxins containing saporin and dianthin‐30 and found a good correlation between SA1641 augmentation of efficacy and the amino acid sequence of the toxin moiety. The effect was not based on an influence of SA1641 on the plasma membrane permeability but surface plasmon resonance measurements indicated a direct interaction of SA1641 and the toxin moieties.
According to the amino acid sequence of the toxin, the SA1641‐mediated efficacy increase can be possibly predicted and allows the specific design of Saponin‐Sensitized Targeted Toxins (SSTT). Based on the results of this study it can be concluded that the SA1641‐mediated efficacy increase is a specific effect that depends on the nature of toxin used for the construction of the targeted toxin.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the Supplementary data related to this article:
Supplementary data
Acknowledgment
This work was funded by the Deutsche Forschungsgemeinschaft (WE 4748/1‐1). The authors acknowledge David J. Flavell, University of Southampton Medical School for language editing the manuscript.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found online at doi:10.1016/j.molonc.2012.01.004.
Weng Alexander, Thakur Mayank, Beceren-Braun Figen, Bachran Diana, Bachran Christopher, Riese Sebastian B., Jenett-Siems Kristina, Gilabert-Oriol Roger, Melzig Matthias F., Fuchs Hendrik, (2012), The toxin component of targeted anti‐tumor toxins determines their efficacy increase by saponins, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.01.004.
References
- Bachran, C. , Durkop, H. , Sutherland, M. , Bachran, D. , Muller, C. , Weng, A. , Melzig, M.F. , Fuchs, H. , 2009. Inhibition of tumor growth by targeted toxins in mice is dramatically improved by saponinum album in a synergistic way. J. Immunother.. 32, 713–725. [DOI] [PubMed] [Google Scholar]
- Bachran, C. , Heisler, I. , Fuchs, H. , Sutherland, M. , 2005. Influence of protein transduction domains on target-specific chimeric proteins. Biochem. Biophys. Res. Commun.. 337, 602–609. [DOI] [PubMed] [Google Scholar]
- Bachran, C. , Sutherland, M. , Bachran, D. , Fuchs, H. , 2007. Quantification of diphtheria toxin mediated ADP-ribosylation in a solid-phase assay. Clin. Chem.. 53, 1676–1683. [DOI] [PubMed] [Google Scholar]
- Bachran, D. , Schneider, S. , Bachran, C. , Urban, R. , Weng, A. , Melzig, M.F. , Hoffmann, C. , Kaufmann, A.M. , Fuchs, H. , 2010. Epidermal growth factor receptor expression affects the efficacy of the combined application of saponin and a targeted toxin on human cervical carcinoma cells. Int. J. Cancer. 127, 1453–1461. [DOI] [PubMed] [Google Scholar]
- Caron, N.J. , Quenneville, S.P. , Tremblay, J.P. , 2004. Endosome disruption enhances the functional nuclear delivery of tat-fusion proteins. Biochem. Biophys. Res. Commun.. 319, 12–20. [DOI] [PubMed] [Google Scholar]
- Collier, R.J. , Kandel, J. , 1971. Structure and activity of diphtheria toxin. I. Thiol-dependent dissociation of a fraction of toxin into enzymically active and inactive fragments. J. Biol. Chem.. 246, 1496–1503. [PubMed] [Google Scholar]
- Fermani, S. , Tosi, G. , Farini, V. , Polito, L. , Falini, G. , Ripamonti, A. , Barbieri, L. , Chambery, A. , Bolognesi, A. , 2009. Structure/function studies on two type 1 ribosome inactivating proteins: bouganin and lychnin. J. Struct. Biol.. 168, 278–287. [DOI] [PubMed] [Google Scholar]
- Frankel, A.E. , Powell, B.L. , Lilly, M.B. , 2002. Diphtheria toxin conjugate therapy of cancer. Cancer Chemother. Biol. Response Modif.. 20, 301–313. [PubMed] [Google Scholar]
- Fuchs, H. , Bachran, C. , 2009. Targeted tumor therapies at a glance. Curr. Drug Targets. 10, 89–93. [DOI] [PubMed] [Google Scholar]
- Heisler, I. , Keller, J. , Tauber, R. , Sutherland, M. , Fuchs, H. , 2003. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer. 103, 277–282. [DOI] [PubMed] [Google Scholar]
- Hsuchen, C.C. , Feingold, D.S. , 1973. Selective membrane toxicity of the polyene antibiotics: studies on lecithin membrane models (liposomes). Antimicrob. Agents Chemother.. 4, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari, J. , Helenius, A. , 2011. Endosome maturation. EMBO J.. 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman, R.J. , 2006. Immunotoxins for targeted cancer therapy. AAPS J.. 8, E532–E551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann, T. , 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65, 55–63. [DOI] [PubMed] [Google Scholar]
- Pirie, C.M. , Hackel, B.J. , Rosenblum, M.G. , Wittrup, K.D. , 2010. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem.. 286, 4165–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund, S. , Argenzio, E. , Tosoni, D. , Cavallaro, E. , Polo, S. , Di Fiore, P.P. , 2008. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell. 15, 209–219. [DOI] [PubMed] [Google Scholar]
- Silverstein, A.M. , 2002. The collected papers of Paul Ehrlich: why was volume 4 never published?. Bull. Hist. Med.. 76, 335–339. [DOI] [PubMed] [Google Scholar]
- Stirpe, F. , Battelli, M.G. , 2006. Ribosome-inactivating proteins: progress and problems. Cell Mol. Life Sci.. 63, 1850–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkouhi, A.K. , Scholte, M. , Storm, G. , Haisma, H.J. , 2010. Endosomal escape pathways for delivery of biologicals. J. Control Release. [DOI] [PubMed] [Google Scholar]
- Weng, A. , Bachran, D. , Gorick, C. , Bachran, C. , Fuchs, H. , Melzig, M.F. , 2009. A simple method for isolation of Gypsophila saponins for the combined application of targeted toxins and saponins in tumor therapy. Planta Med.. 75, 1421–1422. [DOI] [PubMed] [Google Scholar]
- Weng, A. , Gorick, C. , Melzig, M.F. , 2009. Enhancement of toxicity of saporin-based toxins by Gypsophila saponins–kinetic of the saponin. Exp. Biol. Med. (Maywood). 234, 961–966. [DOI] [PubMed] [Google Scholar]
- Weng, A. , Jenett-Siems, K. , Schmieder, P. , Bachran, D. , Bachran, C. , Gorick, C. , Thakur, M. , Fuchs, H. , Melzig, M.F. , 2010. A convenient method for saponin isolation in tumour therapy. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci.. 878, 713–718. [DOI] [PubMed] [Google Scholar]
- Woo, J.H. , Lee, Y.J. , Neville, D.M. , Frankel, A.E. , 2010. Pharmacology of anti-CD3 diphtheria immunotoxin in CD3 positive T-cell lymphoma trials. Methods Mol. Biol.. 651, 157–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the Supplementary data related to this article:
Supplementary data