

Abstract
The genetic and epigenetic material originating from tumour that can be found in body fluids of individuals with cancer harbours tumour‐specific alterations and represents an attractive target for biomarker discovery. Epigenetic changes (DNA methylation, histone modifications and non‐coding RNAs) are present ubiquitously in virtually all types of human malignancies and may appear in early cancer development, and thus they provide particularly attractive markers with broad applications in diagnostics. In addition, because changes in the epigenome may constitute a signature of specific exposure to certain risk factors, they have the potential to serve as highly specific biomarkers for risk assessment. While reliable detection of cancer‐specific epigenetic changes has proven to be technically challenging, a substantial progress has been made in developing the methodologies that allow an efficient and sensitive detection of epigenomic changes using the material originating from body fluids. In this review we discuss the application of epigenomics as a tool for biomarker research, with the focus on the analysis of DNA methylation in biofluids.
Keywords: Biomarkers, DNA methylation, Body fluids, Early cancer detection, Epigenomics
Highlights
Validation of epigenetic biomarkers in clinics and epidemiology is in its infancy.
Recent progress allows reliable detection of cancer‐specific epigenetic changes in body fluids.
New advances in epigenomics hold promise for generation of powerful cancer biomarkers.
1. Introduction
Epigenetics refers to heritable changes in gene expression that are not attributable to alterations in the sequence of DNA. The predominant epigenetic mechanisms are DNA methylation, modifications to chromatin structure, loss of imprinting, and noncoding RNA (Gibney and Nolan, 2010). Epigenetic modifications are known to be heritable between mother and daughter cells (mitotic inheritance) and between generations (meiotic inheritance). Epigenetic mechanisms are fundamental in regulating a multitude of cellular processes. In tumorgenesis, epigenetic aberrations are believed to play key roles in tumor suppressor gene (TSG) inactivation, oncogene activation, and chromosomal instability, all of which are involved in the deregulation of critical cellular pathways and steps of carcinogenesis including tumor initiation, invasion and plasticity (Carmona and Esteller, 2010; Jones, 2012; Jones and Baylin, 2002; Sincic and Herceg, 2011). Epigenetic regulation is also proposed as being potentially influenced by diet and environmental exposures, accounting for the growing interest in associating epigenetic regulation with lifestyle and risk of disease (Feil and Fraga, 2012; Jirtle and Skinner, 2007). Moreover, the reversibility of epigenetic alterations stimulates the development of novel therapeutic approaches with an open field for development of early cancer detection and prevention, namely through chemoprevention.
The current review discusses the application of epigenomics as a tool for biomarker research, focusing on the analysis of DNA methylation in biofluidss and the potential of generating biomarker panels for early cancer detection and screening.
2. Aberrant DNA methylation and cancer
DNA methylation, defined as the covalent addition of a methyl group to a cytosine nucleotide in a sequence of DNA, is an epigenetic event that affects cell function through the alteration of gene expression. In humans, DNA methylation occurs at the 5′ position of the pyrimidine ring of the cytosine residues within CpG dinucleotides through the addition of a methyl moiety to form 5‐methylcytosines and is catalyzed by a family of specific enzymes known as DNA methyltransferases (DNMTs; DNMT1, DNMT3A, and DNMT3B) using the cofactor S‐adenosyl‐methionine (SAM) (Gibney and Nolan, 2010). Regions of the genome rich in sequences of a cytosine preceding a guanine are known as CpG islands (CGIs). In fact, CGIs exist in the promoter regions of approximately half of all genes and are usually unmethylated in normal differentiated cells, while CGIs located in intergenic regions are methylated (Esteller, 2007; Taby and Issa, 2010). In the particular case of cancer, promoter CGIs of numerous TSGs are found to be densely methylated, resulting in transcriptional silencing, with these “epimutations” being cancer type‐specific and tumor stage‐specific (Rodriguez‐Paredes and Esteller, 2011).
The aberrant DNA methylation patterns found in cancer can be distinguished as hypomethylation and hypermethylation. Hypomethylation is characterized as a genome‐wide decrease in methylation and is extremely frequent in CpG sites in repetitive regions of the genome and CGIs outside of promoter regions (Jones, 2012). It is proposed that hypomethylation in coding regions of genes is associated with carcinogenesis through the favoring of mitotic recombination which may lead to deletions, translocations, chromosomal rearrangements as well as alterations in mRNA levels. Also, hypomethylation is associated with alterations to signaling cascades influencing proto‐oncogenes, such as c‐Jun, c‐Myc, and c‐H‐Ras (Calvisi et al., 2007). In addition, repetitive elements spread across the genome and while normally heavily methylated tend to loose methylation. Generally, tumours are characterized by DNA hypomethylation, which increases according to tumour progression, and sporadic gene‐specific hypermethylation (Roberts and Gores, 2005). For example, in hepatocarcinogenesis, it was shown that hypomethylation of repetitive DNA elements such as SAT2, LINE1, and ALU occurs in a multistep manner and correlates with poor prognosis (Lee et al., 2009).
Hypermethylation is characterized by the addition of methyl groups and, if highly specific to the CGIs in the promoter region of a particular gene, may lead to transcriptional silencing of the gene, with subsequent loss of protein expression. This mechanism is currently recognized as an alternative to mutations or allelic loss for gene‐silencing in TSGs (Herman and Baylin, 2003). A number of TSGs and other cancer‐related genes, including the retinoblastoma gene, RASSF1A, VHL, MLH1, CDH1, LKB1, p16 gene (CDKN2A), GSTP1 and MGMT, were found to be silenced by promoter hypermethylation (Feinberg and Tycko, 2004; Jones and Baylin, 2002). Our group showed that specific genes such as RASSF1A, GSTP1, CHRNA3, and DOK1 were aberrantly hypermethylated in hepatocellularcarcinoma (HCC) compared to control cirrhotic or normal liver tissues, suggesting that aberrant hypermethylation exhibits non‐random and tumor‐specific patterns in HCC. Furthermore, an association between hypermethylation of GSTP1 and HBV infection was also demonstrated (Lambert et al., 2011). Also in the context of changes to methylation profiles and HCC, it was shown that several CpG sites in the HBV genome are recurrently methylated in cancer but not in chronic hepatitis (Kaur et al., 2010). Hypermethylation of genes has been implicated in carcinogenesis due to its involvement in cell cycle, DNA repair, angiogenesis, metabolism of carcinogens, apoptosis, and cell–cell interaction (Figure 1). Thus, methylation patterns can be considered as biomarkers for early detection, diagnosis, prognosis, prediction and monitoring of therapy response. The identification of these cancer‐associated methylation signatures may also provide the foundations for cancer prevention strategies with DNA hypermethylation being proposed as a source of potential early event biomarkers in carcinogenesis that may precede the neoplastic process (Belinsky, 2004; Esteller, 2007; Nephew and Huang, 2003; Taby and Issa, 2010).
Figure 1.
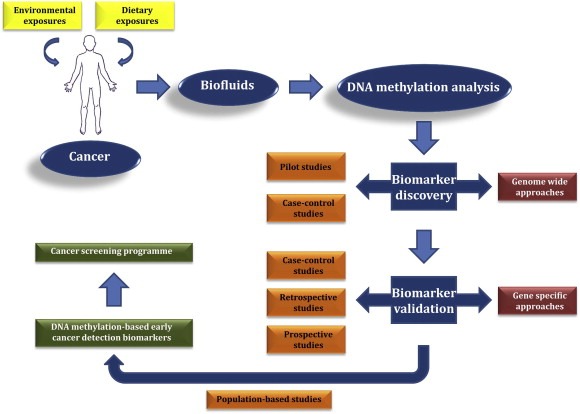
Development of DNA methylation‐based early cancer detection biomarkers, amidst environmental and dietary exposures.
3. Environmental effects on the epigenome
The possible impact of the environment on epigenetic regulation has attracted considerable interest, with environmentally induced changes in gene expression being associated with altered DNA methylation patterns or with altered histone modifications. The interest in DNA methylation relates to its involvement in key developmental and mechanistic pathways as well as its possible association with phenotypical changes due to the interplay between environmental exposures and epigenetics (Borgel et al., 2010; Feil and Fraga, 2012; Law and Jacobsen, 2010; Sincic and Herceg, 2011). Numerous environmental exposures have linked altering epigenetic patterns during a lifetime, and subsequent risk of disease. An interesting study showed how different environmental exposures may alter the epigenome by looking at the methylation and histone modifications in monozygotic twins at different stages in life. It was shown that, early in life, their epigenetic profiles were nearly identical but by age 50 considerable changes were detected (Feinberg, 2008). In general terms, environmental and dietary carcinogens, also known as epimutagens, that are capable of inducing tumoral development by deregulating the epigenenome can be divided into two groups: (a) those that induce, directly and indirectly, changes to genomic DNA and (b) those that affect key cellular processes such as gene transcription, DNA damage and repair, cell cycle control and apoptosis (Herceg, 2007; Herceg and Vaissiere, 2011). Specific examples of these epimutagens include tobacco smoke, alcohol, viruses and bacteria and dietary contaminants such as aflatoxin B1 (Balassiano et al., 2011; Begum et al., 2011; Belinsky et al., 1998; Hernandez‐Vargas et al., 2010; Hutajulu et al., 2011; James et al., 2002; Kaur et al., 2010; Lambert et al., 2011; Li and Minarovits, 2003; Liu et al., 2008; Paliwal et al., 2010; Pulling et al., 2004; Vaissiere et al., 2009b; Zhang et al., 2012).
Viral and dietary epimutagens are of our particular interest and have provided considerable evidence documenting the relationship between environmental carcinogens and changes to the epigenome. With regard to viral infections, it was documented that DNA methylation and chromatin modifications are known to regulate viral gene expression (Zheng and Baker, 2006). In cervical cancer, human papiloma virus (HPV) DNA methylation was implicated in the development and progression of the disease and was shown to inhibit the transcription of the most viral genes (Kalantari et al., 2004). Epstein–Barr virus (EBV) and hepatitis B virus infections were associated with promoter hypermethylation of several genes in several cancers including hepatocellular carcinoma, gastric and nasopharyngeal cancer (Zazula et al., 2006; Zhou et al., 2005). Our group showed that in HCC tumors differentially methylated promoters were found when comparing: (i) early‐stage with later‐stage tumours; (ii) well‐differentiated with poorly differentiated tumours, and (iii) cirrhotic with non‐cirrhotic liver tissue (Hernandez‐Vargas et al., 2010). With regard to dietary carcinogens, several studies have addressed whether AFB1, which is a natural secondary metabolite of the fungus Aspergillus, can exhibit its carcinogenic effects through an epigenetic mechanism. As a result, a strong relationship between AFB1 exposure and methylation of cancer related genes such as RASSF1A, MGMT and p16 in tumour tissues and plasma DNA of HCC patients was established (Herceg, 2007; Herceg and Vaissiere, 2011). Utilizing in vitro models it was demonstrated that AFB1 strongly induces the expression of the SNCG gene, a known target of epigenetic changes in early stages of HCC, while in HCC tissue it was shown that AFB1 exposure is associated with DNA global hypomethylation (Zhang et al., 2006, 2012). The precise mechanisms by which dietary epimutagens induce changes to epigenetic profiles are still unclear. Nevertheless, it is proposed that these epimutagens may bind preferentially to methylated CpG sites, thus inducing DNA damage, as was proposed for carcinogen‐DNA adduct formation at specific codons in the K‐ras gene (Hu et al., 2003).
Although extensive and novel studies were conducted in the past half a decade, there is still no clear‐cut casual relationship between epimutagens and changes to epigenetic signatures (Bollati and Baccarelli, 2010; Christensen and Marsit, 2011; Herceg and Paliwal, 2011; Herceg and Vaissiere, 2011; Hou et al., 2012; Jirtle and Skinner, 2007; Perera and Herbstman, 2011; Stein, 2012). The main limitation of these studies still reside in the fact that the epigenetic changes are thought of as being mostly subtle, cumulative and requiring a long “timeframe” until full manifestation is detectable.
4. Technological tools for DNA methylation‐based epigenomics
The measurement of DNA methylation can be done with a wide range of methods using various types of biological material such as tissue, plasma, serum, sputum, and urine, among others (Figure 2).
Figure 2.
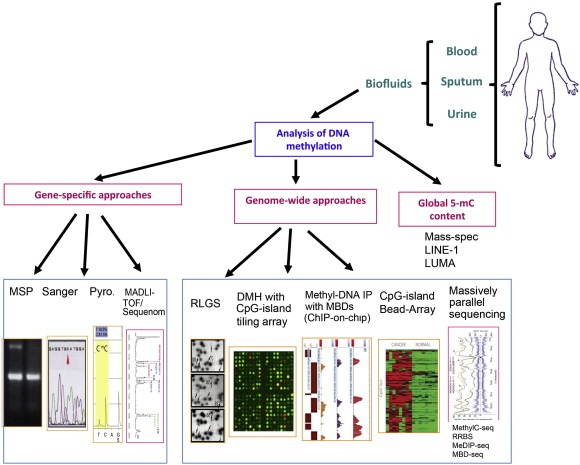
Schematic of methodologies applied to epigenomics.
The inherent stability of DNA is one of the major advantages of detecting methylation. Through the years, methodology for DNA methylation measurement has progressed gradually with the first breakthroughs being based on the use of isoschizomers with different methylation sensitivities. From the early 1990s, the method exploiting an initial bisulfate conversion of DNA which converts unmethylated but not methylated cytosines to uracils has become the “gold standard” for analysis of DNA methylation changes. The bisulfate conversion of DNA followed by PCR amplification allows gene‐specific methlyation analysis (methylation specific PCR, MSP), which is based on using primers and probes specific to the corresponding methylated DNA sequence (Herman et al., 1996). The quantitative MSP is also called MethyLight and has become more commonly used as it is based on the bisulfite converted DNA being amplified by methylation state specific primers and TaqMan® probes (Eads et al., 2000). The major disadvantages of MSP are that it is not a quantitative assay, it monitors only a few CpG sites and that it is prone to false positives. Another PCR‐based approach is called combined bisulfite conversion restriction analysis (COBRA) which relies on the principle of loss or retention of a restriction enzyme site after bisulfite treatment, depending on the methylation status of the targeted cytosine. The major disadvantage of this approach is the limited number of restriction enzymes that can be used.
The bisulfate treatment can also be applied to DNA sequencing in order to determine specific regions of hypo or hypermethylation, namely in the promoter regions of genes. This is a particularly useful technique to determine regions of differential methylation, and aid in primer and probe design for the more specific MSP. For example, bisulfite sequencing (BS) of DNA using Sanger chemistry is based on PCR amplification of bisulfite converted DNA using specific primers, followed by cloning. Clones are then randomly selected and sequenced. This approach will further benefit greatly from the new developments in massive parallel sequencing and its high‐throughput will most likely be useful for target region analysis (Frommer et al., 1992; Taylor et al., 2007).
Alternatively, pyrosequencing, a method of real‐time DNA sequencing, utilizes PCR technology to provide a high‐throughput platform that analyses the multiple CpGs of a given region based on the activity of DNA polymerase. The basis for this technological approach relies on the luminometric detection of pyrophosphate release after nucleotide incorporation (Dupont et al., 2004). Another important strength of pyrosequencing is its robustness with built‐in internal controls for completeness of bisulphite treatment, thus it produces no false positives. Although bisulfate pyrosequencing is one of the most widely used methods for quantitative determination of methylation, it is limited by a few drawbacks, such as only analyzing relatively short fragments and designing of a robust pyrosequencing assay. Overall, the major limitation with these PCR‐based technologies is their targeted nature providing analysis only of specific candidate genes of interest.
Several methods for analyzing the global methylation pattern of tumors have also been developed. One example is the global estimation of 5‐methylcytidine (5‐mC) by high performance capillary electrophoresis or high precision liquid chromatography coupled with Electrospray Ionization/Mass Spectrometry to separate individual nucleosides, generated by enzymatic hydrolysis of DNA, and separate unmethylated cytosines from the methylated ones (Berdasco et al., 2009). Other examples include a luminometric method for global DNA methylation quantification (LUMA) or the measurement of the degree of methylation at repetitive elements, that are spread across the genome and are normally heavily methylated, and which are directly proportional to global methylation content (Karimi et al., 2006; Yang et al., 2004). Recently, in order to address the interest in characterizing and defining the methylation pattern on a genome‐wide scale, microarray platforms were developed. These include the more recently developed immunological approach known as methylated DNA immunoprecipitation (MeDIP). In this approach the first step is enriching methylated DNA based on the principle that genomic DNA is randomly sheared by sonication and immunoprecipitated with an antibody that specifically targets 5‐mC. This technique provides a platform to generate comprehensive, genomic DNA methylation profiles and to identify abnormally (hyper‐ or hypo‐) methylated genomic regions (Weber et al., 2005). A subsequent example is the methylated CpG island amplification (MCA) method which is based on the digestion of genomic DNA with a methylation‐sensitive restriction enzyme that only targets unmethylated sites, SmaI. Here, the DNA, which now has blunted ends between the cytosine and guanine, is digested with the methylation‐ insensitive, Sma I isoschizomer Xma I, leaving a four‐base overhang. Lastly, a ligation of adaptors to the four‐base overhang occurs and adaptor‐specific PCR amplification is performed (Toyota et al., 1999). This method results in the enrichment and amplification of methylated DNA fragments only and can then be used in microarray platforms.
The development of microarray platforms that are compatible with methylation analysis provides a step forward in addressing the challenges of analyzing multiple regions of interest simultaneously in a high‐throughput manner. Several platforms have been used for DNA methylome analysis ranging from CGI or promoter region specific platforms to oligonucleotide tiling arrays virtually covering the whole genome with high resolution. The first platforms were based on restriction enzymes and an example of which is the differential methylation hybridization platform which combined restriction digestion with downstream hybridization to microarrays (Huang et al., 1999). The major limitation of this approach is the relatively small portion of methylome that can be studied. To overcome this limitation, affinity enrichment of DNA was proposed and is now widely used combined with microarrays. An example is MeDIP‐chip (Weber et al., 2005). Also, techniques based on coupling bisulfite treatment with array hybridization are also commonly found and these are normally based on commercially available arrays namely Illumina GoldenGate® BeadArray™ and Infinium®.
With the development of massive parallel sequencing there is now the opportunity to sequence a whole methylome at a single base. These new technological developments provide now the possibility of generating a methylome map of a specific cell type relatively rapid at constantly decreasing costs (Lister and Ecker, 2009). Whole genome shotgun sequencing of bisulphite‐converted (WGSBS or MethylC‐Seq) DNA is now achievable by directly determining the methylation status of all cytosines in a genome. The MethylC‐seq of a whole genome was reported in Arabidopsis and later the methylome at single‐nucleotide resolution in human cells followed quickly thereafter (Cokus et al., 2008; Emes and Farrell, 2012; Lister et al., 2009). The major advantage of massive parallel sequencing is gathering highly detailed information with less DNA input (Emes and Farrell, 2012; Lister and Ecker, 2009). Therefore, methods of enrichment, such as MeDIP‐seq and methylated DNA binding domain sequencing (MDB‐seq), based on antibodies against 5mC or immobilized recombinant methylated CpG binding proteins have been developed for enrichment of methylated fragments of the genome prior to sequencing (Jacinto et al., 2008; Serre et al., 2010). The enriched fragment is then sequenced using massive parallel sequencing and methylated regions of the genome can then be determined by identifying regions of high‐sequence density.
A significant challenge of analyzing DNA methylation in body fluids is the minute amounts of DNA available. Our group developed a strategy that allows for the quantitative and sensitive detection of DNA methylation in minute amounts of DNA present in body fluids, entitled quantitative Methylation Analysis of Minute DNA amounts after whole Bisulfitome Amplification (qMAMBA) (Paliwal et al., 2009; Vaissiere et al., 2009a). This method involves genome‐wide amplification of bisulphate‐modified DNA template followed by quantitative methylation detection using pyrosequencing which then allows for the analysis of multiple genes from a small amount of starting DNA, such as that found in blood, urine and sputum. This method has already been applied to the quantitative profiling of DNA methylation states in a panel of five cancer‐associated genes in a large case–control study of lung cancer. This study allowed for the identification of aberrant DNA methylation patterns associated with the mechanisms by which environmental factors may interact with key genes involved in tumor suppression and contribute to lung cancer (Vaissiere et al., 2009b). Although optimization will be required, qMAMBA could be modified for use in conjunction with other quantitative methods for methylation analysis, such as in conjunction with microarrays or massive parallel sequencing. As an example, one part of the bisulfitome‐amplified product obtained in the second step of the method could be applied to the Illumina Infinium® methylation platform to obtain a signature of aberrant methylation for a large panel of cancer‐associated genes. Subsequently, the other part of the amplified product could be used for validating the observed aberrant methylation for the most promising individual candidate genes. Also, the recently proposed strategies for 5‐hydroxylmethylcytosines (5hmC) detection and measurement provide a novel and attractive approach for study of DNA methylation in biofluids. This method is based on a Tet‐assisted bisulfite sequencing (TABSeq) strategy, which provides a method for single‐base resolution detection of 5hmC amenable to both genome‐wide and loci‐specific sequencing (Yu et al., 2012). The development of these base‐resolution maps of 5hmC provide more accurate estimates of both 5hmC and 5mC levels at each modified cytosine than previous whole‐genome bisulfite sequencing approaches and provide further insights into the understanding of the biological roles of 5hmC as well as gene regulation (Booth et al., 2012; Yu et al., 2012). Although this method was developed and applied for in vitro strategies and is proposed for application in tissue specimen, there is a significant potential of application in biofluids.
5. Early cancer detection and DNA methylation in biofluids
Epigenetic modifications of DNA offer hope and the promise of novel biomarkers for early cancer detection, prediction, prognosis, and response to treatment. Furthermore, reversal of epigenetic changes represents a potential target of novel preventive as well as therapeutic strategies and medication design (Selaru et al., 2009).
Although using primary tissue to study methylation profiles and generate new potential biomarkers is of interest, acquiring this biological material is only achievable by means of invasive techniques which greatly limit the potential of early cancer detection and screening programmes (Hitchins et al., 2011; Hutajulu et al., 2011). The ideal strategy should rely on non‐invasive or minimally invasive techniques which will still allow the study of DNA methylation patterns. To this end, one looks for biofluids, such as blood, sputum, and urine, among others, as surrogate tissues. Nevertheless, the choice of which surrogate tissue to utilize is associated with the type of cancer being investigated.
The most commonly investigated surrogate tissue, and to a certain extent the ideal surrogate, is blood. Acquiring blood requires minimally invasive techniques and it can be applied to all patients, both those at minimal or high risk. In the blood, DNA is found in the form of extracellular DNA and is commonly designated as circulating free DNA (CFDNA). These DNA fragments have distinctly lower molecular weights compared to genomic DNA. The CFDNA fragments apparently circulate as nucleoprotein complexes; however, in healthy individuals, the main part of CFDNA is found adsorbed in the surface of blood cells (Skvortsova et al., 2006). It is postulated that this DNA, in healthy individuals, is primarily of hematopoietic origin while in cancer patients it also results from apoptotic and necrotic processes characteristic of tumor cells with high cellular turnover (Gormally et al., 2007; Li et al., 2003; Ziegler et al., 2002). Several studies showed that all cells may actively release DNA fragments and that CFDNA corresponds to the so‐called metabolic DNA fraction, which is proposed to control transcription into RNA, among other cellular processes (Li et al., 2003; Stroun et al., 2001; Ziegler et al., 2002).
It was also reported that cancer patients have a higher level of circulating free DNA (CFDNA), in plasma or serum, than non‐cancer subjects and that both genetic and epigenetic alterations can be detected in CFDNA (Begum et al., 2011, 2008, 2011, 2010, 2011, 1983, 2011, 2012, 2003, 2005). Furthermore, similar specific alterations of DNA such as mutations or strand stability found both in the tumor and in the CFDNA prove the tumoral origin of the CFDNA (Anker et al., 1997, 2007, 2011, 2012). The proportion of tumor CFDNA in relation to the total CFDNA is influenced by several factors, such as the type of cancer, its stage, grade, size, and location. The use of CFDNA as a potential cancer biomarker can be analyzed in terms of changes to total CFDNA concentration or as tumor‐specific changes to CFDNA. The first was shown to have the potential for cancer diagnosis, if used as part of a diagnosis panel, as well as for prognosis and monitoring of recurrence, as demonstrated in surgically successful treated cancer patients upon follow up (Diehl et al., 2008; Frattini et al., 2008; Huang et al., 2006; Perego et al., 2008; Talamonti et al., 2007; Zhang et al., 2010). The latter was shown to have an even greater potential for cancer diagnosis, and subsequent early detection and screening, based on its higher specificity (Anglim et al., 2008; Baryshnikova et al., 2008; Begum et al., 2011; Borgel et al., 2010; Chang et al., 2008; Herceg and Hainaut, 2007; Schrump and Nguyen, 2005; Skvortsova et al., 2006). This is the particular case of analyzing CFDNA in relation to mutations of oncogenes and TSGs and to alterations in methylation patterns found in cancers.
With regard to studies on CFDNA and mutations of oncogenes and TSGs, a concordance between tumor DNA and CFDNA namely, in K‐ras and p53 mutations, was shown. Both genes were investigated in a wide range of cancers such as bladder, breast, colon, lung, liver, pancreas, endometrial, and ovarian cancer (Dabritz et al., 2005, 2009, 2010, 2008, 2007, 2004, 2005, 2002, 2009, 2011, 2012). Of note is the percentage of tissue samples with identified K‐ras and p53 mutations ranging from 23% to 64% and 17% to 54%, respectively, and the concordance between the mutations found in the tumor and CFDNA samples for these two mutations ranged between 0% to 56% and 14% to 65%, respectively.
With regard to studies on CFDNA and alterations to methylation patterns, much has been investigated, notably in the past five years. After the promising results with CFDNA studies on oncogenes and TSGs, investigating methylation changes became an attractive field due to their key regulatory processes in various cellular pathways related to cancer. For example, it was shown that Vimentin methylation may represent a useful biomarker in gastric cancer due to its higher sensitivity compared to currently used biomarkers and that Septin 9 methylation may prove to be a sensitive and specific biomarker for early colorectal cancer detection (Shirahata et al., 2012; Warren et al., 2011).
Although several studies identified potential biomarkers, sensitivity of individual CFDNA biomarkers is a significant problem. To overcome this, numerous studies combined several methylation biomarkers in a panel or the methylation markers were additionally combined with conventional diagnostic parameters. For example, for bladder cancer the combination of methylated APC and GSTP1 increased the diagnostic sensitivity from 59% for the single biomarker to 80% for the combined ones (Ellinger et al., 2008). In prostate cancer diagnosis, the sensitivity was improved from 29% with the methylated biomarkers of GSTP1, RARB2, and RASSF1A to 89% when these biomarkers were used in combination with the standard parameter PSA (Sunami et al., 2009). In breast cancer, the combination of RARB2 and RASSF1A provided a sensitivity of 95% and specificity of 100% in diagnosing the disease. In esophageal squamous cell carcinoma, the diagnostic accuracy was increased when methylation of multiple genes (RAR‐β, DAPK, CDH1, p16 and RASSF1A) was analyzed in combination, providing an increase to 82.2% sensitivity and 100% specificity in distinguishing healthy and esophageal squamous cell carcinoma patients (Li et al., 2011). In another study, the methylation levels of APC, GSTP1, RASSF1A, and SFRP1 were found significantly higher in HCCs compared to normal or benign liver controls and the combination of these genes as a panel of biomarkers for HCC diagnosis was pursued (Huang et al., 2011). The discrimination of HCCs from controls was achieved with 92.7% sensitivity, 81.9% specificity, 90.5% positive predictive value (PPV), and 87.2% negative predictive value (NPV), and the discrimination of HCC from benign liver controls was achieved with 84.7% sensitivity, 81.1% specificity, 89.7% PPV, and 73.2% NPV (Huang et al., 2011). To increase through‐put in diagnosis, a study in ovarian cancer applied a microarray‐based multiplex assay for CFDNA methylation analysis (MethDet56 technique), which analyzed a total of 56 genes, and identified five unmethylated genes as informative cancer classifiers in CFDNA samples (Melnikov et al., 2009).
Of increasing interest is the analysis of methylation profiles in primary tumors and their corresponding plasma or serum to assess whether there are potential DNA methylation biomarkers that can be found in both materials and therefore provide a panel of highly sensitive and specific non‐invasive biomarkers. To this end, studies showed that methylation in blood can be associated with patients in which the primary tumor also exhibited methylation (Board et al., 2008, 1999, 2002, 2010, 2007, 2009, 2006). In particular, our group has shown that a high frequency of aberrant hypermethylation of MTHFR, RASSF1A, and CDKN2A in lung tumors as compared with control blood samples was consistent with the notion that aberrant DNA methylation occurs in a tumor‐specific and gene‐specific manner (Vaissiere et al., 2009b). We also showed a strong association between MTHFR hypermethylation in lung cancer and tobacco smoking, with methylation of RASSF1A being influenced by sex, with males showing higher levels of methylation (Vaissiere et al., 2009b). The use of methylation array chips may provide a global picture of methylation in CpG sites that can be further investigated in CFDNA (Hernandez‐Vargas et al., 2010). In this work, the global methylation profile was studied in 30 HCC tumours and 124 CpG sites among 94 genes were differentially methylated when compared with surrounding non‐cancerous liver tissue. Of interest are genes such as DNMT1, FAT, MYLK, FLT1, CDKN1C, TFPI2, PDE1B, MME, IGF1R, COL1A2 and TP73, which may be further investigated in CFDNA to improve the diagnosis, early detection and screening of HCC by means of a non‐invasive approach. The major limitation with all the studies on CFDNA conducted to date is the limited number of individuals used in these studies.
Other surrogate tissues have also been investigated as potential methylation biomarkers, albeit to a lesser extent. One example is sputum. It is produced by increased bronchial secretions, and is commonly found in smokers, therefore it can be used to screen populations at high‐risk of lung cancer. Studies showed that aberrant methylation could be found in MAGE A3 and p16 genes in lung cancer patients and these changes to methylation could distinguish cancer from non‐cancer subjects; that the methylation of HOXA9 in lung cancer patients' tissue and sputum was significantly higher compared with control and benign lung disease tissues or sputum, that HOXA9 was hypermethylated especially in early stages of lung cancer; and that individuals exposed to smoky coal emissions in Xuan Mei (China) harbored frequent promoter methylation of p16, MGMT, RASSF1A and DAPK in their sputum (Hwang et al., 2011; Liu et al., 2008; Shin et al., 2012). A complementary study using 50 matched tumor, plasma and sputum samples showed that CDKN2A/p16 hypermethylation is detected in 84% of tumors, and 76% of sputum samples from the same patients, demonstrating the potential use of these biomarkers for early detection of lung cancer in a surrogate tissue (Liu et al., 2003). The main advantages of sputum as a tool for early cancer detection and screening include its non‐invasiveness, and the fact that it contains cells from the lungs and lower respiratory tract. Nevertheless, the material in sputum is from the center of the lungs, and it may not be as useful for the detection of adenocarcinoma, which generally occurs at the periphery.
Another example is urine. This surrogate tissue is of significant interest for bladder cancer investigation and has seen several studies investigating potential DNA methylation biomarkers in the last couple of years. A study looking at the global methylation pattern in bladder cancer identified differential methylation of ZNF154, POU4F2, HOXA9, and EOMES, as overlapping between tissue and urine with the panel of biomarkers achieving 84% sensitivity and 96% specificity in distinguishing healthy subjects from bladder cancer patients (Reinert et al., 2011). Another study proposed a panel of biomarkers composed of RARβ2 and APC genes where both sensitivities and specificities of the methylated genes for bladder cancer detection were superior to urine cytology (Eissa et al., 2011). When combined altogether, the sensitivities improved to (91.8%), (93.5%), (91.9%), and (80.9%) in detection of: bladder cancer, non‐muscle invasive bladder cancer, low‐grade tumors, and bilharzial associated bladder cancer, respectively (Eissa et al., 2011). Another study also identified a panel of methylated genes composed of GDF15, TMEFF2, and VIM that was found to be aberrantly methylated in both tissue and urine and which achieved 100% sensitivity and specificity in tissue and a sensitivity of 94% and specificity of 100% in urine when distinguishing bladder cancer patients from healthy subjects (Costa et al., 2010). The methylated genes proposed above may all be considered promising cancer biomarkers for early bladder cancer detection. Interestingly, although several studies have been conducted from discovery to validation, very few have highlighted common aberrant methylation patterns. Urine was also investigated as a surrogate for distinguishing healthy subjects from prostate cancer patients. An example is a study that analyzed the methylation of RASSF1 and RARB in urine sediments from patients with early stage prostate cancer and showed the high sensitivity of the DNA methylation biomarkers in distinguishing healthy subjects from early and late prostate cancer (Daniunaite et al., 2011).
Although DNA methylation is being analyzed in several biofluids and a plethora of biomarkers being proposed, the main question remains on which should be chosen for further validation and introduced into screening programmes. A screening strategy may have four potential outcomes: true‐positive (the test is positive and the subject actually has cancer), true‐negative (the test is negative and the subject actually does not have cancer), false‐negative (the test is negative but the subject has cancer) and false‐positive (the test is positive but the subject does not have cancer). While the two first outcomes are the most desired in a screening programme, the two latter have the potential of delaying diagnosis of disease and endangering the patient or affecting the patient's quality of life (Brawley and Kramer, 2005). Specificity and sensitivity of any biomarker are key for the “go‐no‐go” decision on further pursuing it. As with any biomarker research strategy, the ideal biomarker would be highly sensitive and specific in different populations, regardless of different cancer etiologies, the patients' age, gender or tumor stage. To meet this objective, which is commonly difficult due to the differences in cancer etiologies or subtypes, the ideal strategy is to consider the use of a panel of biomarkers instead of a sole biomarker and therefore increasing the sensitivity and specificity of early detection, diagnosis, prevention or prediction of cancer. This guiding principle should be applied to potential DNA methylation‐based biomarkers that can be found in surrogate tissues such as biofluids. Furthermore, according to the Early Detection Research Network (EDRN, USA) guidelines, a biomarker panel should follow several steps before being implemented “in‐the‐field”: a preclinical and exploratory investigation (phase 1), establishment of a clinical assay and its validation for the disease of interest (phase 2), retrospective and longitudinal assessment of the performance of the biomarker for early detection of the disease (phase 3), prospective and screening assessment of the performance of the biomarker (phase 4) and impact of screening with a biomarker panel on reducing the burden of cancer in the population (phase 5). The phases of research are generally ordered according to the strength of evidence that each provides in favor of the biomarker, from weakest to strongest, and the results of earlier phases are generally necessary to design later phases. These guidelines were proposed for gene expression microarrays, proteomics, and immunology approaches for early cancer detection and screening and provide a platform for the development of novel cancer‐related DNA‐methylation biomarkers (Pepe et al., 2001; Srivastava and Kramer, 2000).
6. Concluding remarks and future perspectives
Recent progress in the field of epigenetics and epigenomics has provided valuable insights into the mechanisms of carcinogenesis, complementing those gained from the genetic standpoint. The new insights hold great potential for the generation of biomarker panels that may be suited to cancer prevention, early detection, screening, diagnosis, risk assessment and treatment of a multitude of cancers. Particularly in the field of DNA methylation, studies in the last decade have provided significantly increased our knowledge regarding the modulation of cancer‐related key cellular pathways through aberrant methylation that may be induced by epimutagens present in the environment and in the diet. Furthermore, the proposal of DNA methylation‐based biomarkers has grown exponentially in the past half a decade providing a source of mechanism‐based biomarkers as well as potential biomarkers of exposure and, importantly, biomarkers for early cancer detection and screening. Nevertheless, the validation of such biomarker panels is still in its infancy. Several discovery studies have been conducted in both primary tissue and in surrogate tissues such as plasma/serum, urine or sputum but the majority are limited by the small sample sizes. Also, the need to correlate the potential biomarker panels and their profiles in paired tissue‐biofluid is essential to increase the level of specificity of the panels being investigated. To further address these issues, future studies should have in mind a multicentre design based on utilizing paired samples from healthy subjects, at‐risk subjects and cancer patients for a combined discovery‐validation strategy or a large scale validation, in settings where discovery was already conducted. In addition, large cohort and case–control studies offer the most exciting opportunities for the generation or validation of biomarker panels that may be associated with cellular events that may or may not be induced by the diet and environment related to human cancer. These validations studies should also take into account the guidelines of the EDRN in order to provide a solid platform for implementation of early cancer detection and screening programmes. These programmes are particularly important in low and medium resource regions of the world where cancer incidence is high and resources are low. As an example, liver cancer is the is the fifth most common cancer in men and the seventh in women, and most of the burden is in developing countries, where almost 85% of the cases occur (Ferlay et al., 2010). Due to the lack of early detection and screening biomarkers as well as due to the highly invasive approaches for HCC diagnosis, the potential development of a panel of DNA methylation biomarkers in a surrogate tissue such as blood is of extreme interest and necessity. A panel that can successfully differentiate individuals with chronic liver disease from those with HCC through the use of a biofluid, ensuring high‐throughput and that does not require significantly complex protocols and logistical settings, may prove highly valuable. A platform such as the one mentioned above was recently tested and proven successful when differentiating patients with inflammatory pancreatic disease and pancreatic cancer, where rates of sensitivity and specificity were reported to be high using CFDNA from plasma (Liggett et al., 2010).
Overall, the recent progress in the field of epigenetics suggests that further advancements in the characterization of the methylome will bring about new strategies and technologies to ultimately improve the detection, diagnosis and treatment of cancer.
Acknowledgements
We apologize to authors whose relevant publications were not cited due to space limitation. The work of the IARC Epigenetics Group is supported by grants from the National Cancer Institute (NIH), United States; l'Association pour la Recherche sur le Cancer (ARC), France; la Ligue Nationale Contre le Cancer, France; the Swiss Bridge Award; and the Bill and Melinda Gates Foundation (to ZH). ANdC is a recipient of a Postdoctoral Fellowship from the IARC, partially supported by the EC FP7 Marie Curie Actions – People – Co‐funding of regional, national and international programmes (COFUND).
Nogueira da Costa André, Herceg Zdenko, (2012), Detection of cancer-specific epigenomic changes in biofluids: Powerful tools in biomarker discovery and application, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.07.005.
References
- Anglim, P.P. , Alonzo, T.A. , Laird-Offringa, I.A. , 2008. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Molecular Cancer. 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anker, P. , Lefort, F. , Vasioukhin, V. , Lyautey, J. , Lederrey, C. , Chen, X.Q. , Stroun, M. , Mulcahy, H.E. , Farthing, M.J.G. , 1997. K-ras mutations are found in DNA extracted from the plasma of patients with colorectal cancer. Gastroenterology. 112, 1114–1120. [DOI] [PubMed] [Google Scholar]
- Balassiano, K. , Lima, S. , Jenab, M. , Overvad, K. , Tjonneland, A. , Boutron-Ruault, M.C. , Clavel-Chapelon, F. , Canzian, F. , Kaaks, R. , Boeing, H. , Meidtner, K. , Trichopoulou, A. , Laglou, P. , Vineis, P. , Panico, S. , Palli, D. , Grioni, S. , Tumino, R. , Lund, E. , Bueno-de-Mesquita, H.B. , Numans, M.E. , Peeters, P.H.M. , Ramon Quiros, J. , Sanchez, M.-J. , Navarro, C. , Ardanaz, E. , Dorronsoro, M. , Hallmans, G. , Stenling, R. , Ehrnstrom, R. , Regner, S. , Allen, N.E. , Travis, R.C. , Khaw, K.-T. , Offerhaus, G.J.A. , Sala, N. , Riboli, E. , Hainaut, P. , Scoazec, J.-Y. , Sylla, B.S. , Gonzalez, C.A. , Herceg, Z. , 2011. Aberrant DNA methylation of cancer-associated genes in gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Cancer Letters. 311, 85–95. [DOI] [PubMed] [Google Scholar]
- Baryshnikova, E. , Destro, A. , Infante, M.V. , Cavuto, S. , Cariboni, U. , Alloisio, M. , Ceresoli, G.L. , Lutman, R. , Brambilla, G. , Chiesa, G. , Ravasi, G. , Roncalli, M. , 2008. Molecular alterations in spontaneous sputum of cancer-free heavy smokers: results from a large screening program. Clinical Cancer Research. 14, 1913–1919. [DOI] [PubMed] [Google Scholar]
- Begum, S. , Brait, M. , Dasgupta, S. , Ostrow, K.L. , Zahurak, M. , Carvalho, A.L. , Califano, J.A. , Goodman, S.N. , Westra, W.H. , Hoque, M.O. , Sidransky, D. , 2011. An epigenetic marker panel for detection of lung cancer using cell-free serum DNA. Clinical Cancer Research. 17, 4494–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky, S.A. , 2004. Gene-promoter hypermethylation as a biomarker in lung cancer. Nature Reviews Cancer. 4, 707–717. [DOI] [PubMed] [Google Scholar]
- Belinsky, S.A. , Nikula, K.J. , Palmisano, W.A. , Michels, R. , Saccomanno, G. , Gabrielson, E. , Baylin, S.B. , Herman, J.G. , 1998. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proceedings of the National Academy of Sciences of the United States of America. 95, 11891–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco, M. , Fraga, M.F. , Esteller, M. , 2009. Quantification of global DNA methylation by capillary electrophoresis and mass spectrometry. In Tost J., Methods in Molecular Biology. 23–34. [DOI] [PubMed] [Google Scholar]
- Board, R.E. , Knight, L. , Greystoke, A. , Blackhall, F.H. , Hughes, A. , Dive, C. , Ranson, M. , 2008. DNA methylation in circulating tumour DNA as a biomarker for cancer. Biomarker Insights. 2, 307–319. [PMC free article] [PubMed] [Google Scholar]
- Bollati, V. , Baccarelli, A. , 2010. Environmental epigenetics. Heredity. 105, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth, M.J. , Branco, M.R. , Ficz, G. , Oxley, D. , Krueger, F. , Reik, W. , Balasubramanian, S. , 2012. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 336, 934–937. [DOI] [PubMed] [Google Scholar]
- Borgel, J. , Guibert, S. , Li, Y. , Chiba, H. , Schuebeler, D. , Sasaki, H. , Forne, T. , Weber, M. , 2010. Targets and dynamics of promoter DNA methylation during early mouse development. Nature Genetics. 42, 1090–1093. [DOI] [PubMed] [Google Scholar]
- Brawley, O.W. , Kramer, B.S. , 2005. Cancer screening in theory and in practice. Journal of Clinical Oncology. 23, 293–300. [DOI] [PubMed] [Google Scholar]
- Calvisi, D.F. , Ladu, S. , Gorden, A. , Farina, M. , Lee, J.-S. , Conner, E.A. , Schroeder, I. , Factor, V.M. , Thorgeirsson, S.S. , 2007. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. Journal of Clinical Investigation. 117, 2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona, F.J. , Esteller, M. , 2010. Epigenomics of human colon cancer. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 693, 53–60. [DOI] [PubMed] [Google Scholar]
- Chang, H. , Yi, B. , Li, L. , Zhang, H.-Y. , Sun, F. , Dong, S.-Q. , Cao, Y. , 2008. Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Experimental and Molecular Pathology. 85, 96–100. [DOI] [PubMed] [Google Scholar]
- Christensen, B.C. , Marsit, C.J. , 2011. Epigenomics in environmental health. Frontiers in Genetics. 2, 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus, S.J. , Feng, S. , Zhang, X. , Chen, Z. , Merriman, B. , Haudenschild, C.D. , Pradhan, S. , Nelson, S.F. , Pellegrini, M. , Jacobsen, S.E. , 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 452, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, V.L. , Henrique, R. , Danielsen, S.A. , Duarte-Pereira, S. , Eknaes, M. , Skotheim, R.I. , Rodrigues, A. , Magalhaes, J.S. , Oliveira, J. , Lothe, R.A. , Teixeira, M.R. , Jeronimo, C. , Lind, G.E. , 2010. Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clinical Cancer Research. 16, 5842–5851. [DOI] [PubMed] [Google Scholar]
- Dabritz, J. , Hanfler, J. , Preston, R. , Stieler, J. , Oettle, H. , 2005. Detection of Ki-ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. British Journal of Cancer. 92, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daebritz, J. , Preston, R. , Haenfler, J. , Oettle, H. , 2009. Follow-up study of K-ras mutations in the plasma of patients with pancreatic cancer correlation with clinical features and carbohydrate antigen 19-9. Pancreas. 38, 534–541. [DOI] [PubMed] [Google Scholar]
- Daniunaite, K. , Berezniakovas, A. , Jankevicius, F. , Laurinavicius, A. , Lazutka, J.R. , Jarmalaite, S. , 2011. Frequent methylation of RASSF1 and RARB in urine sediments from patients with early stage prostate cancer. Medicina–Lithuania. 47, 147–153. [PubMed] [Google Scholar]
- Diehl, F. , Schmidt, K. , Choti, M.A. , Romans, K. , Goodman, S. , Li, M. , Thornton, K. , Agrawal, N. , Sokoll, L. , Szabo, S.A. , Kinzler, K.W. , Vogelstein, B. , Diaz, L.A. , 2008. Circulating mutant DNA to assess tumor dynamics. Nature Medicine. 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzycka, B. , Terlikowski, S.J. , Mazurek, A. , Kowalczuk, O. , Niklinska, W. , Chyczewski, L. , Kulikowski, M. , 2010. Circulating free DNA, p53 antibody and mutations of KRAS gene in endometrial cancer. International Journal of Cancer. 127, 612–621. [DOI] [PubMed] [Google Scholar]
- Dupont, J.M. , Tost, J. , Jammes, H. , Gut, N.G. , 2004. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Analytical Biochemistry. 333, 119–127. [DOI] [PubMed] [Google Scholar]
- Eads, C.A. , Danenberg, K.D. , Kawakami, K. , Saltz, L.B. , Blake, C. , Shibata, D. , Danenberg, P.V. , Laird, P.W. , 2000. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Research. 28, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa, S. , Swellam, M. , El-Khouly, I.M. , Kassim, S.K. , Shehata, H. , Mansour, A. , Esmat, M. , Nossier, A.I. , Hamdy, M.A. , Awad, N.M. , El-Ahmady, O. , 2011. Aberrant methylation of RAR beta(2) and APC genes in voided urine as molecular markers for early detection of bilharzial and nonbilharzial bladder cancer. Cancer Epidemiology Biomarkers & Prevention. 20, 1657–1664. [DOI] [PubMed] [Google Scholar]
- Ellinger, J. , El Kassem, N. , Heukamp, L.C. , Matthews, S. , Cubukluoz, F. , Kahl, P. , Perabo, F.G. , Mueller, S.C. , von Ruecker, A. , Bastian, P.J. , 2008. Hypermethylation of cell-free serum DNA indicates worse outcome in patients with bladder cancer. Journal of Urology. 179, 346–352. [DOI] [PubMed] [Google Scholar]
- Emes, R.D. , Farrell, W.E. , 2012. Make way for the ’next generation’: application and prospects for genome-wide, epigenome-specific technologies in endocrine research. Journal of Molecular Endocrinology. 49, R19–R27. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , 2007. Epigenetic gene silencing in cancer: the DNA hypermethylome. Human Molecular Genetics. 16, R50–R59. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , Sanchez-Cespedes, M. , Rosell, R. , Sidransky, D. , Baylin, S.B. , Herman, J.G. , 1999. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Research. 59, 67–70. [PubMed] [Google Scholar]
- Feil, R. , Fraga, M.F. , 2012. Epigenetics and the environment: emerging patterns and implications. Nature Reviews Genetics. 13, 97–109. [DOI] [PubMed] [Google Scholar]
- Feinberg, A.P. , 2008. Epigenetics at the epicenter of modern medicine. JAMA – Journal of the American Medical Association. 299, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Feinberg, A.P. , Tycko, B. , 2004. Timeline - The history of cancer epigenetics. Nature Reviews Cancer. 4, 143–153. [DOI] [PubMed] [Google Scholar]
- Ferlay, J. , Shin, H.R. , Bray, F. , Forman, D. , Mathers, C. , Parkin, D.M. , 2010. GLOBOCAN 2008 v1.2, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10 (Internet) International Agency for Research on Cancer; Lyon, France: [Google Scholar]
- Frattini, M. , Gallino, G. , Signoroni, S. , Balestra, D. , Lusa, L. , Battaglia, L. , Sozzi, G. , Bertario, L. , Leo, E. , Pilotti, S. , Pierotti, M.A. , 2008. Quantitative and qualitative characterization of plasma DNA identifies primary and recurrent colorectal cancer. Cancer Letters. 263, 170–181. [DOI] [PubMed] [Google Scholar]
- Frommer, M. , McDonald, L.E. , Millar, D.S. , Collis, C.M. , Watt, F. , Grigg, G.W. , Molloy, P.L. , Paul, C.L. , 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proceedings of the National Academy of Sciences of the United States of America. 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromont-Hankard, G. , Philippe-Chomette, P. , Delezoide, A.L. , Nessmann, C. , Aigrain, Y. , Peuchmaur, M. , 2002. Glial cell-derived neurotrophic factor expression in normal human lung adenomatoid and congenital cystic malformation. Archives of Pathology & Laboratory Medicine. 126, 432–436. [DOI] [PubMed] [Google Scholar]
- Gibney, E.R. , Nolan, C.M. , 2010. Epigenetics and gene expression. Heredity. 105, 4–13. [DOI] [PubMed] [Google Scholar]
- Gormally, E. , Caboux, E. , Vineis, P. , Hainaut, P. , 2007. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutation Research-Reviews in Mutation Research. 635, 105–117. [DOI] [PubMed] [Google Scholar]
- Herceg, Z. , 2007. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis. 22, 91–103. [DOI] [PubMed] [Google Scholar]
- Herceg, Z. , Hainaut, P. , 2007. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Molecular Oncology. 1, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herceg, Z. , Paliwal, A. , 2011. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutation Research-Reviews in Mutation Research. 727, 55–61. [DOI] [PubMed] [Google Scholar]
- Herceg, Z. , Vaissiere, T. , 2011. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics. 6, 804–819. [DOI] [PubMed] [Google Scholar]
- Herman, J.G. , Baylin, S.B. , 2003. Mechanisms of disease: gene silencing in cancer in association with promoter hypermethylation. New England Journal of Medicine. 349, 2042–2054. [DOI] [PubMed] [Google Scholar]
- Herman, J.G. , Graff, J.R. , Myohanen, S. , Nelkin, B.D. , Baylin, S.B. , 1996. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America. 93, 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Vargas, H. , Lambert, M.-P. , Le Calvez-Kelm, F. , Gouysse, G. , McKay-Chopin, S. , Tavtigian, S.V. , Scoazec, J.-Y. , Herceg, Z. , 2010. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. Plos One. 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchins, M.P. , Owens, S.E. , Kwok, C.T. , Godsmark, G. , Algar, U.F. , Ramesar, R.S. , 2011. Identification of new cases of early-onset colorectal cancer with an MLH1 epimutation in an ethnically diverse South African cohort. Clinical Genetics. 80, 428–434. [DOI] [PubMed] [Google Scholar]
- Hou, L. , Zhang, X. , Wang, D. , Baccarelli, A. , 2012. Environmental chemical exposures and human epigenetics. International Journal of Epidemiology. 41, 79–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, W.W. , Feng, Z.H. , Tang, M.S. , 2003. Preferential carcinogen-DNA adduct formation at codons 12 and 14 in the human K-ras gene and their possible mechanisms. Biochemistry. 42, 10012–10023. [DOI] [PubMed] [Google Scholar]
- Huang, T.H.M. , Perry, M.R. , Laux, D.E. , 1999. Methylation profiling of CpG islands in human breast cancer cells. Human Molecular Genetics. 8, 459–470. [DOI] [PubMed] [Google Scholar]
- Huang, Z.-H. , Hu, Y. , Hua, D. , Wu, Y.-Y. , Song, M.-X. , Cheng, Z.-H. , 2011. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Experimental and Molecular Pathology. 91, 702–707. [DOI] [PubMed] [Google Scholar]
- Huang, Z.H. , Li, L.H. , Hua, D. , 2006. Quantitative analysis of plasma circulating DNA at diagnosis and during follow-up of breast cancer patients. Cancer Letters. 243, 64–70. [DOI] [PubMed] [Google Scholar]
- Hutajulu, S.H. , Indrasari, S.R. , Indrawati, L.P.L. , Harijadi, A. , Duin, S. , Haryana, S.M. , Steenbergen, R.D.M. , Greijer, A.E. , Middeldorp, J.M. , 2011. Epigenetic markers for early detection of nasopharyngeal carcinoma in a high risk population. Molecular Cancer. 10, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, S.-H. , Kim, K.U. , Kim, J.-E. , Kim, H.-H. , Lee, M.K. , Lee, C.H. , Lee, S.-Y. , Oh, T. , An, S. , 2011. Detection of HOXA9 gene methylation in tumor tissues and induced sputum samples from primary lung cancer patients. Clinical Chemistry and Laboratory Medicine. 49, 699–704. [DOI] [PubMed] [Google Scholar]
- Iyer, P. , Zekri, A.-R. , Hung, C.-W. , Schiefelbein, E. , Ismail, K. , Hablas, A. , Seifeldin, I.A. , Soliman, A.S. , 2010. Concordance of DNA methylation pattern in plasma and tumor DNA of Egyptian hepatocellular carcinoma patients. Experimental and Molecular Pathology. 88, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto, F.V. , Ballestar, E. , Esteller, M. , 2008. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques. 44, 35 [DOI] [PubMed] [Google Scholar]
- James, S.J. , Melnyk, S. , Pogribna, M. , Pogribny, I.P. , Caudill, M.A. , 2002. Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. Journal of Nutrition. 132, 2361S–2366S. [DOI] [PubMed] [Google Scholar]
- Jirtle, R.L. , Skinner, M.K. , 2007. Environmental epigenomics and disease susceptibility. Nature Reviews Genetics. 8, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.A. , 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics. 13, 484–492. [DOI] [PubMed] [Google Scholar]
- Jones, P.A. , Baylin, S.B. , 2002. The fundamental role of epigenetic events in cancer. Nature Reviews Genetics. 3, 415–428. [DOI] [PubMed] [Google Scholar]
- Kalantari, M. , Calleja-Macias, I.E. , Tewari, D. , Hagmar, B. , Lie, K. , Barrera-Saldana, H.A. , Wiley, D.J. , Bernard, H.U. , 2004. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. Journal of Virology. 78, 12762–12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi, M. , Johansson, S. , Ekstrom, T.J. , 2006. Using LUMA: a luminometric-based assay for global DNA-methylation. Epigenetics. 1, 45–48. [DOI] [PubMed] [Google Scholar]
- Kaur, P. , Paliwal, A. , Durantel, D. , Hainaut, P. , Scoazec, J.-Y. , Zoulim, F. , Chemin, I. , Herceg, Z. , 2010. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. Journal of Infectious Diseases. 202, 700–704. [DOI] [PubMed] [Google Scholar]
- Kimura, T. , Holland, W.S. , Kawaguchi, T. , Williamson, S.K. , Chansky, K. , Crowley, J.J. , Doroshow, J.H. , Lenz, H.J. , Gandara, D.R. , Gumerlock, P.H. , 2004. Mutant DNA in plasma of lung cancer patients - Potential for monitoring response to therapy. In Hoon D.S.B.T.B., Circulating Nucleic Acids in Plasma/Serum Iii and Serum Proteomics. 55–60. [DOI] [PubMed] [Google Scholar]
- Lambert, M.-P. , Paliwal, A. , Vaissiere, T. , Chemin, I. , Zoulim, F. , Tommasino, M. , Hainaut, P. , Sylla, B. , Scoazec, J.-Y. , Tost, J. , Herceg, Z. , 2011. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. Journal of Hepatology. 54, 705–715. [DOI] [PubMed] [Google Scholar]
- Law, J.A. , Jacobsen, S.E. , 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics. 11, 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H.S. , Kim, B.-H. , Cho, N.-Y. , Yoo, E.J. , Choi, M. , Shin, S.-H. , Jang, J.-J. , Suh, K.-S. , Kim, Y.S. , Kang, G.H. , 2009. Prognostic implications of and relationship between CpG island hypermethylation and repetitive DNA hypomethylation in hepatocellular carcinoma. Clinical Cancer Research. 15, 812–820. [DOI] [PubMed] [Google Scholar]
- Li, B. , Wang, B. , Niu, L.-J. , Jiang, L. , Qiu, C.-C. , 2011. Hypermethylation of multiple tumor-related genes associated with DMNT3b upregulation served as a biomarker for early diagnosis of esophageal squamous cell carcinoma. Epigenetics. 6, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C.N. , Hsu, H.L. , Wu, T.L. , Tsao, K.C. , Sun, C.F. , Wu, J.T. , 2003. Cell-free DNA is released from tumor cells upon cell death: a study of tissue cultures of tumor cell lines. Journal of Clinical Laboratory Analysis. 17, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Minarovits, J. , 2003. Host cell-dependent expression of latent Epstein-Barr virus genomes: regulation by DNA methylation. Advances in Cancer Research. 89, 133–156. [DOI] [PubMed] [Google Scholar]
- Liggett, T. , Melnikov, A. , Yi, Q.-l. , Replogle, C. , Brand, R. , Kaul, K. , Talamonti, M. , Abrams, R.A. , Levenson, V. , 2010. Differential methylation of cell-free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer. 116, 1674–1680. [DOI] [PubMed] [Google Scholar]
- Lindforss, U. , Zetterquist, H. , Papadogiannakis, N. , Olivecrona, H. , 2005. Persistence of K-ras mutations in plasma after colorectal tumor resection. Anticancer Research. 25, 657–661. [PubMed] [Google Scholar]
- Lister, R. , Ecker, J.R. , 2009. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Research. 19, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, R. , Pelizzola, M. , Dowen, R.H. , Hawkins, R.D. , Hon, G. , Tonti-Filippini, J. , Nery, J.R. , Lee, L. , Ye, Z. , Ngo, Q.-M. , Edsall, L. , Antosiewicz-Bourget, J. , Stewart, R. , Ruotti, V. , Millar, A.H. , Thomson, J.A. , Ren, B. , Ecker, J.R. , 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.-B. , Zhang, Y.-X. , Zhou, S.-H. , Shi, M.-X. , Cai, J. , Liu, Y. , Chen, K.-P. , Qiang, F.-L. , 2011. CpG island methylator phenotype in plasma is associated with hepatocellular carcinoma prognosis. World Journal of Gastroenterology. 17, 4718–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , An, Q. , Li, L. , Zhang, D.C. , Huang, J.F. , Feng, X.L. , Cheng, S.J. , Gao, Y.N. , 2003. Hypermethylation of p16(INK4a) in Chinese lung cancer patients: biological and clinical implications. Carcinogenesis. 24, 1897–1901. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Lan, Q. , Shen, M. , Jin, J. , Mumford, J. , Ren, D. , Keohavong, P. , 2008. Aberrant gene promoter methylation in sputum from individuals exposed to smoky coal emissions. Anticancer Research. 28, 2061–2066. [PMC free article] [PubMed] [Google Scholar]
- Melnikov, A. , Scholtens, D. , Godwin, A. , Levenson, V. , 2009. Differential methylation profile of ovarian cancer in tissues and plasma. Journal of Molecular Diagnostics. 11, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza, S. , Sharma, G. , Parshad, R. , Srivastava, A. , Gupta, S.D. , Ralhan, R. , 2010. Clinical significance of Stratifin, ER alpha and PR promoter methylation in tumor and serum DNA in Indian breast cancer patients. Clinical Biochemistry. 43, 380–386. [DOI] [PubMed] [Google Scholar]
- Mirza, S. , Sharma, G. , Prasad, C.P. , Parshad, R. , Srivastava, A. , Gupta, S.D. , Ralhan, R. , 2007. Promoter hypermethylation of TMS1, BRCA1, ER alpha and PRB in serum and tumor DNA of invasive ductal breast carcinoma patients. Life Sciences. 81, 280–287. [DOI] [PubMed] [Google Scholar]
- Nephew, K.P. , Huang, T.H.M. , 2003. Epigenetic gene silencing in cancer initiation and progression. Cancer Letters. 190, 125–133. [DOI] [PubMed] [Google Scholar]
- Paliwal, A. , Vaissiere, T. , Herceg, Z. , 2009. Quantitative detection of DNA methylation states in minute amounts of DNA from body fluids. Methods. 52, 242–247. [DOI] [PubMed] [Google Scholar]
- Paliwal, A. , Vaissiere, T. , Krais, A. , Cuenin, C. , Cros, M.-P. , Zaridze, D. , Moukeria, A. , Boffetta, P. , Hainaut, P. , Brennan, P. , Herceg, Z. , 2010. Aberrant DNA methylation links cancer susceptibility locus 15q25.1 to apoptotic regulation and lung cancer. Cancer Research. 70, 2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe, M.S. , Etzioni, R. , Feng, Z.D. , Potter, J.D. , Thompson, M.L. , Thornquist, M. , Winget, M. , Yasui, Y. , 2001. Phases of biomarker development for early detection of cancer. Journal of the National Cancer Institute. 93, 1054–1061. [DOI] [PubMed] [Google Scholar]
- Perego, R.A. , Corizzato, M. , Brambilla, P. , Ferrero, S. , Bianchi, C. , Fasoli, E. , Signorini, S. , Torsello, B. , Invernizzi, L. , Bombelli, S. , Angeloni, V. , Pitto, M. , Battaglia, C. , Proserpio, V. , Magni, F. , Galasso, G. , Mocarelli, P. , 2008. Concentration and microsatellite status of plasma DNA for monitoring patients with renal carcinoma. European Journal of Cancer. 44, 1039–1047. [DOI] [PubMed] [Google Scholar]
- Perera, F. , Herbstman, J. , 2011. Prenatal environmental exposures, epigenetics, and disease. Reproductive Toxicology. 31, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulling, L.C. , Vuillemenot, B.R. , Hutt, J.A. , Devereux, T.R. , Belinsky, S.A. , 2004. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Research. 64, 3844–3848. [DOI] [PubMed] [Google Scholar]
- Reinert, T. , Modin, C. , Castano, F.M. , Lamy, P. , Wojdacz, T.K. , Hansen, L.L. , Wiuf, C. , Borre, M. , Dyrskjot, L. , Orntoft, T.F. , 2011. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clinical Cancer Research. 17, 5582–5592. [DOI] [PubMed] [Google Scholar]
- Roberts, L.R. , Gores, G.J. , 2005. Hepatocellular carcinoma: molecular pathways and new therapeutic targets. Seminars in Liver Disease. 25, 212–225. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paredes, M. , Esteller, M. , 2011. Cancer epigenetics reaches mainstream oncology. Nature Medicine. 17, 330–339. [DOI] [PubMed] [Google Scholar]
- Schrump, D.S. , Nguyen, D.M. , 2005. Targeting the epigenome for the treatment and prevention of lung cancer. Seminars in Oncology. 32, 488–502. [DOI] [PubMed] [Google Scholar]
- Selaru, F.M. , David, S. , Meltzer, S.J. , Hamilton, J.P. , 2009. Epigenetic events in gastrointestinal cancer. American Journal of Gastroenterology. 104, 1910–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre, D. , Lee, B.H. , Ting, A.H. , 2010. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Research. 38, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, B. , Chakrabarty, M. , Cohn, E.M. , Leon, S.A. , 1983. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 51, 2116–2120. [DOI] [PubMed] [Google Scholar]
- Shin, K.-C. , Lee, K.-H. , Lee, C.-H. , Shin, I.-H. , Suh, H.-S. , Jeon, C.-H. , 2012. MAGE A1-A6 RT-PCR and MAGE A3 and p16 methylation analysis in induced sputum from patients with lung cancer and non-malignant lung diseases. Oncology Reports. 27, 911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahata, A. , Sakuraba, K. , Kitamura, Y. , Yokomizo, K. , Gotou, T. , Saitou, M. , Kigawa, G. , Nemoto, H. , Sanada, Y. , Hibi, K. , 2012. Detection of vimentin methylation in the serum of patients with gastric cancer. Anticancer Research. 32, 791–794. [PubMed] [Google Scholar]
- Silva, J.M. , Garcia, J.M. , Dominguez, G. , Silva, J. , Miralles, C. , Cantos, B. , Coca, S. , Provencio, M. , Espana, P. , Bonilla, F. , 2002. Persistence of tumor DNA in plasma of breast cancer patients after mastectomy. Annals of Surgical Oncology. 9, 71–76. [DOI] [PubMed] [Google Scholar]
- Sincic, N. , Herceg, Z. , 2011. DNA methylation and cancer: ghosts and angels above the genes. Current Opinion in Oncology. 23, 69–76. [DOI] [PubMed] [Google Scholar]
- Skvortsova, T.E. , Rykova, E.Y. , Tamkovich, S.N. , Bryzgunova, O.E. , Starikov, A.v. , Kuznetsova, N.P. , Vlassov, V.V. , Laktionov, P.P. , 2006. Cell-free and cell-bound circulating DNA in breast tumours: DNA quantification and analysis of tumour-related gene methylation. British Journal of Cancer. 94, 1492–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. , Kramer, B.S. , 2000. Early detection cancer research network. Laboratory Investigation. 80, 1147–1148. [DOI] [PubMed] [Google Scholar]
- Stein, R.A. , 2012. Epigenetics and environmental exposures. Journal of Epidemiology and Community Health. 66, 8–13. [DOI] [PubMed] [Google Scholar]
- Stroun, M. , Lyautey, J. , Lederrey, C. , Mulcahy, H.E. , Anker, P. , 2001. Alu repeat sequences are present in increased proportions compared to a unique gene in plasma/serum DNA - Evidence for a preferential release from viable cells?. In Lo Y.M.D.C.R.W.K.J.P.J., Circulating Nucleic Acids in Plasma or Serum Ii. 258–264. [DOI] [PubMed] [Google Scholar]
- Sunami, E. , Shinozaki, M. , Higano, C.S. , Wollman, R. , Dorff, T.B. , Tucker, S.J. , Martinez, S.R. , Singer, F.R. , Hoon, D.S.B. , 2009. Multimarker circulating DNA assay for assessing blood of prostate cancer patients. Clinical Chemistry. 55, 559–567. [DOI] [PubMed] [Google Scholar]
- Szymanska, K. , Chen, J.-G. , Cui, Y. , Gong, Y.Y. , Turner, P.C. , Villar, S. , Wild, C.P. , Parkin, D.M. , Hainaut, P. , 2009. TP53 R249S Mutations, exposure to aflatoxin, and occurrence of hepatocellular carcinoma in a cohort of chronic hepatitis B virus carriers from Qidong, China. Cancer Epidemiology Biomarkers & Prevention. 18, 1638–1643. [DOI] [PubMed] [Google Scholar]
- Taby, R. , Issa, J.-P.J. , 2010. Cancer epigenetics. Ca-a Cancer Journal for Clinicians. 60, 376–392. [DOI] [PubMed] [Google Scholar]
- Talamonti, M. , Banki, Peters, J.H. , Schwesinger, W.H. , Jolley, S.G. , Easter, D.W. , Sticca, R. , 2007. Plasma DNA as a molecular marker for completeness of resection and recurrent disease in patients with esophageal cancer – discussion. Archives of Surgery. 142, 538–539. [DOI] [PubMed] [Google Scholar]
- Taylor, K.H. , Kramer, R.S. , Davis, J.W. , Guo, J. , Duff, D.J. , Xu, D. , Caldwell, C.W. , Shi, H. , 2007. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Research. 67, 8511–8518. [DOI] [PubMed] [Google Scholar]
- Toyota, M. , Ho, C. , Ahuja, N. , Jair, K.W. , Li, Q. , Ohe-Toyota, M. , Baylin, S.B. , Issa, J.P.J. , 1999. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Research. 59, 2307–2312. [PubMed] [Google Scholar]
- Ulivi, P. , Zoli, W. , Calistri, D. , Fabbri, F. , Tesei, A. , Rosetti, M. , Mengozzi, M. , Amadori, D. , 2006. p16(INK4A) and CDH13 hypermethylation in tumor and serum of non-small cell lung cancer patients. Journal of Cellular Physiology. 206, 611–615. [DOI] [PubMed] [Google Scholar]
- Vaissiere, T. , Cuenin, C. , Paliwal, A. , Vineis, P. , Hainaut, P. , Herceg, Z. , Genair, E.I. , 2009. Quantitative analysis of DNA methylation after whole bisulfitome amplification of a minute amount of DNA from body fluids. Epigenetics. 4, 221–230. [DOI] [PubMed] [Google Scholar]
- Vaissiere, T. , Hung, R.J. , Zaridze, D. , Moukeria, A. , Cuenin, C. , Fasolo, V. , Ferro, G. , Paliwal, A. , Hainaut, P. , Brennan, P. , Tost, J. , Boffetta, P. , Herceg, Z. , 2009. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Research. 69, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar, S. , Le Roux-Goglin, E. , Gouas, D.A. , Plymoth, A. , Ferro, G. , Boniol, M. , Lereau, M. , Bah, E. , Hall, A.J. , Wild, C.P. , Mendy, M. , Norder, H. , van der Sande, M. , Whittle, H. , Friesen, M.D. , Groopman, J.D. , Hainaut, P. , 2011. Seasonal variation in TP53 R249S-mutated serum DNA with aflatoxin exposure and hepatitis B virus infection. Environmental Health Perspectives. 119, 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar, S. , Ortiz-Cuaran, S. , Abedi-Ardekani, B. , Gouas, D. , Nogueira da Costa, A. , Plymoth, A. , Khuhaprema, T. , Kalalak, A. , Sangrajrang, S. , Friesen, M.D. , Groopman, J.D. , Hainaut, P. , 2012. Aflatoxin-Induced TP53 R249S mutation in hepatocellular carcinoma in Thailand: association with tumors developing in the absence of liver cirrhosis. Plos One. 7, e37707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, J.D. , Xiong, W. , Bunker, A.M. , Vaughn, C.P. , Furtado, L.V. , Roberts, W.L. , Fang, J.C. , Samowitz, W.S. , Heichman, K.A. , 2011. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Medicine. 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, M. , Davies, J.J. , Wittig, D. , Oakeley, E.J. , Haase, M. , Lam, W.L. , Schubeler, D. , 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genetics. 37, 853–862. [DOI] [PubMed] [Google Scholar]
- Wong, I.H.N. , Zhang, J. , Lai, P.B.S. , Lau, W.Y. , Lo, Y.M.D. , 2003. Quantitative analysis of tumor-derived methylated p161NK4a sequences in plasma, serum, and blood cells of hepatocellular carcinoma patients. Clinical Cancer Research. 9, 1047–1052. [PubMed] [Google Scholar]
- Yang, A.S. , Estecio, M.R.H. , Doshi, K. , Kondo, Y. , Tajara, E.H. , Issa, J.P.J. , 2004. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Research. 32, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, W. , Wong, N. , Wong, W.L. , Lai, P.B.S. , Zhong, S. , Johnson, P.J. , 2005. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver International. 25, 266–272. [DOI] [PubMed] [Google Scholar]
- Yu, M. , Hon, G.C. , Szulwach, K.E. , Song, C.-X. , Zhang, L. , Kim, A. , Li, X. , Dai, Q. , Shen, Y. , Park, B. , Min, J.-H. , Jin, P. , Ren, B. , He, C. , 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 149, 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zazula, M. , Ferreira, A.M. , Czopek, J.P. , Kolodziejczyk, P. , Sinczak-Kuta, A. , Klimkowska, A. , Wojcik, P. , Okon, K. , Bialas, M. , Kulig, J. , Stachura, J. , 2006. CDH1 gene promoter hypermethylation in gastric cancer - Relationship to Goseki grading, microsatellite instability status, and EBV invasion. Diagnostic Molecular Pathology. 15, 24–29. [DOI] [PubMed] [Google Scholar]
- Zhang, R. , Shao, F. , Wu, X. , Ying, K. , 2010. Value of quantitative analysis of circulating cell free DNA as a screening tool for lung cancer: a meta-analysis. Lung Cancer. 69, 225–231. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.-J. , Rossner, P. , Chen, Y. , Agrawal, M. , Wang, Q. , Wang, L. , Ahsan, H. , Yu, M.-W. , Lee, P.-H. , Santella, R.M. , 2006. Aflatoxin B-1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. International Journal of Cancer. 119, 985–991. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.-J. , Wu, H.-C. , Yazici, H. , Yu, M.-W. , Lee, P.-H. , Santella, R.M. , 2012. Global hypomethylation in hepatocellular carcinoma and its relationship to aflatoxin B(1) exposure. World Journal of Hepatology. 4, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Z.M. , Baker, C.C. , 2006. Papillomavirus genome structure, expression, and post-transcriptional regulation. Frontiers in Bioscience. 11, 2286–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Jiang, W.H. , Ren, C.P. , Yin, Z.H. , Feng, X.L. , Liu, W.D. , Tao, O. , Yao, K.T. , 2005. Frequent hypermethylation of RASSF1A and TSLC1, and high viral load of Epstein-Barr virus DNA in nasopharyngeal carcinoma and matched tumor-adjacent tissues. Neoplasia. 7, 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler, A. , Zangemeister-Wittke, U. , Stahel, R.A. , 2002. Circulating DNA: a new diagnostic gold mine?. Cancer Treatment Reviews. 28, 255–271. [DOI] [PubMed] [Google Scholar]