

Abstract
Abnormal epigenetic control is a common early event in tumour progression, and aberrant acetylation in particular has been implicated in tumourigenesis. One of the most promising approaches towards drugs that modulate epigenetic processes has been seen in the development of inhibitors of histone deacetylases (HDACs). HDACs regulate the acetylation of histones in nucleosomes, which mediates changes in chromatin conformation, leading to regulation of gene expression. HDACs also regulate the acetylation status of a variety of other non‐histone substrates, including key tumour suppressor proteins and oncogenes. Histone deacetylase inhibitors (HDIs) are potent anti‐proliferative agents which modulate acetylation by targeting histone deacetylases. Interest is increasing in HDI‐based therapies and so far, two HDIs, vorinostat (SAHA) and romidepsin (FK228), have been approved for treating cutaneous T‐cell lymphoma (CTCL). Others are undergoing clinical trials. Treatment with HDIs prompts tumour cells to undergo apoptosis, and cell‐based studies have shown a number of other outcomes to result from HDI treatment, including cell‐cycle arrest, cell differentiation, anti‐angiogenesis and autophagy. However, our understanding of the key pathways through which HDAC inhibitors affect tumour cell growth remains incomplete, which has hampered progress in identifying malignancies other than CTCL which are likely to respond to HDI treatment.
Keywords: HDAC, Drugs epigenetics, Acetylation, Biomarkers, Cancer
Highlights
HDAC is a ubiquitous family of enzymes involved in many aspects of cancer biology.
HDAC inhibitors are potent anti‐proliferative agents and anti‐cancer drugs.
HDAC inhibitors vorinostat and romidepsin are approved for treating T‐cell lymphoma.
The key question is how to identify responsive tumours and relevant biomarkers.
Understanding the biology and key pathways influenced by HDACs is required.
1. Histone deacetylase classes
The vast amount of genomic DNA in eukaryotic cells is assembled onto histone proteins to allow it to be accommodated within the nucleus. An octamer of histones, consisting of an H3–H4 tetramer and two H2A–H2B dimers makes up a nucleosome, around which around 146 bp of DNA is wrapped (Bentley et al., 1984). Nucleosomes interact with the linker histone H1 and other chromatin‐associated proteins to achieve further compaction of DNA to form chromatin (Inche and La Thangue, 2006).
The N‐terminal tail regions of histones undergo a wide variety of enzyme modifications, including acetylation, methylation, phosphorylation and ubiquitination which have significant effects on gene expression, and are referred to as the “histone code” (Moniot et al., 2012). Core histone proteins are reversibly and dynamically acetylated at multiple lysine residues in their N‐terminal tails, and histone acetylation and deacetylation play a key role in regulating gene expression in eukaryotes and modifying chromatin structure. This takes place both by affecting the intrinsic properties of histones and their interactions with other proteins and DNA (Khan and La Thangue, 2008). Histone acetylation is controlled by histone acetyl transferases (HATs) which transfer acetyl groups to the side chain of lysine residues, and histone deacetylases (HDACs), which deacetylate lysine residues and counterbalance activity of HATs (Khan and La Thangue, 2008).
The HDAC family of enzymes regulates not only the acetylation level of histones in chromatin, but also a variety of non‐histone substrates, which include many proteins that are involved with tumour progression, cell cycle control, apoptosis, angiogenesis and cell invasion (Witt et al., 2009). The HDAC family contains 18 genes which are grouped into classes I, II, III and IV (Figure 1) based on their homology to respective yeast orthologues Rpd3, HdaI and Sir2 (Gregoretti et al., 2004).
Figure 1.

Schematic organisation of classes I, II and IV HDACs showing their domain composition, size, cellular localization, target proteins and the cellular processes which are consequently regulated. The HDAC deacetylase catalytic domains are shown in purple, nuclear localization targeting sequences in yellow, and the ubiquitin‐binding BUZ domain of HDAC6 is in orange. A single substrate example is given alongside each enzyme, as well as the process in which this substrate participates.
HDAC inhibitors typically target the “classical” classes I, II and IV HDACs, which comprise 11 family members in humans (Witt et al., 2009). Class III HDACs are named sirtuins (SIRT1‐7) and differ from classical HDACs in that they use NAD+ as the essential cofactor, whereas classical HDACs contain a Zn2+ catalytic ion in their active site (Liu et al., 2009). There is increasing evidence for class III HDACs being important in transcriptional regulation, for example, SIRTI upregulation in cancer cells induces CpG island methylation and aberrant silencing of tumour suppressor genes such as E‐cadherin (Liu et al., 2009). However, this class of HDACs will not be covered in detail in this review (reviewed in Haigis and Sinclair, 2010).
1.1. Class I HDACs
Class I HDACs consist of HDAC1, HDAC2, HDAC3 and HDAC8. HDACs 1, 2 and 3 are located in the nucleus, whereas HDAC8 is located both in the nucleus and the cytoplasm. HDACs 1, 2 and 3 are found as components of multi‐protein complexes involved in transcriptional repression. For example, Mi2 and Co‐REST contain the HDAC1–HDAC2 dimer, whilst the NCoR complex forms around HDAC3 (Yang and Seto, 2008). HDAC8 has not been found in any transcriptional repressor complex so far (de Ruijter et al., 2003).
Class I HDACs are ubiquitously expressed throughout all cells and are likely to be important in regulating proliferation. For example, the pRb tumour suppressor recruits HDAC1 into a complex with E2F (Satoh et al., 2011), to result in HDAC1 binding E2F target promoters during the early G1 phase of the cell cycle to cause decreased histone H4 acetylation of specific lysines to result in gene silencing and cell cycle arrest (Chalkiadaki and Guarente, 2012). The DNA damage response is another process in which class I HDACs are involved in. HDAC1 and HDAC2 are recruited to sites of DNA damage to promote hypoacetylation of histone H3 K56, which is likely to repress transcription and thus allow DNA repair to take place, as well as potentially recruiting non‐homologous end joining factors (Miller et al., 2010). Cells in which HDACs 1 and 2 are depleted are hypersensitive to DNA damaging agents, due to faulty double‐strand break repair by non‐homologous end joining (Miller et al., 2010).
The importance of class I HDACs is highlighted by the fact that global deletion of HDAC1 in mice results in death by embryonic day 9.5, whereas cardiac‐specific deletion of HDAC1 and 2 together, but not each individually, causes neonatal lethality due to cardiac arrhythmia, suggesting HDACs 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility (Montgomery et al., 2007). HDAC2 also has also been shown to regulate chromatin compaction and its inhibition results in chromatin decondensation and tumour cell sensitization to chemotherapy (Marchion et al., 2009). Inactivation of HDAC3 resulted in compromized DNA repair, with DNA damage observed particularly in cycling cells, which may lead to tumour cells with defective cell‐cycle checkpoints being especially sensitive to HDAC3 inhibition (Bhaskara et al., 2008). HDAC8 has recently been shown to be required for transcription of wild‐type and mutant tumour suppressor p53, suggesting that HDAC8 inhibition may be a way to prevent mutant p53 expression in tumours (Yan et al., in press).
1.2. Class II HDACs
Class II HDACs are present in either the cytoplasm or nucleus, and can be shuttled between these compartments. Their functions are typically tissue specific: for example HDACs 4, 5 and 7 regulate muscle cell differentiation by changing localization in response to specific signal which results in changed to gene expression (de Ruijter et al., 2003). Class II HDACs are divided into two subclasses: class IIA members are HDACs 4, 5, 7, and 9 and class IIB members are HDACs 6 and 10.
All class IIA members possess a conserved N‐terminal domain, which is homologous to yeast yHDA1 (Verdin et al., 2003) and regulates nuclear‐cytoplasmic shuttling and DNA‐binding. Class IIA members also contain three conserved sites for binding of 14‐3‐3 proteins, which regulates cellular trafficking of class IIA HDACs in a phosphorylation dependent manner. 14‐3‐3 binding site phosphorylation is dependent on the action of a number of signalling pathways. Whether these HDACs are retained in the cytoplasm or transported to the nucleus regulates activity of transcription factors such as myocyte enhancing factor‐2 (MEF2) and, consequently, myocyte development. In the nucleus, class IIA HDAC, such as HDAC9, are bound to MEF2 proteins in a number of gene promoters, repressing transcription until a myogenic differentiation signal causes their export into the cytoplasm (Verdin et al., 2003).
HDAC4 regulates chondrocyte hypertrophy and endochondral bone formation by inhibiting the Runx2 transcription factor (Sun et al., 2009), its importance highlighted by the fact that HDAC4 knockout mice display premature ossification of developing bones and aberrant skeletogenesis (Vega et al., 2004). HDAC9 has a role in cardiomyocyte differentiation and mice lacking HDAC9 are sensitized to stress signals that induce cardiac hypertrophy (Montgomery et al., 2007).
HDAC7 regulates apoptosis in developing thymocytes, in part by inhibiting the Nur77 receptor via the transcription factor MEF2D. During T‐cell receptor activation, HDAC7 is exported from the nucleus, permitting expression of Nur77 can be expressed and abolishing TCR‐mediated apoptosis (Dequiedt et al., 2003). HDAC5 is also required for Nur77 repression although it occurs at a lower level in the human thymus (Dequiedt et al., 2003). HDAC5 also represses angiogenic genes such as FGF2 and Slit2 in endothelial cells (Urbich et al., 2009).
The two class IIB members HDAC6 and HDAC10 are structurally related in that they both contain two catalytic domains and a C‐terminal zinc finger, although HDAC10 also possesses an extra catalytically inactive domain. HDAC6 has a number of functions both dependent and independent of its histone deacetylase activity. HDAC6 deacetylates α‐tubulin and heat shock protein‐90 (HSP‐90) amongst other proteins, and thereby regulates cell‐motility and adhesion, as well as the response to misfolded proteins (see also Section 4.3). HDAC6 is a unique enzyme in that it has two catalytic sites and the ubiquitin binding BUZ domain, which allows it to target cargo proteins for subsequent processing. The zinc‐finger ubiquitin binding domain of HDAC6 is important in aggresome formation (Kawaguchi et al., 2003), autophagy (Lee et al., 2010), heat shock factor‐1 activation in response to cytotoxic protein aggregates (Boyault et al., 2007) and platelet derived growth factor (PDGF) function (Gao et al., 2007).
HDAC10 has fewer established roles than HDAC6, although it may also regulate HSP‐90 acetylation and thus vascular endothelial growth factor receptor (VEGFR) degradation by the proteasome. HDAC10 inhibition therefore is also able to increase HSP90 acetylation, and causes VEGFR degradation (Park et al., 2008). Interestingly, HDAC10 together with HDAC9 have been shown to be necessary for homologous recombination activity, although it is not yet clear whether this is by direct participation or transcriptional control (Kotian et al., 2011).
1.3. Class IV
HDAC11 is the only HDAC in class IV and very little is known about it at present, although it has been implicated in immune system regulation via its role in interleukin‐10 expression (Lian et al., 2012) and OX40L surface expression in Hodgkin lymphoma (Buglio et al., 2011).
2. HDACs and cancer
Altered expression, deregulation and mutations of HDAC genes are linked to tumour development during cancer in two ways: through modification of gene transcription and via the non‐histone HDAC substrates. Although somatic mutations of HDAC genes are rare, HDAC4 mutations have been identified in breast cancer samples during a large scale sequencing study (Sjoblom et al., 2006). Additionally, mutations which result in HDAC2 protein truncation have been described in human epithelial cancer cell lines (Ropero et al., 2006). The altered expression profiles of various HDACs in different cancer types observed in studies so far and their potential prognostic relevance is summarized in Table 1.
Table 1.
HDAC expression in various cancer types and possible prognostic relevance.
| Levels in cancer types | Prognostic relevance | |
|---|---|---|
| Class I HDACs | ||
| 1 | Upregulated in gastric, colorectal, esophageal and pancreatic cancer | High expression in pancreatic cancer associated with poor prognosisIn hepatocellular carcinoma, high HDAC1 associated with invasion into the portal veinIn lung cancer, high HDAC1 associated with later stages of diseaseBetter disease‐free survival in breast cancer |
| 2 | Upregulated during early colorectal cancer at the polyp stageUpregulated in cervical dysplasia and invasive carcinoma | High levels in colorectal cancer associated with poor patient survivalAssociated with advanced stage disease in gastric, colorectal and prostate cancerAssociated with aggressive CTCL |
| 3 | Upregulated in lung cancer, prostate and colon cancer | High levels in colorectal cancer associated with poor patient survivalAssociated with advanced stage disease in gastric, colorectal and prostate cancerPoor prognostic indicator for post‐liver transplantation HCC |
| 8 | Upregulated in neuroblastoma | Higher expression associated with poorer outcome and advanced stage disease in paediatric neuroblastoma |
| Class IIA HDACs | ||
| 4 | Upregulated in breast cancer samples compared with renal, bladder and colorectal cancer | No data available |
| 5 | Upregulated in colorectal cancer in contrast to renal, bladder and breast cancerDownregulated in cancer and acute myeloid leukaemia | Low expression associated with poor prognosis in lung cancer |
| 7 | Upregulated in colorectal cancer in contrast to bladder, renal and breast cancer | May present an anti‐angiogenesis target |
| 9 | Overexpressed in medulloblastoma/astrocytoma | High levels associated with poor prognosis and survival in childhood acute lymphoblastic leukaemia |
| Class IIB HDACs | ||
| 6 | High in oral squamous cell carcinoma | Associated with favourable outcome in CTCL in higher survival in breast cancer |
| 10 | Overexpressed in hepatocellular carcinoma | Poor prognostic indicator in lung cancer |
| Class IV HDACs | ||
| 11 | Overexpressed in breast cancer | No data available |
HDACs frequently induce aberrant transcription of key genes regulating cell proliferation, cell‐cycle and apoptosis. This takes place by HDACs acting as components of multi‐protein complexes containing corepressor DNA‐binding proteins such as NcoR, SMRT and Sin3A (de Ruijter et al., 2003; Khan and La Thangue, 2008). The consequent transcriptional repression of tumour suppressor genes then promotes carcinogenesis. However, microarray analysis has indicated that only 2–5% of genes are influenced by HDAC inhibition (Glaser et al., 2003) and approximately equal numbers of genes are induced and repressed (Xu et al., 2007). This suggests that mechanisms other than regulation of transcription are important in HDAC effects on cellular processes. For example, regulation of the acetylation status of the diverse range of non‐histone HDAC substrate proteins is likely to be important.
The function of many transcription factors, chaperones and structural proteins depends on their acetylation state, which in turn impacts on a multitude of physiological pathways. For example, HDAC6 deacetylates HSP‐90 which enhances ATP binding and thus promotes assembly of functional HSP‐90 chaperone complexes (Kovacs et al., 2005). β‐catenin is another HDAC6 substrate which is frequently mutated in anaplastic thyroid cancer. Epidermal growth factor (EGF) induces HDAC6 translocation, after which HDAC6 associates with β‐catenin and deacetylates it, causing nuclear localisation of β‐catenin which promotes Wnt signalling and c‐myc activation (Li et al., 2008). This implicates HDAC6 as a link between EGF and Wnt signalling during tumour progression, and suggests that inactivation of HDAC6 could be beneficial in tumours where such signalling is deregulated (Li et al., 2008). HDACs 1 and 2 are also implicated in Wnt signalling via disruption of the β‐catenin–TCF interaction (Ye et al., 2009). Also, Wnt‐dependent increased HDAC2 expression as a consequence of the loss of the adenomatosis polyposis coli tumour suppressor gene is found in the majority of human colon cancers, where HDAC2 is thought to prevent apoptosis (Zhu et al., 2004).
Acetylation also influences key tumour suppressor proteins, for example p53 and retinoblastoma (pRb) and oncoproteins such as c‐Myc and E2F1. Recruitment of histone deacetylase activity has been implicated in repression of transcription by the E2F–Rb complex in the G1 phase of the cell cycle. The lysine residues on which E2F1 is acetylated are highly conserved and adjacent to the DNA‐binding domain, and their acetylation increases E2F1 DNA‐binding activity, trans‐activation potential and half‐life (Martinez‐Balbas et al., 2000).
3. HDAC inhibitors
HDIs are potent anti‐proliferative agents which have been observed to cause apoptosis in cell‐based studies, hence there has been great interest to develop these drugs as potential anti‐cancer agents, as well as agents to dissect HDAC roles (Khan et al., 2010). Most inhibitors being developed as anti‐cancer agents target classes I, II and IV enzymes, although interest in the class III sirtuin family is increasing (Stimson et al., 2009). Many compounds are in clinical trials or other stages of preclinical development. HDIs can be classified according to their chemical structure into hydroxamates, cyclic peptides, benzamides and fatty acids (Marks, 2010), or according to their specificity for various HDAC classes (summarized in Table 2). More recently, chemoproteomic approaches combining affinity capture and quantitative mass spectrometry have been useful in identifying which HDAC complexes are targeted by HDIs, rather than isolated HDACs. For example, valproate affects the Sin3 complex to a lesser degree than other class I complexes with the same catalytic subunit (Bantscheff et al., 2011).
Table 2.
HDI classification by chemical structure and clinical trial use.
| Classification by chemical structure | Named examples (INN) | HDAC specificity | Clinical trial stage |
|---|---|---|---|
| Hydroxamates | SAHA (vorinostat) | Pan‐inhibitor | Approved for CTCL, phase III alone or in combination |
| PXD101 (belinostat) | Pan‐inhibitor | Phase II alone or in combination | |
| LBH589 (panobinostat) | Classes I and II | Phase III alone or in combination | |
| ITF2357 (givinostat) | Pan‐inhibitor | Phase II alone or in combination | |
| 4SC‐201 (resminostat) | Pan‐inhibitor | Phase II alone or in combination | |
| PCI 24781 (abexinostat) | Classes I and II | Phase II alone or in combination | |
| Cyclic peptides | Depsipeptide/FK228 (romidepsin) | Class I | Approved for CTCL and PCTL, phase III alone or in combination |
| Benzamides | MS‐275 (entinostat) | Class I | Phase II alone or in combination |
| MGCD0103 (mocetinostat) | Class I | Phase II alone or in combination | |
| Aliphatic fatty acids | Valproic acid | Classes I and IIa | Phase II alone or in combination |
| Butyrate | Classes I and IIa | Phase II alone or in combination |
The inhibitory effect of a large number of HDAC inhibitors depends on the Zn2+ dependency of HDAC enzymes (Stimson et al., 2009). A classic example of this is trichostatin A (TSA), which was one of the first HDIs to be identified. As the crystal structure of TSA bound to an HDAC analogue reveals, the conserved deacetylase active site contains a tubular pocket, a zinc‐binding site and two Asp–His charge relay systems necessary for the enzymatic activity. The long aliphatic chain of TSA fits into the tubular pocket, with the hydroxamic group at the end of the aliphatic chain coordinating the zinc through its carbonyl and hydroxyl groups (Finnin et al., 1999). The hydroxamic group of SAHA chelates the zinc ion in the same way, but SAHA's aliphatic chain makes fewer van‐der‐Waals contacts to the HDAC tubular pocket compared to TSA, explaining the weaker inhibitory activity of SAHA relative to TSA (Finnin et al., 1999). A number of other HDIs have a similar mechanism of action including belinostat, givinostat, panobinostat and dacinostat (Khan and La Thangue, 2012).
4. Mechanisms of action to induce cell death: targeting pleiotropic activities of HDACs
HDACs act not only on histones, but rather have many different cellular substrates and target proteins, both in the nucleus and in the cytoplasm. On the one hand, a redundant activity of some HDACs has been reported; on the other hand, some HDACs are very specific to client proteins and consequently cause distinct effects. For example, HDAC6 displays pleiotropic functions and regulates many genes and proteins. Therefore, they influence the entire acetylome (Choudhary et al., 2009). HDACs are involved in regulation of the cell cycle and mitosis, the DNA damage response, cellular stress response, protein degradation, cytokine signalling, immunity and inflammation, angiogenesis and cell survival. Because of this plethora of different functions in the cell, HDAC inhibition can cause a broad spectrum of cellular changes, including apoptosis in cancer cells (Figure 2).
Figure 2.
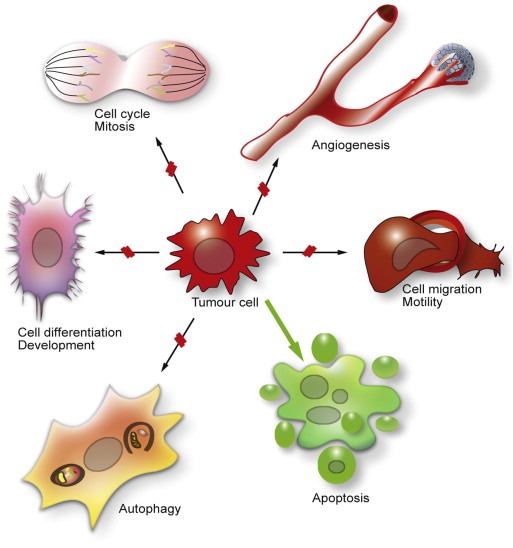
Influence of HDACs on different cell biological processes and consequences of HDAC inhibition. HDACs are involved in many cellular processes, including progression through the cell cycle, cell differentiation and development, cell migration and motility, angiogenesis and autophagy. These processes could promote tumour cell survival, proliferation and metastasis. HDAC inhibition blocks some of these processes, indicated by red lines, or favours other processes such as apoptosis, here indicated by the green arrow.
4.1. Interference with the cell cycle and mitosis
It is well known that HDACs, including HDACs 1, 2, 3, 4 and 6, are involved in cell cycle progression and cell proliferation, especially in cancer cells (Glaser et al., 2003; Haberland et al., 2009). HDAC1 is seen as a positive regulator of proliferation, demonstrated with HDAC1 knockout mice which display developmental abnormalities followed by elevated levels of the cyclin‐dependent kinase inhibitors (CKI) p21WAF1/CIP1 and p27/KIP1 and lethality. In support of these results, HDAC1 targets not only genes involved in proliferation, but also in developmental processes (Zupkovitz et al., 2006). An upregulation of HDAC2 was observed after downregulation of HDAC1, but could not rescue the defects in proliferation and development in mice. Although HDAC1 and HDAC2 share a protein sequence identity of 86%, they appear to have only partially overlapping functions and cannot rescue loss of each other. Experiments with certain differentiated cell types, e.g. cells from the haematopoietic system or fibroblasts, suggest that HDAC1 and HDAC2 have some functions in common, but knockdown of both enzymes causes abnormalities in development as well as aberrant cell cycle progression (Wilting et al., 2010; Yamaguchi et al., 2010). When both HDAC1 and HDAC2 were absent, progression to G1 and proliferation was blocked in several cell types leading to the hypothesis that the concerted action of HDAC1 and HDAC2 promote G1–S progression (Yamaguchi et al., 2010). Similar results were obtained with experiments in tumour cells lacking both HDAC1 and HDAC2 (Haberland et al., 2009). Inhibitors with potential to block both HDAC1 and HDAC2 could therefore be important in the anti‐cancer drug development.
HDAC4 was also able to influence cell proliferation and to repress p21 WAF1/CIP1 in cancer cells. Conversely, reduced function of HDAC4 increases p21 WAF1/CIP1 in some cancer cell lines and inhibits cell proliferation and tumour growth (Mottet et al., 2009; Wilson et al., 2008). Furthermore, HDAC3 is involved in cell cycle arrest in the G2/M phase, but also in aberrant mitosis. S10 phosphorylation in histone 3 is mediated by the Aurora B kinase. It was shown that inhibition of HDAC3 or reduced levels of HDAC3 lead to reduced phosphorylation at S10 and aberrant mitosis (Wilson et al., 2006). Another explanation of the G2/M arrest after administration of HDAC inhibitors, especially during the inhibition of HDAC3 and HDAC6, is increased degradation of Aurora A and B kinases, highlighting the importance of HDAC6 during protein degradation (Cha et al., 2009). Additional evidence for the role of HDACs in mitosis was provided by experiments where HDAC inhibitors were able to interfere with spindle checkpoint proteins, hinder spindle formation (Stevens et al., 2008) and disrupt reversible pericentric heterochromatin and centromere function (Taddei et al., 2001).
4.2. DNA damage response
The DNA damage response (DDR) consists of several processes and interconnected macromolecular complexes which are able to sense and repair DNA damage. There are two major types of DNA breaks: DNA double strand breaks (DSB) and DNA single strand breaks. In order to allow repair of DSBs, cells can activate cell cycle arrest before the DNA replication machinery processes the site. The DNA damage is sensed by a complex consisting of Mre11, Rad50 and NBS1, abbreviated as MRN, and the human single strand DNA binding protein 1 (hSSB1). Further downstream, ataxia telangiectasia mutated kinase (ATM) becomes activated in the case of DSBs. With DNA single strand breaks, the ataxia telangiectasia Rad3‐related (ATR) kinase recognizes and amplifies the signal (Bolderson et al., 2009). Both kinases transmit the signal downstream to the checkpoint proteins 1 and 2 (CHK1 and CHK2), which are kinases that amplify the signal and activate the executionery proteins of the DDR. Normal cell functions are re‐established, providing the DDR was successful, otherwise cell death is initiated (Jackson and Bartek, 2009).
There are several reports that HDAC inhibitors negatively influence these processes, but also activate the DDR (Bakkenist and Kastan, 2003). Several possibilities have been suggested by which HDAC inhibitors could promote the DDR. One explanation could be that due to increased acetylation, the chromatin becomes relaxed and is more susceptible to harmful agents, such as radiation, DNA damaging drugs or reactive oxygen species (ROS). It was demonstrated that elevated ROS levels were present after treatment with HDAC inhibitors and HDAC inhibitors were able to activate cell death after ROS‐induced DNA damage (Eot‐Houllier et al., 2009). Elevated levels of ROS species after HDAC inhibitor treatment may force cells into a stress response followed by apoptosis (Bhalla et al., 2009; Petruccelli et al., 2011). Similar outcomes were observed in combination with a proteasome inhibitor (Bhalla et al., 2009; Dasmahapatra et al., 2011; Feng et al., 2007) and the combination of these drugs led to synergistic effects (see also section 5.5).
However, other mechanisms might be involved in the DDR after administration of HDAC inhibitors to human cells. For example, it has been reported that HDAC inhibitors suppress RAD51 expression, inhibit homologous recombination (Adimoolam et al., 2007) and additionally downregulate other repair genes through the modulation of E2F1 activity (Kachhap et al., 2010). HDAC 9 and HDAC10, both class II HDAC members, are required for the process of homologous recombination (Kotian et al., 2011). A direct inactivation of DDR components at the level of post‐translational modification via blocking of HDAC1 and HDAC2 action is also likely to contribute to the HDAC inhibitor actions (Miller et al., 2010). Other suppressed proteins involved in the DDR include DNA‐PK, RAD50, Ku80, and Ku86 which sensitize the HDAC inhibitor response to radiation in different cell types (Blattmann et al., 2010; Deorukhkar et al., 2010; Mueller et al., 2011). Moreover, HDAC inhibitors are able to act synergistically with other agents such as anti‐cancer drugs or radiation which cause DNA damage directly or indirectly (Camphausen and Tofilon, 2007; Eot‐Houllier et al., 2009; Richon et al., 2009). A recent report showed that the HDAC inhibitor sodium butyrate sensitizes E1A‐Ras‐transformed cells to DNA damaging agents by facilitating formation and persistence of so‐called γH2AX foci which are markers of DSBs (Eot‐Houllier et al., 2009). Several other studies emphasize the occurrence and prolonged duration of phosphorylated histone H2AX (γH2AX) nuclear foci after treatment with HDAC inhibitors and DNA damage causing agents (Eot‐Houllier et al., 2009; Mueller et al., 2011; Zhang et al., 2009a).
4.3. Influence on protein quality control mechanisms
Cells and therefore whole organisms have to permanently control the integrity of all their proteins, the proteome. In order to react to developmental changes, senescence or exogenous influences, they adjust the concentration, localisation, conformation and interaction of proteins to maintain the status quo. These adjustments and maintenance of the proteome is named protein homeostasis or proteostasis (Balch et al., 2008; Hartl et al., 2011). Cells achieve this crucial proteostasis through concerted action of several protein quality control systems, namely protein biogenesis, protein folding, posttranslational modifications and protein degradation. HDACs, especially HDAC6, play an important role in the different protein quality control systems, particularly in the initial steps of the stress response, post‐translational modifications and protein degradation (Figure 3).
Figure 3.
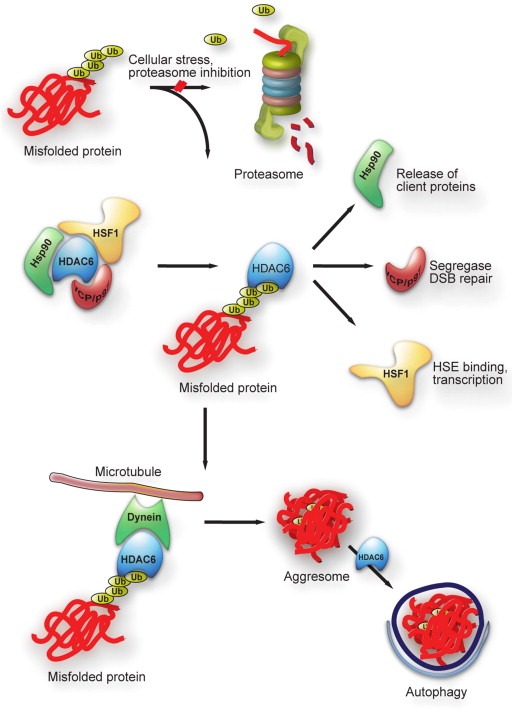
Pleiotropic activities of HDAC6 in protein quality control. Misfolded proteins accumulate upon cellular stress or proteasome inhibition. These ubiquitinated misfolded proteins are recognized by HDAC6 and displace basal interactors of HDAC, in particular Hsp90, HSF1 and VCP. Subsequently, client proteins can be released from Hsp90, HSF1 can trimerize and act as a transcription factor on HSE determined proteins, e.g. Hsp70. VCP exerts its segregase activity and helps in DSB repair upon cellular stress. HDAC6 targets misfolded proteins to the MTOC via microtubules and is also involved in the process of autophagy.
Heat shock factor 1 (HSF1) is a transcription factor that activates transcription of genes coding for proteins which protect the cell against harmful stresses. In HDAC6‐deficient cells re‐constituted with either wild‐type HDAC6 or mutant HDAC6, VCP/p97 is recruited by HDAC6 and associates with the heat‐shock protein 90/heat‐shock factor 1 (Hsp90/HSF1) complex under unstressed conditions (Boyault et al., 2007). Hsp90 and HDAC6 dissociate under stress conditions from HSF1 which trimerizes and locates to the nucleus to act as transcription factor. This mechanism also offers the intriguing possibility that HDAC6 senses misfolded proteins and initiates the stress response by releasing HSF1 together with Hsp90. Both HSF1 and HDAC6 are relevant in the process of carcinogenesis, since they are involved in the RAS/MAPK signalling cascade (Dai et al., 2007; Lee et al., 2008). Pan‐HDAC inhibitors or inhibitors showing specificity for HDAC6 could contribute to cell death by inhibiting these pathways. Sirt1, belonging to the HDAC class III, prevents HSF1 acetylation at K80 and thus inhibits DNA binding (Westerheide et al., 2009). It was also shown that administration of HDIs results in reversible hyperacetylation of Hsp90 and, following release of Hsp90 client proteins, inhibition of the Hsp90 chaperone function and subsequent proteasome degradation of the Hsp90 client proteins. Amongst the released proteins, some oncogenic proteins such as Akt, Bcr‐Abl, c‐Raf and ErB2 were found which are then more susceptible to polyubiquitination and proteasomal degradation (Bali et al., 2005; Nimmanapalli et al., 2003).
Proteins can be degraded via the ubiquitin–proteasome system (UPS) or via autophagocytic pathways both of which act as key protein quality control mechanisms. To defend the cell against misfolded proteins and aggregation, caused by heat stress, chemical substances or mutations, the UPS and autophagy are also possible routes of degradation. A specialized form of aggregates in the cytoplasm of mammalian cells, the aggresome, is located close to the microtubule‐organisation centre (MTOC) (Johnston et al., 1998). HDAC6 is involved in the recognition and transport of aggregated proteins to aggresomes (Figure 3). One hypothesis suggests that HDAC6 acts as an adaptor protein that recognizes polyubiquitin chains of substrates and also the motor protein dynein. It then transports the ubiquitinated substrates along the dynein to the MTOC, enabling formation of aggresomes (Kawaguchi et al., 2003). K63‐linked polyubiquitination could present a recognition site for HDAC6‐dependent transport of misfolded proteins towards the aggresome, which has also been observed after inhibition of the UPS (Olzmann et al., 2007; Tan et al., 2008). HDAC6 inhibition could lead to destruction of the HDAC6‐dynein‐substrate complex and impair the formation of larger aggresomes (Kawaguchi et al., 2003).
A genome‐wide loss‐of‐function screen identified HR23B, which shuttles ubiquitinated cargo proteins to the proteasome, as a sensitivity determinant for HDAC inhibitor‐induced apoptosis (Fotheringham et al., 2009). HR23B is involved in nucleotide excision repair on the one hand, but it is also known to function as a shuttling factor for proteins targeted for proteasomal degradation (Verma et al., 2004). The level of HR23B influences the response of tumour cells to HDAC inhibitors, and HR23B is at high levels in cutaneous T‐cell lymphoma (CTCL) in situ, a malignancy that responds favourably to HDAC inhibitor‐based therapy. These results suggest that deregulated proteasome activity contributes to the anti‐cancer activity of HDAC inhibitors (Fotheringham et al., 2009). In a following study, it was demonstrated that HR23B governs the sensitivity of CTCL cells to HDAC inhibitors. Furthermore, proteasome activity is deregulated in HDAC inhibitor‐treated CTCL cells through a mechanism dependent on HR23B, and HDAC inhibitors sensitize CTCL cells to the effects of proteasome inhibitors (Khan et al., 2010).
Autophagy, and especially the process of macroautophagy (referred to as autophagy in the following sections), uses specialized double‐membrane structures that engulf substrates and form autophagic vesicles that later fuse with lysosomes (He and Klionsky, 2009). Autophagy is strongly upregulated in response to stress such as starvation, growth factor withdrawal or oxidative damage (White and DiPaola, 2009). During autophagy, whole organelles such as mitochondria can be degraded, as well as misfolded protein aggregates (Ravikumar et al., 2004; Rubinsztein, 2006). It is hypothesized that autophagy is a salvage pathway to complement the UPS when it is overwhelmed or impaired by undegradable substrates, such as aggregating proteins (Webb et al., 2003).
Similar to its role in the formation of aggresomes, it turned out that HDAC6 is also a key component in the turnover of misfolded proteins by autophagy (Figure 3). HDAC6 controls the fusion of the autophagosomes with lysosomes and recruits the actin‐remodelling machinery (Lee et al., 2010). A missing link to couple ubiquitinated proteins and proteins on the autophagosome membrane could be the p62 protein, acting as a cargo receptor. It has been shown that p62 can bind ubiquitinated proteins with its ubiquitin‐associated (UBA) domain and a component of autophagic vesicle LC3 (Bjorkoy et al., 2005).
There are contradictory results for whether autophagy has a tumour‐suppressor role or whether it has a protecting role for tumour cells which defends against the influences of cancer drugs (Guo et al., 2011; White and DiPaola, 2009; Yang et al., 2011). This is because autophagy is also used to obtain nutrients and ATP for the cells which could prolong cell survival of tumour cells (Hippert et al., 2006). It has been demonstrated that several anti‐cancer drugs induce autophagy in tumour cells (Rodriguez‐Rocha et al., 2011), and a number of studies have shown that autophagy can be induced by HDAC inhibitors in normal cells and in cancer cells (Rikiishi, 2011; Shao et al., 2004). Impairment of autophagy with the antimalarial drug chloroquine increases HDAC inhibitor‐mediated apoptosis in colon cancer cells (Carew et al., 2010), whereas in another study, SAHA‐induced autophagy leads to autophagy‐associated cell death (Yamamoto et al., 2008). In further studies, it was shown that HDAC inhibitors can trigger mitochondrial‐mediated apoptosis and caspase‐independent autophagic cell death. HeLa cells with APAF‐1 knocked out or Bcl‐xL over‐expressed underwent autophagic cell death in the presence of HDAC inhibitors (Shao et al., 2004). The mammalian target of rapamycin (mTOR) plays an important role in the initiation of autophagy and displays an inhibitory effect on autophagy (Janku et al., 2011). mTOR and phosphorylated mTOR levels decreased after the treatment of endometrial stromal sarcomas with SAHA (Hrzenjak et al., 2008). The connection between the UPS and autophagy lead to new combination therapies with HDAC and proteasomal inhibitors (see section 5.5).
4.4. HDAC inhibitors induce apoptosis
HDAC inhibitors are able to activate apoptosis, either via the extrinsic (death receptor) pathway or intrinsic (mitochondrial) pathway or both in many cancer types and models (Rosato et al., 2003). The extrinsic pathway is activated through ligands, such as TRAIL or FASL, binding to their death receptors. The pan‐HDAC inhibitor SAHA has been shown to induce TRAIL expression in breast cancer cells, and enhanced the cytotoxicity induced by other anti‐cancer drugs (Xu et al., 2008). The adaptor protein FADD could be recruited to the initial signalling complex, followed by caspase‐8 activation. A study using both HDAC inhibitors and death receptor agonists in cancer cells demonstrated synergistic effects in this combination approach (Frew et al., 2008). Various stress stimuli can activate the intrinsic pathway characterized by the disruption of the mitochondrial membrane, causing release of pro‐death proteins such as cytochrome C and SMAC. Cytochrome C release initiates apoptosome formation, which is followed by the activation of caspase‐9. Caspase‐9 and the aforementioned caspase‐8 can activate the effector caspases −3, −6, and −7, which execute the apoptotic response (Maiuri et al., 2007), and in leukaemia cells, SAHA can induce mitochondrial dysfunction and apoptosis through enhanced production of ROS, downregulation of XIAP and activation of JNK (Dai et al., 2005). It has been demonstrated in various solid and haematological cancer cell lines, cancer models and patient material that HDAC inhibitors can upregulate death receptors and ligands, whereby the UPS is involved (Borbone et al., 2010; Nebbioso et al., 2005). There are many other proteins which tightly regulate proapoptotic (e.g. Bak, Bax, Bim, Bid) and antiapoptotic (e.g. Bcl‐2, Bcl‐xL, XIAP) stimuli. Most HDAC inhibitors, including SAHA and TSA, decrease the antiapoptotic proteins Bcl‐2, Bcl‐xL and XIAP, but increase the proapototic proteins, followed by a TRAIL‐mediated apoptotic response in many cancer cell models. There is evidence that both the extrinsic and the intrinsic apoptotic pathway is affected (Fulda, 2008) and further that crosstalk between autophagy and apoptosis exists. However, the complexity of the apoptotic and autophagic pathways make this relationship difficult to investigate, since it varies between different tissues, cell types and stimuli (Behrends et al., 2010; Zhang et al., 2009b). Autophagy and apoptosis are not only connected through various key proteins playing a role in both processes, such as beclin‐1 (Wang et al., 2007), JNK (Lorin et al., 2009), p53 (Morselli et al., 2008) and Atg5 (Bommareddy et al., 2009), but autophagy might also act as an alternative mechanism in rescuing the cell or the surrounding tissues when cancer cells undergo apoptosis.
4.5. Anti‐angiogenesis
The formation of new blood vessels from pre‐existing blood vessels is the process of angiogenesis. Angiogenesis is important for progression and metastasis of both solid tumours and haematological malignancies (Carmeliet, 2005; Moehler et al., 2001) to supply the tumour with oxygen and nutrients and remove CO2 and waste products. As hypoxia and deficiency of nutrients in the tumour microenvironment leads to a growth delay, it is an important therapeutic goal to identify proteins involved in angiogenic pathways. The tightly regulated hypoxia‐inducible factor 1α (HIF‐1α) is one of the key modulators in angiogenesis (Semenza, 2003), although constitutively expressed HIF‐1β is required to build transcriptionally active heterodimers. HIF‐1α is acetylated and hydroxylated under normoxic conditions, interacting with the von‐Hippel‐Lindau (pVHL) ubiquitin E3 ligase complex, which triggers rapid degradation via the UPS (Maxwell et al., 1999). Hypoxic conditions and therefore lack of hydroxylation of HIF‐1α leads to a stabilisation of this protein and an increased activity as a transcription factor. HIF‐1α regulates the expression of several genes involved in angiogenesis and other cellular signalling pathways, including the vascular endothelial growth factor (VEGF) (Forsythe et al., 1996; Maxwell et al., 1997). HDACs can influence the response to hypoxic conditions; HDAC1, HDAC2 and HDAC3 seem to be involved in the process of angiogenesis as under hypoxic conditions, an increased expression of HDACs 1, 2 and 3 has been reported (Kim et al., 2001). Increased expression of HDAC1 leads to a decrease in pVHL, followed by increases in levels of HIF‐1α and VEGF, whilst HDAC inhibitors abrogate these effects (Kim et al., 2001). Additionally, the class II HDACs 4 and 6 are able to influence the process of angiogenesis, and HDAC inhibition may contribute to their function. HDAC4 and HDAC6 can probably bind directly to HIF‐1α and influence its stability, independent from VHL (Qian et al., 2006). There is also evidence that HDAC3 and possibly HDAC1 can directly interact with HIF‐1α, and reduction in the expression levels of HDACs can trigger the degradation of HIF‐1α (Kim et al., 2007), a mechanism which may play a role also for drug‐induced HDAC inhibition. Another VHL independent pathway of HIF‐1α degradation involves the enhanced interaction of HIF‐1α with HSP70, mediated by HDAC‐6 (Kong et al., 2006). Taken together, there is increasing evidence that class I and class II HDAC are useful therapeutic agents for anti‐angiogenesis.
4.6. Effects on immunity and inflammation
There are many studies demonstrating the important role of HDACs both in innate and adaptive immunity. In particular Toll‐like receptor (TLR) and interferon (IFN) signalling pathways are modulated and regulated by HDACs. HDACs, and in an opposite manner HDAC inhibitors, can act on the expression of TLR target genes (Halili et al., 2010). Among the genes and proteins which are positively regulated by HDACs are pro‐inflammatory candidates such as cytokines (e.g. IL‐6, IL‐12, TNF), chemokines (e.g. CCL2, CCL7 and CXCL) and other mediators (e.g. MMP‐9 and endothelin‐1) (Bode et al., 2007; Roger et al., 2011). As discussed in Section 4.5, HDACs modulate HIF‐1α stability and manner of action. HIF‐1α is a proinflammatory transcription factor in TLR4‐dependent inflammatory responses in macrophages (Peyssonnaux et al., 2007), and is important for myeloid‐dependent inflammation in vivo (Cramer et al., 2003).
However, HDACs are also involved in repression or silencing of TLR‐target genes, for example they can affect TLR4‐target genes through chromatin remodelling (Foster et al., 2007). HDAC1 has been shown to modulate TLR‐induced activities of several inflammatory gene promoters, including IL‐12 (Lu et al., 2005) and IFN‐β (Nusinzon and Horvath, 2006). Additionally, class II HDACs are involved in the regulation of TLR responses. Interestingly, HDAC6 is able to reduce the formation of the MyD88 complex and initiates ubiquitin‐dependent MyD88 aggregation, which consequently abrogates the TLR4 response (Into et al., 2010).
HDACs also play various roles in the adaptive immune response, and therefore, HDAC inhibitors could be used as modulators of these processes. HDAC inhibition has been shown to reduce the production of cytokines involved in Th cell differentiation and chemokines in Th cell migration (Brogdon et al., 2007). A negative regulation of antigen‐presentation is initiated by the deacetylation of CIITA through HDAC2 followed by CIITA degradation (Kong et al., 2009; Zika et al., 2003).
HDACs are important enzymes in regulating T and B cell development and modulating their activities. HDAC7 is exported from the nucleus into the cytoplasm after a stimulus by T cell receptors which facilitates the expression of proteins involved in T cell selection (Dequiedt et al., 2005). It has been demonstrated that deletion of both HDAC1 and HDAC2 in B cells impairs B cell development; however, the deletion of either HDAC1 or HDAC2 has no strong effect on B cell development (Yamaguchi et al., 2010). HDAC6 has been shown to influence the levels of IgG and IgM to antigen exposure: HDAC6−/− mice have lower IgG and IgM levels and a reduced antibody response (Zhang et al., 2008), recapitulating what has been observed with HDAC inhibition.
4.7. Effects on IFN signalling
HDACs are involved in cytokine signalling, especially IFN signalling, where HDAC inhibition predominantly targets Janus kinases (JAKs) and STATs. Aberrant STAT3 signalling has been demonstrated in haematological malignancies such as T‐cell lymphomas, resulting in increased cell survival, p53 inhibition and induction of angiogenesis (Yu and Jove, 2004; Yu et al., 2009). HDACs are also able to modulate the acetylation status of the IFN receptors directly. The IFN‐α/β receptor 2 subunit of the type I IFN receptor is acetylated after stimulation with its ligand, followed by recruitment and acetylation of interferon‐stimulated gene factor (ISGF)3 (Tang et al., 2007). The underlying transcription factor complex consists of STAT1 and STAT2 in addition to IRF9. Down‐regulation of HDACs or HDAC inhibition leads to a hyperacetylated status of STAT signalling proteins, and as a result, cells are more prone to undergo apoptosis (Kramer et al., 2009). There are indications that HDACs positively regulate the IFN response and it has been reported that cytoplasmic HDACs are able to modulate STAT1 and promote reactivation in response to its ligand (Kramer et al., 2009). There are other reports underlying the modulating activities of HDACs in type I IFN induced gene expression (Chang et al., 2004) and IFN‐γ induced gene expression (Klampfer et al., 2004). The impact of HDACs and their inhibitors on IFN signalling is emphasized by results showing that HDAC inhibitors are able to increase the susceptibility to pathogens in mice (Roger et al., 2011).
5. Clinical application
Two HDAC inhibitors have been approved by the FDA so far to treat cancer: SAHA (vorinostat) and FK228 (romidepsin) were licensed for the treatment of advanced cutaneous T‐cell lymphoma whilst in addition romidepsin for the treatment of peripheral T‐cell lymphoma. However, histone deacetylase inhibitors have been used in neurology for many years as mood stabilizers and anti‐epileptic drugs, valproic acid being the most prominent amongst them. To act on neurons, it is important for HDIs to cross the blood–brain barrier, which is the case for valproic acid, SAHA, MS‐275, sodium butyrate and phenyl butyrate (Grayson et al., 2010). Here, we will provide an overview of the clinical applications of HDAC inhibitors in cancer therapy (Table 2).
5.1. Hydroxamic acid derivatives
Initial clinical studies with SAHA demonstrated that the substance is well tolerated and promising anti‐cancer activity was observed in different types of cancer, e.g. Hodgkin lymphoma, diffuse large B‐cell lymphoma and cutaneous T‐cell lymphoma (O'Connor et al., 2006). There were also correlative studies to investigate the underlying mechanism of SAHA action in CTCL and to identify potential predictive biomarkers (see also section 6.3). As mentioned in section 4.7, it was demonstrated in clinical samples that the JAK/STAT pathway is involved which could explain some of the resistance observed to SAHA (Fantin et al., 2008). A low response with SAHA was observed in multiple myelomas, AML and Hodgkin lymphoma (Garcia‐Manero et al., 2008; Kirschbaum et al., 2007a; Richardson et al., 2008), whereas more promising results came out in a phase II study in non‐Hodgkin lymphoma patients (Kirschbaum et al., 2007b). Although some of the results are auspicious, no clinical response was observed in AML, CLL or CML.
Other hydroxamic acids or cinnamic hydroxamic acids derivatives currently in clinical trials are belinostat (PXD101), panobinostat (LBH589) and givinostat (ITF2375). Belinostat has been reported to have a good safety profile and patients showed clinical response both during preclinical trials and a phase II trial (Plumb et al., 2003; Pohlman et al., 2009). Additionally, the pan‐HDAC inhibitor givinostat showed some promising results for haematological malignancies, e.g. multiple myeloma or Hodgkin lymphoma, but has also been shown to reduce the production of proinflammatory cytokines in vitro and systemic inflammation in vivo (Leoni et al., 2005).
5.2. Benzamide derivatives
Mocetinostat and entinostat are two examples of benzamide derivatives currently in clinical studies. The latter is an example of a selective HDAC inhibitor for class I HDACs, and is tolerated well both alone and in combination with other drugs (Pili et al., 2012). Phase II clinical studies in patients with relapsed or refractory Hodgkin's lymphoma are underway. Mocetinostat is a benzamide derivative with specificity for the classes I and IV HDACs (Fournel et al., 2008). The drug was in “partial clinical hold”, now removed, by the FDA because it caused pericardial effusion in patients (Boumber et al., 2011). However, the use of mocetinostat showed favourable clinical data in a recent phase II study in patients with Hodgkin's lymphoma (Younes et al., 2010).
5.3. Romidepsin (depsipeptide)
The cyclic peptide romidepsin (FK228 or FR901228), also named depsipeptide, was approved in 2009 by the FDA for the treatment of CTCL. It represents an atypical HDAC inhibitor, a natural product which has to be metabolized in order to become activated in human cells (Konstantinopoulos et al., 2006). Besides its positive effects in CTCL, several clinical trials have been conducted to determine its activity in other cancer types. Clinical responses were seen in peripheral T‐cell lymphoma (Coiffier et al., 2011; Piekarz et al., 2011). Limited antitumour activity has been reported in AML, CLL and MM patients (Byrd et al., 2005; Klimek et al., 2008; Niesvizky et al., 2011), as well as in many solid tumours such as prostate, colorectal and renal cancer (Molife et al., 2010; Stadler et al., 2006; Whitehead et al., 2009). The results of two multicentre phase II studies lead to the approval of romidepsin for the treatment of CTCL patients who have undergone at least one previous systemic therapy and 6% of the patients had complete responses (FDA, 2009).
5.4. Short chain fatty acids
One member of the short chain fatty acids which has been shown to inhibit HDACs is valproic acid (VPA), also known to raise the levels of gamma‐amino butyric acid (GABA). Besides its use in treatment of epilepsy for more than two decades, it also displays HDAC inhibition for class I HDACs (Gottlicher et al., 2001). VPA leads to differentiation of carcinoma cells as well as haematopoietic progenitor cells from AML patients. However, it was subsequently shown that this substance, alone or in combination therapy, has also beneficial effects in solid tumours, including neuroblastomas (Hrebackova et al., 2010), neuroendocrine carcinomas (Mohammed et al., 2011), glioblastomas under certain conditions (Tsai et al., 2011) and in cervical cancer (Chavez‐Blanco et al., 2005).
5.5. HDAC inhibitors in combination therapy with other anti‐cancer agents
Several preclinical studies and early clinical trials gave evidence that HDAC inhibitors can be used in combination with other targeted anti‐cancer drugs, anti‐angiogenesis drugs or radiation (Nolan et al., 2008). It has been demonstrated in many preclinical studies that HDAC inhibitors have synergistic effects with other anti‐cancer or cytotoxic agents.
One example is the combination of HDAC inhibitor and all‐trans retinoic acid in the treatment of acute promyelocytic leukaemia, which was seen to be effective both in vitro and in vivo preclinical models (Kuendgen et al., 2006). It is also known that DNA hypermethylation can lead to a more compact chromatin status which is more resistant to acetylation. This is one of the reasons for the ongoing trials of combination therapy with HDAC inhibitors and DNA demethylating agents or methyltransferase inhibitors. According to these results, sequential administration of demethylating agents or methyltransferase inhibitors seems to be important. For example, the substances 5‐azacytidine or 20‐deoxycytidine gave promising results in various cancer types, such as myeloid neoplasms (Gore et al., 2006). Other anti‐cancer agents such as carboplatinum (Ramalingam et al., 2010), taxanes (Dowdy et al., 2006; Ramalingam et al., 2010), topoisomerase inhibitors (Munster and Daud, 2011), nucleoside analogues (De Schutter et al., 2009; Kalac et al., 2011), tyrosine kinase inhibitors (Bruzzese et al., 2011; Huang et al., 2011; Nguyen et al., 2011), the Hsp90‐inhibitors analogue to geldanamycin (Rao et al., 2008; Yu et al., 2011), as well as radiation therapy (Chinnaiyan et al., 2005), displayed synergistic effects in combination with HDAC inhibitors and lead to promising results in various cancer types. An interesting aspect emerging from these combination therapies is the development of dual targeting agents that could target two or more target groups in cells. An example of such a bivalent agent, combining two complementary chemo‐active groups within a single molecular architecture, is the dual‐acting histone deacetylase and topoisomerase II inhibitor. These dual‐acting agents are derived from SAHA and the anthracycline daunorubicin, prototypical histone deacetylase (HDAC) and topoisomerase II (Topo II) inhibitors, respectively (Guerrant et al., 2012).
There are also results which show evidence of beneficial effects of the combination of HDAC inhibitors with proteasome inhibitors. Phase I and phase II studies are ongoing with the proteasome inhibitor bortezomib in combination with SAHA or romidepsin, for the treatment of multiple myeloma (Badros et al., 2009; Harrison et al., 2008; Weber et al., 2008). A possible explanation for these beneficial outcomes and synergistic effects could be that HDACs are also involved in protein quality control, influencing both the UPS (Fotheringham et al., 2009) and alternative ways of proteasome inhibition. Taken together, these studies imply that HDAC inhibition is more efficient in certain types of cancer when used in combination with other anti‐cancer agents.
6. Biomarker development
One can distinguish between prognostic, predictive, pharmacodynamic and surrogate markers. Prognostic biomarkers anticipate the likelihood of the clinical outcome and give indications whether a particular therapy may be required. Pharmacodynamic biomarkers measure the effect of a drug on the disease and are therefore closely related to surrogate markers which measure the effect of a treatment that can correlate with a clinical endpoint. Predictive biomarkers give the probability of a certain disease, such as a tumour, responding to the treatment (La Thangue and Kerr, 2011). An ideal biomarker should be able to give information not only on the selection of the treatment, but also its possible outcome and response.
6.1. Surrogate markers
Histone acetylation, especially H3 and H4 acetylation, can be used as a surrogate marker. The advantage of using the histones as biomarkers is that they can be measured in solid tumours as well as in peripheral blood mononuclear cells (Stimson and La Thangue, 2009). PXD101 has been shown to promote acetylation of H4 in a mouse model at concentrations which can cause tumour regression (Plumb et al., 2003). A phase I study of the histone deacetylase inhibitor SAHA in patients with advanced leukaemia and myelodysplastic syndromes measured hyperacetylation in peripheral blood mononuclear cells, which was induced in the patients regardless of the response and a deacetylation of histones, was observed after the drug treatment (Garcia‐Manero et al., 2008). Histone acetylation appears to be a useful surrogate marker for pan‐HDAC inhibition at least, but one cannot predict the treatment and tumour response from its outcome (Steele et al., 2008).
As discussed earlier, there are many other proteins both in the cytosol and in the nucleus which are deacetylated by HDACs and therefore affected by HDAC inhibition. It has been demonstrated that many HDAC inhibitors including SAHA, PXD101 and MS‐275 increased the levels of p21, so that p21 could act as a general biomarker of pan‐HDAC inhibition (Arts et al., 2007; Noro et al., 2010). Components of the JAK/STAT pathways are also susceptible to HDAC inhibition and could be considered as targets for HDI use in patients (see also Section 4.7). A phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor, indicated that Hsp90 client proteins including c‐Raf as well as Hsp70 could act as surrogate markers in patients with solid tumours (de Bono et al., 2008).
6.2. Prognostic biomarkers
Possible prognostic biomarkers for the outcome of different types of cancers are the HDACs themselves. Expression profiles of class I histone deacetylases in human cancer tissues revealed that these HDACs are over‐expressed in oesophageal and prostate cancer (Nakagawa et al., 2007). Class I HDAC overexpression seems to correlate with poor prognosis in oesophageal, pancreatic, colon, gastric and lung cancer (Khan and La Thangue, 2012; Krusche et al., 2005; Minamiya et al., 2011; Miyake et al., 2008; Sasaki et al., 2004; Sudo et al., 2011; Theocharis et al., 2011). Overexpression of other HDACs such as HDAC3 (Wu et al., 2010) and HDAC8 (Oehme et al., 2009) seem to correlate with poor outcome. Another study shed light on the prognostic significance of the therapeutic targets HDACs 1, 2, and 6 and acetylated histone H4 in CTCL (Marquard et al., 2008). It was evident that over‐expression of HDAC2 and histone H4 correlates with aggressive forms of CTCL, whereas the class II enzyme HDAC6 showed a different relationship: HDAC6 overexpression correlated with a better prognosis regardless of the CTCL type. Overexpression of the class II members HDAC4, HDAC5 and HDAC7 has been observed in human solid cancers (Ozdag et al., 2006), whereas low expression profiles of HDAC5 and HDAC10 correlate with a poor prognosis in patients with lung cancer (Osada et al., 2004).
In summary, HDACs are often over‐expressed in human cancers, but the different expression profiles between different tissues and cancer types make it difficult to use them routinely as prognostic biomarkers. Advanced techniques, such as array systems and high throughput analyses, could give a more differentiated picture of the expression profiles within tissues and tumours.
6.3. Response specific biomarkers
There are not many predictive biomarkers known regarding HDAC inhibition in tumours. As already mentioned in section 4.3, one promising candidate could be HR23B, one of the identified genes in a loss of function screen (Fotheringham et al., 2009). It has been demonstrated that CTCL correlates with high HR23B expression (Khan et al., 2010). This study also revealed correlation between reduced HR23B expression in CTCL biopsies and lack of clinical response. This is an indication for HR23B as a predictive biomarker for HDAC inhibitor therapy. It remains to be tested whether this will be the case also for other tissues and malignancies, but it has been reported in a recent study that down‐regulation of HR23B significantly inhibited SAHA‐induced apoptosis in 1/6 SAHA‐sensitive MPM cell lines (Hurwitz et al., 2012).
7. Summary and conclusions
HDAC inhibitors represent a promising group of anti‐cancer agents, with two of them now approved for cutaneous T‐cell lymphoma and one for peripheral T‐cell lymphoma, and many are in clinical trials for various cancer types, both haematological and solid malignancies. Besides the promising effects on haematological cancer cells and the use of VPA in epilepsy and bipolar disorders, there is growing evidence that HDAC inhibitors have other beneficial effects when used as anti‐inflammatory drugs or as therapies for neurodegenerative disorders.
There are many reports of different biological functions of HDACs and therefore consequences of HDAC inhibition. However, many of the underlying mechanisms and functions of HDACs in human cells are poorly understood and remain unclear. Different HDACs are involved in different pathways and functions in the cell. They often occur in complexes and are embedded in a network of interactions, and the possibility of crosstalk between the different pathways is high. With this in mind, more studies are needed to reveal different functions of HDACs and determine their interactors and critical substrates in the cell. This should allow rationally designed therapy in different types of cancers, possibly in combination with other anti‐cancer agents, when the targeted pathway is known for each type of HDAC and each type of cancer. The discovery of biomarkers, both response specific and prognostic, will allow a more personalized treatment and consequently reduce side effects. Methods such as gene arrays or laser dissection in combination with proteomic analyses could lead to more extensive biomarker resolution. Another key question is whether the pan‐HDAC inhibitors or the selective HDAC inhibitors will be more beneficial, alone and in combination with other therapies. The many ongoing clinical studies in combination with basic biological research should enable us to resolve some of these questions soon.
Acknowledgements
The research was supported by MRC, CRUK and LLR. Heidi Olzscha was supported by an EMBO Long‐Term Fellowship and Maria New by a Gordon Piller Studentship from the LLR. We would like to thank Simon Carr for critical reading of the manuscript.
New Maria, Olzscha Heidi, La Thangue Nicholas B., (2012), HDAC inhibitor‐based therapies: Can we interpret the code?, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.09.003.
References
- Adimoolam, S. , Sirisawad, M. , Chen, J. , Thiemann, P. , Ford, J.M. , Buggy, J.J. , 2007. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. U S A. 104, 19482–19487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts, J. , Angibaud, P. , Marien, A. , Floren, W. , Janssens, B. , King, P. , van Dun, J. , Janssen, L. , Geerts, T. , Tuman, R.W. , Johnson, D.L. , Andries, L. , Jung, M. , Janicot, M. , van Emelen, K. , 2007. R306465 is a novel potent inhibitor of class I histone deacetylases with broad-spectrum antitumoral activity against solid and haematological malignancies. Br. J. Cancer. 97, 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badros, A. , Burger, A.M. , Philip, S. , Niesvizky, R. , Kolla, S.S. , Goloubeva, O. , Harris, C. , Zwiebel, J. , Wright, J.J. , Espinoza-Delgado, I. , Baer, M.R. , Holleran, J.L. , Egorin, M.J. , Grant, S. , 2009. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 15, 5250–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist, C.J. , Kastan, M.B. , 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Balch, W.E. , Morimoto, R.I. , Dillin, A. , Kelly, J.W. , 2008. Adapting proteostasis for disease intervention. Science. 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Bali, P. , Pranpat, M. , Bradner, J. , Balasis, M. , Fiskus, W. , Guo, F. , Rocha, K. , Kumaraswamy, S. , Boyapalle, S. , Atadja, P. , Seto, E. , Bhalla, K. , 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280, 26729–26734. [DOI] [PubMed] [Google Scholar]
- Bantscheff, M. , Hopf, C. , Savitski, M.M. , Dittmann, A. , Grandi, P. , Michon, A.M. , Schlegl, J. , Abraham, Y. , Becher, I. , Bergamini, G. , Boesche, M. , Delling, M. , Dumpelfeld, B. , Eberhard, D. , Huthmacher, C. , Mathieson, T. , Poeckel, D. , Reader, V. , Strunk, K. , Sweetman, G. , Kruse, U. , Neubauer, G. , Ramsden, N.G. , Drewes, G. , 2011. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265. [DOI] [PubMed] [Google Scholar]
- Behrends, C. , Sowa, M.E. , Gygi, S.P. , Harper, J.W. , 2010. Network organization of the human autophagy system. Nature. 466, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, G.A. , Finch, J.T. , Lewit-Bentley, A. , Roth, M. , 1984. The crystal structure of the nucleosome core particle by contrast variation. Basic Life Sci. 27, 105–117. [DOI] [PubMed] [Google Scholar]
- Bhalla, S. , Balasubramanian, S. , David, K. , Sirisawad, M. , Buggy, J. , Mauro, L. , Prachand, S. , Miller, R. , Gordon, L.I. , Evens, A.M. , 2009. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin. Cancer Res. 15, 3354–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara, S. , Chyla, B.J. , Amann, J.M. , Knutson, S.K. , Cortez, D. , Sun, Z.W. , Hiebert, S.W. , 2008. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell. 30, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy, G. , Lamark, T. , Brech, A. , Outzen, H. , Perander, M. , Overvatn, A. , Stenmark, H. , Johansen, T. , 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattmann, C. , Oertel, S. , Ehemann, V. , Thiemann, M. , Huber, P.E. , Bischof, M. , Witt, O. , Deubzer, H.E. , Kulozik, A.E. , Debus, J. , Weber, K.J. , 2010. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 78, 237–245. [DOI] [PubMed] [Google Scholar]
- Bode, K.A. , Schroder, K. , Hume, D.A. , Ravasi, T. , Heeg, K. , Sweet, M.J. , Dalpke, A.H. , 2007. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology. 122, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolderson, E. , Richard, D.J. , Zhou, B.B. , Khanna, K.K. , 2009. Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin. Cancer Res. 15, 6314–6320. [DOI] [PubMed] [Google Scholar]
- Bommareddy, A. , Hahm, E.R. , Xiao, D. , Powolny, A.A. , Fisher, A.L. , Jiang, Y. , Singh, S.V. , 2009. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 69, 3704–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbone, E. , Berlingieri, M.T. , De Bellis, F. , Nebbioso, A. , Chiappetta, G. , Mai, A. , Altucci, L. , Fusco, A. , 2010. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene. 29, 105–116. [DOI] [PubMed] [Google Scholar]
- Boumber, Y. , Younes, A. , Garcia-Manero, G. , 2011. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin. Investig. Drugs. 20, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault, C. , Zhang, Y. , Fritah, S. , Caron, C. , Gilquin, B. , Kwon, S.H. , Garrido, C. , Yao, T.P. , Vourc'h, C. , Matthias, P. , Khochbin, S. , 2007. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 21, 2172–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogdon, J.L. , Xu, Y. , Szabo, S.J. , An, S. , Buxton, F. , Cohen, D. , Huang, Q. , 2007. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 109, 1123–1130. [DOI] [PubMed] [Google Scholar]
- Bruzzese, F. , Leone, A. , Rocco, M. , Carbone, C. , Piro, G. , Caraglia, M. , Di Gennaro, E. , Budillon, A. , 2011. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. J. Cell. Physiol. 226, 2378–2390. [DOI] [PubMed] [Google Scholar]
- Buglio, D. , Khaskhely, N.M. , Voo, K.S. , Martinez-Valdez, H. , Liu, Y.J. , Younes, A. , 2011. HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood. 117, 2910–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd, J.C. , Marcucci, G. , Parthun, M.R. , Xiao, J.J. , Klisovic, R.B. , Moran, M. , Lin, T.S. , Liu, S.J. , Sklenar, A.R. , Davis, M.E. , Lucas, D.M. , Fischer, B. , Shank, R. , Tejaswi, S.L. , Binkley, P. , Wright, J. , Chan, K.K. , Grever, M.R. , 2005. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 105, 959–967. [DOI] [PubMed] [Google Scholar]
- Camphausen, K. , Tofilon, P.J. , 2007. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J. Clin. Oncol. 25, 4051–4056. [DOI] [PubMed] [Google Scholar]
- Carew, J.S. , Medina, E.C. , Esquivel, J.A. , Mahalingam, D. , Swords, R. , Kelly, K. , Zhang, H. , Huang, P. , Mita, A.C. , Mita, M.M. , Giles, F.J. , Nawrocki, S.T. , 2010. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 14, 2448–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P. , 2005. Angiogenesis in life, disease and medicine. Nature. 438, 932–936. [DOI] [PubMed] [Google Scholar]
- Cha, T.L. , Chuang, M.J. , Wu, S.T. , Sun, G.H. , Chang, S.Y. , Yu, D.S. , Huang, S.M. , Huan, S.K. , Cheng, T.C. , Chen, T.T. , Fan, P.L. , Hsiao, P.W. , 2009. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin. Cancer Res. 15, 840–850. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki, A. , Guarente, L. , 2012. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 8, 287–296. [DOI] [PubMed] [Google Scholar]
- Chang, H.M. , Paulson, M. , Holko, M. , Rice, C.M. , Williams, B.R. , Marie, I. , Levy, D.E. , 2004. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc. Natl. Acad. Sci. U S A. 101, 9578–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Blanco, A. , Segura-Pacheco, B. , Perez-Cardenas, E. , Taja-Chayeb, L. , Cetina, L. , Candelaria, M. , Cantu, D. , Gonzalez-Fierro, A. , Garcia-Lopez, P. , Zambrano, P. , Perez-Plasencia, C. , Cabrera, G. , Trejo-Becerril, C. , Angeles, E. , Duenas-Gonzalez, A. , 2005. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Mol. Cancer. 4, 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan, P. , Vallabhaneni, G. , Armstrong, E. , Huang, S.M. , Harari, P.M. , 2005. Modulation of radiation response by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 62, 223–229. [DOI] [PubMed] [Google Scholar]
- Choudhary, C. , Kumar, C. , Gnad, F. , Nielsen, M.L. , Rehman, M. , Walther, T.C. , Olsen, J.V. , Mann, M. , 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Coiffier, B. , Pro, B. , Prince, H.M. , Foss, F.M. , Sokol, L. , Greenwood, M. , Caballero, D. , Borchmann, P. , Morschhauser, F. , Wilhelm, M. , Pinter-Brown, L. , Padmanabhan, S. , Shustov, A.R. , Nichols, J. , Carroll, S. , Balser, J. , Horwitz, S.M. , 2011. Analysis of patients with common peripheral T-cell lymphoma subtypes from a phase 2 study of romidepsin in relapsed or refractory peripheral T-cell lymphoma. Blood. 118, 272 [Google Scholar]
- Cramer, T. , Yamanishi, Y. , Clausen, B.E. , Forster, I. , Pawlinski, R. , Mackman, N. , Haase, V.H. , Jaenisch, R. , Corr, M. , Nizet, V. , Firestein, G.S. , Gerber, H.P. , Ferrara, N. , Johnson, R.S. , 2003. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 112, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, C. , Whitesell, L. , Rogers, A.B. , Lindquist, S. , 2007. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 130, 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Rahmani, M. , Dent, P. , Grant, S. , 2005. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell. Biol. 25, 5429–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasmahapatra, G. , Lembersky, D. , Son, M.P. , Attkisson, E. , Dent, P. , Fisher, R.I. , Friedberg, J.W. , Grant, S. , 2011. Carfilzomib interacts synergistically with histone deacetylase inhibitors in mantle cell lymphoma cells in vitro and in vivo. Mol. Cancer Ther. 10, 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- de Bono, J.S. , Kristeleit, R. , Tolcher, A. , Fong, P. , Pacey, S. , Karavasilis, V. , Mita, M. , Shaw, H. , Workman, P. , Kaye, S. , Rowinsky, E.K. , Aherne, W. , Atadja, P. , Scott, J.W. , Patnaik, A. , 2008. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin. Cancer Res. 14, 6663–6673. [DOI] [PubMed] [Google Scholar]
- de Ruijter, A.J. , van Gennip, A.H. , Caron, H.N. , Kemp, S. , van Kuilenburg, A.B. , 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schutter, H. , Kimpe, M. , Isebaert, S. , Nuyts, S. , 2009. A systematic assessment of radiation dose enhancement by 5-Aza-2'-deoxycytidine and histone deacetylase inhibitors in head-and-neck squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 73, 904–912. [DOI] [PubMed] [Google Scholar]
- Deorukhkar, A. , Shentu, S. , Park, H.C. , Diagaradjane, P. , Puduvalli, V. , Aggarwal, B. , Guha, S. , Krishnan, S. , 2010. Inhibition of radiation-induced DNA repair and prosurvival pathways contributes to vorinostat-mediated radiosensitization of pancreatic cancer cells. Pancreas. 39, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequiedt, F. , Kasler, H. , Fischle, W. , Kiermer, V. , Weinstein, M. , Herndier, B.G. , Verdin, E. , 2003. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 18, 687–698. [DOI] [PubMed] [Google Scholar]
- Dequiedt, F. , Van Lint, J. , Lecomte, E. , Van Duppen, V. , Seufferlein, T. , Vandenheede, J.R. , Wattiez, R. , Kettmann, R. , 2005. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J. Exp. Med. 201, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdy, S.C. , Jiang, S. , Zhou, X.C. , Hou, X. , Jin, F. , Podratz, K.C. , Jiang, S.W. , 2006. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 5, 2767–2776. [DOI] [PubMed] [Google Scholar]
- Eot-Houllier, G. , Fulcrand, G. , Magnaghi-Jaulin, L. , Jaulin, C. , 2009. Histone deacetylase inhibitors and genomic instability. Cancer Lett. 274, 169–176. [DOI] [PubMed] [Google Scholar]
- Fantin, V.R. , Loboda, A. , Paweletz, C.P. , Hendrickson, R.C. , Pierce, J.W. , Roth, J.A. , Li, L. , Gooden, F. , Korenchuk, S. , Hou, X.S. , Harrington, E.A. , Randolph, S. , Reilly, J.F. , Ware, C.M. , Kadin, M.E. , Frankel, S.R. , Richon, V.M. , 2008. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 68, 3785–3794. [DOI] [PubMed] [Google Scholar]
- FDA, 2009. FDA Approves Drug Treatment for Rare Cancer http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2009/ucm189629.htm [Google Scholar]
- Feng, R. , Oton, A. , Mapara, M.Y. , Anderson, G. , Belani, C. , Lentzsch, S. , 2007. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br. J. Haematol. 139, 385–397. [DOI] [PubMed] [Google Scholar]
- Finnin, M.S. , Donigian, J.R. , Cohen, A. , Richon, V.M. , Rifkind, R.A. , Marks, P.A. , Breslow, R. , Pavletich, N.P. , 1999. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Forsythe, J.A. , Jiang, B.H. , Iyer, N.V. , Agani, F. , Leung, S.W. , Koos, R.D. , Semenza, G.L. , 1996. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, S.L. , Hargreaves, D.C. , Medzhitov, R. , 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 447, 972–978. [DOI] [PubMed] [Google Scholar]
- Fotheringham, S. , Epping, M.T. , Stimson, L. , Khan, O. , Wood, V. , Pezzella, F. , Bernards, R. , La Thangue, N.B. , 2009. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 15, 57–66. [DOI] [PubMed] [Google Scholar]
- Fournel, M. , Bonfils, C. , Hou, Y. , Yan, P.T. , Trachy-Bourget, M.C. , Kalita, A. , Liu, J. , Lu, A.H. , Zhou, N.Z. , Robert, M.F. , Gillespie, J. , Wang, J.J. , Ste-Croix, H. , Rahil, J. , Lefebvre, S. , Moradei, O. , Delorme, D. , Macleod, A.R. , Besterman, J.M. , Li, Z. , 2008. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 7, 759–768. [DOI] [PubMed] [Google Scholar]
- Frew, A.J. , Lindemann, R.K. , Martin, B.P. , Clarke, C.J. , Sharkey, J. , Anthony, D.A. , Banks, K.M. , Haynes, N.M. , Gangatirkar, P. , Stanley, K. , Bolden, J.E. , Takeda, K. , Yagita, H. , Secrist, J.P. , Smyth, M.J. , Johnstone, R.W. , 2008. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. U S A. 105, 11317–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda, S. , 2008. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets. 8, 132–140. [DOI] [PubMed] [Google Scholar]
- Gao, Y.S. , Hubbert, C.C. , Lu, J. , Lee, Y.S. , Lee, J.Y. , Yao, T.P. , 2007. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell. Biol. 27, 8637–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero, G. , Yang, H. , Bueso-Ramos, C. , Ferrajoli, A. , Cortes, J. , Wierda, W.G. , Faderl, S. , Koller, C. , Morris, G. , Rosner, G. , Loboda, A. , Fantin, V.R. , Randolph, S.S. , Hardwick, J.S. , Reilly, J.F. , Chen, C. , Ricker, J.L. , Secrist, J.P. , Richon, V.M. , Frankel, S.R. , Kantarjian, H.M. , 2008. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 111, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Glaser, K.B. , Staver, M.J. , Waring, J.F. , Stender, J. , Ulrich, R.G. , Davidsen, S.K. , 2003. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2, 151–163. [PubMed] [Google Scholar]
- Gore, S.D. , Baylin, S. , Sugar, E. , Carraway, H. , Miller, C.B. , Carducci, M. , Grever, M. , Galm, O. , Dauses, T. , Karp, J.E. , Rudek, M.A. , Zhao, M. , Smith, B.D. , Manning, J. , Jiemjit, A. , Dover, G. , Mays, A. , Zwiebel, J. , Murgo, A. , Weng, L.J. , Herman, J.G. , 2006. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 66, 6361–6369. [DOI] [PubMed] [Google Scholar]
- Gottlicher, M. , Minucci, S. , Zhu, P. , Kramer, O.H. , Schimpf, A. , Giavara, S. , Sleeman, J.P. , Lo Coco, F. , Nervi, C. , Pelicci, P.G. , Heinzel, T. , 2001. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson, D.R. , Kundakovic, M. , Sharma, R.P. , 2010. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders?. Mol. Pharmacol. 77, 126–135. [DOI] [PubMed] [Google Scholar]
- Gregoretti, I.V. , Lee, Y.M. , Goodson, H.V. , 2004. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31. [DOI] [PubMed] [Google Scholar]
- Guerrant, W. , Patil, V. , Canzoneri, J.C. , Oyelere, A.K. , 2012. Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. J. Med. Chem. 55, 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J.Y. , Chen, H.Y. , Mathew, R. , Fan, J. , Strohecker, A.M. , Karsli-Uzunbas, G. , Kamphorst, J.J. , Chen, G. , Lemons, J.M. , Karantza, V. , Coller, H.A. , Dipaola, R.S. , Gelinas, C. , Rabinowitz, J.D. , White, E. , 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland, M. , Johnson, A. , Mokalled, M.H. , Montgomery, R.L. , Olson, E.N. , 2009. Genetic dissection of histone deacetylase requirement in tumor cells. Proc. Natl. Acad. Sci. U S A. 106, 7751–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis, M.C. , Sinclair, D.A. , 2010. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halili, M.A. , Andrews, M.R. , Labzin, L.I. , Schroder, K. , Matthias, G. , Cao, C. , Lovelace, E. , Reid, R.C. , Le, G.T. , Hume, D.A. , Irvine, K.M. , Matthias, P. , Fairlie, D.P. , Sweet, M.J. , 2010. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 87, 1103–1114. [DOI] [PubMed] [Google Scholar]
- Harrison, S.J. , Quach, H. , Yuen, K. , Strayer, A. , Copeman, M.C. , Peinert, S. , Bishton, M. , Wolf, M. , Januszewicz, H. , Kencaly, M. , Herbert, K. , Westerman, D. , Carney, D. , Seymour, J.F. , Johnstone, R.W. , Ritchie, D. , Prince, M. , 2008. High response rates with the combination of bortezomib, dexamethasone and the pan-histone deacetylase inhibitor romidepsin in patients with relapsed or refractory multiple myeloma in a phase I/II clinical trial. Blood. 112, 1267 [Google Scholar]
- Hartl, F.U. , Bracher, A. , Hayer-Hartl, M. , 2011. Molecular chaperones in protein folding and proteostasis. Nature. 475, 324–332. [DOI] [PubMed] [Google Scholar]
- He, C. , Klionsky, D.J. , 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippert, M.M. , O'Toole, P.S. , Thorburn, A. , 2006. Autophagy in cancer: good, bad, or both?. Cancer Res. 66, 9349–9351. [DOI] [PubMed] [Google Scholar]
- Hrebackova, J. , Hrabeta, J. , Eckschlager, T. , 2010. Valproic acid in the complex therapy of malignant tumors. Curr. Drug Targets. 11, 361–379. [DOI] [PubMed] [Google Scholar]
- Hrzenjak, A. , Kremser, M.L. , Strohmeier, B. , Moinfar, F. , Zatloukal, K. , Denk, H. , 2008. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 216, 495–504. [DOI] [PubMed] [Google Scholar]
- Huang, X. , Wang, S. , Lee, C.K. , Yang, X. , Liu, B. , 2011. HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett. 307, 72–79. [DOI] [PubMed] [Google Scholar]
- Hurwitz, J.L. , Stasik, I. , Kerr, E.M. , Holohan, C. , Redmond, K.M. , McLaughlin, K.M. , Busacca, S. , Barbone, D. , Broaddus, V.C. , Gray, S.G. , O'Byrne, K.J. , Johnston, P.G. , Fennell, D.A. , Longley, D.B. , 2012. Vorinostat/SAHA-induced apoptosis in malignant mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. Eur. J. Cancer. 48, 1096–1107. [DOI] [PubMed] [Google Scholar]
- Inche, A.G. , La Thangue, N.B. , 2006. Chromatin control and cancer-drug discovery: realizing the promise. Drug Discov. Today. 11, 97–109. [DOI] [PubMed] [Google Scholar]
- Into, T. , Inomata, M. , Niida, S. , Murakami, Y. , Shibata, K. , 2010. Regulation of MyD88 aggregation and the MyD88-dependent signaling pathway by sequestosome 1 and histone deacetylase 6. J. Biol. Chem. 285, 35759–35769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, S.P. , Bartek, J. , 2009. The DNA-damage response in human biology and disease. Nature. 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku, F. , McConkey, D.J. , Hong, D.S. , Kurzrock, R. , 2011. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 8, 528–539. [DOI] [PubMed] [Google Scholar]
- Johnston, J.A. , Ward, C.L. , Kopito, R.R. , 1998. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachhap, S.K. , Rosmus, N. , Collis, S.J. , Kortenhorst, M.S. , Wissing, M.D. , Hedayati, M. , Shabbeer, S. , Mendonca, J. , Deangelis, J. , Marchionni, L. , Lin, J. , Hoti, N. , Nortier, J.W. , DeWeese, T.L. , Hammers, H. , Carducci, M.A. , 2010. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS One. 5, e11208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalac, M. , Scotto, L. , Marchi, E. , Amengual, J. , Seshan, V.E. , Bhagat, G. , Ulahannan, N. , Leshchenko, V.V. , Temkin, A.M. , Parekh, S. , Tycko, B. , O'Connor, O.A. , 2011. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 118, 5506–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, Y. , Kovacs, J.J. , McLaurin, A. , Vance, J.M. , Ito, A. , Yao, T.P. , 2003. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 115, 727–738. [DOI] [PubMed] [Google Scholar]
- Khan, O. , Fotheringham, S. , Wood, V. , Stimson, L. , Zhang, C. , Pezzella, F. , Duvic, M. , Kerr, D.J. , La Thangue, N.B. , 2010. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc. Natl. Acad. Sci. U S A. 107, 6532–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, O. , La Thangue, N.B. , 2008. Drug insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 5, 714–726. [DOI] [PubMed] [Google Scholar]
- Khan, O. , La Thangue, N.B. , 2012. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 90, 85–94. [DOI] [PubMed] [Google Scholar]
- Kim, M.S. , Kwon, H.J. , Lee, Y.M. , Baek, J.H. , Jang, J.E. , Lee, S.W. , Moon, E.J. , Kim, H.S. , Lee, S.K. , Chung, H.Y. , Kim, C.W. , Kim, K.W. , 2001. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 7, 437–443. [DOI] [PubMed] [Google Scholar]
- Kim, S.H. , Jeong, J.W. , Park, J.A. , Lee, J.W. , Seo, J.H. , Jung, B.K. , Bae, M.K. , Kim, K.W. , 2007. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 17, 647–651. [PubMed] [Google Scholar]
- Kirschbaum, M.H. , Goldman, B.H. , Zain, J.M. , Cook, J.R. , Rimsza, L.M. , Forman, S.J. , Fisher, R.I. , 2007. Vorinostat (suberoylanilide hydroxamic acid) in relapsed or refractory Hodgkin lymphoma: SWOG 0517. Blood. 110, 759a [Google Scholar]
- Kirschbaum, M.H. , Zain, J.M. , Popplewell, L. , Pullarkat, V. , Nademanee, A.P. , Delioukina, M.L. , Pulone, B. , Matsuoka, D. , Frankel, P. , James, Z.A. , Forman, S.J. , Newman, E. , Gandara, D. , 2007. A phase 2 study of vorinostat (suberoylanilide hydroxamic acid, SAHA) in relapsed or refractory indolent non Hodgkin lymphoma. A California cancer consortium study. Blood. 110, 757a [Google Scholar]
- Klampfer, L. , Huang, J. , Swaby, L.A. , Augenlicht, L. , 2004. Requirement of histone deacetylase activity for signaling by STAT1. J. Biol. Chem. 279, 30358–30368. [DOI] [PubMed] [Google Scholar]
- Klimek, V.M. , Fircanis, S. , Maslak, P. , Guernah, I. , Baum, M. , Wu, N. , Panageas, K. , Wright, J.J. , Pandolfi, P.P. , Nimer, S.D. , 2008. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin. Cancer Res. 14, 826–832. [DOI] [PubMed] [Google Scholar]
- Kong, X. , Fang, M. , Li, P. , Fang, F. , Xu, Y. , 2009. HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J. Mol. Cell. Cardiol. 46, 292–299. [DOI] [PubMed] [Google Scholar]
- Kong, X. , Lin, Z. , Liang, D. , Fath, D. , Sang, N. , Caro, J. , 2006. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 26, 2019–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos, P.A. , Vandoros, G.P. , Papavassiliou, A.G. , 2006. FK228 (depsipeptide): a HDAC inhibitor with pleiotropic antitumor activities. Cancer Chemother. Pharmacol. 58, 711–715. [DOI] [PubMed] [Google Scholar]
- Kotian, S. , Liyanarachchi, S. , Zelent, A. , Parvin, J.D. , 2011. Histone deacetylases 9 and 10 are required for homologous recombination. J. Biol. Chem. 286, 7722–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, J.J. , Murphy, P.J. , Gaillard, S. , Zhao, X. , Wu, J.T. , Nicchitta, C.V. , Yoshida, M. , Toft, D.O. , Pratt, W.B. , Yao, T.P. , 2005. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell. 18, 601–607. [DOI] [PubMed] [Google Scholar]
- Kramer, O.H. , Knauer, S.K. , Greiner, G. , Jandt, E. , Reichardt, S. , Guhrs, K.H. , Stauber, R.H. , Bohmer, F.D. , Heinzel, T. , 2009. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 23, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusche, C.A. , Wulfing, P. , Kersting, C. , Vloet, A. , Bocker, W. , Kiesel, L. , Beier, H.M. , Alfer, J. , 2005. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res. Treat. 90, 15–23. [DOI] [PubMed] [Google Scholar]
- Kuendgen, A. , Schmid, M. , Schlenk, R. , Knipp, S. , Hildebrandt, B. , Steidl, C. , Germing, U. , Haas, R. , Dohner, H. , Gattermann, N. , 2006. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 106, 112–119. [DOI] [PubMed] [Google Scholar]
- La Thangue, N.B. , Kerr, D.J. , 2011. Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat. Rev. Clin. Oncol. 8, 587–596. [DOI] [PubMed] [Google Scholar]
- Lee, J.Y. , Koga, H. , Kawaguchi, Y. , Tang, W. , Wong, E. , Gao, Y.S. , Pandey, U.B. , Kaushik, S. , Tresse, E. , Lu, J. , Taylor, J.P. , Cuervo, A.M. , Yao, T.P. , 2010. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y.S. , Lim, K.H. , Guo, X. , Kawaguchi, Y. , Gao, Y. , Barrientos, T. , Ordentlich, P. , Wang, X.F. , Counter, C.M. , Yao, T.P. , 2008. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 68, 7561–7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni, F. , Fossati, G. , Lewis, E.C. , Lee, J.K. , Porro, G. , Pagani, P. , Modena, D. , Moras, M.L. , Pozzi, P. , Reznikov, L.L. , Siegmund, B. , Fantuzzi, G. , Dinarello, C.A. , Mascagni, P. , 2005. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhang, X. , Polakiewicz, R.D. , Yao, T.P. , Comb, M.J. , 2008. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 283, 12686–12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, Z.R. , Xu, Y.F. , Wang, X.B. , Gong, J.P. , Liu, Z.J. , 2012. Suppression of histone deacetylase 11 promotes expression of IL-10 in kupffer cells and induces tolerance following orthotopic liver transplantation in rats. J. Surg. Res. 174, 359–368. [DOI] [PubMed] [Google Scholar]
- Liu, T. , Liu, P.Y. , Marshall, G.M. , 2009. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 69, 1702–1705. [DOI] [PubMed] [Google Scholar]
- Lorin, S. , Borges, A. , Ribeiro Dos Santos, L. , Souquere, S. , Pierron, G. , Ryan, K.M. , Codogno, P. , Djavaheri-Mergny, M. , 2009. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res. 69, 6924–6931. [DOI] [PubMed] [Google Scholar]
- Lu, J. , Sun, H. , Wang, X. , Liu, C. , Xu, X. , Li, F. , Huang, B. , 2005. Interleukin-12 p40 promoter activity is regulated by the reversible acetylation mediated by HDAC1 and p300. Cytokine. 31, 46–51. [DOI] [PubMed] [Google Scholar]
- Maiuri, M.C. , Zalckvar, E. , Kimchi, A. , Kroemer, G. , 2007. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752. [DOI] [PubMed] [Google Scholar]
- Marchion, D.C. , Bicaku, E. , Turner, J.G. , Schmitt, M.L. , Morelli, D.R. , Munster, P.N. , 2009. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol. Cancer Ther. 8, 794–801. [DOI] [PubMed] [Google Scholar]
- Marks, P.A. , 2010. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs. 19, 1049–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquard, L. , Gjerdrum, L.M. , Christensen, I.J. , Jensen, P.B. , Sehested, M. , Ralfkiaer, E. , 2008. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology. 53, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas, M.A. , Bauer, U.M. , Nielsen, S.J. , Brehm, A. , Kouzarides, T. , 2000. Regulation of E2F1 activity by acetylation. EMBO J. 19, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell, P.H. , Dachs, G.U. , Gleadle, J.M. , Nicholls, L.G. , Harris, A.L. , Stratford, I.J. , Hankinson, O. , Pugh, C.W. , Ratcliffe, P.J. , 1997. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. U S A. 94, 8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell, P.H. , Wiesener, M.S. , Chang, G.W. , Clifford, S.C. , Vaux, E.C. , Cockman, M.E. , Wykoff, C.C. , Pugh, C.W. , Maher, E.R. , Ratcliffe, P.J. , 1999. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 399, 271–275. [DOI] [PubMed] [Google Scholar]
- Miller, K.M. , Tjeertes, J.V. , Coates, J. , Legube, G. , Polo, S.E. , Britton, S. , Jackson, S.P. , 2010. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiya, Y. , Ono, T. , Saito, H. , Takahashi, N. , Ito, M. , Mitsui, M. , Motoyama, S. , Ogawa, J. , 2011. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer. 74, 300–304. [DOI] [PubMed] [Google Scholar]
- Miyake, K. , Yoshizumi, T. , Imura, S. , Sugimoto, K. , Batmunkh, E. , Kanemura, H. , Morine, Y. , Shimada, M. , 2008. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas. 36, e1–e9. [DOI] [PubMed] [Google Scholar]
- Moehler, T.M. , Neben, K. , Ho, A.D. , Goldschmidt, H. , 2001. Angiogenesis in hematologic malignancies. Ann. Hematol. 80, 695–705. [DOI] [PubMed] [Google Scholar]
- Mohammed, T.A. , Holen, K.D. , Jaskula-Sztul, R. , Mulkerin, D. , Lubner, S.J. , Schelman, W.R. , Eickhoff, J. , Chen, H. , Loconte, N.K. , 2011. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist. 16, 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molife, L.R. , Attard, G. , Fong, P.C. , Karavasilis, V. , Reid, A.H. , Patterson, S. , Riggs, C.E. , Higano, C. , Stadler, W.M. , McCulloch, W. , Dearnaley, D. , Parker, C. , de Bono, J.S. , 2010. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 21, 109–113. [DOI] [PubMed] [Google Scholar]
- Moniot, S. , Weyand, M. , Steegborn, C. , 2012. Structures, substrates, and regulators of Mammalian sirtuins – opportunities and challenges for drug development. Front. Pharmacol. 3, 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery, R.L. , Davis, C.A. , Potthoff, M.J. , Haberland, M. , Fielitz, J. , Qi, X. , Hill, J.A. , Richardson, J.A. , Olson, E.N. , 2007. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli, E. , Tasdemir, E. , Maiuri, M.C. , Galluzzi, L. , Kepp, O. , Criollo, A. , Vicencio, J.M. , Soussi, T. , Kroemer, G. , 2008. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 7, 3056–3061. [DOI] [PubMed] [Google Scholar]
- Mottet, D. , Pirotte, S. , Lamour, V. , Hagedorn, M. , Javerzat, S. , Bikfalvi, A. , Bellahcene, A. , Verdin, E. , Castronovo, V. , 2009. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 28, 243–256. [DOI] [PubMed] [Google Scholar]
- Mueller, S. , Yang, X. , Sottero, T.L. , Gragg, A. , Prasad, G. , Polley, M.Y. , Weiss, W.A. , Matthay, K.K. , Davidoff, A.M. , DuBois, S.G. , Haas-Kogan, D.A. , 2011. Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: efficacy and underlying mechanisms. Cancer Lett. 306, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster, P.N. , Daud, A.I. , 2011. Preclinical and clinical activity of the topoisomerase I inhibitor, karenitecin, in melanoma. Expert Opin. Investig. Drugs. 20, 1565–1574. [DOI] [PubMed] [Google Scholar]
- Nakagawa, M. , Oda, Y. , Eguchi, T. , Aishima, S. , Yao, T. , Hosoi, F. , Basaki, Y. , Ono, M. , Kuwano, M. , Tanaka, M. , Tsuneyoshi, M. , 2007. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 18, 769–774. [PubMed] [Google Scholar]
- Nebbioso, A. , Clarke, N. , Voltz, E. , Germain, E. , Ambrosino, C. , Bontempo, P. , Alvarez, R. , Schiavone, E.M. , Ferrara, F. , Bresciani, F. , Weisz, A. , de Lera, A.R. , Gronemeyer, H. , Altucci, L. , 2005. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 11, 77–84. [DOI] [PubMed] [Google Scholar]
- Nguyen, T. , Dai, Y. , Attkisson, E. , Kramer, L. , Jordan, N. , Nguyen, N. , Kolluri, N. , Muschen, M. , Grant, S. , 2011. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin. Cancer Res. 17, 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesvizky, R. , Ely, S. , Mark, T. , Aggarwal, S. , Gabrilove, J.L. , Wright, J.J. , Chen-Kiang, S. , Sparano, J.A. , 2011. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 117, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmanapalli, R. , Fuino, L. , Bali, P. , Gasparetto, M. , Glozak, M. , Tao, J. , Moscinski, L. , Smith, C. , Wu, J. , Jove, R. , Atadja, P. , Bhalla, K. , 2003. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 63, 5126–5135. [PubMed] [Google Scholar]
- Nolan, L. , Johnson, P.W. , Ganesan, A. , Packham, G. , Crabb, S.J. , 2008. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential?. Br. J. Cancer. 99, 689–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noro, R. , Miyanaga, A. , Minegishi, Y. , Okano, T. , Seike, M. , Soeno, C. , Kataoka, K. , Matsuda, K. , Yoshimura, A. , Gemma, A. , 2010. Histone deacetylase inhibitor enhances sensitivity of non-small-cell lung cancer cells to 5-FU/S-1 via down-regulation of thymidylate synthase expression and up-regulation of p21(waf1/cip1) expression. Cancer Sci. 101, 1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinzon, I. , Horvath, C.M. , 2006. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol. Cell. Biol. 26, 3106–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, O.A. , Heaney, M.L. , Schwartz, L. , Richardson, S. , Willim, R. , MacGregor-Cortelli, B. , Curly, T. , Moskowitz, C. , Portlock, C. , Horwitz, S. , Zelenetz, A.D. , Frankel, S. , Richon, V. , Marks, P. , Kelly, W.K. , 2006. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 24, 166–173. [DOI] [PubMed] [Google Scholar]
- Oehme, I. , Deubzer, H.E. , Lodrini, M. , Milde, T. , Witt, O. , 2009. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin. Investig. Drugs. 18, 1605–1617. [DOI] [PubMed] [Google Scholar]
- Olzmann, J.A. , Li, L. , Chudaev, M.V. , Chen, J. , Perez, F.A. , Palmiter, R.D. , Chin, L.S. , 2007. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada, H. , Tatematsu, Y. , Saito, H. , Yatabe, Y. , Mitsudomi, T. , Takahashi, T. , 2004. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer. 112, 26–32. [DOI] [PubMed] [Google Scholar]
- Ozdag, H. , Teschendorff, A.E. , Ahmed, A.A. , Hyland, S.J. , Blenkiron, C. , Bobrow, L. , Veerakumarasivam, A. , Burtt, G. , Subkhankulova, T. , Arends, M.J. , Collins, V.P. , Bowtell, D. , Kouzarides, T. , Brenton, J.D. , Caldas, C. , 2006. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 7, 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J.H. , Kim, S.H. , Choi, M.C. , Lee, J. , Oh, D.Y. , Im, S.A. , Bang, Y.J. , Kim, T.Y. , 2008. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 368, 318–322. [DOI] [PubMed] [Google Scholar]
- Petruccelli, L.A. , Dupere-Richer, D. , Pettersson, F. , Retrouvey, H. , Skoulikas, S. , Miller, W.H. , 2011. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One. 6, e20987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyssonnaux, C. , Cejudo-Martin, P. , Doedens, A. , Zinkernagel, A.S. , Johnson, R.S. , Nizet, V. , 2007. Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 178, 7516–7519. [DOI] [PubMed] [Google Scholar]
- Piekarz, R.L. , Frye, R. , Prince, H.M. , Kirschbaum, M.H. , Zain, J. , Allen, S.L. , Jaffe, E.S. , Ling, A. , Turner, M. , Peer, C.J. , Figg, W.D. , Steinberg, S.M. , Smith, S. , Joske, D. , Lewis, I. , Hutchins, L. , Craig, M. , Fojo, A.T. , Wright, J.J. , Bates, S.E. , 2011. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 117, 5827–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pili, R. , Salumbides, B. , Zhao, M. , Altiok, S. , Qian, D. , Zwiebel, J. , Carducci, M.A. , Rudek, M.A. , 2012. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer. 106, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb, J.A. , Finn, P.W. , Williams, R.J. , Bandara, M.J. , Romero, M.R. , Watkins, C.J. , La Thangue, N.B. , Brown, R. , 2003. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2, 721–728. [PubMed] [Google Scholar]
- Pohlman, B. , Advani, R. , Duvic, M. , Hymes, K.B. , Intragumtornchai, T. , Lekhakula, A. , Shpilberg, O. , Lerner, A. , Ben-Yehuda, D. , Beylot-Barry, M. , Hillen, U. , Fagerberg, J. , Foss, F.M. , 2009. Final results of a phase II trial of belinostat (PXD101) in patients with recurrent or refractory peripheral or cutaneous T-cell lymphoma. Blood. 114, 379 [DOI] [PubMed] [Google Scholar]
- Qian, D.Z. , Kachhap, S.K. , Collis, S.J. , Verheul, H.M. , Carducci, M.A. , Atadja, P. , Pili, R. , 2006. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 66, 8814–8821. [DOI] [PubMed] [Google Scholar]
- Ramalingam, S.S. , Maitland, M.L. , Frankel, P. , Argiris, A.E. , Koczywas, M. , Gitlitz, B. , Thomas, S. , Espinoza-Delgado, I. , Vokes, E.E. , Gandara, D.R. , Belani, C.P. , 2010. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 28, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, R. , Fiskus, W. , Yang, Y. , Lee, P. , Joshi, R. , Fernandez, P. , Mandawat, A. , Atadja, P. , Bradner, J.E. , Bhalla, K. , 2008. HDAC6 inhibition enhances 17-AAG – mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood. 112, 1886–1893. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B. , Vacher, C. , Berger, Z. , Davies, J.E. , Luo, S. , Oroz, L.G. , Scaravilli, F. , Easton, D.F. , Duden, R. , O'Kane, C.J. , Rubinsztein, D.C. , 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. [DOI] [PubMed] [Google Scholar]
- Richardson, P. , Mitsiades, C. , Colson, K. , Reilly, E. , McBride, L. , Chiao, J. , Sun, L. , Ricker, J. , Rizvi, S. , Oerth, C. , Atkins, B. , Fearen, I. , Anderson, K. , Siegel, D. , 2008. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk. Lymphoma. 49, 502–507. [DOI] [PubMed] [Google Scholar]
- Richon, V.M. , Garcia-Vargas, J. , Hardwick, J.S. , 2009. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 280, 201–210. [DOI] [PubMed] [Google Scholar]
- Rikiishi, H. , 2011. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J. Biomed. Biotechnol. 2011, 830260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rocha, H. , Garcia-Garcia, A. , Panayiotidis, M.I. , Franco, R. , 2011. DNA damage and autophagy. Mutat. Res. 711, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger, T. , Lugrin, J. , Le Roy, D. , Goy, G. , Mombelli, M. , Koessler, T. , Ding, X.C. , Chanson, A.L. , Reymond, M.K. , Miconnet, I. , Schrenzel, J. , Francois, P. , Calandra, T. , 2011. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 117, 1205–1217. [DOI] [PubMed] [Google Scholar]
- Ropero, S. , Fraga, M.F. , Ballestar, E. , Hamelin, R. , Yamamoto, H. , Boix-Chornet, M. , Caballero, R. , Alaminos, M. , Setien, F. , Paz, M.F. , Herranz, M. , Palacios, J. , Arango, D. , Orntoft, T.F. , Aaltonen, L.A. , Schwartz, S. , Esteller, M. , 2006. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 38, 566–569. [DOI] [PubMed] [Google Scholar]
- Rosato, R.R. , Almenara, J.A. , Dai, Y. , Grant, S. , 2003. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2, 1273–1284. [PubMed] [Google Scholar]
- Rubinsztein, D.C. , 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 443, 780–786. [DOI] [PubMed] [Google Scholar]
- Sasaki, H. , Moriyama, S. , Nakashima, Y. , Kobayashi, Y. , Kiriyama, M. , Fukai, I. , Yamakawa, Y. , Fujii, Y. , 2004. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer. 46, 171–178. [DOI] [PubMed] [Google Scholar]
- Satoh, A. , Stein, L. , Imai, S. , 2011. The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb. Exp. Pharmacol. 206, 125–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza, G.L. , 2003. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 3, 721–732. [DOI] [PubMed] [Google Scholar]
- Shao, Y. , Gao, Z. , Marks, P.A. , Jiang, X. , 2004. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U S A. 101, 18030–18035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom, T. , Jones, S. , Wood, L.D. , Parsons, D.W. , Lin, J. , Barber, T.D. , Mandelker, D. , Leary, R.J. , Ptak, J. , Silliman, N. , Szabo, S. , Buckhaults, P. , Farrell, C. , Meeh, P. , Markowitz, S.D. , Willis, J. , Dawson, D. , Willson, J.K. , Gazdar, A.F. , Hartigan, J. , Wu, L. , Liu, C. , Parmigiani, G. , Park, B.H. , Bachman, K.E. , Papadopoulos, N. , Vogelstein, B. , Kinzler, K.W. , Velculescu, V.E. , 2006. The consensus coding sequences of human breast and colorectal cancers. Science. 314, 268–274. [DOI] [PubMed] [Google Scholar]
- Stadler, W.M. , Margolin, K. , Ferber, S. , McCulloch, W. , Thompson, J.A. , 2006. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer. 5, 57–60. [DOI] [PubMed] [Google Scholar]
- Steele, N.L. , Plumb, J.A. , Vidal, L. , Tjornelund, J. , Knoblauch, P. , Rasmussen, A. , Ooi, C.E. , Buhl-Jensen, P. , Brown, R. , Evans, T.R. , DeBono, J.S. , 2008. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res. 14, 804–810. [DOI] [PubMed] [Google Scholar]
- Stevens, F.E. , Beamish, H. , Warrener, R. , Gabrielli, B. , 2008. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 27, 1345–1354. [DOI] [PubMed] [Google Scholar]
- Stimson, L. , La Thangue, N.B. , 2009. Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett. 280, 177–183. [DOI] [PubMed] [Google Scholar]
- Stimson, L. , Wood, V. , Khan, O. , Fotheringham, S. , La Thangue, N.B. , 2009. HDAC inhibitor-based therapies and haematological malignancy. Ann. Oncol. 20, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Sudo, T. , Mimori, K. , Nishida, N. , Kogo, R. , Iwaya, T. , Tanaka, F. , Shibata, K. , Fujita, H. , Shirouzu, K. , Mori, M. , 2011. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 26, 777–782. [DOI] [PubMed] [Google Scholar]
- Sun, X. , Wei, L. , Chen, Q. , Terek, R.M. , 2009. HDAC4 represses vascular endothelial growth factor expression in chondrosarcoma by modulating RUNX2 activity. J. Biol. Chem. 284, 21881–21890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei, A. , Maison, C. , Roche, D. , Almouzni, G. , 2001. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat. Cell Biol. 3, 114–120. [DOI] [PubMed] [Google Scholar]
- Tan, J.M. , Wong, E.S. , Kirkpatrick, D.S. , Pletnikova, O. , Ko, H.S. , Tay, S.P. , Ho, M.W. , Troncoso, J. , Gygi, S.P. , Lee, M.K. , Dawson, V.L. , Dawson, T.M. , Lim, K.L. , 2008. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439. [DOI] [PubMed] [Google Scholar]
- Tang, X. , Gao, J.S. , Guan, Y.J. , McLane, K.E. , Yuan, Z.L. , Ramratnam, B. , Chin, Y.E. , 2007. Acetylation-dependent signal transduction for type I interferon receptor. Cell. 131, 93–105. [DOI] [PubMed] [Google Scholar]
- Theocharis, S. , Klijanienko, J. , Giaginis, C. , Rodriguez, J. , Jouffroy, T. , Girod, A. , Alexandrou, P. , Sastre-Garau, X. , 2011. Histone deacetylase-1 and -2 expression in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients survival. J. Oral Pathol. Med. 40, 706–714. [DOI] [PubMed] [Google Scholar]
- Tsai, H.C. , Wei, K.C. , Tsai, C.N. , Huang, Y.C. , Chen, P.Y. , Chen, S.M. , Lu, Y.J. , Lee, S.T. , 2011. Effect of valproic acid on the outcome of glioblastoma multiforme. Br. J. Neurosurg. 26, 347–354. [DOI] [PubMed] [Google Scholar]
- Urbich, C. , Rossig, L. , Kaluza, D. , Potente, M. , Boeckel, J.N. , Knau, A. , Diehl, F. , Geng, J.G. , Hofmann, W.K. , Zeiher, A.M. , Dimmeler, S. , 2009. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 113, 5669–5679. [DOI] [PubMed] [Google Scholar]
- Vega, R.B. , Matsuda, K. , Oh, J. , Barbosa, A.C. , Yang, X. , Meadows, E. , McAnally, J. , Pomajzl, C. , Shelton, J.M. , Richardson, J.A. , Karsenty, G. , Olson, E.N. , 2004. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 119, 555–566. [DOI] [PubMed] [Google Scholar]
- Verdin, E. , Dequiedt, F. , Kasler, H.G. , 2003. Class II histone deacetylases: versatile regulators. Trends Genet. 19, 286–293. [DOI] [PubMed] [Google Scholar]
- Verma, R. , Oania, R. , Graumann, J. , Deshaies, R.J. , 2004. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell. 118, 99–110. [DOI] [PubMed] [Google Scholar]
- Wang, Z.H. , Xu, L. , Duan, Z.L. , Zeng, L.Q. , Yan, N.H. , Peng, Z.L. , 2007. Beclin 1-mediated macroautophagy involves regulation of caspase-9 expression in cervical cancer HeLa cells. Gynecol. Oncol. 107, 107–113. [DOI] [PubMed] [Google Scholar]
- Webb, J.L. , Ravikumar, B. , Atkins, J. , Skepper, J.N. , Rubinsztein, D.C. , 2003. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. [DOI] [PubMed] [Google Scholar]
- Weber, D. , Badros, A.Z. , Jagannath, S. , Siegel, D. , Richon, V. , Rizvi, S. , Garcia-Vargas, J. , Reiser, D. , Anderson, K.C. , 2008. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: early clinical experience. Blood. 112, 322 [Google Scholar]
- Westerheide, S.D. , Anckar, J. , Stevens, S.M. , Sistonen, L. , Morimoto, R.I. , 2009. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 323, 1063–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, E. , DiPaola, R.S. , 2009. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 15, 5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead, R.P. , Rankin, C. , Hoff, P.M. , Gold, P.J. , Billingsley, K.G. , Chapman, R.A. , Wong, L. , Ward, J.H. , Abbruzzese, J.L. , Blanke, C.D. , 2009. Phase II trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: a Southwest Oncology Group study (S0336). Invest. New Drugs. 27, 469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, A.J. , Byun, D.S. , Nasser, S. , Murray, L.B. , Ayyanar, K. , Arango, D. , Figueroa, M. , Melnick, A. , Kao, G.D. , Augenlicht, L.H. , Mariadason, J.M. , 2008. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell. 19, 4062–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, A.J. , Byun, D.S. , Popova, N. , Murray, L.B. , L'Italien, K. , Sowa, Y. , Arango, D. , Velcich, A. , Augenlicht, L.H. , Mariadason, J.M. , 2006. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 281, 13548–13558. [DOI] [PubMed] [Google Scholar]
- Wilting, R.H. , Yanover, E. , Heideman, M.R. , Jacobs, H. , Horner, J. , van der Torre, J. , DePinho, R.A. , Dannenberg, J.H. , 2010. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 29, 2586–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt, O. , Deubzer, H.E. , Milde, T. , Oehme, I. , 2009. HDAC family: what are the cancer relevant targets?. Cancer Lett. 277, 8–21. [DOI] [PubMed] [Google Scholar]
- Wu, L.M. , Yang, Z. , Zhou, L. , Zhang, F. , Xie, H.Y. , Feng, X.W. , Wu, J. , Zheng, S.S. , 2010. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS One. 5, e14460 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu, J. , Zhou, J.Y. , Wei, W.Z. , Philipsen, S. , Wu, G.S. , 2008. Sp1-mediated TRAIL induction in chemosensitization. Cancer Res. 68, 6718–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W.S. , Parmigiani, R.B. , Marks, P.A. , 2007. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 26, 5541–5552. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, T. , Cubizolles, F. , Zhang, Y. , Reichert, N. , Kohler, H. , Seiser, C. , Matthias, P. , 2010. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 24, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, S. , Tanaka, K. , Sakimura, R. , Okada, T. , Nakamura, T. , Li, Y. , Takasaki, M. , Nakabeppu, Y. , Iwamoto, Y. , 2008. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 28, 1585–1591. [PubMed] [Google Scholar]
- Yan, W., Liu, S., Xu, E., Zhang, J., Zhang, Y., Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene, in press. [DOI] [PMC free article] [PubMed]
- Yang, S. , Wang, X. , Contino, G. , Liesa, M. , Sahin, E. , Ying, H. , Bause, A. , Li, Y. , Stommel, J.M. , Dell'antonio, G. , Mautner, J. , Tonon, G. , Haigis, M. , Shirihai, O.S. , Doglioni, C. , Bardeesy, N. , Kimmelman, A.C. , 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X.J. , Seto, E. , 2008. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, F. , Chen, Y. , Hoang, T. , Montgomery, R.L. , Zhao, X.H. , Bu, H. , Hu, T. , Taketo, M.M. , van Es, J.H. , Clevers, H. , Hsieh, J. , Bassel-Duby, R. , Olson, E.N. , Lu, Q.R. , 2009. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 12, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes, A. , Bociek, R.G. , Kuruvilla, J. , Laneuville, P. , Fung, H.C. , Drouin, M. , Patterson, T. , Besterman, J. , Martell, R.E. , 2010. Mocetinostat (MGCD0103), an isotype-selective histone deacetylase (HDAC) inhibitor, produces clinical responses in relapsed/refractory Hodgkin lymphoma (HL): update from a phase II clinical study. Blood. 116, 735 [Google Scholar]
- Yu, H. , Jove, R. , 2004. The STATs of cancer – new molecular targets come of age. Nat. Rev. Cancer. 4, 97–105. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Pardoll, D. , Jove, R. , 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, W. , Wang, J. , Jin, J. , Qian, W. , Qian, J. , Cheng, Y. , Wang, L. , 2011. Heat shock protein 90 inhibition results in altered downstream signaling of mutant KIT and exerts synergistic effects on Kasumi-1 cells when combining with histone deacetylase inhibitor. Leuk. Res. 35, 1212–1218. [DOI] [PubMed] [Google Scholar]
- Zhang, F. , Zhang, T. , Teng, Z.H. , Zhang, R. , Wang, J.B. , Mei, Q.B. , 2009. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol. Ther. 8, 823–831. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Kong, X. , Kang, J. , Su, J. , Li, Y. , Zhong, J. , Sun, L. , 2009. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol. Sci. 110, 376–388. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Kwon, S. , Yamaguchi, T. , Cubizolles, F. , Rousseaux, S. , Kneissel, M. , Cao, C. , Li, N. , Cheng, H.L. , Chua, K. , Lombard, D. , Mizeracki, A. , Matthias, G. , Alt, F.W. , Khochbin, S. , Matthias, P. , 2008. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 28, 1688–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, P. , Martin, E. , Mengwasser, J. , Schlag, P. , Janssen, K.P. , Gottlicher, M. , 2004. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 5, 455–463. [DOI] [PubMed] [Google Scholar]
- Zika, E. , Greer, S.F. , Zhu, X.S. , Ting, J.P. , 2003. Histone deacetylase 1/mSin3A disrupts gamma interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol. Cell. Biol. 23, 3091–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupkovitz, G. , Tischler, J. , Posch, M. , Sadzak, I. , Ramsauer, K. , Egger, G. , Grausenburger, R. , Schweifer, N. , Chiocca, S. , Decker, T. , Seiser, C. , 2006. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell. Biol. 26, 7913–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]