

Abstract
Reversible histone methylation has emerged in the last few years as an important mechanism of epigenetic regulation. Histone methyltransferases and demethylases have been identified as contributing factors in the development of several diseases, especially cancer. Therefore, they have been postulated to be new drug targets with high therapeutic potential. Here, we review histone demethylases with a special focus on their potential role in oncology drug discovery. We present an overview over the different classes of enzymes, their biochemistry, selected data on their role in physiology and already available inhibitors.
Keywords: Epigenetics, Histone demethylases, LSD1, JMJC, Lysine demethylase
Highlights
Histone demethylation is a major factor of epigenetic regulation.
It has been implicated in the pathogenesis of cancer in many studies.
First inhibitors for the histone demethylases are available and have shown great promise for drug development.
Structural data on demethylases and enzyme–inhibitor complexes are available and may greatly aid in inhibitor optimization.
1. Introduction
Histone methylation had long been thought to be an irreversible process but since 2004 (Metzger et al., 2005; Shi et al., 2004) it is known that histones, but also other proteins (Huang et al., 2007a; Nicholson and Chen, 2009), are also subject to active enzymatic demethylation (Agger et al., 2008). Reversible histone methylation has been shown to be involved in gene regulation and hence is interesting as a target for therapeutic intervention (Shi, 2007; Yoshimi and Kurokawa, 2011). Very rapidly inhibitors of these enzymes were identified and already show promise for drug development (Lohse et al., 2011a; Spannhoff et al., 2009a). Here, we present an overview over the different classes of histone demethylases, their biochemistry, selected evidence for their role in oncogenesis and inhibitor studies.
2. Reversible histone methylation
Methylation of histones occurs posttranslationally both on lysines as well as arginines (Trievel, 2004). Methyltransferases use the cofactor S‐adenosyl methionine (SAM) to transfer a methyl group onto the basic side chains of these amino acids within proteins. The involvement of histone methyltransferases in disease has been documented in many cases and first inhibitors studies have shown a lot of promise (Albert and Helin, 2010; Bissinger et al., 2010; Nimura et al., 2010; Spannhoff et al., 2009b). Regarding demethylation, the N‐methyl bond is chemically very stable and needs an oxidative mechanism to be broken. There are two classes of histone demethylases: the amine‐oxidase type Lysine specific demethylases (LSD1) (Metzger et al., 2005; Shi et al., 2004) and LSD2 (Karytinos et al., 2009) and a whole group of so‐called JmJC‐domain containing histone demethylases. Initial reports described a new family of JmjC‐domain containing proteins (JHDM1) that are able to demethylate mono‐, di‐ and tri‐methylated lysine residues on histone tails (H3K9me2/me3 and H3K36me2/me3) (Klose et al., 2006a; Tsukada et al., 2006). In the following years several new subfamilies all containing the catalytic active JmjC domain were identified (Christensen et al., 2007; Clifton et al., 2006; Cloos et al., 2006; Eissenberg et al., 2007; Fodor et al., 2006; Iwase et al., 2007; Klose et al., 2007a, 2006b, 2007b; Lee et al., 2007a, 2007b; Liang et al., 2007; Schneider and Shilatifard, 2006; Secombe et al., 2007; Seward et al., 2007; Shin and Janknecht, 2007; Tsukada et al., 2006; Whetstine et al., 2006; Wissmann et al., 2007; Yamane et al., 2006); Yamane et al., 2006). There is ample evidence on active lysine demethylation but limited data on arginine demethylation (Chang et al., 2007). The subtype responsible for the latter reaction (JmJD6) was later on found to be involved rather in lysine side chain hydroxylation (Webby et al., 2009). Table 1 provides an overview over histone demethylases, their targets and involvement in disease.
Table 1.
Overview over histone demethylases, their targets and links to cancer.
| Name | Synonyms | Targets | Links to Cancer |
|---|---|---|---|
| KDM1A | LSD1, AOF2 | H3K4me2/me1, H3K9me2/me1 | Overexpressed in prostate carcinoma (Kahl et al., 2006), bladder cancer (Kauffman et al., 2011), ER‐negative breast cancer (Lim et al., 2010), neuroblastoma (Schulte et al., 2009)Inhibition in animal models of engrafted acute myeloid leukemia (Schenk et al., 2012) |
| KDM1B | LSD2, AOF1 | H3K4me2/me1 | |
| KDM2A | FBXL11A, JHDM1A | H3K36me2/me1 | |
| KDM2B | FBXL10B, JHDM1B | H3K36me2/me1, H3K4me3 | Required for initiation and maintenance of acute myeloid leukemia (Li et al., 2011a) |
| KDM3A | JMJD1A, JHDM2A | H3K9me2/me1 | High expression correlated with bad prognosis in colorectal cancer (Uemura et al., 2010), overexpressed in renal cell carcinoma (Guo et al., 2011) |
| KDM3B | JMJD1B, JHDM2B | H3K9me2/me1 | |
| KDM4A | JMJD2A, JHDM3A | H3K9me3/me2, H3K36me3/me2 | Required for proliferation of breast cancer cells (Lohse et al., 2011a), attenuated expression in bladder cancer (Kauffman et al., 2011), required for latency and replication of viruses that cause cancer (Chang et al., 2011b) |
| KDM4B | JMJD2B | H3K9me3/me2, H3K36me3/me2 | Overexpressed in gastric cancer (Li et al., 2011b), required for proliferation and formation of metastasis in breast cancer cells (Kawazu et al., 2011) |
| KDM4C | JMJD2C, GASC1 | H3K9me3/me2, H3K36me3/me2 | Overexpressed in breast cancer (Liu et al., 2009), esophageal cancer (Yang et al., 2000), MALT lymphoma (Vinatzer et al., 2008), acute myeloid leukemia (Helias et al., 2008) and lung sarcomatoid carcinoma (Italiano et al., 2006) |
| KDM4D | JMJD2D | H3K9me3/me2/me1, H3K36me3/me2 | Required for cell proliferation and survival in colon carcinoma cells (Kim et al., 2012) |
| KDM4E | JMJD2E | H3K9me3/me2 | |
| KDM5A | Jarid1A, RBP2 | H3K4me3/me2 | Involved in drug resistance (Sharma et al., 2010) |
| KDM5B | Jarid1B, PLU1 | H3K4me3/me2 | Tumor‐suppressive function in metastatic melanoma cells (Roesch et al., 2006), pro‐proliferative in breast cancer (Mitra et al., 2011) and overexpressed in prostate cancer (Xiang et al., 2007) |
| KDM5C | Jarid1C, SMCX | H3K4me3/me2 | |
| KDM5D | Jarid1D, SMCY | H3K4me3/me2 | |
| KDM6A | UTX, MGC141941 | H3K27me3/me2 | Tumor‐suppressive function (Tsai et al., 2010) |
| KDM6B | JMJD3, KIAA0346 | H3K27me3/me2 | Overexpressed in Hodgkin's Lymphoma (Anderton et al., 2011) |
| PHF8, KIAA1111, ZNF422 | H3K9me2/me1, H4K20me1 | ||
| KDM7 | KIAA1718 | H3K9me2/me1, H3K27me2/me1 | |
| KDM8 | JMJD5, FLJ13798 | H3K36me2 |
Adapted from Kooistra and Helin (2012) and Lohse et al. (2011a).
3. Effects of histone methylation
Methylation of histone proteins can be associated with enhanced or repressed transcription of individual genes as well as the global establishment of a loose euchromatic or condensed heterochromatic chromatin state depending on the localization of the modification. It is under debate whether histone modifications are the reason of these effects in the form of a “histone code” or whether there is no such strict causality (Henikoff and Shilatifard, 2011). In general, methylation of H3K4, H3K36 and H3K79 is associated with activation of transcription, whereas methylation of H3K9, H3K27 and H4K20 is associated with repression of transcription (Kouzarides, 2007).
Methylated histone residues are specifically recognized by proteins containing chromo‐like domains or plant homeo domains. These proteins partly distinguish between the different methylation states, mono‐, di‐ and trimethylation for lysine and mono‐, symmetrical di‐ and asymmetrical dimethylation for arginine residues, and are also specific for an individual lysine or arginine residue. They carry certain functions to chromatin including enzymatic functions and the ability to recruit other proteins, which link individual histone marks to a certain output.
Methylated H3K9 recruits Heterochromatin protein 1 (HP1) to chromatin, which leads to repression of transcription of euchromatic genes and the formation of heterochromatin (Bannister et al., 2001). The effect multiplies as HP1 recruits further HP1 molecules via self‐association as well as a methyltransferase targeting H3K9, Suv39h1, which in turn increases the abundance of methylated H3K9 and thus again the recruitment of HP1 (Lee et al., 2005a).
Methylation of H3K27 is brought about by the methyltransferase EZH2, which is part of the Polycomb repressive complex 2 (PRC2). It leads to the recruitment of PRC1, which contains a ubiquitin ligase that targets H2AK119. Ubiquitinylated H2AK119 is associated with repression of transcription in a number of ways (Sauvageau and Sauvageau, 2010).
There is ample evidence that links methylation of H3K4 and H3K36 to global formation of euchromatin and enhanced expression of individual genes. Methyltransferases responsible for these activating methyl marks in promoter regions are recruited by phosphorylated RNA polymerase II (Ng et al., 2003). It has been shown that H3K4 methyltransferases are required for the expression of a number of genes (Sedkov et al., 2003), however, the mechanisms underlying the link between methylated H3K4 and H3K36 and active transcription are unclear.
Like methylated H3K4 and H3K36, methylated H3K79 is associated with the formation of euchromatin and enhanced expression of individual genes. It is thought to hinder the binding of histone deacetylases to chromatin, thereby preventing the formation of heterochromatin (Lee et al., 2005a).
4. Histone demethylases
As pointed out above, there are two classes of histone demethylases. One are the amine oxidases LSD1 and 2 and the other the iron‐ and α‐ketoglutarate dependent JmJC‐domain containing proteins.
4.1. Catalytic mechanism of amine‐oxidase like histone demethylases (LSD1/2)
LSD1 is a FAD dependent amine oxidase which was first characterized by Shi et al. in 2004. The enzyme is able to remove methyl groups from histone 3 lysine 4 when mono‐ or dimethylated (H3K4me1/2) at the ϵ‐position, but not the trimethylated lysine (Meyer et al., 2007; Shi et al., 2004). Later, it was shown that the substrate specificity of LSD1 can change to mono‐ or dimethyl H3K9 (H3K9me1/2) when co‐localizing with the androgen receptor (AR) (Metzger et al., 2005) and it demethylates lysine 370 of p53 as a non‐histone substrate (Huang et al., 2007a).
LSD1 is a highly conserved enzyme with three important domains. The amine oxidase‐like domain at its C‐terminal end is homologous to other FAD‐dependent oxidases and consists of a FAD‐binding domain and a substrate binding domain (Anand and Marmorstein, 2007). The catalytic site of the enzyme is built by these two subdomains. LSD1 has, in contrast to other FAD‐dependent enzymes, a wide binding pocket which can, additionally to the active site cavity, interact with the substrate to improve substrate–enzyme interactions (Shi et al., 2004).
The reason why LSD1 is not able to demethylate a trimethyl lysine is caused by the FAD dependent catalytic mechanism. This is limited by the initial step, the oxidation of the ϵ‐amine to form an iminium intermediate with the concurrent reduction of the flavin. The intermediate can then be attacked by a water molecule close to the iminium ion to react to an unstable hemiaminal intermediate which decomposes into formaldehyde and the demethylated H3K4. The reduced FADH2 is reoxidized by oxygen in a subsequent step to form FAD and hydrogen peroxide. Thus, the lysine residue requires a lone electron pair that can be protonated and then form the iminium intermediate. Thus, a trimethylated lysine cannot be demethylated by LSD1/2 (Spannhoff et al., 2009a; Yang et al., 2007b) (see Figure 1).
Figure 1.
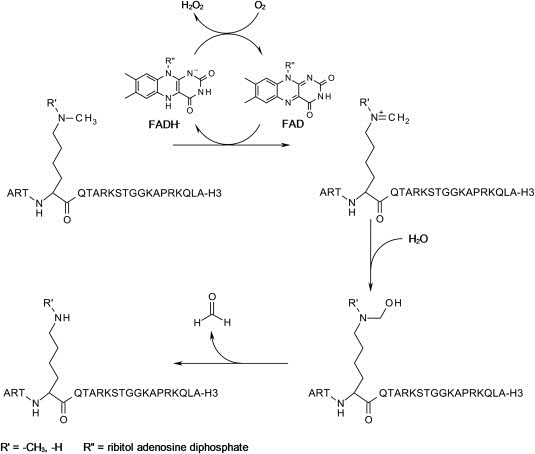
Mechanism of demethylation by LSD1. Details can be found in the text.
4.2. Catalytic mechanism of JumonjiC domain‐containing demethylases
In contrast to the flavin‐dependent demethylases LSD1 and LSD2, which require the presence of an intermediate iminium cation in their respective catalytic cycle, the demethylases containing a JmjC domain act via a fundamentally different reaction mechanism. This also allows for removal of a trimethyl mark, which is not possible for LSD1‐type demethylases for chemical reasons (Arrowsmith et al., 2012; Tsukada et al., 2006; Whetstine et al., 2006).
Jumonji‐type demethylases belong to the cupin superfamily of oxygenases and contain an Fe2+ ion in their catalytic domain and use α‐ketoglutarate as co‐substrate (Arrowsmith et al., 2012; Hou and Yu, 2010; Klose et al., 2006b; Klose and Zhang, 2007; Kooistra and Helin, 2012; Mosammaparast and Shi, 2010; Schneider and Shilatifard, 2006; Shi and Whetstine, 2007; Whetstine et al., 2006).
The demethylation proceeds via C–H bond activation and hydroxylation of the methyl group to hydroxymethyllysine, an unstable hemiaminal, which readily decomposes into formaldehyde and the lysine residue lacking one methyl group. The oxidizing agent responsible for the hydroxylation reaction is molecular dioxygen, of which one oxygen atom is transferred onto the methyl group giving the intermediate hemiaminal, while the other is transferred onto the co‐substrate α‐ketoglutarate, which undergoes spontaneous decarboxylation. Succinate and carbon dioxide are formed as the other products of the reaction (Sippl and Jung, 2009; Tsukada et al., 2006; Whetstine et al., 2006).
The catalytic domain of JMJD2A has been crystallized with shortened peptides of its natural substrate, histone H3 with di‐ and trimethylation at lysine 9 and 36 (H3K9me3, H3K36me2, and H3K36me3) as well as the co‐substrate α‐ketoglutarate giving a detailed view of the coordination sphere around the central metal atom, in this case Ni(II) substituting for Fe(II) originally present in JmjC domains (Couture et al., 2007).
Substrate specificity and recognition of the respective methylation status (me3, me2, or me1) is determined by a methylammonium‐binding pocket directly adjacent to the Fe(II) center. This positions the methyl group carbon atom into close proximity to the Fe(II)‐binding site, enabling its direct hydroxylation (Couture et al., 2007; Hou and Yu, 2010; Lee et al., 2008).
The proposed mechanism of action of JumonjiC domain‐containing demethylases is commonly assumed to be similar to that of other Fe(II)‐containing and α‐ketoglutarate‐dependent hydroxylases (Clausen et al., 2009; Hausinger, 2004). A plausible sequence of reaction steps is outlined in Figure 2. The catalytically active iron ion in the unbound state is in the +2 oxidation state and coordinated by two histidine residues, a glutamate residue, and three molecules of water. Initially, the co‐substrate α‐ketoglutarate as well as dioxygen are coordinated onto the iron center, displacing the water ligands (step 1). Most likely, a single electron transfer occurs next from the iron(II) ion onto the coordinated oxygen yielding a reactive peroxide radical anion species (step 2). This can, in turn, attack the α‐ketoglutarate ligand leading to a mixed anhydride bound to the remaining iron(III)‐hydroxyl radical anion (step 3). This highly reactive hydroxyl radical is able to activate the stable C–H bond in the adjacent methyl group of the substrate methyllysine and to form hydroxymethyllysine via abstraction of a proton and subsequent transfer of the hydroxyl group onto the methyl carbon atom (step 4) leaving behind a coordinative gap on the central iron(II) ion. Upon dissociation of the bound mixed anhydride from the central iron, it decomposes into carbon dioxide and succinate as by‐products (step 5). Binding of three molecules of water regenerates the original catalytic species (Clausen et al., 2009; Lohse et al., 2011a; Shi and Whetstine, 2007).
Figure 2.
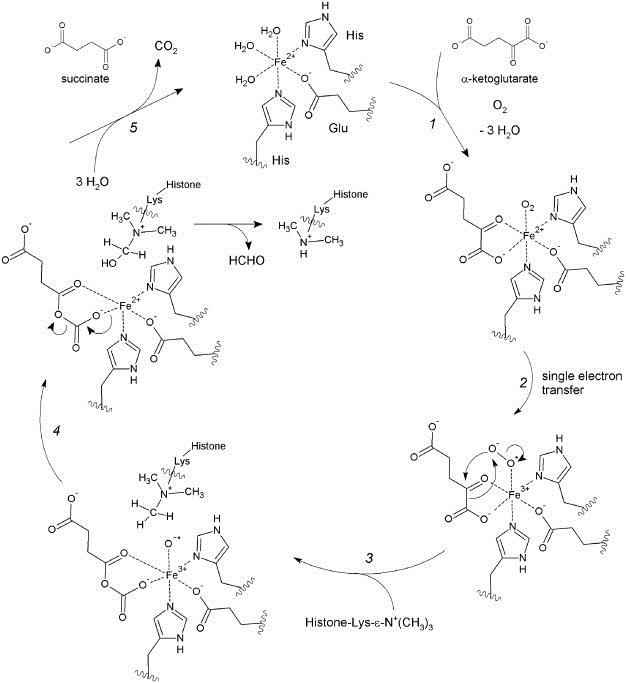
Mechanism of oxidative demethylation by JMJD. Details can be found in the text.
Alternative mechanisms involving an intermediate oxo‐ferryl(IV) species are also being discussed (Couture et al., 2007; Klose et al., 2006b; Ng et al., 2007; Suzuki and Miyata, 2011).
This reaction cycle could, in principle, be repeated resulting in removal of one methyl group after the other. The number of demethylation reactions that a particular Jumonji‐type demethylase performs and the required methylation state of the substrate are, however, determined by the specificity of its methylammonium‐binding pocket (Couture et al., 2007; Ng et al., 2007).
5. Involvement of histone demethylases in disease
The physiological role of histone demethylases is poorly understood. They play an important role in embryonic development and in controlling the balance between self‐renewal and differentiation of stem cells. Knockout of JMJD5 leads to transcriptional upregulation of p53 in mice embryos and results in active resorption at an early stage of embryonic development (Oh and Janknecht, 2012). JMJD2A is required for the expression of neural crest specifier genes and knockdown leads to a significant decrease in the expression of these genes in embryonic chicken (Strobl‐Mazzulla et al., 2010). LSD1 is essential for embryonic stem cells to retain their properties. Knockdown of LSD1 in human embryonic stem cells results in an increase of H3K4 monomethylation and dimethylation of target genes ultimately leading to differentiation of the cells (Adamo et al., 2011).
Pathophysiologically, there are strong links between the expression of certain histone demethylases and initiation and maintenance of various malignant tumors. There are several examples of overexpression of histone demethylases in tumor tissues. In fact, the possible oncogenic potential of a number of demethylases was described long before their demethylase activity became known. JMJD2C, for example, was long termed ‘gene amplified in squamous cell carcinoma 1 (GASC1)’, because of the relatively early description of its amplification in esophageal cancer cell lines (Yang et al., 2000).
One pathway by which individual histone demethylases are overexpressed in various cancers is their induction under hypoxic conditions. Hypoxia is common in tumor tissues as their blood vessel systems are often inadequate to provide cells with the required amount of oxygen. Several demethylases from the JMJD1 and JMJD2 protein families are direct targets of hypoxia‐inducible factor (HIF), which is activated under hypoxic conditions and leads to increased expression. Demethylase activity is hardly affected by low oxygen concentrations, making histone demethylases a functional part of the cellular response to hypoxic stress (Yang et al., 2009).
The importance of histone demethylases for the maintenance of the aggressive phenotype of cancer cells has been shown in several knockdown experiments: Bladder carcinomas known to overexpress LSD1 display greatly reduced proliferation when treated with small interfering RNA corresponding to the LSD1 gene product. A rescue with exogenous LSD1 is possible (Hayami et al., 2011). Similarly, knockdown of JMJD2B in gastric cancer cells strongly affects clonogenicity. The growth of xenograft tumors in mice is reduced after knockdown and in some cases, tumors fail to form altogether (Li et al., 2011b).
Histone demethylases also present a target of interest in hormone‐dependent cancers. LSD1 and JMJD2C are coactivators of the androgen receptor. In complex, they collaboratively demethylate trimethylated H3K9 with JMJD2C producing dimethylated H3K9 and LSD1 removing the remaining methyl groups. They cooperate in the removal of a repressive histone mark, which results in enhanced transcription of androgen dependent genes (Wissmann et al., 2007). Of note is the changed substrate specificity of LSD1 in complex with the androgen receptor. Whereas it normally demethylates dimethylated H3K4, it accepts dimethylated H3K9 as a substrate when bound to the androgen receptor (AR) (Metzger et al., 2005). This shift is in part mediated by phosphorylation of H3T6 by PKCβ1 (Metzger et al., 2010). In prostate cancer cells, AR activation is initially required for cell proliferation. Tumor growth can be suppressed by treatment with anti‐androgens. However, at a later stage, tumor cells grow androgen‐independently and no longer respond to treatment with anti‐androgens. Treatment with LSD1 inhibitors possibly in combination with JMJD2C inhibitors might be a future option and first experiments with a novel LSD1 inhibitor in a mouse model gave positive results (see inhibitor section) (Willmann et al., 2012). Similarly, LSD1 has also been shown to be a coactivator of the estrogen receptor alpha (Garcia‐Bassets et al., 2007). Both the proliferation of estrogen receptor alpha positive and negative breast cancer cells is affected by inhibition of LSD1, raising the question of its other important roles in these cells (Pollock et al., 2012).
Histone demethylases have also been linked to drug resistance in cancer cells. It is a common phenomenon that patients who undergo extended chemotherapy respond well to it initially, but a subpopulation of cancer cells becomes drug‐resistant and resumes growth eventually. The histone demethylase Jarid1A is elevated in such drug tolerant persisters in the prostate carcinoma cell line PC9. Jarid1A knockdown in these cells reduces the number of drug tolerant cells and conversely, overexpression of Jarid1A in PC9 cells decreases the sensitivity of the whole population (Sharma et al., 2010).
Similarly to other chromatin modifying enzymes, histone demethylases play a role in viral diseases. JMJD2A is crucial for the replication and regulation of gene expression of Kaposi's sarcoma‐associated herpesvirus (KSHV). Immunosuppressed patients infected with this virus often develop Kaposi's sarcoma or primary effusion lymphoma. JMJD2A is required for KSHV replication and to maintain virus latency. In cells in which virus DNA is transcribed JMJD2A is inhibited by a viral protein, K‐bZIP (Chang et al., 2011b). These findings show that a fine balance between JMJD2A activation and inhibition is necessary for progression of the viral disease and suggest that treatment with JMJD2A inhibitors might help prevent the development of virus‐associated cancers in infected patients.
Underlining the different functions of individual demethylases, some enzymes have been reported to act as tumor suppressors. Overexpression of Jarid1B in melanoma cells leads to a reduction in cell proliferation and DNA replication. It attenuates the transcription of melanoma‐progression‐related genes, showing Jarid1B to have a tumor suppressive function (Roesch et al., 2006).
There are very few reports that link histone demethylases to diseases other than cancer. LSD1 is involved in the ‘hyperglycemic memory’ of cells, a series of mechanisms that causes negative effects of hyperglycemia to persist for an extended period of time after return to normoglycemia. LSD1 shows enhanced recruitment to monomethylated H3K9 in hyperglycemic cells and thus contributes to an increased expression of pro‐inflammatory genes such as NFκB. The expression of these genes is elevated in mice during and after periods of transient hyperglycemia (Brasacchio et al., 2009). There is also a link between histone demethylases and neurodevelopmental disorders, which matches the physiological function of demethylases as described above. For example, several point mutations are known for Jarid1C that lead to X‐linked mental retardation, a cognitive disorder affecting males (Iwase et al., 2007).
6. Inhibitors of histone demethylases
For both classes of histone demethylases, inhibitors have already been identified and we will highlight the most important examples below.
6.1. Inhibitors of LSD1/2
As LSD1 is an amino‐oxidase, a comparison with other well known amine oxidases was the first approach to identify new inhibitors. The well studied MAO‐A (monoamine oxidase A) and MAO‐B show high sequence homology to LSD1. The MAO enzymes remove amine groups from neurotransmitters in a FAD‐dependent mechanism similar to that of LSD1. The crystal structures also indicate a very similar binding mode for the cofactor FAD in the active site of the enzymes (Stavropoulos et al., 2006). Since MAO inhibitors are in use as antidepressant drugs and against Parkinson's disease for decades, they were interesting candidates for testing, esp. with regard to a potential clinical application. Therefore, available MAO inhibitors were tested in their potency against LSD1. Especially the approved antidepressant drug trans‐2‐phenylcyclopropylamine (tranylcypromine, TCP, PCPA, (1)) could be identified as a potent LSD1 inhibitor in the micromolar range (IC50 ∼ 2–21 μM, depends on assay design). Other MAO inhibitors like pargyline (2), clorgiline or phenelzine (3) showed also inhibition, yet with greatly decreased potency (Lee et al., 2006; Schmidt and McCafferty, 2007) (see Figure 3). The inhibitory potency of PCPA (1) against MAO‐B is slightly higher than against LSD1. Structural analysis of the co‐crystalized inhibitor‐MAO complexes showed covalent binding of PCPA to the C(4a) atom of FAD (Binda et al., 2003), while the PCPA‐FAD bond in LSD1 is formed at nitrogen 5 (N5) of the flavin part of the molecule (Mimasu et al., 2008; Ueda et al., 2009; Yang et al., 2007b).
Figure 3.
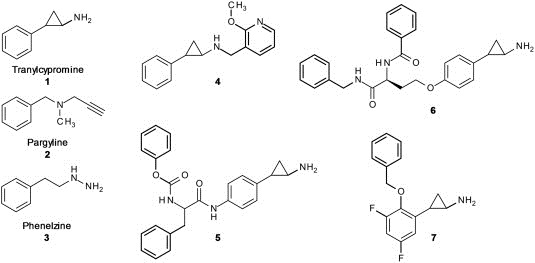
Covalent LSD1 inhibitors Tranylcypromine (1, PCPA), Pargyline (2), Phenelzine (3) and various PCPA analogs.
An overexpression of LSD1 could be found in several cancer cell lines and tumors (Hayami et al., 2011; Kahl et al., 2006; Schildhaus et al., 2011), so the focus of cellular inhibitor screening and testing was and mainly is on cancer cells. For AML, all‐trans retinoic acid (ATRA) is used as an agent in therapy. Treatment with PCPA (1) and all‐trans retinoic acid could resensitize ATRA‐insensitive TEX cells as well as enhance the ATRA effect on myeloid differentiation of ATRA‐sensitive HL‐60 cells. In ChIP seq experiments, the myeloid‐differentiation‐associated gene promoters showed an increase in H3K4 dimethylation, a transcription activation mark and LSD1 substrate indicating an efficient inhibition of LSD1 activity by PCPA. Remarkably, treatment with ATRA plus PCPA significantly diminished the engraftment of primary human AML cells in vivo in nonobese diabetic (NOD)‐severe combined immunodeficient (SCID) mice, suggesting that ATRA in combination with PCPA may target leukemia‐initiating cells (Schenk et al., 2012). Since LSD1 co‐localizes together with other epigenetic enzymes in a complex with the androgen receptor a potential use of inhibitors for treatment of androgen‐dependent cancers is under special consideration (Metzger et al., 2005). Androgen‐sensitive human prostate adenocarcinoma cells (LNCap) were growth inhibited by pargyline, showing an increase in mono‐ and dimethylation level of H3K9 (Metzger et al., 2005). The more potent p‐bromo‐ and p‐phenyl‐analogs of PCPA inhibited growth of LNCap cells in the micromolar range (Benelkebir et al., 2011). In bladder carcinoma cells, a suppression of AR dependent transcription was observed after inhibition of LSD1 activity by PCPA. But due to the fact that AR‐negative cells also responded to PCPA treatment, aspects other than AR‐dependent transcription are discussed for bladder cancers (Kauffman et al., 2011). These impressive data make LSD1 inhibitors promising targets for future drug discovery, but more potent and selective inhibitors than PCPA (1) and pargyline (2) were and will probably be needed avoid of potential off‐target effects.
Two patented analogs, one being compound (4), from Oryzon Genomics (Guibourt, 2010) were tested in an in vitro assay (IC50 670 nM and 96 nM (4), IC50 (PCPA): 15.7 μM) and in an ex vivo experiment with MLL‐AF9 human acute myeloid leukemia cell lines. In these cell lines, the authors showed significant reduction of the colony‐forming cells in the range of the in vitro IC50 of the compounds (IC50 270 nM and 50 nM (4), IC50 (PCPA): 8 μM). Thus, theses two analogs were, compared to PCPA, 23‐fold and 57‐fold more active in this biological setup. The compounds were able to induce a loss of clonogenic potential and induction of differentiation in both murine and primary human MLL leukemia cells, both in vitro and in vivo (Harris et al., 2012). The authors conclude that LSD1 is required to sustain the expression of the MLL‐AF9‐associated oncogenic program and that LSD1 is a promising target for other AML subtypes that overexpress LSD1 and for future AML drug discovery.
X‐ray studies with PCPA analogs (5) synthesized in the Mai lab showed, in analogy to PCPA, covalent inhibition of LSD1 (see Figure 3). Like PCPA, these substances also bind to N5 of the FAD molecule, suggesting that other PCPA analogs will have the same inhibition mechanism (Binda et al., 2010). The first published analogs were PCPA‐lysine hybrid compounds (6) (Ueda et al., 2009). These compounds were selective inhibitors of LSD1 over MAO‐A and ‐B due to their bulky peptidomimetic substituents in the ortho‐ and para‐position of the phenyl ring of PCPA. X‐ray structures of MAO‐A and MAO‐B indicate that the active site cavity is not voluminous enough to accommodate these compounds. Western blot analysis of HEK293 cells showed global accumulation of H3K4(me2) which is in line with cellular inhibition of LSD1. Further cell growth studies showed inhibition of HEK293 cells and five other cancer cell lines with GI50 values ranging from 6 μM to 67 μM. Since Wang et al. demonstrated that HEK293 cells have low LSD1 but high LSD2 expression, it is of interest to see if these compounds are able to inhibit both enzymes and if the cell growth inhibition could be explained by both, LSD1 and/or LSD2 inhibition (Wang et al., 2011).
Other PCPA analogs (7) synthesized in the Yokoyama lab inhibit LSD1 even stronger with IC50 values down to 1 μM in vitro and selectivity over MAO‐A and MAO‐B (Mimasu et al., 2010). In cellular Western blot experiments in HEK293T cells with a H3K4(me2) antibody, they could show an approximately 50‐fold higher LSD1 inhibition of (7) as compared to PCPA inhibition. The PCPA derivatives from the Mai lab (5) are selective over MAO‐A but not MAO‐B, with a K i (LSD1) of 1.1 μM (K i (PCPA): 271 μM) (Binda et al., 2010). The LSD2 inhibition was weaker than the LSD1 inhibition. The compound enhances the efficacy of retinoic acid on growth inhibition and differentiation of acute promyelocytic leukemia (NB4) cells, including primary murine APL blasts.
p‐Phenolethers of PCPA, inhibitors in the range of potency of PCPA (Ki(PCPA): 550μM ± 150 μM vs. Ki(compounds): 173–756 μM) and with little selectivity against MAO‐B (Ki(PCPA): 13.6 μM ± 3.0 μM vs. Ki(compounds): 63–106 μM), were recently published to inhibit tumor cell growth of ERα‐positive (MCF‐7) and ERα–negative (MDA‐MB‐231) cell lines (Pollock et al., 2012). LSD1 had been shown to be necessary for estrogen‐dependent ERα‐mediated transcription (Hu et al., 2008) by ChIP analysis but here the authors could show that the proliferation of both ERα‐positive and ERα–negative breast cancer cells responds to inhibition of LSD1 with small molecules.
Another group of LSD1 inhibitors was found due to the similarity of the LSD1 catalytic domain to FAD‐dependent polyamine oxidases, including spermine oxidase. Since they have a sequence identity of more than 60%, the LSD1 inhibition of spermine oxidase inhibitors like biguanidines (8) and bisguanidines (9) was predicted (Figure 4) (Huang et al., 2007b). Indeed, they were demonstrated to be noncompetitive LSD1 inhibitors in vitro. In HCT116 human colon carcinoma cells and RKO colon carcinoma cells, treatment with the two most potent inhibitors (8) and (9) lead to H3K4 hypermethylation as seen in a Western blot. Additionally, RT‐PCR demonstrated reexpression of epigenetically silenced genes. Further ChIP analyses showed that H3K4 hypermethylation appears at the promoter regions of the affected genes. The same group published other oligoamine inhibitors, PG‐11144 (10) and PG‐11150, with similar effects on HCT116 cells. In this case the inhibition mechanisms at LSD1 in vitro seemed to be reversible and substrate competitive for these compounds (Huang et al., 2009). Athymic nude mice bearing HCT116 xenografts were analyzed for tumor growth and weight loss when treated with (9), (10) or 5‐Azacytidine, a DNA methyltransferase (DNMT) inhibitor, alone or in combination. Single treated mice showed moderate, but significant reduced tumor growth, the combination of DNMT and LSD1 inhibitor showed synergistic effects with higher tumor growth reduction than treatment with one substance alone. This points out a general potential for synergistic anticancer therapy using a combination of LSD1 and DNMT inhibitors.
Figure 4.
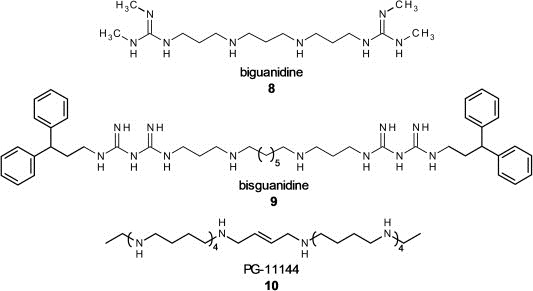
Non‐covalent LSD1 inhibitors with biguanidine (8), bisguanidine (9) and oligoamine (PG‐11144, 10) scaffold.
Recently, new small molecules (e.g. CBB1007 (11), see Figure 5) were reported to inhibit cancer cells with pluripotent stem cell properties but not non‐stem cell lineages. These guanidinium groups carrying compounds are described as reversible, allosteric inhibitors selective for LSD1 with in vitro IC50 values down to 5.27 μM. MAO inhibition was not studied. Cell growth inhibition could be shown for F9, NCCIT and NTERA‐2 cells which have a high expression of LSD1 and carry the pluripotent stem cell markers Oct4 and Sox2. No influence was reported for cells without these stem cell markers and low LSD1 expression, e.g. HeLa and HEK293 cells (Wang et al., 2011).
Figure 5.
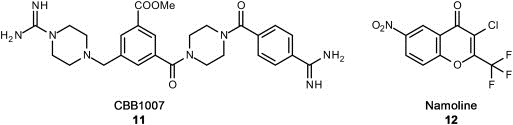
Novel substrate competitive LSD1 inhibitors with basic guanidinium structure (11) and the in vivo active γ‐pyrone Namoline (12).
The most recent reported inhibitor for LSD1 is Namoline (12), a non‐covalent and selective inhibitor with a γ‐pyrone scaffold and in vitro activity of 51 μM and proven in vivo activity (Willmann et al., 2012). Namoline (12) treated LNCap prostate cancer cells were analyzed for their H3K9(me2) levels, a marker for LSD1 activity in prostate cancer cells due to its substrate switch in the co‐activator complex with androgen receptor. H3K9(me2) levels increased after treatment with more than 20 μM compound, indicating cell permeability and LSD1 inactivation in tumor cells. In LNCap xenografted mice, Namoline stopped tumor cell growth. This is the first example of a non‐covalent LSD1 inhibitor with anticancer activity in animals.
6.2. Inhibitors of JumonjiC domain‐containing demethylases
The availability of structural information and more insight into the catalytic mechanism and methylation mark selectivity of Jumonji domain‐containing demethylases has spurred an interest in the development of novel inhibitors for this class of enzymes. Since it has only been known for less than a decade, assays to assess the inhibitory potency of compounds are only starting to emerge. Some inhibitors have been discovered in random screening campaigns, while others were designed and developed as analogs of the co‐substrate, the substrate, or a combination of the two. Furthermore, a number of inhibitors have been proposed that are thought to directly bind to and chelate the catalytically active iron(II) ion in the catalytic core and thereby inhibit the demethylation reaction.
Quite a number of potential inhibitors of, at least in part, considerable potency have been described in the literature as recently reviewed (Lohse et al., 2011a; Suzuki and Miyata, 2011). However, there is often a lack of investigations into their subtype selectivity and specificity for Jumonji demethylases and most compounds can be expected to also show activity on other related, yet undesired targets. This may be the reason that none of the inhibitors have advanced to further investigations beyond cell culture experiments, let alone clinical studies.
A first collection of potential inhibitor scaffolds was published in 2008 by the Schofield and Oppermann groups (Rose et al., 2008; Schofield et al., 2010) based on similar compounds that were known inhibitors of other iron(II) and α‐ketoglutarate‐dependent enzymes as summarized in 6, 7, 8, 9. They used an enzyme‐coupled assay to determine the inhibitory effect of compounds employing formaldehyde dehydrogenase (FDH), which oxidizes the demethylation reaction product formaldehyde under concomitant conversion of NAD+ to NADH. The fluorescence of NADH can be measured and correlated to the amount of formaldehyde formed during the reaction and, thus, the activity of the Jumonji JMJD2E enzyme (Couture et al., 2007; Rose et al., 2008).
Figure 6.
Chemical structures of Jumonji demethylase inhibitor scaffolds based on other iron‐dependent enzymes and their optimized derivatives. See text for details and references.
Figure 7.
Chemical structures of a disulfiram derivative and hydroxyglutarate as well as Jumonji demethylase inhibitors found from screening natural products libraries and a diverse chemical library. See text for details and references.
Figure 8.
Chemical structures of more recently discovered small‐molecule inhibitors of Jumonji domain‐containing demethylases. See text for details and references.
Figure 9.
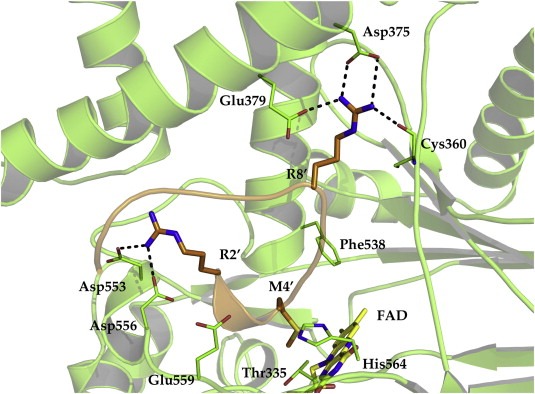
Crystal structure of LSD1‐histone H3 peptide complex (2V1D.pdb). The co‐crystalized substrate analog contains a methionine instead of the methylated Lys4 of H3. The peptide side chains are displayed as dark orange sticks, whereas the FAD cofactor is shown in yellow. Only relevant amino acids are displayed. Amino acids of the histone peptide are labeled using single letter code.
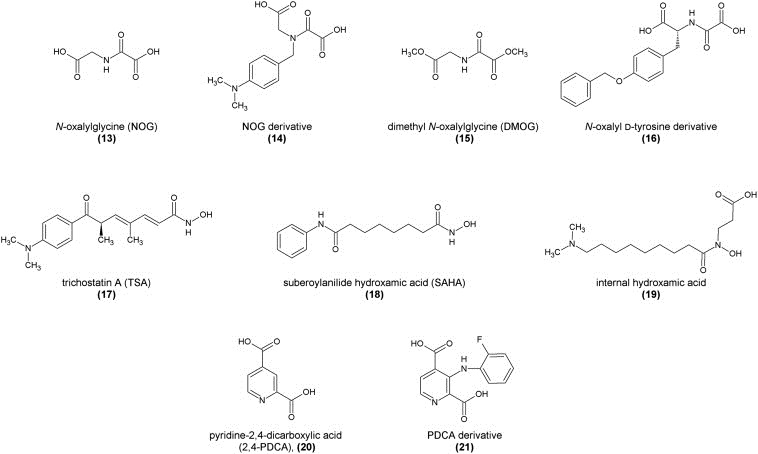
Using this assay, N‐oxalylglycine (13), an amide analog of the co‐substrate α‐ketoglutarate, was shown to be a mildly active in vitro inhibitor by competitively replacing the co‐substrate from the enzyme's active site. It inhibited JMJD2E with an IC50 value of 78 μM or only 24 μM after a pre‐incubation period of 30 min. However, compounds of this type are very drug‐unlike due to their very polar and charged nature and have also been shown previously to be inhibitors of other enzymes such as the human hypoxia‐inducible factor (HIF) hydroxylases (PHD1‐3) and factor inhibiting hypoxia‐inducible factor (FIH) (Ivan et al., 2002; McDonough et al., 2005). Derivatives using other amino acids such as phenylalanine have been synthesized and tested. While they were less potent (IC50 values between 100 and 600 μM), a certain selectivity over HIF proteins was observed due to the aromatic side chains enabling binding into a hydrophobic pocket only present in the Jumonji active site.
A crystal structure of a truncated version of JMJD2A in complex with NOG could be obtained using nickel(II) substituting for the iron(II) ion of the catalytic centre (Ng et al., 2007). This revealed the binding mode of NOG to be identical to the original co‐substrate α‐ketoglutarate with NOG binding to the catalytic iron(II) ion via its carbonyl and adjacent carboxylate function, whilst the other carboxylate group is engaged in a hydrogen bond with a tyrosine residue of the catalytic pocket.
The structural template for NOG derivatives as Jumonji inhibitors was further elaborated by Hamada et al. using a variety of other non‐natural amino acids coupled to an oxalyl residue introducing a positively charged dimethylamino group, anticipating that it would interact with hydroxyl groups of tyrosine and serine residues in the active pocket. Using an antibody‐based in vitro demethylation assay with JMJD2A, JMJD2C, and JMJD2D, the NOG derivative (14) was discovered as the most potent, yet unselective inhibitor, showing activity at millimolar concentrations (Hamada et al., 2009). This study also introduced a prodrug concept for these highly polar inhibitor compounds by preparing their respective methyl esters for improved cell permeability. The esters are thought to be cleaved intracellularly, releasing the diacid as the active drug molecule. In an in vivo assay using cells overexpressing JMJD2C, the levels of H3K9me3 and H3K36me3 were monitored using specific antibodies. While the methyl ester prodrug of hit (14) did not show any activity, the methyl ester prodrug of the lead compound NOG (dimethyl oxalylglycine, DMOG, (15)) restored trimethylation levels at a concentration of 2.5 mM by inhibiting the demethylation activity of JMJD2C (Hamada et al., 2009).
In order to obtain selectivity of NOG‐type compounds over other iron(II) and α‐ketoglutarate‐dependent enzymes, a crystallographic study revealed that a hydrophobic subpocket adjacent to the active site might be addressed by substituted derivatives. This subpocket is significantly larger in the case of the Jumonji domain proteins than for FIH proteins. A series of derivatives was prepared and evaluated using a combination of non‐denaturing mass spectrometric screening, FDH‐coupled enzymatic assays, as well as crystallization and structural modeling techniques. Derivatives of the natural amino acid d‐tyrosine with substituents connected either via a sulfate or ether bridge showed higher potencies and selectivities. One of the most active inhibitors (16) showed an IC50 value of 37.1 μM in the in vitro FDH‐coupled assay against JMJD2E and was also crystallized in complex with JMJD2A. Structural analysis revealed the binding mode to be as predicted, interfering with both α‐ketoglutarate and substrate binding, while the aromatic rings occupy a large hydrophobic pocket adjacent to the substrate binding cleft (Rose et al., 2010).
In the same study by the Oppermann and Schofield groups (Rose et al., 2008; Schofield et al., 2010), compounds of the hydroxamic acid type such as trichostatin A (TSA, (17)) and a simplified version suberoylanilide hydroxamic acid (SAHA, (18)), known to be good inhibitors of histone deacetylase proteins (HDACs) due to their zinc‐binding capabilities, were tested and proved to be potential inhibitors of JMJD2E in an in vitro MALDI‐TOF mass spectrometric assay. This action is probably due to a chelation of iron ions in the active site of Jumonji proteins (Rose et al., 2008). As these compounds are known to be active on HDACs with much higher potencies (Marks and Breslow, 2007), side effects are probable and makes future development of this compound class less promising. A very recent report demonstrated, however, that treatment of LNCaP cells with trichostatin A (TSA, 2) led to downregulation of JMJD2B and, eventually, apoptosis (Zhu et al., 2012).
Nonetheless, the potential of hydroxamic acids to function as iron chelators and, thus, inhibitors of Jumonji‐type demethylases initiated a study by Hamada et al. into this structure class, developing inhibitors with internal hydroxamic acids and long aminoalkyl chains. This structure‐guided drug design yielded a series of novel inhibitor compounds as tested in an FDH‐coupled enzymatic assay with the optimized compound (19) carrying a dimethylamino group connected via an octyl chain linker. This compound exhibited an in vitro IC50 value of 1.0 μM on JMJD2C and 3.0 μM on JMJD2A, but a greater than 100‐fold selectivity over other Fe(II)‐binding enzymes such as PHD1 and PHD2. Molecular modeling revealed the binding mode to be similar to that of other hydroxamic acids, i.e. complexation of the iron(II) ion in a bidentate manner, but also the relevance of the charged dimethylamino group, which is involved in hydrophilic interactions with charged amino acids. The correct length of the linker unit is required to optimally fill a hydrophobic channel. A methyl ester prodrug was designed for better cell permeability. However, it did not show any in vivo activity on LNCaP cells expressing JMJD2C when administered alone, but only in synergistic action together with NCL‐2, an LSD1‐specific inhibitor, in a cell‐type specific manner (Hamada et al., 2010).
A series of pyridine dicarboxylic acids was also tested in the original study by the Oppermann and Schofield groups with pyridine 2,4‐dicarboxylic acid (2,4‐PDCA, also 2,4‐lutidinic acid, (20)) being the most potent JMJD2E inhibitor with an IC50 value of only 1.4 μM in the in vitro FDH‐coupled assay. It showed clear competitive inhibition with the co‐substrate α‐ketoglutarate. This was also supported by a crystal structure that could be obtained using JMJD2A and nickel(II) in the active site in complex with 2,4‐PDCA revealing its binding mode as a bidentate complexation of the central metal ion via the pyridine nitrogen atom as well as the adjacent carboxylic acid (Rose et al., 2008). The 4‐carboxylic acid, in this case, is involved in an electrostatic interaction with a lysine residue of the active site cavity. Compounds of this structural class are, however, also known to potently inhibit other enzymes such as collagen and HIF prolyl hydroxylases (Tiainen et al., 2008).
A number of different Jumonji protein subtypes have been crystallized in complex with various inhibitors and the crystal structures solved enabling insight into their respective binding modes as recently reviewed (Lohse et al., 2011a). These could lay the groundwork for upcoming structure‐based drug design campaigns.
By comparing the crystal structure of JMJD2A in complex with 2,4‐PDCA (20) (Rose et al., 2008) to that of hypoxia inducible factor (HIF) PHD2, it became clear that addition of larger substituents to the pyridine ring system might enable further interactions with the active site pocket and induce selectivity for the Jumonji proteins, whose active site pocket is larger than that found in PHD2. 3‐Substituted pyridine 2,4‐dicarboxylic acids were, hence, synthesized with different substituents of varying sizes and evaluated for their inhibitory effect in an FDH‐coupled spectrophotometric assay. While the inhibitory potency generally decreased, almost all compounds showed complete selectivity for JMJD2E over PHD2 with the ortho‐fluorophenyl derivative (21) having the highest in vitro potency (IC50 = 2.5 μM), comparable to that of unsubstituted 2,4‐PDCA (Thalhammer et al., 2011).
Several reports have determined other inhibitors based on the catalytic mechanism of Jumonji domain‐containing enzymes. It was shown, for example, that JMJD1A could be inhibited in vitro in an antibody‐based assay by the presence of nickel chloride with an IC50 value of 2.5 μM or 25 μM in the presence of iron(II) ions, indicating that nickel displaced the catalytically relevant iron(II) from the active site. As nickel(II) ions cannot be oxidized further, they cannot take part in the reaction cycle by formation of intermediate higher‐valent species. This effect could even be reproduced in vivo using human embryonic 293T cells overexpressing JMJD1A that were exposed to 1 mM nickel chloride (Chen et al., 2010).
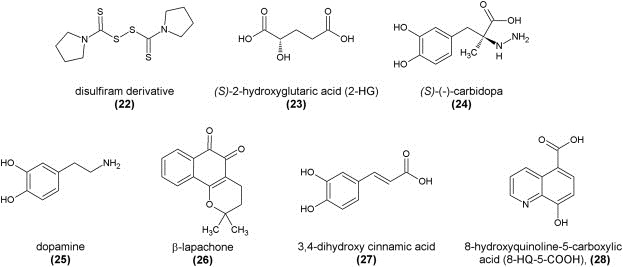
In a similar manner, compounds that are known to complex zinc(II) ions can be considered inhibitors since several Jumonji‐type demethylases contain structural Zn(II)‐binding domains located close to the methyllysine‐binding pocket. This has been shown in a proof‐of‐concept study using, amongst others, derivatives of the anti‐alcohol abuse drug disulfiram. The most potent zinc‐ejecting inhibitor of JMJD2A with an in vitro IC50 value of 3.3 μM in a MALDI‐TOF mass spectrometric assay turned out to be the pyrrolidinyl derivative (22) (Schofield et al., 2011; Sekirnik et al., 2009). This strategy might have potential for the development of inhibitors for subtypes of Jumonji proteins containing a structural zinc(II) ion (JmJD2) over the others that do not have that.
Furthermore, compounds with high structural similarity to the co‐substrate α‐ketoglutarate such as its reduced metabolite 2‐hydroxyglutarate (23) can competitively inhibit Jumonji demethylases and other α‐ketoglutarate‐dependent dioxygenases in the millimolar range in vitro as demonstrated in a recent study. 2‐Hydoxyglutarate is a metabolite that commonly accumulates in low grade gliomas and secondary glioblastoma multiforme as well as acute myeloid leukemia which might be due to excessive JmJ‐domain demethylase activity. An in vitro mass spectrometric assay using histone demethylase CeKDM7A from C. elegans showed the (S) enantiomer to be more active than the (R) enantiomer. A crystal structure of CeKDM7A complexed with the (R) enantiomer revealed that, upon binding, it occupies the same space as α‐ketoglutarate normally would, the iron(II) ion being coordinated by one oxygen atom from the carboxylate group as well as the hydroxyl group. The inhibitory effect could also be demonstrated for human KDM2A (JHDM1A), establishing 2‐HG as a weak antagonist of α‐ketoglutarate (Xu et al., 2011).
In their seminal study, Simeonov et al. redesigned, optimized, simplified, and miniaturized the FDH‐coupled enzymatic assay for Jumonji demethylases, making it available for large‐scale high‐throughput screening purposes to readily identify potential small molecule inhibitors (Sakurai et al., 2010). This assay was used to screen a natural product substance library in order to develop novel lead structures, which resulted in the discovery of several naturally occurring flavonoids and catechols as inhibitors, which presumably act as unspecific iron(II) ion chelators either in the active site of the enzyme or in solution. These compounds were counter‐screened against FDH inhibition and validated in a secondary MALDI‐TOF mass spectrometric assay. Examples include (S)‐(−)‐carbidopa (24), dopamine (25), and β‐lapachone (26), with in vitro IC50 values against JMJD2E of 8.0 μM, 7.1 μM, and 3.6 μM, respectively (Sakurai et al., 2010).
Only recently, a new study identified several more catechols as inhibitors of Jumonji demethylases from a natural product library screening. The most potent inhibitor of this series was 3,4‐dihydroxy cinnamic acid (27) with an in vitro IC50 value of 13.7 μM against JMJD2C (KDM4C) and 5.5 μM against KDM6A, but no inhibition against the unrelated iron(II)‐dependent enzyme PHF8 (Nielsen et al., 2012).
However, these natural products also have pronounced other physiological effects or are even administered as drugs as in the case of carbidopa, so that development of these interesting in vitro results into actual medicines will most likely not be possible.
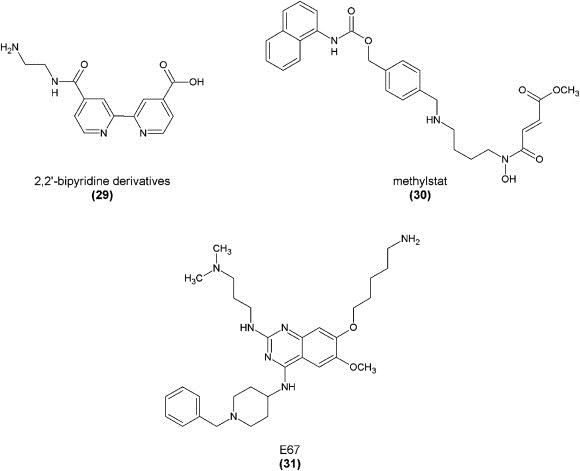
Using their high‐throughput FDH‐coupled in vitro assay, the Simeonov group also screened a larger diverse compound library, identifying a new compound class as potential demethylase inhibitors, the 8‐hydroxyquinolines (8‐HQs). Hits were confirmed in a secondary MALDI‐TOF mass spectrometric assay. Structure‐activity relationships were studied and the most potent compound was found to be the derivative with an additional carboxylic acid function (28) with an IC50 value of 0.2 μM against JMJD2E. A crystallographic study with JMJD2A showed this compound to bind to the central metal ion in a bidentate manner via its quinoline‐nitrogen and the 8‐hydroxyl group. The carboxylic acid moiety is positioned towards basic amino acid residues of the active site pocket. In an antibody‐based cellular demethylase assay, in vivo activity of these compounds could also be shown with an IC50 value of 86.5 μM, without the need for prior derivatization or synthesis of a prodrug (King et al., 2010).
The hydroxyquinoline template was also recently used as the selectivity‐inducing group in the construction of a small‐molecule probe with a photoactive cross‐linking group coupling it to a linker moiety and an affinity purification tag. This makes this compound suitable for identification of oxygenases in crude cell extracts, inhibitor profiling, or substrate identification studies (Rotili et al., 2011).
Another compound class with iron(II)‐binding capabilities is the group of substituted 2,2′‐bipyridines with at least one carboxylic acid group, as already mentioned in the first report about inhibitor scaffolds (Rose et al., 2008; Schofield et al., 2010). The structure space for this compound class has recently been investigated. Compounds were screened using the in vitro FDH‐coupled assay as well as a non‐denaturing ESI mass spectrometric assay. One of the most potent inhibitors of this type (29) (IC50 against JMJD2E of 180 nM) contained an additional positively charged amino group that is involved in an electrostatic interaction with an aspartate residue of the active site pocket. While an aromatic interaction in this position might also play a role, this compound was crystallized with JMJD2A, confirming its binding mode as a bidentate metal complexation via the two ring nitrogen atoms, an ionic interaction of the free carboxylate group to a catalytic pocket lysine residue, and the salt bridge mediated by the amine group (Chang et al., 2011a).
Another small‐molecule inhibitor was suggested that inhibited Jumonji demethylase JMJD2C in an in vitro ESI‐TOF‐based mass spectrometric assay and a DEFLIA‐based assay with an IC50 value of 4.3 μM. The compound, however, is not selective for any subtype and also comparably active against other enzymes of this family such as PHF8 and prolyl hydroxylases. Its methyl ester, methylstat (30), based on the aforementioned prodrug concept is described to be able to penetrate into cells and inhibit demethylase activity in vivo. Proliferation of an esophageal carcinoma cell line was inhibited with a GI50 of 5.1 μM, yet possibly also due to effects other than Jumonji demethylase inhibition (Luo et al., 2011).
Moreover, it was recently observed that an inhibitor of G9a methyltransferase (BIX‐01294) and its analog E67 could also inhibit human H3K9me2 demethylase KIAA1718 (JHDM1D). Both enzymes can recognize methyllysine residues either as product or as substrate. Compound E67 (31) was shown to inhibit KIAA1718 with an IC50 value of 3.7 μM in an in vitro mass spectrometric demethylation assay. E67 was also shown to be active against the related demethylase PHF8, but inactive against JARID1C. It did, however, exhibit cytotoxicity at concentrations around 50 μM against various cell types. A crystal structure confirmed binding of this compound to the active site of the enzyme (Upadhyay et al., 2012).
With the great amount of structural knowledge and insight into the catalytic mechanism of Jumonji demethylases available to date, structure‐ and mechanism‐based approaches for inhibitor design can be envisioned.
Subtype‐selective inhibitors were designed based on the peptidic substrates of Jumonji demethylases by thoroughly investigating the kinetics and binding affinities of different peptides to the different enzymes of the Jumonji family and coupling them to a small molecule inhibitor at the H3K9‐lysine ϵ‐amino group. Based on the best substrate length of a truncated histone tail peptide that would still be recognized by each subtype, inhibitors were synthesized carrying a uracil or bromouracil group and evaluated in vitro using the FDH‐coupled assay as well as MALDI‐TOF techniques. This yielded, for example, the 5‐mer peptide H3(7–11) with a bromouracil moiety ARK(BrU)ST as an inhibitor of JMJD2C (K i = 27 μM) and a four‐fold selectivity over the related subtype JMJD2A (Lohse et al., 2011b).
In a similar approach, a novel inhibitor was recently designed by investigating the crystal structure of Jumonji demethylases. In a two‐component approach, a mimic of the peptidic substrate was chemically connected to an analog of the co‐substrate α‐ketoglutarate via a disulfide bridge. This compound was able to bind to both binding sites in the enzyme simultaneously with high affinity and chelate the active center iron(II) ion, inhibiting the enzymatic reaction. The binding mode was confirmed through crystal structure analyses of the enzyme in complex with the two‐component inhibitor. A chemically stabilized analog potently inhibited JMJD2E (IC50 = 90 nM) and JMJD2A (IC50 = 270 nM) in vitro, but not any of the other JmjC domain‐containing enzymes investigated in this study (Woon et al., 2012).
While these two studies revealed potent and in part highly selective inhibitors, their peptidic nature can be expected to be an obstacle for further development of these compounds into potential drugs with regard to cell permeability, intracellular stability, and other pharmacokinetic parameters.
Even though Jumonji demethylases were discovered only quite recently, a number of different inhibitors have already been reported. Nonetheless, most of their scaffolds derive from other structurally or mechanistically related enzymes and these compounds are, therefore, oftentimes also active against other enzyme families. Most inhibitors are substrate and co‐substrate mimics and so far have only very limited or undetermined specificity. To date, there is no reported inhibitor with proven selectivity for any one subfamily or even among closely related isoforms. It will, thus, be a paramount objective for the near future to discover more potent and, especially important, more selective inhibitors with the ability to target specific subtypes and isoforms in order to develop them into useful pharmacologic tools. They can then be used to further the biochemical understanding of the roles of these enzymes and to develop new drugs. This might be achieved by a combination of structure‐guided design, virtual screening, high‐throughput screening, further structural insight into the differences of the enzyme isoforms and the development of novel Jumonji‐specific inhibitor compound scaffolds.
7. Structural aspects of the demethylase LSD1 with implications to inhibitor design
As a FAD‐dependant amino oxidase (Lohse et al., 2011a; Shi et al., 2004; Spannhoff et al., 2009a) LSD1 shares structural similarity and sequence identity with other amino oxidase enzymes including MAO A/B (∼20% sequence identity) (Yang et al., 2007b) and polyaminoxidases (PAO ∼26% sequence identity) (Forneris et al., 2009). LSD1 possesses a catalytic site closely related to the other amino oxidases. However despite similarities, LSD1 possesses a unique peptide substrate binding pocket that is much larger than that of MAO A and MAO B (Forneris et al., 2009). Several X‐ray structures of LSD1 have been solved within the last few years (Aravind and Iyer, 2002; Chen et al., 2006a; Forneris et al., 2007; Lee et al., 2005b; Mimasu et al., 2008; Stavropoulos et al., 2006; Yang et al., 2007, 2007, 2006). These 3D structures show that LSD1 contains three domains: an N‐terminal SWIRM domain (Aravind and Iyer, 2002; Stavropoulos et al., 2006), a tower domain which is formed by two long α‐helices and it is a sort of “binding platform” for interacting proteins such as CoREST (Forneris et al., 2007; Lee et al., 2005b; Stavropoulos et al., 2006; Yang et al., 2006), and an amino oxidase domain which hosts the catalytic site. The catalytic site is formed by two different lobes, the first one recognizes and binds substrates, whereas the second one binds the flavin adenine dinucleotide cofactor (Stavropoulos et al., 2006; Yang et al., 2006).
From crystal structures of LSD1 co‐crystalized with histone H3 peptides, several interactions were described as important binding hotspots (pdb codes: 2V1D (Forneris et al., 2007), 2UXN (Yang et al., 2007a) and 2Y48 (Baron et al., 2011)). In one of these peptides, encompassing the terminal 12 residues of H3, the exchange of Lys4 to a methionine resulted in a potent LSD1 inhibitor with a K i of 0.05 μM. The X‐ray structures of the peptide‐LSD1 complexes showed that the peptide adopts three consecutive γ‐turns that resulted in a perfect fit with the binding pocket. The analysis further revealed that only three residues of the histone tail can be hosted in the binding pocket (Figure 10). Major contributions to peptide binding are coming from hydrogen bond interaction of H3‐Arg8 with Asp375, Glu379, and the backbone of Cys360 as well as the interaction of H3‐Arg2 with Asp553 and Asp556 of LSD1 (Figure 9) (Baron et al., 2011; Forneris et al., 2007; Yang et al., 2007a).
Figure 10.
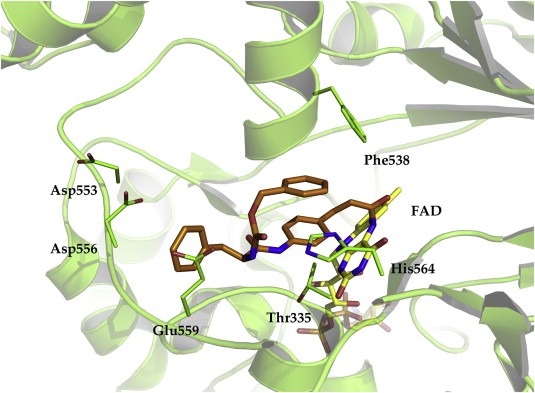
Crystal structure of LSD1 in complex with a covalently bound tranylcypromine derivative (2XAS.pdb). The inhibitor is colored orange whereas the cofactor is colored yellow. The protein backbone is displayed as green ribbon and only relevant amino acids for the interaction are shown.
The first inhibitors that were crystallized with LSD1 were tranylcypromine and analogs (Binda et al., 2010; Mimasu et al., 2010; Ueda et al., 2009). Eight crystal structures with these sort of inhibitors were solved within the last few years (pdb codes: 2XAS (Binda et al., 2010), 2XAH (Binda et al., 2010), 2XAG (Binda et al., 2010), 2XAF (Binda et al., 2010), 2XAJ (Binda et al., 2010), 2XAQ (Binda et al., 2010), 3ABT (Mimasu et al., 2010) and 3ABU (Mimasu et al., 2010)) (Binda et al., 2010; Mimasu et al., 2010). It was observed that tranylcypromine, which is a suicide inhibitor, does not seem to interact strongly with the active site of LSD1. The phenyl ring of the FAD‐tranylcypromine adduct makes only weak interactions with the methyl groups of Thr335 and Thr810. Interestingly, the co‐crystallization of tranylcypromine with LSD1 resulted in two different reaction adducts. In one complex, a five‐membered ring linking C‐4a and N‐5 of FAD is formed. The other reaction product shows a 3‐phenylpropanol substituent attached to the N‐5 atom of FAD. Increasing the size of the substitutent at the phenyl ring of tranylcypromine resulted in more bulky derivatives (in 2XAS.pdb, Figure 10) that could be cocrystallized with LSD1. It shows that the “head” of the inhibitor is covalently attached to the FAD cofactor whereas the “tail” of the inhibitor fills the central part of the substrate binding pocket. One of the aromatic rings present in the ligand is even facing out of the binding pocket. This shows that the fit between inhibitor and LSD1 is far from being perfect (Binda et al., 2010). In order to fully understand the relevance of different binding modes and to design more active inhibitors, the solution of new crystal structures with non‐covalent LSD1 inhibitors is still mandatory.
8. Structural information to guide further inhibitor design: JmjC‐domain containing histone demethylases
This class comprises 28 members and can be categorized based on their phylogeny into seven different subfamilies (KDM2; KDM3; KDM4; KDM5, JARID2; KDM6; KDM7; KDM8) (Arrowsmith et al., 2012; Lohse et al., 2011a; Mosammaparast and Shi, 2010). The difference between them consists of a number of different domains for DNA‐binding, substrate recognition, stabilization and yet uncharacterized domains (Lohse et al., 2011a).
JmjC‐domain containing proteins are members of the Cupin superfamily and form a jelly‐role like all β‐fold 3D structure that includes Fe(II) and α‐ketoglutarate (αKG) as co‐factors (Hou and Yu, 2010; Lohse et al., 2011a). Nowadays different crystal structures are available which give insights into the binding modes of cofactor (αKG) and mono‐, di‐ or tri‐methylated lysine substrates of different JmjC‐subfamilies (Chen et al., 2006b; Couture et al., 2007). Also several crystal structures of inhibitor‐complexes are resolved. The inhibitors N‐oxalylglycine (NOG, pdb: 2Q8E, 2XML, 3DXU, 3AVR, 3KV4, 3PU3) (Couture et al., 2007; Horton et al., 2011, 2009; Sengoku and Yokoyama, 2011), 8‐hydroxyquinoline‐5‐carboxylic acid (pdb: 3NJY) (Shin and Janknecht, 2007), pyridine‐2,4‐dicarboxylic acid (pdb: 2VD7, 2W2I) (Hillringhaus et al., 2011), 8‐hydroxy‐3‐(piperazin‐1‐yl)quinolone‐5‐carboxylic acid (pdb: 3RVH) and diaminozide (pdb: 4DO0) (Rose et al., 2012) target the cofactor binding pocket and try to interact with the Fe(II) ion. Another inhibitor complex is available which targets the substrate ammonium binding pocket (pdb: 3U78) (Upadhyay et al., 2012). A full list of all available human JmjC domain containing structures is given in the supporting material (Table S 1).
The superposition of JmjC‐domain of subfamilies KDM2A, KDM4A, KDM6A and KDM7 shows a high similarity of the catalytic domain among different families (Figure 11). The iron ion (nickel ion: 2WWJ, 3AVR, 3U78) is octahedrally coordinated by two histidine residues that are conserved in all KDMs (H212, H188, H1146, H282) and (H284, H276, H1226, H354) the order corresponds to Figure 11. The third coordination position differs within different families ‐ aspartic acid in KDM2/6 and glutamic acid in KDM4/7, D214, E190, E1148, D284 respectively. The cofactor αKG and a water molecule occupy the remaining three coordination positions.
Figure 11.
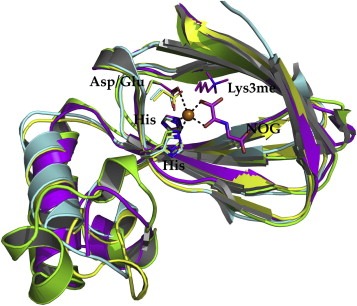
Superposition of KDM2A (yellow, PDB ID 2YU1), KDM4A (green, PDB ID 2WWJ), KDM6A (magenta, PDB ID 3AVR) and KDM7 (cyan, PDB ID 3U78) of the JmjC domain only (RMSD CA: 1.1Å). The residues chelating the iron ion are represented in sticks and color coded equivalent to their ribbon. The inhibitor N‐oxalylglycine (NOG) and the trimethylated lysine of 3AVR are shown in sticks and colored in magenta.
The residues of the cofactor binding site are more distinct among the subfamilies and offer therefore the potential for a selective structure based drug design. Figure 12 shows the differences of key amino acids of the cofactor binding site. N198 (2WWJ, KDM4A) and N1156 (3AVR, KDM6A) establish a hydrogen bond at the back of the pocket with the carboxylic moiety of NOG and O‐benzyl‐N‐(carboxycarbonyl)‐d‐tyrosine next to the Fe(II) ion. In the structures 2YU1 (KDM2A) and 3U78 (KDM7) the aspargine is replaced by a tyrosine (Y222 and Y292 respectively), which causes a different Fe(II) coordination of the carboxylic moiety of αKG and a loss of the hydrogen bond. The co‐crystallized inhibitor O‐benzyl‐N‐(carboxycarbonyl)‐d‐tyrosine of 2WWJ offers a π–π stacking interaction with F185 at the front part of the pocket, that is only conserved within the KDM4 and KDM5 subfamily (Lohse et al., 2011a). KDM2A/6A/7 show a threonine at this location (T209, T1143, T279 respectively), which prohibit a π‐ring system at this position (Figure 12).
Figure 12.
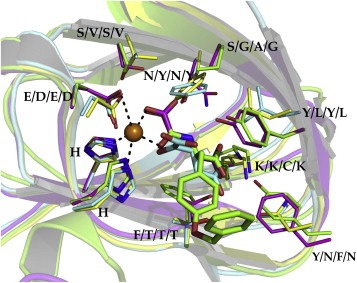
Close‐up view of the active sites corresponding to Figure 11. Additionally, the αKG cofactor (cyan, 3U78) and the inhibitor O‐benzyl‐N‐(carboxycarbonyl)‐d‐tyrosine (green, 2WWJ) are depicted. The residue labels are ordered according to the different subfamilies KDM2A, KDM4A, KDM6A and KDM7, respectively.
As more and particularly better resolved crystal structures become available especially from yet unresolved subfamilies (e.g. KMD3, KDM5) will lead to a better understanding and generation of new selective drug candidates.
9. Conclusion
The two classes of histone demethylases, Lysine specific demethylases and JumonjiC domain containing demethylases, present emerging targets for drug discovery. They play a role in various malignant tumors and leukemia as shown by analysis of patient samples and overexpression and knockdown experiments as well as inhibitor studies. For some advanced LSD1 inhibitors, promising data exists for treatment of xenograft tumors in animal models and. The search for inhibitors has yielded potent and specific LSD1 inhibitors, whereas the existing inhibitors of JMJD yet fail to distinguish between the different families of JMJD and often also recognize similar enzymes from unrelated families. Structural studies of histone demethylases will help in the design of inhibitors with increased potency and selectivity.
Note added in proof
A selective inhibitors for the demethylase JMJD3 has been described after acceptance of our manuscript: Kruidenier, L. et al. Nature (2012) doi:10.1038/nature11262.
Supporting information
The following is/are the supplementary data related to this article:
Supplementary data
Acknowledgments
We thank the Deutsche Forschungsgemeinschaft for funding of research of inhibitors of LSD1 (M. Jung: Ju295/7‐1; W. Sippl: Si868/7‐1) and JmJC‐domain containing demethylases (Jung: Project A04 within SFB992 Medical Epigenetics "MEDEP").
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2012.07.004.
Hoffmann Inga, Roatsch Martin, Schmitt Martin L., Carlino Luca, Pippel Martin, Sippl Wolfgang, Jung Manfred, (2012), The role of histone demethylases in cancer therapy, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.07.004.
References
- Adamo, A. , Sese, B. , Boue, S. , Castano, J. , Paramonov, I. , Barrero, M.J. , Izpisua Belmonte, J.C. , 2011. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell. Biol. 13, 652–659. [DOI] [PubMed] [Google Scholar]
- Agger, K. , Christensen, J. , Cloos, P.A. , Helin, K. , 2008. The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 18, 159–168. [DOI] [PubMed] [Google Scholar]
- Albert, M. , Helin, K. , 2010. Histone methyltransferases in cancer. Semin. Cell. Dev. Biol. 21, 209–220. [DOI] [PubMed] [Google Scholar]
- Anand, R. , Marmorstein, R. , 2007. Structure and mechanism of lysine-specific demethylase enzymes. J. Biol. Chem. 282, 35425–35429. [DOI] [PubMed] [Google Scholar]
- Anderton, J.A. , Bose, S. , Vockerodt, M. , Vrzalikova, K. , Wei, W. , Kuo, M. , Helin, K. , Christensen, J. , Rowe, M. , Murray, P.G. , Woodman, C.B. , 2011. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin's Lymphoma. Oncogene. 30, 2037–2043. [DOI] [PubMed] [Google Scholar]
- Aravind, L. , Iyer, L.M. , 2002. The SWIRM domain: a conserved module found in chromosomal proteins points to novel chromatin-modifying activities. Genome. Biol. 3, Research 0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith, C.H. , Bountra, C. , Fish, P.V. , Lee, K. , Schapira, M. , 2012. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Bannister, A.J. , Zegerman, P. , Partridge, J.F. , Miska, E.A. , Thomas, J.O. , Allshire, R.C. , Kouzarides, T. , 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Baron, R. , Binda, C. , Tortorici, M. , McCammon, J.A. , Mattevi, A. , 2011. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure. 19, 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benelkebir, H. , Hodgkinson, C. , Duriez, P.J. , Hayden, A.L. , Bulleid, R.A. , Crabb, S.J. , Packham, G. , Ganesan, A. , 2011. Enantioselective synthesis of tranylcypromine analogues as lysine demethylase (LSD1) inhibitors. Bioorg. Med. Chem. 19, 3709–3716. [DOI] [PubMed] [Google Scholar]
- Binda, C. , Li, M. , Hubalek, F. , Restelli, N. , Edmondson, D.E. , Mattevi, A. , 2003. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. U S A. 100, 9750–9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda, C. , Valente, S. , Romanenghi, M. , Pilotto, S. , Cirilli, R. , Karytinos, A. , Ciossani, G. , Botrugno, O.A. , Forneris, F. , Tardugno, M. , Edmondson, D.E. , Minucci, S. , Mattevi, A. , Mai, A. , 2010. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132, 6827–6833. [DOI] [PubMed] [Google Scholar]
- Bissinger, E.M. , Heinke, R. , Sippl, W. , Jung, M. , 2010. Histone methyltransferase inhibitors. Med. Chem. Commun. 1, 114–124. [Google Scholar]
- Brasacchio, D. , Okabe, J. , Tikellis, C. , Balcerczyk, A. , George, P. , Baker, E.K. , Calkin, A.C. , Brownlee, M. , Cooper, M.E. , El-Osta, A. , 2009. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 58, 1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, B. , Chen, Y. , Zhao, Y. , Bruick, R.K. , 2007. JMJD6 is a histone arginine demethylase. Science. 318, 444–447. [DOI] [PubMed] [Google Scholar]
- Chang, K.H. , King, O.N. , Tumber, A. , Woon, E.C. , Heightman, T.D. , McDonough, M.A. , Schofield, C.J. , Rose, N.R. , 2011. Inhibition of histone demethylases by 4-carboxy-2,2'-bipyridyl compounds. Chem. Med. Chem. 6, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, P.C. , Fitzgerald, L.D. , Hsia, D.A. , Izumiya, Y. , Wu, C.Y. , Hsieh, W.P. , Lin, S.F. , Campbell, M. , Lam, K.S. , Luciw, P.A. , Tepper, C.G. , Kung, H.J. , 2011. Histone demethylase JMJD2A regulates Kaposi's sarcoma-associated herpes virus replication and is targeted by a viral transcriptional factor. J. Virol. 85, 3283–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Giri, N.C. , Zhang, R. , Yamane, K. , Zhang, Y. , Maroney, M. , Costa, M. , 2010. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J. Biol. Chem. 285, 7374–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Yang, Y. , Wang, F. , Wan, K. , Yamane, K. , Zhang, Y. , Lei, M. , 2006. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. U S A. 103, 13956–13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Zang, J. , Whetstine, J. , Hong, X. , Davrazou, F. , Kutateladze, T.G. , Simpson, M. , Mao, Q. , Pan, C.-H. , Dai, S. , Hagman, J. , Hansen, K. , Shi, Y. , Zhang, G. , 2006. Structural insights into histone demethylation by JMJD2 family members. Cell. 125, 691–702. [DOI] [PubMed] [Google Scholar]
- Christensen, J. , Agger, K. , Cloos, P.A.C. , Pasini, D. , Rose, S. , Sennels, L. , Rappsilber, J. , Hansen, K.H. , Salcini, A.E. , Helin, K. , 2007. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 128, 1063–1076. [DOI] [PubMed] [Google Scholar]
- Clausen, R.P. , Pedersen, M.T. , Helin, K. , 2009. Histone demethylases. In Sippl W., Jung M.(Eds.), Epigenetic Targets in Drug Discovery. Wiley-VCH; Weinheim: 269–290. [Google Scholar]
- Clifton, I.J. , McDonough, M.A. , Ehrismann, D. , Kershaw, N.J. , Granatino, N. , Schofield, C.J. , 2006. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J. Inorg. Biochem. 100, 644–669. [DOI] [PubMed] [Google Scholar]
- Cloos, P.A. , Christensen, J. , Agger, K. , Maiolica, A. , Rappsilber, J. , Antal, T. , Hansen, K.H. , Helin, K. , 2006. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 442, 307–311. [DOI] [PubMed] [Google Scholar]
- Couture, J.F. , Collazo, E. , Ortiz-Tello, P.A. , Brunzelle, J.S. , Trievel, R.C. , 2007. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat. Struct. Mol. Biol. 14, 689–695. [DOI] [PubMed] [Google Scholar]
- Eissenberg, J.C. , Lee, M.G. , Schneider, J. , Ilvarsonn, A. , Shiekhattar, R. , Shilatifard, A. , 2007. The trithorax-group gene in Drosophila little imaginal discs encodes a trimethylated histone H3 Lys4 demethylase. Nat. Struct. Mol. Biol. 14, 344–346. [DOI] [PubMed] [Google Scholar]
- Fodor, B.D. , Kubicek, S. , Yonezawa, M. , O'Sullivan, R.J. , Sengupta, R. , Perez-Burgos, L. , Opravil, S. , Mechtler, K. , Schotta, G. , Jenuwein, T. , 2006. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 20, 1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris, F. , Battaglioli, E. , Mattevi, A. , Binda, C. , 2009. New roles of flavoproteins in molecular cell biology: histone demethylase LSD1 and chromatin. FEBS J. 276, 4304–4312. [DOI] [PubMed] [Google Scholar]
- Forneris, F. , Binda, C. , Adamo, A. , Battaglioli, E. , Mattevi, A. , 2007. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 282, 20070–20074. [DOI] [PubMed] [Google Scholar]
- Garcia-Bassets, I. , Kwon, Y.S. , Telese, F. , Prefontaine, G.G. , Hutt, K.R. , Cheng, C.S. , Ju, B.G. , Ohgi, K.A. , Wang, J. , Escoubet-Lozach, L. , Rose, D.W. , Glass, C.K. , Fu, X.D. , Rosenfeld, M.G. , 2007. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 128, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guibourt, N., 2010. Phenylcyclopropylamine derivatives and their medical use. International Patent, WO2010/084160.
- Guo, X. , Shi, M. , Sun, L. , Wang, Y. , Gui, Y. , Cai, Z. , Duan, X. , 2011. The expression of histone demethylase JMJD1A in renal cell carcinoma. Neoplasma. 58, 153–157. [DOI] [PubMed] [Google Scholar]
- Hamada, S. , Kim, T.D. , Suzuki, T. , Itoh, Y. , Tsumoto, H. , Nakagawa, H. , Janknecht, R. , Miyata, N. , 2009. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg. Med. Chem. Lett. 19, 2852–2855. [DOI] [PubMed] [Google Scholar]
- Hamada, S. , Suzuki, T. , Mino, K. , Koseki, K. , Oehme, F. , Flamme, I. , Ozasa, H. , Itoh, Y. , Ogasawara, D. , Komaarashi, H. , Kato, A. , Tsumoto, H. , Nakagawa, H. , Hasegawa, M. , Sasaki, R. , Mizukami, T. , Miyata, N. , 2010. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of Jumonji domain-containing protein 2 histone demethylase inhibitors. J. Med. Chem. 53, 5629–5638. [DOI] [PubMed] [Google Scholar]
- Harris, W.J. , Huang, X. , Lynch, J.T. , Spencer, G.J. , Hitchin, J.R. , Li, Y. , Ciceri, F. , Blaser, J.G. , Greystoke, B.F. , Jordan, A.M. , Miller, C.J. , Ogilvie, D.J. , Somervaille, T.C. , 2012. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 21, 473–487. [DOI] [PubMed] [Google Scholar]
- Hausinger, R.P. , 2004. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68. [DOI] [PubMed] [Google Scholar]
- Hayami, S. , Kelly, J.D. , Cho, H.S. , Yoshimatsu, M. , Unoki, M. , Tsunoda, T. , Field, H.I. , Neal, D.E. , Yamaue, H. , Ponder, B.A. , Nakamura, Y. , Hamamoto, R. , 2011. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer. 128, 574–586. [DOI] [PubMed] [Google Scholar]
- Helias, C. , Struski, S. , Gervais, C. , Leymarie, V. , Mauvieux, L. , Herbrecht, R. , Lessard, M. , 2008. Polycythemia vera transforming to acute myeloid leukemia and complex abnormalities including 9p homogeneously staining region with amplification of MLLT3, JMJD2C, JAK2, and SMARCA2. Cancer Genet. Cytogen. 180, 51–55. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. , Shilatifard, A. , 2011. Histone modification: cause or cog?. Trends Genet. 27, 389–396. [DOI] [PubMed] [Google Scholar]
- Hillringhaus, L. , Yue, W.W. , Rose, N.R. , Ng, S.S. , Gileadi, C. , Loenarz, C. , Bello, S.H. , Bray, J.E. , Schofield, C.J. , Oppermann, U. , 2011. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 286, 41616–41625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, J.R. , Upadhyay, A.K. , Hashimoto, H. , Zhang, X. , Cheng, X. , 2011. Structural basis for human PHF2 Jumonji domain interaction with metal ions. J. Mol. Biol. 406, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, J.R. , Upadhyay, A.K. , Qi, H.H. , Zhang, X. , Shi, Y. , Cheng, X. , 2009. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 17, 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, H. , Yu, H. , 2010. Structural insights into histone lysine demethylation. Curr. Opin. Struct. Biol. 20, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Q. , Kwon, Y.S. , Nunez, E. , Cardamone, M.D. , Hutt, K.R. , Ohgi, K.A. , Garcia-Bassets, I. , Rose, D.W. , Glass, C.K. , Rosenfeld, M.G. , Fu, X.D. , 2008. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. U S A. 105, 19199–19204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Sengupta, R. , Espejo, A.B. , Lee, M.G. , Dorsey, J.A. , Richter, M. , Opravil, S. , Shiekhattar, R. , Bedford, M.T. , Jenuwein, T. , Berger, S.L. , 2007. p53 is regulated by the lysine demethylase LSD1. Nature. 449, 105–U180. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Greene, E. , Murray Stewart, T. , Goodwin, A.C. , Baylin, S.B. , Woster, P.M. , Casero, R.A. , 2007. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. U S A. 104, 8023–8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , Stewart, T.M. , Wu, Y. , Baylin, S.B. , Marton, L.J. , Perkins, B. , Jones, R.J. , Woster, P.M. , Casero, R.A. , 2009. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin. Cancer Res. 15, 7217–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano, A. , Attias, R. , Aurias, A. , Perot, G. , Burel-Vandenbos, F. , Otto, J. , Venissac, N. , Pedeutour, F. , 2006. Molecular cytogenetic characterization of a metastatic lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genet. Cytogen. 167, 122–130. [DOI] [PubMed] [Google Scholar]
- Ivan, M. , Haberberger, T. , Gervasi, D.C. , Michelson, K.S. , Gunzler, V. , Kondo, K. , Yang, H. , Sorokina, I. , Conaway, R.C. , Conaway, J.W. , Kaelin, W.G. , 2002. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. U S A. 99, 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase, S. , Lan, F. , Bayliss, P. , de la Torre-Ubieta, L. , Huarte, M. , Qi, H.H. , Whetstine, J.R. , Bonni, A. , Roberts, T.M. , Shi, Y. , 2007. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 128, 1077–1088. [DOI] [PubMed] [Google Scholar]
- Kahl, P. , Gullotti, L. , Heukamp, L.C. , Wolf, S. , Friedrichs, N. , Vorreuther, R. , Solleder, G. , Bastian, P.J. , Ellinger, J. , Metzger, E. , Schule, R. , Buettner, R. , 2006. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 66, 11341–11347. [DOI] [PubMed] [Google Scholar]
- Karytinos, A. , Forneris, F. , Profumo, A. , Ciossani, G. , Battaglioli, E. , Binda, C. , Mattevi, A. , 2009. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 284, 17775–17782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, E.C. , Robinson, B.D. , Downes, M.J. , Powell, L.G. , Lee, M.M. , Scherr, D.S. , Gudas, L.J. , Mongan, N.P. , 2011. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol. Carcinogen. 50, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazu, M. , Saso, K. , Tong, K.I. , McQuire, T. , Goto, K. , Son, D.O. , Wakeham, A. , Miyagishi, M. , Mak, T.W. , Okada, H. , 2011. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS ONE. 6, e17830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, T.D. , Oh, S. , Shin, S. , Janknecht, R. , 2012. Regulation of tumor suppressor p53 and HCT116 cell physiology by histone demethylase JMJD2D/KDM4D. PLoS ONE. 7, e34618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, O.N. , Li, X.S. , Sakurai, M. , Kawamura, A. , Rose, N.R. , Ng, S.S. , Quinn, A.M. , Rai, G. , Mott, B.T. , Beswick, P. , Klose, R.J. , Oppermann, U. , Jadhav, A. , Heightman, T.D. , Maloney, D.J. , Schofield, C.J. , Simeonov, A. , 2010. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS ONE. 5, e15535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose, R.J. , Gardner, K.E. , Liang, G. , Erdjument-Bromage, H. , Tempst, P. , Zhang, Y. , 2007. Demethylation of histone H3K36 and H3K9 by Rph1: a Vestige of an H3K9 methylation system in Saccharomyces cerevisiae?. Mol. Cell. Biol. 27, 3951–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose, R.J. , Kallin, E.M. , Zhang, Y. , 2006. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7, 715–727. [DOI] [PubMed] [Google Scholar]
- Klose, R.J. , Yamane, K. , Bae, Y. , Zhang, D. , Erdjument-Bromage, H. , Tempst, P. , Wong, J. , Zhang, Y. , 2006. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 442, 312–316. [DOI] [PubMed] [Google Scholar]
- Klose, R.J. , Yan, Q. , Tothova, Z. , Yamane, K. , Erdjument-Bromage, H. , Tempst, P. , Gilliland, D.G. , Zhang, Y. , Kaelin, W.G. , 2007. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 128, 889–900. [DOI] [PubMed] [Google Scholar]
- Klose, R.J. , Zhang, Y. , 2007. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell. Biol. 8, 307–318. [DOI] [PubMed] [Google Scholar]
- Kooistra, S.M. , Helin, K. , 2012. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell. Biol. 13, 297–311. [DOI] [PubMed] [Google Scholar]
- Kouzarides, T. , 2007. Chromatin modifications and their function. Cell. 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Lee, D.Y. , Teyssier, C. , Strahl, B.D. , Stallcup, M.R. , 2005. Role of protein methylation in regulation of transcription. Endocr. Rev. 26, 147–170. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Thompson, J.R. , Botuyan, M.V. , Mer, G. , 2008. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 15, 109–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. , Norman, J. , Shilatifard, A. , 2007. ScienceDirect.com - Cell - Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a Polycomb-like protein. Cell. 128, 877–887. [DOI] [PubMed] [Google Scholar]
- Lee, M.G. , Wynder, C. , Cooch, N. , Shiekhattar, R. , 2005. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 437, 432–435. [DOI] [PubMed] [Google Scholar]
- Lee, M.G. , Wynder, C. , Schmidt, D.M. , McCafferty, D.G. , Shiekhattar, R. , 2006. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 13, 563–567. [DOI] [PubMed] [Google Scholar]
- Lee, N. , Zhang, J. , Klose, R.J. , Erdjument-Bromage, H. , Tempst, P. , Jones, R.S. , Zhang, Y. , 2007. The trithorax-group protein Lid is a histone H3 trimethyl-Lys4 demethylase. Nat. Struct. Mol. Biol. 14, 341–343. [DOI] [PubMed] [Google Scholar]
- Li, B.X. , Zhang, M.C. , Luo, C.L. , Yang, P. , Li, H. , Xu, H.M. , Xu, H.F. , Shen, Y.W. , Xue, A.M. , Zhao, Z.Q. , 2011. Effects of RNA interference-mediated gene silencing of JMJD2A on human breast cancer cell line MDA-MB-231 in vitro. J. Exp. Clin. Canc. Res. 30, 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Zhao, L. , Zang, W. , Liu, Z. , Chen, L. , Liu, T. , Xu, D. , Jia, J. , 2011. Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is overexpressed in gastric cancer. Biochem. Biophys. Res. Commun. 416, 372–378. [DOI] [PubMed] [Google Scholar]
- Liang, G. , Klose, R.J. , Gardner, K.E. , Zhang, Y. , 2007. Yeast Jhd2p is a histone H3 Lys4 trimethyl demethylase. Nat. Struct. Mol. Biol. 14, 243–245. [DOI] [PubMed] [Google Scholar]
- Lim, S. , Janzer, A. , Becker, A. , Zimmer, A. , Schule, R. , Buettner, R. , Kirfel, J. , 2010. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 31, 512–520. [DOI] [PubMed] [Google Scholar]
- Liu, G. , Bollig-Fischer, A. , Kreike, B. , van de Vijver, M.J. , Abrams, J. , Ethier, S.P. , Yang, Z.Q. , 2009. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 28, 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse, B. , Kristensen, J.L. , Kristensen, L.H. , Agger, K. , Helin, K. , Gajhede, M. , Clausen, R.P. , 2011. Inhibitors of histone demethylases. Bioorg. Med. Chem. 19, 3625–3636. [DOI] [PubMed] [Google Scholar]
- Lohse, B. , Nielsen, A.L. , Kristensen, J.B. , Helgstrand, C. , Cloos, P.A. , Olsen, L. , Gajhede, M. , Clausen, R.P. , Kristensen, J.L. , 2011. Targeting histone lysine demethylases by truncating the histone 3 tail to obtain selective substrate-based inhibitors. Angew. Chem. Int. Ed. 50, 9100–9103. [DOI] [PubMed] [Google Scholar]
- Luo, X. , Liu, Y. , Kubicek, S. , Myllyharju, J. , Tumber, A. , Ng, S. , Che, K.H. , Podoll, J. , Heightman, T.D. , Oppermann, U. , Schreiber, S.L. , Wang, X. , 2011. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 133, 9451–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, P.A. , Breslow, R. , 2007. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90. [DOI] [PubMed] [Google Scholar]
- McDonough, M.A. , McNeill, L.A. , Tilliet, M. , Papamicael, C.A. , Chen, Q.Y. , Banerji, B. , Hewitson, K.S. , Schofield, C.J. , 2005. Selective inhibition of factor inhibiting hypoxia-inducible factor. J. Am. Chem. Soc. 127, 7680–7681. [DOI] [PubMed] [Google Scholar]
- Metzger, E. , Imhof, A. , Patel, D. , Kahl, P. , Hoffmeyer, K. , Friedrichs, N. , Muller, J.M. , Greschik, H. , Kirfel, J. , Ji, S. , Kunowska, N. , Beisenherz-Huss, C. , Gunther, T. , Buettner, R. , Schule, R. , 2010. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 464, 792–796. [DOI] [PubMed] [Google Scholar]
- Metzger, E. , Wissmann, M. , Yin, N. , Muller, J.M. , Schneider, R. , Peters, A.H. , Gunther, T. , Buettner, R. , Schule, R. , 2005. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 437, 436–439. [DOI] [PubMed] [Google Scholar]
- Meyer, R. , Wolf, S.S. , Obendorf, M. , 2007. PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J. Steroid. Biochem. Mol. Biol. 107, 1–14. [DOI] [PubMed] [Google Scholar]
- Mimasu, S. , Sengoku, T. , Fukuzawa, S. , Umehara, T. , Yokoyama, S. , 2008. Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochem. Biophys. Res. Commun. 366, 15–22. [DOI] [PubMed] [Google Scholar]
- Mimasu, S. , Umezawa, N. , Sato, S. , Higuchi, T. , Umehara, T. , Yokoyama, S. , 2010. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry (Mosc). 49, 6494–6503. [DOI] [PubMed] [Google Scholar]
- Mitra, D. , Das, P.M. , Huynh, F.C. , Jones, F.E. , 2011. Jumonji/ARID1 B (JARID1B) protein promotes breast tumor cell cycle progression through epigenetic repression of microRNA let-7e. J. Biol. Chem. 286, 40531–40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosammaparast, N. , Shi, Y. , 2010. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 79, 155–179. [DOI] [PubMed] [Google Scholar]
- Ng, H.H. , Robert, F. , Young, R.A. , Struhl, K. , 2003. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 11, 709–719. [DOI] [PubMed] [Google Scholar]
- Ng, S.S. , Kavanagh, K.L. , McDonough, M.A. , Butler, D. , Pilka, E.S. , Lienard, B.M. , Bray, J.E. , Savitsky, P. , Gileadi, O. , von Delft, F. , Rose, N.R. , Offer, J. , Scheinost, J.C. , Borowski, T. , Sundstrom, M. , Schofield, C.J. , Oppermann, U. , 2007. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 448, 87–91. [DOI] [PubMed] [Google Scholar]
- Nicholson, T.B. , Chen, T. , 2009. LSD1 demethylates histone and non-histone proteins. Epigenetics. 4, 129–132. [DOI] [PubMed] [Google Scholar]
- Nielsen, A.L. , Kristensen, L.H. , Stephansen, K.B. , Kristensen, J.B. , Helgstrand, C. , Lees, M. , Cloos, P. , Helin, K. , Gajhede, M. , Olsen, L. , 2012. Identification of catechols as histone-lysine demethylase inhibitors. FEBS Lett. 586, 1190–1194. [DOI] [PubMed] [Google Scholar]
- Nimura, K. , Ura, K. , Kaneda, Y. , 2010. Histone methyltransferases: regulation of transcription and contribution to human disease. J. Mol. Med. 88, 1213–1220. [DOI] [PubMed] [Google Scholar]
- Oh, S. , Janknecht, R. , 2012. Histone demethylase JMJD5 is essential for embryonic development. Biochem. Biophys. Res. Commun. 420, 61–65. [DOI] [PubMed] [Google Scholar]
- Pollock, J.A. , Larrea, M.D. , Jasper, J.S. , McDonnell, D.P. , McCafferty, D.G. , 2012. Lysine-specific histone demethylase 1 inhibitors control breast cancer proliferation in ERalpha-dependent and -independent manners. ACS Chem. Biol. Article ASAP. 10.1021/cb300108c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch, A. , Becker, B. , Schneider-Brachert, W. , Hagen, I. , Landthaler, M. , Vogt, T. , 2006. Re-expression of the retinoblastoma-binding protein 2-homolog 1 reveals tumor-suppressive functions in highly metastatic melanoma cells. J. Invest. Dermatol. 126, 1850–1859. [DOI] [PubMed] [Google Scholar]
- Rose, N.R. , Ng, S.S. , Mecinovic, J. , Lienard, B.M. , Bello, S.H. , Sun, Z. , McDonough, M.A. , Oppermann, U. , Schofield, C.J. , 2008. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 51, 7053–7056. [DOI] [PubMed] [Google Scholar]
- Rose, N.R. , Woon, E.C. , Kingham, G.L. , King, O.N. , Mecinovic, J. , Clifton, I.J. , Ng, S.S. , Talib-Hardy, J. , Oppermann, U. , McDonough, M.A. , Schofield, C.J. , 2010. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. J. Med. Chem. 53, 1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, N.R. , Woon, E.C. , Tumber, A. , Walport, L.J. , Chowdhury, R. , Li, X.S. , King, O.N. , Lejeune, C. , Ng, S.S. , Krojer, T. , Chan, M.C. , Rydzik, A.M. , Hopkinson, R.J. , Che, K. , Daniel, M. , Strain-Damerell, C. , Gileadi, C. , Kochan, G. , Leung, I.K. , Dunford, J.E. , Yeoh, K.K. , Ratcliffe, P.J. , Burgess-Brown, N. , von Delft, F. , Muller, S. , Marsden, B. , Brennan, P.E. , McDonough, M.A. , Oppermann, U.C. , Klose, R. , Schofield, C.J. , Kawamura, A. , 2012. The plant growth regulator daminozide is a selective inhibitor of the human KDM2/7 histone demethylases. J. Med. Chem. Article ASAP. 10.1021/jm300677j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotili, D. , Altun, M. , Kawamura, A. , Wolf, A. , Fischer, R. , Leung, I.K. , Mackeen, M.M. , Tian, Y.M. , Ratcliffe, P.J. , Mai, A. , Kessler, B.M. , Schofield, C.J. , 2011. A photoreactive small-molecule probe for 2-oxoglutarate oxygenases. Chem. Biol. 18, 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai, M. , Rose, N.R. , Schultz, L. , Quinn, A.M. , Jadhav, A. , Ng, S.S. , Oppermann, U. , Schofield, C.J. , Simeonov, A. , 2010. A miniaturized screen for inhibitors of Jumonji histone demethylases. Mol. BioSyst. 6, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau, M. , Sauvageau, G. , 2010. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 7, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, T. , Chen, W.C. , Gollner, S. , Howell, L. , Jin, L. , Hebestreit, K. , Klein, H.U. , Popescu, A.C. , Burnett, A. , Mills, K. , Casero, R.A. , Marton, L. , Woster, P. , Minden, M.D. , Dugas, M. , Wang, J.C. , Dick, J.E. , Muller-Tidow, C. , Petrie, K. , Zelent, A. , 2012. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildhaus, H.U. , Riegel, R. , Hartmann, W. , Steiner, S. , Wardelmann, E. , Merkelbach-Bruse, S. , Tanaka, S. , Sonobe, H. , Schule, R. , Buettner, R. , Kirfel, J. , 2011. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum. Pathol. 42, 1667–1675. [DOI] [PubMed] [Google Scholar]
- Schmidt, D.M. , McCafferty, D.G. , 2007. Trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry (Mosc). 46, 4408–4416. [DOI] [PubMed] [Google Scholar]
- Schneider, J. , Shilatifard, A. , 2006. Histone demethylation by hydroxylation: chemistry in action. ACS Chem. Biol. 1, 75–81. [DOI] [PubMed] [Google Scholar]
- Schofield, C.J., McDonough, M.A., Rose, N.R., Thalhammer, A., 2010. Histone lysine demethylase inhibitors; Patent No. WO 2010/043866 A2, GB.
- Schofield, C.J., Rose, N.R., Sekirnik, R., 2011. JMJD2 Demethylase Inhibitors; Patent No. WO 2011/030108 A1, GB.
- Schulte, J.H. , Lim, S. , Schramm, A. , Friedrichs, N. , Koster, J. , Versteeg, R. , Ora, I. , Pajtler, K. , Klein-Hitpass, L. , Kuhfittig-Kulle, S. , Metzger, E. , Schule, R. , Eggert, A. , Buettner, R. , Kirfel, J. , 2009. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 69, 2065–2071. [DOI] [PubMed] [Google Scholar]
- Secombe, J. , Li, L. , Carlos, L. , Eisenman, R.N. , 2007. The Trithorax group protein Lid is a trimethyl histone H3K4 demethylase required for dMyc-induced cell growth. Genes Dev. 21, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedkov, Y. , Cho, E. , Petruk, S. , Cherbas, L. , Smith, S.T. , Jones, R.S. , Cherbas, P. , Canaani, E. , Jaynes, J.B. , Mazo, A. , 2003. Methylation at lysine 4 of histone H3 in ecdysone-dependent development of drosophila. Nature. 426, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirnik, R. , Rose, N.R. , Thalhammer, A. , Seden, P.T. , Mecinovic, J. , Schofield, C.J. , 2009. Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(II). Chem. Commun. 6376–6378. [DOI] [PubMed] [Google Scholar]
- Sengoku, T. , Yokoyama, S. , 2011. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 25, 2266–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward, D.J. , Cubberley, G. , Kim, S. , Schonewald, M. , Zhang, L. , Tripet, B. , Bentley, D.L. , 2007. Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat. Struct. Mol. Biol. 14, 240–242. [DOI] [PubMed] [Google Scholar]
- Sharma, S.V. , Lee, D.Y. , Li, B. , Quinlan, M.P. , Takahashi, F. , Maheswaran, S. , McDermott, U. , Azizian, N. , Zou, L. , Fischbach, M.A. , Wong, K.K. , Brandstetter, K. , Wittner, B. , Ramaswamy, S. , Classon, M. , Settleman, J. , 2010. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y. , 2007. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat. Rev. Genet. 8, 829–833. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Lan, F. , Matson, C. , Mulligan, P. , Whetstine, J.R. , Cole, P.A. , Casero, R.A. , 2004. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Whetstine, J.R. , 2007. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell. 25, 1–14. [DOI] [PubMed] [Google Scholar]
- Shin, S. , Janknecht, R. , 2007. Diversity within the JMJD2 histone demethylase family. Biochem. Biophys. Res. Commun. 353, 973–977. [DOI] [PubMed] [Google Scholar]
- Sippl, W. , Jung, M. , 2009. Epigenetic Targets in Drug Discovery Wiley-VCH; Weinheim: [Google Scholar]
- Spannhoff, A. , Hauser, A.T. , Heinke, R. , Sippl, W. , Jung, M. , 2009. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. Chem. Med. Chem. 4, 1568–1582. [DOI] [PubMed] [Google Scholar]
- Spannhoff, A. , Sippl, W. , Jung, M. , 2009. Cancer treatment of the future: inhibitors of histone methyltransferases. Int. J. Biochem. Cell. Biol. 41, 4–11. [DOI] [PubMed] [Google Scholar]
- Stavropoulos, P. , Blobel, G. , Hoelz, A. , 2006. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 13, 626–632. [DOI] [PubMed] [Google Scholar]
- Strobl-Mazzulla, P.H. , Sauka-Spengler, T. , Bronner-Fraser, M. , 2010. Histone demethylase JmjD2A regulates neural crest specification. Dev. Cell. 19, 460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Miyata, N. , 2011. Lysine demethylases inhibitors. J. Med. Chem. 54, 8236–8250. [DOI] [PubMed] [Google Scholar]
- Thalhammer, A. , Mecinovic, J. , Loenarz, C. , Tumber, A. , Rose, N.R. , Heightman, T.D. , Schofield, C.J. , 2011. Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org. Biomol. Chem. 9, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiainen, P. , Pasanen, A. , Sormunen, R. , Myllyharju, J. , 2008. Characterization of recombinant human prolyl 3-hydroxylase isoenzyme 2, an enzyme modifying the basement membrane collagen IV. J. Biol. Chem. 283, 19432–19439. [DOI] [PubMed] [Google Scholar]
- Trievel, R.C. , 2004. Structure and function of histone methyltransferases. Crit. Rev. Eukaryot. Gene Expr. 14, 147–169. [DOI] [PubMed] [Google Scholar]
- Tsai, M.C. , Wang, J.K. , Chang, H.Y. , 2010. Tumor suppression by the histone demethylase UTX. Cell Cycle. 9, 2043–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada, Y. , Fang, J. , Erdjument-Bromage, H. , Warren, M.E. , Borchers, C.H. , Tempst, P. , Zhang, Y. , 2006. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 439, 811–816. [DOI] [PubMed] [Google Scholar]
- Ueda, R. , Suzuki, T. , Mino, K. , Tsumoto, H. , Nakagawa, H. , Hasegawa, M. , Sasaki, R. , Mizukami, T. , Miyata, N. , 2009. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J. Am. Chem. Soc. 131, 17536–17537. [DOI] [PubMed] [Google Scholar]
- Uemura, M. , Yamamoto, H. , Takemasa, I. , Mimori, K. , Hemmi, H. , Mizushima, T. , Ikeda, M. , Sekimoto, M. , Matsuura, N. , Doki, Y. , Mori, M. , 2010. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin. Cancer Res. 16, 4636–4646. [DOI] [PubMed] [Google Scholar]
- Upadhyay, A.K. , Rotili, D. , Han, J.W. , Hu, R. , Chang, Y. , Labella, D. , Zhang, X. , Yoon, Y.S. , Mai, A. , Cheng, X. , 2012. An analog of BIX-01294 selectively inhibits a family of histone H3 lysine 9 Jumonji demethylases. J. Mol. Biol. 416, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinatzer, U. , Gollinger, M. , Mullauer, L. , Raderer, M. , Chott, A. , Streubel, B. , 2008. Mucosa-associated lymphoid tissue lymphoma: novel translocations including rearrangements of ODZ2, JMJD2C, and CNN3. Clin. Cancer Res. 14, 6426–6431. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Lu, F. , Ren, Q. , Sun, H. , Xu, Z. , Lan, R. , Liu, Y. , Ward, D. , Quan, J. , Ye, T. , Zhang, H. , 2011. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 71, 7238–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webby, C.J. , Wolf, A. , Gromak, N. , Dreger, M. , Kramer, H. , Kessler, B. , Nielsen, M.L. , Schmitz, C. , Butler, D.S. , Yates, J.R. , Delahunty, C.M. , Hahn, P. , Lengeling, A. , Mann, M. , Proudfoot, N.J. , Schofield, C.J. , Bottger, A. , 2009. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science. 325, 90–93. [DOI] [PubMed] [Google Scholar]
- Whetstine, J.R. , Nottke, A. , Lan, F. , Huarte, M. , Smolikov, S. , Chen, Z. , Spooner, E. , Li, E. , Zhang, G. , Colaiacovo, M. , Shi, Y. , 2006. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 125, 467–481. [DOI] [PubMed] [Google Scholar]
- Willmann, D. , Lim, S. , Wetzel, S. , Metzger, E. , Jandausch, A. , Wilk, W. , Jung, M. , Forne, I. , Imhof, A. , Janzer, A. , Kirfel, J. , Waldmann, H. , Schule, R. , Buettner, R. , 2012. Impairment of prostate cancer cell growth by a selective and reversible LSD1 inhibitor. Int. J. Cancer. [DOI] [PubMed] [Google Scholar]
- Wissmann, M. , Yin, N. , Muller, J.M. , Greschik, H. , Fodor, B.D. , Jenuwein, T. , Vogler, C. , Schneider, R. , Gunther, T. , Buettner, R. , Metzger, E. , Schule, R. , 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell. Biol. 9, 347–353. [DOI] [PubMed] [Google Scholar]
- Woon, E.C. , Tumber, A. , Kawamura, A. , Hillringhaus, L. , Ge, W. , Rose, N.R. , Ma, J.H. , Chan, M.C. , Walport, L.J. , Che, K.H. , Ng, S.S. , Marsden, B.D. , Oppermann, U. , McDonough, M.A. , Schofield, C.J. , 2012. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew. Chem. Int. Ed. 51, 1631–1634. [DOI] [PubMed] [Google Scholar]
- Xiang, Y. , Zhu, Z. , Han, G. , Ye, X. , Xu, B. , Peng, Z. , Ma, Y. , Yu, Y. , Lin, H. , Chen, A.P. , Chen, C.D. , 2007. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. P. Natl. Acad. Sci. USA. 104, 19226–19231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Yang, H. , Liu, Y. , Yang, Y. , Wang, P. , Kim, S.H. , Ito, S. , Yang, C. , Xiao, M.T. , Liu, L.X. , Jiang, W.Q. , Liu, J. , Zhang, J.Y. , Wang, B. , Frye, S. , Zhang, Y. , Xu, Y.H. , Lei, Q.Y. , Guan, K.L. , Zhao, S.M. , Xiong, Y. , 2011. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane, K. , Toumazou, C. , Tsukada, Y. , Erdjument-Bromage, H. , Tempst, P. , Wong, J. , Zhang, Y. , 2006. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 125, 483–495. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Ledaki, I. , Turley, H. , Gatter, K.C. , Montero, J.C. , Li, J.L. , Harris, A.L. , 2009. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. NY Acad. Sci. 1177, 185–197. [DOI] [PubMed] [Google Scholar]
- Yang, M. , Culhane, J.C. , Szewczuk, L.M. , Gocke, C.B. , Brautigam, C.A. , Tomchick, D.R. , Machius, M. , Cole, P.A. , Yu, H. , 2007. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 14, 535–539. [DOI] [PubMed] [Google Scholar]
- Yang, M. , Culhane, J.C. , Szewczuk, L.M. , Jalili, P. , Ball, H.L. , Machius, M. , Cole, P.A. , Yu, H. , 2007. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry (Mosc). 46, 8058–8065. [DOI] [PubMed] [Google Scholar]
- Yang, M. , Gocke, C.B. , Luo, X. , Borek, D. , Tomchick, D.R. , Machius, M. , Otwinowski, Z. , Yu, H. , 2006. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell. 23, 377–387. [DOI] [PubMed] [Google Scholar]
- Yang, Z.Q. , Imoto, I. , Fukuda, Y. , Pimkhaokham, A. , Shimada, Y. , Imamura, M. , Sugano, S. , Nakamura, Y. , Inazawa, J. , 2000. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 60, 4735–4739. [PubMed] [Google Scholar]
- Yoshimi, A. , Kurokawa, M. , 2011. Key roles of histone methyltransferase and demethylase in leukemogenesis. J. Cell. Biochem. 112, 415–424. [DOI] [PubMed] [Google Scholar]
- Zhu, S. , Li, Y. , Zhao, L. , Hou, P. , Shangguan, C. , Yao, R. , Zhang, W. , Zhang, Y. , Tan, J. , Huang, B. , Lu, J. , 2012. TSA-induced JMJD2B downregulation is associated with cyclin B1-dependent survivin degradation and apoptosis in LNCap cells. J. Cell. Biochem.r. 113, 2375–2382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is/are the supplementary data related to this article:
Supplementary data