

Abstract
Background
Metabolomics, a global study of metabolites and small molecules, is a novel expanding field. In this pilot study, metabolomics has been applied to serum samples from women with metastatic breast cancer to explore outcomes and response to treatment.
Patients and methods
Pre‐treatment and serial on‐treatment serum samples were available from an international clinical trial in which 579 women with metastatic breast cancer were randomized to paclitaxel plus either a targeted anti‐HER2 treatment (lapatinib) or placebo. Serum metabolomic profiles were obtained using 600 MHz nuclear magnetic resonance spectroscopy. Profiles were compared with time to progression, overall survival and treatment toxicity.
Results
Pre‐ and on‐treatment serum samples were assessed for over 500 patients. Unbiased metabolomic profiles in the biologically unselected overall trial population did not correlate with outcome or toxicity. In a subgroup of patients with HER2‐positive disease treated with paclitaxel plus lapatinib, metabolomic profiles from patients in the upper and lower thirds of the dataset showed significant differences for time to progression (N = 22, predictive accuracy = 89.6%) and overall survival (N = 16, predictive accuracy = 78.0%).
Conclusions
In metastatic breast cancer, metabolomics may play a role in sub selecting patients with HER2 positive disease with greater sensitivity to paclitaxel plus lapatinib.
Keywords: Metabolomics, Breast cancer, Metastatic, HER2 positive, Prediction
Highlights
In advanced breast cancer, differential serum NMR metabolite profiles exist.
In HER2+ breast cancer, ‘on treatment’ serum profiles correlate with outcome.
Metabolomics may identify HER2+ patients with greater sensitivity to treatment.
1. Introduction
Breast cancer is a common disease, occurring in one in every eight women (Howlader et al., 2011). Breast cancers exhibit marked heterogeneity in biology and disease behavior, and individuals with seemingly similar disease can have substantially different outcomes, response to treatment and treatment‐related toxicity. It is a clinical priority that individuals are spared ineffective treatment and excessive toxicity. However, the current management approach in individuals with metastatic breast cancer is limited by a lack of markers for prediction, or early detection, of outcomes and treatment effects.
In patients with metastatic breast cancer, there are few prognostic tools. Poor prognostic features include: multiple metastatic sites, presence of visceral disease, lack of estrogen receptor (ER)/progesterone receptor (PgR) expression, and high numbers of circulating tumor cells (Andreopoulou and Hortobagyi, 2008). The only clinical predictive biomarkers are expression of the estrogen receptor for sensitivity to endocrine therapy, and amplification of the HER2 gene and/or overexpression of the HER2 protein for sensitivity to anti‐HER2 therapy. To determine response to treatment, standard therapeutic practice is to measure clinical, radiological and/or serum markers of disease 6–9 weeks after treatment initiation. Regarding toxicity, there are no biomarkers to predict toxicity. Toxicity is often not detected until there are significant symptoms and/or end‐organ deregulation. Concern about toxicity may lead to arbitrary dose reductions, perhaps at the expense of efficacy. New clinical tools are required to unmask diversity in disease behavior, and predict outcomes and treatment effects in individuals.
Metabolomics has established itself in the past decade as a useful complement to the characterization of several physiological and pathological conditions and offers promise as a clinical tool (Nicholson, 2006; Shockcor and Holmes, 2002). The extensive analysis of metabolites in a specific biological specimen, i.e. its ‘metabolome’, can be considered the downstream end product of the complex interaction of genome, transcriptome and proteome. Usually, a metabolomic analysis does not rely on the measurement of a single metabolite‐associated peak but it considers the spectrum as a whole (Nicholson and Lindon, 2008). An 1H‐nuclear magnetic resonance (NMR) metabolomic profile contains qualitative and quantitative information on the hundreds of different small molecules present in the sample (Aranibar et al., 2006; Serkova and Niemann, 2006; Wishart et al., 2007). This approach offers evident advantages with respect to knowledge‐guided search of metabolites, since it does not need to make any assumptions on the identity of the metabolites that are relevant for the selected pathology.
The metabolome is affected by perturbations due to physiology, pathology and iatrogenic factors (Griffin, 2003; Mendes and Kell, 1996; Raamsdonk et al., 2001; Urbanczyk‐Wochniak et al., 2003). Advanced approaches for statistical reduction and analysis of the raw data obtained in the NMR profiles may provide information on the metabolite pattern alterations and their association with pathology (Bathen et al., 2000; Bertini et al., 2009; Lanza et al., 2010; Sreekumar et al., 2009).
Cancer may be associated with profound alterations in metabolic systems, particularly induction of cell membrane phospholipids biosynthesis and breakdown, and preferential use of glucose through non oxidative pathways (Aboagye and Bhujwalla, 1999; Boros et al., 2002; Chan et al., 2009; Griffin and Shockcor, 2004; Sreekumar et al., 2009; Warburg, 1956). In breast cancer, studies using cell‐lines and breast tissue have revealed distinct metabolomic patterns in normal breast, benign disease, carcinoma in situ and invasive carcinoma (Aboagye and Bhujwalla, 1999; Katz‐Brull et al., 2002; Mackinnon et al., 1997; Oakman et al., 2011a). Furthermore, changes in cell metabolism have been correlated with breast cancer cell response to therapy (Beloueche‐Babari et al., 2006; Sterin et al., 2001).
In early breast cancer, tumor tissue – either a tumor biopsy or operative specimen – can be directly assessed. Many studies have employed high resolution magic angle spinning resonance spectroscopy to assess breast cancer tissue (Li et al., 2011, 2010, 2006). In metastatic breast cancer, access to tumor tissue usually necessitates an invasive biopsy. Analysis of serum is much more attractive for ease of collection and the potential for serial assessments. Our group and others have reported differential serum metabolomic fingerprints between healthy individuals and those with cancer (Asiago et al., 2010; Ikeda et al., 2011; Oakman et al., 2011b; OuYang et al., 2011). Another appealing aspect of assessment of serum rather than tumor tissue is that the captured metabolomic signal derives from both the tumor and the individual. In the setting of cancer, in which there is a critical role played by both the tumor and the host in tumor behavior and treatment efficacy, this incorporation of host features may be valuable.
Application of 1H‐NMR spectroscopy to serum metabolomic analysis in the quest for novel predictive biomarkers depends on breast cancer metabolic phenotypes causing systemic and 1H‐NMR detectable signals. In this pilot study, we have used 1H‐NMR spectroscopy metabolite profiling and advanced statistical methods to analyze serum in patients with metastatic breast cancer. We are interested in whether metabolomics may play a role in prediction of overall survival (OS), response to systemic treatment and/or treatment toxicity. The clinical drive for this study is that response and toxicity prediction by a pre‐treatment analysis might guide choice of treatment, while an early on‐treatment analysis might provide an early indication of response and/or toxicity and guide continuation or early discontinuation of treatment.
2. Patients and methods
This project was undertaken by the Department of Medical Oncology, Hospital of Prato, and the Centre for Magnetic Resonance (CERM), University of Florence, Italy, in collaboration with GlaxoSmithKline.
2.1. Patients
This project is a translational research substudy of the multinational, phase III randomized, double‐blind trial, EGF30001, which enrolled patients between January 2004 and July 2005 (Di Leo et al., 2008) This study compared taxane chemotherapy, paclitaxel 175 mg/m2, every three weeks plus lapatinib 1500 mg/day or placebo in 579 women with advanced breast cancer. Lapatinib is an oral dual tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR/ErbB1/HER1) and human epidermal growth factor receptor (ErbB2/HER2). Patient eligibility for trial participation specified that HER2 status was negative or unknown. Other eligibility criteria included patient age over 18 years, histological confirmation of stage III or IV breast cancer, good functional status, adequate organ function, and at least six months separating completion of adjuvant chemotherapy and disease relapse. The primary endpoint was time to progression (TTP), which was defined as the time from randomization until disease progression or death from breast cancer. As part of the trial, a central database documented outcomes (objective response rates as per RECIST criteria, TTP, OS) and toxicity (as per NCI CTC criteria).
The study was approved by the institutional review board of each participating centre. Patients provided written informed consent for trial participation. The study was sponsored by GlaxoSmithKline.
Efficacy and safety results for the total trial population have been published, reporting no additional benefit from the combination of lapatinib and paclitaxel over paclitaxel alone (Di Leo et al., 2008). A preplanned retrospective centralized evaluation of HER2 status using fluorescence insitu hybridization (FISH) and immunohistochemistry (IHC) identified 86 patients with HER2‐positive disease. Benefit from the addition of lapatinib over paclitaxel alone was restricted to patients with HER2‐positive disease. The commonest adverse effects from combination therapy were alopecia, diarrhea and rash.
A serum bank was created during the trial, with samples for each patient programmed pre‐treatment at baseline, and then on‐treatment or post‐treatment at week 9, week 21, and beyond. The total number of samples for any one patient in the trial varied, depending on the duration of trial participation by the individual and site compliance with sample collection. Samples were received from 89 hospitals in 24 countries.
Follow‐up data for TTP and OS used in this pilot study was updated from the data used in the original efficacy and safety report (Di Leo et al., 2008). The original data lock was March 2007. In this study, TTP and OS data derived from the second survival update of August 2007.
Toxicity data was recorded in the central database as per NCI CTC criteria. As per these criteria, toxicity is graded ranging from 0 to 5, in which grade 0 is no toxicity and grade 5 is toxic death. The analyses were undertaken for general toxicity and specific toxicity. General toxicity was defined as any toxicity ≥ grade 3 (excluding haematological toxicity and alopecia). Patients with toxicity ≥ grade 3 were compared to patients without any toxicity ≥ grade 3. Analysis was undertaken in all patients, and by treatment arm (paclitaxel plus lapatinib; paclitaxel plus placebo). Specific toxicities, felt to be clinically important in the management of patients treated with paclitaxel and/or lapatinib, were neurotoxicity, skin toxicity and diarrhea. Patients with ≥ grade 3 neurotoxicity were compared to patients without ≥ grade 3 neurotoxicity. Analysis was undertaken using all patients, and then by treatment arm (paclitaxel plus lapatinib; paclitaxel plus placebo). This analysis was repeated for skin toxicity and for diarrhea.
2.2. Sample preparation
Non‐fasting peripheral blood samples were collected according to criteria in the trial protocol. Samples were frozen within 1 h at −20 °C, followed by long term storage at −80 °C. Stored frozen serum samples – baseline (pre‐treatment) and serial samples (Week 9 and 21) – were transferred to CERM in September 2008. Serum samples and accompanying clinical data were de‐identified prior to transfer.
2.3. NMR analysis
Frozen serum samples were thawed at room temperature and shaken before use. A phosphate buffer (70 mM, pH 7.4) was added in a 1:1 ratio before analysis. One dimensional 1H‐NMR spectra were measured on a spectrometer operating at 600 MHz proton Larmor frequency using standardized protocols (NOESY1D and CPMG sequences). The CPMG sequences were devoid of signals from macromolecules. Each spectrum in the region 10.00–0.02 ppm was segmented into 0.02‐ppm chemical shifts bins (buckets) prior to any statistical analysis. Bucketing is a means to reduce the number of total variables and to compensate for small shifts in the spectra. The size of 0.02 ppm was chosen as it is a sort of standard size for metabolomics, and in general it gives a good compromise between resolution and robustness of the analysis. To check for the impact of binning size on the classification accuracy, further analyses were undertaken using different sized bins (Supplement Tables 1–12). Except for very high resolution buckets (0.0001 ppm or lower), comparable results were found, indicating that binning size is not a critical parameter in this study. The poor performances observed with higher resolution bucketing are potentially attributable to the presence of pH induced shifts in peak position which could not be compensated for by narrow bin size.
For subsequent statistical analysis, non‐normalized data was used in order to not introduce spurious effects due to large peaks intensities. Normalization can be avoided, especially for serum samples, if spectra are collected at the same magnetic field and with the same instrumental parameters, as was the case in this study. In fact, some recent publications avoid this normalization step (Kinross et al., 2011). In the current study, several trials were performed changing bucket size, normalization and scaling (Supplement Tables 1–12). In order to not overcomplicate the analysis, we chose the simplest model which provided the best performance.
2.4. Prediction analyses
Data reduction and classification were obtained by means of orthogonal partial least square discriminant analysis (OPLS‐DA) (Stenlund et al., 2008) on the centered data matrix, without scaling, using the algorithm implemented in the R‐library “kopls” (Bylesjo et al., 2008) and a linear kernel.
The accuracy for classification was assessed by means of a cross‐validation scheme. The original data set was split into a training set (80%) and a test set (20%). An OPLS‐DA model was built using the training set and its predictive accuracy was assessed on the test set. The whole procedure was repeated 50 times inside a Monte Carlo cross‐validation scheme and the results averaged.
For classification purposes the groups were created by dividing patient samples into two classes represented by the highest and the lowest values (33% in most cases) relative to the target variable. The focus was on classification analysis, rather than on survival analysis, because of its simplicity and because, although there are some attempts in statistical literature to extend the survival analysis for multivariate problem, this remains not a trivial task when the number of variables is more than the number of samples (Li and Luan, 2005). To assess which buckets were significantly different between different groups a univariate Wilcoxon test was used. A P‐value ≤0.05 was considered statistically significant. Univariate analysis was performed because univariate models look at one metabolite‐at‐a‐time and are easy to interpret, while multivariate models provide a full view but often lack interpretability, especially in high‐dimensional data cases (Hageman et al., 2008). Thus, multivariate and univariate analysis are complementary.
All calculations were made using home‐made scripts written in our lab using the R language (Ihaka and Gentleman, 1996).
3. Results
Of the 579 patients in the EGF30001 trial, 509 patients (88%) had an assessable baseline sample, 418 (72%) had an assessable week 9 sample and 236 (41%) had an assessable week 21 sample. Reasons for incomplete sample assessment were multiple, including poor site compliance with sample collection, sample collection but inadequate quantity and/or quality for current study, patient withdrawal from study, and patient death. Censored data was excluded. Regarding OS, in the original publication, 311 of the entire 579 patients (54%) were censored and 49 of 86 HER2 positive patients (57%) were censored. With updated OS follow‐up, 259 of 579 patients (45%) were censored and 35 of 86 HER2 positive patients (41%) were censored.
The analyzed samples were received from 89 hospitals in 24 countries. Inter‐hospital variability was evident in the data with a clear separation in the OPLS score plot between samples from different hospitals (Supplementary Figure 1). Variation in sample collection and/or storage procedures might have contributed to this variability, despite the strict criteria detailed in the trial protocol. Other possible confounders, not explored further in the current study, are race and diet. It was not possible to overcome this interhospital variability by restricting the analysis to one hospital site as patient numbers per site were too limited: of the 89 hospitals only 7 had at least 10 patients with assessable baseline serum samples. Thus, samples from all hospitals were subsequently analyzed together.
Evaluation of unbiased metabolomic serum profiles did not identify robust associations with response to treatment, TTP, OS or toxicity (general toxicity and specific toxicity). Multiple analyses with diverse chemometric approaches were undertaken comparing (i) all baseline samples (N = 509), (ii) all week 9 samples (N = 418), and (iii) all week 21 samples (N = 236). The same analyses were undertaken with patients divided by treatment arm (paclitaxel plus lapatinib; paclitaxel plus placebo). Substantial differences were noted in the metabolomic profiles. However, while some trends emerged, there were no clinically meaningful differences between metabolomic profiles and the clinical outcomes of interest.
The availability of serial samples for individuals allowed novel analyses comparing serial metabolomic profiles for an individual. Intra‐individual metabolomic profiles were compared, on the basis that the best control for any individual would indeed be their own serum sample/s. For a subset of individuals, serial metabolomic profiles were compared: (i) baseline vs. week 9 (N = 295), and (ii) baseline vs. week 9 vs. week 21 (N = 179). The closest profile to any sample was another sample from the same individual, in keeping with well known tight physiological homeostatic control. However, intra‐individual profile alterations did not correlate with clinical outcomes of interest.
The study focus was refined using biologically defined subgroups. The most sound biological subgroup was the group of patients with HER2‐positive disease who received paclitaxel plus the HER2‐targeted treatment, lapatinib. Such an analysis homogenized the potential ‘metabolomic noise’ of HER2 status and lapatinib, and allowed exploration of a targeted HER2 treatment in the target HER2‐positive population. Eighty‐six patients had HER2‐positive disease. Forty‐nine of the HER2‐positive patients had received paclitaxel and lapatinib.
Using baseline samples from these 49 patients, there was no differential metabolomic signal for OS, TTP, response to treatment, or toxicity (general toxicity and specific toxicity). Using the on‐treatment week 9 samples, differential metabolomic profiles were observed for TTP and OS, but not for response to treatment or toxicity. Of the 49 patients with paclitaxel/lapatinib‐treated HER2‐positive disease, 34 patients had an assessable week 9 sample and data for TTP. These patients were divided into 3 groups based on duration of TTP. A model was built using the patients with maximally divergent TTP – i.e. the lower tertile (TTP range: 62–175 days) and the upper tertile (TTP range: 367–821 days). The rationale for this separation was that if detectable metabolomic differences were present, they would be most clearly seen in the extremes. Using the lower and upper tertiles (N = 11 vs. 11), metabolomic distinction was noted between patients. See Figure 1 and Table 1. This analysis had a sensitivity of 87.3% and specificity of 91.9%. The predictive accuracy was very high at 89.6%. Significant differential expression (p < 0.05) of the following metabolites was noted: glucose, higher in the patients with longer TTP, and glutamate and phenylalanine, both higher in the patients with shorter TTP.
Figure 1.
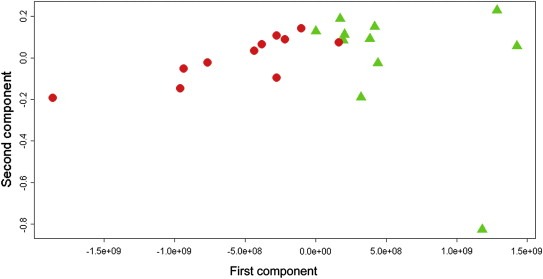
Time to progression. HER2 positive patients treated with paclitaxel‐lapatinib (n = 22). Orthogonal partial least squares score plot: red circles: short TTP; green triangles: long TTP.
Table 1.
Paclitaxel plus lapatinib‐treated patients with HER2‐positive disease (N = 22). On‐treatment week 9 serum samples. TTP prediction
| Outcome as predicted by metabolomics | Predictive accuracy | |||
|---|---|---|---|---|
| Short TTP | Long TTP | |||
| Actual outcome | Short TTP | 87.3% | 12.7% | |
| Long TTP | 8.1% | 91.9% | 89.6% | |
For analysis of OS, 24 of 49 patients with paclitaxel/lapatinib‐treated HER2‐positive disease had an assessable week 9 sample and OS data. These patients were divided into 3 groups based on duration of OS. In the lower group OS ranged from 105 to 470 days, while the upper group's OS ranged from 664 to 843 days. Using the upper and lower groups (N = 9 vs. 7), metabolomic distinction was noted between patients. See Figure 2 and Table 2. This analysis had a sensitivity of 73.0% and specificity of 84.4%. The predictive accuracy was 78.0%.
Figure 2.
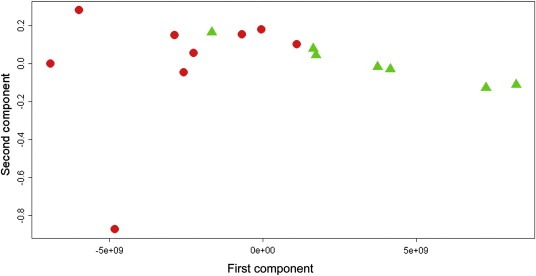
Overall survival. HER2 positive patients treated with paclitaxel‐lapatinib (n = 16). Orthogonal partial least squares score plot: red circles: short OS; green triangles: long OS.
Table 2.
Paclitaxel plus lapatinib‐treated patients with HER2‐positive disease (N = 16). On‐treatment week 9 serum samples. OS prediction
| Outcome as predicted by metabolomics | Predictive accuracy | |||
|---|---|---|---|---|
| Short OS | Long OS | |||
| Actual outcome | Short OS | 73.0% | 27.0% | |
| Long OS | 15.6% | 84.4% | 78.0% | |
Unfortunately due to the small number of patients with paclitaxel/lapatinib‐treated HER2‐positive disease treated within the EGF 30001 trial, there was not the opportunity within this pilot study to validate the on‐treatment TTP and OS models in a biologically similar validation set of patients with week 9 samples and paclitaxel/lapatinib‐treated HER2‐positive disease. Therefore, week 9 samples from all patients not used in the training set were used as independent validation sets. The week 9 TTP validation set included 261 patients with heterogeneity in HER2 status and treatment: 26 patients with paclitaxel/placebo‐treated HER2‐positive disease and 235 with HER2‐negative disease. In this set (N = 261), the model was unable to predict TTP. For the upper (N = 87) and lower (N = 87) TTP tertiles, sensitivity was 58.5%, specificity was 51.1% and predictive accuracy was 54.8%. A smaller validation was tested in only the HER2 positive patients not included in the training set (N = 26). These patients differed from the training set in that they received paclitaxel and placebo. In this smaller validation set, despite greater disease homogeneity, the model was unable to predict for TTP (data not shown).
Similarly, in the week 9 OS validation set which included 261 patients, the model was unable to predict OS. For the upper (N = 87) and lower (N = 87) OS tertiles, sensitivity was 61.3%, specificity was 52.7% and predictive accuracy was 56.6%. As with TTP, a smaller validation set restricted to patients with HER2 positive disease was tried, but was unable to predict for OS (data not shown).
4. Discussion
The quest in this study was to employ NMR analysis and advanced statistics to identify clinically meaningful differential serum metabolite profiles in advanced breast cancer patients. The study demonstrates that serum metabolomic profiles might play a clinical role in HER2‐positive patients in detecting activity of the targeted anti‐HER2 therapy lapatinib plus paclitaxel. Baseline profiles did not correlate with the outcomes of interest. However, in the HER2‐positive subgroup of patients treated with paclitaxel and lapatinib, the on‐treatment week 9 samples showed a high predictive accuracy for TTP and OS.
The HER2 gene encodes for a 185‐kDa tyrosine kinase glycoprotein transmembrane receptor which is overexpressed in 15–25% of breast cancers. HER2‐positivity is a negative prognostic marker, associated with aggressive disease and high rates of disease relapse. HER2‐positivity is a positive predictive marker for identification of individuals with potential sensitivity to HER2‐targeted treatments, the human monoclonal antibody, trastuzumab, and the EGFR/HER2 tyrosine kinase inhibitor, lapatinib. However, within HER2‐positive disease not all patients will respond to HER2 targeted therapy. There is a need to distinguish between individuals with HER2‐positive disease who will and will not respond to anti‐HER2 treatment.
In this pilot study, use of the serum bank linked with the phase III trial EGF30001 allowed access to on‐treatment samples for a large number of patients. The first on‐treatment sample was drawn 9 weeks after treatment commencement. Unfortunately there were no earlier on‐treatment blood draws. By week 9, response to treatment may already be evident clinically by changes in patient symptoms, clinical examination and/or radiological imaging. To improve upon current practice and current timeframes for response assessment, the optimal timing of a metabolomic profile would be earlier in the treatment course, for example 2–4 weeks after treatment initiation. Such an early assessment might spare potential toxicity in patients with no signal of efficacy and allow a prompt switch to other potentially efficacious treatment. Despite the unavailability of a week 2–4 sample in the current trial, analysis of the week 9 samples was useful on the basis that if there was no signal detected at week 9 when responses are overt, no changes would be expected at an earlier time point.
As a downstream measure, metabolomics may be a very sensitive measure of phenotype. A number of metabolomic‐based studies have shown altered metabolite profiles in different cancer types (Ikeda et al., 2011; Oakman et al., 2011b; OuYang et al., 2011; Slupsky et al., 2010; Sreekumar et al., 2009). Several studies in breast cancer, particularly assessing breast cancer cells and tumor tissue, have reported correlations between malignancy and numerous metabolites, including choline and choline derivatives, amino acids and glucose (Aboagye and Bhujwalla, 1999; Katz‐Brull et al., 2002). Use of the profile rather than specific metabolites offers a global analysis. A study using high resolution magic angle spinning spectroscopy in intact breast cancer specimens found that a multi‐metabolite profile was more powerful in predicting outcome than single metabolites (Sitter et al., 2010).
In the clinical setting, metabolomics may add to information provided by other clinical tools. A recent study suggested that tumor tissue metabolomic profiles offered refined classification of molecularly defined luminal‐A breast cancers (Borgan et al., 2010). Another study reported detection of recurrent breast cancer using serum metabolite profiling by a combination of NMR and 2 dimensional gas chromatography–mass spectroscopy (2D GC–MS) (Asiago et al., 2010). Eleven key metabolites were identified and used to create a predictive model for recurrence, which allowed detection of metastatic breast cancer earlier than detection by the serum tumor marker Ca 27.29.
Many metabolites have been shown to correlate with breast cancer, including choline, lipids, glucose, lactate and amino acids. In contrast to most metabolomic studies to date in breast cancer which have compared metabolite profiles between breast cancer (early and/or advanced) and no breast cancer, all patients in the current study had metastatic breast cancer and were receiving treatment, and the goal was to identify differential metabolite profiles for outcome and response to therapy. The baseline samples did not predict for outcome (i.e. there was no detectable prognostic signal). However, on‐treatment metabolomic profiles in a biologically sound subgroup showed promise for early detection of response and survival. The promising serum profiles presumably derive not from tumor alone, but represent a composite metabolomic signal from the tumor, the host and the treatment effect.
Clinical interpretation of serum metabolomic profiles is very challenging, as the search is for a clinically meaningful signal amongst the complex physiological serum metabolomic fingerprint for an individual. Significant differential expression of the metabolites was noted: in the TTP analysis, patients with longer TTP had higher glucose and lower glutamate and phenylalanine compared with patients with shorter TTP. Due to the complexity of the serum signal, it is hard to attribute specific key metabolites to a specific tumor, host and/or treatment metabolic pathway. Indeed, we have focused on analyses on the complete metabolomic profile, or metabolomic fingerprint, rather than on singled out individual metabolites.
Until recently, research in breast cancer has largely focused on features of the tumor. There is now increasing recognition of the importance of host tissue hypoxia, tumor microenvironment and host immunity in tumor behavior and treatment activity which has stimulated much research into host features as potentially valuable clinical biomarkers. Concurrent signal detection from host and tumor is an appealing feature of serum metabolomic approaches.
A strength of 1H‐NMR spectroscopy is that it is both qualitative and quantitative. This allows analysis of profiles and comparison between profiles based on both the presence and quantity of metabolites in samples. Serum metabolomic approaches are rapid, minimally invasive and cost efficient. However, use of serum samples also poses several challenges. One of the greatest challenges is sample quality. Metabolomics by NMR is extremely sensitive to minimal variation in sample conditions and even a small effect can greatly affect the results. Even within the confines of a tightly regulated phase III trial, there was substantial heterogeneity in sample quality, in part attributable to inconsistent, poor sample handling and conservation.
5. Conclusion
In this pilot analysis, high predictive accuracy in discriminating between long and short OS and long and short TTP is extremely encouraging, especially considering the complexity of the task. Unfortunately due to the small number of patients with paclitaxel/lapatinib‐treated HER2‐positive disease treated within the EGF30001 trial, there was not the opportunity within this pilot study to validate the TTP and OS models in a biologically similar validation set of patients. The training set was unable to predict TTP and OS in week 9 samples in other patients – paclitaxel/placebo‐treated HER2‐positive disease and HER2 negative disease – however, it is unclear if this lack of validation is due to biological heterogeneity between training and validation sets. If the differential metabolomic signals in the training set derive from lapatinib in the presence of the target HER2, this signal would not be reproducible in the absence of HER2 and lapatinib. The results presented herein highlight an exciting potential translation of the evolving field of metabolomics into clinical practice.
Conflict of interest
None.
Supporting information
Supplementary data
Supplementary data
Acknowledgements
The authors wish to acknowledge the contributions of the GlaxoSmithKline, PA, USA; the Breast Cancer Research Foundation (BCRF), New York, USA; and Associazione “Sandro Pitigliani”, Prato, Italy.
Supplementary material 1.
1.1.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molonc.2012.05.003.
Tenori Leonardo, Oakman Catherine, Claudino Wederson M., Bernini Patrizia, Cappadona Silvia, Nepi Stefano, Biganzoli Laura, Arbushites Michael C., Luchinat Claudio, Bertini Ivano, Di Leo Angelo, (2012), Exploration of serum metabolomic profiles and outcomes in women with metastatic breast cancer: A pilot study, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.05.003.
References
- Aboagye, E.O. , Bhujwalla, Z.M. , 1999. Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Res.. 59, 80–84. [PubMed] [Google Scholar]
- Andreopoulou, E. , Hortobagyi, G.N. , 2008. Prognostic factors in metastatic breast cancer: successes and challenges toward individualized therapy. J. Clin. Oncol.. 26, 3660–3662. [DOI] [PubMed] [Google Scholar]
- Aranibar, N. , 2006. Metabolomic analysis using optimized NMR and statistical methods. Anal. Biochem.. 355, 62–70. [DOI] [PubMed] [Google Scholar]
- Asiago, V.M. , 2010. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res.. 70, 8309–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathen, T.F. , 2000. Analysis and classification of proton NMR spectra of lipoprotein fractions from healthy volunteers and patients with cancer or CHD. Anticancer Res.. 20, 2393–2408. [PubMed] [Google Scholar]
- Beloueche-Babari, M. , 2006. Identification of magnetic resonance detectable metabolic changes associated with inhibition of phosphoinositide 3-kinase signaling in human breast cancer cells. Mol. Cancer Ther.. 5, 187–196. [DOI] [PubMed] [Google Scholar]
- Bertini, I. , 2009. The metabonomic signature of celiac disease. J. Proteome Res.. 8, 170–177. [DOI] [PubMed] [Google Scholar]
- Borgan, E. , 2010. Merging transcriptomics and metabolomics–advances in breast cancer profiling. BMC Cancer. 10, 628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros, L.G. , 2002. Metabolic profiling of cell growth and death in cancer: applications in drug discovery. Drug Discov. Today. 7, 364–372. [DOI] [PubMed] [Google Scholar]
- Bylesjo, M. , 2008. K-OPLS package: kernel-based orthogonal projections to latent structures for prediction and interpretation in feature space. BMC. Bioinform.. 9, 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, E.C. , 2009. Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J. Proteome Res.. 8, 352–361. [DOI] [PubMed] [Google Scholar]
- Di Leo, A. , 2008. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J. Clin. Oncol.. 26, 5544–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, J.L. , 2003. Metabonomics: NMR spectroscopy and pattern recognition analysis of body fluids and tissues for characterisation of xenobiotic toxicity and disease diagnosis. Curr. Opin. Chem. Biol.. 7, 648–654. [DOI] [PubMed] [Google Scholar]
- Griffin, J.L. , Shockcor, J.P. , 2004. Metabolic profiles of cancer cells. Nat. Rev. Cancer. 4, 551–561. [DOI] [PubMed] [Google Scholar]
- Hageman, J.A. , 2008. Simplivariate models: ideas and first examples. PLoS One. 3, e3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader, N. , 2011. SEER Cancer Statistics Review, 1975–2008 National Cancer Institute; Bethesda: http://seer.cancer.gov/csr/1975_2008/ [Google Scholar]
- Ihaka, R. , Gentleman, R. , 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat.. 5, 299–314. [Google Scholar]
- Ikeda, A. , 2011. Serum metabolomics as a novel diagnostic approach for gastrointestinal cancer. Biomed. Chromatogr.. [DOI] [PubMed] [Google Scholar]
- Katz-Brull, R. , 2002. Metabolic markers of breast cancer: enhanced choline metabolism and reduced choline-ether-phospholipid synthesis. Cancer Res.. 62, 1966–1970. [PubMed] [Google Scholar]
- Kinross, J.M. , 2011. Global metabolic phenotyping in an experimental laparotomy model of surgical trauma. J. Proteome Res.. 10, 277–287. [DOI] [PubMed] [Google Scholar]
- Lanza, I.R. , 2010. Quantitative metabolomics by H-NMR and LC-MS/MS confirms altered metabolic pathways in diabetes. PLoS One. 5, e10538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Luan, Y. , 2005. Boosting proportional hazards models using smoothing splines, with applications to high-dimensional microarray data. Bioinformatics. 21, 2403–2409. [DOI] [PubMed] [Google Scholar]
- Li, M. , 2011. An HR-MAS MR metabolomics study on breast tissues obtained with core needle biopsy. PLoS One. 6, e25563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon, W.B. , 1997. Fine-needle biopsy specimens of benign breast lesions distinguished from invasive cancer ex vivo with proton MR spectroscopy. Radiology. 204, 661–666. [DOI] [PubMed] [Google Scholar]
- Mendes, P. , Kell, D.B. , 1996. On the analysis of the inverse problem of metabolic pathways using artificial neural networks. Biosystems. 38, 15–28. [DOI] [PubMed] [Google Scholar]
- Nicholson, J.K. , 2006. Global systems biology, personalized medicine and molecular epidemiology. Mol. Syst. Biol.. 2, 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, J.K. , Lindon, J.C. , 2008. Systems biology: metabonomics. Nature. 455, 1054–1056. [DOI] [PubMed] [Google Scholar]
- Oakman, C. , 2011. Uncovering the metabolomic fingerprint of breast cancer. Int. J. Biochem. Cell Biol.. 43, 1010–1020. [DOI] [PubMed] [Google Scholar]
- Oakman, C. , 2011. Identification of a serum-detectable metabolomic fingerprint potentially correlated with the presence of micrometastatic disease in early breast cancer patients at varying risks of disease relapse by traditional prognostic methods. Ann. Oncol.. 22, 1295–1301. [DOI] [PubMed] [Google Scholar]
- OuYang, D. , 2011. Metabolomic profiling of serum from human pancreatic cancer patients using 1H NMR spectroscopy and principal component analysis. Appl. Biochem. Biotechnol.. 165, 148–154. [DOI] [PubMed] [Google Scholar]
- Raamsdonk, L.M. , 2001. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol.. 19, 45–50. [DOI] [PubMed] [Google Scholar]
- Serkova, N.J. , Niemann, C.U. , 2006. Pattern recognition and biomarker validation using quantitative 1H-NMR-based metabolomics. Expert. Rev. Mol. Diagn.. 6, 717–731. [DOI] [PubMed] [Google Scholar]
- Shockcor, J.P. , Holmes, E. , 2002. Metabonomic applications in toxicity screening and disease diagnosis. Curr. Top. Med. Chem.. 2, 35–51. [DOI] [PubMed] [Google Scholar]
- Sitter, B. , 2010. Quantification of metabolites in breast cancer patients with different clinical prognosis using HR MAS MR spectroscopy. NMR Biomed.. 23, 424–431. [DOI] [PubMed] [Google Scholar]
- Sitter, B. , 2006. Comparison of HR MAS MR spectroscopic profiles of breast cancer tissue with clinical parameters. NMR Biomed.. 19, 30–40. [DOI] [PubMed] [Google Scholar]
- Slupsky, C.M. , 2010. Urine metabolite analysis offers potential early diagnosis of ovarian and breast cancers. Clin. Cancer Res.. 16, 5835–5841. [DOI] [PubMed] [Google Scholar]
- Sreekumar, A. , 2009. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 457, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Stenlund, H. , 2008. Orthogonal projections to latent structures discriminant analysis modeling on in situ FT-IR spectral imaging of liver tissue for identifying sources of variability. Anal. Chem.. 80, 6898–6906. [DOI] [PubMed] [Google Scholar]
- Sterin, M. , 2001. Levels of phospholipid metabolites in breast cancer cells treated with antimitotic drugs: a 31P-magnetic resonance spectroscopy study. Cancer Res.. 61, 7536–7543. [PubMed] [Google Scholar]
- Urbanczyk-Wochniak, E. , 2003. Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Rep.. 4, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg, O. , 1956. On the origin of cancer cells. Science. 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. , 2007. HMDB: the human metabolome database. Nucleic Acids Res.. 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data