

Abstract
It is a time of great promise and expectation for the applications of knowledge about mechanisms of cancer toward more effective and enduring therapies for human disease. Conceptualizations such as the hallmarks of cancer are providing an organizing principle with which to distill and rationalize the abject complexities of cancer phenotypes and genotypes across the spectrum of the human disease. A countervailing reality, however, involves the variable and often transitory responses to most mechanism‐based targeted therapies, returning full circle to the complexity, arguing that the unique biology and genetics of a patient's tumor will in the future necessarily need to be incorporated into the decisions about optimal treatment strategies, the frontier of personalized cancer medicine. This perspective highlights considerations, metrics, and methods that may prove instrumental in charting the landscape of evaluating individual tumors so to better inform diagnosis, prognosis, and therapy. Integral to the consideration is remarkable heterogeneity and variability, evidently embedded in cancer cells, but likely also in the cell types composing the supportive and interactive stroma of the tumor microenvironment (e.g., leukocytes and fibroblasts), whose diversity in form, regulation, function, and abundance may prove to rival that of the cancer cells themselves. By comprehensively interrogating both parenchyma and stroma of patients' cancers with a suite of parametric tools, the promise of mechanism‐based therapy may truly be realized.
Keywords: Personalized medicine, Targeted therapy, Tumor microenvironment
1. Introduction
The complexity and variability of cancer as a disease has long been recognized. It is manifested in the dramatically different time courses of disease inception, progression and pathogenic impact that are evident across the gamut of tissues susceptible to aberrant proliferative growths. Thus, it is well established that solid tumors (and diffuse proliferations of leukemias) differ dramatically from one organ to another, as well as from one transformed cell type to another that are susceptible to cancer within a particular tissue or organ. This variability is unambiguous at multiple levels: genetically, chromosomally, histologically, physiologically, pathologically, and in terms of prognosis. Different oncogenic mutations drive particular cellular proliferations leading to given cancer types, and different tumor suppressors are characteristically lost in function, often by distinctive mechanisms. Tumors in different organs can therefore look much different in terms of gross‐ and histo‐pathology, and have dramatically different effects on the patient. As knowledge about cancers at all of these levels increased in the 1980s and 1990s, building an encyclopedia of dizzying complexity and variability, the conceptual landscape has been increasingly obscured by this overwhelming wealth of detail and difference. The hallmarks of cancer elaborated by Hanahan and Weinberg (2000) presented an organize principle from which to consider this complexity, as differing ways to a similar end: the proposition was that most all cancers acquire, by one means or another, a similar set of capabilities necessary to manifest malignant disease. In the recently updated conceptualization (Hanahan and Weinberg, 2011), there are six core and two emerging hallmark capabilities: sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis, along with deregulating cellular energetics (and metabolism), and avoiding immune destruction. Virtually all tumors evidence these capabilities, albeit to varying degrees. Thus, one imagines, as an individual tumor develops, that it faces a series of barriers and roadblocks that must be surmounted or circumvented, some of which are tissue specific, thus requiring tissue‐specific solutions if cancer cells are to grow profusely and progress to malignancy and other pathologic states. But even in facing the same challenge, different tumors may solve the problem in different ways, developing distinctive mutations and adaptations that allow it to acquire these 6–8 capabilities (Hanahan and Weinberg, 2011). Therefore, the notion arises that the ostensibly confusing individual variability underlays a profound commonality, of acquiring the same hallmark capabilities. The variable means of acquiring such functional capabilities are embedded in two enabling characteristics that each has intrinsic variability – genome instability with (random but genetically selected) mutation, and tumor‐promoting inflammation; both demonstrably facilitate acquisition of hallmark capabilities while at the same time being inherently diverse in their manifestation (Hanahan and Weinberg, 2011). It is apparent that the cancer hallmarks conceptualization has proved to be a useful heuristic tool for the cancer research community, with its overarching view of the landscape, albeit one that is viewed from a high altitude with consequently low resolution mechanistically for a given tumor type and subtype, and for individual tumors therein.
In the decade since the introduction of the hallmarks concept (Hanahan and Weinberg, 2000), the encyclopedia of cancer has continued to expand in scope and detail, adding more facts and more depth to the molecular genetic and histo‐pathologic descriptions of cancers in organs throughout the body. Adding to that complexity is the increasing awareness that there is remarkable variability even within a given tumor type, above and beyond the differences in that tumor type from other types. Thus we are now in an era of molecular subtypes, where particular tumor types can be subdivided into molecular subtypes with significantly different features, ranging from signature oncogenic mutations to genome‐wide transcriptional profiles, to histo‐pathological subtleties (for example in inflammatory cell profiles). Concurrently, highly heralded targeted therapies have been introduced into clinical practice, with the anticipation that such mechanism‐based drugs would significantly improve prognosis (Sawyers, 2004; Weinstein, 2002). Yet, although cancer treatments have shown incremental efficacy over the last two decades, the overall survival rate of patients with advanced and/or metastatic cancer (including cancers of the prostate or breast, lung, and colon‐rectum, which are responsible for approximately half of cancer incidence in the United States) remains extremely poor. The sobering reality is that the efficacy of most targeted therapies is transitory for most patients, with almost inevitable relapse to progressive disease (de Bono and Ashworth, 2010). Moreover, a common pattern is emerging: targeted therapies are only producing appreciable therapeutic responses in a fraction of patients of a particular type, and even within a specific subtype. Some tumors, while envisioned to respond, do not, and instead continue to grow and progress to states of heightened malignancy. Yet others, often a rare fraction, show enduring responses (de Bono and Ashworth, 2010).
Thus, despite the overarching clarity imparted by the hallmarks conceptualization, we are back full circle to facing the stark reality of patient/tumor heterogeneity. The net result is that we will need to understand the specific complexity and variability of tumors in the context of the individual, so as to determine, based on the characteristics of their disease, how to best treat it so as to produce beneficial responses and enduring efficacy. This realization has opened the frontier of personalized cancer medicine, where individual difference will not be ignored but rather recognized and characterized, and then leveraged for the benefit of that patient, providing better advice on prognosis and therapeutic options, in particular identifying drugs and treatment regimens (and combinations) that are likely to produce beneficial responses for that person in particular. A number of recent reviews and commentaries have discussed aspects of this new frontier (de Bono and Ashworth, 2010; Haber et al., 2011; Luo et al., 2009; Martini et al., 2011), as do articles in the current special issue of Molecular Oncology. Herein we reflect further on the cancer biology of personalized cancer medicine, posing questions and suggesting approaches anchored nevertheless in the notion of the hallmarks of cancer as an integrative tool that may help address the challenges of personalized cancer medicine.
2. Variable responses to targeted therapies reveal and motivate personalized cancer medicine
By targeting molecular pathways that are both specific and essential to a given cancer type or subtype, targeted therapies have been envisioned to provide more effective and less toxic treatment options than conventional therapies such as chemotherapy and radiotherapy. The concept of targeted therapies (‘magic bullets’) raised high expectations, which were initially fueled by the clinical efficacy of imatinib in chronic myeloid leukemia (CML) patients (Druker et al., 1996, 2006; Weinstein, 2002). Tempering such optimism, however, have been subsequent observations that many mechanism‐targeted drugs have relatively modest clinical benefit, in terms of overall survival, and often in only a fraction of patients with tumors expressing the drug target and hence predicted to benefit (de Bono and Ashworth, 2010; Haber et al., 2011; Martini et al., 2011; Sawyers, 2009). It is an emerging theme that the variable penetrance and typically transitory response profiles of many current targeted therapies reflects on the difficulty (and evidently, the necessity) to match these drugs with the molecular characteristics – both pre‐existing, and evolving during therapy – of individual tumors, as elaborated below.
2.1. Redundancy in proliferative signaling pathways variably limits the efficacy of targeted therapies in different patients
The molecular complexity and heterogeneity of most human malignancies (including tumor type, subtype and individual patient‐specific variation) – together with the redundancy and multi‐layered control of proliferative signaling pathways in cancer cells – is such that tumors only rarely depend on a single regulatory pathway for growth and survival. Such redundancies may underlay the lack of response in some patients whose tumors express oncogenic targets and therefore were treated with targeted drugs in anticipation of therapeutic benefit, and yet there was none.
The existence of intrinsic resistance despite signature oncogenic mutations is exemplified by the clinical experience with HER2‐positive breast cancer. HER2 (ERBB2) is an epidermal growth factor receptor (EGFR) family protein whose overexpression in breast cancer is associated with increased disease recurrence and a worse prognosis (Esteva et al., 2010). Trastuzumab, an anti‐HER2 monoclonal antibody, is prescribed for patients whose breast cancer cells show HER2 overexpression, either by gene amplification or markedly increased protein level. However, only 30% of such HER2‐positive tumors respond to trastuzumab therapy, indicating that the majority of these tumors are not strictly dependent on HER2‐mediated signaling for their growth. Many non‐responsive HER2‐positive breast tumors harbor mutations that evidently circumvent tumor dependency on HER2 signaling and mediate refractoriness to trastuzumab. These include parallel activation of insulin growth factor 1 (IGF1) receptor signaling, overexpression of EGF family ligands, or mutations that activate the PI3K pathway; in fact, PI3K pathway activation in HER2‐positive breast cancer provides a biomarker to identify patients who are unlikely to respond to trastuzumab therapy (Esteva et al., 2010).
Similarly, only 10% of metastatic colorectal cancer (mCRC) patients respond to cetuximab, a monoclonal antibody that blocks the EGFR (ERBB1), which is broadly expressed in mCRC (Walther et al., 2009). Both retrospective and prospective clinical studies have shown that mCRC patients with KRAS mutations – or mutations in NRAS, BRAF, PIK3CA and HER2 – show negligible response to cetuximab, irrespective of elevated expression of EGFR (De Roock et al., 2010). It should be noted that both RAS and PI3K lie downstream in the HER2 and EGFR signaling pathways, which may explain why the lack of constitutively‐activated RAS and PI3K pathways is necessary (albeit not sufficient) for trastuzumab and cetuximab to be effective.
2.2. Emergence of secondary resistance to growth‐inhibitory drugs variably limits the efficacy of targeted therapies in different patients
In addition to drug refractoriness ab initio, the emergence of secondary (or acquired) resistance may limit the duration of clinical benefit from targeted therapies. Secondary drug resistance occurs in a fraction of imatinib‐treated CML patients, generally after a prolonged remission phase, typically as a result of secondary mutations in the BCR‐ABL fusion gene (Weisberg et al., 2007). For several types of solid tumors, the occurrence of secondary drug resistance to the targeted therapy represents a very frequent event. For instance, non‐small cell lung carcinomas (NSCLCs) that harbor EGFR kinase‐activating mutations are broadly sensitive to the EGFR tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib, but almost invariably relapse after an initial response phase; in half of the cases, secondary resistance is caused by novel secondary mutations in the EGFR coding sequence (Workman and Clarke, 2011). Similarly, metastatic melanomas that harbor the activating BRAF(V600E) mutation regularly relapse after a window of therapeutic response provided by the mutant‐BRAF inhibitor, vemurafenib (Flaherty et al., 2011); recent data suggest that resistance to vemurafenib may be mediated – at least in part – by the appearance of aberrantly spliced BRAF(V600E) isoforms (Poulikakos et al., 2011). Much like intrinsic resistance, acquired resistance mutations can also affect “parallel” signaling pathways. For example, 20% of NSCLC specimens that had developed resistance to gefitinib or erlotinib show amplification of the MET oncogene (Engelman et al., 2007). Furthermore, recent studies have annotated PIK3CA mutations in NSCLC patients that developed resistance to these TKIs (Sequist et al., 2011; Turke et al., 2010). Thus, both drug‐resistant target mutations and indirect, target‐circumventing resistance mutations can undermine the long‐term efficacy of the targeted drugs.
In summary, both intrinsic redundancy in proliferative signaling pathways and therapy‐induced selection of drug‐resistant mutations evidently limit the efficacy of targeted therapies. Defining the complexity of activated proliferative signals – both at diagnosis and during therapy – in individual tumors carrying a common and druggable oncogenic target is therefore required to predict and harness the potential of such targeted therapies. Furthermore, monitoring the early emergence of acquired mutations during the response phase may help to identify the most suitable drug for subsequent treatment(s) aimed to elicit long‐lasting cancer control.
3. Addressing tumor heterogeneity and complexity
Despite the simplicity imparted by the hallmarks conceptualization and the availability of targeted drugs that can interfere with several of the hallmark capabilities of cancer, the remarkable diversity, discussed above, in therapeutic responses among patients with the same cancer presents a need for deeper knowledge about the biology and genetics of an individual's tumor. Although distinctive genetic or epigenetic signatures reflecting particular natural histories of tumorigenesis and progression undoubtedly bear upon the broad spectrum of tumor phenotypes (and variable responses to therapies) observed amongst individuals with the same tumor type or subtype, it is evident that such phenotypic variation is also impacted by several other factors. Thus, in addition to inter‐tumoral (or patient specific) variation, significant intra‐tumoral variation is evident in many tumor types, in forms spanning histological to genetic heterogeneity. Such variability may be reflective of regional foci of clonal evolution (Yachida et al., 2010), as well as tumor self‐seeding from disseminated cancer cells (Comen et al., 2011). Moreover, the heterogeneity of putative tumor‐initiating cells (or cancer stem cells) may contribute to subtype variation in certain solid tumor types, as suggested by studies employing mouse models of glioblastoma, intestinal, and prostate cancer (Visvader, 2011). Additionally, specific traits of the accessory cells that make up the tumor‐supportive stroma may influence tumor phenotypes and response to therapy (see below). Collectively, such intra‐tumoral variation – often reflecting the existence of multiple subclonal tumor populations – might represent an important yet unrecognized determinant for the emergence of secondary drug resistance.
Addressing the complexity and variability of individual tumors and the identification of clinically useful biomarkers will therefore be fundamental to determine how to best treat cancer patients and produce responses of enduring efficacy. Furthermore, the emergence of secondary resistance after initially effective tumor responses calls for repeated tumor profiling analysis during the course of therapy. These realizations motivate the development and implementation of tractable methodology for efficiently auditing the specific complexities of cancer patients, both via tumor biopsies and various forms of blood‐borne tumor‐derived exfoliates (Figure 1). We highlight below potentially applicable technologies for unveiling patient‐specific characteristics so as to guide, optimize, and manage personalized therapeutic regimens.
Figure 1.
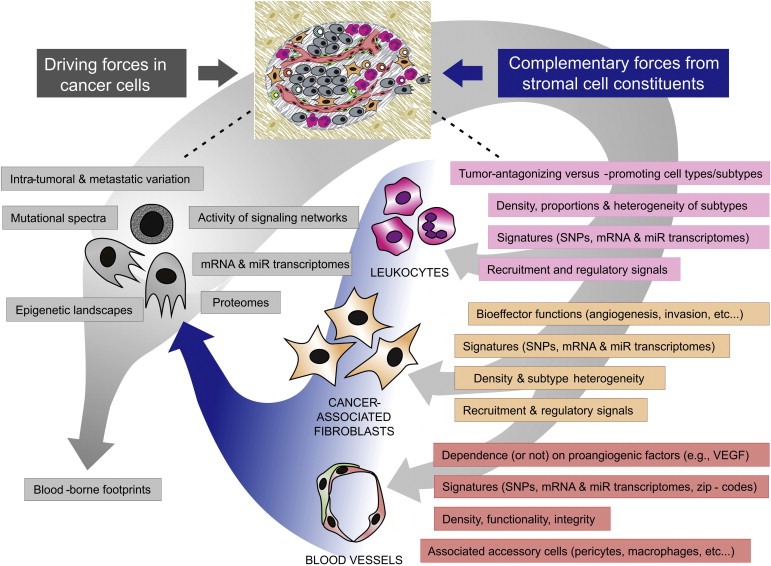
Dimensions and key parameters of personalized cancer biology. The complexity and variability of cancer should be interrogated in individual patients. Heterogeneity of both cancer cells (left) and stromal cells (right) can be audited through molecular analysis (including genomics, epigenomics and transcriptomics); immunohistochemistry and flow cytometry; noninvasive imaging; and examination of re‐biopsy specimens obtained from both primary/metastatic tumors and tumor cell exfoliates, before and during treatment, as well as upon treatment failure. The constituent types, relative abundance and dynamics of cancer‐associated leukocytes and fibroblasts (CAFs) should be determined before, during and after treatment; stromal cell signatures may be predictive of patient's prognosis and represent biomarkers of tumor response to therapy. Molecular analysis of whole tumor tissue should be extended to fractionated cancer‐associated stromal cells, including tumor‐derived endothelial cells; molecular signatures comprising single nucleotide polymorphisms (SNPs) and mRNA/miR transcriptomes may be predictive of patient's response to therapy (e.g., antiangiogenic drugs), and thereby provide an encyclopedia of potential new targets of therapy.
3.1. Cancer genomes
Cancer genome sequencing efforts have revealed the remarkable genetic complexity of individual cancers, while raising expectations for rapid advances in cancer classification and diagnosis, prognosis and therapeutic design thanks to the identification of “gene signatures” that recur in certain cancer types.
The Cancer Genome Atlas, a catalogue of the DNA mutations found in various human tumors, is revealing that hundreds of coding genes may be mutated in each cancer type/subtype. This figure is suspected to be an underestimation of the mutational landscape of individual tumors, as the analytic techniques that have been used most frequently, such as PCR‐based sequencing of DNA extracted from total tumor lysates, may not detect mutations occurring in subclonal tumor cell populations (Loeb, 2011; Sellers, 2011). This is illustrated, for example, by exomic sequencing of separate, histologically distinct regions micro‐dissected from individual human pancreatic ductal carcinomas (PDACs): each showed a specific mutational repertoire indicative of multiple subclonal populations within the same primary tumor (Yachida et al., 2010). Consistent with these findings, recent whole‐genome deep (or high‐throughput) sequencing analysis of individual tumors has revealed that a particular tumor specimen may contain up to 50,000 different non‐synonymous mutations affecting several hundred genes, with mutation rates of 0.5–20 per megabase (Loeb, 2011; Sellers, 2011). As opposed to exomic sequencing, unbiased whole‐genome deep sequencing should in principle be able to detect complex genetic rearrangements, insertions and deletions, which may enact pivotal gain‐ and loss‐of‐function driver events not manifested by point mutations, as shown recently in prostate cancer (Berger et al., 2011). The remarkable number of mutations found in cancer genomes supports the notion that cancers express a “mutator phenotype” that is an enabling characteristic for acquisition of hallmark capabilities, caused for example by loss‐of‐function mutations in genes that maintain genomic integrity (Hanahan and Weinberg, 2011; Loeb, 2011).
Whole‐genome deep sequencing, ideally of multiple regions spanning a tumor, holds promise to markedly improve the annotation of the mutational landscape of individual tumors. Although the aforementioned subclonal mutations in focal tumor regions are unlikely to provide growth advantage to the tumor at large (and thus unlikely to be clinically useful targets of first‐line therapy), they might convert into alternative driver mutations upon functional blockade of the original driver mutation, and thus contribute to relapse concomitant with drug resistance. It remains to be ascertained, for example by re‐sequencing tumors before therapy and following therapeutic resistance, whether deep sequencing can identify such latent mutations and potentially predict the likely forms of adaptive resistance, thereby guiding second‐line treatment strategies.
3.2. Cancer epigenomes
The past decade has witnessed an explosion of knowledge about the epigenetic regulatory changes that occur in cancer cells, leading to the concept that cancer is also a disease of the “epigenome” (Baylin and Jones, 2011). Specific changes in histone acetylation and methylation patterns are being observed in various cancer types. DNA methylation and other covalent, somatically heritable chromatin modifications are also being found. These epigenetic changes are evidently relevant to gene expression and may have an important role in driving tumorigenesis. Excessive DNA methylation of normally unmethylated gene promoter regions is observed in many cancer types, leading to suppression of gene function; for example, the tumor suppressor gene CDKN2A is frequently inactivated by DNA hypermethylation in various tumor types (Baylin and Jones, 2011). DNA hypermethylation and consequent gene promoter inactivation may also decrease the expression of microRNAs (miRNAs, or miRs) that function to finely tune proliferative signaling in normal cells (see below).
Specific DNA hypermethylation signatures have emerged as potential biomarkers to assess cancer risk, aid early detection, and predict therapeutic responses, at least for some cancer types (Baylin and Jones, 2011). Furthermore, there is increasing evidence that certain epigenetic marks may associate with drug resistance. A recent study showed proof‐of‐concept that cancer cells may employ a dynamic survival strategy to protect the population from eradication by potentially lethal drugs (Sharma et al., 2010). According to this model, individual cancer cells transiently assume a reversible, drug‐tolerant state that requires the activity of histone demethylases; remarkably, the drug‐tolerant cancer cells can be selectively ablated by treatment with histone deacetylase (HDAC) inhibitors. Thus, “epigenetic therapy” using inhibitors of HDACs or DNA methyltransferases could be used in combination with targeted drugs to improve their efficacy and/or limit the emergence of secondary resistance. Of note, two DNA methyltransferase inhibitors, azacitidine and decitabine, have been approved recently for myelodysplastic syndrome (MDS) and are currently being tested in acute myeloid leukemia (AML) patients (Baylin and Jones, 2011).
Importantly, the discussion thus far has focused on the cancer cells, and largely ignored the other constituent cell types composing the tumor microenvironment. It can be envisioned that epigenetic changes will prove important also in defining regulatory states in the tumor stroma, for example in the cancer associated fibroblasts (CAFs) that are evidently reprogrammed (or educated) to adopt stable aberrant phenotypes.
3.3. Cancer transcriptomes
Genome‐wide transcriptional analysis of the expressed mRNAs, initially using microarray technologies and more recently RNA deep sequencing, has been widely used to audit cancer cell genomes in whole tumor lysates from both human tumors and mouse models of human cancers. Increasingly sophisticated bioinformatics tools have been applied to such databases, revealing gene signatures of coordinately up‐ and down‐regulated gene transcripts that correlate with and can thereby identify particular tumor subtypes and stages of progression, and in some cases predict prognosis (Golub et al., 1999; Quackenbush, 2006; Reis‐Filho and Pusztai, 2011; Sotiriou and Piccart, 2007, 2009; van't Veer and Bernards, 2008; Walther et al., 2009). Moreover, cross‐filtering of cognate mouse and human tumor transcriptome databases is proving useful in reducing complexity and potentially identifying functionally significant alterations from amongst the “white noise” of non‐specific consequential variation (Ding et al., 2011; Zender and Lowe, 2008).
An added dimension to this approach has come from assessing alterations in the expression profile of non‐coding RNAs, micro‐RNAs in particular. miRNAs are small RNAs that finely tune the expression of multiple target genes; altered miRNA expression has been observed in a variety of human pathological conditions, including cancer (Bartel, 2009). The function of miRNAs may be deregulated in cancer in several manners, including miRNA gene mutation, deletion or promoter hypermethylation, but also as a consequence of mutations in their target sequences (Esquela‐Kerscher and Slack, 2006; Nana‐Sinkam and Croce, 2011; Palmero et al., 2011). Although attenuated expression of the miRNA transcriptome is frequently observed (Kumar et al., 2007; Lu et al., 2005), various miRNAs are overexpressed in specific tumors compared to their healthy tissue of origin (Ma et al., 2010a,b; Volinia et al., 2006), and thus are termed “oncomiRs”. Furthermore, the predicted targets for several cancer‐associated miRNAs include protein‐coding tumor suppressors and oncogenes, supporting a role for miRNAs in cancer pathogenesis (Esquela‐Kerscher and Slack, 2006; Nana‐Sinkam and Croce, 2011; Palmero et al., 2011). The rapid implementation of miRNA profiling techniques (such as miRNA SAGE, microarray and deep‐sequencing) has helped in defining “miRNA gene signatures” for many cancer types, and several miRNAs now represent robust disease‐specific predictive and prognostic biomarkers (Kasinski and Slack, 2011). miRNA signatures can also be identified in the peripheral circulation of cancer patients (Mitchell et al., 2008), and several tumor‐associated miRNAs have been isolated from circulating microvesicles and exosomes (Rani et al., 2011). Of note, miRNAs are relatively stable compared to mRNAs and can be profiled from paraffin‐embedded tissues and body fluids such as blood and urine (Palmero et al., 2011; Wittmann and Jack, 2010).
In addition to cancer cells, miRNAs regulate the biology and functions of diverse tumor‐associated stromal cells, including CAFs, tumor endothelial cells, and inflammatory/immune cells (Anand and Cheresh, 2011; Aprelikova and Green, 2012; O'Connell et al., 2010; Schetter et al., 2010). There is also evidence that miRNAs can be horizontally transferred between tumor and stromal cells via microvesicles or exosomes (Simpson et al., 2009; Valadi et al., 2007; Yang et al., 2011). It can be envisioned that both miRNA and protein‐coding RNA profiling of fractionated constituent cells of the tumor stroma will prove instructive about tumor transcriptomes ranging from prognosis to potential intrinsic resistance mechanisms.
3.4. Cancer proteomes
Whereas DNA sequencing and microarray techniques may help identifying mutated or misregulated genes that could represent druggable targets, mass spectrometry‐based cancer proteomics may enable the identification of tumor‐derived proteins that can serve as biomarkers of disease and response to therapy (Hanash and Taguchi, 2010; Ludwig and Weinstein, 2005). Indeed, noninvasive proteomic analysis of body fluids (particularly serum or plasma) has identified potential biomarker signatures for several types of cancers, including asymptomatic or radiologically undetectable tumors. However, proteomic‐based biomarker discovery faces major technical hurdles that will need to be surmounted, namely: (i) the low abundance of most protein biomarkers in blood and other body fluids, particularly in early stage tumors; (ii) the difficulty to establish the relevance of a putative biomarker for tumor biology in experimental models; and (iii) the difficulty to validate the relevance of candidate biomarkers in well‐controlled clinical trials (Liotta and Petricoin, 2011; Sawyers, 2008).
Nevertheless, by way of illustrating the potential of this technology, it is worth considering a recent study that investigated the utility of mass‐spectrometry to identify plasma protein gene signatures reflective of pathways driving the development of specific lung tumor subtypes, both in human subjects and mouse models of the disease (Taguchi et al., 2011). To this aim, the authors employed three NSCLC models that harbor human‐disease causing mutations (e.g., EGFR and KRAS/p53 mutations), and compared proteomic profiles of such lung cancer models with those of mouse models of unrelated tumors or inflammation. Notably, model‐specific protein signatures were identified, such as an EGFR signature that was specific to the EGFR mutant mouse model. Importantly, selected protein signatures could be validated in human lung cancer cell lines as well as sera from yet asymptomatic lung cancer patients (Taguchi et al., 2011).
Thus, together with highly sensitive and noninvasive imaging techniques, blood‐based cancer biomarkers may improve early diagnosis of certain cancer subtypes, inform on therapy options, or enable early discovery of secondary resistance in treated patients. Toward that end, better‐standardized procedures for collecting and analyzing biological samples will be required to increase the reach of proteomics research and establish its ultimate clinical utility. In this regard, isolation of blood circulating exosomes might provide a mean to enrich for tumor‐derived proteins (Simpson et al., 2009). It remains to be seen, however, whether the sensitivity and bandwidth of proteomic profiling will be sufficient to interrogate patient‐specific differences that could guide individualized treatment strategies.
The challenge now is to integrate the increasing wealth of information provided by RNA and DNA profiling, as well as proteomics, to increase our understanding of cancer heterogeneity and devise personalized therapies that can effectively exploit such information. A cross‐disciplinary systems biology approach will be needed to translate the information contained in multidimensional data sets into molecular pathways and networks that may represent potential targets for therapy or useful predictive and prognostic biomarkers in individual cancer patients.
4. Variability and dynamics of stromal cell components
Much of the frontier of personalized cancer medicine is being charted from the perspective of the cancer cell and its individual patient‐specific alterations. Yet, as already alluded above, there is good reason to predict that heterologous cell types recruited to assemble the tumor microenvironment will also prove to be significantly variable in their abundance and functional characteristics, in ostensibly similar tumors of the same organ types, and even within subtypes and stages of progression. It is well established that multiple cell types and subtypes beyond the cancer cells – themselves variable in regards to differentiation, epithelial‐to‐mesenchymal (EMT) status, and stem cell phenotypes – contribute to the pathologic manifestation of tumors, including endothelial cells and pericytes of the tumor vasculature, CAFs, and a multitude of hematopoietic (immune inflammatory) cell types (Figure 1), amongst which are both tumor‐promoting and tumor‐antagonizing constituencies (Hanahan and Weinberg, 2011). We comment below on the major classes of stromal cells in the tumor microenvironment in regard to knowledge and implications about their biology that is relevant to the concept of personalized cancer medicine.
4.1. Tumor blood vessels
Both human and mouse tumors show considerable variation in the density, morphology and functionality of blood vessels (Langenkamp and Molema, 2009; Nagy et al., 2010). Furthermore, the endothelial cells of tumor blood vessels can display features –fenestration patterns, pericyte and inflammatory‐cell association, proliferation and apoptosis rates, and gene expression profiles – that vary not only among different tumor types or individual tumors, but also in a local–regional manner within a given tumor. Such heterogeneity may be influenced by the site in which a tumor arises (i.e., the specific organ or tissue microenvironment), the tumor growth stage, the biophysical properties of the surrounding stroma (e.g., interstitial pressure, collagen cross‐linking and extra‐cellular matrix tension), and many other ill‐defined spatiotemporal differences such as angiogenic gene expression by tumor and stromal cells (Carmeliet and Jain, 2011a; Chung and Ferrara, 2011; Hanahan and Weinberg, 2011; Kerbel, 2008; Leite de Oliveira et al., 2011; McDonald and Choyke, 2003; Nagy et al., 2010; Potente et al., 2011; Weis and Cheresh, 2011). As a consequence of these many variables affecting vascular phenotypes, experimental tumors growing subcutaneously in mice may differ significantly from spontaneous tumors in terms of vascular density, functionality, phenotype, and gene expression. For example, human PDACs are characteristically poorly vascularized, with a prevalence of non‐perfused blood vessels. These features are not recapitulated by subcutaneously growing PDACs, which lack the abundant desmoplastic stroma and high interstitial pressure that limits blood vessel functionality in autochthonous (or orthotopic) tumors. Even orthotopic xenotransplant PDAC tumors growing in the pancreas are hypervascular in comparison to genetically engineered mouse models (GEMMs) and human PDACs (Olive et al., 2009); for other tumor types, however, orthotopic transplants may more accurately reflect the bona fide angiogenic and stromal components of the tumor microenvironment, and as such present an important experimental alternative to subcutaneous tumor xenografts (Francia et al., 2011).
Although human tumors display type‐specific vascular patterns, our understanding of such variability is mostly limited to histo‐pathological features (Langenkamp and Molema, 2009; Nagy et al., 2010). Different human cancers are variably sensitive to antiangiogenic drugs (e.g., inhibitors of endothelial TK receptors or antibodies that block vascular growth factors); furthermore, tumors of a generally refractory type may sporadically show dramatic responses to antiangiogenic drugs (Carmeliet and Jain, 2011b; Chung et al., 2010; Goel et al., 2011; Leite de Oliveira et al., 2011). It can be argued that tumor type or subtype‐specific aspects of the biology of associated endothelial cells (e.g., gene expression programs) may have a role in determining such different responses. Studies in GEMMs of cancer have begun elucidating the molecular bases of such variation. It has emerged, for example, that certain tumor types are less dependent on VEGF (and hence resistant to VEGF blockade) than others, as they can use noncanonical (e.g., non‐sprouting) modalities of vascular growth or alterative proangiogenic pathways for sprouting angiogenesis (Abdullah and Perez‐Soler, 2011; Bergers and Hanahan, 2008; Leite de Oliveira et al., 2011). For instance, some tumors can grow along pre‐existing blood vessels without evoking an angiogenic response (and indeed are refractory to VEGF blockade); this process is referred to as vascular co‐option (Carmeliet and Jain, 2011a) and is specially observed in well‐vascularized tissues such as the brain (Holash et al., 1999). Other tumors may display vasculogenic mimicry, a process by which tumor cells alter their gene expression profile toward an undifferentiated phenotype, and gain the ability to form vascular‐like structures that do not depend on VEGF for their growth (Maniotis et al., 1999; Soda et al., 2011). If such vascular subtleties are not recapitulated in transplant tumor models representing a particular organ‐specific cancer, then targeted antiangiogenic drugs may fail to accurately demonstrate their effects and limitations. Additionally, there is clinical evidence for intrinsic resistance (Bergers and Hanahan, 2008) to pharmacological inhibition of the predominant VEGF signaling pathway, seen for example in the progression‐free and overall survival plots of renal cancer patients receiving the VEGF pathway inhibitors bevacizumab (Yang et al., 2003), sunitinib (Motzer et al., 2007) and sorafenib (Escudier et al., 2007), wherein some ostensibly similar patients do not respond at all to the antiangiogenic therapy, and rather continue progressing, apparently refractory to the anti‐angiogenic therapies (Albiges et al., 2011; Garcia et al., 2010; Rini et al., 2008).
GEMMs of cancer, and potentially orthotopic patient‐derived xenotransplants (PDXs; see below), may serve as instructive experimental tools for auditing the molecular biology of tumor‐associated blood vessels in the context of genetically defined tumor types that satisfactorily recapitulate the human disease. Studies that employed peptide libraries displayed on the surface of bacteriophages (“phage display”) indeed identified several tumor type and stage‐specific vascular markers (termed “vascular zip codes”) in defined GEMMs of cancer (Hoffman et al., 2003; Joyce et al., 2003). But how will these findings in mouse tumor models translate into a better understanding of the molecular biology of angiogenic processes in human cancer? And to what extent does genetic variation in individual human cancers affect angiogenic programs and tumor responses to antiangiogenic and other anticancer therapies? Gene expression signatures of human tumor endothelial cells, obtained by either micro‐dissection of fixed tissues or fluorescence‐activated cell sorting of fresh tumor biopsies, could be analyzed using the aforementioned techniques, both retrospectively and prospectively from tumors showing variable degrees of response to antiangiogenic therapy, in order to identify molecular signatures that associate with better responses. Technological advances in laser capture microscopy and in expression profiling by genome wide RNA‐sequencing from paraffin‐embedded formalin fixed tissues are beginning to render this approach feasible. Furthermore, it is expected that the advancing capabilities of genetic tools for mapping polymorphisms in the human genome (and that of mice) will allow the genes comprising the various regulatory networks controlling angiogenesis to be audited for polymorphic variations, which might affect angiogenic phenotypes by altering the strength and integration of signals impacting the state of the system. Indeed it is emerging that polymorphic variation in relevant antiangiogenic targets may provide useful predictive biomarkers of response to therapy. For example, there is proving to be considerable genetic variability within the VEGF gene, evidenced by multiple single nucleotide polymorphisms (SNPs). Notably, certain VEGF SNPs were shown to predict tumor responses and overall survival of metastatic breast cancer patients receiving combination of chemo‐ and anti‐VEGF therapy (bevacizumab) (Jain et al., 2009; Schneider et al., 2008). These provocative observations, while lacking mechanistic details, may help explaining the puzzling therapeutic heterogeneity of patient's responses to anti‐VEGF therapy, and call for auditing genetic variation and gene expression signatures in vascular cells.
4.2. Fibroblasts
Cancer‐associated fibroblasts represent another important stromal cell population in most tumor types (Kalluri and Zeisberg, 2006; Tlsty and Coussens, 2006; Pietras and Ostman, 2010). While often referred to generically as CAFs, there are evidently a number of distinctive fibroblastic cell types encompassed by this designation, including recruited myofibroblasts expressing smooth muscle actin, and activated (‘reactive’) resident tissue fibroblasts (Pietras and Ostman, 2010). The functionality of CAFs has been demonstrated for example by co‐injection of CAFs together with tumor cells, which enhances tumor growth by promoting ECM synthesis and stiffening, inducing angiogenesis, and recruiting growth‐promoting inflammatory cells such as macrophages (Erez et al., 2010; Kalluri and Zeisberg, 2006; Orimo et al., 2005; Tlsty and Coussens, 2006). CAFs are particularly abundant in certain human cancers, such as PDAC, and in various carcinomas at advanced stages of progression (e.g., breast and colorectal cancer). In such tumors, CAFs produce copious fibrotic tissue – referred to as “desmoplastic stroma” – particularly at the boundaries between the invasive cancer and the host tissue. The presence of desmoplastic stroma is associated with enhanced tumor cell invasiveness and is a major determinant of tumor cell malignancy (Egeblad et al., 2010; Levental et al., 2009). The high interstitial pressure measured in the desmoplastic stroma limits tumor blood vessel perfusion and the delivery of chemo and other forms of therapy. Furthermore, CAFs may also mediate resistance to anti‐VEGF therapy, likely via their production of stromal‐cell derived factor‐1 (SDF1, or CXCL12) – a potent chemotactic signal for proangiogenic myeloid cells (Orimo et al., 2005) – or by directly stimulating angiogenesis via their secretion of platelet‐derived growth factor‐C (PDGF‐C) (Crawford et al., 2009) and basic fibroblast growth factor (FGF2) (Pietras et al., 2008).
As noted above, CAFs are likely to comprise distinct subpopulations, whose relative abundance may vary in different tumor types (Kalluri and Zeisberg, 2006; Pietras and Ostman, 2010). However, little is known about potential patient‐specific CAF heterogeneity in human tumors of the same type/subtype. Does their composition and ‘education/induction/recruitment’ by the cancer cells and the tumor microenvironment vary in functionally significant ways across cohorts of individuals with the same cancer type and subtype? And, do their resultant gene expression signatures and functions vary, or might constitutional genetic polymorphisms affect their involvement? Elucidating the prevalence of CAF variability within tumor types and subtypes could reveal important determinants, new individualized therapeutic targets, and predictive biomarkers for tumor responses to anticancer therapies.
4.3. Leukocytes
Leukocytes make up a large proportion of the tumor‐associated stromal cells in most human and mouse tumor types (de Visser et al., 2006). Subsets of these cells, such as CD8+ cytotoxic T‐cells and natural killer (NK) cells, may play roles in restricting tumor development and progressive growth (de Visser et al., 2006; Dunn et al., 2006). On the other hand, a growing body of research in mouse cancer models has now implicated multiple leukocyte species as causal players in cancer initiation and progression. For example, macrophages and granulocytes are known to generate an “inflammatory” tumor microenvironment that promotes angiogenesis and ECM remodeling, enhances tumor cell motility and invasion, and suppresses antitumor adaptive immune responses (Mantovani et al., 2008; Motz and Coukos, 2011; Murdoch et al., 2008; Qian and Pollard, 2010; Squadrito and De Palma, 2011). For these reasons, tumor‐promoting inflammation is now regarded as an enabling characteristic of cancer (Hanahan and Weinberg, 2011).
It is increasingly evident that variable degrees of infiltration by tumor antagonizing inflammatory cells, principally CD8+ cytotoxic T‐cells, can be prognostic for outcome amongst patients with ostensibly similar tumors in terms of type and grade, with greater infiltration predicting better outcome (Fridman et al., 2011; Galon et al., 2006; Koebel et al., 2007). On the other hand, the relative abundance of immunosuppressive T‐regulatory cells (Tregs) (Bates et al., 2006; Pages et al., 2010) or CD4+ effector T‐cells (Denardo et al., 2011; Kohrt et al., 2005; Zhou et al., 2009) can be prognostic of comparatively poor outcome, as can the abundance of various myeloid cell types, including macrophages, neutrophils and immature myeloid cells (Qian and Pollard, 2010; Steidl et al., 2011; Zhou et al., 2009), a subset of which are referred to as myeloid‐derived suppressor cells on account of their ability to inhibit T‐cell functions (Sica and Bronte, 2007). Furthermore, studies in mouse models of cancer have revealed that profuse myeloid cell infiltration correlates with resistance to antiangiogenic therapies targeting the VEGF‐signaling axis (Bergers and Hanahan, 2008; Ferrara, 2010; Squadrito and De Palma, 2011), or accelerated tumor re‐growth following local tumor irradiation (Ahn and Brown, 2008; Kioi et al., 2010; Kozin et al., 2010). Additionally, there is considerable phenotypic and functional heterogeneity among macrophage and neutrophil subtypes, most simply reflected in the amalgam of conventionally or alternatively activated macrophages found in tumors, as well as in inflamed or healing tissues (Biswas and Mantovani, 2010; Coffelt et al., 2010; Gordon and Martinez, 2010; Nucera et al., 2011; Piccard et al., 2011). While less advanced in regard to appreciating individual variability, one can envision that the abundance and characteristics of these and other types of inflammatory cells will indeed prove to be deterministic and hence important parameters for personalized diagnosis. Moreover, there is increasing evidence that the bioeffector functions and hence pro‐ versus antitumor activities of both lymphoid and myeloid cell types can vary significantly in a organ‐ and tumor type dependent manner, as suggested by studies employing sophisticated mouse models of cancer engineered to develop in distinctive immunological backgrounds (Andreu et al., 2010; Ciampricotti et al., 2011; Daniel et al., 2005; DeNardo et al., 2009). Such organ‐ and tumor type peculiarities of infiltrating leukocytes may underlay the puzzling controversy surrounding the general prognostic value of macrophage and neutrophil infiltration in human tumors (Azambuja et al., 2011; Qualls and Murray, 2011; Steidl et al., 2011). Moreover, apparent contradictions in prognostic value between studies in the same tumor type may reflect differences in the inflammatory dynamics in tumors of distinctive subtype, grade, stage, and size, as well as patient‐to‐patient variability. These complexities call for better methods with which to audit the complexity of tumor‐associated inflammatory cells in mouse and human. With the advent and continuing refinement of automated flow cytometry and tissue immunohistochemistry processing devices enabling 8–10 antigens to be audited in parallel, one can envision that tumor biopsies will in the future be screened to assess their particular constitution of inflammatory cell types and subtypes in a comprehensive and standardized manner. Such knowledge may possibly contribute to informed decisions about treatment strategies. The premise of this approach is exemplified by a recent study that analyzed multiple leukocyte populations in a large cohort of breast cancer patients treated by surgery alone, which identified “leukocyte signatures” – i.e. different compositions of infiltrating leukocytes in individual tumors – that can predict the patient's likelihood to respond favorably to adjuvant chemotherapy (Denardo et al., 2011). In particular, the combination of high macrophage and low cytotoxic T‐cell abundance – but not of either cell population alone – identifies a subset of patients at high risk for developing chemoresistant, metastatic disease. These intriguing findings call for the systematic analysis of leukocyte infiltrates in tumors of different types and subtypes. Defining the molecular profiles of “high‐risk leukocytes” (either by immunophenotyping or gene expression profiling) may also enable the validation of novel tumor biomarkers. Furthermore, specific leukocyte signatures might be used to stratify patients carrying defined genetic lesions, in order to explore how such signatures correlate with the individual patient's response to the targeted therapy and the emergence of secondary resistance. If such studies demonstrate the prognostic value of specific leukocyte signatures for individual tumor responses to targeted therapies, then further characterization of leukocyte complexity and function in mouse tumor models may contribute toward the development of more effective, fully personalized cancer therapies, that for example target the inflammatory component of the tumor by reprogramming leukocyte infiltrates from a tumor‐promoting to an anti‐tumor function (Hagemann et al., 2008; Squadrito and De Palma, 2011; Stout et al., 2009).
Finally, it should be mentioned that certain targeted drugs may indirectly activate inflammatory cells to modulate therapeutic responses in both mouse tumor models and cancer patients (Beatty et al., 2011). For example, the anti‐HER2 antibody trastuzumab is thought to mediate tumor regression by both interrupting oncogenic signaling in tumor cells and inducing immunoglobulin (IgG) receptor (FcR)‐mediated cytotoxicity (Park et al., 2010). Indeed, FcR receptors expressed on the surface of certain inflammatory cells can recognize IgG‐coated tumor cells, and the interaction between FcRs and IgGs can in turn activate the inflammatory cells to release cytotoxic molecules that kill the tumor cells. The therapeutic effect of trastuzumab is in fact decreased in the absence of FcR signaling in mice (Clynes et al., 2000), and specific FcR polymorphisms are associated with clinical outcome in breast cancer patients (Musolino et al., 2008). These interesting observations also imply that SNPs in genes expressed by the stromal components of the tumor may influence tumor responses to targeted therapies.
4.4. Population dynamics of stromal cell constituents
The above depictions of the phenotypic diversity of stromal subtypes have largely ignored another facet to the challenge of understanding the heterogeneity of tumor biology: population dynamics. How do individual tumors of a particular subtype vary in the proportional abundance and organization of the various stromal cell types and subtypes? How do population dynamics of an individual's tumor change during malignant progression and metastasis, and in response to therapy? And, what is the implication of regional variation in stromal cell composition within a solid tumor – another dimension to the mutational subclones discussed above – in terms of prognosis and predicted response or resistance to different therapies? Addressing these questions is an important agenda for the field of personalized cancer medicine.
5. Harnessing mouse models of cancer to investigate individualized therapies recognizing and exploiting tumor heterogeneity in cancer patients
Transplant tumor models – in particular human tumor cell lines that are propagated in vitro and grown in immunodeficient mice as tumor xenografts – have the potential to model important aspects of the individual variability of human cancer, typically the heterogeneity intrinsic to the cancer cells of an individual's tumor (Sharpless and Depinho, 2006). There are, however, notable limitations to such models in terms of tumor progression and microenvironment. In fact, transplant tumor models do not in general recapitulate several key features of spontaneous, organ‐specific tumorigenesis, such as: premalignant to malignant progression; angiogenic switching; acquisition of defined histo‐pathological marks; and organ‐specific and progression‐stage dependent interplay with the immune system and other components of the tumor microenvironment. Because they do not fully recapitulate tumor microenvironmental (and histo‐pathological) complexity, transplant tumor models have often failed to predict drug responses (Sharpless and Depinho, 2006). We envision increasing refinement of transplant tumor models, mostly through (i) the use of orthotopic sites of inoculation to generate tumors growing in the parental organs; (ii) the implantation of primary patient‐derived material – either tumor fragments or briefly cultured cancer cells – to replicate and propagate individual variability; and (iii) co‐inoculation of cancer cells (or primary epithelial cells engineered ex vivo to express defined oncogenes) together with human stromal cell types, e.g., patient‐derived fibroblasts. As discussed below, such refinements of transplant tumor models – along with the implementation of both germline and somatic GEMMs – may help in developing tumor models that better recapitulate the features of individual cancers and their responses to targeted therapies.
5.1. Patient‐derived xenotransplants (PDXs)
Recent studies have demonstrated that small tumor biopsies inoculated in immunodeficient mice can retain the morphological and genetic heterogeneity of the parental tumor, including individual diversity, even when propagated ectopically, e.g., subcutaneously (Bertotti et al., 2011). This successful recapitulation may be due to the “supportive” role of the parental tumor stroma (primarily CAFs but also vascular and hematopoietic cells), which is co‐transplanted with the cancer cells and is possibly propagated with the latter even through a few serial passages. Indeed, small human lung cancer biopsies inoculated subcutaneously in severely immunodeficient NOD‐SCID/IL2Rγnull (NSG) mice were found to retain intra‐tumoral populations of human T‐cells, which could also expand and colonize peripheral lymphoid organs for prolonged periods (Simpson‐Abelson et al., 2008). These findings suggest that some stromal cell constituents of non‐disrupted human cancer biopsies can engraft long‐term in situ and even systemically in PDX models. It remains to be seen whether terminally differentiated human cells such as macrophages or vascular cells behave similarly in such xenografted tumors.
While the subcutaneous site is convenient and often the choice for logistical reasons, most tumors growing subcutaneously lack the full manifestation of the fibroblastic stroma of their parental tumors, and the tumor vasculature is often different, both in quantity and quality of tumor blood vessels, as noted above. Furthermore, subcutaneous PDX models may fail to reflect the patterns of tumor growth and spreading that are observed in cancer patients; and, while many tumor biopsies fail to engraft, others may produce xenografts with large areas of necrotic tissue due to inadequate re‐vascularization of the tumor mass. Implantation of such tumor biopsies (or perhaps fractionated cancer cells and admixed CAFs following brief culture without serial passaging) at orthotopic sites may circumvent some of the limitations of ectopic injection, particularly for those tumor types that typically contain abundant fibrotic stroma (e.g., pancreatic and breast cancer) or hematopoietic cell infiltrates (e.g., ovarian tumors). For example, breast cancer biopsies xenografted orthotopically together with primary human mesenchymal cells in NOD‐SCID mice retain the diversity and essential features of the original human breast cancer subtypes, including organ‐specific (e.g., lung or bone) metastasis (Derose et al., 2011). Furthermore, human ovarian tumors can be successfully established as orthotopic xenografts in NSG mice, recapitulating growth and progression patterns observed in ovarian cancer patients, while retaining critical human stromal cell components such as CAFs and T‐cells (Bankert et al., 2011). It is anticipated, however, that human stromal cells will be progressively lost or diluted by mouse‐derived cells during tumor growth and serial transplants in either ectopic or orthotopic sites, raising challenges for biobank preservation to allow replication of results and additional analyses.
5.2. Primary cells genetically modified with oncogenic signature mutations
Defined human tumor types may also be recapitulated in mouse models by genetically engineering primary human epithelial cells (Fan et al., 1997; Heyer et al., 2010; Khavari, 2006). This approach was illustrated, e.g., by using mammary (Duss et al., 2007) or epidermal (Fan et al., 1997; Khavari, 2006) epithelial cells obtained from reduction mammoplasties or skin biopsies, respectively. The isolated cells were propagated in vitro and then transduced with integrative viral vectors (e.g., γ‐retro or lenti‐viruses) that carried the desired combination of signature mutations that are characteristic of the human tumor being modeled. Orthotopic injection of these genetically modified primary epithelial cells, particularly when made in combination with human primary stromal cells such as fibroblasts, can generate human tumors that quite faithfully recapitulate the histo‐pathological features of genotype‐specific human disease (Fan et al., 1997; Heyer et al., 2010; Khavari, 2006). These transplant models have an important advantage over germline GEMMs of cancer, in that they allow for testing of multiple mutations in multiplex combinations, in a time and cost effective manner (Wu and Robinson, 2009).
5.3. Non‐germline (somatic) GEMMs of cancer
Recent studies have demonstrated the utility of non‐germline (or somatic) GEMMs of cancer. This approach is based on the injection of genetically modified embryonic stem (ES) cells into pre‐implantation mouse embryos (Heyer et al., 2010; Zhou et al., 2010). The ES cells are engineered to contain multiple chromosomal integrations of inducible, cell type‐specific human signature mutations; the mouse chimeras that are derived from injection of the ES cells into mouse blastocysts can therefore carry multiple cancer‐enabling alleles that are expressed only in the cells derived from the genetically modified ES cells, which will constitute a variably small fraction of the total cells in any given tissue. These spontaneous tumor models, as opposed to germline GEMMs, more faithfully model the stochastic nature of tumorigenesis, as the tumor arises sporadically due to the chimeric nature of the cancer‐enabled cells. The non‐germline nature of such models obviously prohibits propagation of the mice, but allows for generating libraries of ES cells that carry oncogenes relevant to human disease and that can be conveniently stored (Heyer et al., 2010). Tumors that arise in such chimeric mice can be serially transplanted to develop secondary cohorts of mice for experimental therapeutic trials. However, the potential immunogenicity of human oncogenic products typically requires the secondary transplants to be propagated in immunodeficient mice, in order to avoid interference or abject rejection by the adaptive immune system.
5.4. Limitations of transplant tumor models: immunological deficiencies and cross‐species incompatibilities
An unavoidable limitation of the aforementioned transplant tumor models lies in the immunodeficiency of the transplant host, leaving the inflammatory response seriously biased, invariably lacking T‐ and B‐lymphocytes, which amongst their repertoire include not only tumor antagonizing but also tumor promoting subtypes. This will remain an important and perhaps unsolvable qualification to xenotransplant models, which argues for parallel efforts toward continuing refinement of GEMMs of cancer that can reflect aspects of individual tumor heterogeneity. Another limitation of xenotransplant models is that human tumor cells may fail to appropriately respond to mouse‐derived signals (e.g., growth factors and hormones) because of cross‐species biological incompatibility. This was shown, for example, in a study demonstrating that human breast cancer cells inoculated in mice, while expressing prolactin receptors, are insensitive to mouse prolactin and are selected in vivo for growth independence of circulating prolactin (Utama et al., 2006). These findings suggest that the biology (and consequent drug‐response profiles) of human cancers propagated in mice may be influenced by the mouse (endocrine) environment.
A strategy to circumvent some of the limitations of xenotranplantation while preserving the flexibility of transplant tumor model may involve the injection of genetically modified mouse cells, including lineage‐restricted stem/progenitor cells, orthotopically into syngeneic, immunocompetent mice (Bachoo et al., 2002; Heyer et al., 2010). This “mouse‐in‐mouse” transplant model has the advantage over human xenotransplant models to expose tumor development to the same immunological barriers that are present in GEMMs of cancer, albeit without the genetic heterogeneity that distinguishes otherwise apparently similar human tumors.
5.5. Germline GEMMs of cancer
Conventional (or germline) GEMMs of cancer undeniably provide a powerful means to study tumor biology and response to therapy (Hanahan et al., 2007; van Miltenburg and Jonkers, 2012). Such tumor models can in many cases recapitulate the histo‐pathological and genetic features of their human disease counterparts, originating from “normal cells” that reside in tissue‐ or organ‐specific microenvironments and are subjected, at least in part, to the biological barriers to tumorigenesis that are erected by the particular normal tissue microenvironment. GEMMs of cancer can be generated to harbor the specific molecular lesions that are causally associated with human cancer. However, tissue‐specific expression of oncogenic proteins may induce epithelial transformation throughout the targeted tissue, often represented by diffuse hyperplasias that may cause significant morbidity and even mortality in mice (Heyer et al., 2010; Zhou et al., 2010). The implementation of inducible systems in which signature mutations are activated only in a small proportion of the cells of a defined tissue and/or in a temporally defined manner (e.g., by tamoxifen‐inducible CRE recombinases that are delivered topically or activated by suboptimal doses of tamoxifen) may better recapitulate the stochastic nature of tumorigenesis (Jonkers and Berns, 2002; Meylan et al., 2009). Yet, even the most sophisticated GEMMs of cancer – such as mice that carry multiple genetic lesions reflecting a defined cancer gene signature – may lack the individual variation that is commonly observed within each cancer type/subtype. This aspect can be potentially addressed by employing outbred strains of mice, or mice that are prone to genetic instability due to deficiencies in the genes that maintain genomic integrity.
Another limitation of GEMMs of cancer is that such models often show a low incidence and/or narrow spectrum of visceral metastases. Because metastatic disease represents the ultimate target of anticancer therapies, preclinical models that can recapitulate organ‐specific metastasis should be developed and employed to more realistically assay the therapeutic potential of candidate drugs. Orthotopic transplants (including PDX models) may enhance the incidence of distant metastatic spread compared with subcutaneous transplants (Derose et al., 2011), especially if the primary tumor is surgically removed to allow the development of macroscopic metastases from the disseminated tumor cells (Francia et al., 2011). Highly metastatic human tumor variants can also be generated through multiple rounds of in vivo growth of tumor cell lines in immunodeficient mice, each involving orthotopic tumor cell transplantation/growth/resection, and isolation of metastatic cells from the visceral organs (Francia et al., 2011; Kerbel et al., 1984). This approach has been used to select highly metastatic tumor cell lines that may be representative of various human cancer types/subtypes (Francia et al., 2011); in some cases, genetic modification of the tumor cells was used to introduce relevant driver oncogenic mutations in the tumor cells (du Manoir et al., 2006). However, this approach is not easily applicable to the derivation of patient‐specific tumor cell lines that can inform on treatment options for the individual, as the in vivo selection procedure is laborious and lengthy (Francia et al., 2011).
In sum, we view the future of personalized cancer research to incorporate both germline and somatic GEMMs in concert with refined (preferably orthotopic) PDX models that will each be selectively informative about the biology and genetics of variation amongst individual tumors of a given type.
6. Concluding remarks
The importance of individual diversity in the biology of cancer amongst patients with ostensibly similar tumors (and in particular with common driver mutations) is compellingly revealed in the variable responses and even intrinsic resistance to driver‐ and mechanism‐targeted therapies, arguing that we need to develop a deeper understanding of their differences, so as to fine‐tune therapies to be efficacious in each individual by recognizing particular drug sensitivities as well as the evident insensitivities.
Thus, for example, the identification of EGFR and ALK mutations in NSCLC and their association with sensitivity to EGFR and ALK TKIs is revolutionizing the treatment of this cancer (Gaughan and Costa, 2011). In fact, treatment strategy can now be decided on a truly individual basis, and will produce beneficial responses in the majority of the patients carrying the respective mutation. Although most patients eventually develop secondary resistance and relapse, re‐biopsy can establish the molecular basis of resistance in some cases, allowing physicians to offer further treatment options, e.g., irreversible EGFR TKIs (Kwak et al., 2005), which are a suitable option for half of the relapsing patients. The NSCLC experience thus offers a compelling case for refining clinical trial designs so as to identify patient sub‐populations that are most likely to benefit from the therapy. Of course, even in this tumor type there is much to be learned, about how to best treat those patients lacking such driver mutations; perhaps deeper mining of the cancer cell and of the tumor stroma using the evolving armamentarium of analytic technologies combined with advancing knowledge will reveal functionally important molecular and cellular (stromal) targets amenable to therapeutic modulation to the benefit of particular patients.
The grand challenge now is to generalize and expand this conceptual awakening across the spectrum of human cancers, exploiting the complexity and variability within a particular type/subtype to therapeutic benefit, rather than failing because of it. The heterogeneity of cancer cells (and of cancer stem cells) as well as of stromal components – tumor vascularization, tumor‐promoting and tumor‐antagonizing immune cell infiltrates, CAF subtypes, etc. – should be interrogated through molecular screening, immunohistochemistry, better noninvasive imaging, and also careful examination of re‐biopsy specimens obtained during treatment as well as at treatment failure (Figure 1). Quantitative immunohistochemistry or immunofluorescence assays are being developed that can accurately validate on tissue microarrays (TMAs) the expression of DNA/RNA signatures identified by whole‐tumor profiling; these techniques have the advantage of being highly reproducible and easily applicable for standardized tumor typing/grading, inter‐patient comparisons, and the dissection of intra‐tumoral heterogeneity. Recent applications of such TMA‐based technologies have validated prognostic gene signatures for several tumor types, including prostate and lung cancer (Anagnostou et al., 2011; Dimou et al., 2011; Ding et al., 2011; Zender and Lowe, 2008). Furthermore, the development of sophisticated computer‐based methodologies for TMA analysis (Beck et al., 2011) may facilitate better standardization of the results and also quantitatively assess clinically significant morphometric parameters (e.g., epithelial–stromal ratio, multiple nuclear pleomorphisms, etc…) that would escape conventional pathological examination.
It has been proposed that a synchronized “GEMM‐to‐human” strategy should be implemented to accelerate preclinical and clinical data collection and promote rationally designed clinical trials that address individual variation and secondary resistance (Nardella et al., 2011). According to this strategy, each phase I/II clinical trial should be conducted in parallel with appropriate (and possibly diversified) GEMMs of cancer, so that relevant information obtained from patients and mice is integrated to facilitate patient stratification and identification of biomarkers of disease and response to therapy; among the correlative metrics that can be envisioned are analysis of genome mutational status and constitutional polymorphic variation, transcriptome and proteome profiles, effects of the therapeutic regimens(s), noninvasive imaging of responses, and assessment of relapse consequent to adaptive resistance. This strategy will likely be complemented by analyzing the spectrum of tumor phenotypes in cohorts PDX mice reflecting the diversity of particular human cancers (Bankert et al., 2011; Derose et al., 2011). In the future, PDX models may evolve to involve severely immunodeficient mice (e.g., NSG mice) engineered to express defined human cytokines and histocompatibility genes along with engrafted human hematopoietic stem cells, so as to recapitulate and sustain upon serial passage more authentic tumor microenvironments involving both tumor‐antagonizing and tumor‐promoting immune inflammatory cells of human origin. Data obtained from GEMMs and PDX models of cancer and from patient biopsies and blood borne materials (proteins, microRNA, exosomes, circulating tumor‐derived cells) hold promise to inform hypothesis‐testing and personalized “GEMM/PDX‐to‐human” trials that may address individual variation in tumor responses to the drug(s) and/or mechanisms of acquired resistance. Such coordinated therapeutic trials in mouse models and humans, designed to recognize, reflect, and leverage patient‐to‐patient variability, are envisioned to help pioneer the frontier of personalized cancer medicine toward more broadly efficacious therapies for human cancers.
Acknowledgements
We thank Etienne Meylan for comments and suggestions on the manuscript. We note that not all relevant results have been discussed and cited, owing to space limitations. We thank Mario Leonardo Squadrito for preparing the Figure. MDP is supported by grants from the European Research Council (Starting Grant 243128/TIE2+Monocytes) and the Associazione Italiana per la Ricerca sul Cancro (IG‐2010).
De Palma Michele, Hanahan Douglas (2012), The biology of personalized cancer medicine: Facing individual complexities underlying hallmark capabilities, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.01.011.
Contributor Information
Michele De Palma, Email: michele.depalma@epfl.ch.
Douglas Hanahan, Email: douglas.hanahan@epfl.ch.
References
- Abdullah, S.E. , Perez-Soler, R. , 2011. Mechanisms of resistance to vascular endothelial growth factor blockade. Cancer. 10.1002/cncr.26540 [DOI] [PubMed] [Google Scholar]
- Ahn, G.O. , Brown, J.M. , 2008. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 13, 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albiges, L. , Salem, M. , Rini, B. , Escudier, B. , 2011. Vascular endothelial growth factor-targeted therapies in advanced renal cell carcinoma. Hematol. Oncol. Clin. North Am.. 25, 813–833. [DOI] [PubMed] [Google Scholar]
- Anagnostou, V.K. , Dimou, A.T. , Botsis, T. , Killiam, E.J. , Gustavson, M.D. , Homer, R.J. , Boffa, D. , Zolota, V. , Dougenis, D. , Tanoue, L. , 2011. Molecular classification of nonsmall-cell lung cancer using a 4-protein quantitative assay. Cancer. 10.1002/cncr.26450 [DOI] [PubMed] [Google Scholar]
- Anand, S. , Cheresh, D.A. , 2011. MicroRNA-mediated regulation of the angiogenic switch. Curr. Opin. Hematol.. 18, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu, P. , Johansson, M. , Affara, N.I. , Pucci, F. , Tan, T. , Junankar, S. , Korets, L. , Lam, J. , Tawfik, D. , DeNardo, D.G. , 2010. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 17, 121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprelikova, O. , Green, J.E. , 2012. MicroRNA regulation in cancer-associated fibroblasts. Cancer Immunol. Immunother. 61, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azambuja, D. , Natkunam, Y. , Biasoli, I. , Lossos, I.S. , Anderson, M.W. , Morais, J.C. , Spector, N. , 2011. Lack of association of tumor-associated macrophages with clinical outcome in patients with classical Hodgkin's lymphoma. Ann. Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo, R.M. , Maher, E.A. , Ligon, K.L. , Sharpless, N.E. , Chan, S.S. , You, M.J. , Tang, Y. , DeFrances, J. , Stover, E. , Weissleder, R. , 2002. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 1, 269–277. [DOI] [PubMed] [Google Scholar]
- Bankert, R.B. , Balu-Iyer, S.V. , Odunsi, K. , Shultz, L.D. , Kelleher, R.J. , Barnas, J.L. , Simpson-Abelson, M. , Parsons, R. , Yokota, S.J. , 2011. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS One. 6, e24420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel, D.P. , 2009. MicroRNAs: target recognition and regulatory functions. Cell. 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, G.J. , Fox, S.B. , Han, C. , Leek, R.D. , Garcia, J.F. , Harris, A.L. , Banham, A.H. , 2006. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol.. 24, 5373–5380. [DOI] [PubMed] [Google Scholar]
- Baylin, S.B. , Jones, P.A. , 2011. A decade of exploring the cancer epigenome – Biological and translational implications. Nat. Rev. Cancer. 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty, G.L. , Chiorean, E.G. , Fishman, M.P. , Saboury, B. , Teitelbaum, U.R. , Sun, W. , Huhn, R.D. , Song, W. , Li, D. , Sharp, L.L. , 2011. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 331, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, A.H. , Sangoi, A.R. , Leung, S. , Marinelli, R.J. , Nielsen, T.O. , van de Vijver, M.J. , West, R.B. , van de Rijn, M. , Koller, D. , 2011. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci. Transl. Med.. 3, 108 ra113 [DOI] [PubMed] [Google Scholar]
- Berger, M.F. , Lawrence, M.S. , Demichelis, F. , Drier, Y. , Cibulskis, K. , Sivachenko, A.Y. , Sboner, A. , Esgueva, R. , Pflueger, D. , Sougnez, C. , 2011. The genomic complexity of primary human prostate cancer. Nature. 470, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers, G. , Hanahan, D. , 2008. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer. 8, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti, A. , Migliardi, G. , Galimi, F. , Sassi, F. , Torti, D. , Isella, C. , Corà, D. , Di Nicolantonio, F. , Buscarino, M. , 2011. A molecularly annotated platform of patient-derived xenografts (“Xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discovery. 1, 508–523. [DOI] [PubMed] [Google Scholar]
- Biswas, S.K. , Mantovani, A. , 2010. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol.. 11, 889–896. [DOI] [PubMed] [Google Scholar]
- de Bono, J.S. , Ashworth, A. , 2010. Translating cancer research into targeted therapeutics. Nature. 467, 543–549. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. , Jain, R.K. , 2011. Molecular mechanisms and clinical applications of angiogenesis. Nature. 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P. , Jain, R.K. , 2011. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov.. 10, 417–427. [DOI] [PubMed] [Google Scholar]
- Chung, A.S. , Ferrara, N. , 2011. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol.. 27, 563–584. [DOI] [PubMed] [Google Scholar]
- Chung, A.S. , Lee, J. , Ferrara, N. , 2010. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat. Rev. Cancer. 10, 505–514. [DOI] [PubMed] [Google Scholar]
- Ciampricotti, M. , Vrijland, K. , Hau, C.S. , Pemovska, T. , Doornebal, C.W. , Speksnijder, E.N. , Wartha, K. , Jonkers, J. , de Visser, K.E. , 2011. Development of metastatic HER2(+) breast cancer is independent of the adaptive immune system. J. Pathol.. 224, 56–66. [DOI] [PubMed] [Google Scholar]
- Clynes, R.A. , Towers, T.L. , Presta, L.G. , Ravetch, J.V. , 2000. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med.. 6, 443–446. [DOI] [PubMed] [Google Scholar]
- Coffelt, S.B. , Lewis, C.E. , Naldini, L. , Brown, J.M. , Ferrara, N. , De Palma, M. , 2010. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am. J. Pathol.. 176, 1564–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comen, E. , Norton, L. , Massague, J. , 2011. Clinical implications of cancer self-seeding. Nat. Rev. Clin. Oncol.. 8, 369–377. [DOI] [PubMed] [Google Scholar]
- Crawford, Y. , Kasman, I. , Yu, L. , Zhong, C. , Wu, X. , Modrusan, Z. , Kaminker, J. , Ferrara, N. , 2009. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 15, 21–34. [DOI] [PubMed] [Google Scholar]
- Daniel, D. , Chiu, C. , Giraudo, E. , Inoue, M. , Mizzen, L.A. , Chu, N.R. , Hanahan, D. , 2005. CD4+ T cell-mediated antigen-specific immunotherapy in a mouse model of cervical cancer. Cancer Res.. 65, 2018–2025. [DOI] [PubMed] [Google Scholar]
- De Roock, W. , Claes, B. , Bernasconi, D. , De Schutter, J. , Biesmans, B. , Fountzilas, G. , Kalogeras, K.T. , Kotoula, V. , Papamichael, D. , Laurent-Puig, P. , 2010. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol.. 11, 753–762. [DOI] [PubMed] [Google Scholar]
- DeNardo, D.G. , Barreto, J.B. , Andreu, P. , Vasquez, L. , Tawfik, D. , Kolhatkar, N. , Coussens, L.M. , 2009. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 16, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denardo, D.G. , Brennan, D.J. , Rexhepaj, E. , Ruffell, B. , Shiao, S.L. , Madden, S.F. , Gallagher, W.M. , Wadhwani, N. , Keil, S.D. , Junaid, S.A. , 2011. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov.. 1, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derose, Y.S. , Wang, G. , Lin, Y.C. , Bernard, P.S. , Buys, S.S. , Ebbert, M.T. , Factor, R. , Matsen, C. , Milash, B.A. , Nelson, E. , 2011. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med.. 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou, A. , Agarwal, S. , Anagnostou, V. , Viray, H. , Christensen, S. , Gould Rothberg, B. , Zolota, V. , Syrigos, K. , Rimm, D.L. , 2011. Standardization of epidermal growth factor receptor (EGFR) measurement by quantitative immunofluorescence and impact on antibody-based mutation detection in non-small cell lung cancer. Am. J. Pathol.. 179, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Z. , Wu, C.J. , Chu, G.C. , Xiao, Y. , Ho, D. , Zhang, J. , Perry, S.R. , Labrot, E.S. , Wu, X. , Lis, R. , 2011. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker, B.J. , Tamura, S. , Buchdunger, E. , Ohno, S. , Segal, G.M. , Fanning, S. , Zimmermann, J. , Lydon, N.B. , 1996. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med.. 2, 561–566. [DOI] [PubMed] [Google Scholar]
- Druker, B.J. , Guilhot, F. , O'Brien, S.G. , Gathmann, I. , Kantarjian, H. , Gattermann, N. , Deininger, M.W. , Silver, R.T. , Goldman, J.M. , Stone, R.M. , 2006. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med.. 355, 2408–2417. [DOI] [PubMed] [Google Scholar]
- Dunn, G.P. , Koebel, C.M. , Schreiber, R.D. , 2006. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol.. 6, 836–848. [DOI] [PubMed] [Google Scholar]
- Duss, S. , Andre, S. , Nicoulaz, A.L. , Fiche, M. , Bonnefoi, H. , Brisken, C. , Iggo, R.D. , 2007. An oestrogen-dependent model of breast cancer created by transformation of normal human mammary epithelial cells. Breast Cancer Res.. 9, R38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad, M. , Nakasone, E.S. , Werb, Z. , 2010. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell. 18, 884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Erez, N. , Truitt, M. , Olson, P. , Arron, S.T. , Hanahan, D. , 2010. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 17, 135–147. [DOI] [PubMed] [Google Scholar]
- Escudier, B. , Eisen, T. , Stadler, W.M. , Szczylik, C. , Oudard, S. , Siebels, M. , Negrier, S. , Chevreau, C. , Solska, E. , Desai, A.A. , 2007. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med.. 356, 125–134. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher, A. , Slack, F.J. , 2006. Oncomirs – microRNAs with a role in cancer. Nat. Rev. Cancer. 6, 259–269. [DOI] [PubMed] [Google Scholar]
- Esteva, F.J. , Yu, D. , Hung, M.C. , Hortobagyi, G.N. , 2010. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat. Rev. Clin. Oncol.. 7, 98–107. [DOI] [PubMed] [Google Scholar]
- Fan, H. , Oro, A.E. , Scott, M.P. , Khavari, P.A. , 1997. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat. Med.. 3, 788–792. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. , 2010. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr. Opin. Hematol.. 17, 219–224. [DOI] [PubMed] [Google Scholar]
- Flaherty, K.T. , Yasothan, U. , Kirkpatrick, P. , 2011. Vemurafenib. Nat. Rev. Drug Discov.. 10, 811–812. [DOI] [PubMed] [Google Scholar]
- Francia, G. , Cruz-Munoz, W. , Man, S. , Xu, P. , Kerbel, R.S. , 2011. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat. Rev. Cancer. 11, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman, W.H. , Galon, J. , Pages, F. , Tartour, E. , Sautes-Fridman, C. , Kroemer, G. , 2011. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res.. 71, 5601–5605. [DOI] [PubMed] [Google Scholar]
- Galon, J. , Costes, A. , Sanchez-Cabo, F. , Kirilovsky, A. , Mlecnik, B. , Lagorce-Pages, C. , Tosolini, M. , Camus, M. , Berger, A. , Wind, P. , 2006. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- Garcia, J.A. , Hutson, T.E. , Elson, P. , Cowey, C.L. , Gilligan, T. , Nemec, C. , Dreicer, R. , Bukowski, R.M. , Rini, B.I. , 2010. Sorafenib in patients with metastatic renal cell carcinoma refractory to either sunitinib or bevacizumab. Cancer. 116, 5383–5390. [DOI] [PubMed] [Google Scholar]
- Gaughan, E.M. , Costa, D.B. , 2011. Genotype-driven therapies for non-small cell lung cancer: focus on EGFR, KRAS and ALK gene abnormalities. Ther. Adv. Med. Oncol.. 3, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel, S. , Duda, D.G. , Xu, L. , Munn, L.L. , Boucher, Y. , Fukumura, D. , Jain, R.K. , 2011. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev.. 91, 1071–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub, T.R. , Slonim, D.K. , Tamayo, P. , Huard, C. , Gaasenbeek, M. , Mesirov, J.P. , Coller, H. , Loh, M.L. , Downing, J.R. , Caligiuri, M.A. , 1999. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 286, 531–537. [DOI] [PubMed] [Google Scholar]
- Gordon, S. , Martinez, F.O. , 2010. Alternative activation of macrophages: mechanism and functions. Immunity. 32, 593–604. [DOI] [PubMed] [Google Scholar]
- Haber, D.A. , Gray, N.S. , Baselga, J. , 2011. The evolving war on cancer. Cell. 145, 19–24. [DOI] [PubMed] [Google Scholar]
- Hagemann, T. , Lawrence, T. , McNeish, I. , Charles, K.A. , Kulbe, H. , Thompson, R.G. , Robinson, S.C. , Balkwill, F.R. , 2008. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med.. 205, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Wagner, E.F. , Palmiter, R.D. , 2007. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev.. 21, 2258–2270. [DOI] [PubMed] [Google Scholar]
- Hanash, S. , Taguchi, A. , 2010. The grand challenge to decipher the cancer proteome. Nat. Rev. Cancer. 10, 652–660. [DOI] [PubMed] [Google Scholar]
- Heyer, J. , Kwong, L.N. , Lowe, S.W. , Chin, L. , 2010. Non-germline genetically engineered mouse models for translational cancer research. Nat. Rev. Cancer. 10, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, J.A. , Giraudo, E. , Singh, M. , Zhang, L. , Inoue, M. , Porkka, K. , Hanahan, D. , Ruoslahti, E. , 2003. Progressive vascular changes in a transgenic mouse model of squamous cell carcinoma. Cancer Cell. 4, 383–391. [DOI] [PubMed] [Google Scholar]
- Holash, J. , Maisonpierre, P.C. , Compton, D. , Boland, P. , Alexander, C.R. , Zagzag, D. , Yancopoulos, G.D. , Wiegand, S.J. , 1999. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 284, 1994–1998. [DOI] [PubMed] [Google Scholar]
- Jain, L. , Vargo, C.A. , Danesi, R. , Sissung, T.M. , Price, D.K. , Venzon, D. , Venitz, J. , Figg, W.D. , 2009. The role of vascular endothelial growth factor SNPs as predictive and prognostic markers for major solid tumors. Mol. Cancer Ther.. 8, 2496–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers, J. , Berns, A. , 2002. Conditional mouse models of sporadic cancer. Nat. Rev. Cancer. 2, 251–265. [DOI] [PubMed] [Google Scholar]
- Joyce, J.A. , Laakkonen, P. , Bernasconi, M. , Bergers, G. , Ruoslahti, E. , Hanahan, D. , 2003. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 4, 393–403. [DOI] [PubMed] [Google Scholar]
- Kalluri, R. , Zeisberg, M. , 2006. Fibroblasts in cancer. Nat. Rev. Cancer. 6, 392–401. [DOI] [PubMed] [Google Scholar]
- Kasinski, A.L. , Slack, F.J. , 2011. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer. 11, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel, R.S. , Man, M.S. , Dexter, D. , 1984. A model of human cancer metastasis: extensive spontaneous and artificial metastasis of a human pigmented melanoma and derived variant sublines in nude mice. J. Natl. Cancer Inst.. 72, 93–108. [DOI] [PubMed] [Google Scholar]
- Kerbel, R.S. , 2008. Tumor angiogenesis. N. Engl. J. Med.. 358, 2039–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavari, P.A. , 2006. Modelling cancer in human skin tissue. Nat. Rev. Cancer. 6, 270–280. [DOI] [PubMed] [Google Scholar]
- Kioi, M. , Vogel, H. , Schultz, G. , Hoffman, R.M. , Harsh, G.R. , Brown, J.M. , 2010. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest.. 120, 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebel, C.M. , Vermi, W. , Swann, J.B. , Zerafa, N. , Rodig, S.J. , Old, L.J. , Smyth, M.J. , Schreiber, R.D. , 2007. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 450, 903–907. [DOI] [PubMed] [Google Scholar]
- Kohrt, H.E. , Nouri, N. , Nowels, K. , Johnson, D. , Holmes, S. , Lee, P.P. , 2005. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med.. 2, e284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin, S.V. , Kamoun, W.S. , Huang, Y. , Dawson, M.R. , Jain, R.K. , Duda, D.G. , 2010. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res.. 70, 5679–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M.S. , Lu, J. , Mercer, K.L. , Golub, T.R. , Jacks, T. , 2007. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet.. 39, 673–677. [DOI] [PubMed] [Google Scholar]
- Kwak, E.L. , Sordella, R. , Bell, D.W. , Godin-Heymann, N. , Okimoto, R.A. , Brannigan, B.W. , Harris, P.L. , Driscoll, D.R. , Fidias, P. , Lynch, T.J. , 2005. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. U S A. 102, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenkamp, E. , Molema, G. , 2009. Microvascular endothelial cell heterogeneity: general concepts and pharmacological consequences for anti-angiogenic therapy of cancer. Cell Tissue Res.. 335, 205–222. [DOI] [PubMed] [Google Scholar]
- Leite de Oliveira, R. , Hamm, A. , Mazzone, M. , 2011. Growing tumor vessels: more than one way to skin a cat – Implications for angiogenesis targeted cancer therapies. Mol. Aspects Med.. 32, 71–87. [DOI] [PubMed] [Google Scholar]
- Levental, K.R. , Yu, H. , Kass, L. , Lakins, J.N. , Egeblad, M. , Erler, J.T. , Fong, S.F. , Csiszar, K. , Giaccia, A. , Weninger, W. , 2009. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta, L.A. , Petricoin, E. , 2011. Cancer biomarkers: closer to delivering on their promise. Cancer Cell. 20, 279–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb, L.A. , 2011. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer. 11, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Getz, G. , Miska, E.A. , Alvarez-Saavedra, E. , Lamb, J. , Peck, D. , Sweet-Cordero, A. , Ebert, B.L. , Mak, R.H. , Ferrando, A.A. , 2005. MicroRNA expression profiles classify human cancers. Nature. 435, 834–838. [DOI] [PubMed] [Google Scholar]
- Ludwig, J.A. , Weinstein, J.N. , 2005. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer. 5, 845–856. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Solimini, N.L. , Elledge, S.J. , 2009. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. , Reinhardt, F. , Pan, E. , Soutschek, J. , Bhat, B. , Marcusson, E.G. , Teruya-Feldstein, J. , Bell, G.W. , Weinberg, R.A. , 2010. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol.. 28, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. , Young, J. , Prabhala, H. , Pan, E. , Mestdagh, P. , Muth, D. , Teruya-Feldstein, J. , Reinhardt, F. , Onder, T.T. , Valastyan, S. , 2010. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol.. 12, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis, A.J. , Folberg, R. , Hess, A. , Seftor, E.A. , Gardner, L.M. , Pe'er, J. , Trent, J.M. , Meltzer, P.S. , Hendrix, M.J. , 1999. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am. J. Pathol.. 155, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Manoir, J.M. , Francia, G. , Man, S. , Mossoba, M. , Medin, J.A. , Viloria-Petit, A. , Hicklin, D.J. , Emmenegger, U. , Kerbel, R.S. , 2006. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin. Cancer Res.. 12, 904–916. [DOI] [PubMed] [Google Scholar]
- Mantovani, A. , Allavena, P. , Sica, A. , Balkwill, F. , 2008. Cancer-related inflammation. Nature. 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Martini, M. , Vecchione, L. , Siena, S. , Tejpar, S. , Bardelli, A. , 2011. Targeted therapies: how personal should we go?. Nat. Rev. Clin. Oncol. 9, 87–97. [DOI] [PubMed] [Google Scholar]
- McDonald, D.M. , Choyke, P.L. , 2003. Imaging of angiogenesis: from microscope to clinic. Nat. Med.. 9, 713–725. [DOI] [PubMed] [Google Scholar]
- Meylan, E. , Dooley, A.L. , Feldser, D.M. , Shen, L. , Turk, E. , Ouyang, C. , Jacks, T. , 2009. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 462, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, P.S. , Parkin, R.K. , Kroh, E.M. , Fritz, B.R. , Wyman, S.K. , Pogosova-Agadjanyan, E.L. , Peterson, A. , Noteboom, J. , O'Briant, K.C. , Allen, A. , 2008. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. U S A. 105, 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz, G.T. , Coukos, G. , 2011. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat. Rev. Immunol.. 11, 702–711. [DOI] [PubMed] [Google Scholar]
- Motzer, R.J. , Hutson, T.E. , Tomczak, P. , Michaelson, M.D. , Bukowski, R.M. , Rixe, O. , Oudard, S. , Negrier, S. , Szczylik, C. , Kim, S.T. , 2007. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med.. 356, 115–124. [DOI] [PubMed] [Google Scholar]
- Murdoch, C. , Muthana, M. , Coffelt, S.B. , Lewis, C.E. , 2008. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer. 8, 618–631. [DOI] [PubMed] [Google Scholar]
- Musolino, A. , Naldi, N. , Bortesi, B. , Pezzuolo, D. , Capelletti, M. , Missale, G. , Laccabue, D. , Zerbini, A. , Camisa, R. , Bisagni, G. , 2008. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol.. 26, 1789–1796. [DOI] [PubMed] [Google Scholar]
- Nagy, J.A. , Chang, S.H. , Shih, S.C. , Dvorak, A.M. , Dvorak, H.F. , 2010. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost.. 36, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nana-Sinkam, S.P. , Croce, C.M. , 2011. Non-coding RNAs in cancer initiation and progression and as novel biomarkers. Mol. Oncol.. 5, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella, C. , Lunardi, A. , Patnaik, A. , Cantley, L.C. , Pandolfi, P.P. , 2011. The APL paradigm and the “Co-clinical trial” project. Cancer Discov.. 2011, 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucera, S. , Biziato, D. , De Palma, M. , 2011. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int. J. Dev. Biol.. 55, 495–503. [DOI] [PubMed] [Google Scholar]
- O'Connell, R.M. , Rao, D.S. , Chaudhuri, A.A. , Baltimore, D. , 2010. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol.. 10, 111–122. [DOI] [PubMed] [Google Scholar]
- Olive, K.P. , Jacobetz, M.A. , Davidson, C.J. , Gopinathan, A. , McIntyre, D. , Honess, D. , Madhu, B. , Goldgraben, M.A. , Caldwell, M.E. , Allard, D. , 2009. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo, A. , Gupta, P.B. , Sgroi, D.C. , Arenzana-Seisdedos, F. , Delaunay, T. , Naeem, R. , Carey, V.J. , Richardson, A.L. , Weinberg, R.A. , 2005. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 121, 335–348. [DOI] [PubMed] [Google Scholar]
- Pages, F. , Galon, J. , Dieu-Nosjean, M.C. , Tartour, E. , Sautes-Fridman, C. , Fridman, W.H. , 2010. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 29, 1093–1102. [DOI] [PubMed] [Google Scholar]
- Palmero, E.I. , de Campos, S.G. , Campos, M. , de Souza, N.C. , Guerreiro, I.D. , Carvalho, A.L. , Marques, M.M. , 2011. Mechanisms and role of microRNA deregulation in cancer onset and progression. Genet. Mol. Biol.. 34, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Jiang, Z. , Mortenson, E.D. , Deng, L. , Radkevich-Brown, O. , Yang, X. , Sattar, H. , Wang, Y. , Brown, N.K. , Greene, M. , 2010. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 18, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccard, H. , Muschel, R.J. , Opdenakker, G. , 2011. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit. Rev. Oncol. Hematol. [DOI] [PubMed] [Google Scholar]
- Pietras, K. , Pahler, J. , Bergers, G. , Hanahan, D. , 2008. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med.. 5, e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras, K. , Ostman, A. , 2010. Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res.. 316, 1324–1331. [DOI] [PubMed] [Google Scholar]
- Potente, M. , Gerhardt, H. , Carmeliet, P. , 2011. Basic and therapeutic aspects of angiogenesis. Cell. 146, 873–887. [DOI] [PubMed] [Google Scholar]
- Poulikakos, P.I. , Persaud, Y. , Janakiraman, M. , Kong, X. , Ng, C. , Moriceau, G. , Shi, H. , Atefi, M. , Titz, B. , Gabay, M.T. , 2011. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 480, 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, B.Z. , Pollard, J.W. , 2010. Macrophage diversity enhances tumor progression and metastasis. Cell. 141, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush, J. , 2006. Microarray analysis and tumor classification. N. Engl. J. Med.. 354, 2463–2472. [DOI] [PubMed] [Google Scholar]
- Qualls, J.E. , Murray, P.J. , 2011. Tumor macrophages protective and pathogenic roles in cancer development. Curr. Top Dev. Biol.. 94, 309–328. [DOI] [PubMed] [Google Scholar]
- Rani, S. , O'Brien, K. , Kelleher, F.C. , Corcoran, C. , Germano, S. , Radomski, M.W. , Crown, J. , O'Driscoll, L. , 2011. Isolation of exosomes for subsequent mRNA, microRNA, and protein profiling. Methods Mol. Biol.. 784, 181–195. [DOI] [PubMed] [Google Scholar]
- Reis-Filho, J.S. , Pusztai, L. , 2011. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 378, 1812–1823. [DOI] [PubMed] [Google Scholar]
- Rini, B.I. , Michaelson, M.D. , Rosenberg, J.E. , Bukowski, R.M. , Sosman, J.A. , Stadler, W.M. , Hutson, T.E. , Margolin, K. , Harmon, C.S. , DePrimo, S.E. , 2008. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J. Clin. Oncol.. 26, 3743–3748. [DOI] [PubMed] [Google Scholar]
- Sawyers, C. , 2004. Targeted cancer therapy. Nature. 432, 294–297. [DOI] [PubMed] [Google Scholar]
- Sawyers, C.L. , 2008. The cancer biomarker problem. Nature. 452, 548–552. [DOI] [PubMed] [Google Scholar]
- Sawyers, C.L. , 2009. Shifting paradigms: the seeds of oncogene addiction. Nat. Med.. 15, 1158–1161. [DOI] [PubMed] [Google Scholar]
- Schetter, A.J. , Heegaard, N.H. , Harris, C.C. , 2010. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 31, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, B.P. , Wang, M. , Radovich, M. , Sledge, G.W. , Badve, S. , Thor, A. , Flockhart, D.A. , Hancock, B. , Davidson, N. , Gralow, J. , 2008. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J. Clin. Oncol.. 26, 4672–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers, W.R. , 2011. A blueprint for advancing genetics-based cancer therapy. Cell. 147, 26–31. [DOI] [PubMed] [Google Scholar]
- Sequist, L.V. , Waltman, B.A. , Dias-Santagata, D. , Digumarthy, S. , Turke, A.B. , Fidias, P. , Bergethon, K. , Shaw, A.T. , Gettinger, S. , Cosper, A.K. , 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med.. 3, 75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, S.V. , Lee, D.Y. , Li, B. , Quinlan, M.P. , Takahashi, F. , Maheswaran, S. , McDermott, U. , Azizian, N. , Zou, L. , Fischbach, M.A. , 2010. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless, N.E. , Depinho, R.A. , 2006. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov.. 5, 741–754. [DOI] [PubMed] [Google Scholar]
- Sica, A. , Bronte, V. , 2007. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest.. 117, 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, R.J. , Lim, J.W. , Moritz, R.L. , Mathivanan, S. , 2009. Exosomes: proteomic insights and diagnostic potential. Expert Rev. Proteomics. 6, 267–283. [DOI] [PubMed] [Google Scholar]
- Simpson-Abelson, M.R. , Sonnenberg, G.F. , Takita, H. , Yokota, S.J. , Conway, T.F. , Kelleher, R.J. , Shultz, L.D. , Barcos, M. , Bankert, R.B. , 2008. Long-term engraftment and expansion of tumor-derived memory T cells following the implantation of non-disrupted pieces of human lung tumor into NOD-scid IL2Rgamma(null) mice. J. Immunol.. 180, 7009–7018. [DOI] [PubMed] [Google Scholar]
- Soda, Y. , Marumoto, T. , Friedmann-Morvinski, D. , Soda, M. , Liu, F. , Michiue, H. , Pastorino, S. , Yang, M. , Hoffman, R.M. , Kesari, S. , 2011. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. U S A. 108, 4274–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou, C. , Piccart, M.J. , 2007. Taking gene-expression profiling to the clinic: when will molecular signatures become relevant to patient care?. Nat. Rev. Cancer. 7, 545–553. [DOI] [PubMed] [Google Scholar]
- Sotiriou, C. , Pusztai, L. , 2009. Gene-expression signatures in breast cancer. N. Engl. J. Med.. 360, 790–800. [DOI] [PubMed] [Google Scholar]
- Squadrito, M.L. , De Palma, M. , 2011. Macrophage regulation of tumor angiogenesis: implications for cancer therapy. Mol. Aspects Med.. 32, 123–145. [DOI] [PubMed] [Google Scholar]
- Steidl, C. , Farinha, P. , Gascoyne, R.D. , 2011. Macrophages predict treatment outcome in Hodgkin's lymphoma. Haematologica. 96, 186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout, R.D. , Watkins, S.K. , Suttles, J. , 2009. Functional plasticity of macrophages: in situ reprogramming of tumor-associated macrophages. J. Leukoc. Biol.. 86, 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi, A. , Politi, K. , Pitteri, S.J. , Lockwood, W.W. , Faca, V.M. , Kelly-Spratt, K. , Wong, C.H. , Zhang, Q. , Chin, A. , Park, K.S. , 2011. Lung cancer signatures in plasma based on proteome profiling of mouse tumor models. Cancer Cell. 20, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty, T.D. , Coussens, L.M. , 2006. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol.. 1, 119–150. [DOI] [PubMed] [Google Scholar]
- Turke, A.B. , Zejnullahu, K. , Wu, Y.L. , Song, Y. , Dias-Santagata, D. , Lifshits, E. , Toschi, L. , Rogers, A. , Mok, T. , Sequist, L. , 2010. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 17, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utama, F.E. , LeBaron, M.J. , Neilson, L.M. , Sultan, A.S. , Parlow, A.F. , Wagner, K.U. , Rui, H. , 2006. Human prolactin receptors are insensitive to mouse prolactin: implications for xenotransplant modeling of human breast cancer in mice. J. Endocrinol.. 188, 589–601. [DOI] [PubMed] [Google Scholar]
- Valadi, H. , Ekstrom, K. , Bossios, A. , Sjostrand, M. , Lee, J.J. , Lotvall, J.O. , 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol.. 9, 654–659. [DOI] [PubMed] [Google Scholar]
- van Miltenburg, M.H. , Jonkers, J. , 2012. Using genetically engineered mouse models to validate candidate cancer genes and test new therapeutic approaches. Curr. Opin. Genet. Dev.. 10.1016/j.gde.2012.01.004 [DOI] [PubMed] [Google Scholar]
- van't Veer, L.J. , Bernards, R. , 2008. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 452, 564–570. [DOI] [PubMed] [Google Scholar]
- de Visser, K.E. , Eichten, A. , Coussens, L.M. , 2006. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer. 6, 24–37. [DOI] [PubMed] [Google Scholar]
- Visvader, J.E. , 2011. Cells of origin in cancer. Nature. 469, 314–322. [DOI] [PubMed] [Google Scholar]
- Volinia, S. , Calin, G.A. , Liu, C.G. , Ambs, S. , Cimmino, A. , Petrocca, F. , Visone, R. , Iorio, M. , Roldo, C. , Ferracin, M. , 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U S A. 103, 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther, A. , Johnstone, E. , Swanton, C. , Midgley, R. , Tomlinson, I. , Kerr, D. , 2009. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer. 9, 489–499. [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , 2002. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Weis, S.M. , Cheresh, D.A. , 2011. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med.. 17, 1359–1370. [DOI] [PubMed] [Google Scholar]
- Weisberg, E. , Manley, P.W. , Cowan-Jacob, S.W. , Hochhaus, A. , Griffin, J.D. , 2007. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer. 7, 345–356. [DOI] [PubMed] [Google Scholar]
- Wittmann, J. , Jack, H.M. , 2010. Serum microRNAs as powerful cancer biomarkers. Biochim. Biophys. Acta. 1806, 200–207. [DOI] [PubMed] [Google Scholar]
- Workman, P. , Clarke, P.A. , 2011. Resisting targeted therapy: fifty ways to leave your EGFR. Cancer Cell. 19, 437–440. [DOI] [PubMed] [Google Scholar]
- Wu, M. , Robinson, M.O. , 2009. Human-in-mouse breast cancer model. Cell Cycle. 8, 2317–2318. [DOI] [PubMed] [Google Scholar]
- Yachida, S. , Jones, S. , Bozic, I. , Antal, T. , Leary, R. , Fu, B. , Kamiyama, M. , Hruban, R.H. , Eshleman, J.R. , Nowak, M.A. , 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 467, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J.C. , Haworth, L. , Sherry, R.M. , Hwu, P. , Schwartzentruber, D.J. , Topalian, S.L. , Steinberg, S.M. , Chen, H.X. , Rosenberg, S.A. , 2003. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med.. 349, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. , Chen, J. , Su, F. , Yu, B. , Lin, L. , Liu, Y. , Huang, J.D. , Song, E. , 2011. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer. 10, 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender, L. , Lowe, S.W. , 2008. Integrative oncogenomic approaches for accelerated cancer-gene discovery. Curr. Opin. Oncol.. 20, 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Ding, T. , Pan, W. , Zhu, L.Y. , Li, L. , Zheng, L. , 2009. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer. 125, 1640–1648. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Rideout, W.M. , Zi, T. , Bressel, A. , Reddypalli, S. , Rancourt, R. , Woo, J.K. , Horner, J.W. , Chin, L. , Chiu, M.I. , 2010. Chimeric mouse tumor models reveal differences in pathway activation between ERBB family- and KRAS-dependent lung adenocarcinomas. Nat. Biotechnol.. 28, 71–78. [DOI] [PubMed] [Google Scholar]