

Abstract
Epidermal growth factor receptor (EGFR) is a validated target in different human malignancies. EGFR tyrosine kinase inhibitors (TKIs) are known to contribute considerably to the extension of progression‐free survival in EGFR‐mutant non‐small cell lung cancer and monoclonal antibodies (mAbs) targeting EGFR have also improved the efficacy outcomes in KRAS wild‐type colorectal cancer. Nevertheless, a significant percentage of lung and colorectal cancer patients do not respond to anti‐EGFR agents and secondary resistance after initial benefit is a challenging reality faced by clinicians. Extensive preclinical work on the potential mechanisms of resistance to EGFR inhibitors in different disease settings has guided the development of second‐generation irreversible EGFR TKIs, more efficient anti‐EGFR mAbs, and combination strategies with agents targeting other receptors and downstream effectors. In order to elucidate the role of the multiple therapeutic strategies under investigation to overcome EGFR inhibitors‐resistance, rational drug development based on stringent preclinical data, biomarker validation and proper selection of patients in the ongoing clinical trials are of paramount importance. Preliminary results of clinical trials evaluating these approaches will be discussed in this manuscript, with emphasis on TKIs in lung cancer and mAbs in advanced colorectal cancer.
Keywords: Biomarker, Epidermal growth factor receptor, Monoclonal antibody, Resistance, Tyrosine kinase inhibitor
Abbreviations
- ADCC
Antibody‐dependent cell‐mediated cytotoxicity
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- FGFR
Fibroblast growth factor receptor
- HGF
Hepatocyte growth factor
- IGF‐1R
Insulin‐like growth factor 1 receptor
- mAbs
Monoclonal antibodies
- MET
Mesenchymal–epithelial transition receptor
- mTOR
Mammalian target of rapamycin
- NSCLC
Non‐small cell lung cancer
- OS
Overall survival
- PFS
Progression‐free survival
- PI3K
Phosphatidylinositol 3‐kinase
- RR
Response rate
- TGF‐α
Transforming growth factor alpha
- TKI
Tyrosine kinase inhibitors
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
1. Introduction
Targeted therapies represent the most promising agents in development for the treatment of cancer. Epidermal growth factor receptor (EGFR), the founding member of a family of four ErbB receptor tyrosine kinases – EGFR/(human epidermal growth factor receptor 1 [HER1]/ErbB1), HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4 – has been a focus of intense research in the last two decades. This pathway is importantly implicated in tumor cell growth, local invasion, angiogenesis, metastasis, protein translation and cell metabolism. Binding of ligands/growth factors, such as EGF (epidermal growth factor), amphiregulin and TGF‐α (transforming growth factor alpha), to the extracellular domain of ErbB receptors stabilizes them in a conformation that allows dimerization, an essential requirement for transactivation of the tyrosine kinase portion of the dimer moiety, leading to phosphorylation and downstream signaling. Dimerization can occur between two different ErbB receptors (heterodimerization) or between two molecules of the same receptor (homodimerization). The ErbB dimers have different signaling potencies; homodimers weakly perpetuate signals as compared to heterodimers. The most important pathways activated in solid tumors are RAS/RAF/MEK/ERK and phosphatidylinositol 3‐kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), as seen in Figure 1. Interestingly, EGFR mainly activates the MEK pathway and signaling through HER3 heterodimers strongly activates the PI3K component (Scaltriti and Baselga, 2006; Baselga and Swain, 2009).
Figure 1.
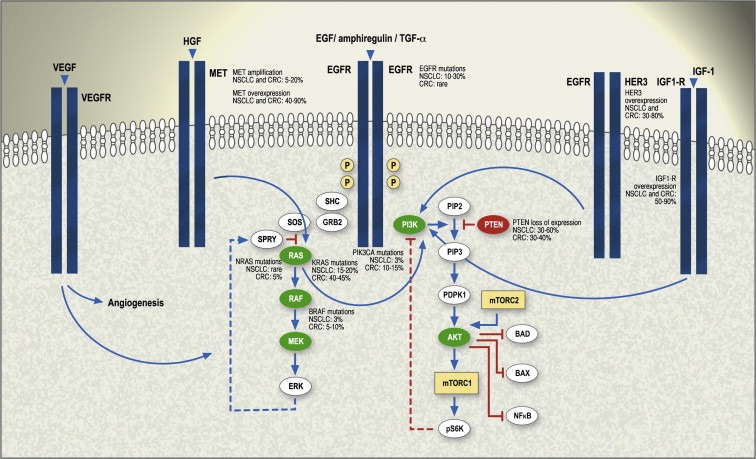
Simplified diagram illustrating relationships of EGFR homo‐ or heterodimers (EGFR‐HER3) and other growth factor receptors (IGF‐1R, MET), which activate the critical downstream effectors RAS/RAF/MEK/ERK and PI3K/AKT/mTOR in cancer cells. Parallel pathways, including VEGFR, also play a role in the resistance to EGFR inhibition. The frequency of mutations, gene amplifications and protein overexpression of the main players in these signaling pathways is depicted, both in non‐small cell lung cancer and in colorectal cancer. CRC: colorectal cancer; EGFR: epidermal growth factor receptor; HGF: hepatocyte growth factor; IGF‐1R: insulin‐like growth factor 1 receptor; MET: mesenchymal–epithelial transition receptor; NSCLC: non‐small cell lung cancer; TGF‐α: transforming growth factor alpha.
EGFR plays a critical role in the biology of many different tumors. Gain of function genetic alterations in EGFR are observed in many epithelial tumors and certain malignancies are clearly dependent on the pathway – “oncogene‐addicted” – such as lung cancer cells presenting mutations in the EGFR kinase domain. This makes EGFR an attractive target for cancer therapy (Salomon et al., 1995; Weinstein, 2002). Small molecules tyrosine kinase inhibitors (TKIs) of EGFR, like erlotinib and gefitinib, or monoclonal antibodies (mAbs) targeting the extracellular domain of EGFR, such as cetuximab and panitumumab, are validated therapeutic strategies. In addition to the receptor, TKIs of other targets within the EGFR pathway, such as RAF, MEK, PI3K, AKT and mTOR, are also in clinical development.
In this context, molecular aberrations on the EGFR pathway are the most commonly studied predictive biomarkers of response/resistance to targeted agents in cancer. Mutations in exons 19 and 21 of the kinase domain of EGFR, particularly deletions in exon 19 and exon 21 point mutations in codon 858 (L858R), which are the most frequent mutations in non‐small cell lung cancer (NSCLC), confer tumor cell dependency on EGFR signaling and are predictive of response to the first‐generation reversible EGFR TKIs (Lynch et al., 2004). On the other hand, the genetic background of some tumors simply precludes a function for EGFR inhibition, what is called inherited resistance. For instance, the presence of mutant KRAS is a strong predictor of lack of response to EGFR mAbs in patients with CRC (Liévre et al., 2006) and to EGFR TKIs in patients with NSCLC (Pao et al., 2005). Interestingly, KRAS mutations do not seem to identify patients who do not benefit from anti‐EGFR mAbs in NSCLC, although definitive conclusions cannot be made due to the limited number of studies (O'Byrne et al., 2009; Khambata‐Ford et al., 2010).
Importantly, the oncogenic pathways driven by EGFR are interconnected in a complex network involving both negative and positive feedback loops that regulate the activity of their components in response to stimuli. The antitumor effects on oncoprotein inhibitors may be attenuated by relief of these feedback loops, rescuing the tumor cells from targeted inhibition of a driver oncogene. In this manuscript we summarize the extensive preclinical work on primary and acquired resistance to EGFR inhibitors. Drug development to overcome resistance to TKIs in NSCLC and mAbs in CRC will be discussed. A mechanism‐centered review focusing on two different disease settings allows the reader to have a broader picture of the therapeutic development challenge. The preliminary results of clinical trials evaluating second‐generation EGFR TKIs, more efficient anti‐EGFR mAbs, and combination strategies with agents targeting other receptors and downstream effectors will be presented.
2. Overcoming resistance to EGFR TKIs – the NSCLC story
Prospective trials comparing standard platinum‐based chemotherapy with first‐generation EGFR TKIs in patients with and without activating EGFR mutations confirmed the predictive value of molecular selection of patients for first‐line treatment of advanced NSCLC (Mok et al., 2009; Maemondo et al., 2010; Mitsudomi et al., 2010; Zhou et al., 2010; Rosell et al., 2011a). Clinical data with erlotinib and gefitinib in patients with NSCLC whose tumors harbor activating EGFR mutations indicate that these patients eventually develop resistance to reversible EGFR TKIs, which may result from secondary acquired EGFR mutations or other mechanisms not directly related to the EGFR genotype.
Tumors become resistant when they reactivate downstream signaling despite the presence of the EGFR inhibitor. Primary resistance is typically caused by mutations in the EGFR gene that are not associated with sensitivity to first‐generation EGFR TKIs, such as insertion mutations in exon 20, or by other somatic mutations in genes that have an impact on the EGFR signaling pathway, such as KRAS. Acquired resistance, defined as documented disease progression after objective clinical benefit with an EGFR TKI (radiologic response or stable disease for more than 6 months), may be caused by additional mutations in the EGFR gene obtained during the course of treatment that change the protein‐coding sequence or by amplification of another oncogene signaling pathway (Jackman et al., 2010). The most commonly identified mechanism of resistance is a gatekeeper mutation at position 790 (EGFR T790M), which abrogates the ability of gefitinib or erlotinib to inhibit EGFR. This mutation has been found in 50% of the tissue samples from patients with acquired gefitinib resistance (Kosaka et al., 2006; Arcila et al., 2011; Sequist et al., 2011a). However, the T790M mutation may also be present prior to treatment with erlotinib or gefitinib and, therefore, may also contribute to primary resistance. It has been demonstrated that some patients who respond may have T790M mutations in a small percentage of tumor cells before EGFR TKI therapy and clonal selection may allow the T790M‐expressing cells to assume an increasingly larger percentage of the tumor over time (Inukai et al., 2006; Rosell et al., 2011b).
2.1. Irreversible EGFR inhibitors
In preclinical studies, irreversible EGFR TKIs have been shown to effectively inhibit T790M‐mutant cancers (Kwak et al., 2005; Engelman et al., 2007a; Li et al., 2008). These agents may prevent and overcome primary and acquired resistance to first‐generation reversible EGFR TKIs. In addition, they may increase TKI effectiveness by prolonging the inhibition of EGFR signaling and delaying the acquisition or growth of T790M‐mutant clones. The first agent explored in this setting was the irreversible TKI neratinib (Table 1). Clinical studies have not shown significant activity against EGFR‐mutant lung cancers with acquired resistance to erlotinib/gefitinib, likely because of toxicity that limited administration of necessary doses to attain therapeutic drug concentrations (Sequist et al., 2010). Afatinib and dacomitinib are potent drugs with a more favorable pharmacokinetic and toxicity profile. In the LUX‐Lung 2 study, 129 patients with activating EGFR mutations and no previous EGFR TKI therapy received afatinib as single agent. Overall response rate (RR) was 60%, with a promising progression‐free survival (PFS) of 14 months (Yang et al., 2010). This drug has shown activity in patients whose tumors harbored less common EGFR mutations (Shih et al., 2010). Its efficacy was also evaluated as a rescue treatment after failure to erlotinib or gefitinib in a population clinically enriched for the presence of EGFR‐activating mutations (Miller et al., 2010). At interim analysis of the randomized phase III trial LUX‐Lung 1 (358 events in 585 patients), a significant increase in PFS was seen in patients that received the study drug as compared to placebo (3.3 versus 1.1 months). However, median overall survival (OS) was not different in the two arms. Afatinib was also associated with significant improvements in the secondary endpoints of confirmed disease control rate of at least 8 weeks (58% versus 19%) and confirmed objective RR (11% versus 0.5%). Further phase III studies are looking at the activity of this agent as compared to standard chemotherapy in patients with NSCLC tumors harboring EGFR‐activating mutations (ClinicalTrials.gov identifier NCT00949650 and NCT01121393).
Table 1.
Mechanism of action of agents tested in the EGFRa‐inhibitor refractory setting
| Agent | Mechanism of action |
|---|---|
| Afatinib | Irreversible TKIb of EGFRa and HER2 |
| Bevacizumab | mAbc targeting VEGFd |
| BMS‐690514 | Multitargeted TKIb of EGFR,a HER2 and VEGFRe |
| Brivanib | Multitargeted TKIb of VEGFRe and FGFRf |
| Cabozantinib | Multitargeted TKIb of MET,g VEGFR,e RET, c‐KIT, FLT‐3 |
| Cixutumumab | mAbc targeting IGF‐1Rh |
| Dacomitinib | Irreversible TKIb of EGFR,a HER2 and HER4 |
| Dalotuzumab | mAbc targeting IGF‐1Rh |
| Figitumumab | mAbc targeting IGF‐1Rh |
| Ganitumab | mAbc targeting IGF‐1Rh |
| Neratinib | Irreversible TKIb of EGFRa and HER2 |
| Onartuzumab | mAbc targeting METg |
| Rilotumumab | mAbc targeting HGFi |
| Sorafenib | Multitargeted TKIb of VEGFR,e PDGFR,j c‐KIT, FLT‐3, RAF |
| Sunitinib | Multitargeted TKIb of VEGFR,e PDGFR,j c‐KIT, FLT‐3 |
| Tivantinib | TKIb of METg |
| Vandetanib | Multitargeted TKIb of EGFRa and VEGFRe |
Epidermal growth factor receptor.
Tyrosine kinase inhibitor.
Monoclonal antibody.
Vascular endothelial growth factor.
Vascular endothelial growth factor receptor.
Fibroblast growth factor receptor.
Mesenchymal–epithelial transition.
Insulin‐like growth factor 1 receptor.
Hepatocyte growth factor.
Platelet derived growth factor receptor.
Regarding the pan‐HER TKI dacomitinib (PF‐00299804), preclinical studies have shown activity in NSCLC cell lines harboring the EGFR T790M, but not in those with KRAS mutations (Engelman et al., 2007a). Preliminary data from a phase II randomized trial showed that PFS was superior when compared with erlotinib in the general population of patients with chemotherapy‐refractory NSCLC, not selected according to EGFR mutation status (12.4 weeks versus 8.3 weeks) (Mok et al., 2010). As first‐line treatment of patients with known EGFR mutations or clinically selected (Asians with adenocarcinoma and non‐ or light smoking history), dacomitinib showed encouraging efficacy, with 6‐month PFS rate of 67% (85% in those with EGFR mutations). This drug was evaluated in patients with KRAS wild‐type NSCLC refractory to at least one chemotherapy regimen and erlotinib in another phase II study (Campbell et al., 2010). Only 3 partial responses were seen in 62 evaluable patients. Additional trials with this agent started accrual, including a direct comparison with erlotinib as second‐line treatment of advanced NSCLC (ClinicalTrials.gov identifier NCT01360554).
Recently, preliminary results of the phase 1 trial assessing the combination of afatinib and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib were presented (Janjigian et al., 2011b). In preclinical studies, this drug combination efficiently depleted both phosphorylated and total EGFR and neutralized the increased EGFR ligand production in the tumor microenvironment. In addition, near complete responses were seen in T790M transgenic murine models (Regales et al., 2009). Different from the negative trial evaluating the addition of cetuximab to erlotinib in patients with lung adenocarcinoma and clinically defined acquired resistance to first‐generation EGFR TKIs (Janjigian et al., 2011a), the combined use of afatinib and cetuximab was associated with over 30% confirmed partial responses in 22 evaluable patients, including those with documented T790M mutations (Janjigian et al., 2011b). These results validate preclinical data suggesting that many tumors are still addicted to the EGFR signaling pathway despite development of acquired resistance. In addition, they encourage further investigation of irreversible EGFR TKIs in combination approaches, taking into account the limited benefit seen when these agents were used as monotherapy in the erlotinib/gefitinib‐refractory setting.
2.2. Targeting additional pathways
2.2.1. Proangiogenic growth factors
Another strategy to circumvent resistance to EGFR inhibitors in lung cancer is by targeting parallel signaling pathways that contribute to the malignant phenotype. Based on preclinical studies suggesting that vascular endothelial growth factor (VEGF) pathway activation may contribute to resistance to first‐generation EGFR TKIs (Viloria‐Petit et al., 2001), many clinical trials have evaluated combined inhibition of EGFR and VEGF pathways, albeit with poor results. The addition of the antiangiogenic agents bevacizumab or sunitinib to erlotinib in unselected patients with chemorefractory NSCLC was associated with mild improvement in PFS but no benefit in terms of OS in randomized phase III trials – BeTA and Sun 1087 (Herbst et al., 2011; Scagliotti et al., 2010). On the other hand, in a randomized phase II trial of erlotinib in combination with sorafenib in chemorefractory advanced NSCLC, a benefit with the use of dual targeted therapy was observed in EGFR wild‐type tumors. In this subgroup of 67 patients, median PFS was 3.38 months for sorafenib/erlotinib versus 1.77 months for placebo/erlotinib (p = 0.018); median OS was 8 versus 4.5 months, respectively (p = 0.019) (Spigel et al., 2011a). Considering the small sample size, additional studies in patients selected according to mutation status are warranted.
Multitargeted TKIs of EGFR and VEGFR have also been studied in NSCLC. Vandetanib was directly compared with erlotinib in the large ZEST trial (Natale et al., 2009). No significant differences in objective RR, PFS or OS were observed in an unselected patient population. In a phase II trial with BMS‐690514, 7 out of 32 erlotinib‐resistant patients had prolonged disease stabilization (including 2 with documented EGFR T790M mutation) and one erlotinib‐naive patient whose tumor harbored a KRAS mutation had a partial response (Bahleda et al., 2009). BMS‐690514 has been compared with upfront erlotinib in a randomized phase II trial and the final results are still pending. Nevertheless, the large studies conducted so far indicate that dual targeting of EGFR and VEGFR is not a promising strategy to overcome resistance to EGFR inhibitors in NSCLC.
2.2.2. MET pathway
Downstream activation of RAS/RAF/MEK/ERK and PI3K/AKT/mTOR via alternative pathways is an important mechanism of resistance to EGFR TKIs. Dual input to signaling has been described with mesenchymal–epithelial transition factor (MET) amplification (Bean et al., 2007; Engelman et al., 2007b). There is preclinical evidence showing that coupling of the MET receptor tyrosine kinase to HER3 leads to sustained activation of the PI3K/AKT/mTOR pathway, bypassing the inhibited EGFR (Engelman et al., 2007b). In this situation, combined inhibition of EGFR and the alternative pathway may prevent the emergence of resistance that eventually occurs following initial response to EGFR TKIs and also increase the proportion of EGFR wild‐type patients that respond. MET amplification occurs in 5%–20% of EGFR mutation positive NSCLC patients who develop resistance after an initial response to erlotinib or gefitinib (Engelman et al., 2007b; Arcila et al., 2011; Sequist et al., 2011a). Importantly, both MET and EGFR are on chromosome 7, and polysomy of chromosome 7 is common in NSCLC, particularly in those samples harboring EGFR mutations (Cappuzzo et al., 2005). It has also been identified in a small subpopulation of tumor cells prior to drug exposure, thus suggesting that clonal selection of MET amplification in EGFR‐mutants may occur (Turke et al., 2010).
Agents targeting the MET receptor are undergoing clinical evaluation in patients with advanced NSCLC. Results of a phase II randomized trial that compared erlotinib plus placebo versus erlotinib plus tivantinib (ARQ‐197) were recently published (Sequist et al., 2011b). Progression‐free survival was higher with the dual inhibition approach (3.8 versus 2.3 months), although not statistically significant (hazard ratio 0.81, p = 0.24). Exploratory analysis revealed that the small cohort with KRAS mutations achieved higher benefit (hazard ratio for PFS 0.18, p = 0.006). A phase III trial testing this combination is underway (ClinicalTrials.org Identifier NCT01244191).
Promising results were also seen in clinical studies with an anti‐MET receptor mAb. In a phase II randomized trial evaluating the benefit onartuzumab added to erlotinib, the subgroup of patients high MET expression by IHC analysis (n = 66) had significantly increased PFS (2.9 versus 1.5 months, hazard ratio 0.53, p = 0.05) and overall survival (12.6 versus 3.8 months, hazard ratio 0.37, p = 0.002) with the combination approach as compared to erlotinib and placebo (Spigel et al., 2011b). Phase III trial will start recruitment soon (ClinicalTrials.org Identifier NCT01456325). Another agent already tested in the refractory setting is cabozantinib (XL184). In the phase 1 trial of this potent VEGFR and MET TKI in combination with erlotinib, 3 patients previously exposed to EGFR TKIs had ≥30% reduction in tumor measurements, including one with MET amplification (Wakelee et al., 2010). Additional trials have not started accrual so far.
2.2.3. IGF‐1R and HER3
Parallel signaling pathways that interact with EGFR as a result of heterodimer formation, such as Insulin‐like Growth Factor 1 Receptor (IGF‐1R), may contribute to first‐generation EGFR TKI resistance (Morgillo et al., 2006; Guix et al., 2008). The IGF‐1R is highly expressed in lung cancer cells, mainly squamous cell carcinomas (Ouban et al., 2003). Despite preclinical data showing that IGF‐1R signaling counteracts the antitumor action of erlotinib, clinical trials testing combination approaches have been negative. A phase III trial investigating figitumumab with erlotinib versus erlotinib alone in the chemotherapy‐refractory setting was closed after an interim analysis because of the potential futility of the combination regimen. In addition, results of phase II randomized trial evaluating dalotuzumab added to erlotinib in unselected NSCLC patients do not support further investigation of this strategy (median PFS of 2.5 versus 1.6 months, hazard ratio 0.86, p = 0.26) (Rosell et al., 2011c).
Recent publications suggest that co‐targeting EGFR and HER3 in NSCLC might be an interesting approach, especially in adenocarcinomas and EGFR‐mutant tumors, which are known to highly express HER3 (Felip et al., 2011). As shown in Table 2, clinical development of anti‐HER3 mAbs in NSCLC is still in an early stage.
Table 2.
Selected trials combining EGFRa TKIsb and other targeted agents with focus on NSCLC.c
| EGFR inhibitor | Combination agent | Target and class | Phase | Indication | ClinicalTrials.gov identifier |
|---|---|---|---|---|---|
| Erlotinib | GDC‐0941 | PI3K/mTOR TKIb | Ib | Solid tumors | NCT00975182 |
| MK‐2206 | AKT TKIb | II | NSCLCc | NCT01294306 | |
| AZD6244 | MEK TKIb | II | NSCLCc | NCT01229150 | |
| GSK1120212 | MEK TKIb | Ib | Solid tumors | NCT01192165 | |
| IMC‐A12 | IGF‐1RdmAbe | I‐II | NSCLCc | NCT00778167 | |
| MM‐121 | HER3 mAbe | I‐II | NSCLCc | NCT00994123 | |
| Gefitinib | MK‐2206 | AKT TKIb | I | NSCLCc | NCT01147211 |
| Dacomitinib | PF‐02341066 | METf TKIb | I | NSCLCc | NCT01121575 |
Epidermal growth factor receptor.
Tyrosine kinase inhibitor.
Non‐small cell lung cancer.
Insulin‐like growth factor 1 receptor.
Monoclonal antibody.
Mesenchymal–epithelial transition receptor.
2.2.4. Downstream effectors
Based on promising results of preclinical studies, drugs that block signaling molecules downstream of the EGFR, such as MEK and PI3K/AKT/mTOR inhibitors, are also being investigated in advanced lung cancer. The mTOR inhibitor everolimus, when added to erlotinib in chemorefractory NSCLC patients, did not significantly improve response and survival endpoints when compared to the EGFR TKI alone (Leighl et al., 2010). Nevertheless, in the phase I trial combining the PI3K/mTOR inhibitor XL765 with erlotinib in patients with lung cancer refractory to anti‐EGFR therapies, prolonged disease stabilization in 4 out of 21 patients was observed (Cohen et al., 2010). As seen in Table 2, the results of multiple phase I/II trials of combined targeted therapies are expected soon.
Furthermore, the combination of MEK inhibitors and PI3K pathway inhibitors is a promising treatment strategy for KRAS mutant NSCLC. Early signs of antitumor activity were observed in the ongoing phase I trials that evaluate the combinations of the MEK inhibitor GDC‐0973 plus the PI3K inhibitor GDC‐0941 and the MEK inhibitor AZD6244 plus the AKT inhibitor MK‐2206 (Shapiro et al., 2011a; Tolcher et al., 2011). This approach will certainly move forward to phase II trials in the near future.
3. Overcoming resistance to EGFR mAbs – the CRC story
Different activating mutations in receptor tyrosine kinases and molecular aberrations in downstream components of the EGFR pathway are found in CRC, making it a very heterogeneous disease. Therefore, efficacy of targeted therapies is likely related to the specific genetic background of the tumor. Mutations in KRAS, for example, have been shown to be strongly associated with primary resistance to anti‐EGFR mAbs. Nevertheless, cetuximab and panitumumab are not effective in all patients with KRAS wild‐type CRC. Other potential molecular markers downstream of EGFR, namely mutations in NRAS, BRAF, PIK3CA and PTEN loss of function, may also predict lack of response to anti‐EGFR therapies in CRC (Dienstmann et al., 2011a).
Patients with advanced CRC that initially respond to EGFR mAbs eventually become resistant to these drugs. As in lung cancer, acquired resistance might occur through the selection of clones that are already not sensitive at the start of treatment, or the development of secondary resistance in cancer cells that are initially sensitive to EGFR targeted therapy. Various molecular mechanisms are likely to have a role in acquired resistance to EGFR mAbs and research in this field guided important advances in drug development. These innovative approaches consist in developing more efficient mAbs, targeting simultaneously different receptors and other members of the EGFR family, and the combination of mAbs and TKIs of key components of the downstream signaling cascade.
3.1. More efficient anti‐EGFR mAbs
The mechanism of action of anti‐EGFR mAbs involves: (a) the inhibition of ligand binding and receptor dimerization, with the consequent inhibition of downstream signaling; (b) internalization of the EGFR and its subsequent degradation, without receptor phosphorylation and activation; (c) immune effects, exerting their activity through fragment c (Fc)‐mediated mechanisms, such as antibody‐dependent cell‐mediated cytotoxicity (ADCC) and complement‐dependent cytotoxicity. The latter effect appears to be more intense with IgG1 mAbs such as cetuximab, as compared to the IgG2 mAb panitumumab (Campoli et al., 2010).
Development of innovative mAbs with more efficient binding ability to the EGFR is a promising strategy to overcome resistance to the agents in clinical use. One example is Sym004, a combination of two chimeric mAbs targeting non‐overlapping epitopes of domain III of the EGFR. Each antibody is manufactured separately as an individual drug and Sym004 is prepared and released as a 1:1 ratio mixture. Both antibodies bind simultaneously to the extracellular domain of EGFR, inducing highly efficient internalization and degradation of the receptor on cancer cells. Preclinical studies demonstrate stronger tumor suppression as compared to cetuximab and panitumumab in a wide range of cancer types, with a clear dose–response relationship (Pedersen et al., 2010). Furthermore, Sym004 inhibits growth and proliferation of cancer cells that have acquired resistance to anti‐EGFR antibody therapy induced by increased EGFR ligand production in the tumor microenvironment. The first‐in‐human trial did not show unexpected toxicities and, based on preliminary signs of clinical activity, Sym004 is being tested as monotherapy in selected patients with KRAS wild‐type CRC progressing to previous cetuximab or panitumumab‐based therapy (Dienstmann et al., 2011b) (ClinicalTrials.gov Identifier NCT01117428).
Another strategy to enhance efficacy of anti‐EGFR mAbs is by increasing immune‐mediated citotoxicity. RG7160 (also GA201) is a humanized glycoengineered anti‐EGFR IgG1 mAb exhibiting increased binding affinity for all FcγRIIIa variants expressed on immune effector cells. This significantly improves cell killing in ADCC‐based assays and is associated with greater activity in preclinical models when compared with cetuximab and panitumumab (Gerdes et al., 2008). Specifically, the structural modification potentiates not only the activity of receptor inhibition, but also the ability of inducing cell death mediated by immune cells. Encouraging results have been observed in the phase I trial of RG7160 in EGFR‐positive refractory CRC, with partial responses and prolonged disease stabilization observed both in the KRAS mutant and wild‐type subgroups (Paz‐Ares et al., 2011). Preliminary analysis of RG7160‐related pharmacodynamic effects has identified significant reduction of natural killer cells in peripheral blood and immune cell infiltration in paired tumor biopsies. This agent is currently being tested in a randomized phase II trial in combination with standard irinotecan and fluorouracil‐based second‐line chemotherapy in advanced CRC (ClinicalTrials.gov Identifier NCT01326000).
3.2. Targeting additional pathways
3.2.1. Proangiogenic growth factors
The first approach to increase the efficacy of anti‐EGFR mAbs tested in the clinic was the combination with VEGF pathway inhibitors. In the BOND‐2 phase II trial, the activity seen with the addition of bevacizumab to cetuximab plus irinotecan seemed to be favorable when compared with historical controls in chemorefractory patients who were bevacizumab naive (Saltz et al., 2007). Nevertheless, when the anti‐EGFR therapy was added to bevacizumab‐based first‐line chemotherapy in advanced CRC, no added benefit was observed in large phase III trials (Hecht et al., 2009; Tol et al., 2009). In the PACCE trial, the addition of panitumumab to bevacizumab and oxaliplatin‐based chemotherapy was associated with shortening of the PFS interval among patients with tumors carrying wild‐type KRAS (9.8 versus 11.5 months) (Hecht et al., 2009). Similarly, in the CAIRO‐2 study, addition of cetuximab to XELOX plus bevacizumab regimen had no effect on PFS among those with KRAS wild‐type tumors (10.5 versus 10.6 months) (Tol et al., 2009). These data raises the possibility of a negative interaction (pharmacokinetic and/or pharmacodynamic) between these two types of agents when combined with chemotherapy, even in the setting where anti‐EGFR therapy can effectively inhibit EGFR signaling. Another possible explanation that may have contributed to this finding is the increased toxicity of the combination, thus leading to frequent delays in chemotherapy and reduced dose‐intensity of the regimens.
Additionally, in order to overcome resistance, anti‐EGFR mAbs have been combined with small molecules targeting proangiogenic factors, such as brivanib. A phase III trial comparing cetuximab/placebo with cetuximab/brivanib in KRAS wild‐type chemotherapy‐refractory CRC patients recently completed accrual (ClinicalTrials.gov Identifier NCT00640471). The correlative studies planned in this trial will bring important information on double EGFR/VEGFR targeting in cancer (Shapiro et al., 2011b).
3.2.2. MET pathway
MET overexpression is found in most CRCs, both KRAS wild‐type and KRAS mutant tumors, and MET signaling appears to cooperate with EGFR pathway signaling to promote the growth of CRC cells (Seiden‐Long et al., 2006). Higher expression levels of the MET receptor and of its ligand (hepatocyte growth factor – HGF) have been correlated with advanced stage and poor survival in CRC (Kammula et al., 2007). Both mAbs targeting the MET receptor or HGF ligand and TKIs targeting MET are in clinical investigation in advanced CRC. Preliminary results of a randomized phase I/II trial in KRAS wild‐type chemorefractory patients demonstrated significant improvement in overall RR with the addition of rilotumumab to panitumumab (31% in the two mAbs arm versus 21% in the panitumumab/placebo arm) (Eng et al., 2011). As seen in Table 3, the combination of irinotecan, cetuximab and tivantinib is also being explored in advanced CRC as a way of overcoming resistance to anti‐EGFR mAbs.
Table 3.
Selected combination trials of cetuximab and other targeted agents with focus on CRC.a
| Combination agent | Target and class | Phase | Indication | ClinicalTrials.gov identifier |
|---|---|---|---|---|
| Ridaforolimus | mTOR TKIb | Ib | CRCa (H&Nc and NSCLCd) | NCT01212627 |
| Tensirolimus | mTOR TKIb | Ib | CRCa | NCT00593060 |
| Everolimus | mTOR TKIb | Ib‐II | CRCa (plus irinotecan) | NCT00522665 |
| PX‐866 | PI3K TKIb | II | CRCa (H&Nc) | NCT01252628 |
| XL281 (BMS‐908662) | RAF TKIb | I | CRCa (RAS or RAF mutant) | NCT01086267 |
| AZD6244 | MEK TKIb | Ib | Solid tumors | NCT01217450 |
| BMS‐754807 | IGF‐1Re/IRf TKIb | I‐II | CRCa (H&Nc) | NCT00908024 |
| Tivantinib | METg TKIb | Ib‐II | CRCa (plus irinotecan) | NCT01075048 |
| Ramucirumab | VEGFR2i mAbh | II | CRCa | NCT01079780 |
| EMD 525797 | alpha v integrin mAbh | Ib‐II | CRCa (plus irinotecan) | NCT01008475 |
Colorectal cancer.
Tyrosine kinase inhibitor.
Head and neck cancer.
Non‐small cell lung cancer.
Insulin‐like growth factor 1 receptor.
Insulin receptor.
Mesenchymal–epithelial transition receptor.
monoclonal antibody.
Vascular endothelial growth factor receptor 2.
3.2.3. IGF‐1R
IGF‐1R is upregulated in 50–90% of CRC, being a poor prognostic factor (Weber et al., 2002). Several preclinical studies demonstrate that IGF‐1R activation in CRC cells leads to stimulation of EGFR, most likely related to IGF‐1‐induced release of TGF‐α, one of the EGFR ligands (Scartozzi et al., 2010). Moreover, in presence of altered IGF‐1R pathway, cancer cells can escape anti‐EGFR mediated cell death through continued activation of the PI3K pathway, which is related to the ability of IGF‐1R to heterodimerize with EGFR. More importantly, overexpression of IGF‐1 has been correlated with resistance to cetuximab in KRAS wild‐type CRC (Cappuzzo et al., 2008; Scartozzi et al., 2010).
A synergistic antitumor effect of combinations of anti‐EGFR and anti‐IGF‐1R agents has also been shown in CRC cell lines (Hu et al., 2008). Nevertheless, the clinical studies that evaluated the addition of anti‐IGF‐1R mAbs to cetuximab or panitumumab in CRC have not demonstrated improvements in response or disease control rates. The first agent tested in CRC patients refractory anti‐EGFR agents was cixutumumab (IMC‐A12) (Reidy et al., 2010). In a randomized phase II trial, clinical benefit was seen only in one out of 64 patients treated with cixutumumab in combination with cetuximab. No additional antitumor activity was identified in a selected population of patients (KRAS wild‐type who experienced prolonged response to a previous anti‐EGFR targeted therapy). Additionally, the combination of panitumumab and ganitumab also failed to improve response rates as compared to panitumumab plus placebo in KRAS wild‐type metastatic CRC patients without previous anti‐EGFR therapy (Eng et al., 2011). The third agent investigated in a large randomized phase II trial was dalotuzumab. Remarkably, when added to cetuximab and irinotecan in patients with chemorefractory KRAS wild‐type CRC, survival endpoints were inferior as compared to standard therapy (Watkins et al., 2011). Predictive biomarker analysis is ongoing to identify subpopulations.
3.2.4. HER3
The extensive crosstalk among the ErbB family receptors leads to upregulation of parallel pathways after blockade of a particular receptor as a compensatory mechanism. As in lung cancer, one potential mechanism of resistance to anti‐EGFR therapy in CRC is related to the ability of EGFR to form heterodimers with HER3, inducing receptor autophosphorylation and triggering anti‐apoptotic signals. In the clinical scenario, HER3 overexpression, described in 30–80% of primary or metastatic CRC (Lee et al., 2002), has been associated with resistance to therapeutic agents targeting EGFR (Baselga and Swain, 2009; Yonesaka et al., 2010) and its expression correlates with a poor outcome in CRC patients treated with irinotecan plus cetuximab (Scartozzi et al., 2011). Inhibiting EGFR and HER3 could lead to greater efficiency and also overcome resistance to currently available EGFR‐directed therapies. MEHD7945A is a humanized IgG1 mAb with a high affinity to EGFR and HER3. Significantly superior activity was observed in multiple xenograft models when compared with monospecific EGFR targeting agents (Schaefer et al., 2011). Phase I trial is ongoing with a planned expansion at the recommended dose in KRAS wild‐type CRC patients (ClinicalTrials.gov Identifier NCT01207323). In addition, specific anti‐HER3 mAbs such as AMG 888 (ClinicalTrials.gov Identifier NCT00730470) and MM‐121 (ClinicalTrials.gov Identifier NCT00734305) are also under investigation.
3.2.5. Downstream effectors
The most promising approaches to circumvent or reverse resistance to anti‐EGFR targeted therapy and even effectively target tumor cells primarily resistant to these agents (such as KRAS mutant), are rational combinations of targeted treatments that include inhibitors of downstream effectors of the EGFR pathway. At the present time, several drugs that inhibit activated BRAF, MEK, PI3K, AKT and mTOR are available and clinical trials with these agents are actively recruiting patients, some of them selecting therapy based on the genetic profile of the tumor, as shown in Table 3. However, comprehensive understanding of the precise role of these potential drug targets in colorectal tumors and the oncogenic dependence that tumors might have on these components is still lacking (De Roock et al., 2011). As an example, the clinical activity seen with the selective BRAF inhibitor vemurafenib in previously treated metastatic CRC patients with BRAF V600E mutations is less remarkable than what was seen in melanoma patients. In an extension cohort of the phase I study with vemurafenib, only 1 confirmed partial response and 4 minor responses (≥10% shrinkage) were observed in 19 patients (Kopetz et al., 2010). This activity does confirm mutant BRAF as a therapeutic target in this disease but also shows that the biology of BRAF activation in CRC is clearly more heterogeneous than in other tumor types. In addition to a greater understanding of the disease's biology, clinical trials have to incorporate biomarker stratification or enrichment strategies. The negative results seen in the phase 2 trial that evaluated the addition of sorafenib, which also targets RAF, to standard first‐line chemotherapy in unselected patients with advanced CRC is one example of questionable trial design (Tabernero et al., 2011). Subgroup analysis did not lead to meaningful conclusions due to the small sample size of the population.
It is well known that RAS has several downstream effector pathways that are not blocked by inhibiting only MEK. In addition, due to frequent coactivation of the PI3K pathway in CRC and extensive crosstalk between receptor tyrosine kinase cascades, a combination of targeted drugs seems appropriate. Many trials are underway evaluating MEK inhibitors added to PI3K, AKT and mTOR inhibitors. So far, the preliminary clinical results of these phase 1 trials have not shown radiologic responses in the KRAS mutant CRC population, but the number of patients treated is small for definitive conclusions (Shapiro et al., 2011a; Tolcher et al., 2011; Kurzrock et al., 2011). This strategy also needs also further investigation in BRAF and PIK3CA mutant CRC (Wee et al., 2009).
In tumors resistant to anti‐EGFR mAbs because of abnormal PI3K/PTEN status, the addition of PI3K pathway inhibitors to standard treatment is an interesting approach already being explored in multiple phase I/II trials, as shown in Table 3. In addition, the combination of cetuximab and MEK or RAF inhibitors is undergoing clinical testing based on increased antitumor activity in preclinical models with BRAF mutant CRC cell lines (Di Nicolantonio et al., 2008). Definitive results of these trials are eagerly expected.
4. Conclusions
EGFR is situated at the apex of this complex signaling network. This explains why, other than NSCLC patients with activating EGFR mutations, only a small number of patients initially respond to first‐generation EGFR inhibitors. In addition, acquired resistance is very common among those who respond. The most important mechanism of resistance to anti‐EGFR agents in cancer cells is activation of alternative intracellular signaling pathways as an early adoptive response. It also appears that many resistance mechanisms can coexist in the same tumor. Thus, in order to overcome and even preclude resistance to EGFR inhibitors, a combinatorial approach targeting multiple points within these pathways may be necessary.
Regarding EGFR targeting in NSCLC, the irreversible EGFR TKIs have yet to meet expectations in terms of clinical effectiveness. One efficacy‐limiting factor is believed to be the dosage restriction imposed by the toxicity related to concurrent inhibition of wild‐type EGFR. The combination of afatinib and cetuximab is the most promising in the erlotinib‐refractory context and deserves further investigation. Furthermore, the upfront addition of MET inhibitors to erlotinib has also been associated with increased efficacy outcomes as compared to EGFR TKI therapy alone. Concerning CRC, the progresses in drug development to overcome resistance to EGFR targeting agents have been limited. Results of the studies with “second‐generation” anti‐EGFR mAbs are expected soon, as well as the trials evaluating combination approaches with MET/HGF inhibitors and TKIs of key downstream effectors. In both malignancies, combined inhibition of EGFR and VEGFR or EGFR and IGF‐1R has not improved clinical outcomes.
Personalized cancer medicine based on molecular profiling of tumors is the treatment strategy of the future. To achieve the goal of efficient drug development, successful preclinical development and clear understanding of intra‐ and inter‐EGFR signaling pathway connections is essential. Proper selection of patients in the ongoing clinical trials with targeted agents is mandatory, as well as studies focusing on re‐biopsy of patients progressing on EGFR targeting agents. These strategies are feasible and should be a priority in experienced centers in order to improve the efficacy of EGFR inhibitors in cancer patients.
Dienstmann Rodrigo, De Dosso Sara, Felip Enriqueta, Tabernero Josep, (2012), Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer, Molecular Oncology, 6, doi: 10.1016/j.molonc.2011.11.009.
Contributor Information
Rodrigo Dienstmann, Email: rdienstmann@vhebron.net.
Sara De Dosso, Email: Sara.DeDosso@eoc.ch.
Enriqueta Felip, Email: efelip@vhebron.net.
Josep Tabernero, Email: jtabernero@vhebron.net.
References
- Arcila, M.E. , Oxnard, G.R. , Nafa, K. , Riely, G.J. , Solomon, S.B. , Zakowski, M.F. , Kris, M.G. , Pao, W. , Miller, V.A. , Ladanyi, M. , 2011. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res.. 17, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahleda, R. , Soria, J.C. , Harbison, C.T. , Park, J. , Felip, E. , Hanna, N. , Laurie, S.A. , Armand, J. , Shepherd, F.A. , Herbst, R. , 2009. Tumor regression and pharmacodynamic biomarker validation in non-small cell lung cancer patients treated with the ErbB/VEGFR inhibitor BMS-690514. J. Clin. Oncol.. 27, abstract 431s [Google Scholar]
- Baselga, J. , Swain, S.M. , 2009. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer. 9, 463–475. [DOI] [PubMed] [Google Scholar]
- Bean, J. , Brennan, C. , Shih, J.Y. , Riely, G. , Viale, A. , Wang, L. , Chitale, D. , Motoi, N. , Szoke, J. , Broderick, S. , Balak, M. , Chang, W.C. , Yu, C.J. , Gazdar, A. , Pass, H. , Rusch, V. , Gerald, W. , Huang, S.F. , Yang, P.C. , Miller, V. , Ladanyi, M. , Yang, C.H. , Pao, W. , 2007. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. U. S. A.. 104, 20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A. , Reckamp, K.L. , Camidge, D.R. , Giaccone, G. , Gadgeel, S.M. , Khuri, F.R. , Engelman, J.A. , Denis, L.J. , O'Connell, J.P. , Jänne, P.A. , 2010. PF-00299804 (PF299) patient reported outcomes and efficacy in adenocarcinoma and non-adeno non-small cell lung cancer: a phase II trial in advanced NSCLC after failure of chemotherapy and erlotinib. J. Clin. Oncol.. 28, abstract 7596 [Google Scholar]
- Campoli, M. , Ferris, R. , Ferrone, S. , Wang, X. , 2010. Immunotherapy of malignant disease with tumor antigen-specific monoclonal antibodies. Clin. Cancer Res.. 16, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappuzzo, F. , Hirsch, F.R. , Rossi, E. , Bartolini, S. , Ceresoli, G.L. , Bemis, L. , Haney, J. , Witta, S. , Danenberg, K. , Domenichini, I. , Ludovini, V. , Magrini, E. , Gregorc, V. , Doglioni, C. , Sidoni, A. , Tonato, M. , Franklin, W.A. , Crino, L. , Bunn, P.A. , Varella-Garcia, M. , 2005. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J. Natl. Cancer Inst.. 97, 643–655. [DOI] [PubMed] [Google Scholar]
- Cappuzzo, F. , Varella-Garcia, M. , Finocchiaro, G. , Skokan, M. , Gajapathy, S. , Carnaghi, C. , Rimassa, L. , Rossi, E. , Ligorio, C. , Di Tommaso, L. , Holmes, A.J. , Toschi, L. , Tallini, G. , Destro, A. , Roncalli, M. , Santoro, A. , Jänne, P.A. , 2008. Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br. J. Cancer. 99, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, R.B. , Jänne, P.A. , Engelman, J.A. , Martínez, P. , Nishida, Y. , Gendreau, S. , Wu, B. , Felip, E. , 2010. A phase I safety and pharmacokinetic study of PI3K/TORC1/ TORC2 inhibitor XL765 (SAR245409) in combination with erlotinib in patients with advanced solid tumors. J. Clin. Oncol.. 28, abstract 3015 [Google Scholar]
- De Roock, W. , Claes, B. , Bernasconi, D. , De Schutter, J. , Biesmans, B. , Fountzilas, G. , Kalogeras, K.T. , Kotoula, V. , Papamichael, D. , Laurent-Puig, P. , Penault-Llorca, F. , Rougier, P. , Vincenzi, B. , Santini, D. , Tonini, G. , Cappuzzo, F. , Frattini, M. , Molinari, F. , Saletti, P. , De Dosso, S. , Martini, M. , Bardelli, A. , Siena, S. , Sartore-Bianchi, A. , Tabernero, J. , Macarulla, T. , Di Fiore, F. , Gangloff, A.O. , Ciardiello, F. , Pfeiffer, P. , Qvortrup, C. , Hansen, T.P. , Van Cutsem, E. , Piessevaux, H. , Lambrechts, D. , Delorenzi, M. , Tejpar, S. , 2011. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol.. 12, 594–603. [DOI] [PubMed] [Google Scholar]
- Di Nicolantonio, F. , Martini, M. , Molinari, F. , Sartore-Bianchi, A. , Arena, S. , Saletti, P. , De Dosso, S. , Mazzucchelli, L. , Frattini, M. , Siena, S. , Bardelli, A. , 2008. BRAF wild-type is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol.. 26, 5705–5712. [DOI] [PubMed] [Google Scholar]
- Dienstmann, R. , Tolcher, A.W. , Papadopoulos, K.P. , Rasco, D.W. , Tabernero, J. , Brana, I. , Piera, A. , Skartved, N.J. , Aladdin, H. , Petersen, J. , Patnaik, A. , 2011. Phase I trial of the first-in-class EGFR antibody mixture, Sym004, in patients with advanced solid tumors. J. Clin. Oncol.. 29, abstract 3089 [Google Scholar]
- Dienstmann, R. , Vilar, E. , Tabernero, J. , 2011. Molecular predictors of response to chemotherapy in colorectal cancer. Cancer J.. 17, 114–126. [DOI] [PubMed] [Google Scholar]
- Eng, C. , Van Cutsem, E. , Nowara, E. , Swieboda-Sadlej, A. , Tebbutt, N.C. , Mitchell, E.P. , Davidenko, I. , Oliner, K. , Chen, L. , Huang, J. , McCaffery, I. , Loh, E. , Smethurst, D. , Tabernero, J. , 2011. A randomized, phase Ib/II trial of rilotumumab (AMG 102; ril) or ganitumab (AMG 479; gan) with panitumumab (pmab) versus pmab alone in patients with wild-type KRAS metastatic colorectal cancer. J. Clin. Oncol.. 29, abstract 3500 [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Gale, C.M. , Lifshits, E. , Gonzales, A.J. , Shimamura, T. , Zhao, F. , Vincent, P.W. , Naumov, G.N. , Bradner, J.E. , Althaus, I.W. , Gandhi, L. , Shapiro, G.I. , Nelson, J.M. , Heymach, J.V. , Meyerson, M. , Wong, K.K. , Jänne, P.A. , 2007. PF00299804, an irreversible pan- ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res.. 67, 11924–11932. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Jänne, P.A. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Felip, E. , Salcedo, M. , Murtra-Garrell, N. , Navarro, A. , Teixido, C. , Hernandez-Losa, J. , Cedres, S. , Martinez, P. , Lopez, E. , Montero, M.A. , Freixinos, V. , Argiles, G. , Nuñez, I. , Peg, V. , Pallisa, E. , Canela, M. , Tabernero, J. , Ramon y Cajal, S. , Tallada, N. , 2011. Expression of ErbB2 and ErbB3 in resected non-small cell lung cancer patients. J. Clin. Oncol.. 29, abstract 7037 [Google Scholar]
- Gerdes, C. , Patre, M. , Nicolini, V. , Moessner, E. , Klingner, G. , Bruenker, P. , Moser, S. , Grau, R. , Schmidt, C. , Guarino, B. , Sondermann, P. , Jaegar, C. , Preiss, S. , van Puijenbroek, E. , Bossenmaier, B. , Ries, C. , Friess, T. , Umana, P. , 2008. GA201, a novel humanized, glycoengineered EGFR antibody with enhanced ADCC and superior in vivo efficacy in xenograft models. Proc. Am. Assoc. Cancer Res.. abstract 3973 [Google Scholar]
- Guix, M. , Faber, A.C. , Wang, S.E. , Olivares, M.G. , Song, Y. , Qu, S. , Rinehart, C. , Seidel, B. , Yee, D. , Arteaga, C.L. , Engelman, J.A. , 2008. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest.. 18, 2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht, J.R. , Mitchell, E. , Chidiac, T. , Scroggin, C. , Hagenstad, C. , Spigel, D. , Marshall, J. , Cohn, A. , McCollum, D. , Stella, P. , Deeter, R. , Shahin, S. , Amado, R.G. , 2009. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J. Clin. Oncol.. 27, 672–680. [DOI] [PubMed] [Google Scholar]
- Herbst, R.S. , Ansari, R. , Bustin, F. , Flynn, P. , Hart, L. , Otterson, G.A. , Vlahovic, G. , Soh, C.H. , O'Connor, P. , Hainsworth, J. , 2011. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet. 377, 1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y.P. , Patil, S.B. , Panasiewicz, M. , Li, W. , Hauser, J. , Humphrey, L.E. , Brattain, M.G. , 2008. Heterogeneity of receptor function in colon carcinoma cells determined by cross-talk between type I insulin-like growth factor receptor and epidermal growth factor receptor. Cancer Res.. 68, 8004–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inukai, M. , Toyooka, S. , Ito, S. , Asano, H. , Ichihara, S. , Soh, J. , Suehisa, H. , Ouchida, M. , Aoe, K. , Aoe, M. , Kiura, K. , Shimizu, N. , Date, H. , 2006. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res.. 66, 7854–7858. [DOI] [PubMed] [Google Scholar]
- Jackman, D. , Pao, W. , Riely, G.J. , Engelman, J.A. , Kris, M.G. , Jänne, P.A. , Lynch, T. , Johnson, B.E. , Miller, V.A. , 2010. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol.. 28, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjigian, Y.Y. , Azzoli, C.G. , Krug, L.M. , Pereira, L.K. , Rizvi, N.A. , Pietanza, M.C. , Kris, M.G. , Ginsberg, M.S. , Pao, W. , Miller, V.A. , Riely, G.J. , 2011. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res.. 17, 2521–2527. [DOI] [PubMed] [Google Scholar]
- Janjigian, Y.Y. , Groen, H.J. , Horn, L. , Smit, E.F. , Fu, Y. , Wang, F. , Shahidi, M. , Denis, L.J. , Pao, W. , Miller, V.A. , 2011. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J. Clin. Oncol.. 29, abstract 7525^ [Google Scholar]
- Kammula, U.S. , Kuntz, E.J. , Francone, T.D. , Zeng, Z. , Shia, J. , Landmann, R.G. , Paty, P.B. , Weiser, M.R. , 2007. Molecular coexpressionof the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett.. 248, 219–228. [DOI] [PubMed] [Google Scholar]
- Khambata-Ford, S. , Harbison, C.T. , Hart, L.L. , Awad, M. , Xu, L.A. , Horak, C.E. , Dakhil, S. , Hermann, R.C. , Lynch, T.J. , Weber, M.R. , 2010. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J. Clin. Oncol.. 28, 918–927. [DOI] [PubMed] [Google Scholar]
- Kopetz, S. , Desai, J. , Chan, E. , Hecht, J.R. , O'Dwyer, P.J. , Lee, R.J. , Nolop, K.B. , Saltz, L. , 2010. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J. Clin. Oncol.. 28, abstract 3534 [Google Scholar]
- Kosaka, T. , Yatabe, Y. , Endoh, H. , Yoshida, K. , Hida, T. , Tsuboi, M. , Tada, H. , Kuwano, H. , Mitsudomi, T. , 2006. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res.. 12, 5764–5769. [DOI] [PubMed] [Google Scholar]
- Kurzrock, R. , Patnaik, A. , Rosenstein, A. , Fu, S. , Papadopoulos, K.P. , Smith, D.A. , Falchook, G.S. , Chambers, G. , Gauvin, J.L. , Naing, A. , Smith, L.S. , Gonzalez, T. , Tsimberidou, A.M. , Mays, T.A. , Cox, D.S. , Hong, D.S. , DeMarini, D.J. , Le, N.T. , Morris, S.R. , Tolcher, A.W. , 2011. Phase I dose-escalation of the oral MEK1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral AKT inhibitor GSK2141795 (GSK795). J. Clin. Oncol.. 29, abstract 3085 [Google Scholar]
- Kwak, E.L. , Sordella, R. , Bell, D.W. , Godin-Heymann, N. , Okimoto, R.A. , Brannigan, B.W. , Harris, P.L. , Driscoll, D.R. , Fidias, P. , Lynch, T.J. , Rabindran, S.K. , McGinnis, J.P. , Wissner, A. , Sharma, S.V. , Isselbacher, K.J. , Settleman, J. , Haber, D.A. , 2005. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. U. S. A.. 102, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.C. , Wang, S.T. , Chow, N.H. , Yang, H.B. , 2002. Investigation of the prognostic value of coexpressed erbB family members for the survival of colorectal cancer patients after curative surgery. Eur. J. Cancer. 38, 1065–1071. [DOI] [PubMed] [Google Scholar]
- Leighl, N.B. , Soria, J.C. , Bennouna, J. , Blais, N. , Traynor, A.M. , Papadimitrakopoulou, V. , Klimovsky, J. , Jappe, A. , Jehl, V. , Johnson, B.E. , 2010. Phase II study of everolimus plus erlotinib in previously treated patients with advanced non-small cell lung cancer. J. Clin. Oncol.. 28, abstract 7524 [DOI] [PubMed] [Google Scholar]
- Li, D. , Ambrogio, L. , Shimamura, T. , Kubo, S. , Takahashi, M. , Chirieac, L.R. , Padera, R.F. , Shapiro, G.I. , Baum, A. , Himmelsbach, F. , Rettig, W.J. , Meyerson, M. , Solca, F. , Greulich, H. , Wong, K.K. , 2008. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 27, 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévre, A. , Bachet, J.B. , Le Corre, D. , Boige, V. , Landi, B. , Emile, J.F. , Côté, J.F. , Tomasic, G. , Penna, C. , Ducreux, M. , Rougier, P. , Penault-Llorca, F. , Laurent-Puig, P. , 2006. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res.. 66, 3992–3995. [DOI] [PubMed] [Google Scholar]
- Lynch, T.J. , Bell, D.W. , Sordell, R. , Gurubhagavatula, S. , Okimoto, R.A. , Brannigan, B.W. , Harris, P.L. , Haserlat, S.M. , Supko, J.G. , Haluska, F.G. , Louis, D.N. , Christiani, D.C. , Settleman, J. , Haber, D.A. , 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med.. 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Maemondo, M. , Inoue, A. , Kobayashi, K. , Sugawara, S. , Oizumi, S. , Isobe, H. , Gemma, A. , Harada, M. , Yoshizawa, H. , Kinoshita, I. , Fujita, Y. , Okinaga, S. , Hirano, H. , Yoshimori, K. , Harada, T. , Ogura, T. , Ando, M. , Miyazawa, H. , Tanaka, T. , Saijo, Y. , Hagiwara, K. , Nukiwa, T. , North-East Japan Study Group, 2010. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med.. 362, 2380–2388. [DOI] [PubMed] [Google Scholar]
- Miller, V.A. , Hirsh, V. , Cadranel, J. , Chen, Y.-M. , Park, K. , Kim, S.W. , Caicun, Z. , Oberdick, M. , Shahidi, M. , Yang, C.H. , 2010. Phase IIB/ III double-blind randomized trial of afatinib (BIBW 2992, an irreversible inhibitor of the EGFR/HER1 and HER2) + best supportive care (BSC) versus placebo + BSC in patients with NSCLC failing 1–2 lines of chemotherapy and erlotinib or gefitinib (LUX-LUNG 1). Ann. Oncol.. 21, viii122–viii161. abstract LBA1 [Google Scholar]
- Mitsudomi, T. , Morita, S. , Yatabe, Y. , Negoro, S. , Okamoto, I. , Tsurutani, J. , Seto, T. , Satouchi, M. , Tada, H. , Hirashima, T. , Asami, K. , Katakami, N. , Takada, M. , Yoshioka, H. , Shibata, K. , Kudoh, S. , Shimizu, E. , Saito, H. , Toyooka, S. , Nakagawa, K. , Fukuoka, M. , West Japan Oncology Group, 2010. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol.. 11, 121–128. [DOI] [PubMed] [Google Scholar]
- Mok, T.S. , Wu, Y.L. , Thongprasert, S. , Yang, C.H. , Chu, D.T. , Saijo, N. , Sunpaweravong, P. , Han, B. , Margono, B. , Ichinose, Y. , Nishiwaki, Y. , Ohe, Y. , Yang, J.J. , Chewaskulyong, B. , Jiang, H. , Duffield, E.L. , Watkins, C.L. , Armour, A.A. , Fukuoka, M. , 2009. Gefitinib or paclitaxel-carboplatin in pulmonary adenocarcinoma. N. Engl. J. Med.. 361, 947–957. [DOI] [PubMed] [Google Scholar]
- Mok, T. , Spigel, D.R. , Park, K. , Socinski, M.A. , Tung, S.Y. , Kim, D.-W. , Ou, S.-H.I. , Zhang, H. , O'Connell, J.P. , Jänne, P. , 2010. Efficacy and safety of PF299804 as first-line treatment of patients with advanced NSCLC selected for activating mutation of epidermal growth factor receptor (EGFR). Ann. Oncol.. 21, viii122–viii161. abstract LBA18 [Google Scholar]
- Morgillo, F. , Woo, J.K. , Kim, E.S. , Hong, W.K. , Lee, H.Y. , 2006. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res.. 66, 10100–10111. [DOI] [PubMed] [Google Scholar]
- Natale, R.B. , Thongprasert, S. , Greco, F.A. , Thomas, M. , Tsai, C.M. , Sunpaweravong, P. , Ferry, D. , Mulatero, C. , Whorf, R. , Thompson, J. , Barlesi, F. , Langmuir, P. , Gogov, S. , Rowbottom, J.A. , Goss, G.D. , 2009. Vandetanib versus erlotinib in patients with advanced non-small cell lung cancer after failure of at least one prior cytotoxic chemotherapy: a randomized, double-blind phase III trial (ZEST). J. Clin. Oncol.. 27, abstract 409s [DOI] [PubMed] [Google Scholar]
- O'Byrne, K.J. , Bondarenko, I. , Barrios, C. , Eschbach, C. , Martens, U. , Hotko, Y. , Kortsik, C. , Celik, I. , Stroh, C. , Pirker, R. , 2009. Molecular and clinical predictors of outcome for cetuximab in non-small cell lung cancer (NSCLC): data from the FLEX study. J. Clin. Oncol.. 27, abstract 8007 [Google Scholar]
- Ouban, A. , Muraca, P. , Yeatman, T. , Coppola, D. , 2003. Expression and distribution of insulin-like growth factor-1 receptor in human carcinomas. Hum. Pathol.. 34, 803–808. [DOI] [PubMed] [Google Scholar]
- Pao, W. , Wang, T.Y. , Riely, G.J. , Miller, V.A. , Pan, Q. , Ladanyi, M. , Zakowski, M.F. , Heelan, R.T. , Kris, M.G. , Varmus, H.E. , 2005. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med.. 2, e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares, L.G. , Gomez-Roca, C. , Delord, J.P. , Cervantes, A. , Markman, B. , Corral, J. , Soria, J.C. , Bergé, Y. , Roda, D. , Russell-Yarde, F. , Hollingsworth, S. , Baselga, J. , Umana, P. , Manenti, L. , Tabernero, J. , 2011. Phase I pharmacokinetic and pharmacodynamic dose-escalation study of RG7160 (GA201), the first glycoengineered monoclonal antibody against the epidermal growth factor receptor, in patients with advanced solid tumors. J. Clin. Oncol.. 29, 3783–3790. [DOI] [PubMed] [Google Scholar]
- Pedersen, M.W. , Jacobsen, H.J. , Koefoed, K. , Hey, A. , Pyke, C. , Haurum, J.S. , Kragh, M. , 2010. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res.. 70, 588–597. [DOI] [PubMed] [Google Scholar]
- Regales, L. , Gong, Y. , Shen, R. , de Stanchina, E. , Vivanco, I. , Goel, A. , Koutcher, J.A. , Spassova, M. , Ouerfelli, O. , Mellinghoff, I.K. , Zakowski, M.F. , Politi, K.A. , Pao, W. , 2009. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J. Clin. Invest.. 119, 3000–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy, D.L. , Vakiani, E. , Fakih, M.G. , Saif, M.W. , Hecht, J.R. , Goodman-Davis, N. , Hollywood, E. , Shia, J. , Schwartz, J. , Chandrawansa, K. , Dontabhaktuni, A. , Youssoufian, H. , Solit, D.B. , Saltz, L.B. , 2010. Randomized, phase II study of the Insulin-Like Growth Factor-1 Receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J. Clin. Oncol.. 28, 4240–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell, R. , Gervais, R. , Vergnenegre, A. , Massuti, B. , Felip, E. , Cardenal, F. , Garcia Gomez, R. , Pallares, R. , Sanchez, J.M. , Porta, R. , Cobo, M. , Di Seri, M. , Garrido Lopez, P. , Insa, A. , De Marinis, F. , Corre, R. , Carreras, M. , Carcereny, E. , Taron, M. , Paz-Ares, L.G. , 2011. Erlotinib versus chemotherapy in advanced non-small cell lung cancer patients with epidermal growth factor receptor mutations: interim results of the European Erlotinib Versus Chemotherapy (EURTAC) phase III randomized trial. J. Clin. Oncol.. 29, abstract 7503 [Google Scholar]
- Rosell, R. , Molina, M.A. , Costa, C. , Simonetti, S. , Gimenez-Capitan, A. , Bertran-Alamillo, J. , Mayo, C. , Moran, T. , Mendez, P. , Cardenal, F. , Isla, D. , Provencio, M. , Cobo, M. , Insa, A. , Garcia-Campelo, R. , Reguart, N. , Majem, M. , Viteri, S. , Carcereny, E. , Porta, R. , Massuti, B. , Queralt, C. , de Aguirre, I. , Sanchez, J.M. , Sanchez-Ronco, M. , Mate, J.L. , Ariza, A. , Benlloch, S. , Sanchez, J.J. , Bivona, T.G. , Sawyers, C.L. , Taron, M. , 2011. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin. Cancer Res.. 17, 1160–1168. [DOI] [PubMed] [Google Scholar]
- Rosell, R., Moran, T., Felip, E., Sanchez Torres, J., Borghaei, H., Guan, S., Brown, H., Fitzgerald, T., Clark, J., Sathyanarayanan, S., Ayers, M., Hardwick, J., Lu, B., Yan, L., Johnson, D., 2011c. An open label, randomized Ph II study evaluating dalotuzumab combined with erlotinib in patients with non-small cell lung cancer following failure of prior chemotherapy. 14th World Conference on Lung Cancer, abstract O19.02.
- Salomon, D.S. , Brandt, R. , Ciardiello, F. , Normanno, N. , 1995. Epidermal growth factor related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol.. 19, 183–232. [DOI] [PubMed] [Google Scholar]
- Saltz, L.B. , Lenz, H.J. , Kindler, H.L. , Hochster, H.S. , Wadler, S. , Hoff, P.M. , Kemeny, N.E. , Hollywood, E.M. , Gonen, M. , Quinones, M. , Morse, M. , Chen, H.X. , 2007. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J. Clin. Oncol.. 25, 4557–4561. [DOI] [PubMed] [Google Scholar]
- Scagliotti, G.V. , Krzakowski, M. , Szczesn, A. , Strausz, J. , Makhson, A. , Reck, M. , Tye, L. , Selaru, P. , Chao, R.C. , Govindan, R. , 2010. Sunitinib in combination with erlotinib for the treatment of advanced/metastatic non-small cell lung cancer: a phase III study. Ann. Oncol.. 21, viii3 abstract LBA6 [Google Scholar]
- Scaltriti, M. , Baselga, J. , 2006. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin. Cancer Res.. 12, 5268–5272. [DOI] [PubMed] [Google Scholar]
- Scartozzi, M. , Mandolesi, A. , Giampieri, R. , Pierantoni, C. , Loupakis, F. , Zaniboni, A. , Galizia, E. , Giustini, L. , Silva, R.R. , Bisonni, R. , Berardi, R. , Biagetti, S. , Menzo, S. , Falcone, A. , Bearzi, I. , Cascinu, S. , 2010. Insulin-like growth factor 1 expression correlates with clinical outcome in K-RAS wild type colorectal cancer patients treated with cetuximab and irinotecan. Int. J. Cancer. 127, 1941–1947. [DOI] [PubMed] [Google Scholar]
- Scartozzi, M. , Mandolesi, A. , Giampieri, R. , Bittoni, A. , Pierantoni, C. , Zaniboni, A. , Galizia, E. , Giustini, L. , Silva, R.R. , Bisonni, R. , Berardi, R. , Biscotti, T. , Biagetti, S. , Bearzi, I. , Cascinu, S. , 2011. The role of HER-3 expression in the prediction of clinical outcome for advanced colorectal cancer patients receiving irinotecan/cetuximab. Oncologist. 16, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, G. , Haber, L. , Crocker, L.M. , Shia, S. , Shao, L. , Dowbenko, D. , Totpal, K. , Wong, A. , Lee, C.V. , Stawicki, S. , Clark, R. , Fields, C. , Lewis Phillips, G.D. , Prell, R.A. , Danilenko, D.M. , Franke, Y. , Stephan, J.P. , Hwang, J. , Wu, Y. , Bostrom, J. , Sliwkowski, M.X. , Fuh, G. , Eigenbrot, C. , 2011. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 20, 472–486. [DOI] [PubMed] [Google Scholar]
- Seiden-Long, I.M. , Brown, K.R. , Shihn, W. , Wigle, D.A. , Radulovich, N. , Jurisica, I. , Tsao, M.S. , 2006. Transcriptional targets of hepatocyte growth factor signaling and K-ras oncogene activation in colorectal cancer. Oncogene. 25, 91–102. [DOI] [PubMed] [Google Scholar]
- Sequist, L.V. , Besse, B. , Lynch, T.J. , Miller, V.A. , Wong, K.K. , Gitlitz, B. , Eaton, K. , Zacharchuk, C. , Freyman, A. , Powell, C. , Ananthakrishnan, R. , Quinn, S. , Soria, J.C. , 2010. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol.. 28, 3076–3083. [DOI] [PubMed] [Google Scholar]
- Sequist, L.V. , Waltman, B.A. , Dias-Santagata, D. , Digumarthy, S. , Turke, A.B. , Fidias, P. , Bergethon, K. , Shaw, A.T. , Gettinger, S. , Cosper, A.K. , Akhavanfard, S. , Heist, R.S. , Temel, J. , Christensen, J.G. , Wain, J.C. , Lynch, T.J. , Vernovsky, K. , Mark, E.J. , Lanuti, M. , Iafrate, A.J. , Mino-Kenudson, M. , Engelman, J.A. , 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med.. 3, 75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist, L.V. , von Pawel, J. , Garmey, E.G. , Akerley, W.L. , Brugger, W. , Ferrari, D. , Chen, Y. , Costa, D.B. , Gerber, D.E. , Orlov, S. , Ramlau, R. , Arthur, S. , Gorbachevsky, I. , Schwartz, B. , Schiller, J.H. , 2011. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J. Clin. Oncol.. 29, 3307–3315. [DOI] [PubMed] [Google Scholar]
- Shapiro, G. , LoRusso, P. , Kwak, E.L. , Cleary, J.M. , Musib, L. , Jones, C. , de Crespigny, A. , Belvin, M. , McKenzie, M. , Gates, M.R. , Chan, I.T. , Bendell, J.C. , 2011. Clinical combination of the MEK inhibitor GDC-0973 and the PI3K inhibitor GDC-0941: a first-in-human phase Ib study testing daily and intermittent dosing schedules in patients with advanced solid tumors. J. Clin. Oncol.. 29, abstract 3005^ [Google Scholar]
- Shapiro, J.D. , Siu, L.L. , Zalcberg, J.R. , Moore, M.J. , Ringash, J. , Mittmann, N. , Simes, J. , O'Callaghan, C.J. , Tu, D. , Walters, I.B. , Magoski, N. , Smith, P. , Nomikos, D. , Zhu, L. , Savoie, M. , Virk, S. , El-Tahche, F. , Gill, R. , Price, T.J. , Jonker, D.J. , 2011. A phase III study of cetuximab plus either brivanib alaninate versus placebo in patients with chemotherapy-refractory KRAS wild-type advanced colorectal cancer: the NCIC CTG/AGITG CO.20 trial. J. Clin. Oncol.. 29, abstract TPS163 [DOI] [PubMed] [Google Scholar]
- Shih, J. , Yu, C. , Su, W. , Hsia, T. , Graziano, S.L. , Calvo, R. , Cong, X.J. , Shahidi, M. , Yang, C. , Miller, V. , 2010. Activity of BIBW 2992, an irreversible EGFR/HER1 and HER2 TKI, in lung adenocarcinoma patients harbouring less common EGFR mutations. Ann. Oncol.. 21, viii122–viii161. abstract 415P [Google Scholar]
- Spigel, D.R. , Burris, H.A. , Greco, F.A. , Shipley, D.L. , Friedman, E.K. , Waterhouse, D.M. , Whorf, R.C. , Mitchell, R.B. , Daniel, D.B. , Zangmeister, J. , Bass, J.D. , Hainsworth, J.D. , 2011. Randomized, double-blind, placebo-controlled, phase II trial of sorafenib and erlotinib or erlotinib alone in previously treated advanced non-small-cell lung cancer. J. Clin. Oncol.. 29, 2582–2589. [DOI] [PubMed] [Google Scholar]
- Spigel, D.R. , Ervin, T.J. , Ramlau, R. , Daniel, D.B. , Goldschmidt, J.H. , Blumenschein, G.R. , Krzakowski, M.J. , Robinet, G. , Clement-Duchene, C. , Barlesi, F. , Govindan, R. , Patel, T. , Orlov, S.V. , Wertheim, M.S. , Zha, J. , Pandita, A. , Yu, W. , Yauch, R.L. , Patel, P.H. , Peterson, A.C. , 2011. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J. Clin. Oncol.. 29, abstract 7505 [Google Scholar]
- Tabernero, J., Garcia-Carbonero, R., Köhne, C.H., O'Dwyer, P.J., Sobrero, A., Van Cutsem, E., Gladkov, O., Davidenko, I., Salazar, R., Cassidy, J., 2011. A phase 2b, double-blind, randomized study evaluating the efficacy and safety of sorafenib compared with placebo when administered in combination with chemotherapy (modified FOLFOX6) for first-line treatment of patients with metastatic colorectal cancer (mCRC): the RESPECT trial. ESMO/ ECCO Conference Stockholm, abstract LBA19.
- Tol, J. , Koopman, M. , Cats, A. , Rodenburg, C.J. , Creemers, G.J. , Schrama, J.G. , Erdkamp, F.L. , Vos, A.H. , van Groeningen, C.J. , Sinnige, H.A. , Richel, D.J. , Voest, E.E. , Dijkstra, J.R. , Vink-Börger, M.E. , Antonini, N.F. , Mol, L. , van Krieken, J.H. , Dalesio, O. , Punt, C.J. , 2009. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N. Engl. J. Med.. 360, 563–572. [DOI] [PubMed] [Google Scholar]
- Tolcher, A.W. , Baird, R.D. , Patnaik, A. , Moreno Garcia, V. , Papadopoulos, K.P. , Garrett, C.R. , Olmos, D. , Shannon, K.A. , Zazulina, V. , Rubin, E.H. , Smith, I.C. , Ryan, J. , Smith, P.D. , Taylor, A. , Learoyd, M. , Lupinacci, L. , Yan, L. , De Bono, J.S. , 2011. A phase I dose-escalation study of oral MK-2206 (allosteric AKT inhibitor) with oral selumetinib (AZD6244; MEK inhibitor) in patients with advanced or metastatic solid tumors. J. Clin. Oncol.. 29, abstract 3004 [Google Scholar]
- Turke, A.B. , Zejnullahu, K. , Wu, Y.L. , Song, Y. , Dias-Santagata, D. , Lifshits, E. , Toschi, L. , Rogers, A. , Mok, T. , Sequist, L. , Lindeman, N.I. , Murphy, C. , Akhavanfard, S. , Yeap, B.Y. , Xiao, Y. , Capelletti, M. , Iafrate, A.J. , Lee, C. , Christensen, J.G. , Engelman, J.A. , Jänne, P.A. , 2010. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 17, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viloria-Petit, A. , Crombet, T. , Jothy, S. , Hicklin, D. , Bohlen, P. , Schlaeppi, J.M. , Rak, J. , Kerbel, R.S. , 2001. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res.. 61, 5090–5101. [PubMed] [Google Scholar]
- Wakelee, H.A. , Gettinger, S.N. , Engelman, J.A. , Jänne, P.A. , West, H.J. , Subramaniam, D.S. , Leach, J.W. , Wax, M.B. , Yaron, Y. , Lara, P. , 2010. A phase Ib/II study of XL184 (BMS 907351) with and without erlotinib in patients with non-small cell lung cancer. J. Clin. Oncol.. 28, abstract 3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, D.J. , Tabernero, J. , Schmoll, H. , Trarbach, T. , Ramos, F.J. , Howe, J. , Brown, H.M. , Clark, J. , Hsu, K. , Lu, B.D. , Cunningham, D. , 2011. A randomized phase II/III study of the anti-IGF-1R antibody MK-0646 (dalotuzumab) in combination with cetuximab and irinotecan in the treatment of chemorefractory metastatic colorectal cancer with wild-type KRAS status. J. Clin. Oncol.. 29, abstract 3501 [Google Scholar]
- Weber, M.M. , Fottner, C. , Liu, S.B. , Jung, M.C. , Engelhardt, D. , Baretton, G.B. , 2002. Overexpression of the insulin-like growth factor I receptor in human colon carcinomas. Cancer. 95, 2086–2095. [DOI] [PubMed] [Google Scholar]
- Wee, S. , Jagani, Z. , Xiang, K.X. , Loo, A. , Dorsch, M. , Yao, Y.M. , Sellers, W.R. , Lengauer, C. , Stegmeier, F. , 2009. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res.. 69, 4286–4293. [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , 2002. Addiction to oncogenes – the achilles heal of cancer. Science. 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Yang, C. , Shih, J. , Su, W. , Hsia, T. , Sequist, L.V. , Chang, G. , Calvo, R. , Cong, X.J. , Shahidi, M. , Miller, V. , 2010. A phase II study of BIBW 2992 in patients with adenocarcinoma of the lung and activating EGFR/HER1 mutations (LUX-LUNG 2). Ann. Oncol.. 21, viii122–viii161. abstract 367PD [Google Scholar]
- Yonesaka, K. , Okamoto, I. , Satoh, T. , Takeda, K. , Takada, M. , Nishio, K. , Fukuoka, M. , Saijo, N. , Jänne, P.A. , Nakagawa, K. , 2010. Heregulin as a novel cetuximab-resistance factor in colorectal cancer. J. Clin. Oncol.. 28, abstract e14044 [Google Scholar]
- Zhou, C. , Wu, Y.-L. , Chen, G. , Feng, J. , Liu, X. , Wang, C. , Zhang, S. , Wang, J. , Zhou, S. , Ren, S. , 2010. Efficacy results from the randomised phase III OPTIMAL (CTONG 0802) study comparing first-line erlotinib versus carboplatin plus gemcitabine, in Chinese advanced non-small-cell lung cancer patients with EGFR activating mutations. Ann. Oncol.. 21, viii122–viii161. abstract LBA13 [Google Scholar]