

Abstract
Androgen receptor (AR) activity is associated with cancer development and progression. In hepatocellular carcinoma (HCC), AR contributes to HCC incidence, but the role of AR in HCC cell migration and invasion remains largely unknown. In this study, we found that AR was expressed at high levels in a subgroup of HCC cell lines with high metastatic potential. Experiments using lentiviral overexpression or small hairpin RNA knockdown of AR as well as activation of AR by its ligand indicated that AR activation promoted HCC cell migration and invasion. We also found that AR activation enhanced the expression of a metastasis‐promoting gene, ID1, which led to increased HCC cell migration and invasion. An AR antagonist was able to block this process, suggesting that AR activation in AR‐positive HCC may be therapeutically inhibited as a potential intervention strategy.
Keywords: Androgen receptor, Hepatocellular carcinoma, Metastasis, ID1, Antagonist, Cell migration and invasion
Highlights
AR was expressed at high levels in a subgroup of HCC cell lines with high metastatic potential.
AR activation promoted HCC cell migration and invasion.
AR activation induced a metastasis‐promoting gene ID1.
AR‐activation‐promoted HCC cell migration and invasion was ID1‐dependent.
AR antagonist was able to block AR‐activation‐promoted HCC cell migration and invasion.
Abbreviations
- HCC
hepatocellular carcinoma
- AR
Androgen receptor
- HBV
hepatitis B virus
- AFP
alpha-fetoprotein
- CDX
casodex
- mRNA
messenger RNA
- PCR
polymerase chain reaction
- shRNA
small hairpin RNA
- SD
standard deviations
1. Introduction
Hepatocellular carcinoma (HCC) is one of the most common causes of cancer‐related death. Although liver cancer is known to be associated with hepatitis B virus (HBV), hepatitis C virus, and alcohol abuse, other risk factors that are genetic and physiological also contribute to the etiology of HCC (De Maria et al., 2002; Tang et al., 2004; Gomaa et al., 2008; Kalra et al., 2008; Yeh and Chen, 2010).
Androgen receptor (AR) is a ligand‐activated nuclear receptor and regulates gene expression in a variety of tissues during normal development, reproduction, other sexually dimorphic processes, and disease stages including cancers (Ramachandran et al., 2011; Tsai and O'Malley, 1994; Mangelsdorf et al., 1995; Shang et al., 2002). AR is well‐known to promote to prostate cancer development and progression (Heinlein and Chang, 2004), and HCC onset (Li et al., 2012; Naugler et al., 2007). Androgens and androgen receptor also contribute to the sex disparity in HCC incidence (Tejura et al., 1989; Nagasue et al., 1995). The use of anabolic androgenic steroids has been reported to be associated with malignant HCC transformation (Socas et al., 2005; Gorayski et al., 2008; Martin et al., 2008). Additional animal studies revealed that HCC incidence was lower and later in mice lacking hepatic AR than in their wild‐type littermates (Ma et al., 2008). However, little is known about the role of androgens and AR in HCC progression and metastasis.
In this study we elucidated the relationship between AR activity and HCC cell migration and invasion, and found that activation of AR induced ID1 and promoted HCC cell migration and invasion. This process was blocked by anti‐androgen casodex (CDX), suggesting that AR may be a potential therapeutic target for HCC development.
2. Methods and Materials
2.1. Cell culture
HCC cells (ATCC, Manassas, VA, USA) were cultured under the following conditions: SNU‐182, SNU‐387, SNU‐398, SNU‐423, SNU‐449, and SNU‐475 cells were cultured using 10% fetal bovine serum (Cat#10099‐141, Invitrogen, Carlsbad, CA) in either RPMI‐1640 (Cat#C11875, Invitrogen) or SMMC7721 media. Huh7, Hep3B, HepG2, HepG2.2.15, BEL7402, SK‐Hep‐1, Hepa1‐6, PLC/PRF/5, C3A, MHCC‐97H, MHCC‐97L, and LM3 cells were cultured using 10% fetal bovine serum (Invitrogen) in Dulbecco's modified Eagle medium (Cat#C11965, Invitrogen). THLE‐2 cells were cultured using bronchial epithelial cell growth media (BEGM Bullet Kit, Cat#CC3170, Clonetics Corporation, Walkersville, MD, USA). SNU‐387 and NCI‐H1755 were from female HCC, and the others were from male HCC. All cell cultures were maintained at 37 °C in 5% CO2. Cells were collected at 80% confluence for corresponding experiments. R1881 and CDX were purchased from Sigma.
2.2. Lentiviral overexpression
The lentiviral expression systems were obtained from System Biosciences (SBI, Mountain View, CA, USA). The overexpression vector was co‐transfected with packaging vectors psPAX and pMD2.G at a ratio of 3:2:1 into 293T cells using LipofectamineM 2000 (Invitrogen, USA) for 3 days before the virus‐ containing media were harvested. When the viruses were used to infect target cells, polybrene (5 μg/ml; Sigma, USA) was added to media. Two days later, puromycin (5 μg/ml; Sigma) was added for selecting the positive cells.
2.3. shRNA
Lentiviral shRNAs targeting AR and ID1 were purchased as the human GIPZ lentiviral shRNAmir target gene set from Openbiosystems (Lafayette, CO). The lentivirus generation and usage procedures were described above. Multiple shRNA clones for each targeted gene were used in the experiments.
2.4. Real‐time RT‐PCR
RNA was isolated from experimental cells using TRIzol (Cat#15596, Invitrogen) and chloroform (AMERSCO, USA) according to the manufacturer's protocol. Isopropanol (Sigma) was used to precipitate the RNA, and the final RNA sample was dissolved in RNase‐free water. Reverse transcription was performed using a PrimeScript reverse transcription kit (Cat# DRR037A, TaKaRa) in a reverse transcription reaction at 37 °C for 15 min.
Real‐time RT‐PCR was performed using the SYBR green method. cDNAs from the reverse transcription above and the SYBR real‐time PCR kit (Cat# DRR041A, TaKaRa) were used for real‐time RT‐PCR in 50‐ul reactions. The ABI 7300 model real‐time PCR system was used for detection (ABI, USA).
The following primers were used in the experiments: AR‐sense: GAACTCGATCGTATCATTGCAT; AR‐anti‐sense: GATGCAGCTCTCTCGCAATA; ID‐1‐sense: GCATGCGTTCCTGCGGACGAT; ID‐1 antisense: TGCAGTTAAGGGCGCAGTGGC; 18S‐sense: GCAATTATTCCCCATGAACG; and 18S‐anti‐sense: GGGACTTAATCAACGCAAGC.
2.5. Western blot analysis
Western blot analysis was carried out as previously described (Li et al., 2008). Cells were washed by PBS buffer three times before being lysed using RIPA buffer. Cell lysates were separated via 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) with 40 μg total protein loaded in each lane of gels. After electrophoresis, proteins were transferred onto nitrocellulose membranes for Western blotting. Phosphate‐buffered saline (PBS) with Tween 20 containing 5% milk was used to block the nitrocellulose membranes for 1 h; then, the membranes were incubated with phosphate‐buffered saline (PBS) with Tween 20 containing 2.5% milk and antibodies as indicated. The ECL + plus Western blot detection system was purchased from Thermo. The antibodies for each target protein were rabbit monoclonal anti‐AR (D6F11) (3H8; Cell Signaling Technology, Danvers, MA); rabbit monoclonal anti‐ID1 (195‐14; BioCheck, Foster City, CA); anti‐Tubulin (BD Biosciences); monoclonal anti‐beta‐Actin‐HRP (Sigma); and anti‐FLAG‐HRP (Sigma).
2.6. Cell migration and invasion assays
Cell migration assays were performed according to the manufacturer's protocols. Cells were trypsinized and resuspended in serum‐free media and subsequently seeded in transwell chambers (BD Falcon, USA). Then, the cells were cultured for 24–48 h followed by PBS washes, fixation with 4% formaldehyde (Sigma), and 0.1% crystal violet staining. The un‐migrated cells were removed using cotton swabs, and the migrated cells were counted from five random fields under a microscope. P values were calculated using an unpaired two‐tailed t test. For invasion assays, transwell chambers were covered with matrigel (BD Falcon, USA) before an experimental procedure similar to that for the migration assays was performed.
For experiments with hormone treatments, cells were first cultured in Dulbecco's modified Eagle medium (DMEM) or RPMI media (GIBCO, USA) with 10% charcoal‐stripped serum (Gemini Bio‐Products, West Sacramento, CA) for 3 days; then, cells were subjected to each hormone treatment (control[DMSO]; R1881 [10−8 M]; R1881 [10−8 M] + CDX [10−5 M]; and CDX [10−5 M]) for 16 h before cell migration or invasion assays were performed as described above.
2.7. Statistical analysis
Error bars in the figures of the migration and invasion assays indicate the means and standard deviations (n = 5). Statistical analysis was performed using a two‐tailed Student's t test, and P < 0.05 was considered statistically significant.
3. Results
3.1. Higher AR mRNA and protein expression levels in 20 HCC cell lines correlate with greater cell metastatic potential
To elucidate the role of AR in HCC progression, we analyzed AR mRNA levels in 20 HCC cell lines (Figure 1A) that can be divided into two subgroups: Group I and II. Most cell lines of Group I have been shown to exhibit less metastatic potential than those of group II (SNU‐398 as an outlier) (Lee and Thorgeirsson, 2002). We found that in most of the cell lines in Group II, AR expression was positive (P = 0.0005), compared with Group I, thus suggesting that high AR expression may contribute to HCC cell migration and invasion (Figure 1A, B).
Figure 1.
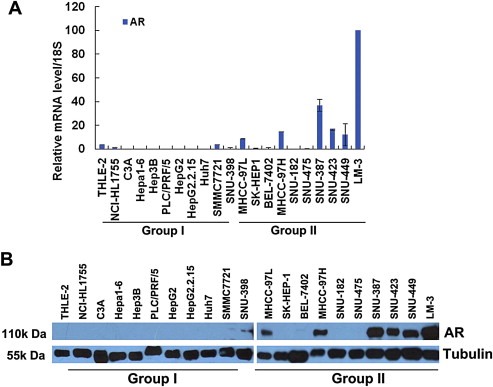
High AR mRNA and protein expression levels in HCC cell lines correlated with high cell metastatic potential. (A) AR mRNA levels in 20 HCC cell lines. AR mRNA levels in each HCC cell line and the normal liver cell line THLE‐2 were evaluated using real‐time RT‐PCR analysis as indicated, and data values were normalized to 18S RNA. Group I and Group II are indicated with low or high metastatic potential, respectively. (B) High AR protein expression levels in 20 HCC cell lines correlated with cell metastatic potential. AR protein expression levels in each HCC cell line and the normal liver cell line THLE‐2 were evaluated using Western blot analysis as indicated. Tubulin served as a loading control.
Moreover, among three metastatic HCC cell lines, MHCC‐97L, MHCC‐97H, and LM‐3, with low, medium, and high metastatic potential, respectively, on the basis of a metastatic HCC model in nude mice (Tang et al., 2004), both AR mRNA and protein levels corresponded: MHCC‐97L < MHCC‐97H < LM‐3 (Figure 1A and B). These results further supported the notion that AR likely contributes to HCC metastatic migration and invasion.
3.2. Migration and invasion rates are higher in AR‐positive HCC cells than in AR‐negative HCC cells
To gain more insight into the role of AR in HCC cell migration and invasion, we used transwell cell migration and invasion assays to examine two AR‐negative HCC cell lines, HepG2 and Hep3B, and two AR‐positive HCC cell lines, SNU‐449 and SNU‐423. Consistent with the results described above, the rates of cell migration and invasion were much higher in SNU‐449 and SNU‐423 than in HepG2 and Hep3B (Figure 2A, B, C and D).
Figure 2.
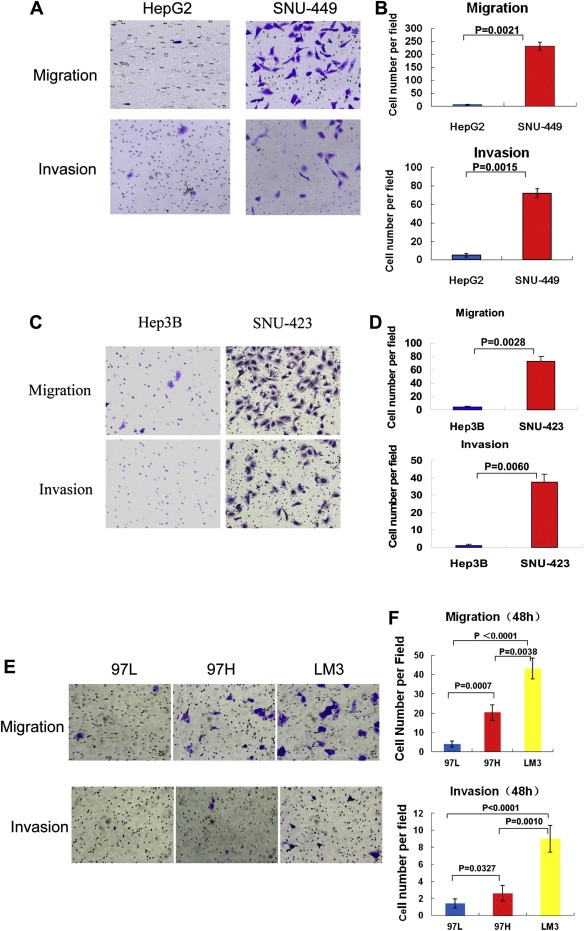
HCC cells with higher AR expression levels had higher cell migration and invasion rates. (A) AR‐positive HCC cell line SNU‐449 had higher migration and invasion rates than AR‐negative cell line HepG2. Transwell cell migration and invasion assays were performed as described in Methods and Materials. Representative cell pictures are shown. (B) Counted cell numbers of the experiment in (A) are shown. Error bars indicate means ± SDs. (C) AR‐positive HCC cell line SNU‐423 had higher migration and invasion rates than AR‐negative cell line Hep3B. Transwell cell migration and invasion assays were performed as described above. (D) Counted cell numbers of the experiment in (C) are shown. Error bars indicate means ± SDs. (E) The higher levels of AR, the higher migration and invasion rates of the HCC cells: 97L < 97H < LM3. Same experiments were performed as in (A) except using MHCC‐97L (97L), MHCC‐97H (97H) and LM‐3 (LM3) cells. (F) Counted cell numbers of the experiment in (E) are shown. Error bars indicate means ± SDs.
We next sought to determine whether the rates of cell migration and invasion were higher in HCC cell lines with high AR expression than in HCC cell lines with low AR expression. In the MHCC‐97L, MHCC‐97H, and LM‐3 cell lines, the stepwise order of migration and invasion rates from low to high was MHCC‐97L < MHCC‐97H < LM‐3 (Figure 2E and F). These results substantiated our notion that higher AR expression levels correlate with greater HCC cell migration and invasion potential.
3.3. Ectopic expression of AR in HepG2.2.15 cells increases cell migration and invasion, whereas shRNA knockdown of AR in SNU‐449 cells decreases cell migration and invasion
To determine whether AR directly contributes to HCC cell migration and invasion, we used a lentiviral overexpression system and AR‐negative HepG2.2.15 cells to generate AR‐positive HepG2.2.15 cells (Figure 3A). Cell migration and invasion assays were performed and revealed that the rates of cell migration and invasion were higher in the AR‐positive HepG2.2.15 cells than in the AR‐negative lentiviral vector control HepG2.2.15 cells (Figure 3B and C), thereby indicating that AR promotes HepG2.2.15 cell migration and invasion.
Figure 3.
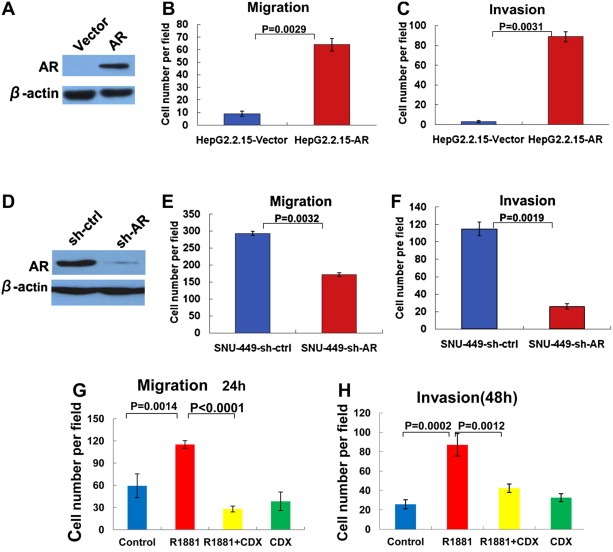
Ectopic expression of AR or activation of AR by androgen, and shRNA knockdown of AR increased and decreased HCC cell migration and invasion, respectively. (A) Ectopic expression of AR in HepG2.2.15 cells. Western blot analysis of AR expression in HepG2.2.15 cells infected by the lentivirus vector as a control (Vector) or the lentivirus expressing AR (AR) using each antibody as indicated. Beta actin served as a loading control. (B) (C) Ectopic expression of AR in HepG2.2.15 cells (HepG2.2.15‐AR) increased cell migration and invasion. Transwell cell migration (B) and invasion (C) assays were performed as described previously. Quantitative cell numbers are shown. HepG2.2.15 cells infected by an empty lentiviral vector (HepG2.2.15‐Vector) served as a control. (D) shRNA knockdown of AR in SNU‐449 cells. Western blot analysis of AR expression in SNU‐449 cells infected by the lentivirus vector as a control (sh‐ctrl) or the lentivirus expressing shRNA against AR (sh‐AR). Beta actin served as a loading control. (E) (F) shRNA knockdown of AR in SNU‐449 cells (SNU‐449‐sh‐AR) decreased cell migration and invasion. Transwell cell migration (E) and invasion (F) assays were performed as described in Methods and Materials. SNU‐449 cells infected by an empty lentiviral vector (SNU‐449‐sh‐ctrl) served as a control. Quantitative cell numbers are shown. (G) Androgen agonist R1881 increased SNU‐449 cell migration. SNU‐449 cells were cultured in 10% charcoal‐stripped serum media for 3 days and then treated with hormone R1881 (10−8 M) (R1881) for 16 h followed by transwell cell migration. Cells treated with vehicle DMSO served as a control (Control). Androgen antagonist CDX alone (CDX) or in combination with R1881 (R1881 + CDX) was used in the experiments to block androgen activation. Cell numbers were counted from five random fields in the experiment. Error bars indicate means ± SDs. (H) Androgen agonist R1881 increased SNU‐449 cell invasion. Similar experiments as in (G) except that cell invasion assays were performed.
We also used a lentiviral system to knock down AR via shRNA in an AR‐positive HCC cell line, SNU‐449. AR protein levels were significantly reduced by shRNA against AR compared with the control shRNA (Figure 3D). We then performed cell migration and invasion assays. The results indicated that shRNA knockdown of AR significantly decreased SNU‐449 cell migration and invasion (Figure 3E and F), thereby further suggesting that AR promotes HCC cell migration and invasion.
3.4. Androgen agonist R1881 increases SNU‐449 cell migration and invasion
We next sought to determine whether activation of AR by its androgen agonist ligand affects HCC cell migration and invasion. Cell migration and invasion assays were performed in the presence or absence of R1881. R1881 markedly increased SNU‐449 cell migration and invasion, and these effects were blocked by androgen antagonist CDX, which is a non‐steroidal anti‐androgen drug, thereby indicating that activation of AR by its ligand promotes cell migration and invasion (Figure 3G and H). Similar results, although to a less extent compared to those in SNU‐449, were obtained using SNU‐387 cells (Fig. S1‐2), which were an AR‐positive HCC cell line generated from a female, suggesting that androgen effect on HCC cell migration and invasion is not sex‐specific.
3.5. Androgen agonist R1881 stimulates metastatic gene ID1 expression
Because activation of AR by its ligand increased HCC cell migration and invasion, we were interested in determining the molecular mechanisms of this activity. Utilizing gene expression microarrays, we analyzed SNU‐449 cells upon treatment with ligand R1881. The metastasis‐promoting gene ID1 (Desprez et al., 2003; Ruzinova and Benezra, 2003; Minn et al., 2005; Lee et al., 2006) was identified to be activated by R1881, and, thus, potentially mediated the enhancement of HCC cell migration and invasion after AR activation. To validate this result, we treated SNU‐449 cells with R1881 for 16 h and used quantitative real‐time reverse transcriptase polymerase chain reaction (RT‐PCR) to examine changes in ID1 mRNA levels. Indeed, ID1 was activated significantly by R1881, and this activation was attenuated by antagonist CDX (Figure 4A). Moreover, ID1 induction by R1881 was determined by a time‐course Western blot, indicating ID1 protein levels were also increased over the 16hr period treatment with a maximal level at 16hr (Figure 4B). We further asked whether AR directly binds to the ID1 promoter in response to R1881. Chromatin immunoprecipitation (ChIP) assays were performed and revealed that AR did not bind directly to the ID1 promoter (Fig. S3). Cycloheximide (CHX), an inhibitor of protein biosynthesis, was also used in an experiment, suggesting that induction of ID1 by R1881 is not direct and requires new protein synthesis. Because if AR directly binds to the ID1 promoter and induces its mRNA, under CHX treatment conditions, ID1 is still induced; in contrast, if activation of AR induces another gene mRNA and protein first which in turn induces ID1 as a secondary response, under CHX conditions, there is no new protein production and no ID1 activation. Our result was in line with the second scenario, suggesting that induction of ID1 expression by R1881 is rather a secondary response (Fig. S4).
Figure 4.
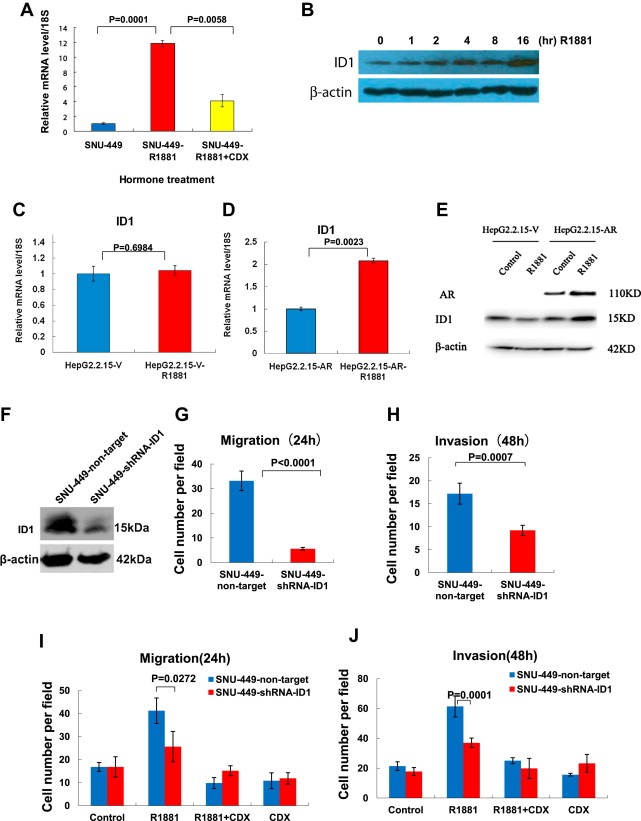
Activation of AR by androgen stimulated expression of the metastasis‐promoting gene ID1, and shRNA knockdown of ID1 attenuated androgen‐stimulated cell migration and invasion. (A) Androgen agonist R1881 stimulated ID1 mRNA expression in SUN‐449 cells. SNU‐449 cells were cultured in 10% charcoal‐stripped serum media for 3 days and then treated with hormone R1881 (10−8 M) for 16 h (SNU‐449‐R1881) followed by RNA isolation and real‐time RT‐PCR analysis of ID1. Vehicle treatment (SNU‐449) served as a control. Androgen antagonist CDX was included in experiments to block androgen activation (SNU‐449‐R1881 + CDX). 18S was used for normalization of mRNA levels. Error bars indicate means ± SDs. (B) R1881 induced ID1 protein levels. Time‐course experiment was performed in a manner similar to (A) above followed by Western blot analysis using indicated antibodies. (C) (D) R1881‐stimulated ID1 mRNA expression depended on the presence of AR. (C) AR‐negative HepG2.2.15 cells infected with the lentiviral‐empty‐vector (HepG2.2.15‐V) were analyzed for ID1 mRNA levels by real time RT‐PCR after treatment with hormone R1881 for 16 h (HepG2.2.15‐V‐R1881). (D) HepG2.2.15 cells infected with the lentiviral‐AR (HepG2.2.15‐AR) were analyzed for ID1 mRNA levels by real time RT‐PCR after treatment with hormone R1881 for 16 h (HepG2.2.15‐AR‐R1881). 18S was used for normalization of mRNA levels. Error bars indicate means ± SDs. P < 0.05 was considered significant. (E) Western blot analysis of AR and ID1 expression in (C) and (D) above. (F) (G) (H) shRNA knockdown of ID1 decreased SNU‐449 cell migration and invasion. (F) Western blot analysis of shRNA knockdown of ID1 in SNU‐449 cells. Lentiviral shRNA for non‐targeting control (SNU‐449‐non‐target) or for ID1 (SNU‐449‐shRNA‐ID1) were used to knockdown ID1 in SNU‐449 cells. Western blot analysis was performed by using the indicated antibodies. (G) shRNA knockdown of ID1 decreased SNU‐449 cell migration. Transwell migration and invasion assays were performed using the SNU‐449 cells in (F). Cell numbers were counted and shown. Error bars indicate means ± SDs. (H) shRNA knockdown of ID1 decreased SNU‐449 cell invasion. Similar experiment as in (G) above except invasion assays was performed. (I) shRNA knockdown of ID1 attenuated androgen‐stimulated cell migration. shRNA non‐targeting control SNU‐449 cells (SNU‐449‐non‐target) or shRNA ID1‐targeting SNU‐449 cells (SNU449‐shRNA‐ID1) were treated R1881 and/or CDX as indicated, followed by cell migration assays. Quantitative cell numbers are shown. Error bars indicate means ± SDs. (J) shRNA knockdown of ID1 attenuated androgen‐stimulated cell invasion. Experiments for cell invasion assays were performed in a fashion similar to those in (I) above. Error bars indicate means ± SDs.
Next, we sought to determine whether ID1 activation by R1881 depends on AR expression. Treatment of AR‐negative HepG2.2.15 control cells with R1881 for 16 h resulted in no change in ID1 mRNA levels; in contrast, treatment of AR‐positive HepG2.2.15 stable cell lines with R1881 (as described above) resulted in significant elevation of the levels of ID1 in both mRNA and protein (Figure 4C, D, E, and S5), thus indicating that ligand induction of ID1 requires AR activation.
3.6. shRNA knockdown of ID1 attenuates androgen‐stimulated cell migration and invasion
To investigate whether ID1 gene activation is critical in androgen‐stimulated cell migration and invasion, we knocked down ID1 expression via shRNA in SNU‐449 cells (Figure 4F). Migration and invasion assays revealed that shRNA knockdown of ID1 reduced the rate of SNU‐449 cell migration and invasion (Figure 4G and H).
We then treated this ID1‐stable‐knockdown cell line with androgen agonist R1881 and performed cell migration and invasion assays. In shRNA non‐targeting control cells, R1881 treatment resulted in significantly elevated cell migration, which was blocked by CDX; in contrast, this ligand‐enhanced cell migration was attenuated markedly in the cells with shRNA knockdown of ID1 (Figure 4I). Similarly, shRNA knockdown of ID1 resulted in attenuation of androgen‐stimulated cell invasion (Figure 4J). Taken together, our data indicated that activation of AR by its ligand enhances the expression of the metastasis‐promoting gene ID1, which, in turn, increases HCC cell migration and invasion and thereby eventually leads to HCC metastasis.
4. Discussion
The sex disparity in HCC incidence is well known, frequently with a male to female ratio of 2:1–4:1. Although sex hormones and receptors attributes to the disparity, the function of androgen and AR in HCC progression and metastasis is still largely unknown. In this study, our data suggested that AR expression and particularly its activation contribute to HCC cell migration and invasion.
In 20 HCC cell lines, AR was expressed at high levels in Group II, which had high metastatic potential (Figure 1A, B and Figure 2A–F); this further links the role of AR expression in promoting HCC progression. In the cell lines in which AR was expressed at high levels and its transcription was active (Fig S6), alpha‐fetoprotein (AFP) was expressed at very low levels (unpublished data). Previous studies have shown that 50–70% of HCC patients produce high levels of AFP (Abelev and Eraiser, 1999), however, high levels of AFP expression alone are not associated with HCC progression and prognosis (Shirabe et al., 1997; Johnson, 1999). Notably, consistent with our results, an HBV‐negative HCC case that was not attributed to alcoholism and had low AFP expression levels was reported to be associated to anabolic androgenic steroid use (Gorayski et al., 2008), thus suggesting a role of male sex hormone signaling in HCC, particularly AFP‐negative HCC, development.
Our experiments using overexpression and shRNA knockdown of AR demonstrated that AR promotes HCC HepG2.2.15 and SNU‐449 cell migration and invasion, respectively (Figure 3A–F). AR knockdown or treatment with casodex decreased SNU‐449 cell migration and invasion, but did not reach the levels of cells not expressing AR, suggesting that, in addition to AR signaling, other cell migration and invasion stimulatory factors or cell signaling may also exist and contribute to the HCC cell migration and invasion. HepG2.2.15 is an HBV‐positive cell line. Previous studies have shown that HBV and AR cooperate to increase the risk of HCC (Chiu et al., 2007; Wang et al., 2009). In HepG2.2.15 cells, ectopic expression of AR and endogenous HBV may enhance each other's activity and promote cell migration and invasion.
To explore the molecular mechanisms of the promotion of HCC cell migration and invasion via activation of AR, we studied androgen‐stimulated gene expression and identified ID1 as a key target gene during this process. ID1 has been demonstrated to inhibit cell differentiation and promote metastasis in multiple cancers (Minn et al., 2005; Lee et al., 2006; Ciarrocchi et al., 2011). Our data indicated that ID1 was an essential metastatic gene in that it mediated androgen‐stimulated HCC cell migration and invasion (Figure 4G–J). More importantly, androgen‐stimulated HCC cell migration and invasion were blocked in the presence of anti‐androgen CDX (Figure 3G and H, Figure 4I and J). These results suggested that targeting AR by inhibiting its activation in a subclass of AR‐positive HCC using antagonists may be a useful strategy for therapeutic intervention of metastatic HCC. Previous randomized clinical trials evaluating androgen deprivation and AR antagonists in HCC treatment did not show beneficial effects (GRETCH, 2004). Our results suggested that such interventions would have a greater benefit in patients pre‐selected based on AR‐positive and AFP/virus‐negative staining.
Collectively, AR activation is likely one of important risk factors for HCC mortality. Further investigation of molecular mechanisms of AR in the metastatic HCC subtypes may have an application in combating HCC‐related death.
Novelty & impact Statements
Androgen receptor (AR) contributes to hepatocellular carcinoma (HCC) incidence, but the role of AR in HCC cell migration and invasion remains largely unknown. In this study, we found that AR activation promoted HCC cell migration and invasion via activation of a metastasis‐promoting gene ID1. An AR antagonist was able to block this process, suggesting that targeting AR in AR‐positive HCC may be as a potential therapeutic intervention strategy.
Financial support
This work was supported by funding from the State Key Laboratory of Oncogenes and Related Genes, the Shanghai Cancer Institute, and National Natural Science Foundation of China (81072163).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Supplementary data
Acknowledgments
We thank Drs. Xianghuo He and Yongzhong Liu for providing the HCC cell lines, Bin He for the pCDH‐AR‐expressing plasmid, and Prof. Hongyang Wang for the HepG2.2.15 cell line.
Supplementary material 1.
Supplementary material associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.molonc.2012.06.005.
Ao Junping, Meng Jiao, Zhu Lei, Nie Huizhen, Yang Chenchen, Li Jinjun, Gu Jianren, Lin Qiushi, Long Weiwen, Dong Xiaoqun and Li Chao, (2012), Activation of androgen receptor induces ID1 and promotes hepatocellular carcinoma cell migration and invasion, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.06.005.
References
- Groupe d'Etude et de Traitement du Carcinome H'epatocellulaire (GRETCH), 2004. Randomized trial of leuprorelin and flutamide in male patients with hepatocellular carcinoma treated with tamoxifen. Hepatology. 40, (6) 1361–1369. [DOI] [PubMed] [Google Scholar]
- Abelev, G.I. , Eraiser, T.L. , 1999. Cellular aspects of alpha-fetoprotein reexpression in tumors. Semin. Cancer Biol.. 9, (2) 95–107. [DOI] [PubMed] [Google Scholar]
- Chiu, C.M. , Yeh, S.H. , 2007. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc. Natl. Acad. Sci. USA. 104, (8) 2571–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarrocchi, A. , Piana, S. , 2011. Inhibitor of DNA binding-1 induces mesenchymal features and promotes invasiveness in thyroid tumour cells. Eur. J. Cancer. 47, (6) 934–945. [DOI] [PubMed] [Google Scholar]
- De Maria, N. , Manno, M. , 2002. Sex hormones and liver cancer. Mol. Cell Endocrinol.. 193, (1–2) 59–63. [DOI] [PubMed] [Google Scholar]
- Desprez, P.Y. , Sumida, T. , 2003. Helix-loop-helix proteins in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia. 8, (2) 225–239. [DOI] [PubMed] [Google Scholar]
- Gomaa, A.I. , Khan, S.A. , 2008. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J. Gastroenterol.. 14, (27) 4300–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorayski, P. , Thompson, C.H. , 2008. Hepatocellular carcinoma associated with recreational anabolic steroid use. Br. J. Sports Med.. 42, (1) 74–75. discussion 75 [DOI] [PubMed] [Google Scholar]
- Heinlein, C.A. , Chang, C. , 2004. Androgen receptor in prostate cancer. Endocr. Rev.. 25, (2) 276–308. [DOI] [PubMed] [Google Scholar]
- Johnson, P.J. , 1999. Role of alpha-fetoprotein in the diagnosis and management of hepatocellular carcinoma. J. Gastroenterol. Hepatol.. 14, (Suppl.) S32–S36. [DOI] [PubMed] [Google Scholar]
- Kalra, M. , Mayes, J. , 2008. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol.. 14, (39) 5945–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.S. , Thorgeirsson, S.S. , 2002. Functional and genomic implications of global gene expression profiles in cell lines from human hepatocellular cancer. Hepatology. 35, (5) 1134–1143. [DOI] [PubMed] [Google Scholar]
- Lee, T.K. , Poon, R.T. , 2006. Regulation of angiogenesis by Id-1 through hypoxia-inducible factor-1alpha-mediated vascular endothelial growth factor up-regulation in hepatocellular carcinoma. Clin. Cancer Res.. 12, (23) 6910–6919. [DOI] [PubMed] [Google Scholar]
- Li, C. , Liang, Y.Y. , 2008. Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol. Cell. 31, (6) 835–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Tuteja, G. , 2012. Foxa1 and foxa2 are essential for sexual dimorphism in liver cancer. Cell. 148, (1–2) 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, W.L. , Hsu, C.L. , 2008. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 135, (3) 947–955. 955 e941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf, D.J. , Thummel, C. , 1995. The nuclear receptor superfamily: the second decade. Cell. 83, (6) 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, N.M. , Abu Dayyeh, B.K. , 2008. Anabolic steroid abuse causing recurrent hepatic adenomas and hemorrhage. World J. Gastroenterol.. 14, (28) 4573–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn, A.J. , Gupta, G.P. , 2005. Genes that mediate breast cancer metastasis to lung. Nature. 436, (7050) 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasue, N. , Yu, L. , 1995. Androgen and oestrogen receptors in hepatocellular carcinoma and surrounding liver parenchyma: impact on intrahepatic recurrence after hepatic resection. Br. J. Surg.. 82, (4) 542–547. [DOI] [PubMed] [Google Scholar]
- Naugler, W.E. , Sakurai, T. , 2007. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 317, (5834) 121–124. [DOI] [PubMed] [Google Scholar]
- Ramachandran, B. , Stell, A. , 2011. Novel insights on imaging sex-hormone-dependent tumourigenesis in vivo. Endocr. Relat. Cancer. 18, (3) R41–R51. [DOI] [PubMed] [Google Scholar]
- Ruzinova, M.B. , Benezra, R. , 2003. Id proteins in development, cell cycle and cancer. Trends Cell Biol.. 13, (8) 410–418. [DOI] [PubMed] [Google Scholar]
- Shang, Y. , Myers, M. , 2002. Formation of the androgen receptor transcription complex. Mol. Cell. 9, (3) 601–610. [DOI] [PubMed] [Google Scholar]
- Shirabe, K. , Takenaka, K. , 1997. Significance of alpha-fetoprotein levels for detection of early recurrence of hepatocellular carcinoma after hepatic resection. J. Surg. Oncol.. 64, (2) 143–146. [DOI] [PubMed] [Google Scholar]
- Socas, L. , Zumbado, M. , 2005. Hepatocellular adenomas associated with anabolic androgenic steroid abuse in bodybuilders: a report of two cases and a review of the literature. Br. J. Sports Med.. 39, (5) e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z.Y. , Ye, S.L. , 2004. A decade's studies on metastasis of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol.. 130, (4) 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejura, S. , Rodgers, G.R. , 1989. Sex-steroid receptors in the diethylnitrosamine model of hepatocarcinogenesis: modifications by gonadal ablation and steroid replacement therapy. J. Mol. Endocrinol.. 3, (3) 229–237. [DOI] [PubMed] [Google Scholar]
- Tsai, M.J. , O'Malley, B.W. , 1994. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem.. 63, 451–486. [DOI] [PubMed] [Google Scholar]
- Wang, S.H. , Yeh, S.H. , 2009. Identification of androgen response elements in the enhancer I of hepatitis B virus: a mechanism for sex disparity in chronic hepatitis B. Hepatology. 50, (5) 1392–1402. [DOI] [PubMed] [Google Scholar]
- Yeh, S.H. , Chen, P.J. , 2010. Gender disparity of hepatocellular carcinoma: the roles of sex hormones. Oncology. 78, (Suppl. 1) 172–179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data