

Abstract
CDC25 (cell division cycle 25) phosphatases are essential for cell cycle control under normal conditions and in response to DNA damage. They are represented by three isoforms, CDC25A, B and C, each of them being submitted to an alternative splicing mechanism. Alternative splicing of many genes is affected in response to genotoxic stress, but the impact of such a stress on CDC25 splicing has never been investigated. In this study, we demonstrate that genotoxic agents (doxorubicin, camptothecin, etoposide and cisplatin), alter the balance between CDC25C splice variants in human breast cancer cell lines both at the mRNA and protein levels. This modulation occurs during the response to moderate, sub‐lethal DNA damage. Our results also suggest that the CDC25C splice variants expression shift induced by a genotoxic stress is dependent on the ATM/ATR signaling but not on p53. This study highlights the modulation of CDC25C alternative splicing as an additional regulatory event involved in cellular response to DNA damage in breast cancer cells.
Keywords: CDC25 phosphatases, Alternative splicing, Genotoxic stress, Breast cancer cells
Highlights
We report that DNA‐damaging agents alter CDC25C splicing in breast cancer cells.
CDC25C splicing modulation occurs during the response to moderate DNA damage.
This modulation is dependent on ATM/ATR but not on p53.
We highlight an additional regulatory event involved in response to DNA damage.
Abbreviations
- ATM
ataxia telangiectasia-mutated
- ATR
ATM- and Rad3-related
- CDC25
cell division cycle 25
- CDK
cyclin-dependent kinase
- CPT
camptothecin
- DAPI
4′,6-diamidino-2-phenylindole
- Dox
doxorubicin
- DSB
double strand breaks
- Eto
etoposide
- hnRNP
heterogeneous ribonucleoproteins
- PARP
poly (ADP-ribose) polymerase
- PFT-α
pifithrin-α
- SR
ser/arg rich protein
1. Introduction
CDC25 (cell division cycle 25) phosphatases play a critical role in the eukaryotic cell cycle progression during normal cell division and checkpoint response following DNA damage (Boutros et al., 2007; Karlsson‐Rosenthal and Millar, 2006). They allow the activation of cyclin/cyclin‐dependent kinases (CDK) complexes by dephosphorylating the Thr14 and Tyr15 residues located in the ATP binding site of CDK. In humans, CDC25 belong to a multigene family consisting of three isoforms: CDC25A, CDC25B and CDC25C. CDC25A appears to be preferentially required during the G1/S transition (Jinno et al., 1994), whereas CDC25B and CDC25C are essentially involved in the entry into mitosis (Lammer et al., 1998; Millar et al., 1991). However, recent evidence suggests that all CDC25 isoforms can regulate both G1/S and G2/M transitions (Boutros et al., 2006). In order to assure a controlled progression through each cell cycle phase and thus maintain genomic integrity, a tight regulation of CDC25 phosphatases activity is needed, both in unperturbed cell cycle and in response to DNA damage checkpoints. This regulation depends upon post‐translational mechanisms such as phosphorylations, sub‐cellular relocalization and proteasome‐mediated degradation (Aressy and Ducommun, 2008), together with p53‐dependent transcriptional repression of the three CDC25 isoforms (Dalvai et al., 2011; Demidova et al., 2009; St Clair et al., 2004).
Overexpression of CDC25A and CDC25B was reported in a wide variety of human malignancies including breast cancer and was commonly associated with both tumor aggressiveness and poor prognosis for the patients (Kristjánsdóttir and Rudolph, 2004). With regard to CDC25C, only a few studies showed an overexpression of this isoform in cancers (Hernández et al., 2001; Ozen and Ittmann, 2005; Wang et al., 2010). However, growing evidence suggests that the overexpression of CDC25C could be underrated because of the non‐consideration of its alternative splicing (Albert et al., 2011; Kristjánsdóttir and Rudolph, 2004).
In fact, all CDC25 genes generate several transcripts from an alternative splicing mechanism: at least two variants were identified so far for CDC25A (Wegener et al., 2000) and five for CDC25B (Baldin et al., 1997; Forrest et al., 1999) and CDC25C (Bureik et al., 2000; Wegener et al., 2000) (Figure 1A). Although the knowledge is so far limited concerning the role of each CDC25 splice variant, it appears that some variants are preferentially up‐regulated in cancers. For instance, CDC25B3 variant was found to be overexpressed in pancreatic cancer (Guo et al., 2004), whereas CDC25B2 variant overexpression is correlated with the grade of differentiation in colorectal cancer (Hernández et al., 2001) and with tumor aggressiveness in non‐Hodgkin's lymphoma (Hernández et al., 2000). Interestingly, this last variant possesses a higher activity in fission yeast (Baldin et al., 1997) and is more stable during mitosis than other CDC25B isoforms (Kieffer et al., 2007). Additionally, CDC25C5 mRNA level is increased in prostate cancer in correlation with prostate‐specific antigen recurrence (Ozen and Ittmann, 2005).
Figure 1.

Modulation of CDC25C splicing by doxorubicin in human breast cancer cell lines. (A) Schematic representation of CDC25A, CDC25B and CDC25C pre‐mRNA and alternatively spliced transcripts sequences. Colored rectangles correspond to alternatively spliced exons. Arrows show the localization of primers used to amplify the different CDC25A, CDC25B and CDC25C variants by semi‐quantitative RT‐PCR (see Materials and Methods for primers sequences). Amplicon length expected for each splice variant appears at the right of the figure. (B) MCF‐7 cells were treated with 1 μM of doxorubicin at the indicated times. RNA was subjected to semi‐quantitative RT‐PCR to detect CDC25A, CDC25B and CDC25C splice variants. PCR products are identified on the left and molecular weight markers are indicated on the right. The β‐actin gene was used as a standardizing control. (C), top, RNA from MCF‐7 cells treated with doxorubicin as indicated were subjected to Real‐Time quantitative RT‐PCR to evaluate the ratio between CDC25C5 and C1 variants. The results are expressed as C5/ C1 (mean ± S.D.) of three independent experiments. 18S rRNA was used as an endogenous reference gene. ** Shows significant difference from control at p < 0.01 (Student's t test); bottom, schematic representation of primers (arrows) and TaqMan® probe (bold line) used to detect C1 and C5 variants. (D) CDC25C protein expression from MCF‐7 cells treated as indicated was examined by immunoblotting. α‐tubulin was used as a loading control. Molecular weights for each protein are indicated on the right. (E) The MCF‐7 multidrug‐resistant counterpart cell line Vcr‐R was treated with 2 μM of doxorubicin for 12 h. CDC25C splicing was studied by semi‐quantitative RT‐PCR as described above.
Alternative pre‐mRNA splicing is an essential mechanism contributing to proteome diversity. Indeed, a recent study revealed that more than 90% of human genes may undergo alternative splicing (Wang et al., 2008a). It is now clear that it contributes with transcriptional regulation to the control of gene expression in various conditions (Chen and Manley, 2009). Interestingly, alteration of splicing events has been reported in a variety of diseases, including cancer (Pajares et al., 2007). In cancer cells, this alteration leads to the apparition of new transcripts or to the modification of the ratio between normally existing splice variants which can potentially play a role in tumor progression. In addition, alternative splicing of many genes is modulated in response to cellular stresses such as oxidative stress, heat shock or hypoxia (Biamonti and Caceres, 2009). Particularly, a growing number of studies highlight the crucial role of alternative splicing in response to genotoxic stress (Busà and Sette, 2010). Of particular interest is the effect of DNA‐damaging agents and irradiations on the splicing of genes involved in cellular processes as essential as apoptosis (caspase‐2, Bcl‐x) (Shkreta et al., 2010; Solier et al., 2004), cell motility (CD44) (Filippov et al., 2007) and cell cycle control (cyclin D1, MDM2 and MDM4) (Chandler et al., 2006; Lents et al., 2008; Wang et al., 2008b).
While the transcriptional and post‐translational regulation of CDC25 in response to DNA damage has been widely investigated, the impact of genotoxic stress on CDC25 phosphatases alternative splicing has never been reported to our knowledge. In this study, we show that various genotoxic agents, namely doxorubicin, camptothecin, etoposide and cisplatin, are able to alter the ratio between CDC25C splice variants in human breast cancer cell lines. This regulation only occurs in condition of mild DNA damage, is associated to a cell cycle arrest, and disappears at the onset of apoptosis. Moreover, part of the signaling pathway mediating CDC25C splicing modulation was elucidated. Our results provide new insights concerning the variety of regulation events involved in cellular response to DNA damage.
2. Materials and methods
2.1. Drugs and materials
Doxorubicin, pifithrin‐α, caffeine, camptothecin, etoposide, cisplatin, vinblastin, propidium iodide and 4′,6‐diamidino‐2‐phenylindole were purchased from Sigma–Aldrich (St Quentin Fallavier, France).
2.2. Cell culture conditions and drug treatment
MCF‐7 (ECACC, Salisbury, UK, 86012803) (Soule et al., 1973), Vcr‐R (Whelan et al., 1992) and MDA‐MB‐231 (ATCC, Manassas, USA, HTB‐26) (Cailleau et al., 1974) cell lines were grown in RPMI‐1640 medium (Eurobio, Les Ulis, France) supplemented with 10% (v/v) fetal bovine serum, 2 mM glutamine and 100 UI/mL penicillin/100 μg/mL streptomycin at 37 °C in a humidified atmosphere containing 5% CO2.
Cells were seeded at a density of 1.106 in 100‐mm Petri dishes and allowed to attach for 24 h. Cells were then exposed to the different drugs as described in each figure legend.
2.3. RNA isolation and cDNA synthesis
Total RNA was extracted using TRI Reagent® (Sigma–Aldrich) following the manufacturer's instructions. For subsequent quantitative RT‐PCR experiments, genomic DNA was eliminated by a RNase‐free DNase I treatment (Qiagen, Courtaboeuf, France). RNA was then quantified and checked for integrity by means of agarose gel electrophoresis. Reverse transcription was performed as described in Albert et al. (2011).
2.4. Semi‐quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) analysis
PCRs were performed using the following primer pairs (see Figure 1A for localization): for CDC25A splice variants (Wegener et al., 2000) Av forward (Fw) 5′‐GGGGGACTGTCGCCTGTCACCAACCT‐3′ and Av reverse (Rv) 5′‐GGGGTCTCCTCCTCATTCTTCAGATTC‐3′; for CDC25B splice variants (Baldin et al., 1997) Bv Fw 5′‐GCTTCCTCGCCGGTCACCAC‐3′ and Bv Rv 5′‐CCTGCGGCTGGCCCACTC‐3′; for CDC25C splice variants (Wegener et al., 2000) Cv Fw 5′‐CTCCTGGAGAGAGACACTTCCTTTAC‐3′ and Cv Rv 5′‐CCACTTCTGCTCACCTTTGCTTCTTG‐3′; and for β‐actin as an internal control Fw 5′‐GGACGACATGGAGAAAATCTGG‐3′ and Rv 5′‐TGGATAGCAACGTACATGGCTG‐3′. Amplification conditions were as described in Albert et al. (2011).
2.5. Real‐time quantitative RT‐PCR
CDC25C1 and C5 transcripts level was measured by Real‐Time RT‐PCR with the StepOne Plus System (Applied Biosystems, Courtaboeuf, France) using the following specific primers and TaqMan® probes (see Figure 1C for localization): C1 Fw 5′‐TGGGGAGATAACTGCCACTCA‐3′; C1 Rv 5′‐AGAAGCTGTGCTGGGCTACATT‐3′; C5 Fw 5′‐AGCATTTTGTCTGGGTCACCTG‐3′; C5 Rv 5′‐ GTTTCCATTGTCATCCCAGCTA‐3′; and the C1/C5 probe 5′‐6FAM‐ATTCTTCAGGACTTCAGGAAGTGCATT‐TAMRA‐3′ for detection of both variants. Quantitative PCR (qPCR) reactions were performed using iTaq Supermix with ROX (Bio‐Rad, Marnes‐la‐Coquette, France) as described in Albert et al. (2011). 18S rRNA (Applied Biosystems) was used as an endogenous reference gene in each assay. CDC25C5/C1 ratio was relatively quantified using the C5/ C1 expression where ΔC T = (C T target gene − C T 18S rRNA). Results were normalized to 1.0 in the non‐treated control.
2.6. Protein extraction and immunoblotting
Cells were lysed in ice‐cold lysis buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 0.1% deoxycholate, 0.5% NP‐40, 10% glycerol, supplemented with protease inhibitors cocktail (Sigma–Aldrich), 1 mM phenylmethylsulfonyl fluoride, 25 mM NaF and 2 mM Na2VO4. Cells were then sonicated (2 × 5 s) and harvested at 12,000 g for 15 min at 4 °C. 50 μg of total proteins were separated on 8–14% SDS‐polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Bio‐Rad). Immunoblotting was carried out following standard procedures using primary antibodies directed against CDC25C (1:200, sc‐327), p53 (1:200, sc‐6243), p21 (1:200, sc‐6246) purchased from Santa Cruz Biotechnology (Heidelberg, Germany), antibody specific of the 85 kDa cleaved‐fragment of PARP (1:1000, 214/215, Invitrogen, Cergy‐Pontoise, France) and anti‐γ‐H2AX (1/2000, 05‐636; Upstate/Millipore, Molsheim, France). The equal loading of the proteins was confirmed by probing with α‐tubulin antibody (Sigma–Aldrich). Immunodetection was performed with horseradish peroxidase‐coupled secondary antibodies and Western Blotting Luminol (Santa Cruz). Densitometry of bands was analyzed using ChemiDoc™ (Bio‐Rad). Relative protein expression in non‐treated control was set to 1.0.
2.7. Immunofluorescence
Cells attached on glass coverslips were washed three times with cold PBS and fixed with 4% formaldehyde in PBS for 15 min at room temperature. Then, cells were permeabilized and blocked with 0.1% Triton X‐100 and 3% bovine serum albumin in PBS for 30 min at room temperature. The mouse anti‐γ‐H2AX primary antibody was diluted 1:500 and incubated overnight at 4 °C prior to incubation with Fluorescein isothiocyanate (FITC)‐conjugated (1:100, Argene, Verniolle, France) or AlexaFluor 594‐conjugated (1:500, Molecular Probes/Invitrogen) anti‐mouse IgG for 1 h in the dark. Cells were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) at 50 μg/mL for 5 min, mounted in PermaFluor mounting medium (Immunon, Pittsburg, PA, USA) and visualized with a Nikon Eclipse 80i fluorescence microscope (Nikon, Champigny‐sur‐Marne, France).
2.8. Cell cycle analysis
Floating and adherent treated cells were harvested by trypsinization, washed twice in PBS and fixed in ice‐cold 70% ethanol at −20 °C. The fixed cells were subsequently stained with a solution containing 50 μg/mL propidium iodide, 0.1 mg/mL RNase I and 0.05% Triton X‐100 for 20 min in the dark and analyzed on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). The percentage of cells in each phase of the cell cycle was determined using ModFit LT software (Verity software, Brush Prairie, WA, USA).
2.9. Statistical analysis
All semi‐quantitative RT‐PCR and immunoblot analyses presented are representatives of at least three independent experiments giving similar results. For quantitative RT‐PCR and cell cycle analyses, data are presented as the mean ± S.D. of three independent experiments. Statistical analysis was performed using the Student's t‐test. Differences between treatment and control were considered as statistically significant for a p value <0.05.
3. Results
3.1. Doxorubicin alters CDC25C alternative splicing in human breast cancer cell lines
To test the hypothesis that a genotoxic stress could modify the alternative splicing pattern of CDC25, we first chose to examine the effect of the DNA damage‐inducing agent doxorubicin (Dox), an anthracycline commonly used in breast cancer chemotherapy (Kaklamani and Gradishar, 2005). The human mammary adenocarcinoma MCF‐7 cells were treated with 1 μM of doxorubicin, a concentration corresponding to the IC50 at 24 h of treatment (concentration of drug reducing the cell growth by 50% compared to the control, determined by an MTT test, data not shown). The levels of CDC25 alternatively spliced transcripts were then analyzed by semi‐quantitative RT‐PCR using primers surrounding the region submitted to alternative splicing and generating amplicons of different length for each CDC25 variant (Figure 1A). These primers recognize all CDC25 splice variants currently known, except CDC25B5. CDC25A and CDC25B splicing profiles were not modified following treatment with Dox, although the global transcription level of CDC25A was decreased as reported previously (Demidova et al., 2009) (Figure 1B). Interestingly, Dox induced an up‐regulation of the shortest CDC25C5 splice variant accompanied by a down‐regulation of the full‐length CDC25C1 variant, this shift in CDC25C splicing profile occurring between 6 and 12 h of treatment (Figure 1B). This splicing modulation is likely due to the Dox treatment and not to an eventual cell cycle modification, as we observed no significant difference in cycle repartition between control and Dox‐treated MCF‐7 cells at 6 h and 12 h and no change in CDC25C splicing at these times under non‐stressing conditions (data not shown). The previous results were then confirmed by Real‐Time quantitative RT‐PCR experiments showing an increase of the CDC25C5/C1 ratio reaching a maximal value of 17‐fold at 24 h post‐treatment (Figure 1C). It is worth noting that concentrations of Dox below 1 μM do not induce CDC25C splicing alteration (data not shown), indicating that we selected the minimum optimal concentration.
Using an antibody directed against a domain constant in all CDC25C variants (C‐terminal domain), we were able to distinguish CDC25C1 and C5 proteins on the basis of their molecular weight differences (Figure 1D). Other CDC25C variants were not detected by immunoblotting, probably due to their too low level of expression. Consistent with mRNA results, doxorubicin also induced a modification of the CDC25C5/C1 ratio at the protein level, with a reduction of CDC25C1 and an increase in the expression of the CDC25C5 isoform. However, this modification appeared from 12 h and was accentuated until 24 h, later than that observed at the transcript level (Figure 1C). This time delay could be explained by the relatively high stability of CDC25C1 protein, which half‐life is about 12 h (Aressy and Ducommun, 2008).
Interestingly, the same increase of CDC25C5/C1 ratio was observed in the MCF‐7‐derived multidrug‐resistant cell line Vcr‐R treated with Dox (Figure 1E), but was also accompanied by a higher expression of CDC25C3 and C4 transcripts in these cells.
3.2. Modulation of CDC25C splicing is involved in the response to moderate DNA damage induced by doxorubicin
In order to decipher the link between CDC25C splicing modulation induced by doxorubicin and the DNA damage pathway, we first chose to compare the effect of a sub‐lethal concentration of Dox (1 μM) to that of a higher concentration of this drug (5 μM). As shown in Figure 2A, CDC25C splicing modulation is substantially less pronounced, both at mRNA and protein levels, for the highest concentration of Dox. We considered the possibility that this observation could be linked to differences in the severity of DNA insult induced by the two concentrations of Dox. To address this hypothesis, we evaluated the phosphorylation of the histone H2AX Ser139 (γ‐H2AX), a selective and sensitive marker of DNA damage, especially of DNA double strand breaks (DSB) (Mah et al., 2010). Immunoblot analysis showed an induction of γ‐H2AX with 5 μM of Dox starting from 12 h of treatment (Figure 2B). To study the possibility of a lower induction of DNA damage with 1 μM of Dox that could not be detected by immunoblotting, we used immunofluorescence detection as a more sensitive method. In this context, the intrinsic fluorescence of Dox had to be taken into account. Control images without anti‐γ‐H2AX antibody incubation showed orange‐brown cells characteristic of the overflow of the red fluorescence of Dox in the green channel used for FITC detection (data not shown). Nevertheless, this fluorescence overlap could be set aside, since the own fluorescence of Dox can be clearly distinguished from that of γ‐H2AX staining (green). Figure 2C shows the presence of discrete γ‐H2AX foci in cells treated with Dox during 12 h, the number of positive cells and the fluorescence intensity increasing with the drug concentration. After 24 h of treatment, cells with punctuated γ‐H2AX foci were still present, but also accompanied by cells with a diffuse staining, which were more numerous with 5 μM of Dox (Figure 2C). Altogether, these observations indicate that the formation of DSB depends upon treatment time and drug concentration.
Figure 2.
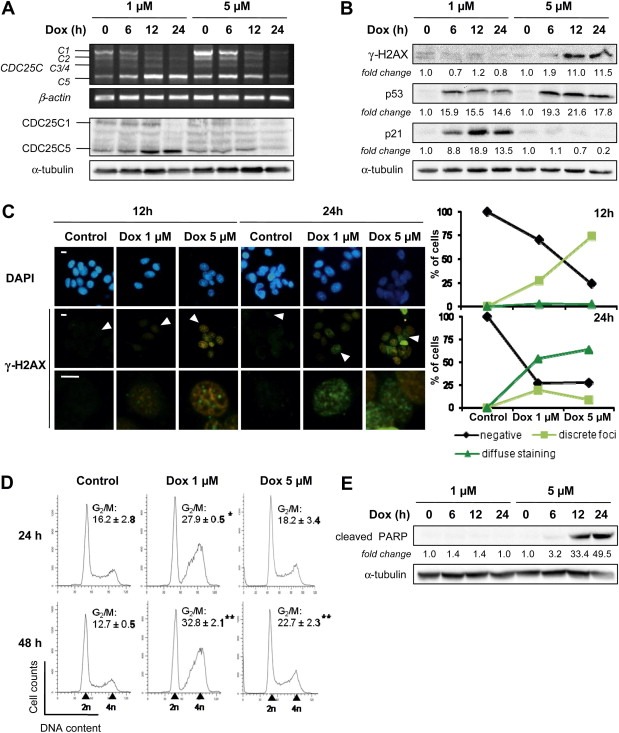
CDC25C splicing modulation induced by doxorubicin is associated with DNA‐damage and cell cycle arrest, independently of apoptosis. MCF‐7 cells were treated with 1 μM and 5 μM of doxorubicin for the indicated times. (A) CDC25C variants expression was evaluated by RT‐PCR (top) and immunoblotting (bottom). β‐actin and α‐tubulin were used as internal controls for the RT‐PCR and immoblotting assays, respectively. (B) Immunoblotting of DNA damage response proteins. The fold‐changes of protein levels between treatment and control, measured by densitometry, are shown beneath the blots. Equal loading was confirmed by α‐tubulin immunoblot. (C) Immunofluorescence analysis of nuclear γ‐H2AX foci formation using anti‐γ‐H2AX specific antibody (green, FITC). Images at the bottom correspond to a magnification of the cell pointed by an arrow in the images above. Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. Graphs on the right represent the count of at least 50 cells that were classified into non‐stained cells (negative), cells with discrete γ‐H2AX foci (discrete foci, at least three foci per cells) and cells with diffuse γ‐H2AX staining (diffuse staining). (D) Cell cycle distribution was monitored by flow cytometry after propidium iodide staining. Shown is the result of a representative FACS experiment and the frequency of cells in G2/M phase. Data are displayed as mean ± S.D. of three independent experiments. * and ** indicate a significant difference between treated and untreated cells with p < 0.05 and p < 0.01, respectively. (E) Analysis of PARP cleavage by immunoblotting. The fold‐changes of protein levels between treatment and control, measured by densitometry, are shown beneath the blots. Equal loading was confirmed by α‐tubulin immunoblot.
Depending on the nature and the intensity of the DNA injury, the DNA damage checkpoint signaling leads either to a cell cycle arrest in order to allow time for the cell to repair its DNA or to the initiation of apoptosis if the repair fails (Jackson and Bartek, 2009). Activation of the checkpoint pathway in MCF‐7 cells was confirmed by the induction of the p53 protein for both concentrations of Dox (Figure 2B). However, the consequence of this activation differs according to the drug concentration. On the one hand, in MCF‐7 cells treated with 1 μM of Dox, the expression of the cell cycle inhibitor p21 is induced (Figure 2B) in association with a blockage in the G2/M phase of the cell cycle (Figure 2D), whereas in presence of 5 μM of Dox, no induction of p21 was observed (Figure 2B), which correlates with a more limited cell cycle arrest (Figure 2D). On the other hand, apoptosis is induced only with the highest concentration of Dox as seen by the cleavage of the poly (ADP‐ribose) polymerase (PARP) (Figure 2E). It is worth noting that concentrations lower than 1 μM of Dox still induce a cell cycle arrest in G2/M phase, along with a p21 induction (data not shown). Therefore, other mechanisms such as topoisomerase II poisoning or DNA intercalation involved in cell cycle arrest may occur before the establishment of CDC25C splicing regulation.
Altogether, these results suggest that the regulation of CDC25C splicing occurs during the response to moderate, sub‐lethal DNA damage induced by doxorubicin.
3.3. Alteration of CDC25C splicing by doxorubicin is dependent on ATM/ATR kinases and independent on p53
Since CDC25C is regulated at the transcriptional level by p53 in response to DNA damage (St Clair et al., 2004) and transcription and splicing are two mechanisms closely connected (Chen and Manley, 2009), we asked whether the regulation of CDC25C splicing required a functional p53. In this context, we treated MCF‐7 cells (with a wild‐type p53) with pifithrin‐α (PFT‐α), a pharmacological inhibitor of p53 transcriptional activity (Komarov et al., 1999). The inhibition of p53 activity was confirmed by the two‐fold decrease of p21 induction, a well‐identified p53 target gene (Figure 3A). This level of p53 inhibition was not enhanced by a higher concentration of PFT‐α (40 μM, data not shown), but still appeared to be sufficient to induce a cell cycle arrest in G1 phase (55.9 ± 3.1% in control and 70.22 ± 1.1% in 20 μM PFT‐α‐treated cells, data not shown). This inhibition as well as the lack of p53 activity in the p53‐mutated breast cancer cell line MDA‐MB‐231 (Bartek et al., 1990) did not prevent the increase of the CDC25C5/C1 ratio after Dox treatment (Figure 3A and C), suggesting that the modulation of CDC25C splicing induced by DNA damage is a p53‐independent mechanism.
Figure 3.
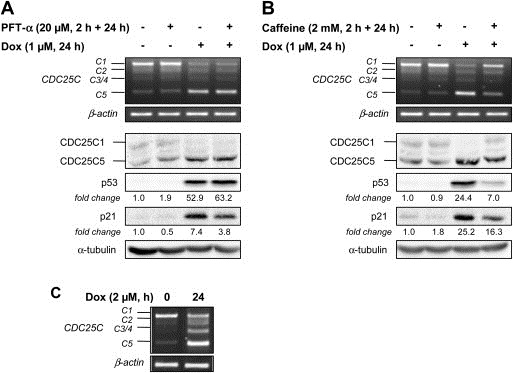
Regulation of CDC25C splicing is dependent on ATM/ATR and independent on p53 pathways. MCF‐7 cells (with wild‐type p53) were pre‐treated with 20 μM of the p53 inhibitor pifithrin‐α (PFT‐α) (A) or with 2 mM of the ATM/ATR kinases inhibitor caffeine (B) for 2 h, followed by a co‐incubation with 1 μM of doxorubicin for 24 h, top, RNA was subjected to RT‐PCR to detect CDC25C splice variants. The β‐actin gene was used as an internal control; bottom, Immunoblots were performed to evaluate CDC25C, p53 and p21 proteins expression. The fold‐changes of protein expression between treatment and control, measured by densitometry, are shown beneath the blots. Staining of the blot with α‐tubulin was used as an internal loading control. (C) MDA‐MB‐231 cells (with mutated‐p53) were treated with 2 μM of doxorubicin for 24 h and CDC25C splice variants expression was assessed by RT‐PCR. The β‐actin gene was used as an internal control.
Upstream of p53 in the checkpoint signaling pathway, ATM and ATR kinases play a crucial role in the response to DNA damage. They are known in particular to activate CHK1 and CHK2 kinases, responsible for CDC25A proteasome‐mediated degradation and CDC25B and CDC25C cytoplasmic sequestration (Karlsson‐Rosenthal and Millar, 2006). Therefore, we asked whether ATM/ATR kinases were also able to affect CDC25C splicing following a treatment with Dox. For this purpose, MCF‐7 cells were treated with caffeine, a well‐known inhibitor of both ATM and ATR kinases activities (Sarkaria et al., 1999). To ensure that caffeine effectively inhibited ATM/ATR signaling pathway, we performed immunoblot analyses and observed the expected decrease of p53 and p21 induction after Dox treatment (Figure 3B). Interestingly, the changes in CDC25C alternative splicing induced by Dox were suppressed in the presence of caffeine (at both RNA and protein levels), indicating that this regulatory process could be dependent on the ATM/ATR signaling pathway. However, the involvement of other pathways cannot be excluded since caffeine is not a specific inhibitor of ATM and ATR.
3.4. Modulation of CDC25C alternative splicing by other DNA‐damaging agents
To confirm the association between CDC25C splicing modulation and the DNA damage response pathway, MCF‐7 cells were also treated with other genotoxic agents, all of them used at a concentration corresponding to the IC50 for 24 h of treatment (determined by an MTT test, data not shown). Camptothecin (CPT) and etoposide (Eto), which respectively inhibit DNA topoisomerase I and II, induce the formation of DNA breaks as seen by γ‐H2AX staining and activation of the p53/p21 checkpoint pathway (Figure 4A and B). Moreover, these drugs provoked a comparable increase of CDC25C5/C1 ratio as observed with Dox (Figure 4A and B). A similar effect on CDC25C alternative splicing was obtained with the chemotherapeutic drug cisplatin, known to react with nucleophilic sites in DNA forming monoadducts as well as intra‐ and interstrand crosslinks (Figure 4C). Conversely, CDC25C splicing was not affected by the anti‐microtubule drug vinblastin which, as expected, did not affect DNA integrity (Figure 4C).
Figure 4.
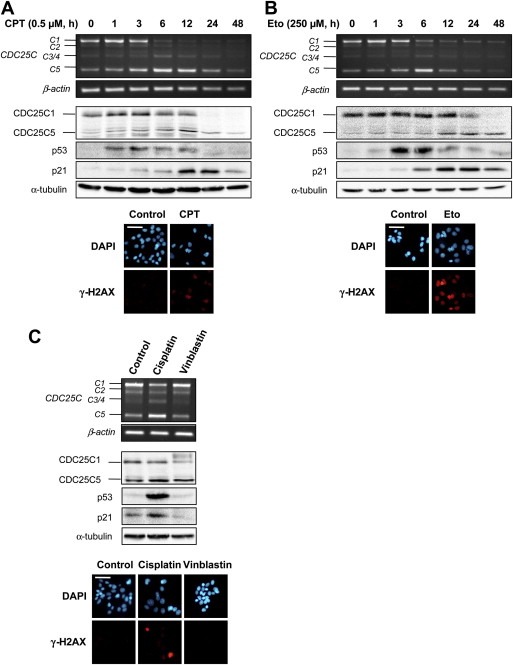
Modulation of CDC25C alternative splicing in response to other DNA‐damaging agents in MCF‐7 cells. Cells were treated with the topoisomerase I inhibitor camptothecin (CPT, 0.5 μM) (A) and the topoisomerase II inhibitor etoposide (Eto, 250 μM) (B) for the indicated times. Cells were also treated with the chemotherapeutic agents cisplatin (50 μM) and vinblastin (1 μM) for 12 h (C). Top, RNA was subjected to semi‐quantitative RT‐PCR to detect CDC25C splice variants. The β‐actin gene was used as a standardizing control; middle, Cell lysates were subjected to immunoblotting. Equal protein loading was confirmed by α‐tubulin immunoblot; bottom, cells were treated with the different compounds for 24 h and immunostained with anti‐γ‐H2AX specific antibodies (red, AlexaFluor 594). Nuclei were counterstained with DAPI (blue). Scale bars, 50 μm.
Finally, it is worth noting that all these compounds, as previously indicated in the case of Dox, did not induce a modification of CDC25A and CDC25B alternative splicing (data not shown).
4. Discussion
CDC25 phosphatases are key players of cell cycle progression. Moreover, they are tightly regulated in response to DNA damage, both by p53‐mediated transcriptional repression of the three CDC25 isoforms (Dalvai et al., 2011; Demidova et al., 2009; St Clair et al., 2004) and by post‐translational processes involving CDC25A proteasome‐dependent degradation and cytoplasmic sequestration of CDC25B and CDC25C through interaction with 14‐3‐3 proteins (Karlsson‐Rosenthal and Millar, 2006). In addition to these regulation events, we provide here the first evidence for the alteration in CDC25C alternative splicing under conditions of genotoxic stress. Indeed, we showed that CDC25C5 splice variant is up‐regulated to the detriment of CDC25C1, both at mRNA and protein levels, following treatment of MCF‐7 mammary adenocarcinoma cell line with IC50 concentrations of various DNA‐damaging agents related to cancer therapy such as the DNA topoisomerase inhibitors doxorubicin, camptothecin and etoposide, and the DNA crosslinker cisplatin. Our results indicate that this regulation is associated to double‐strand breaks as all compounds affecting CDC25C alternative splicing also induce the formation of γ‐H2AX foci. Conversely, CDC25A and CDC25B alternative splicing is not affected in these conditions, suggesting that the pathways involved in the regulation of the three CDC25 isoforms splicing are distinct. A growing number of recent studies showed the existence of a link between the DNA damage response and the modification of alternative splicing of several genes involved, among others, in cell cycle control such as cyclin D1, MDM2 and MDM4 (Chandler et al., 2006; Lents et al., 2008; Markey and Berberich, 2008; Wang et al., 2008b) and in apoptosis such as caspase‐2, Bcl‐x and Fas (Filippov et al., 2008; Shkreta et al., 2010; Solier et al., 2004), suggesting that splicing associated to cell proliferation and death may be disrupted in cells subjected to a genotoxic stress.
Despite an increasing interest in the alternative splicing regulation following DNA damage, the mechanisms underlying this process are yet poorly understood. In order to start to understand the molecular mechanisms engaged in CDC25C splicing modulation, we treated MCF‐7 cells with the p53 transcriptional activity inhibitor pifithrin‐α and used the MDA‐MB‐231 breast cancer cell line with a mutated‐p53 to evaluate the contribution of p53 in CDC25C splicing modifications induced by doxorubicin. Since a CDC25C splice variants expression shift was still observed in these conditions, we concluded that this event is controlled independently of p53, unlike CDC25C transcriptional repression (St Clair et al., 2004). Additionally, we inferred that the shift in CDC25C splicing induced by doxorubicin might be dependent on ATM and ATR kinase activities since turning off these pathways with caffeine blocked this process. Several studies showed that cancerous cells are able to use different ways to modify alternative pre‐mRNA splicing in order to deal with DNA injuries. It has been proposed that genotoxic stress can decrease the rate of transcription via RNA polymerase II phosphorylation (Muñoz et al., 2009) and can also alter the communication between transcription and splicing machineries (Dutertre et al., 2010), both events resulting in increased exon skipping. Moreover, recent evidence suggests that DNA damage‐inducing agents can affect the chromatin structure through acetylation, thereby making some domains more accessible for the splicing machinery (Busà and Sette, 2010). Finally, the activation of signaling pathways in response to various stress conditions may lead to phosphorylation of splicing factors such as hnRNP (heterogeneous ribonucleoproteins) and SR (ser/arg rich) proteins, thereby modifying their activity, sub‐cellular localization or interaction with proteins (Biamonti and Caceres, 2009). Few studies have identified the nature of splicing factors able to trigger the alternative splicing of particular genes following DNA damage. For instance, Filippov et al. (2007, 2008) showed that treatment with the genotoxic agent mitomycin C induces the expression of the SRp55 splicing factor in association with a shift in the splicing pattern of genes encoding the CD44 receptor, the mda7/IL24 cytokine and the KSR1 and ZAK kinases. The splicing factors eventually implicated in CDC25C splicing regulation in response to genotoxic stress remain to be identified. However, according to our results, they might be downstream effectors of the ATM/ATR pathway. This assumption is in agreement with, a large‐scale proteomic screen which identified numerous splicing factors as ATM/ATR phosphorylation targets (Matsuoka et al., 2007).
At this stage, the consequence of CDC25C splicing disruption on cell survival following treatment with genotoxic agents can only be speculated. In this study, we showed that the modification of CDC25C splicing pattern appears to occur in response to moderate DNA damage. Indeed, treatment of MCF‐7 cells with a sub‐lethal concentration of doxorubicin (1 μM), inducing mild DNA damage as observed by the weak γ‐H2AX staining, induced an increase of CDC25C5/C1 ratio in association with a cell cycle arrest. This effect was less pronounced with a strong genotoxic, pro‐apoptotic concentration of doxorubicin (5 μM) inducing PARP cleavage, suggesting that the regulation of CDC25C splicing occurs prior to the onset of apoptosis. While the main response to cytotoxic drugs consists in the activation of death pathways, this effect can be attenuated by the simultaneous induction of protective mechanisms. For instance, splicing modulation of caspase‐2 pre‐mRNA in response to topoisomerases inhibitors leads to the up‐regulation of the anti‐apoptotic short caspase‐2 transcript (Solier et al., 2004) and the modification of Fas receptor splicing profile induced by mitomycin C results in an enrichment of the anti‐apoptotic soluble Fas isoforme (Filippov et al., 2008). These observations together with our results reasonably suggest that the increase of CDC25C5/C1 ratio could help the cells to withstand apoptosis induced by a moderate genotoxic stress. This assumption is reinforced by the existence of the same CDC25C splicing modulation induced by doxorubicin in the multidrug‐resistant Vcr‐R cell line, although CDC25C3 and C4 transcripts were also overexpressed and could play a role still unclear in DNA damage response in these cells. Additionally, we previously showed that CDC25C5/C1 transcripts ratio was higher in the drug‐resistant Vcr‐R and Adr‐R cells compared to their sensitive counterpart MCF‐7 under non‐stressing conditions (Albert et al., 2011). Coincidentally, overexpression of CDC25C5 splice variant has been reported in prostate cancer and correlates with biochemical (prostate‐specific antigen) recurrence (Ozen and Ittmann, 2005). It was also observed that this alternative spliced variant increases uncoupling of mitosis to DNA replication in Schizosaccharomyces pombe compared to CDC25C1 full‐length variant (Bureik et al., 2000). The particular features of CDC25C5 could be linked to the absence of the KEN box, a motif responsible for CDC25 ubiquitin‐mediated degradation, which could confer a higher stability to this protein (Chen et al., 2002), and to the apparition of a new putative CDK phosphorylation site at Ser66. Intriguingly, Varmeh and Manfredi (2008) have shown that adenovirus‐mediated overexpression of CDC25C sensitizes cells to doxorubicin‐induced apoptosis. However, this study was most likely performed with the CDC25C1 isoform. It will be of interest to similarly evaluate the effect of the overexpression of the CDC25C5 isoform, considering that CDC25C1 and CDC25C5 variants may have distinct or even antagonistic functions.
5. Conclusion
This study highlights a higher level of complexity than previously found in the regulation of CDC25C gene expression in response to DNA damage, namely the modulation of its alternative splicing, in addition to its well‐known transcriptional and post‐translational regulations. Further studies are needed to elucidate the exact mechanism by which CDC25C alternative splicing is modulated and the significance of this event in relation to cellular resistance towards genotoxic stress.
Authorship contributions
All authors participated in the conception of this study and in the interpretation of data. Hélène Albert performed all experiments and wrote the first draft of the manuscript. Other authors contributed to the final manuscript.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgment
This work was supported in part by the Ligue Contre le Cancer (committee 57). H.A. is recipient of a fellowship from the French Ministry of Research. We gratefully acknowledge Susana Santos for helpful discussion.
Albert Hélène, Battaglia Eric, Monteiro Carolino and Bagrel Denyse, (2012), Genotoxic stress modulates CDC25C phosphatase alternative splicing in human breast cancer cell lines, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.06.003.
Contributor Information
Hélène Albert, Email: helene.albert@univ-lorraine.fr.
Eric Battaglia, Email: eric.battaglia@univ-lorraine.fr.
Carolino Monteiro, Email: cm@ff.ul.pt.
Denyse Bagrel, Email: denyse.bagrel@univ-lorraine.fr.
References
- Albert, H. , Santos, S. , Battaglia, E. , Brito, M. , Monteiro, C. , Bagrel, D. , 2011. Differential expression of CDC25 phosphatases splice variants in human breast cancer cells. Clin. Chem. Lab. Med.. 49, 1707–1714. [DOI] [PubMed] [Google Scholar]
- Aressy, B. , Ducommun, B. , 2008. Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med. Chem.. 8, 818–824. [DOI] [PubMed] [Google Scholar]
- Baldin, V. , Cans, C. , Superti-Furga, G. , Ducommun, B. , 1997. Alternative splicing of the human CDC25B tyrosine phosphatase. Possible implications for growth control?. Oncogene. 14, 2485–2495. [DOI] [PubMed] [Google Scholar]
- Bartek, J. , Iggo, R. , Gannon, J. , Lane, D.P. , 1990. Genetic and immochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene. 5, 893–899. [PubMed] [Google Scholar]
- Biamonti, G. , Caceres, J.F. , 2009. Cellular stress and RNA splicing. Trends Biochem. Sci.. 34, 146–153. [DOI] [PubMed] [Google Scholar]
- Boutros, R. , Dozier, C. , Ducommun, B. , 2006. The when and wheres of CDC25 phosphatases. Curr. Opin. Cell. Biol.. 18, 185–191. [DOI] [PubMed] [Google Scholar]
- Boutros, R. , Lobjois, V. , Ducommun, B. , 2007. CDC25 phosphatases in cancer cells: key players? Good targets?. Nat. Rev. Cancer. 7, 495–507. [DOI] [PubMed] [Google Scholar]
- Bureik, M. , Rief, N. , Drescher, R. , Jungbluth, A. , Montenarh, M. , Wagner, P. , 2000. An additional transcript of the cdc25C gene from A431 cells encodes a functional protein. Int. J. Oncol.. 17, 1251–1258. [DOI] [PubMed] [Google Scholar]
- Busà, R. , Sette, C. , 2010. An emerging role for nuclear RNA-mediated responses to genotoxic stress. RNA Biol.. 7, 390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailleau, R. , Young, R. , Olivé, M. , Reeves, W.J.J. , 1974. Breast tumor cell lines from pleural effusions. J. Natl. Cancer I. 53, 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler, D.S. , Singh, R.K. , Caldwell, L.C. , Bitler, J.L. , Lozano, G. , 2006. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res.. 66, 9502–9508. [DOI] [PubMed] [Google Scholar]
- Chen, M. , Manley, J.L. , 2009. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell. Biol.. 10, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. , Zhang, Z. , Bower, J. , Lu, Y. , Leonard, S.S. , Ding, M. , Castranova, V. , Piwnica-Worms, H. , Shi, X. , 2002. Arsenite-induced Cdc25C degradation is through the KEN-box and ubiquitin–proteasome pathway. Proc. Natl. Acad. Sci. U.S.A.. 99, 1990–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvai, M. , Mondesert, O. , Bourdon, J.-C. , Ducommun, B. , Dozier, C. , 2011. Cdc25B is negatively regulated by p53 through Sp1 and NF-Y transcription factors. Oncogene. 30, 2282–2288. [DOI] [PubMed] [Google Scholar]
- Demidova, A.R. , Aau, M.Y. , Zhuang, L. , Yu, Q. , 2009. Dual regulation of Cdc25A by Chk1 and p53-ATF3 in DNA replication checkpoint control. J. Biol. Chem.. 284, 4132–4139. [DOI] [PubMed] [Google Scholar]
- Dutertre, M. , Sanchez, G. , De Cian, M.C. , Barbier, J. , Dardenne, E. , Gratadou, L. , Dujardin, G. , Le Jossic-Corcos, C. , Corcos, L. , Auboeuf, D. , 2010. Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct. Mol. Biol.. 17, 1358–1366. [DOI] [PubMed] [Google Scholar]
- Filippov, V. , Filippova, M. , Duerksen-Hughes, P.J. , 2007. The early response to DNA damage can lead to activation of alternative splicing activity resulting in CD44 splice pattern changes. Cancer Res.. 67, 7621–7630. [DOI] [PubMed] [Google Scholar]
- Filippov, V. , Schmidt, E.L. , Filippova, M. , Duerksen-Hughes, P.J. , 2008. Splicing and splice factor SRp55 participate in the response to DNA damage by changing isoform ratios of target genes. Gene. 420, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest, A.R.R. , McCormack, A.K. , DeSouza, C.P.C. , Sinnamon, J.M. , Tonks, I.D. , Hayward, N.K. , Ellem, K.A.O. , Gabrielli, B.G. , 1999. Multiple splicing variants of cdc25B regulate G2/M progression. Biochem. Biophys. Res. Commun.. 260, 510–515. [DOI] [PubMed] [Google Scholar]
- Guo, J. , Kleeff, J. , Li, J. , Ding, J. , Hammer, J. , Zhao, Y. , Giese, T. , Korc, M. , Büchler, M.W. , Friess, H. , 2004. Expression and functional significance of CDC25B in human pancreatic ductal adenocarcinoma. Oncogene. 23, 71–81. [DOI] [PubMed] [Google Scholar]
- Hernández, S. , Bessa, X. , Beà, S. , Hernández, L. , Nadal, A. , Mallofré, C. , Muntane, J. , Castells, A. , Fernández, P.L. , Cardesa, A. , Campo, E. , 2001. Differential expression of cdc25 cell-cycle activating phosphatases in human colorectal carcinoma. Lab. Invest.. 81, 465–473. [DOI] [PubMed] [Google Scholar]
- Hernández, S. , Hernández, L. , Beà, S. , Pinyol, M. , Nayach, I. , Bellosillo, B. , Nadal, A. , Ferrer, A. , Fernández, P.L. , Montserrat, E. , Cardesa, A. , Campo, E. , 2000. Cdc25A and the splicing variant cdc25B2, but not cdc25B1, -B3 or –C, are over-expressed in aggressive human non-Hodgkin's lymphomas. Int. J. Cancer (Pred. Oncol.). 89, 148–152. [PubMed] [Google Scholar]
- Jackson, S.P. , Bartek, J. , 2009. The DNA-damage response in human biology and disease. Nature. 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno, S. , Suto, K. , Nagata, A. , Igarashi, M. , Kanaoka, Y. , Nojima, H. , Okayama, H. , 1994. CDC25A is a novel phosphatase functioning early in the cell cycle. EMBO J.. 13, 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaklamani, V.G. , Gradishar, W.J. , 2005. Adjuvant therapy of breast cancer. Cancer Invest.. 23, 548–560. [DOI] [PubMed] [Google Scholar]
- Karlsson-Rosenthal, C. , Millar, J.B.A. , 2006. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell. Biol.. 16, 285–292. [DOI] [PubMed] [Google Scholar]
- Kieffer, I. , Lorenzo, C. , Dozier, C. , Schmitt, E. , Ducommun, B. , 2007. Differential mitotic degradation of the CDC25B phosphatase variants. Oncogene. 26, 7847–7858. [DOI] [PubMed] [Google Scholar]
- Komarov, P.G. , Komarova, E.A. , Kondratov, R.V. , Christov-Tselkov, K. , Coon, J.S. , Chernov, M.V. , Gudkov, A.V. , 1999. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 285, 1733–1737. [DOI] [PubMed] [Google Scholar]
- Kristjánsdóttir, K. , Rudolph, J. , 2004. Cdc25 phosphatases and cancer. Chem. Biol.. 11, 1043–1051. [DOI] [PubMed] [Google Scholar]
- Lammer, C. , Wagerer, S. , Saffrich, R. , Mertens, D. , Ansorge, W. , Hoffmann, I. , 1998. The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J. Cell. Sci.. 111, 2445–2453. [DOI] [PubMed] [Google Scholar]
- Lents, N.H. , Wheeler, L.W. , Baldassare, J.J. , Dynlacht, B.D. , 2008. Identification and characterization of a novel Mdm2 splice variant acutely induced by the chemotherapeutic agents adriamycin and actinomycin D. Cell Cycle. 7, 1580–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, L.-J. , El-Osta, A. , Karagiannis, T.C. , 2010. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 24, 679–686. [DOI] [PubMed] [Google Scholar]
- Markey, M. , Berberich, S.J. , 2008. hdmX transcripts decrease following genotoxic stress. Oncogene. 27, 6657–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, S. , Ballif, B.A. , Smogorzewska, A. , McDonald, E.R. , Hurov, K.E. , Luo, J. , Bakalarsk, I.C.E. , Zhao, Z. , Solimini, N. , Lerenthal, Y. , Shiloh, Y. , Gygi, S.P. , Elledge, S.J. , 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- Millar, J.B.A. , Blevitt, J. , Gerace, L. , Sadhu, K. , Featherstone, C. , Russell, P. , 1991. p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc. Natl. Acad. Sci. U.S.A.. 88, 10500–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz, M.J. , Pérez, S.M.S. , Paronetto, M.P. , de la Mata, M. , Pelisch, F. , Boireau, S. , Glover-Cutter, K. , Ben-Dov, C. , Blaustein, M. , Lozano, J.J. , Bird, G. , Bentley, D. , Bertrand, E. , Kornblihtt, A.R. , 2009. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 137, 708–720. [DOI] [PubMed] [Google Scholar]
- Ozen, M. , Ittmann, M. , 2005. Increased expression and activity of CDC25C phosphatase and an alternatively spliced variant in prostate cancer. Clin. Cancer Res.. 11, 4701–4706. [DOI] [PubMed] [Google Scholar]
- Pajares, M.J. , Ezponda, T. , Catena, R. , Calvo, A. , Pio, R. , Montuenga, L.M. , 2007. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol.. 8, 349–357. [DOI] [PubMed] [Google Scholar]
- Sarkaria, J.N. , Busby, E.C. , Tibbetts, R.S. , Roos, P. , Taya, Y. , Karnitz, L.M. , Abraham, R.T. , 1999. Inhibition of ATM and ATR kinase activities by the radiosansitizing agent, caffeine. Cancer Res.. 59, 4375–4382. [PubMed] [Google Scholar]
- Shkreta, L. , Michelle, L. , Toutant, J. , Tremblay, M.L. , Chabot, B. , 2010. The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x. J. Biol. Chem.. 286, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solier, S. , Lansiaux, A. , Logette, E. , Wu, J. , Soret, J. , Tazi, J. , Bailly, C. , Desoche, L. , Solary, E. , Corcos, L. , 2004. Topoisomerase I and II inhibitors control caspase-2 pre-messenger RNA splicing in human cells. Mol. Cancer Res.. 2, 53–61. [PubMed] [Google Scholar]
- Soule, H.D. , Vasquez, J. , Long, A. , Albert, S. , Brennan, M.A. , 1973. Human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer I. 51, 1409–1416. [DOI] [PubMed] [Google Scholar]
- St. Clair, S. , Giono, L. , Varmeh-Ziaie, S. , Resnick-Silverman, L. , Liu, W. , Padi, A. , Dastidar, J. , DaCosta, A. , Mattia, M. , Manfredi, J.J. , 2004. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol. Cell.. 16, 725–736. [DOI] [PubMed] [Google Scholar]
- Varmeh, S. , Manfredi, J.J. , 2008. Overexpression of the dual specificity phosphatase, Cdc25C, confers sensitivity on tumor cells to doxorubicin-induced cell death. Mol. Cancer Ther.. 7, 3789–3799. [DOI] [PubMed] [Google Scholar]
- Wang, E.T. , Sandberg, R. , Luo, S. , Khrebtukova, I. , Zhang, L. , Mayr, C. , Kingsmore, S.F. , Schroth, G.P. , Burge, C.B. , 2008. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Dean, J.L. , Millar, E.K.A. , Tran, T.H. , McNeil, C.M. , Burd, C.J. , Henshall, S.M. , Utama, F.E. , Witkiewicz, A. , Rui, H. , Sutherland, R.L. , Knudsen, K.E. , Knudsen, E.S. , 2008. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res.. 68, 5628–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Trope, C.G. , Florenes, V.A. , Suo, Z. , Nesland, J.M. , Holm, R. , 2010. Overexpression of CDC25B, CDC25C and phospho-CDC25C (Ser216) in vulvar squamous cell carcinomas are associated with malignant features and aggressive cancer phenotypes. BMC Cancer. 10, 233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener, S. , Hampe, W. , Herrmann, D. , Schaller, H.C. , 2000. Alternative splicing in the regulatory region of the human phosphatases CDC25A and CDC25C. Eur. J. Cell. Biol.. 79, 810–815. [DOI] [PubMed] [Google Scholar]
- Whelan, R.D. , Waring, C.J. , Wolf, C.R. , Hayes, J.D. , Hosking, L.K. , Hill, B.T. , 1992. Overexpression of p-glycoprotein and glutathione S-transferase pi in MCF-7 cells selected for vincristine resistance in vitro. Int. J. Cancer. 52, 241–246. [DOI] [PubMed] [Google Scholar]