

Abstract
Resistance to chemotherapy is a serious problem for the successful treatment of ovarian cancer patients but signalling pathways that contribute to this chemoinsensitivity are largely unknown. We demonstrate that the chemotherapeutic drug doxorubicin induces activation of the HER3‐PI3K‐AKT signalling cascade in ovarian cancer cells. We further show that the induction of this anti‐apoptotic signalling pathway is based on upregulated expression of HER3 ligands, their shedding by the metalloprotease ADAM17, and is dependent on the HER2 receptor. The doxorubicin‐mediated activation of this important survival cascade can be blocked by the kinase inhibitors lapatinib or erlotinib as well as by the therapeutic monoclonal antibody trastuzumab. Inhibition of the doxorubicin‐induced activation of HER3‐PI3K‐AKT signalling significantly increased apoptosis of ovarian cancer cells. Besides doxorubicin, treatment of cells with cisplatin resulted in activation of the HER3 receptor whereas other chemotherapeutics did not show this effect. The increase in HER3 phosphorylation was detected in well‐established ovarian cancer cell lines which originate from patients previously treated with these chemotherapeutic drugs. Based on these results, we postulate that activation of the HER3‐PI3K‐AKT cascade represents a major mechanism of chemoresistance in ovarian cancer.
Keywords: HER3, Doxorubicin, Ovarian cancer, Apoptosis, Chemotherapy
Highlights
Doxorubicin induces activation of HER3‐PI3K‐AKT signalling in ovarian cancer cells.
Inhibition of doxorubicin‐induced activation of HER3 signalling increases apoptosis.
HER3 signalling is effectively blocked by lapatinib, erlotinib, and trastuzumab.
Besides doxorubicin, treatment of cells with cisplatin results in activation of HER3.
Potential new treatment strategy for recurrent ovarian cancer.
1. Introduction
Despite the development of new anti‐cancer therapeutics like tyrosine kinase inhibitors (TKI) and therapeutic monoclonal antibodies, classical chemotherapy still represents the standard treatment for most cancers in the clinic. Although initially effective, chemotherapeutic drug resistance often occurs and the management of multi‐drug resistant and recurrent or refractory tumours represents a difficult problem for clinical oncologists. In this regard, ovarian cancer represents a paramount example. Although first‐line response rates are about 80%, most patients relapse and the 5‐year survival rate of woman with advanced disease is only 30% (Agarwal and Kaye, 2003). Intensive research for better therapeutic treatment strategies administered after standard‐of‐care chemotherapy in different combinations and concentrations, varying conditions of radiotherapy, immunotherapy or biological therapy did not result in a significant survival advantage of ovarian cancer patients (Hennessy et al., 2009).
One mechanism mediating drug resistance relies on cellular changes like an increased repair of DNA damage, alterations in cell cycle and/or reduction of apoptosis by the activation of anti‐apoptotic pathways (McCubrey et al., 2008). In this regard, activation of the PI3K‐AKT pathway is an important requirement of cancer cells to escape cell death upon exposure to toxic stimuli. In esophageal squamous cell carcinoma, phosphorylation of AKT was significantly higher among patients who received chemotherapy and this increase was associated with poor prognosis (Yoshioka et al., 2008). In cervical cancer, significantly increased levels of phosphorylated AKT were detected in a radiation‐resistant versus a radiation‐sensitive group (Kim et al., 2006). Furthermore, the involvement of an activated PI3K‐AKT signalling cascade promoting resistance against several chemotherapeutic drugs was shown in various cell culture model systems (Li et al., 2005; Liu et al., 2007; Winograd‐Katz and Levitzki, 2006; Yu et al., 2008).
The HER3 (ErbB3) receptor is the third member of the EGF receptor family (Kraus et al., 1989; Plowman et al., 1990). The cytoplasmic part of the HER3 receptor possesses six potential PI3K binding sites and represents the preferred dimerisation partner when signalling occurs via the PI3K‐AKT pathway (Schulze et al., 2005). Moreover, the fundamental role of the HER3 receptor as an upstream activator of this pathway as well as its PI3K‐AKT and FOXO dependent feedback mechanisms, mediating insensitivity to TKI treatment, were recently elucidated (Chakrabarty et al., 2011; Chandarlapaty et al., 2011; Engelman et al., 2007; Garrett et al., 2011; Sergina et al., 2007). Dimerisation, preferentially with HER2, can be initiated upon ligand binding to the extracellular domain of the receptor which subsequently results in activation of downstream signalling pathways. The HER2/HER3 heterodimer reflects the most potent mitogenic signalling complex in the HER network despite the fact that both partners are incapable of signalling on their own (Pinkas‐Kramarski et al., 1996; Wallasch et al., 1995). HER3 and HER2 overexpression was associated in breast cancer and it was shown that HER3 cooperates with HER2 to effectively transform NIH 3T3 cells (Alimandi et al., 1995; Witton et al., 2003). Moreover, expression of the HER3 receptor correlated with reduced patient survival in malignant melanoma and formation of metastases (Reschke et al., 2008). Furthermore, HER3 was connected to decreased survival of patients with cancer of the ovary and in colorectal cancer an inverse correlation of high HER3 expression levels and shorter patient survival times was reported quite recently (Beji et al., 2012; Tanner et al., 2006).
In the present study we highlight a novel chemotherapy‐induced activation of the important HER3‐PI3K‐AKT anti‐apoptotic signalling pathway, its role in promoting drug resistance in ovarian cancer cells, and point out potential intervention strategies.
2. Materials and methods
2.1. Cell culture techniques
Cell lines were grown in a humidified 93% air, 7% CO2 incubator at 37 °C and routinely assayed for mycoplasma contamination. The human ovarian cancer cell lines OVCAR‐3, 2780, 2774, and SKOV‐6 were maintained in MEM media supplemented with 10% fetal bovine serum (FBS) and non essential amino acids. Human OVCAR‐4, OVCAR‐5, OVCAR‐8, and SKOV‐8 ovarian cancer cell lines were cultured in RPMI media supplemented with 10% FBS. The SKOV‐3 cell line was maintained in McCoy's media with 10% FBS.
2.2. RNA interference
Transfection of 21‐nucleotide siRNA duplexes (Ambion) was carried out using OligofectAMINE (Invitrogen) or Lipofectamine RNAiMAX (Invitrogen) and OPTI‐MEM media (GIBCO) without FBS. After 4 h media was changed to normal media containing FBS, and after additional 24 h cells were used for further experiments.
2.3. Flow cytometry (propidium‐iodide assay)
Cells were seeded at a density of 2×104 or 3×104 cells/well into 12‐well plates. The next day, media was changed and cells were treated. After 72 h of cultivation in the presence of inhibitors and FBS, the supernatant was collected and combined with the trypsinised cells, centrifuged, and incubated with 0.1% Triton, 0.1% sodium citrate and 0.02 mM propidium‐iodide (Sigma) in the dark at 4 °C. After 2 h, cells were analysed by flow cytometry (FACS‐Calibur, BD Bioscience) using the CellQuest Pro Software. The sub‐G1 population was counted as the apoptotic population and presented as fraction of the total cells counted.
2.4. Caspase 3/7‐Glo assay (Promega)
Cells were seeded at a density of 2000–3000 cells/well into 96 well plates in a volume of 50 μl and treatment of cells was initiated the next day. After additional 24 h, 50 μl of Caspase 3/7‐Glo assay buffer was added and cells were incubated in the dark at room temperature. After 1 h, 50 μl was transferred to a white 96‐microwell plate and measured in a microplate luminometer (LB96V, EG&G Berthold). The Caspase 3/7‐Glo assay is a luminescence assay that measures activities of executioner caspases‐3 and ‐7.
2.5. Western blotting
Cells were cultivated, lysed with buffer containing 1% Triton X‐100, and equal amounts of protein were resolved by SDS‐PAGE. Proteins were transferred to nitrocellulose membrane (Schleicher & Schuell), blocked for several hours in NET‐gelatin and incubated at 4 °C overnight with the corresponding primary antibody in NET‐gelatin. Anti P‐HER3 (Y1289) #4791, P‐HER2 (Y1248) #2247, P‐EGFR (Y1173) #4407, HER4 #4795, EGFR #2232, and P‐AKT (S473) #9271 antibodies were purchased from Cell Signalling. The HER3 (2F12) #05‐390, HER3 blocking Ab (105.5) #05‐471, HER2 #06‐562, and p85 #06‐496 antibodies were bought from Millipore and the anti‐Tubulin #T9026 antibody was purchased from Sigma–Aldrich. The anti‐ADAM17 #AB19027 antibody was purchased from Chemicon. Membranes were washed three times with NET‐gelatin and incubated with a horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse secondary antibody diluted in NET‐gelatin for 1 h at room temperature. After additional washing, detection was done using enhanced chemiluminescence (ECL; Western Lightning, Perkin Elmer) on X‐ray films.
2.6. Immunoprecipitation
Cell lysates were pre‐cleared with 20 μl of protein A‐ or G‐Sepharose for 1 h. Respective antibodies were pre‐coupled to 40 μl Sepharose beads in lysis buffer for 1 h and washed twice with lysis buffer. Lysates and pre‐coupled antibody‐beads were incubated at 4 °C for 4 h and precipitates were washed three times with 700 μl lysis buffer, suspended in 3× Laemmli buffer, boiled for 10 min, and directly subjected to western blot analysis. Antibodies used for IP were purchased from Millipore (HER3, #05‐390) and from Santa Cruz (HER4, #sc‐283).
2.7. Small molecule inhibitors and therapeutic monoclonal antibodies
Lapatinib as well as erlotinib was purchased from Vichem Chemie (Hungary) and LY294002 was bought from Cell Signalling. The therapeutic monoclonal antibodies Herceptin and Erbitux were purchased from the Max‐Planck Pharmacy in Martinsried.
3. Results
3.1. Doxorubicin treatment induces activation of the HER3‐PI3K‐AKT anti‐apoptotic signalling cascade
We detected a doxorubicin‐dependent increase in HER3 (Tyr1289) and AKT (Ser473) phosphorylation upon treatment with 1 μM of doxorubicin for 24 and 48 h in three (OVCAR‐3, OVCAR‐4, SKOV‐6) out of nine tested ovarian cancer cell lines (Figure 1A). All cell lines analysed (OVCAR‐3, OVCAR‐4, OVCAR‐5, OVCAR‐8, SKOV‐3, SKOV‐6, SKOV‐8, 2774, 2780) originate from either untreated or pre‐treated ovarian cancer patients (Freedman et al., 1978; Hamilton et al., 1983; Louie et al., 1986; Provencher et al., 1993; Schilder et al., 1990). The observed, doxorubicin‐mediated phosphorylation of AKT kinase was effectively abrogated when OVCAR‐3 and OVCAR‐4 cells were treated with the PI3K inhibitor LY294002 in addition to doxorubicin (Figure 1B). Moreover, precipitation of the PI3K subunit p85 together with the HER3 receptor was clearly enhanced in cells previously treated with doxorubicin (Figure 1C). We therefore analysed if the cytotoxic effect of doxorubicin can be increased by blocking the doxorubicin‐mediated activation of AKT with LY294002. Cellular apoptosis was detected (72 h after treatment) by measuring the sub‐G1 content by FACS analysis (propidium‐iodide (PI) assay) representing the apoptotic fraction of total cells counted as well as by measuring the activation of the caspase cascade 24 h after addition of drugs (Caspase 3/7‐Glo assay). Interestingly, apoptosis was markedly increased in both cell lines when doxorubicin was combined with the PI3K inhibitor LY294002 (Figure 1D).
Figure 1.
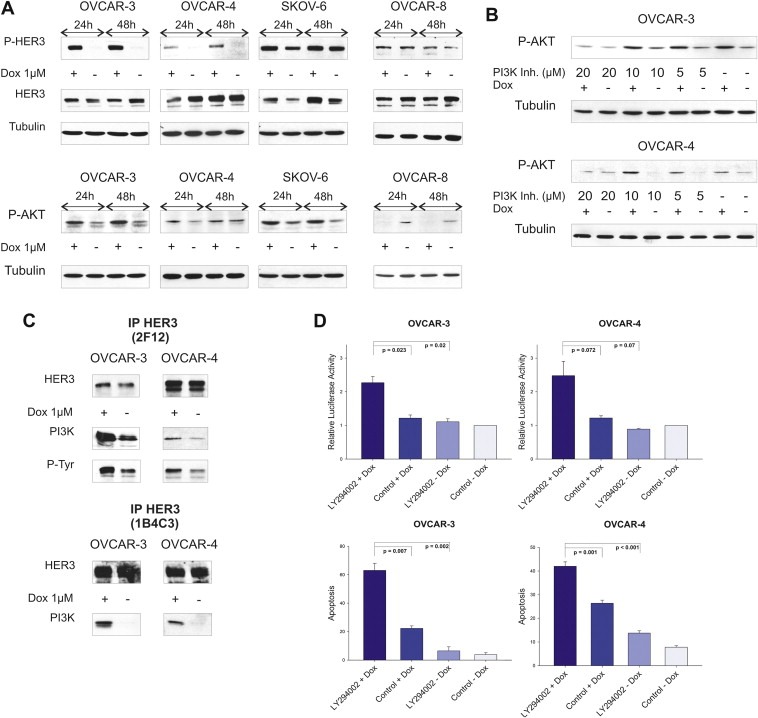
Doxorubicin induces activation of the HER3‐PI3K‐AKT anti‐apoptotic signalling pathway. (A) Phosphorylation of HER3 (P‐tyr 1289) and AKT kinase (P‐ser 473) was analysed 24 h as well as 48 h after addition of 1 μM doxorubicin. Total amounts of HER3 were determined with the HER3 antibody (2F12, Millipore) while Tubulin is shown as loading control. The OVCAR‐8 cell line is displayed as a negative control representing non‐responding cell lines in this context. (B) Cells were treated with or without doxorubicin (1 μM) for 24 h in combination with indicated concentrations of the PI3K inhibitor LY294002 and compared to DMSO treated cells. (C) Cells were treated with or without doxorubicin (1 μM) for 24 h. The HER3 receptor was immunoprecipitated with a commercially available HER3 specific antibody (Millipore, 2F12) which recognises the intracellular part of the receptor as well as with a homemade antibody (1B4C3) detecting the N‐terminal part of HER3. Immunoblots for total HER3, phospho‐tyrosine (P‐tyr) as well as for p85 (regulatory subunit of PI3K) are shown. (D) Cell lines were incubated with 20 μM of LY294002 in combination with doxorubicin (OVCAR‐3 = 1 μM and OVCAR‐4 = 2 μM) and induction of apoptosis (analysed after 24 h) was compared to single treatments and DMSO control. Relative luciferase activity reflects the activation of executioner caspases‐3 and ‐7 (Caspase 3/7‐Glo assay) in relation to caspase activity of DMSO control treated cells. Mean values and SEM of three independent experiments are shown (upper graphs). Sub‐G1 content (%) was analysed 72 h after treatment with 20 μM of LY294002 in combination with doxorubicin (OVCAR‐3 = 1 μM and OVCAR‐4 = 2 μM) and was compared to single treatments and DMSO control. Mean values (n = 3) and SEM are indicated (lower graphs).
3.2. Downregulation of the HER3 receptor increases doxorubicin‐mediated apoptosis
To validate the importance of the HER3 receptor in the context of chemosensitivity HER3 was transiently downregulated by RNAi. Cell lines were either treated with HER3 specific siRNA or with control siRNA against luciferase (Gl2). As visualised in Figure 2A, total amounts of HER3 could be effectively diminished in all three cell lines and the knockdown completely abrogated the doxorubicin‐mediated increase in HER3 phosphorylation in OVCAR‐3 and OVCAR‐4 cells. Apoptosis (sub‐G1 content) was measured 72 h after the addition of doxorubicin to previously siRNA treated cells (Figure 2B). As anticipated, downregulation of HER3 combined with doxorubicin was superior when compared to single treatments. Interestingly, no beneficial effect on apoptosis could be observed in the OVCAR‐8 cell line used as negative control in this experiment (negative control=no increase in P‐HER3 and P‐AKT upon addition of doxorubicin).
Figure 2.
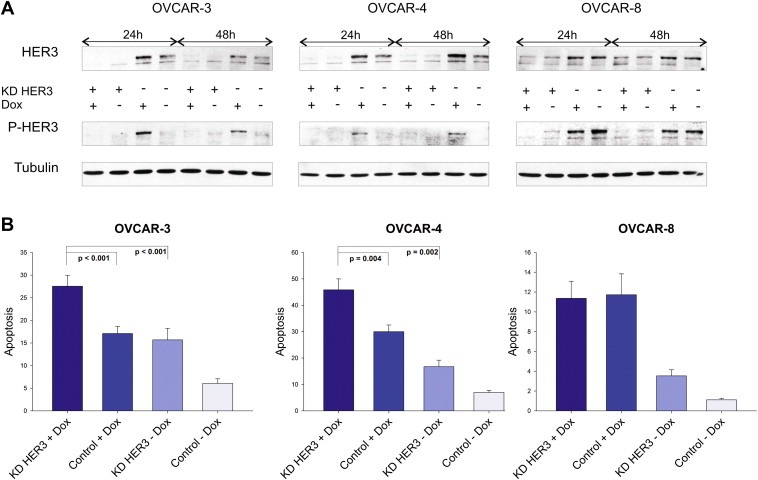
Doxorubicin‐induced apoptosis is increased upon downregulation of HER3. (A) Doxorubicin‐mediated phosphorylation of HER3 was analysed after siRNA‐mediated downregulation of HER3 and compared to siRNA control. Cells were additionally treated with or without doxorubicin (1 μM) for 24 h and 48 h. Immunoblot analysis of total HER3 as well as phospho‐HER3 (P‐tyr 1289) levels are demonstrated while Tubulin serves as loading control. (B) Apoptosis (sub‐G1 content in %) was analysed 72 h after addition of doxorubicin (OVCAR‐3 = 1 μM, OVCAR‐4 = 2 μM, OVCAR‐8 = 1 μM). Mean values and SEM for the HER3 knockdown (KD HER3) combined with or without doxorubicin (Dox), as well as for siRNA control (control) treated cells are illustrated. All experiments were performed at least five times. Knockdown experiments in OVCAR‐3 and OVCAR‐4 cells were separately performed with two different HER3 specific siRNAs with the Oligofectamine transfection reagent (Invitrogen) and results were then validated by using a mixture of three HER3 siRNAs with the RNAiMax transfection reagent (Invitrogen).
3.3. Lapatinib and erlotinib block doxorubicin‐mediated phosphorylation of HER3 and AKT and enhance cellular apoptosis
It is widely accepted that the HER3 receptor has an impaired kinase activity or at least high substrate selectivity (Guy et al., 1994; Jura et al., 2009; Pinkas‐Kramarski et al., 1996; Sierke et al., 1997; van der Horst et al., 2005). Nevertheless, it represents an important heterodimerisation partner for other members of the EGFR family like the HER2 receptor. Based on this information, we aimed to identify the potential dimerisation partner of HER3 in our ovarian cancer cell line model system. We targeted two well known HER3 interaction partners namely the EGFR and HER2 by using two “specific” tyrosine kinase inhibitors, lapatinib (Tykerb) and erlotinib (Tarceva). Lapatinib, a dual kinase inhibitor, is approved by the FDA for the treatment of breast cancer and is capable of inhibiting the kinase activity of EGFR and HER2. Erlotinib is a “selective” inhibitor of the EGFR and is approved for treatment of non‐small cell lung cancer and pancreatic cancer. Interestingly, both inhibitors have the potential to inhibit other members of the EGFR family. In this regard, erlotinib also blocks HER2 activity at submicromolar concentrations by direct interaction with the receptor, whereas lapatinib binds to the HER4 receptor as well (Qiu et al., 2008; Schaefer et al., 2007; Wood et al., 2004). Both inhibitors can therefore be considered as potential HER2 inhibitors at concentrations used in our experiments (Suppl. Figure 1A). Applying these inhibitors, we analysed whether a combinatorial treatment of either lapatinib or erlotinib with doxorubicin abrogates the detectable increase in HER3 phosphorylation as well as activation of AKT. Interestingly, both inhibitors effectively blocked the doxorubicin‐induced increase in HER3 and AKT phosphorylation with lapatinib being superior to erlotinib (Figure 3A). In addition, we analysed the efficacy of this combinatorial treatment on the level of apoptosis induction in OVCAR‐3 and OVCAR‐4 cells to further evaluate a potential clinical application of this combinatorial approach. Cell lines were treated with lapatinib or erlotinib in combination with doxorubicin and activation of caspases was measured 24 h later. Treatment with lapatinib as well as erlotinib resulted in about two fold increased caspase activation when compared to untreated cells (Figure 3B; upper graphs). Single administration of these inhibitors or doxorubicin revealed only minor effects. Moreover, DNA fragmentation was analysed 72 h after treatment and an increase in apoptosis could be detected (Figure 3B; lower graphs). As opposed to this, neither the addition of lapatinib nor erlotinib in combination with doxorubicin resulted in a significant increase in apoptosis of the “control” cell line OVCAR‐8 when compared to doxorubicin single treatment (Figure 3C; upper and lower graph). These results are remarkable, because both cell lines exhibit only a moderate expression of the EGFR and HER2 receptors (Suppl. Figure 2A).
Figure 3.
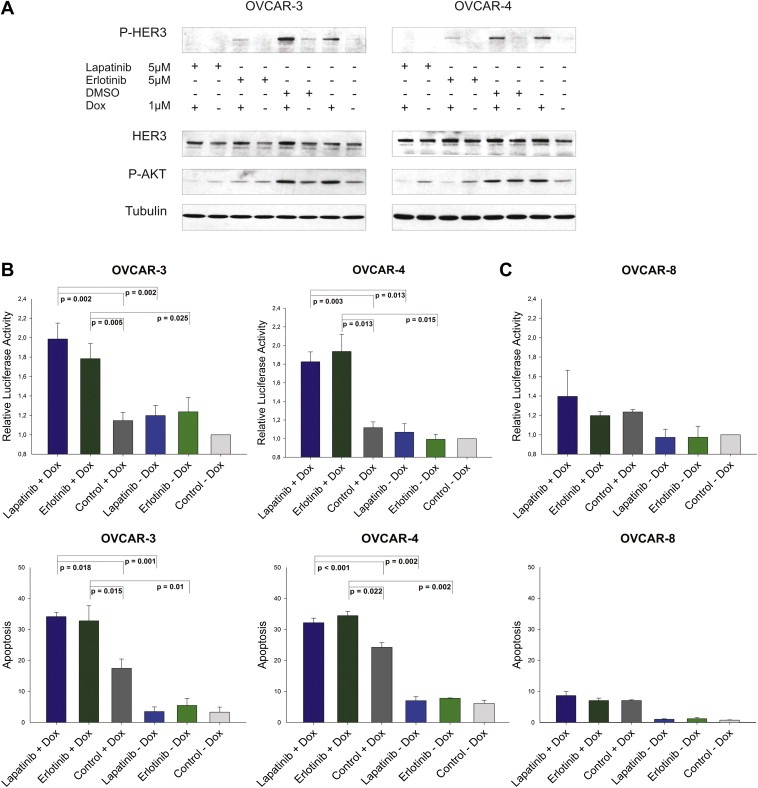
Lapatinib and erlotinib block the doxorubicin‐mediated induction of HER3 and AKT phosphorylation and increase apoptosis. (A) OVCAR‐3 and OVCAR‐4 cells were treated with lapatinib (5 μM), erlotinib (5 μM) or DMSO in combination with or without 1 μM of doxorubicin. Cells were lysed 24 h after addition of drugs and 50 μg (total protein amount) was subjected to SDS‐PAGE, blotted and probed with corresponding antibodies. Representative immunoblots for P‐HER3, P‐AKT, total HER3, and Tubulin are shown. (B) Relative luciferase activity representing the increase in caspase activity (Caspase 3/7‐Glo assay) of cells treated with inhibitor and doxorubicin relative to DMSO control treated cells. Mean values and SEM (n = 5 for OVCAR‐3 and n = 4 for OVCAR‐4) are shown. Cells were incubated for 24 h with lapatinib (10 μM), erlotinib (10 μM) or DMSO in combination with 1 μM (OVCAR‐3) and 2 μM (OVCAR‐4) of doxorubicin (upper graphs). Mean values and SEM (n = 3) of the cellular sub‐G1 content (%) upon treatment with lapatinib or erlotinib (5 μM) in combination with doxorubicin (1 μM = OVCAR‐3, 2 μM = OVCAR‐4) for 72 h (lower graphs). (C) OVCAR‐8 served as a “negative” control in this experiment (upper and lower graph). Caspase activity and sub‐G1 content was analysed as described for the OVCAR‐3 and OVCAR‐4 cell line.
3.4. Doxorubicin‐induced activation of HER3 is ligand‐mediated and dependent on ADAM17 metalloprotease activity
We next examined if the doxorubicin‐mediated increase in HER3 phosphorylation is elicited by an autocrine or paracrine activation loop. Therefore, we analysed NRG1 (HER3 ligand) expression upon treatment with doxorubicin. Cell lines were incubated with doxorubicin for 14 and 24 h, total RNA was isolated, reverse‐transcribed and isoform‐specific primers were used for PCR mediated amplification of NRG1 isoforms. As anticipated, expression of NRG1 β1 and β2 was upregulated in both cell lines upon doxorubicin treatment (Figure 4A).
Figure 4.
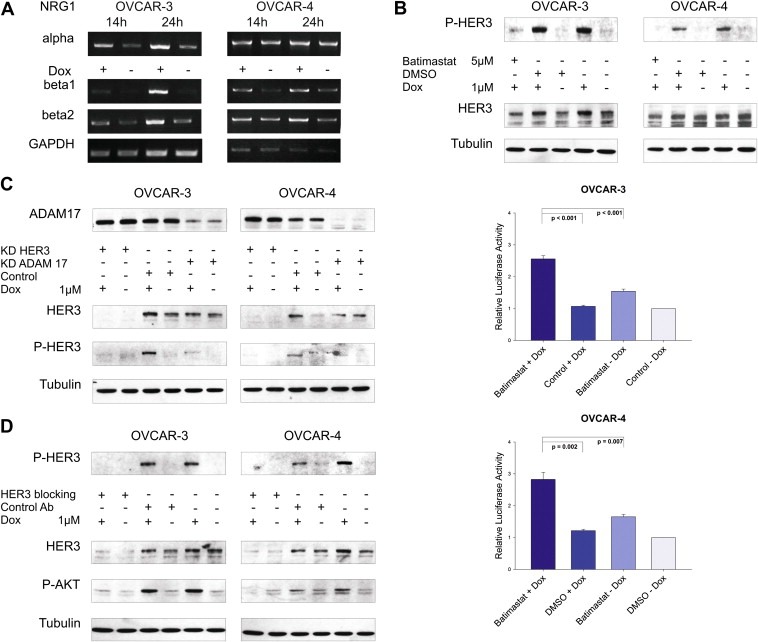
Doxorubicin‐induced activation of HER3 is ligand‐mediated and dependent on ADAM17 metalloprotease activity. (A) RNA was extracted from OVCAR‐3 and OVCAR‐4 and reverse transcribed after cells were treated with or without doxorubicin (1 μM) for indicated time. Expression of ligands was analysed by semiquantitative PCR using NRG1α, NRG1β1 and NRG1β2 specific primers. GAPDH serves as loading control. (B) Representative immunoblots of cells treated with batimastat (5 μM) in combination with doxorubicin (1 μM) for 24 h compared to control treated cells (upper graphs). Caspase activity (Caspase 3/7‐Glo assay) was measured upon treatment of cells with batimastat and doxorubicin and compared to single treatments and DMSO control. Mean values and SEM (n = 6 for OVCAR‐3 and n = 5 for OVCAR‐4). Cells were seeded in 96‐well plates, treated for 24 h with or without batimastat (5 μM) and/or doxorubicin (OVCAR‐3 = 1 μM, OVCAR‐4 = 2 μM), and were analysed afterwards (lower graphs). (C) Western blot analysis of OVCAR‐3 and OVCAR‐4 cells upon downregulation of ADAM17 or HER3 in combination with or without doxorubicin (1 μM) for 24 h. Representative immunoblots for ADAM17, P‐HER3, total HER3, and Tubulin are shown. (D) OVCAR‐3 and OVCAR‐4 were incubated with or without doxorubicin (1 μM) for 24 h and incubated with the HER3 blocking antibody 105.5 (Millipore) at a concentration of 10 μg/ml for 2 h before lysis of cells. Representative immunoblots of P‐HER3, total HER3, P‐AKT, and Tubulin are visualised.
Ligands of the EGFR family are generated as membrane‐anchored precursor proteins that can be proteolytically cleaved by metalloproteases and are thereby released from the cell (Blobel, 2005). Batimastat is a broadband inhibitor of the ADAM family of metalloproteases and blocks shedding of EGFR ligands (Borrell‐Pages et al., 2003; Dong et al., 1999). Batimastat was used in combination with doxorubicin to block the potential shedding of HER3 ligands and subsequent activation of the HER3 receptor. Interestingly, the doxorubicin‐mediated increase of the HER3 phospho‐signal was completely abrogated when cells were incubated with doxorubicin in combination with batimastat (Figure 4B; upper graphs). Moreover, a strong induction of caspase activation was measured in both cell lines for the combinatorial drug setting, whereas only a marginal apoptotic effect was detectable upon single treatment with batimastat or doxorubicin (Figure 4B; lower graphs).
ADAM17 deficient cells are defective in shedding several EGFR ligands like TGFα, HB‐EGF and amphiregulin (Merlos‐Suarez et al., 2001; Peschon et al., 1998; Sunnarborg et al., 2002). Recently, the major role of ADAM17 in the cleavage of the three EGFR ligands mentioned above and epiregulin as a new substrate has been strengthened by Sahin and colleagues (Sahin et al., 2004). In NSCLC, activation of the HER3 receptor correlated with the expression of ADAM17 but not with ADAM10 and only the downregulation of ADAM17 but not ADAM9, ADAM10 or ADAM15 had an effect on HER3 and AKT activity in A549 lung cancer cells (Zhou et al., 2006). Therefore, we investigated whether ADAM17 is also involved in shedding of HER3 ligands in our system. As anticipated, the doxorubicin‐induced activation of the HER3 receptor was completely blocked in both cell lines upon knockdown of ADAM17 with a comparable decline in signal intensity as observed for the downregulation of HER3 (Figure 4C).
3.5. Exogenous addition of recombinant HER3 ligands partially reverses the apoptotic effect of batimastat plus doxorubicin
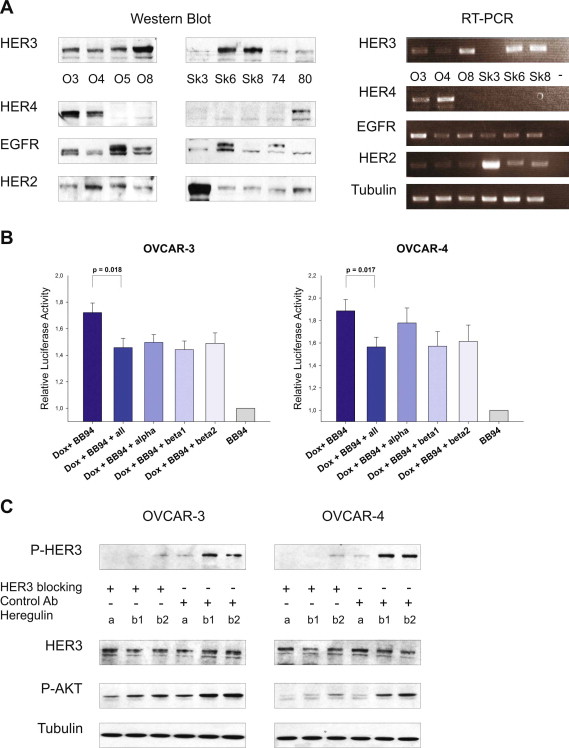
We next tried to reverse the induction of apoptosis apparent upon combined treatment with batimastat and doxorubicin. If our considerations were right, the exogenous induced re‐activation of the HER3‐PI3K‐AKT signalling cascade, upon stimulation with HER3 ligands, should result in a decrease in apoptosis of cells previously treated with doxorubicin and batimastat. Cells were incubated with doxorubicin in combination with batimastat and different NRG1 isoforms were added after 14 and 19 h. Activation of the caspase cascade was monitored after 24 h. Interestingly, with the exception of NRG1 α in OVCAR‐4 cells, the addition of exogenous HER3 ligands partially reversed doxorubicin‐induced apoptosis either when administered as single ligands or as combination (Suppl. Figure 2B). However, no complete reduction of apoptosis was observable by the exogenous addition of ligands, which might be due to the artificial character of this experiment.
3.6. HER3 blocking antibody treatment effectively abrogates the doxorubicin‐mediated activation of HER3 and AKT
Therapeutic monoclonal antibodies targeting the HER3 receptor are in clinical trials and might soon become available as a novel cancer therapy. We utilised the highly specific and commercially available HER3 blocking antibody 105.5 (Millipore) for which inhibition of ligand‐mediated HER3 phosphorylation was previously shown (Chen et al., 1996). Initially, we tested whether this monoclonal HER3 antibody is capable of blocking the NRG1‐mediated increase in HER3 and AKT phosphorylation in OVCAR‐3 and OVCAR‐4 cells, and whether it abrogates receptor activation induced by NRG1 isoforms (NRG1 α, NRG1 β1 and NRG1 β2). As expected, a remarkable reduction in HER3 and AKT phosphorylation was detected (Suppl. Figure 2C). Based on these promising results, we slightly modified the experimental set up. Instead of stimulating cells with exogenous ligands, they were now incubated with doxorubicin for 24 h and the blocking antibody was added for an additional time of 2 h before lysis. Notably, the HER3 blocking antibody completely inhibited the doxorubicin‐induced increase of HER3 and AKT phospho‐signals in both cell lines whereas no effect was detected with an isotype control antibody (Figure 4D).
3.7. Supernatant of doxorubicin treated cells increases HER3 phosphorylation in MCF7 breast cancer cells
Tumours are characterised by genetically unstable, heterogeneous cell populations carrying DNA abnormalities that initiated the malignant behaviour as well as many additional genetic lesions that emerged during tumorigenesis due to selective pressure in diverse microenvironments. In this respect, we wanted to know whether the doxorubicin‐induced expression of HER3 ligands and their assumed secretion has the potential to activate the HER3‐PI3K‐AKT survival pathway in surrounding tumour cells. For the detection of these ligands the well‐established breast cancer cell line MCF7, whose HER3 receptor can be effectively phosphorylated by different NRG1 isoforms, was used. Media from OVCAR‐3 and OVCAR‐4 cells, cultivated in the presence of doxorubicin for 24 h, was removed and utilised for the stimulation of MCF7 cells. As assumed, the supernatant potently activated the HER3 receptor in MCF7 cells whereas media from untreated cells did not (Suppl. Figure 3A). This data further indicates that the expressed HER3 ligands, after being cleaved by metalloproteases, are soluble and capable to induce activation of the HER3‐PI3K‐AKT pathway in an autocrine and/or paracrine manner.
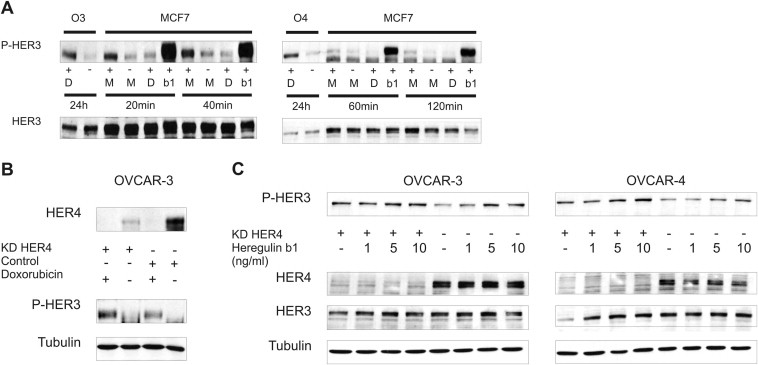
3.8. The HER4 receptor can be immunoprecipitated with HER3 in the doxorubicin‐untreated state and this interaction is lost upon treatment with doxorubicin
With the exception of HER2, which exists in an “open” and therefore active conformation, members of the EGF receptor family normally exist as monomers in a “closed” inactive conformation (Cho et al., 2003; Garrett et al., 2003). Therefore, the HER2 receptor is constitutively ready for dimerisation. In contrast, this conformation has to be stabilised by ligand binding to the extracellular domain of the receptor in other EGFR family members. It has been frequently reported that the HER3 receptor lacks an active kinase function due to sequence substitutions. In addition, it is not capable to form homodimers upon activation but nevertheless seems to be the preferred dimerisation partner of other EGFR family members (Berger et al., 2004; Pinkas‐Kramarski et al., 1996). To elucidate the doxorubicin‐mediated HER3 receptor activation in more detail we employed a mass spectrometry‐based approach where we pulled down the receptor by immunoprecipitation to identify potential HER3 interactors (data not shown). We identified the HER4 receptor as HER3 dimerisation partner with a HER3/HER4 interaction in the “unstimulated” (no doxorubicin) state and observed an apparent decline of this interaction upon treatment with doxorubicin which seems to be due to a decrease of total HER4 protein (Figure 5A). Nevertheless, the residual HER3/HER4 interaction could still be responsible for the doxorubicin‐mediated increase in HER3 phosphorylation. In this regard, we tested whether the doxorubicin‐induced phosphorylation of the HER3 receptor can be blocked by RNAi mediated downregulation of HER4 but no reduction of the doxorubicin‐induced HER3 phosphorylation was observed (Suppl. Figure 3B). Surprisingly, we detected a clear increase in HER3 activation when cells were depleted of HER4 (siRNA mediated) and subsequently stimulated with different amounts of Heregulin β1 (Suppl. Figure 3C).
Figure 5.
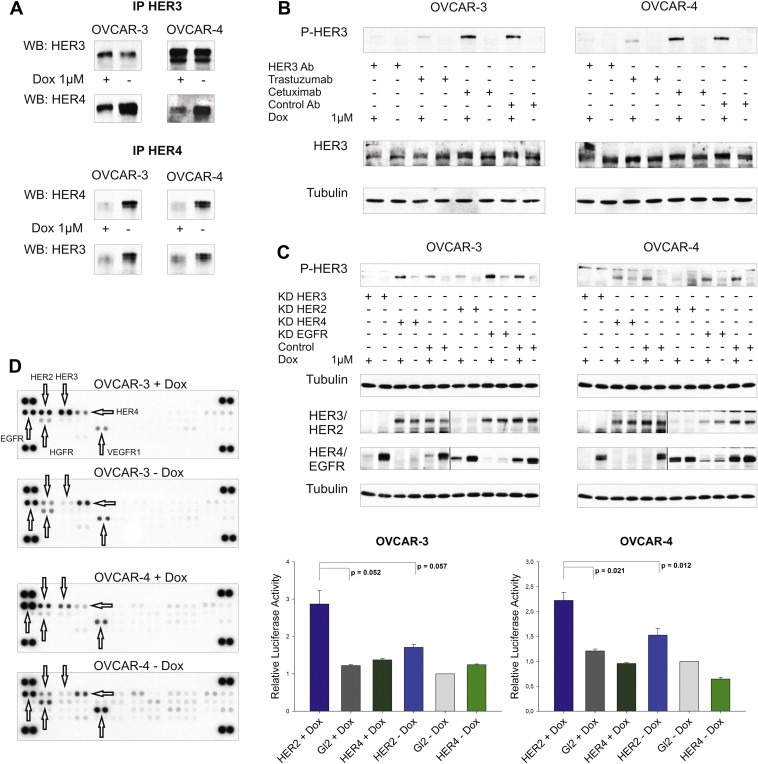
Doxorubicin‐induced activation of HER3 is mediated by the HER2 receptor. (A) HER3 was immunoprecipitated with a HER3 specific antibody (Millipore, 2F12) and the HER4 receptor was co‐precipitated. Vice versa, HER3 was co‐immunoprecipitated with the HER4 receptor (Santa Cruz, sc‐283). Immunoblots of HER3 and HER4 after immunoprecipitation are displayed. (B) OVCAR‐3 and OVCAR‐4 cells were incubated with or without doxorubicin (1 μM) for 24 h and were then treated with the 105.5 (Millipore) HER3 blocking antibody (HER3 Ab), trastuzumab (HER2 Ab), cetuximab (EGFR Ab) or isotype control (control Ab) antibody at a concentration of 10 μg/ml for an additional time of 2 h before cells were lysed. Representative immunoblots of P‐HER3 (P‐tyr 1289), HER3, and Tubulin are shown. (C) Representative immunoblots of the siRNA‐mediated downregulation of EGFR, HER2, HER3, and HER4 compared to siRNA control (Gl2) in OVCAR‐3 and OVCAR‐4 cells. Cells were additionally stimulated with or without doxorubicin (1 μM) and lysed after 24 h (48 h after the knockdown was initiated). 50 μg (total protein amount) was subjected to SDS‐PAGE, blotted and probed with corresponding antibodies (upper graphs). Two separate gels were used to analyse differences in HER3 phosphorylation as well as to probe efficacy of EGFR, HER2, HER3, and HER4 knockdown. In addition, the nitrocellulose membrane (knockdown) was cut in two halfs and separately probed with the different antibodies as indicated. Mean values and SEM (n = 3) of caspase activation (Caspase 3/7‐Glo assay) upon downregulation of HER2 or HER4 in combination with doxorubicin relative to cells treated with siRNA control. Cells were transfected with specific siRNAs for HER2 or HER4 and incubated with or without doxorubicin (1 μM OVCAR‐3 and 2 μM OVCAR‐4) for additional 24 h (lower graphs). (D) Phosphorylation of 42 different human RTKs analysed with the “human phospho‐RTK‐Array Kit” (R&D Systems). OVCAR‐3 and OVCAR‐4 cells were incubated with or without 1 μM of doxorubicin (24 h) and phosphorylation of RTKs was analysed according to the manufacturer instructions.
3.9. Trastuzumab but not cetuximab blocks doxorubicin‐mediated phosphorylation of HER3
Paradoxically, it was not possible to detect the potential HER3 interaction/dimerisation partner in the doxorubicin‐induced state responsible for the activation of HER3 by mass spectrometry. However, co‐treatment with either lapatinib or erlotinib in combination with doxorubicin completely abrogated the induced HER3 phosphorylation (Figure 3A). While these inhibitors were developed against the EGFR only (erlotinib) or the EGFR and HER2 (lapatinib), it has been reported that erlotinib also inhibits the HER2 receptor by direct interaction whereas lapatinib binds to HER4 as well (Qiu et al., 2008; Schaefer et al., 2007; Wood et al., 2004). Based on this TKI unspecificity, we tried to identify the potential HER3 dimerisation partner by using two FDA approved and highly specific therapeutic monoclonal antibodies namely trastuzumab (Herceptin) and cetuximab (Erbitux). Cell lines were incubated with doxorubicin for 24 h and treated with either trastuzumab or cetuximab 2 h before lysis. The potential decline in HER3 phospho‐signal was compared to the signal observed with the HER3 blocking antibody. Notably, the doxorubicin‐mediated increase of HER3 phosphorylation was completely abolished in trastuzumab treated cells (Figure 5B). The detectable decline of HER3 phosphorylation was comparable to the effect we observed with the HER3 blocking antibody. In contrast, there was no effect on the HER3 phospho‐signal intensity to be observed with the EGFR blocking antibody cetuximab or with the isotype control antibody, as expected. Interestingly, the mechanism of action resulting in inhibition of the doxorubicin‐induced HER3 phosphorylation seems to differ between trastuzumab and the kinase inhibitors lapatinib or erlotinib (Suppl. Figures 1A and 1B).
3.10. HER2 is responsible for the doxorubicin‐induced activation of the HER3 receptor
To further validate the results we obtained with trastuzumab, we transiently downregulated the EGFR as well as HER2 in both cell lines and subsequently treated them with doxorubicin. Both cell lines were incubated with specific siRNAs against EGFR, HER2, HER3, or the HER4 receptor (downregulation of HER4 and HER3 served as negative and positive controls in this experiment), treated with doxorubicin, and phosphorylation of HER3 was analysed. As anticipated, only the knockdown of HER2 and HER3 abrogated the doxorubicin‐mediated increase of the HER3 phospho‐signal (Figure 5C; upper graphs). Moreover, a strong increase in apoptosis was detected upon downregulation of HER2 and subsequent treatment with doxorubicin (Figure 5C; lower graphs). In contrast, knockdown of the HER4 receptor combined with the addition of doxorubicin did neither result in a beneficial effect in regard to apoptosis nor did it abolish the phosphorylation of HER3.
3.11. Doxorubicin affects phosphorylation of EGFR family members whereas no activation of other RTKs was detected
To further evaluate if other RTKs might be affected by the addition of doxorubicin we used a “Human Phospho‐RTK Array” to analyse the phosphorylation pattern of 42 different human RTKs. OVCAR‐3 and OVCAR‐4 cells were treated with or without doxorubicin for 24 h and differences in phosphorylation were analysed. Confirming our previous data, we detected a strong increase in HER3 phosphorylation, a moderate activation of HER2, and a clear reduction of HER4 signal intensity upon addition of doxorubicin (Figure 5D). Additionally, a minor induction of EGFR phosphorylation could be detected in the OVCAR‐4 cell line. Treatment with doxorubicin did not result in an increased signal of any other RTK analysed with the “Human Phospho‐RTK Array Kit”.
3.12. Cisplatin and doxorubicin increase activation of HER3 while other chemotherapeutic drugs do not
Doxorubicin is currently used as second line treatment for recurrent ovarian cancer and the administration of a platinum‐based compound in combination with a taxane represents the standard first‐line treatment. Back in the 80s, when most of the well characterised ovarian cancer cell lines were established, the chemotherapeutic drug regimen for ovarian cancer was different (Agarwal and Kaye, 2003). Interestingly, OVCAR‐3 and OVCAR‐4 cell lines were previously established from cancer patients who have been pre‐treated with a combination of doxorubicin, cyclophosphamide, and cisplatin (Hamilton et al., 1983; Schilder et al., 1990) (Figure 6B). Based on this information, we further analysed whether the observed activation of HER3 might represent a generalised mechanism for chemotherapeutic drug insensitivity or whether this response is restricted to doxorubicin only. Cell lines were treated with cisplatin, etoposide, fluorouracil, cyclophosphamide, dacarbazine, and doxorubicin as positive control. Interestingly, out of these six chemotherapeutic drugs tested, only the addition of doxorubicin and cisplatin resulted in a clear increase in HER3 phosphorylation (Figure 6A). As anticipated, no increase in P‐HER3 was detected in the negative control cell line OVCAR‐8.
Figure 6.
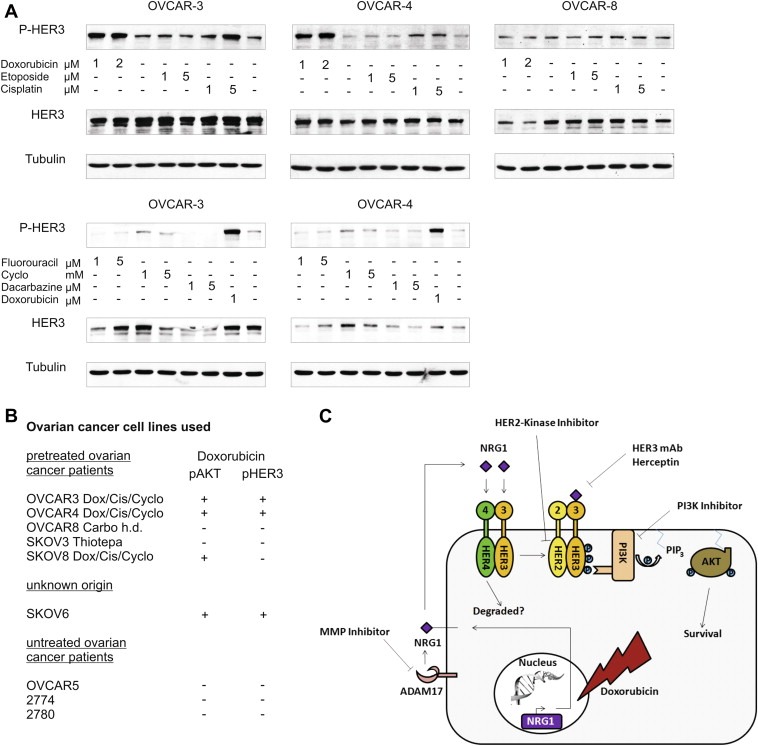
Doxorubicin and cisplatin induce phosphorylation of HER3 while other chemotherapeutic drugs do not. (A) Cell lines were treated with indicated concentrations of different chemotherapeutic drugs for 24 h. Phosphorylation of HER3 was analysed with the P‐HER3 specific antibody (P‐tyr 1289). Tubulin serves as loading control. (B) Nine different ovarian cancer cell lines are grouped based on chemotherapeutic pre‐treatment of cancer patients from where cell lines have originally been established (Dox = doxorubicin; Cis = cisplatin; Cyclo = cyclophosphamide; Carbo = carboplatin). The doxorubicin‐induced increase in HER3 and AKT phosphorylation is indicated. (C) Hypothetical cell signalling model representing the pathway which is induced upon addition of doxorubicin.
4. Discussion
Most studies on chemotherapeutic drug resistance are based on chemoinsensitive cancer cell lines generated by continuous cellular exposure with the respective chemotherapeutic drug. Many of these reports focus on differences in the cellular expression levels of important anti‐apoptotic proteins between insensitive cell lines and their respective sensitive counterparts (Hui et al., 2008; Liu et al., 2007; Servidei et al., 2008). We examined nine ovarian cancer cell lines which originate from either untreated or pre‐treated patients and resemble the natural occurrence of chemotherapeutic resistance (Freedman et al., 1978; Hamilton et al., 1983; Louie et al., 1986; Provencher et al., 1993; Schilder et al., 1990). In this regard, the detected and doxorubicin‐mediated increase in HER3 and AKT phosphorylation could only be observed in cell lines which were originally established from patients previously treated with a combination of chemotherapeutic drugs. In contrast, it was not possible to detect an increase in phosphorylation in cell lines established from previously untreated patients. Notably, the doxorubicin‐induced long‐term increase in AKT phosphorylation was dependent on activation of the HER3 receptor. We assumed and confirmed that the increase in HER3 phosphorylation correlates with an upregulated expression of HER3 ligands and that they bind to the HER3 receptor in an autocrine or paracrine way. All ligands of the EGFR family are generated as membrane‐anchored precursor proteins that can be proteolytically cleaved by metalloproteases and are thereby released from cells (Blobel, 2005). We demonstrated the involvement of a metalloprotease by successfully abrogating the doxorubicin‐mediated phosphorylation of HER3 with the broadband metalloprotease inhibitor batimastat. The importance of ADAM17‐mediated shedding of several ligands of the EGFR family like TGFα, HB‐EGF, amphiregulin and epiregulin has been elucidated by several groups (Merlos‐Suarez et al., 2001; Peschon et al., 1998; Sahin et al., 2004; Sunnarborg et al., 2002). In accordance to this, downregulation of ADAM17 completely abrogated the doxorubicin‐induced increase in HER3 phosphorylation in our experiments. Therefore, these results fully support our assumption that the doxorubicin‐induced activation of the anti‐apoptotic HER3‐PI3K‐AKT pathway is based on an upregulated expression of HER3 ligands as well as the involvement of metalloprotease ADAM17.
We further aimed to identify the dimerisation partner of the HER3 receptor responsible for the doxorubicin‐mediated activation of the HER3‐PI3K‐AKT axis in our system. Two approved TKIs, lapatinib and erlotinib, targeting HER2 and EGFR or only EGFR respectively were used in combination with doxorubicin (Pollack et al., 1999; Xia et al., 2002). Interestingly, treatment with either lapatinib or erlotinib resulted in the complete abrogation of the doxorubicin‐mediated increase in HER3 phosphorylation. Considering the complete loss of HER3 phosphorylation and the significant increase in apoptosis observed with both inhibitors one might assume that the EGFR is responsible for the doxorubicin‐induced increase in HER3 phosphorylation. However, lapatinib as well as erlotinib have the potential to inhibit other members of the EGFR family besides their main targets (Guix et al., 2008; Qiu et al., 2008; Schaefer et al., 2007). In a mass spectrometry experiment, HER4 was detected as heterodimerisation partner of HER3 in the untreated state (data not shown). However, siRNA‐mediated downregulation of HER4 did not abolish the doxorubicin‐induced activation of HER3. Interestingly, a recent report provides a possible explanation for the occurrence and importance of the HER3/HER4 interaction in the untreated state (Jura et al., 2009). Based on a structural analysis of the HER3 kinase domain, Jura and colleagues postulated that the formation of active heterodimers with other members of the EGFR family is restricted by the formation of heterooligomers between the HER3 and the HER4 receptor. Notably, this observation is in agreement with the increased, ligand‐induced phosphorylation of HER3 upon downregulation of HER4. Moreover, a tremendous reduction of total HER4 protein was observed in our experiments upon cellular exposure to doxorubicin. Therefore, the model proposed by Jura et al. represents an interesting explanation. Further experiments will be necessary to put more light on the functional role of this interaction.
However, the HER2 dependency of the doxorubicin‐induced activation of the HER3‐PI3K‐AKT pathway was further elucidated by the use of specific blocking antibodies. Precisely, positive results which were obtained by using trastuzumab (HER2 Ab) to block the doxorubicin‐induced phosphorylation of HER3 as well as the ineffectivity of cetuximab (EGFR Ab) supported the important role of the HER2 receptor in this context. Therefore, the observed abrogation of HER3 phosphorylation upon doxorubicin treatment and subsequent addition of trastuzumab might be due to a decrease in HER2/HER3 interaction although we could not detect this heterodimer directly.
By inhibiting the doxorubicin‐mediated activation of the anti‐apoptotic HER3‐PI3K‐AKT signalling cascade (see Figure 6C) we significantly increased the sensitivity of ovarian cancer cells to chemotherapy‐induced apoptosis. According to our results, trastuzumab and/or lapatinib could represent an interesting option for clinicians for the treatment of recurrent ovarian cancer when combined with doxorubicin. In addition, therapeutic monoclonal antibodies directly targeting the HER3 receptor hold tremendous potential for treatment of ovarian or possibly other cancers when administered in combination with doxorubicin or platinum‐based drugs.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the Supplementary data related to this article:
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
Acknowledgements
We thank Renate Gautsch and Bianca Sperl for the excellent technical assistance.
Supplementary data 1.
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2012.07.001.
Bezler Martin, Hengstler Jan G. and Ullrich Axel, (2012), Inhibition of doxorubicin‐induced HER3‐PI3K‐AKT signalling enhances apoptosis of ovarian cancer cells, Molecular Oncology, 6, doi: 10.1016/j.molonc.2012.07.001.
References
- Agarwal, R. , Kaye, S.B. , 2003. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer. 3, 502–516. [DOI] [PubMed] [Google Scholar]
- Alimandi, M. , Romano, A. , Curia, M.C. , Muraro, R. , Fedi, P. , Aaronson, S.A. , Di Fiore, P.P. , Kraus, M.H. , 1995. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 10, 1813–1821. [PubMed] [Google Scholar]
- Beji, A. , Horst, D. , Engel, J. , Kirchner, T. , Ullrich, A. , 2012. Toward the prognostic significance and therapeutic potential of HER3 receptor tyrosine kinase in human colon cancer. Clin. Cancer Res.. 18, 956–968. [DOI] [PubMed] [Google Scholar]
- Berger, M.B. , Mendrola, J.M. , Lemmon, M.A. , 2004. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett.. 569, 332–336. [DOI] [PubMed] [Google Scholar]
- Blobel, C.P. , 2005. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol.. 6, 32–43. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages, M. , Rojo, F. , Albanell, J. , Baselga, J. , Arribas, J. , 2003. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J.. 22, 1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty, A. , Sanchez, V. , Kuba, M.G. , Rinehart, C. , Arteaga, C.L. , 2011. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. U.S.A.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty, S. , Sawai, A. , Scaltriti, M. , Rodrik-Outmezguine, V. , Grbovic-Huezo, O. , Serra, V. , Majumder, P.K. , Baselga, J. , Rosen, N. , 2011. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 19, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Levkowitz, G. , Tzahar, E. , Karunagaran, D. , Lavi, S. , Ben-Baruch, N. , Leitner, O. , Ratzkin, B.J. , Bacus, S.S. , Yarden, Y. , 1996. An immunological approach reveals biological differences between the two NDF/heregulin receptors, ErbB-3 and ErbB-4. J. Biol. Chem.. 271, 7620–7629. [PubMed] [Google Scholar]
- Cho, H.S. , Mason, K. , Ramyar, K.X. , Stanley, A.M. , Gabelli, S.B. , Denney, D.W. , Leahy, D.J. , 2003. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 421, 756–760. [DOI] [PubMed] [Google Scholar]
- Dong, J. , Opresko, L.K. , Dempsey, P.J. , Lauffenburger, D.A. , Coffey, R.J. , Wiley, H.S. , 1999. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc. Natl. Acad. Sci. U.S.A.. 96, 6235–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Janne, P.A. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Freedman, R.S. , Pihl, E. , Kusyk, C. , Gallager, H.S. , Rutledge, F. , 1978. Characterization of an ovarian carcinoma cell line. Cancer. 42, 2352–2359. [DOI] [PubMed] [Google Scholar]
- Garrett, J.T. , Olivares, M.G. , Rinehart, C. , Granja-Ingram, N.D. , Sanchez, V. , Chakrabarty, A. , Dave, B. , Cook, R.S. , Pao, W. , McKinely, E. , Manning, H.C. , Chang, J. , Arteaga, C.L. , 2011. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. U.S.A.. 108, 5021–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett, T.P. , McKern, N.M. , Lou, M. , Elleman, T.C. , Adams, T.E. , Lovrecz, G.O. , Kofler, M. , Jorissen, R.N. , Nice, E.C. , Burgess, A.W. , Ward, C.W. , 2003. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell. 11, 495–505. [DOI] [PubMed] [Google Scholar]
- Guix, M. , Granja Nde, M. , Meszoely, I. , Adkins, T.B. , Wieman, B.M. , Frierson, K.E. , Sanchez, V. , Sanders, M.E. , Grau, A.M. , Mayer, I.A. , Pestano, G. , Shyr, Y. , Muthuswamy, S. , Calvo, B. , Krontiras, H. , Krop, I.E. , Kelley, M.C. , Arteaga, C.L. , 2008. Short preoperative treatment with erlotinib inhibits tumor cell proliferation in hormone receptor-positive breast cancers. J. Clin. Oncol.. 26, 897–906. [DOI] [PubMed] [Google Scholar]
- Guy, P.M. , Platko, J.V. , Cantley, L.C. , Cerione, R.A. , Carraway, K.L. , 1994. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A.. 91, 8132–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, T.C. , Young, R.C. , McKoy, W.M. , Grotzinger, K.R. , Green, J.A. , Chu, E.W. , Whang-Peng, J. , Rogan, A.M. , Green, W.R. , Ozols, R.F. , 1983. Characterization of a human ovarian carcinoma cell line (NIH: OVCAR-3) with androgen and estrogen receptors. Cancer Res.. 43, 5379–5389. [PubMed] [Google Scholar]
- Hennessy, B.T. , Coleman, R.L. , Markman, M. , 2009. Ovarian cancer. Lancet. 374, 1371–1382. [DOI] [PubMed] [Google Scholar]
- Hui, R.C. , Francis, R.E. , Guest, S.K. , Costa, J.R. , Gomes, A.R. , Myatt, S.S. , Brosens, J.J. , Lam, E.W. , 2008. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther.. 7, 670–678. [DOI] [PubMed] [Google Scholar]
- Jura, N. , Shan, Y. , Cao, X. , Shaw, D.E. , Kuriyan, J. , 2009. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. U.S.A.. 106, 21608–21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, T.J. , Lee, J.W. , Song, S.Y. , Choi, J.J. , Choi, C.H. , Kim, B.G. , Lee, J.H. , Bae, D.S. , 2006. Increased expression of pAKT is associated with radiation resistance in cervical cancer. Br. J. Cancer. 94, 1678–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus, M.H. , Issing, W. , Miki, T. , Popescu, N.C. , Aaronson, S.A. , 1989. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc. Natl. Acad. Sci. U.S.A.. 86, 9193–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Lu, Y. , Liang, K. , Liu, B. , Fan, Z. , 2005. Differential responses to doxorubicin-induced phosphorylation and activation of Akt in human breast cancer cells. Breast Cancer Res.. 7, R589–R597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L.Z. , Zhou, X.D. , Qian, G. , Shi, X. , Fang, J. , Jiang, B.H. , 2007. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res.. 67, 6325–6332. [DOI] [PubMed] [Google Scholar]
- Louie, K.G. , Hamilton, T.C. , Winker, M.A. , Behrens, B.C. , Tsuruo, T. , Klecker, R.W. , McKoy, W.M. , Grotzinger, K.R. , Myers, C.E. , Young, R.C. , 1986. Adriamycin accumulation and metabolism in adriamycin-sensitive and -resistant human ovarian cancer cell lines. Biochem. Pharmacol.. 35, 467–472. [DOI] [PubMed] [Google Scholar]
- McCubrey, J.A. , Sokolosky, M.L. , Lehmann, B.D. , Taylor, J.R. , Navolanic, P.M. , Chappell, W.H. , Abrams, S.L. , Stadelman, K.M. , Wong, E.W. , Misaghian, N. , Horn, S. , Basecke, J. , Libra, M. , Stivala, F. , Ligresti, G. , Tafuri, A. , Milella, M. , Zarzycki, M. , Dzugaj, A. , Chiarini, F. , Evangelisti, C. , Martelli, A.M. , Terrian, D.M. , Franklin, R.A. , Steelman, L.S. , 2008. Alteration of Akt activity increases chemotherapeutic drug and hormonal resistance in breast cancer yet confers an achilles heel by sensitization to targeted therapy. Adv. Enzym. Regul.. 48, 113–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlos-Suarez, A. , Ruiz-Paz, S. , Baselga, J. , Arribas, J. , 2001. Metalloprotease-dependent protransforming growth factor-alpha ectodomain shedding in the absence of tumor necrosis factor-alpha-converting enzyme. J. Biol. Chem.. 276, 48510–48517. [DOI] [PubMed] [Google Scholar]
- Peschon, J.J. , Slack, J.L. , Reddy, P. , Stocking, K.L. , Sunnarborg, S.W. , Lee, D.C. , Russell, W.E. , Castner, B.J. , Johnson, R.S. , Fitzner, J.N. , Boyce, R.W. , Nelson, N. , Kozlosky, C.J. , Wolfson, M.F. , Rauch, C.T. , Cerretti, D.P. , Paxton, R.J. , March, C.J. , Black, R.A. , 1998. An essential role for ectodomain shedding in mammalian development. Science. 282, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Pinkas-Kramarski, R. , Soussan, L. , Waterman, H. , Levkowitz, G. , Alroy, I. , Klapper, L. , Lavi, S. , Seger, R. , Ratzkin, B.J. , Sela, M. , Yarden, Y. , 1996. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J.. 15, 2452–2467. [PMC free article] [PubMed] [Google Scholar]
- Plowman, G.D. , Whitney, G.S. , Neubauer, M.G. , Green, J.M. , McDonald, V.L. , Todaro, G.J. , Shoyab, M. , 1990. Molecular cloning and expression of an additional epidermal growth factor receptor-related gene. Proc. Natl. Acad. Sci. U.S.A.. 87, 4905–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack, V.A. , Savage, D.M. , Baker, D.A. , Tsaparikos, K.E. , Sloan, D.E. , Moyer, J.D. , Barbacci, E.G. , Pustilnik, L.R. , Smolarek, T.A. , Davis, J.A. , Vaidya, M.P. , Arnold, L.D. , Doty, J.L. , Iwata, K.K. , Morin, M.J. , 1999. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J. Pharmacol. Exp. Ther.. 291, 739–748. [PubMed] [Google Scholar]
- Provencher, D.M. , Finstad, C.L. , Saigo, P.E. , Rubin, S.C. , Hoskins, W.J. , Federici, M.G. , Stockert, E. , Lloyd, K.O. , Lewis, J.L. , 1993. Comparison of antigen expression on fresh and cultured ascites cells and on solid tumors of patients with epithelial ovarian cancer. Gynecol. Oncol.. 50, 78–83. [DOI] [PubMed] [Google Scholar]
- Qiu, C. , Tarrant, M.K. , Choi, S.H. , Sathyamurthy, A. , Bose, R. , Banjade, S. , Pal, A. , Bornmann, W.G. , Lemmon, M.A. , Cole, P.A. , Leahy, D.J. , 2008. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 16, 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reschke, M. , Mihic-Probst, D. , van der Horst, E.H. , Knyazev, P. , Wild, P.J. , Hutterer, M. , Meyer, S. , Dummer, R. , Moch, H. , Ullrich, A. , 2008. HER3 is a determinant for poor prognosis in melanoma. Clin. Cancer Res.. 14, 5188–5197. [DOI] [PubMed] [Google Scholar]
- Sahin, U. , Weskamp, G. , Kelly, K. , Zhou, H.M. , Higashiyama, S. , Peschon, J. , Hartmann, D. , Saftig, P. , Blobel, C.P. , 2004. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol.. 164, 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, G. , Shao, L. , Totpal, K. , Akita, R.W. , 2007. Erlotinib directly inhibits HER2 kinase activation and downstream signaling events in intact cells lacking epidermal growth factor receptor expression. Cancer Res.. 67, 1228–1238. [DOI] [PubMed] [Google Scholar]
- Schilder, R.J. , Hall, L. , Monks, A. , Handel, L.M. , Fornace, A.J. , Ozols, R.F. , Fojo, A.T. , Hamilton, T.C. , 1990. Metallothionein gene expression and resistance to cisplatin in human ovarian cancer. Int. J. Cancer. 45, 416–422. [DOI] [PubMed] [Google Scholar]
- Schulze, W.X. , Deng, L. , Mann, M. , 2005. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol.. 1, 2005-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergina, N.V. , Rausch, M. , Wang, D. , Blair, J. , Hann, B. , Shokat, K.M. , Moasser, M.M. , 2007. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 445, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servidei, T. , Riccardi, A. , Mozzetti, S. , Ferlini, C. , Riccardi, R. , 2008. Chemoresistant tumor cell lines display altered epidermal growth factor receptor and HER3 signaling and enhanced sensitivity to gefitinib. Int. J. Cancer. 123, 2939–2949. [DOI] [PubMed] [Google Scholar]
- Sierke, S.L. , Cheng, K. , Kim, H.H. , Koland, J.G. , 1997. Biochemical characterization of the protein tyrosine kinase homology domain of the ErbB3 (HER3) receptor protein. Biochem. J.. 322, (Pt 3) 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnarborg, S.W. , Hinkle, C.L. , Stevenson, M. , Russell, W.E. , Raska, C.S. , Peschon, J.J. , Castner, B.J. , Gerhart, M.J. , Paxton, R.J. , Black, R.A. , Lee, D.C. , 2002. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem.. 277, 12838–12845. [DOI] [PubMed] [Google Scholar]
- Tanner, B. , Hasenclever, D. , Stern, K. , Schormann, W. , Bezler, M. , Hermes, M. , Brulport, M. , Bauer, A. , Schiffer, I.B. , Gebhard, S. , Schmidt, M. , Steiner, E. , Sehouli, J. , Edelmann, J. , Lauter, J. , Lessig, R. , Krishnamurthi, K. , Ullrich, A. , Hengstler, J.G. , 2006. ErbB-3 predicts survival in ovarian cancer. J. Clin. Oncol.. 24, 4317–4323. [DOI] [PubMed] [Google Scholar]
- van der Horst, E.H. , Weber, I. , Ullrich, A. , 2005. Tyrosine phosphorylation of PYK2 mediates heregulin-induced glioma invasion: novel heregulin/HER3-stimulated signaling pathway in glioma. Int. J. Cancer. 113, 689–698. [DOI] [PubMed] [Google Scholar]
- Wallasch, C. , Weiss, F.U. , Niederfellner, G. , Jallal, B. , Issing, W. , Ullrich, A. , 1995. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J.. 14, 4267–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winograd-Katz, S.E. , Levitzki, A. , 2006. Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene. 25, 7381–7390. [DOI] [PubMed] [Google Scholar]
- Witton, C.J. , Reeves, J.R. , Going, J.J. , Cooke, T.G. , Bartlett, J.M. , 2003. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J. Pathol.. 200, 290–297. [DOI] [PubMed] [Google Scholar]
- Wood, E.R. , Truesdale, A.T. , McDonald, O.B. , Yuan, D. , Hassell, A. , Dickerson, S.H. , Ellis, B. , Pennisi, C. , Horne, E. , Lackey, K. , Alligood, K.J. , Rusnak, D.W. , Gilmer, T.M. , Shewchuk, L. , 2004. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res.. 64, 6652–6659. [DOI] [PubMed] [Google Scholar]
- Xia, W. , Mullin, R.J. , Keith, B.R. , Liu, L.H. , Ma, H. , Rusnak, D.W. , Owens, G. , Alligood, K.J. , Spector, N.L. , 2002. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 21, 6255–6263. [DOI] [PubMed] [Google Scholar]
- Yoshioka, A. , Miyata, H. , Doki, Y. , Yasuda, T. , Yamasaki, M. , Motoori, M. , Okada, K. , Matsuyama, J. , Makari, Y. , Sohma, I. , Takiguchi, S. , Fujiwara, Y. , Monden, M. , 2008. The activation of Akt during preoperative chemotherapy for esophageal cancer correlates with poor prognosis. Oncol. Rep.. 19, 1099–1107. [PubMed] [Google Scholar]
- Yu, H.G. , Ai, Y.W. , Yu, L.L. , Zhou, X.D. , Liu, J. , Li, J.H. , Xu, X.M. , Liu, S. , Chen, J. , Liu, F. , Qi, Y.L. , Deng, Q. , Cao, J. , Liu, S.Q. , Luo, H.S. , Yu, J.P. , 2008. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int. J. Cancer. 122, 433–443. [DOI] [PubMed] [Google Scholar]
- Zhou, B.B. , Peyton, M. , He, B. , Liu, C. , Girard, L. , Caudler, E. , Lo, Y. , Baribaud, F. , Mikami, I. , Reguart, N. , Yang, G. , Li, Y. , Yao, W. , Vaddi, K. , Gazdar, A.F. , Friedman, S.M. , Jablons, D.M. , Newton, R.C. , Fridman, J.S. , Minna, J.D. , Scherle, P.A. , 2006. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 10, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the Supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data