

Abstract
Despite recent advancements in multidisciplinary treatments, the overall survival and quality of life of patients with advanced head and neck squamous cell carcinoma (HNSCC) have not improved significantly over the past decade. Molecular targeted therapies, which have been addressed and advanced by the concept of “oncogene addiction”, have demonstrated only limited successes so far. To explore a novel clue for clinically effective targeted therapies, we analyzed the molecular circuitry of HNSCC through the lens that HNSCC is an evolving system. In the trajectory of this somatic evolution, HNSCC acquires biological robustness under a variety of selective pressures including genetic, epigenetic, micro‐environmental and metabolic stressors, which well explains the major mechanism of “escaping from oncogene addiction”. On the other hand, this systemic view appears to instruct us approaches to target latent vulnerability of HNSCC that is masked behind the plasticity and evolvability of this complex adaptive system.
Keywords: Head and neck cancer, Oncogene and non-oncogene addiction, Molecular targeting, Cancer evolution, Robustness
Highlights
-
►
There is an urgent need to develop a novel conceptual framework for the treatment of HNSCC.
-
►
The biological robustness of HNSCC was analyzed through a somatic evolution model.
-
►
This model well explains the mechanism of “escaping from oncogene addiction”.
-
►
We discuss about the possible approaches to target vulnerability of evolving HNSCC.
Abbreviations
- HNSCC
head and neck squamous cell carcinoma
- EMT
epithelial–mesenchymal transition
- MET
mescenchymal epithelial transition
- CSC
cancer stem cell
- EGFR
epidermal growth factor receptor
- HPV
human papilloma virus
- CML
chronic myeloid leukemia
- PDGFR
platelet derived growth factor rcepor
- ILGFR
insulin like growth factor receptor
- Stat3
signal tranducers and activators of transcription
- MAPK
mitogen activated protein kinase
- PI3K
phosphainositide-3-kinase
- CIN85
c-Cbl interacting protein of 85 kDa
- OGG
oxoguanine DNA glycosylase
- LOH
loss of heterozygosity
- SNP
single nucleotide polymorphism
- IAP
inhibitor of apoptosis
- CDC
cell division cycle
- COX
cyclooxygenase
- HIF
hypoxia inducible factor
- PDK
pyruvate dehydrogenase kinase
- TKLT
transketolase-like protein
- ZEB1
zink finger E-box-binding homeobox-1
- HMGA2
high-mobility group AT-hook2
- IL
inteleukin
- ALDH
aldehyde dehydrogenase
- eIF
eukaryotic initiation factor
- HSP
heat shock protein
- TGF
transforming growth factor
- HGF
hepatocellular growth factor
- IGF
insulin like growth factor
- PDGF
platelet derived growth factor
- IL
interleukin
- MMP
matrix metalloproteinase
- VEGF
vascular endothelial growth factor
- PDGF
platelet derived growth factor
- IP-10
interferon-gamma inducible 10 kD protein
- TNF
tumor necrosis factor
- IFN
interferon
1. Introduction
Head and neck squamous cell carcinoma (HNSCC), the sixth most common cancer worldwide, often generates from organs including the larynx, pharynx, oral cavity, and tongue that play critical roles in respiratory, nutritional, social and communicative functions (Hunter et al., 2005; Masuda et al., 2010). Surgical intervention in these organs often leads to a considerable impairment of the patient's quality of life, although remarkable progress has been made in reconstructive surgery. Thus, head and neck surgeons and oncologists have often struggled to balance potentially competing goals of therapy, i.e., organ preservation and survival. Mainly to spare surgical intervention, several dose‐intensified novel treatment protocols have been introduced in the last decade, including concurrent chemoradiotherapy (e.g. clinical trials led by the Radiation Therapy Oncology Group), “super‐selective” intra‐arterial chemoradiotherapy (e.g., RADPLAT), and induction chemotherapy followed by radiation or concurrent chemoradiation (e.g., Tax 323 and Tax 324 protocols) reviewed in refs. Alkureishi et al. (2006) and Haddad and Shin (2008). Preliminary results of these treatments appear promising; however, it remains to be seen whether these dose‐intensified types of approaches will have a definitive and positive impact on both long‐term overall survival and “functional” organ preservation. Moreover these treatments can be associated with considerable complications (e.g., requirement for feeding tube following severe laryngeal and pharyngeal dysfunction) and potential treatment‐related death (Corry et al., 2010; Machtay et al., 2008; Rosenthal et al., 2006). Thus, although there have been recent advances in both surgical and non‐surgical therapy, the overall survival and quality of life of patients with HNSCC have not improved significantly over the past decade, especially for patients with advanced stage.
To resolve this dilemma, growing interest has focused on the development of novel treatment strategies based on the HNSCC cell biology. It is generally accepted that the development and progression of HNSCC occur through the stepwise and progressive accumulation of genetic and epigenetic alterations, following direct and repeated exposures to carcinogens including tobacco, alcohol, and viral infections (Hunter et al., 2005; Leemans et al., 2010). Thus, HNSCC represents the classical models of both multistage carcinogenesis and field cancerization (Hunter et al., 2005). Several investigators (Forastiere et al., 2001; Haddad and Shin, 2008; Leemans et al., 2010; Lippman and Hong, 2001) have proposed HNSCC progression models, in which the normal epithelium undergoes an orderly transformation through hyperplasia, dysplasia, carcinoma in situ and invasive carcinoma in accordance with cumulative genetic and epigenetic alterations including loss of 9p21 (p16‐INK4a), 3p21, 17p13, 13q21, 14q24, 4q26–28, 6p and 8p23 and gain of 11q13 (cyclin D1). Epigenetic silencing of p16‐INK4a and loss‐of‐function mutation of TP53 can also be involved in this process. However, these linear progression models based on intrinsic abnormalities may be overly simplistic, because it has become apparent that malignant phenotypes of cancer are maintained by far more complex system. Numerous intrinsic abnormalities have been reported to be involved in HNSCC pathogenesis and the number of these abnormalities is expanding rapidly (Leemans et al., 2010; Matta and Ralhan, 2009; Molinolo et al., 2009). Moreover, tumor development and progression are not autonomous processes that are achieved by cancer cells alone. The tumor microenvironment profoundly modulates tumor cell biology through interactions between the tumor and the surrounding matrix and non‐cancerous cells therein (e.g., bone marrow derived cells), leading to the development of tumor favorable echo‐systems or niches, in which cancer cells acquire peculiar abilities to survive metabolic stressors including acidosis, hypoxia and malnutrition, to circumvent immune surveillance, and to invade and metastasize to distant organs (De Boeck et al., 2010; Hanahan and Weinberg, 2011; Hoogsteen et al., 2007; Joyce and Pollard, 2009; Kroemer and Pouyssegur, 2008). Other concepts that expanded the complexity of HNSCC carcinogenesis include epithelial–mesenchymal transition (EMT) (Polyak and Weinberg, 2009) and cancer stem cell (CSC) (Brabletz et al., 2005; Chaffer and Weinberg, 2011; Rosen and Jordan, 2009; Visvader and Lindeman, 2008), which are mainly caused by drastic reprogramming of gene expression profile. In addition, the heterogenic cells in HNSCC might mutually influence their internal circuitry. In order to organize our current knowledge on HNSCC biology, we have numerated potential biomarkers related to HNSCC pathogenesis, according to their main biological functions in Tables 1 and 2.
Table 1.
Intrinsic and cell surface factors associated with HNSCC pathogenesis.
| Function | Factors | Refs. no in foot note |
|---|---|---|
| 1. Agonist dependent or independent signal transduction | EGFR, erbB2 (HER2), PDGFR, IGFR, c‐Met, src, Ras, Stat3, MAPK, NF‐κB, PI3K‐PTEN‐AktmTOR, Wnt/β‐catenin, TGF‐β/smad, CXCR, p38, CIN85 | 1 |
| 2. DNA replication and repair | p53, nucleotide excision repair (NER), OGG | 2 |
| 3. Genetic and chromosomal instability | chromosomal number variation, DNA aneuploidy, LOH, SNP, chromosomal rearrangemen, | 3 |
| 4. Cell fate, survival, differentiation, senescence, and apoptosis | Telomerase, Survivin, Bcl‐2/XL, Bax, IAPs, p53, Myc | 4 |
| 5. Mitotic regulation | aurora kinase A | 5 |
| 6. Cell cycle and check point control | cyclin D1, p14, p16, p21, p27, pRB, CDC25A,CDC25B | 6 |
| 7. Cell adhesion and motility | integrins, E‐cadherin, CD44, moesin, laminin | 7 |
| 8. Transcription, translation post‐translation regulation and chromatin remodeling | DNA methylation, miRNA, eIF4, S6, HSP, BMI1, EZH2 | 8 |
| 9. Viral infection | human papilloma virus, Epstein–Barr virus | 9 |
| 10. Intracellular metabolism | aerobic glycolysis (Warburg effect), hypoxia, xenobiotics, prostaglandin synthesis, Cox‐2, reactive oxygen species (ROS), HIF‐1, mitochondrial DNA mutation, PDK1, LDH, TKLT‐1, Myc, AktmTOR | 10 |
| 11. Epithelial–mesenchymal transition | Src, BMI1, ZEB1, TrkB, Cox‐2, HMGA2, IL‐1β, Snail, Twist, E‐cadherin, Vimentin | 11 |
| 12. Cancer stemness | CD44, Notch, Wnt, Oct, Nanog, Stat3, CD133, ALDH1 | 12 |
References: 1 ‐ Allen et al. (2007), Barnes et al. (2007), Bran et al. (2009), Hunter et al. (2005), Junttila et al. (2007), Knowles et al. (2009), Masuda et al. (2010), Molinolo et al. (2009), Ruan et al. (2006), Sen et al. (2009), Wakasaki et al. (2010); 2 ‐ Hunter et al. (2005), Paz‐Elizur et al. (2008), Wang et al. (2007); 3 ‐ Chen and Chen (2008), Ha et al. (2009), Hopkins et al. (2008); 4 ‐ Hunter et al. (2005), Masuda et al. (2010); 5 ‐ Guan et al. (2007), Reiter et al. (2006); 6 ‐ Gasparotto et al. (1997), Hunter et al. (2005), Todd et al. (2002); 7 ‐ Gonzalez‐Moles et al. (2003), Gonzalez‐Moles et al. (2007), Hunter et al. (2005), Kuratomi et al. (2006), Masuda et al. (2000), Van Waes et al. (1995); 8 ‐ Kang et al. (2010), Molinolo et al. (2009), Tran et al. (2010), Wang et al. (2011a), Yang et al. (2010); 9 ‐ Chung and Gillison (2009), Lo et al. (2006). 10 ‐ Hoogsteen et al. (2007), Koukourakis et al. (2009); Lin et al. (2002); McFate et al. (2008); Schneider et al. (2008); Sun et al. (2010); Wigfield et al. (2008); Zhou et al. (2007b). 11 ‐ Dohadwala et al. (2010), Grille et al. (2003), Kupferman et al. (2010), Mandal et al. (2008), Miyazawa et al. (2004), Song et al. (2009), St John et al. (2009), Yang et al. (2010). 12 ‐ Agrawal et al. (2011), Chen (2009), Chiou et al. (2008), Clay et al. (2010), Masuda et al. (2010), Prince et al. (2007), Song et al. (2010), Stransky et al. (2011), Yang et al. (2010).
Table 2.
Extrinsic factors associated with HNSCC pathogenesis.
| Function | Factors | Refs no in foot note |
|---|---|---|
| 1. Mitogens, adhesion and motility molecules,and proteases | TGF‐α, Amphiregurlin, HGF, IGF, PDGF, ILs, MMPs, chemokines | [1] |
| 2. Non‐cancerous cells in stroma | Bone marrow derived cells, cancer associated fibroblast, tumor associated macrophage,Treg | [2] |
| 3. Angiogenesis | VEGF, PDGF | [3] |
| 4. Extracellular metabolism | acidosis, hypoxia, ROS | [4] |
| 5. Immune response | Rantes, IP‐10, TNF‐α, IFN‐γ, VEGF | [5] |
References: 1 ‐ Barnes et al. (2007), Bran et al. (2009), Hunter et al. (2005), Knowles et al. (2009), Reuter et al. (2007). 2 ‐ De Boeck et al. (2010), Strauss et al. (2007), Tabachnyk et al. (2012); 3 ‐ Bran et al. (2009), Matta and Ralhan (2009); 4 ‐ Hoogsteen et al. (2007); 5 ‐ Jewett et al. (2006).
In general, this overflowing information does not lead to the identification of the critical molecular target or biomarker of HNSCC, except for the recent finding of human papilloma virus (HPV)‐positive oropharyngeal cancer. Since a majority of these cancers demonstrate quite favorable response to organ‐preserving treatments, they are now recognized as a distinct entity of HNSCC that can be cured within the framework of conventional treatments (Ang et al., 2010; Chung and Gillison, 2009; Leemans et al., 2010). In contrast, ongoing targeted therapy for HPV‐negative HNSCC has had limited success, mainly with the use of epidermal growth factor receptor (EGFR) inhibitors, either used alone or in combination with radiation (Bernier et al., 2009; Specenier and Vermorken, 2012). This is partly because the predominant driver mutation or genetic abnormality in oncogene (e.g., BCR‐ABL in CML or BRAF in malignant melanoma) (Chapman et al., 2011; Hughes et al., 2003) has not been discovered in HNSCC (Agrawal et al., 2011; Stransky et al., 2011). Nevertheless, it is obvious that this limited success mainly results form our paucity of insight into the molecular mechanism that drives HNSCC development and progression. For example, the major mechanism that causes the frequent (>80%) activation of EGFR signaling in HNSCC is not known, although its significant role in HNSCC development and progression has been highlighted for more than two decade (Masuda et al., 2011). The purpose of this perspective review is to illuminate current obstacles to and future directions for clinical effective targeted therapies based on the concept that HNSCC is an evolving system.
2. Molecular circuitry of HNSCC and oncogene addiction
It is now apparent that the development and progression of HNSCC are not a consequence of the simple summation and accumulation of the intrinsic and extrinsic abnormalities mentioned above (Tables 1 and 2). In cancer cells, many of these abnormalities interact with each other through complex networks and form a highly integrated “molecular circuitry” in order to maintain the malignant phenotypes of cancer during tumor progression (Molinolo et al., 2009; Weinstein, 2000; Weinstein et al., 1997). Despite this complexity, reversal of only one or a few genetic abnormalities can profoundly inhibit cancer cell growth and in some cases translate to improved clinical survival rates; a phenomenon termed “oncogene addiction” (as well as tumor suppressor gene hypersensitivity) proposed by our mentor Dr. Weinstein (Weinstein et al., 1997). Examples of this phenomenon were observed in mechanistic studies in human cancer cell lines, genetically engineered mouse models of human cancer, and clinical trials with molecular targeted agents (Weinstein, 2000, 2002; Weinstein and Joe, 2008, 2006). The concept of oncogene addiction has been generally accepted as a rationale for molecular targeted therapy, and an increasing number of studies and reviews on this subject have been published (Baylin and Ohm, 2006; Felsher, 2008; Garraway and Sellers, 2006b; Grant, 2008; Kaelin, 2005; Kroemer and Pouyssegur, 2008; Luo et al., 2009; Masuda et al., 2010; Mills et al., 2001; Sharma and Settleman, 2007). Although the precise mechanism of oncogene addiction is not fully known, Dr. Weinstein speculated that tumor specific homeostasis that is maintained by “bizarre” molecular circuitry might play a fundamental role in the development of this phenomenon (Weinstein, 2000; Weinstein and Joe, 2006). For example, probably to encounter the effects of growth enhancing molecules, paradoxical increases of tumor suppressive molecules are frequently observed in this bizarre circuitry (Weinstein, 2000). This situation would be analogous to a car driving at high speeds with both the accelerator and brake engaged simultaneously. Hence, when the accelerator is released (i.e., the effects of tumor enhancing molecules are inhibited), the car comes to a sudden stop (i.e., cell ceases to proliferate or undergoes apoptosis). In the concepts of “oncogene shock” and “oncogene amnesia”, Sharma and Settleman (2007), and Felsher (2008) also proposed this homeostatic feedback theory to explain the mechanism of oncogene addiction. In addition to this feedback theory, there are several possible explanations for the mechanisms of oncogene addiction such as genetic streamlining, or synthetic lethality (Kaelin, 2005; Mills et al., 2001). Due to the genetic streamlining model, once a cancer cell becomes highly and continuously dependent on a given proliferative signal, other collateral growth signals regress and eventually the remaining signal predominates proliferation (Mills et al., 2001). As a result, if this predominant signal is inhibited, the cancer cell cannot survive any more losing the driving force for proliferation. The synthetic lethality concept describes a condition in which combinational mutations in two genes leads to cell death, whereas mutation in one of two genes is compatible with survival (Kaelin, 2005). A clinical manifestation of this phenomenon was discovered in a recent study (Fong et al., 2009). Inhibition of poly (ADP‐ribose) polymerase (PARP) has been shown specifically kill cancer cells including ovarian, breast and prostate cancer that have mutations in BRCA1 and BRCA2. In this setting, PARP and BRCA, are paralogous genes in their ability to repair DNA damage by the homologous recombinant pathway. Simultaneous inhibition of these two paralogs results in synthetic lethality. The theory of “molecular house of cards” (Kaelin, 2005) also explain the mechanism of synthetic lethality. In the molecular circuitry of cancer that proceeds with accumulative genetic aberrations, each additional mutation must be beneficial (adaptive) or at least neutral to pre‐existing mutations for the maintenance of cellular integrity. Due to this biological restriction, the molecular circuitry of cancer presumably falls into the state of molecular house of cards in accordance with tumor progression. Thus, extraction of jut one card (i.e., reversal of one genetic abnormality) might lead to the collapse of the house. Of note, the clinical example of synthetic lethality has not yet been discovered in HNSCC.
Indeed, examples of bizarre molecular circuitry of HNSCC have been demonstrated in clinical samples (Homma et al., 1999; Lo Muzio et al., 2005; Naresh et al., 2001; Pena et al., 1999; Wilson et al., 2001). In several studies, the increased Bcl‐2 expression or low‐apoptotic index paradoxically associated with favorable outcomes in patients with early stage HNSCC (Homma et al., 1999; Lo Muzio et al., 2005; Naresh et al., 2001; Pena et al., 1999; Wilson et al., 2001). These counterintuitive findings may imply that in a subset of early stage HNSCC, anti‐apoptotic signals protect cancer cells from apoptosis in order to maintain a net volume of tumor mass that is mainly composed of less progressive and therefore slow‐growing cell population. Sathyan et al. (2007) also demonstrated an intriguing finding in 151 cases of oral cancer obtained from patients in India, which were known to have a rare TP53 mutation. They found that in these samples, the activating H‐ras mutations significantly correlated with lower expression of cyclin D1 and CDK4, higher expression of pRB and p16INK4A, and thereby a better prognosis. Although the precise mechanism of these paradoxical associations between oncogenic and tumor suppressive proteins is not clear, this setting, at least in part, mimics the findings observed in HPV‐positive oropharyngeal cancer patients. They demonstrate a significantly favorable response to chemotherapy or chemoradiation due to the distinctive molecular profiling: wild type but inactivated p53 protein by viral E6, inactivated pRB protein by viral E7 and high p16INK4A expression (Ang et al., 2010; Chung and Gillison, 2009; Leemans et al., 2010). It might be also relevant to this favorable outcome that HPV‐positive HNSCC is genetically more stable than HPV‐negative HNSCC: 2.28 vs 4.38 mutations per mega base in whole‐exome sequencing and 19 vs 119 somatic rearrangements in whole‐genome sequencing (Leemans et al., 2010; Stransky et al., 2011). In a recent commentary article to the study by Ang and colleagues (Ang et al., 2010), Lowy and Munger postulated an interesting theory that in HPV‐positive HNSCCs chemoradiation might reactivate the function of wild type p53 and pRB by reducing the levels of viral E6 and E7 protein, and thereby induce G1 check point control and consequent apoptosis, resulting in the better response (Lowy and Munger, 2010). This scenario well exemplify an example of oncogene addiction in HNSCC, thus HPV‐positive HNSCC is highly dependent on (e.g., addicted to) E6 and E7 proteins for the maintenance of malignant phenotype.
3. Cancer evolution and the mechanisms of escaping from “oncogene addiction”
Despite the fact that the concept of oncogene addiction has gained wide acceptance as a rationale for molecular targeting, a majority of these therapies have demonstrated only marginal effects so far on a variety of malignancies including HPV‐negative HNSCC. Sharma and Settleman described this frustrating situation referring to the “one–two punch” – “oncogene addiction has set the stage for the knock‐out by the first punch but is holding the devastating second” (Sharma and Settleman, 2007). Based on the recent conceptual framework on cancer biology, the main reason of this limited success could be attributed to the cancer evolution theory, in which cancer is considered as a peculiar organ or an adaptive system composed of heterogeneous cell population, because this complex machinery can demonstrates amazing plasticity and robustness in this evolving process (Greaves and Maley, 2012; Kitano, 2004; Merlo et al., 2006; Nowell, 1976; Tian et al., 2011). Consistent with the Darwinian principle, this somatic evolution proceeds under harsh selective pressures including a variety of genetic, epigenetic, metabolic, immunologic and micro‐environmental stressors that are primarily deleterious for the maintenance of cellular integrity (Luo et al., 2009). To survive and overcome these stressors, cancer cell must continuously reprogram its internal wiring diagram of molecular circuitry by cunningly orchestrating cancer causing (adaptive), non‐cancer causing (non‐adaptive), and housekeeping maneuvers or nodes to avoid systemic failure (i.e., cell death), and thereby achieve a state of cancer specific homeostasis or equilibrium. This intracellular molecular landscape or gene regulatory network is termed “cancer attractor”, borrowing visual representation of the normal cell differentiation trajectory originally proposed by Waddington in 1957 (Brock et al., 2009; Huang et al., 2009; Marusyk et al., 2012). In Figure 1, we schematically proposed a model of evolutionary trajectory of HNSCC based on the concept of cancer attractor. As opposed to the normal cell differentiation, in which developing cells flow down the surface of rugged slope, cancer evolutionary trajectory is supposed to be uphill movement in terms of biological potential (i.e., fitness). A clone acquired novel driver mutation, epigenetic abnormality, or altered gene expression profile in response to selective pressures can go up to a higher attractor state toward de‐differentiation. In accordance with tumor evolution, it is obvious that each attractor become more complex, because the internal molecular circuitry has to process exponentially growing information, as exemplified by the accumulated number of mutations or drastic change in gene expression profile caused by chromatin remodeling, which is often observed in advanced stage tumor. Due to principals of systems biology (Kitano, 2004; Tian et al., 2011), this complexity provides cancer cells with biological robustness by making the basin of attractor deeper (i.e., stable), although, in parallel, the hurdle to the next attractor becomes higher (Figure 1). Consequently, the molecular circuitry of cancer cells in higher attractor demonstrates remarkable resilience or adaptability to more severe selective pressures (Brock et al., 2009; Gillies et al., 2012; Marusyk et al., 2012), whereas that of lower attractors tends to be unstable. This is consistent with clinical evidence that a majority of HPV‐negative HNSCCs with advanced stage show remarkable resistance to chemo/radiation. It is of note that an individual tumor is composed of heterogeneous cancer cells, which inhabit different attractors. We linked tumor evolutionary trajectory to hallmarks of HNSCC in Figure 1, which is discussed later.
Figure 1.
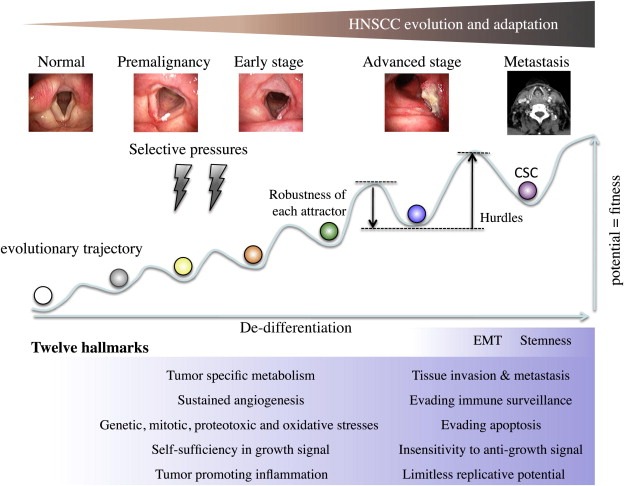
Tumor evolutionary trajectory and twelve hallmarks of HNSCC. HNSCC evolves form normal epithelium to advanced tumors. In this process, intracellular molecular circuitry or gene regulatory network achieves transient equilibrium state, “cancer attractor”, which is depicted as a basin in the trajectory. A variety of genetic, epigenetic, micro‐environmental and metabolic stressors work as selective pressures. To acquire advanced fitness, immature clone goes up to the higher attractor state. In accordance with evolution, the state of each attractor becomes more robust, although, in parallel, the hurdle to the next attractor becomes higher. When compared to the normal cell differentiation trajectory for which visual representation of attractor was originally developed, cancer evolutionary trajectory seems to be the reveres movement, i.e., de‐differentiation. In our opinion (see Figure 2), cancer stem cell (CSC) of HNSCC (purple) lies in the highest attractor. Of note, for simplification we depicted the trajectory of HNSCC in a two‐dimensional format, but it should be multi‐dimensional and the trajectory is not a single route. We postulate that in this evolutionary trajectory HNSCC acquires twelve hallmarks. As discussed in Figure 2 and text, we categorized the epithelial–mesenchymal transition (EMT) and CSC as the distinctive properties that the most advanced HNSCC display.
This scenario well explains the mechanism of oncogene addiction as well as that of “escaping from oncogene addiction” (i.e., tolerance) – a dismal phenomenon that is frequently observed in molecular targeting therapies. As cancer proceeds in its evolutionary trajectory, a tumor mass becomes more heterogeneous involving diverse clones in higher attractors, which demonstrate stronger resistance to molecular targeting and chemo/radiation through either acquired (i.e., pre‐existing) or de novo mechanism achieved by additional mutation or non‐mutagenic adaptation (Brock et al., 2009; Gillies et al., 2012; Marusyk et al., 2012; Stratton, 2011). As a result, even if a given molecular targeting therapy or chemo/radiation can kill a majority of clones, the increased emergence of these refractory clones, particularly in advanced HNSCC, leads to the re‐growth of tumor.
Taken together, it should be emphasized that for evolving cancers, molecular targeting as well as conventional DNA‐damaging treatment might be just another selective pressure for further evolution, because they might promote selective sweep and/or emergence of resilient cells.
4. Targeting hallmarks of HNSCC
4.1. Twelve hallmarks of HNSCC
In view of this evolutionary trait, it seems quite difficult to timely identify the Achilles' heal of HNSCC within the complex and dramatically changing molecular circuitry. However, despite a myriad of intrinsic and extrinsic alterations, cancer evolution proceed with the acquisition of a series of malignant phenotypes that can be boiled down into several properties, as first categorized by Hanahan and Weinberg into six hallmarks of cancer (Hanahan and Weinberg, 2000). To date, several additional traits were proposed as additional hallmarks by four research groups including a recent revision by Hanahan and Weinberg (Colotta et al., 2009; Hanahan and Weinberg, 2011; Kroemer and Pouyssegur, 2008; Luo et al., 2009). Moreover, in view of the impact of two fundamental discoveries, EMT and CSC, we propose a model of the 12 hallmarks of solid tumors that might be applicable to HNSCC (Figure 1): (1) self sufficiency in growth signals, (2) insensitivity to growth inhibitory signals, (3) evasion of programmed cell death, (4) limitless replicative potentials, (5) sustained angiogenesis, (6) tissue invasion and metastasis (7) evading immune surveillance, (8) cancer promoting inflammation, (9) genetic instability, mitotic stress, proteotoxic stress, and oxidative stress, (10) cancer specific metabolism, (11) EMT, and (12) CSC. Rather than the perspective of the reductionist that has governed cancer researches for a long time, these hallmarks‐oriented views might better provide us a clue to identify the Achilles' heal of an evolving system.
4.2. Targeting EMT and CSC for improved chemo/radioselection
In our categorization of HNSCC hallmarks, we ranked the EMT and CSC at the top of the hierarchy (i.e., the most malignant phenotypes) (Figure 1). This view is based on our hypothesis depicted in Figure 2. The CSC population of HNSCC is likely to originate from clonally expanding cancer cells (Figure 1) at a relatively advanced phase of tumorigenesis, because a majority of early stage HNSCCs, in particular, Stage I and II laryngeal and oropharyngeal cancers, are cured by chemoradiation or radiation alone and seldom recur or metastasize (Cohan et al., 2009; Hirasawa et al., 2010; Kumamoto et al., 2002; Selek et al., 2004). Therefore, early stage HNSCC apparently lacks the distinctive properties of CSC: strong resistance to chemoradiation (Baumann et al., 2008; Dean et al., 2005). Hence, the emergence of the CSC in HNSCC appears to occur at a relatively advanced phase of tumor progression. However, this model does not exclude the possibility that CSCs of HNSCC originate from normal stem cells that have experienced genetic and epigenetic alterations (Figure 2). Our definition of CSC in HNSCC is akin to that proposed by Baumann et al (Baumann et al., 2008): a CSC is the cell that has capacity to cause tumor recurrence when left after irradiation in its natural environment. Intriguingly, recurrent and metastatic tumors seldom respond to chemotherapeutic agents and often grow at an amazingly rapid speed leading to the death of the host on the order of months. Thus, these tumors are likely composed of relatively homogeneous cells that have two distinctive traits: insensitivity to DNA‐damaging therapies and ability to rapidly expand gross tumor volume (Figure 2). Presumably recurrent or metastatic tumor cells are mainly composed of advanced cancer cells in the higher cancer attractors, which originate (i.e., fall down) from the CSC attractor (Figure 1). Thus, these cells can grow fast escaping from quiescent state of CSC, but still retain the distinctive feature of “stemness”, strong protection from apoptosis (Visvader and Lindeman, 2008).
Figure 2.
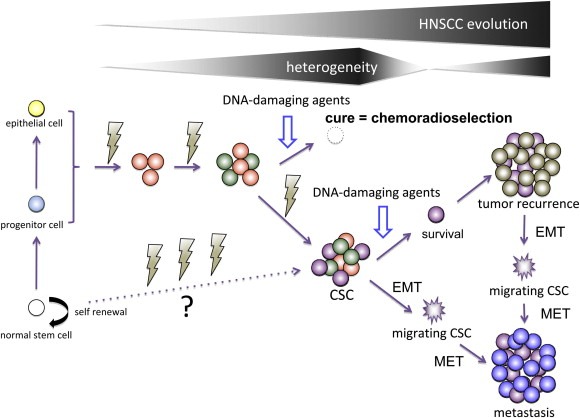
Proposed mechanism of the development of cancer stem cell (CSC) (purple) in HNSCC and its relation to sensitivity to DNA‐damaging agents, and tumor recurrence and metastasis. CSCs of HNSCC appear to originate from cancer cells that have undergone processes of multistage carcinogenesis (orange and green) at the advanced stage of tumor progression, since a majority of early stage tumors are cured by chemoradiation or irradiation alone (i.e., chemoradioselection) lacking the CSC phenotype. Thus, the origin of HNSCC is not likely CSC. However, this model does not exclude the possibility that CSCs of HNSCC originate from normal stem cells (white) that accumulated a series of abnormalities. CSCs can survive treatments with DNA‐damaging agents and propagate recurrent tumor. CSCs that have undergone epithelialߝ;mesenchymal transition (EMT) can migrate as “migrating CSCs” (jagged purple) and propagate metastatic tumors after they have, in turn, undergone mesenchymal epithelial transition (MET). Recurrent and metastatic tumors appear to be composed of relatively homogeneous cell population that that has two distinctive properties: insensitive to DNA‐damaging therapy and ability to rapidly expand gross tumor volume. For more details see text.
As described in recent reviews (Brabletz et al., 2005; Chaffer and Weinberg, 2011; Hanahan and Weinberg, 2011; Polyak and Weinberg, 2009), the concepts of EMT and the CSC are closely related and the dynamically changing phenotypes displayed by individual cancer cells during EMT or MET appear to be compatible with stem cell or multi‐potential progenitor cells. Thus, in this review we support the migrating CSC theory: CSCs that undergo EMT acquire the abilities to migrate, metastasize and propagate metastatic tumors (Figure 2) (Brabletz et al., 2005). Taken together, we consider EMT and stemness as distinctive properties displayed by the most evolved cancer cells that have acquired invulnerability as well as pluripotency to transform into phenotypically diverse cells (Figure 1).
Within the last decade, the mainstay of definitive treatment for advanced HNSCC has shifted from radical surgery plus post‐operative irradiation to organ‐preserving strategies, employing dose‐intensified chemotherapy and/or chemoradiotherapy (Haddad and Shin, 2008). During this shift, the novel concept of chemoselection has emerged as a rationale for organ preservation (Urba et al., 2006; Worden et al., 2008, 2009). This concept highlights the observation that those patients who display favorable responses to induction chemotherapy tend to be cured by additional chemoradiotherapy without surgical intervention, thereby facilitating organ preservation. In line with this concept, we have developed a program of concomitant chemoradiation, including a course of 30–40 Gy, as a means of “selection” (i.e., chemoradioselection) for more than 30 yeas (Kumamoto et al., 2002; Masuda et al., 2012). Accordingly, if we could reverse the EMT and CSC hallmarks in HNSCC, which is now considered to be the major reason for chemo/radio resistance, an increasing number of patients might benefit from chemoradioselection (Figure 2), achieving organ preservation.
Recent studies on CSC and EMT in HNSCC are summarized in Table 1. Several markers that could be used in the isolation of CSCs of HNSCC have been identified including CD44, CD133 and ALDH (Chen, 2009; Clay et al., 2010; Prince and Ailles, 2008). However, it is of note that the reliability of CD44 as a marker of HNSCC stemness was questioned by Mack and Gires (2008), because frequent CD44 expression can be observed in both normal and carcinoma tissues of the head and neck. Consistent with our idea that the process of HNSCC evolution is that of de‐differentiation (Figure 1), recent studies of HNSCC have demonstrated constitutive activation of several embryonic markers or molecules that play critical roles during organogenesis, maintenance of cellular lineage, wound healing, and differentiation. These include Stat3, Oct‐4, Nanog, BMI1, Wnt, and Notch (Agrawal et al., 2011; Chen et al., 2009; Chiou et al., 2008; Masuda et al., 2010; Prince et al., 2007; Song et al., 2010; Stransky et al., 2011; Zhang et al., 2010). In general, our understanding of the molecular mechanisms of CSC development in HNSCC remains limited.
It has been demonstrated that several signaling axes or molecules can promote EMT in HNSCC including, IL‐1β/Cox‐2/ZEB1, src, TrkB/Twist, BMI1/PTEN/PI3K/Akt, and BMI1/Twist, causing the downregulation of E‐cadherin (Table 1) (Dohadwala et al., 2010; Grille et al., 2003; Kupferman et al., 2010; Mandal et al., 2008; Song et al., 2009; St John et al., 2009). Overexpression of the mesenchymal marker, HMGA2 that was observed in more than 70% oral squamous cell carcinoma tissues, was significantly associated with poor outcomes in patients (Miyazawa et al., 2004). A recent study demonstrated Twist1 and BMI1, interacting each other, promote tumor acquisition of both EMT and CSC traits and thereby worsen the prognosis of patients with HNSCC (Yang et al., 2010). Furthermore, the existence of migrating CSCs (Figure 2) in HNSCC was demonstrated by a recent study, in which a highly metastatic subpopulation of cancer cells selected from a xenograft mouse model expressed high levels of CSS markers, including CXCR4 and integrin β1, and altered levels of EMT markers such as E‐cadherin and vimentin (Chen, 2009). Biddle et al. (2011) also revealed that in HNSCC cell line, there is a population of CSC that has undergone EMT. This population, they termed EMT CSCs, display the expression levels of high CD44 and low ESA. These findings suggest that fundamental functions of CSC and EMT indeed overlap in HNSCC.
The processes by which cancer cells acquire the traits of stemness and the ability to dynamically change their morphology and motility through EMT (i.e., formation of migrating CSC) (Figure 2) are presumably a consequence of drastic reprogramming (e.g., chromatin remodeling and transcriptional reprogramming) of molecular circuitry toward a state of de‐differentiation, that may resemble embryonic development and wound healing (Bracken and Helin, 2009). This idea was exemplified in studies with high‐throughput expression profiling analyses that displayed a “lineage addiction” model of human melanoma (Garraway and Sellers, 2006, 2006; Garraway et al., 2005), “wound signature” profile of breast cancer (Chang et al., 2005, 2004; Wong et al., 2008b), and embryonic stem cell‐like gene expression signature in several types of cancer (Ben‐Porath et al., 2008; Wong et al., 2008a). Consistent with these findings, two whole‐exome sequencing studies recently demonstrated that the mechanism of quamous differentiation might be genetically dysregulated in HNSCC (Agrawal et al., 2011; Stransky et al., 2011). They identically discovered frequent loss‐of‐function mutation in Notch, thus demonstrating its tumor suppressive role. Stransky et al. (2011) further analyzed their data and speculated that a maturation arrest (i.e., de‐differentiation), caused by mutations in several genes that regulate squamous differentiation (e.g., Notch and TP63), may play an important role in HNSCC carcinogenesis. The reported roles of Notch in carcinogenesis (i.e., oncogenic or tumor suppressive) have been controversial so far (Ranganathan et al., 2011). However, these reports, taken together with recent similar findings of frequent Notch mutation in SCC of the skin and lung (Wang et al., 2011b), suggest that, at least in SCC, Notch appears to function as a tumor suppressor. Collectively, further insights into the mechanism of the squamous cell differentiation in HNSCC might lead to the effective molecular targeting that can reverse EMT and CSC hallmarks and thereby improve the effects of chemoradioselection (Figures 1 and 2). In addition, it appears to be of great importance to design mechanistic studies to gain insight into the roles of chromatin remodeling in the induction of EMT and CSC in HNSCC.
4.3. Targeting the “CPU” in the molecular circuitry of HNSCC
Despite a myriad of experiments data (Tables 1 and 2) including the two whole‐exome sequencing studies mentioned above (Agrawal et al., 2011; Stransky et al., 2011), predominant gain‐of‐function mutations in oncogenes have not yet been identified in HNSCC. On the other hand, the most prevalent genetic alterations or epigenetically silenced genes are mainly observed in tumor suppressors including TP53, CDKI and Notch (Stransky et al., 2011). These findings raise a fundamental question: who drives HNSCC? A possible explanation may be that throughout the development and progression of HNSCC, the driving force is generated by the dynamic interactions of multiple non‐mutated growth enhancing factors, rather than by a few mutated oncogenes. This distinctive characteristic of HNSCC is apparently a major hurdle that prevents clinically effective molecular targeting. However, this setting might be a double‐wedged sword, because HNSCC has to constantly and stably generate driving force through the contextual stoichiometry of multiple intracellular molecules under a variety of intrinsic and extrinsic selective pressures. Otherwise HNSCC is not able to climb or rather falls down from the evolutionary trajectory (Figure 1). Theoretically, if we could identify a molecule that function as a central processing unit (CPU) to generate the driving force for a relatively long span of HNSCC evolution, inhibition of this molecule might cause considerable damage to HNSCC. As with computer circuitry, the “CPU molecule” must be a processor of multiple incoming information signals (inputs) and as well as an integrator and a transmitter of outgoing signals (i.e., driving force) to the peripheral devices (outputs). In cancer cell biology inputs correspond to a variety of information including mitogenic stimuli, exposure to carcinogens, crosstalk with cells in surrounding stroma, environmental conditions, and genetic and metabolic stressors. On the other hand, outputs are comparable to a variety of biological events including cell cycle progression, check point control, immortalization, cell fate determination: senescence, apoptosis, necrosis and proliferation, modulation of angiogenic switch, and enhancement of cellular motility, which are the fundamental components of cancer hallmarks.
Among the various factors that can drive HNSCC development and progression (Tables 1 and 2), Stat3, NF‐κB and mTOR appear to play the role of potential “CPU” molecules. These three signaling axes are frequently activated in subsets of HNSCC and underlie a variety of input signals mentioned above (i.e., CPU inputs). Moreover, they can promote cell proliferation, cell cycle progression, survival, angiogenesis, motility, immune evasion, tumor‐promoting inflammation, cellular metabolism, EMT and stemness (i.e., CPU outputs), through modulating the expression levels of a variety of effecter proteins (Amornphimoltham et al., 2005; Colotta et al., 2009; Engelman, 2009; Guertin and Sabatini, 2007; Masuda et al., 2010; Molinolo et al., 2007; Pedrero et al., 2005; Ruan et al., 2006; Van Waes, 2007; Yu et al., 2009). As a result, inhibition of Stat3, NF‐κB or mTOR is likely to cause substantial damage to cancer cells in which these signals are constitutively activated. However, it might be optimistic to expect that single inhibition of only one of these signals may lead to complete regression, because HNSCC cells may survive these heavy “blows” by compensating each other as a surrogate CPU. This speculation is based on the findings that these three signaling can modulate a surprisingly overlapping set of downstream molecules (e.g., c‐myc, cyclin D1 and IAPs) and as well there is crosstalk or feedback among them (Colotta et al., 2009; Squarize et al., 2006; Yu et al., 2009; Zhou et al., 2007a). In addition, the effects of the single inhibition might be attenuated by the heterogeneity of HNSCC. However, considering the pivotal roles of these three molecules in the molecular circuitry of HNSCC, dual or triple inhibition might surpass the robustness of HNSCC. Since small molecule inhibitors of Stat3, NF‐κB and mTOR are being developed and clinically available (Engelman, 2009; Masuda et al., 2010; Ruan et al., 2006; Specenier and Vermorken, 2012), the timing might be optimal for the development of multiple CPU targeting on condition that the problem of possible toxicities will be addressed.
4.4. Targeting non‐oncogene addiction – tumor specific metabolism
Another clue for an effective molecular targeting was introduce in a recent review by Luo et al. (2009). They extended the cancer addiction concept from oncogene to non‐oncogene and described its significance in relation to the five stress phenotypes of cancer. Non‐oncogene addiction describes a phenomenon in which cancer cell growth and survival strongly depend on the activities of a wide variety of genes and pathways that are not inherently oncogenic themselves. Intriguingly, activation of these genes or pathways appears to play a far more essential role in the maintenance of cancer specific phenotypes than in that of normal cell viability. This observation seems to be of particular interest in the distinctive molecular circuitry of HNSCC that lacks predominant driver mutation in oncogenes as mentioned above. Among the five stress types of cancer, the tumor specific metabolism seems to be the most attractive target, because this concept provides a distinctive view that cancer is a peculiar living organ in human body (DeBerardinis et al., 2008; Hanahan and Weinberg, 2011; Kroemer and Pouyssegur, 2008; Tennant et al., 2010). In the 1920s, Warburg found that cancer cells consume glucose at a surprisingly high rate and secrete most glucose‐derived carbon as lactate, rather than oxidizing it completely in the tricarboxilic acid (TCA) cycle even under high oxygene condition – a phenomenon known as the “Warburg effect” (Warburg, 1923). The reason why cancer cells employ this inefficient metabolic system as the source of ATP is not clear but must be related to their ability to adapt cancer specific environment. In a recent comprehensive review, Kroemer and Pouyssegur (2008) summarized the attributes of the cancer specific metabolic system into four points. First, in this setting cancer cells can live in conditions of fluctuating oxygen tension. Second, an acidic condition generated by cancer‐derived lactic acid is advantageous for tumor invasion and immune suppression. Moreover lactate produced by cancer cells is taken up by stromal cells to generate pyruvate that, in turn, refuels cancer cells. In this microecosystem, anaerobic cancer cells utilize aerobic stromal cells as a source of energy for survival and proliferation through these complementary metabolic systems. Third, nicotinamide adenine dinucleotide phosphate (NADPH) produced through the pentose phosphate pathway (PPP) protects cancer cells against oxidative damage derived from their microenvironment or chemotherapeutic agents. Fourth and perhaps most importantly, cancer cells obtain high demanding biomaterials for de novo cell replication (i.e., tumor proliferation) via glucose degradation. To this end, intermediates generated in the glycolytic pathway are used for biosynthesis of nucleotides, lipids, and amino acids rather than oxidative phosphorylation. Collectively, tumor specific aerobic glycolysis (i.e., “Warburg effect”) may describe a much more sophisticated setting than Warburg originally anticipated, clearly reflecting the processes of cancer evolution.
Several recent findings demonstrated that HNSCC is indeed dependent on this peculiar glycolysis pathway. The list of dysregulated molecules in HNSCC metabolism is shown in Table 1. In brief, upregulation of PDK1, LDH, TKTL1, HIF‐1α, and ROS, frequent (about 50%) mitochondrial DNA mutations and constitutive activation of the Akt/mTOR pathway were associated with enhanced cellular growth and in some cases with poor prognosis of patients by advancing Warbrurg metabolic phenotype (Engelman, 2009; Hoogsteen et al., 2007; Koukourakis et al., 2009; McFate et al., 2008; Molinolo et al., 2009; Schneider et al., 2008; Sun et al., 2010; Tennant et al., 2010; Wigfield et al., 2008; Zhou et al., 2007b). Of note, there were strong interactions within these factors.
Collectively, “glycolysis addiction” might be a highly exploitable target in HNSCC for three reasons. First, Warburg effect might be a necessary condition for the maintenance of other hallmarks of HNSCC, especially, in advanced stage, because without a method to meet the excessive demand for energy and biomaterials, cancer cannot progress beyond a certain point: probably quite early stage. Second, even for highly adapting cancer cells it seems difficult to establish de novo alternative metabolic pathways independent of excessive glucose supply, so far as they are constituents of human body. Third, taken together, inhibition of aerobic glycolysis might surpass the plasticity of HNSCC and thereby can cause the drastic reversion of HNSCCs to early stage tumors, a majority of which could be cured by conventional DNA‐damaging modalities – the strategy of chemoradioselection mentioned above. A recent finding that addition of 2‐deoxy‐d‐glucose, an inhibitor of glucose metabolism, to cisplatin demonstrated enhanced cytotoxicity in HNSCC cell lines, at least in part, supports this idea (Simons et al., 2007). The development of anti‐Warburg therapy seems promising.
5. Molecular diagnosis for the ideal molecular targeting
In this perspective review, we have discussed about working hypotheses to circumvent the robustness of HNSCC and also to reverse its hallmarks to the curable levels by conventional DNA‐damaging agents form a systemic view. However, it is apparent that he development and progression of HPV‐negative HNSCC is dependent on the distinctive molecular circuitry that function as a “complex adaptive system” (Merlo et al., 2006; Tian et al., 2011). Thus, in parallel with this strategy, the development of the “molecular diagnosis”, a holistic conceptualization of the entire molecular circuitry of HNSCC, appears to be the most powerful solution that enables us to precisely capture the vulnerability of HNSCC and as well to accurately predict the effects of molecular targeting and the consequent probabilities of adaptation (i.e., further evolution). During the last decade, we have witnessed tremendous progress in the development of molecular techniques to obtain genetic, epigenetic, transcriptomic, proteomic, secretomic and metabolic information on an unprecedented scale, although the assessment of dynamic interplay between the tumor and environmental factors is still underway (Gonzalez‐Angulo et al., 2010; Hanahan and Weinberg, 2011; Kreeger and Lauffenburger, 2010). It is expected that by processing this myriad information through powerful and sophisticated methods based on computational biology, including network theory, and systems biology that integrate physics and mathematical approaches into biological and medical insights, we can visualize how cellular behavior of HNSCC is driven by complex molecular circuitry (Gonzalez‐Angulo et al., 2010; Kreeger and Lauffenburger, 2010; Merlo et al., 2006; Tian et al., 2011). Compared to other human malignancies, HNSCC has an apparent advantage that tumor specimens can be relatively easily obtained under direct observation. Thus, HNSCC might provide the ideal platform of “molecular diagnosis” for continued global monitoring of the intrinsic molecular circuitry that will allow the timely identification of the Achilles' heel that is hidden behind the robustness and evolvability of HNSCC.
Conflict of interest
A.K. Joe is an Associate Medical Director, Hoffmann‐La Roche, Nutley, NJ, USA.
Acknowledgement
This review was supported in part by fund from Grants‐in‐Aid for Scientific Research (C): 21592195 and (C): 24592600 to Muneyuki Masuda. This review is fondly dedicated to our mentor Professor I B Weinstein of glorious memory.
Masuda Muneyuki, Toh Satoshi, Wakasaki Takahiro, Suzui Masumi, Joe Andrew K., (2013), Somatic evolution of head and neck cancer — Biological robustness and latent vulnerability, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.10.009.
References
- Agrawal, N. , Frederick, M.J. , Pickering, C.R. , Bettegowda, C. , Chang, K. , Li, R.J. , Fakhry, C. , Xie, T.X. , Zhang, J. , Wang, J. , Zhang, N. , El-Naggar, A.K. , Jasser, S.A. , Weinstein, J.N. , Trevino, L. , Drummond, J.A. , Muzny, D.M. , Wu, Y. , Wood, L.D. , Hruban, R.H. , Westra, W.H. , Koch, W.M. , Califano, J.A. , Gibbs, R.A. , Sidransky, D. , Vogelstein, B. , Velculescu, V.E. , Papadopoulos, N. , Wheeler, D.A. , Kinzler, K.W. , Myers, J.N. , 2011. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 333, 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkureishi, L.W. , de Bree, R. , Ross, G.L. , 2006. RADPLAT: an alternative to surgery?. Oncologist. 11, 469–480. [DOI] [PubMed] [Google Scholar]
- Allen, C.T. , Ricker, J.L. , Chen, Z. , Van Waes, C. , 2007. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 29, 959–971. [DOI] [PubMed] [Google Scholar]
- Amornphimoltham, P. , Patel, V. , Sodhi, A. , Nikitakis, N.G. , Sauk, J.J. , Sausville, E.A. , Molinolo, A.A. , Gutkind, J.S. , 2005. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res.. 65, 9953–9961. [DOI] [PubMed] [Google Scholar]
- Ang, K.K. , Harris, J. , Wheeler, R. , Weber, R. , Rosenthal, D.I. , Nguyen-Tan, P.F. , Westra, W.H. , Chung, C.H. , Jordan, R.C. , Lu, C. , Kim, H. , Axelrod, R. , Silverman, C.C. , Redmond, K.P. , Gillison, M.L. , 2010. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med.. 363, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, C.J. , Ohshiro, K. , Rayala, S.K. , El-Naggar, A.K. , Kumar, R. , 2007. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin. Cancer Res.. 13, 4291–4299. [DOI] [PubMed] [Google Scholar]
- Baumann, M. , Krause, M. , Hill, R. , 2008. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer. 8, 545–554. [DOI] [PubMed] [Google Scholar]
- Baylin, S.B. , Ohm, J.E. , 2006. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction?. Nat. Rev. Cancer. 6, 107–116. [DOI] [PubMed] [Google Scholar]
- Ben-Porath, I. , Thomson, M.W. , Carey, V.J. , Ge, R. , Bell, G.W. , Regev, A. , Weinberg, R.A. , 2008. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet.. 40, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier, J. , Bentzen, S.M. , Vermorken, J.B. , 2009. Molecular therapy in head and neck oncology. Nat. Rev. Clin. Oncol.. 6, 266–277. [DOI] [PubMed] [Google Scholar]
- Biddle, A. , Liang, X. , Gammon, L. , Fazil, B. , Harper, L.J. , Emich, H. , Costea, D.E. , Mackenzie, I.C. , 2011. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res.. 71, 5317–5326. [DOI] [PubMed] [Google Scholar]
- Brabletz, T. , Jung, A. , Spaderna, S. , Hlubek, F. , Kirchner, T. , 2005. Opinion: migrating cancer stem cells – an integrated concept of malignant tumour progression. Nat. Rev. Cancer. 5, 744–749. [DOI] [PubMed] [Google Scholar]
- Bracken, A.P. , Helin, K. , 2009. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer. 9, 773–784. [DOI] [PubMed] [Google Scholar]
- Bran, B. , Bran, G. , Hormann, K. , Riedel, F. , 2009. The platelet-derived growth factor receptor as a target for vascular endothelial growth factor-mediated anti-angiogenetic therapy in head and neck cancer. Int. J. Oncol.. 34, 255–261. [PubMed] [Google Scholar]
- Brock, A. , Chang, H. , Huang, S. , 2009. Non-genetic heterogeneity – a mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet.. 10, 336–342. [DOI] [PubMed] [Google Scholar]
- Chaffer, C.L. , Weinberg, R.A. , 2011. A perspective on cancer cell metastasis. Science. 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Chang, H.Y. , Nuyten, D.S. , Sneddon, J.B. , Hastie, T. , Tibshirani, R. , Sorlie, T. , Dai, H. , He, Y.D. , van't Veer, L.J. , Bartelink, H. , van de Rijn, M. , Brown, P.O. , van de Vijver, M.J. , 2005. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc. Natl. Acad. Sci. U S A. 102, 3738–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H.Y. , Sneddon, J.B. , Alizadeh, A.A. , Sood, R. , West, R.B. , Montgomery, K. , Chi, J.T. , van de Rijn, M. , Botstein, D. , Brown, P.O. , 2004. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol.. 2, E7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, P.B. , Hauschild, A. , Robert, C. , Haanen, J.B. , Ascierto, P. , Larkin, J. , Dummer, R. , Garbe, C. , Testori, A. , Maio, M. , Hogg, D. , Lorigan, P. , Lebbe, C. , Jouary, T. , Schadendorf, D. , Ribas, A. , O'Day, S.J. , Sosman, J.A. , Kirkwood, J.M. , Eggermont, A.M. , Dreno, B. , Nolop, K. , Li, J. , Nelson, B. , Hou, J. , Lee, R.J. , Flaherty, K.T. , McArthur, G.A. , 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med.. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.Y. , Chiou, S.H. , Huang, C.Y. , Jan, C.I. , Lin, S.C. , Tsai, M.L. , Lo, J.F. , 2009. Distinct population of highly malignant cells in a head and neck squamous cell carcinoma cell line established by xenograft model. J. Biomed. Sci.. 16, 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Chen, C. , 2008. DNA copy number variation and loss of heterozygosity in relation to recurrence of and survival from head and neck squamous cell carcinoma: a review. Head Neck. 30, 1361–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z.G. , 2009. The cancer stem cell concept in progression of head and neck cancer. J. Oncol.. 2009, 894064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou, S.H. , Yu, C.C. , Huang, C.Y. , Lin, S.C. , Liu, C.J. , Tsai, T.H. , Chou, S.H. , Chien, C.S. , Ku, H.H. , Lo, J.F. , 2008. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res.. 14, 4085–4095. [DOI] [PubMed] [Google Scholar]
- Chung, C.H. , Gillison, M.L. , 2009. Human papillomavirus in head and neck cancer: its role in pathogenesis and clinical implications. Clin. Cancer Res.. 15, 6758–6762. [DOI] [PubMed] [Google Scholar]
- Clay, M.R. , Tabor, M. , Owen, J.H. , Carey, T.E. , Bradford, C.R. , Wolf, G.T. , Wicha, M.S. , Prince, M.E. , 2010. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck. 32, 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohan, D.M. , Popat, S. , Kaplan, S.E. , Rigual, N. , Loree, T. , Hicks, W.L. , 2009. Oropharyngeal cancer: current understanding and management. Curr. Opin. Otolaryngol. Head Neck Surg.. 17, 88–94. [DOI] [PubMed] [Google Scholar]
- Colotta, F. , Allavena, P. , Sica, A. , Garlanda, C. , Mantovani, A. , 2009. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 30, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Corry, J. , Peters, L.J. , Rischin, D. , 2010. Optimising the therapeutic ratio in head and neck cancer. Lancet Oncol.. 11, 287–291. [DOI] [PubMed] [Google Scholar]
- De Boeck, A. , Narine, K. , De Neve, W. , Mareel, M. , Bracke, M. , De Wever, O. , 2010. Resident and bone marrow-derived mesenchymal stem cells in head and neck squamous cell carcinoma. Oral Oncol.. 46, 336–342. [DOI] [PubMed] [Google Scholar]
- Dean, M. , Fojo, T. , Bates, S. , 2005. Tumour stem cells and drug resistance. Nat. Rev. Cancer. 5, 275–284. [DOI] [PubMed] [Google Scholar]
- DeBerardinis, R.J. , Lum, J.J. , Hatzivassiliou, G. , Thompson, C.B. , 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell. Metab.. 7, 11–20. [DOI] [PubMed] [Google Scholar]
- Dohadwala, M. , Wang, G. , Heinrich, E. , Luo, J. , Lau, O. , Shih, H. , Munaim, Q. , Lee, G. , Hong, L. , Lai, C. , Abemayor, E. , Fishbein, M.C. , Elashoff, D.A. , Dubinett, S.M. , St John, M.A. , 2010. The role of ZEB1 in the inflammation-induced promotion of EMT in HNSCC. Otolaryngol. Head Neck Surg.. 142, 753–759. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , 2009. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer. 9, 550–562. [DOI] [PubMed] [Google Scholar]
- Felsher, D.W. , 2008. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit?. Cancer Res.. 68, 3081–3086. discussion 3086 [DOI] [PubMed] [Google Scholar]
- Fong, P.C. , Boss, D.S. , Yap, T.A. , Tutt, A. , Wu, P. , Mergui-Roelvink, M. , Mortimer, P. , Swaisland, H. , Lau, A. , O'Connor, M.J. , Ashworth, A. , Carmichael, J. , Kaye, S.B. , Schellens, J.H. , de Bono, J.S. , 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med.. 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Forastiere, A. , Koch, W. , Trotti, A. , Sidransky, D. , 2001. Head and neck cancer. N. Engl. J. Med.. 345, 1890–1900. [DOI] [PubMed] [Google Scholar]
- Garraway, L.A. , Sellers, W.R. , 2006. From integrated genomics to tumor lineage dependency. Cancer Res.. 66, 2506–2508. [DOI] [PubMed] [Google Scholar]
- Garraway, L.A. , Sellers, W.R. , 2006. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer. 6, 593–602. [DOI] [PubMed] [Google Scholar]
- Garraway, L.A. , Widlund, H.R. , Rubin, M.A. , Getz, G. , Berger, A.J. , Ramaswamy, S. , Beroukhim, R. , Milner, D.A. , Granter, S.R. , Du, J. , Lee, C. , Wagner, S.N. , Li, C. , Golub, T.R. , Rimm, D.L. , Meyerson, M.L. , Fisher, D.E. , Sellers, W.R. , 2005. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 436, 117–122. [DOI] [PubMed] [Google Scholar]
- Gasparotto, D. , Maestro, R. , Piccinin, S. , Vukosavljevic, T. , Barzan, L. , Sulfaro, S. , Boiocchi, M. , 1997. Overexpression of CDC25A and CDC25B in head and neck cancers. Cancer Res.. 57, 2366–2368. [PubMed] [Google Scholar]
- Gillies, R.J. , Verduzco, D. , Gatenby, R.A. , 2012. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer. 12, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo, A.M. , Hennessy, B.T. , Mills, G.B. , 2010. Future of personalized medicine in oncology: a systems biology approach. J. Clin. Oncol.. 28, 2777–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Moles, M.A. , Bravo, M. , Ruiz-Avila, I. , Esteban, F. , Rodriguez-Archilla, A. , Gonzalez-Moles, S. , Arias, B. , 2003. Adhesion molecule CD44 as a prognostic factor in tongue cancer. Anticancer Res.. 23, 5197–5202. [PubMed] [Google Scholar]
- Gonzalez-Moles, M.A. , Gil-Montoya, J.A. , Ruiz-Avila, I. , Esteban, F. , Delgado-Rodriguez, M. , Bascones-Martinez, A. , 2007. Prognostic significance of p21WAF1/CIP1, p16INK4a and CD44s in tongue cancer. Oncol. Rep.. 18, 389–396. [PubMed] [Google Scholar]
- Grant, S. , 2008. Cotargeting survival signaling pathways in cancer. J. Clin. Invest.. 118, 3003–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves, M. , Maley, C.C. , 2012. Clonal evolution in cancer. Nature. 481, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grille, S.J. , Bellacosa, A. , Upson, J. , Klein-Szanto, A.J. , van Roy, F. , Lee-Kwon, W. , Donowitz, M. , Tsichlis, P.N. , Larue, L. , 2003. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res.. 63, 2172–2178. [PubMed] [Google Scholar]
- Guan, Z. , Wang, X.R. , Zhu, X.F. , Huang, X.F. , Xu, J. , Wang, L.H. , Wan, X.B. , Long, Z.J. , Liu, J.N. , Feng, G.K. , Huang, W. , Zeng, Y.X. , Chen, F.J. , Liu, Q. , 2007. Aurora-A, a negative prognostic marker, increases migration and decreases radiosensitivity in cancer cells. Cancer Res.. 67, 10,436–10,444. [DOI] [PubMed] [Google Scholar]
- Guertin, D.A. , Sabatini, D.M. , 2007. Defining the role of mTOR in cancer. Cancer Cell. 12, 9–22. [DOI] [PubMed] [Google Scholar]
- Ha, P.K. , Chang, S.S. , Glazer, C.A. , Califano, J.A. , Sidransky, D. , 2009. Molecular techniques and genetic alterations in head and neck cancer. Oral Oncol.. 45, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad, R.I. , Shin, D.M. , 2008. Recent advances in head and neck cancer. N. Engl. J. Med.. 359, 1143–1154. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hirasawa, N. , Itoh, Y. , Ishihara, S. , Kubota, S. , Itoh, J. , Fujimoto, Y. , Nakashima, T. , Naganawa, S. , 2010. Radiotherapy with or without chemotherapy for patients with T1–T2 glottic carcinoma: retrospective analysis. Head Neck Oncol.. 2, 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma, A. , Furuta, Y. , Oridate, N. , Nakano, Y. , Kohashi, G. , Yagi, K. , Nagahashi, T. , Yagi, K. , Nagahashi, T. , Fukuda, S. , Inoue, K. , Inuyama, Y. , 1999. Prognostic significance of clinical parameters and biological markers in patients with squamous cell carcinoma of the head and neck treated with concurrent chemoradiotherapy. Clin. Cancer Res.. 5, 801–806. [PubMed] [Google Scholar]
- Hoogsteen, I.J. , Marres, H.A. , Bussink, J. , van der Kogel, A.J. , Kaanders, J.H. , 2007. Tumor microenvironment in head and neck squamous cell carcinomas: predictive value and clinical relevance of hypoxic markers. A review. Head Neck. 29, 591–604. [DOI] [PubMed] [Google Scholar]
- Hopkins, J. , Cescon, D.W. , Tse, D. , Bradbury, P. , Xu, W. , Ma, C. , Wheatley-Price, P. , Waldron, J. , Goldstein, D. , Meyer, F. , Bairati, I. , Liu, G. , 2008. Genetic polymorphisms and head and neck cancer outcomes: a review. Cancer Epidemiol. Biomarkers Prev.. 17, 490–499. [DOI] [PubMed] [Google Scholar]
- Huang, S. , Ernberg, I. , Kauffman, S. , 2009. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell. Dev. Biol.. 20, 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, T.P. , Kaeda, J. , Branford, S. , Rudzki, Z. , Hochhaus, A. , Hensley, M.L. , Gathmann, I. , Bolton, A.E. , van Hoomissen, I.C. , Goldman, J.M. , Radich, J.P. , 2003. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N. Engl. J. Med.. 349, 1423–1432. [DOI] [PubMed] [Google Scholar]
- Hunter, K.D. , Parkinson, E.K. , Harrison, P.R. , 2005. Profiling early head and neck cancer. Nat. Rev. Cancer. 5, 127–135. [DOI] [PubMed] [Google Scholar]
- Jewett, A. , Head, C. , Cacalano, N.A. , 2006. Emerging mechanisms of immunosuppression in oral cancers. J. Dent Res.. 85, 1061–1073. [DOI] [PubMed] [Google Scholar]
- Joyce, J.A. , Pollard, J.W. , 2009. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 9, 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila, M.R. , Ala-Aho, R. , Jokilehto, T. , Peltonen, J. , Kallajoki, M. , Grenman, R. , Jaakkola, P. , Westermarck, J. , Kahari, V.M. , 2007. p38alpha and p38delta mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene. 26, 5267–5279. [DOI] [PubMed] [Google Scholar]
- Kaelin, W.G. , 2005. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 5, 689–698. [DOI] [PubMed] [Google Scholar]
- Kang, S. , Elf, S. , Lythgoe, K. , Hitosugi, T. , Taunton, J. , Zhou, W. , Xiong, L. , Wang, D. , Muller, S. , Fan, S. , Sun, S.Y. , Marcus, A.I. , Gu, T.L. , Polakiewicz, R.D. , Chen, Z.G. , Khuri, F.R. , Shin, D.M. , Chen, J. , 2010. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J. Clin. Invest.. 120, 1165–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano, H. , 2004. Cancer as a robust system: implications for anticancer therapy. Nat. Rev. Cancer. 4, 227–235. [DOI] [PubMed] [Google Scholar]
- Knowles, L.M. , Stabile, L.P. , Egloff, A.M. , Rothstein, M.E. , Thomas, S.M. , Gubish, C.T. , Lerner, E.C. , Seethala, R.R. , Suzuki, S. , Quesnelle, K.M. , Morgan, S. , Ferris, R.L. , Grandis, J.R. , Siegfried, J.M. , 2009. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res.. 15, 3740–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukourakis, M.I. , Giatromanolaki, A. , Winter, S. , Leek, R. , Sivridis, E. , Harris, A.L. , 2009. Lactate dehydrogenase 5 expression in squamous cell head and neck cancer relates to prognosis following radical or postoperative radiotherapy. Oncology. 77, 285–292. [DOI] [PubMed] [Google Scholar]
- Kreeger, P.K. , Lauffenburger, D.A. , 2010. Cancer systems biology: a network modeling perspective. Carcinogenesis. 31, 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer, G. , Pouyssegur, J. , 2008. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 13, 472–482. [DOI] [PubMed] [Google Scholar]
- Kumamoto, Y. , Masuda, M. , Kuratomi, Y. , Toh, S. , Shinokuma, A. , Chujo, K. , Yamamoto, T. , Komiyama, S. , 2002. “FAR” chemoradiotherapy improves laryngeal preservation rates in patients with T2N0 glottic carcinoma. Head Neck. 24, 637–642. [DOI] [PubMed] [Google Scholar]
- Kupferman, M.E. , Jiffar, T. , El-Naggar, A. , Yilmaz, T. , Zhou, G. , Xie, T. , Feng, L. , Wang, J. , Holsinger, F.C. , Yu, D. , Myers, J.N. , 2010. TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene. 29, 2047–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuratomi, Y. , Kumamoto, M. , Kidera, K. , Toh, S. , Masuda, M. , Nakashima, T. , Inokuchi, A. , 2006. Diffuse expression of laminin gamma2 chain in disseminating and infiltrating cancer cells indicates a highly malignant state in advanced tongue cancer. Oral Oncol.. 42, 73–76. [DOI] [PubMed] [Google Scholar]
- Leemans, C.R. , Braakhuis, B.J. , Brakenhoff, R.H. , 2010. The molecular biology of head and neck cancer. Nat. Rev. Cancer. 11, 9–22. [DOI] [PubMed] [Google Scholar]
- Lin, D.T. , Subbaramaiah, K. , Shah, J.P. , Dannenberg, A.J. , Boyle, J.O. , 2002. Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck. 24, 792–799. [DOI] [PubMed] [Google Scholar]
- Lippman, S.M. , Hong, W.K. , 2001. Molecular markers of the risk of oral cancer. N. Engl. J. Med.. 344, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Lo, A.K. , Lo, K.W. , Tsao, S.W. , Wong, H.L. , Hui, J.W. , To, K.F. , Hayward, D.S. , Chui, Y.L. , Lau, Y.L. , Takada, K. , Huang, D.P. , 2006. Epstein-Barr virus infection alters cellular signal cascades in human nasopharyngeal epithelial cells. Neoplasia. 8, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Muzio, L. , Falaschini, S. , Farina, A. , Rubini, C. , Pezzetti, F. , Campisi, G. , De Rosa, G. , Capogreco, M. , Carinci, F. , 2005. Bcl-2 as prognostic factor in head and neck squamous cell carcinoma. Oncol. Res.. 15, 249–255. [DOI] [PubMed] [Google Scholar]
- Lowy, D.R. , Munger, K. , 2010. Prognostic implications of HPV in oropharyngeal cancer. N. Engl. J. Med.. 363, 82–84. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Solimini, N.L. , Elledge, S.J. , 2009. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machtay, M. , Moughan, J. , Trotti, A. , Garden, A.S. , Weber, R.S. , Cooper, J.S. , Forastiere, A. , Ang, K.K. , 2008. Factors associated with severe late toxicity after concurrent chemoradiation for locally advanced head and neck cancer: an RTOG analysis. J. Clin. Oncol.. 26, 3582–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, B. , Gires, O. , 2008. CD44s and CD44v6 expression in head and neck epithelia. PLoS One. 3, e3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, M. , Myers, J.N. , Lippman, S.M. , Johnson, F.M. , Williams, M.D. , Rayala, S. , Ohshiro, K. , Rosenthal, D.I. , Weber, R.S. , Gallick, G.E. , El-Naggar, A.K. , 2008. Epithelial to mesenchymal transition in head and neck squamous carcinoma: association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer. 112, 2088–2100. [DOI] [PubMed] [Google Scholar]
- Marusyk, A. , Almendro, V. , Polyak, K. , 2012. Intra-tumour heterogeneity: a looking glass for cancer?. Nat. Rev. Cancer. 12, 323–334. [DOI] [PubMed] [Google Scholar]
- Masuda, M. , Kamizono, K. , Uryu, H. , Fujimura, A. , Uchi, R. , 2012. Roles of therapeutic selective neck dissection in multidisciplinary treatment. In Kummoona R., Neck Dissection – Clinical Application and Recent Advances. In Tech; [Google Scholar]
- Masuda, M. , Kuratomi, Y. , Shiratsuchi, H. , Nakashima, T. , Kunitake, N. , Komiyama, S. , 2000. Decreased CD44H expression in early-stage tongue carcinoma associates with late nodal metastases following interstitial brachytherapy. Head Neck. 22, 662–665. [DOI] [PubMed] [Google Scholar]
- Masuda, M. , Wakasaki, T. , Suzui, M. , Toh, S. , Joe, A.K. , Weinstein, I.B. , 2010. Stat3 orchestrates tumor development and progression: the Achilles' heel of head and neck cancers?. Curr. Cancer Drug Target.. 10, 117–126. [DOI] [PubMed] [Google Scholar]
- Masuda, M. , Wakasaki, T. , Toh, S. , Shimizu, M. , Adachi, S. , 2011. Chemoprevention of head and neck cancer by green tea extract: EGCG – the role of EGFR signaling and “Lipid Raft”. J. Oncol.. 2011, 540148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta, A. , Ralhan, R. , 2009. Overview of current and future biologically based targeted therapies in head and neck squamous cell carcinoma. Head Neck Oncol.. 1, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFate, T. , Mohyeldin, A. , Lu, H. , Thakar, J. , Henriques, J. , Halim, N.D. , Wu, H. , Schell, M.J. , Tsang, T.M. , Teahan, O. , Zhou, S. , Califano, J.A. , Jeoung, N.H. , Harris, R.A. , Verma, A. , 2008. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem.. 283, 22,700–22,708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo, L.M. , Pepper, J.W. , Reid, B.J. , Maley, C.C. , 2006. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer. 6, 924–935. [DOI] [PubMed] [Google Scholar]
- Mills, G.B. , Lu, Y. , Kohn, E.C. , 2001. Linking molecular therapeutics to molecular diagnostics: inhibition of the FRAP/RAFT/TOR component of the PI3K pathway preferentially blocks PTEN mutant cells in vitro and in vivo. Proc. Natl. Acad. Sci. U S A. 98, 10,031–10,033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa, J. , Mitoro, A. , Kawashiri, S. , Chada, K.K. , Imai, K. , 2004. Expression of mesenchyme-specific gene HMGA2 in squamous cell carcinomas of the oral cavity. Cancer Res.. 64, 2024–2029. [DOI] [PubMed] [Google Scholar]
- Molinolo, A.A. , Amornphimoltham, P. , Squarize, C.H. , Castilho, R.M. , Patel, V. , Gutkind, J.S. , 2009. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol.. 45, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinolo, A.A. , Hewitt, S.M. , Amornphimoltham, P. , Keelawat, S. , Rangdaeng, S. , Meneses Garcia, A. , Raimondi, A.R. , Jufe, R. , Itoiz, M. , Gao, Y. , Saranath, D. , Kaleebi, G.S. , Yoo, G.H. , Leak, L. , Myers, E.M. , Shintani, S. , Wong, D. , Massey, H.D. , Yeudall, W.A. , Lonardo, F. , Ensley, J. , Gutkind, J.S. , 2007. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin. Cancer Res.. 13, 4964–4973. [DOI] [PubMed] [Google Scholar]
- Naresh, K.N. , Lakshminarayanan, K. , Pai, S.A. , Borges, A.M. , 2001. Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: a hypothesis to support this paradoxical association. Cancer. 91, 578–584. [PubMed] [Google Scholar]
- Nowell, P.C. , 1976. The clonal evolution of tumor cell populations. Science. 194, 23–28. [DOI] [PubMed] [Google Scholar]
- Paz-Elizur, T. , Sevilya, Z. , Leitner-Dagan, Y. , Elinger, D. , Roisman, L.C. , Livneh, Z. , 2008. DNA repair of oxidative DNA damage in human carcinogenesis: potential application for cancer risk assessment and prevention. Cancer Lett.. 266, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrero, J.M. , Carracedo, D.G. , Pinto, C.M. , Zapatero, A.H. , Rodrigo, J.P. , Nieto, C.S. , Gonzalez, M.V. , 2005. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int. J. Cancer. 114, 242–248. [DOI] [PubMed] [Google Scholar]
- Pena, J.C. , Thompson, C.B. , Recant, W. , Vokes, E.E. , Rudin, C.M. , 1999. Bcl-xL and Bcl-2 expression in squamous cell carcinoma of the head and neck. Cancer. 85, 164–170. [DOI] [PubMed] [Google Scholar]
- Polyak, K. , Weinberg, R.A. , 2009. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer. 9, 265–273. [DOI] [PubMed] [Google Scholar]
- Prince, M.E. , Ailles, L.E. , 2008. Cancer stem cells in head and neck squamous cell cancer. J. Clin. Oncol.. 26, 2871–2875. [DOI] [PubMed] [Google Scholar]
- Prince, M.E. , Sivanandan, R. , Kaczorowski, A. , Wolf, G.T. , Kaplan, M.J. , Dalerba, P. , Weissman, I.L. , Clarke, M.F. , Ailles, L.E. , 2007. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. U S A. 104, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan, P. , Weaver, K.L. , Capobianco, A.J. , 2011. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat. Rev. Cancer. 11, 338–351. [DOI] [PubMed] [Google Scholar]
- Reiter, R. , Gais, P. , Jutting, U. , Steuer-Vogt, M.K. , Pickhard, A. , Bink, K. , Rauser, S. , Lassmann, S. , Hofler, H. , Werner, M. , Walch, A. , 2006. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin. Cancer Res.. 12, 5136–5141. [DOI] [PubMed] [Google Scholar]
- Reuter, C.W. , Morgan, M.A. , Eckardt, A. , 2007. Targeting EGF-receptor-signalling in squamous cell carcinomas of the head and neck. Br. J. Cancer. 96, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen, J.M. , Jordan, C.T. , 2009. The increasing complexity of the cancer stem cell paradigm. Science. 324, 1670–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal, D.I. , Lewin, J.S. , Eisbruch, A. , 2006. Prevention and treatment of dysphagia and aspiration after chemoradiation for head and neck cancer. J. Clin. Oncol.. 24, 2636–2643. [DOI] [PubMed] [Google Scholar]
- Ruan, H.Y. , Masuda, M. , Ito, A. , Umezawa, K. , Nakashima, T. , Yasumatsu, R. , Kuratomi, Y. , Yamamoto, T. , Weinstein, I.B. , Komune, S. , 2006. Effects of a novel NF-kappaB inhibitor, dehydroxymethylepoxyquinomicin (DHMEQ), on growth, apoptosis, gene expression, and chemosensitivity in head and neck squamous cell carcinoma cell lines. Head Neck. 28, 158–165. [DOI] [PubMed] [Google Scholar]
- Sathyan, K.M. , Nalinakumari, K.R. , Kannan, S. , 2007. H-Ras mutation modulates the expression of major cell cycle regulatory proteins and disease prognosis in oral carcinoma. Mod. Pathol.. 20, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Schneider, A. , Younis, R.H. , Gutkind, J.S. , 2008. Hypoxia-induced energy stress inhibits the mTOR pathway by activating an AMPK/REDD1 signaling axis in head and neck squamous cell carcinoma. Neoplasia. 10, 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selek, U. , Garden, A.S. , Morrison, W.H. , El-Naggar, A.K. , Rosenthal, D.I. , Ang, K.K. , 2004. Radiation therapy for early-stage carcinoma of the oropharynx. Int. J. Radiat. Oncol. Biol. Phys.. 59, 743–751. [DOI] [PubMed] [Google Scholar]
- Sen, B. , Saigal, B. , Parikh, N. , Gallick, G. , Johnson, F.M. , 2009. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res.. 69, 1958–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, S.V. , Settleman, J. , 2007. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev.. 21, 3214–3231. [DOI] [PubMed] [Google Scholar]
- Simons, A.L. , Ahmad, I.M. , Mattson, D.M. , Dornfeld, K.J. , Spitz, D.R. , 2007. 2-Deoxy-d-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res.. 67, 3364–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Chang, I. , Chen, Z. , Kang, M. , Wang, C.Y. , 2010. Characterization of side populations in HNSCC: highly invasive, chemoresistant and abnormal Wnt signaling. PLoS One. 5, e11456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, L.B. , Li, J. , Liao, W.T. , Feng, Y. , Yu, C.P. , Hu, L.J. , Kong, Q.L. , Xu, L.H. , Zhang, X. , Liu, W.L. , Li, M.Z. , Zhang, L. , Kang, T.B. , Fu, L.W. , Huang, W.L. , Xia, Y.F. , Tsao, S.W. , Li, M. , Band, V. , Band, H. , Shi, Q.H. , Zeng, Y.X. , Zeng, M.S. , 2009. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Invest.. 119, 3626–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specenier, P. , Vermorken, J.B. , 2012. Biologic therapy in head and neck cancer: a road with hurdles. ISRN Oncol.. 2012, 163752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarize, C.H. , Castilho, R.M. , Sriuranpong, V. , Pinto, D.S. , Gutkind, J.S. , 2006. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 8, 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John, M.A. , Dohadwala, M. , Luo, J. , Wang, G. , Lee, G. , Shih, H. , Heinrich, E. , Krysan, K. , Walser, T. , Hazra, S. , Zhu, L. , Lai, C. , Abemayor, E. , Fishbein, M. , Elashoff, D.A. , Sharma, S. , Dubinett, S.M. , 2009. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin. Cancer Res.. 15, 6018–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky, N. , Egloff, A.M. , Tward, A.D. , Kostic, A.D. , Cibulskis, K. , Sivachenko, A. , Kryukov, G.V. , Lawrence, M.S. , Sougnez, C. , McKenna, A. , Shefler, E. , Ramos, A.H. , Stojanov, P. , Carter, S.L. , Voet, D. , Cortes, M.L. , Auclair, D. , Berger, M.F. , Saksena, G. , Guiducci, C. , Onofrio, R.C. , Parkin, M. , Romkes, M. , Weissfeld, J.L. , Seethala, R.R. , Wang, L. , Rangel-Escareno, C. , Fernandez-Lopez, J.C. , Hidalgo-Miranda, A. , Melendez-Zajgla, J. , Winckler, W. , Ardlie, K. , Gabriel, S.B. , Meyerson, M. , Lander, E.S. , Getz, G. , Golub, T.R. , Garraway, L.A. , Grandis, J.R. , 2011. The mutational landscape of head and neck squamous cell carcinoma. Science. 333, 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton, M.R. , 2011. Exploring the genomes of cancer cells: progress and promise. Science. 331, 1553–1558. [DOI] [PubMed] [Google Scholar]
- Strauss, L. , Bergmann, C. , Szczepanski, M. , Gooding, W. , Johnson, J.T. , Whiteside, T.L. , 2007. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin. Cancer Res.. 13, 4345–4354. [DOI] [PubMed] [Google Scholar]
- Sun, W. , Liu, Y. , Glazer, C.A. , Shao, C. , Bhan, S. , Demokan, S. , Zhao, M. , Rudek, M.A. , Ha, P.K. , Califano, J.A. , 2010. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin. Cancer Res.. 16, 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabachnyk, M. , Distel, L.V. , Buttner, M. , Grabenbauer, G.G. , Nkenke, E. , Fietkau, R. , Lubgan, D. , 2012. Radiochemotherapy induces a favourable tumour infiltrating inflammatory cell profile in head and neck cancer. Oral Oncol.. 48, 594–601. [DOI] [PubMed] [Google Scholar]
- Tennant, D.A. , Duran, R.V. , Gottlieb, E. , 2010. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer. 10, 267–277. [DOI] [PubMed] [Google Scholar]
- Tian, T. , Olson, S. , Whitacre, J.M. , Harding, A. , 2011. The origins of cancer robustness and evolvability. Integr. Biol. (Camb.). 3, 17–30. [DOI] [PubMed] [Google Scholar]
- Todd, R. , Hinds, P.W. , Munger, K. , Rustgi, A.K. , Opitz, O.G. , Suliman, Y. , Wong, D.T. , 2002. Cell cycle dysregulation in oral cancer. Crit. Rev. Oral Biol. Med.. 13, 51–61. [DOI] [PubMed] [Google Scholar]
- Tran, N. , O'Brien, C.J. , Clark, J. , Rose, B. , 2010. Potential role of micro-RNAs in head and neck tumorigenesis. Head Neck. 32, 1099–1111. [DOI] [PubMed] [Google Scholar]
- Urba, S. , Wolf, G. , Eisbruch, A. , Worden, F. , Lee, J. , Bradford, C. , Teknos, T. , Chepeha, D. , Prince, M. , Hogikyan, N. , Taylor, J. , 2006. Single-cycle induction chemotherapy selects patients with advanced laryngeal cancer for combined chemoradiation: a new treatment paradigm. J. Clin. Oncol.. 24, 593–598. [DOI] [PubMed] [Google Scholar]
- Van Waes, C. , 2007. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin. Cancer Res.. 13, 1076–1082. [DOI] [PubMed] [Google Scholar]
- Van Waes, C. , Surh, D.M. , Chen, Z. , Kirby, M. , Rhim, J.S. , Brager, R. , Sessions, R.B. , Poore, J. , Wolf, G.T. , Carey, T.E. , 1995. Increase in suprabasilar integrin adhesion molecule expression in human epidermal neoplasms accompanies increased proliferation occurring with immortalization and tumor progression. Cancer Res.. 55, 5434–5444. [PubMed] [Google Scholar]
- Visvader, J.E. , Lindeman, G.J. , 2008. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer. 8, 755–768. [DOI] [PubMed] [Google Scholar]
- Wakasaki, T. , Masuda, M. , Niiro, H. , Komune, S. , Jabbarzadeh- Tabriz, S. , Noda, K. , Taniyama, T. , Komune, S. , Akashi, K. , 2010. A critical role of c-Cbl interacting protein of 85 kDa in the development and progression of head and neck squamous cell carcinomas via the Ras–ERK pathway. Neoplasia. 12, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Liu, X. , Chen, Z. , Huang, H. , Jin, Y. , Kolokythas, A. , Wang, A. , Dai, Y. , Wong, D.T. , Zhou, X. , 2011. Polycomb group protein EZH2-mediated E-cadherin repression promotes metastasis of oral tongue squamous cell carcinoma. Mol. Carcinog.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N.J. , Sanborn, Z. , Arnett, K.L. , Bayston, L.J. , Liao, W. , Proby, C.M. , Leigh, I.M. , Collisson, E.A. , Gordon, P.B. , Jakkula, L. , Pennypacker, S. , Zou, Y. , Sharma, M. , North, J.P. , Vemula, S.S. , Mauro, T.M. , Neuhaus, I.M. , Leboit, P.E. , Hur, J.S. , Park, K. , Huh, N. , Kwok, P.Y. , Arron, S.T. , Massion, P.P. , Bale, A.E. , Haussler, D. , Cleaver, J.E. , Gray, J.W. , Spellman, P.T. , South, A.P. , Aster, J.C. , Blacklow, S.C. , Cho, R.J. , 2011. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. U S A. 108, 17,761–17,766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Spitz, M.R. , Lee, J.J. , Huang, M. , Lippman, S.M. , Wu, X. , 2007. Nucleotide excision repair pathway genes and oral premalignant lesions. Clin. Cancer Res.. 13, 3753–3758. [DOI] [PubMed] [Google Scholar]
- Warburg, O. , 1923. Metabolism of tumors. Biochem. Zeitschr.. 142, 317–333. [Google Scholar]
- Weinstein, I.B. , 2000. Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis. 21, 857–864. [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , 2002. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , Begemann, M. , Zhou, P. , Han, E.K. , Sgambato, A. , Doki, Y. , Arber, N. , Ciaparrone, M. , Yamamoto, H. , 1997. Disorders in cell circuitry associated with multistage carcinogenesis: exploitable targets for cancer prevention and therapy. Clin. Cancer Res.. 3, 2696–2702. [PubMed] [Google Scholar]
- Weinstein, I.B. , Joe, A. , 2008. Oncogene addiction. Cancer Res.. 68, 3077–3080. discussion 3080 [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , Joe, A.K. , 2006. Mechanisms of disease: oncogene addiction–a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol.. 3, 448–457. [DOI] [PubMed] [Google Scholar]
- Wigfield, S.M. , Winter, S.C. , Giatromanolaki, A. , Taylor, J. , Koukourakis, M.L. , Harris, A.L. , 2008. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br. J. Cancer. 98, 1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, G.D. , Saunders, M.I. , Dische, S. , Richman, P.I. , Daley, F.M. , Bentzen, S.M. , 2001. bcl-2 expression in head and neck cancer: an enigmatic prognostic marker. Int. J. Radiat. Oncol. Biol. Phys.. 49, 435–441. [DOI] [PubMed] [Google Scholar]
- Wong, D.J. , Liu, H. , Ridky, T.W. , Cassarino, D. , Segal, E. , Chang, H.Y. , 2008. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, D.J. , Nuyten, D.S. , Regev, A. , Lin, M. , Adler, A.S. , Segal, E. , van de Vijver, M.J. , Chang, H.Y. , 2008. Revealing targeted therapy for human cancer by gene module maps. Cancer Res.. 68, 369–378. [DOI] [PubMed] [Google Scholar]
- Worden, F.P. , Kumar, B. , Lee, J.S. , Wolf, G.T. , Cordell, K.G. , Taylor, J.M. , Urba, S.G. , Eisbruch, A. , Teknos, T.N. , Chepeha, D.B. , Prince, M.E. , Tsien, C.I. , D'Silva, N.J. , Yang, K. , Kurnit, D.M. , Mason, H.L. , Miller, T.H. , Wallace, N.E. , Bradford, C.R. , Carey, T.E. , 2008. Chemoselection as a strategy for organ preservation in advanced oropharynx cancer: response and survival positively associated with HPV16 copy number. J. Clin. Oncol.. 26, 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden, F.P. , Moyer, J. , Lee, J.S. , Taylor, J.M. , Urba, S.G. , Eisbruch, A. , Teknos, T.N. , Chepeha, D.B. , Prince, M.E. , Hogikyan, N. , Lassig, A.A. , Emerick, K. , Mukherji, S. , Hadjiski, L. , Tsien, C.I. , Miller, T.H. , Wallace, N.E. , Mason, H.L. , Bradford, C.R. , Wolf, G.T. , 2009. Chemoselection as a strategy for organ preservation in patients with T4 laryngeal squamous cell carcinoma with cartilage invasion. Laryngoscope. 119, 1510–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M.H. , Hsu, D.S. , Wang, H.W. , Wang, H.J. , Lan, H.Y. , Yang, W.H. , Huang, C.H. , Kao, S.Y. , Tzeng, C.H. , Tai, S.K. , Chang, S.Y. , Lee, O.K. , Wu, K.J. , 2010. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell. Biol.. 12, 982–992. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Pardoll, D. , Jove, R. , 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Peng, J. , Zhang, H. , Zhu, Y. , Wan, L. , Chen, J. , Chen, X. , Lin, R. , Li, H. , Mao, X. , Jin, K. , 2010. Notch1 signaling is activated in cells expressing embryonic stem cell proteins in human primary nasopharyngeal carcinoma. J. Otolaryngol. Head Neck Surg.. 39, 157–166. [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Wulfkuhle, J. , Zhang, H. , Gu, P. , Yang, Y. , Deng, J. , Margolick, J.B. , Liotta, L.A. , Petricoin, E. , Zhang, Y. , 2007. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. U S A. 104, 16,158–16,163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, S. , Kachhap, S. , Sun, W. , Wu, G. , Chuang, A. , Poeta, L. , Grumbine, L. , Mithani, S.K. , Chatterjee, A. , Koch, W. , Westra, W.H. , Maitra, A. , Glazer, C. , Carducci, M. , Sidransky, D. , McFate, T. , Verma, A. , Califano, J.A. , 2007. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. U S A. 104, 7540–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]