

Abstract
Lung adenocarcinoma cells harboring epidermal growth factor receptor (EGFR) mutations are sensitive to EGFR tyrosine kinase inhibitors (TKIs), including gefitinib. Acquired resistance to EGFR‐TKIs develops after prolonged treatments. The study was prompt to explore effective strategies against resistance to EGFR‐TKIs. We established gefitinib resistant PC‐9 cells which harbor EGFR exon 19 deletion. Known mechanisms for intrinsic or acquired EGFR‐TKI resistance, including KRAS mutation, HER2 mutation, EGFR T790M mutation and MET gene amplification, were studied, and we did not observe any known mechanisms for intrinsic or acquired resistance to EGFR‐TKIs in the resistant cells. In the parental PC‐9 cells, labeled as PC‐9/wt, gefitinib completely inhibited EGF‐induced phosphorylation of EGFR, AKT and ERK. Gefitinib inhibited EGFR phosphorylation, but was unable to block EGF‐induced phosphorylation of ERK in resistant cells, labeled as PC‐9/gef cells, including PC‐9/gefB4, PC‐9/gefE3, and PC‐9/gefE7 subclones. We detected NRAS Q61K mutation in the PC‐9/gef cells but not the PC‐9/wt cells. MEK inhibitors, either AZD6244 or CI1040, inhibited ERK phosphorylation and sensitized gefitinib‐induced cytotoxicity in PC‐9/gef cells. Whereas MEK inhibitors or gefitinib alone did not activate caspases in PC‐9/gef cells, combination of gefitinib and AZD6244 or CI1040 induced apoptosis. Our in vivo studies showed that gefitinib inhibited growth of PC‐9/wt xenografts but not PC‐9/gef xenografts. Furthermore, combination of a MEK inhibitor and gefitinib inhibited growth of both PC‐9/wt xenografts and PC‐9/gefB4 xenografts. To conclude, persistent activation of ERK pathway contributes to the acquired gefitinib‐resistance. Combined treatment of gefitinib and MEK inhibitors may be therapeutically useful for acquired gefitinib‐resistance lung adenocarcinoma cells harboring EGFR mutations.
Keywords: EGF receptor, Lung cancer, Gefitinib, Reversal of drug resistance, Kinase and phosphatase inhibitors, NRAS
Highlights
-
►
PC9/gef cells are 270‐fold resistant than PC9 cells to gefitinib.
-
►
PC9/gef cells contain EGFR del 19 mutation but not EGFR T790M or MET amplification.
-
►
NRAS Q61K mutation is found in all PC9/gef cells.
-
►
ERK phosphorylation can not be suppressed by gefitinib in PC9/gef cells.
-
►
MEK inhibitors plus gefitinib inhibit growth of PC9/gef cells in vitro and in vivo.
1. Introduction
Lung adenocarcinomas are characterized by frequent aberrancies in driver genes, especially the epidermal growth factor receptor (EGFR) gene (Li et al., 2011). EGFR tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, are highly effective in lung cancer patients with activating EGFR mutations, such as exon 19 deletion and exon 21 L858R mutation, in their tumor samples (Mok et al., 2009; Zhou et al., 2011). EGFR‐TKIs attenuate autophosphorylation of EGFR and subsequent activation of downstream pathways regulating cell survival and growth, including the PI3K‐AKT‐mTOR signal pathway and the MAPK cascade (Ras‐Raf‐MEK‐ERK) (Ono and Kuwano, 2006).
Despite the success of using EGFR‐TKIs in the treatment for lung adenocarcinoma patients harboring specific EGFR mutations, all responding patients eventually developed acquired resistance to EGFR‐TKIs. Several mechanisms underlying acquired resistance to EGFR‐TKIs have been proposed, including a second mutation in EGFR kinase domain, especially T790M mutation (Pao et al., 2005a), and the MET amplification (Engelman et al., 2007). T790M gatekeeper mutation decreases binding of EGFR‐TKI to the mutant EGFR (Sos et al., 2010). MET amplification‐induced persistent activation of PI3K‐AKT‐mTOR pathway through a HER3‐dependent pathway is considered to be the common downstream pathway of most types of acquired resistance mentioned above (Engelman and Janne, 2008). Accordingly, diminished activation of PI3K‐AKT‐mTOR pathway may reverse drug resistance. In support of this notion, both MET inhibitors (Engelman et al., 2007) and PI3K inhibitors (Donev et al., 2011) which reportedly reversed acquired resistance are suggested as effective therapies for drug resistance.
Over‐activation of the MAPK cascade, by KRAS gene mutation for example, has been observed in lung adenocarcinomas primarily resistant to EGFR‐TKI (Pao et al., 2005b); however, the role of the MAPK cascade on acquired resistance has not been extensively examined. In the present study, we demonstrated that the MAPK cascade was upregulated in these gefitinib acquired‐resistant cells. We further demonstrated that MEK inhibitors, such as CI‐1040 and AZD6244, reversed the resistance both in vitro and in vivo.
2. Materials and methods
2.1. Drugs and cell lines
Gefitinib and AZD6244 (selumetinib) were provided from Astrazeneca (Alderley Park, UK); and CI‐1040 from Pfizer (New York, USA). Gefitinib, AZD6244 and CI‐1040 were prepared in dimethyl sulfoxide (DMSO) to obtain a stock solution of 10 mM. PC‐9 cells are a human lung adenocarcinoma cell line harboring a deletion in exon 19 of EGFR (Bean et al., 2007). Gefitinib‐resistant PC‐9 cells were established and maintained as described previously (Chang et al., 2011). In short, parental PC‐9 cells, hereafter as PC‐9/wt cells, were grown in culture media containing escalating concentrations of gefitinib. After 6 months of passages, cells that could grow in micromolar concentrations of gefitinib were kept in drug‐free media for 2 weeks and were labeled as PC‐9/gef cells. Single cell coloning was performed, and several clones were obtained, including PC‐9/gefB4, PC‐9/gefC2, PC‐9/gefC4, PC‐9/gefC7, PC‐9/gefE3 and PC‐9/gefE7, etc. PC‐9/gefB4, PC‐9/gefE3 and PC‐9/gefE7 were selected for the current study and were maintained in regular RPMI media without gefitinib.
2.2. Growth inhibition assay
One thousand five hundred cells were placed in 96‐well flat‐bottomed plates and cultured for 24 h. Various concentrations of gefitinib plus AZD6244 or CI‐1040 were included in the culture medium for 96 h. The cytotoxic effects of gefitinib plus AZD6244 or CI‐1040 were determined by sulforhodamine B assay (Vichai and Kirtikara, 2006). Cell viability was determined by dividing the absorbance values of treated cells to that of untreated cells. IC50 calculated from the dose–response curve was defined as the concentration of gefitinib which 50% growth inhibition was obtained.
2.3. Western blot
To evaluate downstream signaling of PC‐9/wt and gefitinib‐resistant cells, cells were cultured in serum‐free media for 24 h prior to drug treatment. Afterward, cells were treated with gefitinib plus AZD6244 or CI‐1040 at various concentrations for 1 h and then were stimulated with 20 ng/ml of epidermal growth factor (EGF) for 10 min. Treated cells were harvested, washed with phosphate buffered saline, and lysed. Western blot was performed as described elsewhere. Primary antibodies used were as follows: α‐tubulin (Calbiochem, San Diego, CA), EGFR pY1173 and IGF‐1Rβ (Santa Cruz Biotechnology Inc, Santo Cruz, CA), pc‐Met Y1356 (Abgent Inc, San Diego, CA), EGFR and its phosphorylated components pY845, pY992, pY1068, and pY1148 (Cell Signaling Technology, Danvers, MA), AKT and pAKT S473, ERK and pERK, MEK1/2 and pMEK1/2 S217/221, HER2 and pHER2, HER3 and pHER3 Y1289, c‐Met, poly (ADP‐ribose) polymerase (PARP), caspase‐3, and caspase‐9 (Cell Signaling Technology, Danvers, MA).
2.4. Genetic analysis of the EGFR, KRAS, NRAS and HER2 genes
Cultured lung adenocarcinoma cells were scraped and DNA was extracted and purified using QIAamp DNA mini kit (Qiagen, Venlo, the Netherlands). The whole EGFR from cDNA was sequenced to detect possible newly onset secondary mutations. The KRAS, NRAS, and HER2 genes mutation were detected by partial sequencing of the DNA. The primers for the KRAS gene for polymerase chain reaction (PCR) were (5′ to 3′) F‐GAATGGTCCTGCACCAGTAA and R‐GTGTGACATGTTCTAATATAATCA. The primers to sequence exon 2 of the NRAS gene were GTTTTCCCAGTCACGACACCAAATGGAAGGTCACACTAGGGTTT and CAGGAAACAGCTATGACACAGGATCAGGTCAGCGGGC, and for exon 3 of the NRAS gene were GTTTTCCCAGTCACGACTGAGGGACAAACCAGATAGGCA and CAGGAAACAGCTATGACCCCTAGTGTGGTAACCTCATTTCCC‐CA. The primers to detect HER2 exon 20 codon 776 YVMA insertion were GCCATGGCTGTGGTTTGTGATGG and ATCCTAGCCCCTTGTGGACATAGG. The amplified DNA was then sent for direct sequencing.
2.5. Analysis of MET genomic amplification by quantitative RT‐PCR
Quantitative RT‐PCR for the MET gene amplification was performed as described previously (Bean et al., 2007). 5,10‐Methylenetetrahydrofolate reductase (MTHFR) was used as an internal control. The MET gene amplification is defined as the ratio of MET to MTHFR is larger than 5.
2.6. Xenograft mouse model
Five million PC‐9/wt and PC‐9/gefB4 cells were injected subcutaneously into the back 6‐weeks‐old male Balb/c nude mice. Xenograft size was measured daily and tumor volume was determined as (length × width2)/2. When tumors grew to 300 mm3, mice were randomized to 6 groups: vehicle (99.8% ethanol:cremophor EL:5% dextrose = 1:1:8), AZD6244 (25 mg/kg, twice per day), CI‐1040 (200 mg/kg, twice per day), gefitinib (50 mg/kg once per day), gefitinib plus AZD6244 and gefitinib plus CI‐1040. Drugs were administrated through an oral gavage. These animals were maintained in individual ventilated cages according to the guidelines established in "Guide For The Care And Use Of Laboratory Animals" prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources Commission on Life Sciences, National Research Council, U.S.A. (1985). Use of animals has been approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital, Taipei, Taiwan.
2.7. Statistics
Comparisons between PC‐9/wt and gefitinib‐resistant PC‐9 subclones were carried out by student t‐test. p‐value <0.05 was regarded as significant.
3. Results
3.1. Characteristics of gefitinib‐resistant lung adenocarcinoma cells
We established several gefitinib‐resistant PC‐9 subclones and labeled them as PC‐9/gefB4, PC‐9/gefE3, and PC‐9/gefE7. The cell doubling times for PC‐9/wt cells and PC‐9/gefB4 cells were 37.2 ± 1.8 h and 49.0 ± 1.2 h, respectively (p < 0.0001 by t‐test). The IC50 of gefitinib for the PC9/wt cells 18.9 ± 1.9 nM. The PC‐9/gefB4, PC‐9/gefE3, and PC‐9/gefE7 were 273 folds, 355 folds, and 268 folds, respectively, more resistant than the PC‐9/wt cells (Figure 1A and 1B).
Figure 1.
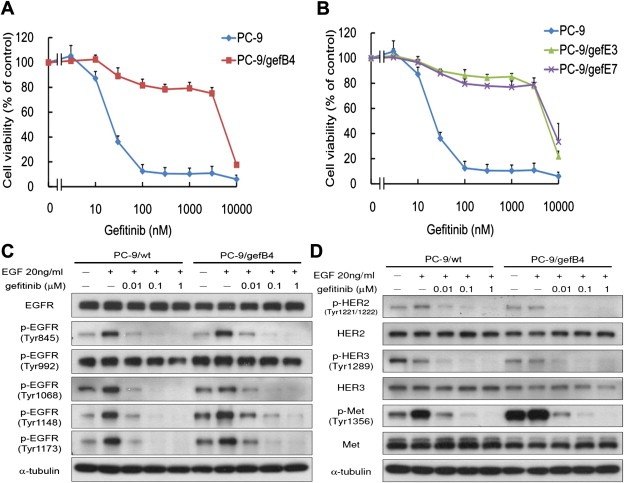
Establishment and characterization of the gefitinib resistant cells. (A) Cell viability of PC‐9/wt and PC‐9/gefB4 cells on various concentrations of gefitinib. (B) Cell viability of PC‐9/wt, PC‐9/gefE3 and PC‐9/gefE7 cells on various concentrations of gefitinib for 96 h. Values are the mean ± S.E.M. (n = 3). (C) Effects of gefitinib on EGF‐induced phosphorylation of EGFR in PC‐9/wt and PC‐9/gef cells (D) Effect of gefitinib on HER2, HER3 and c‐Met as well as related proteins involved in EGF‐EGFR pathways in PC‐9/wt and PC‐9/gef cells.
We explored potential mechanisms relating to the acquired resistance. We sequenced the EGFR gene and the sequence of the EGFR gene in the PC‐9/gefB4 cells exactly matched that of the PC‐9/wt cells; no T790M mutation or other insertional mutation was detected in the PC‐9/gefB4 cells as well as other subclones. The ratio of the copies of the MET gene to the reference gene MTHFR was less than 1, indicating that MET gene was not amplified in the PC‐9/gefB4 cells. We further evaluated several well‐known mechanisms responsible for the intrinsic gefitinib resistance in PC‐9/gefB4 cells. We did not observed HER2 exon 20 insertional mutation as well as the KRAS oncogene mutation. In summary, no known mechanisms responsible for either acquired or intrinsic gefitinib resistance were observed in the PC‐9/gefB4 cells.
To confirm that the resistance resulted from mechanisms other than alterations of receptor tyrosine kinase mentioned above, we evaluated the activities of EGFR, HER2, HER3, and cMET in PC‐9/wt and PC‐9/gef B4 cells. As cross‐activation among receptor tyrosine kinases were reported (Guo et al., 2008; Tang et al., 2008), we observed activation of EGFR and cMET on stimulating cells with EGF (Figure 1C). As HER2 and HER3 may bind to EGFR as heterodimers, we also observed activation of HER2 and HER3 on stimulating cells with EGF (Figure 1C). Considering that there are many tyrosine phosphorylation sites in EGFR, several tyrosine phosphorylation sites on EGFR kinase domain were chosen, including Y845, Y992, Y1068, Y1148 and Y1173 (Figure 1C). Whereas Tyr992 was persistently phosphorylated in the presence of gefitinib, we observed a dose‐dependently attenuationed by gefitinib of EGF‐induced phosphorylation on Y845, Y1068, Y1148 and Y1173 in both PC‐9/wt and PC‐9/gef cells (Figure 1C). Additionally, gefitinib inhibited EGF‐induced phosphorylation of HER2, HER3, and cMET in both cells PC‐9/wt and PC‐9/gefB4 cells (Figure 1D). In summary, the phosphorylation of EGFR, HER2, HER3, and cMET were inhibited in the resistant PC‐9/gef B4 cells, suggesting that the resistance was unrelated to these receptors functionally.
3.2. Persistent activation of the MAPK cascades in the resistant cells and mutation of the NRAS gene
Both AKT‐mTOR and the MAPK cascades are the main downstream pathways of the EGFR signal (Ono and Kuwano, 2006). Whereas gefitinib abolished EGF‐induced phosphorylation of MEK and ERK in the PC‐9/wt cells, we observed persistent EGF‐induced phosphorylation of MEK and ERK despite of gefitinib treatment in the PC‐9/gefB4, PC‐9/gefE3, and PC‐9/gefE7 cells (Figure 2A and B). These data suggest the presence of persistently activated MAPK cascades in the gefitinib‐resistant cells. Despite AKT was dephosphorylated in both PC‐9/wt cell and resistant cells upon gefitinib treatment, we observed a residual phosphor‐AKT signal in the resistant cells upon 0.1 μM of gefitinib treatment (Figure 2A and 2B).
Figure 2.
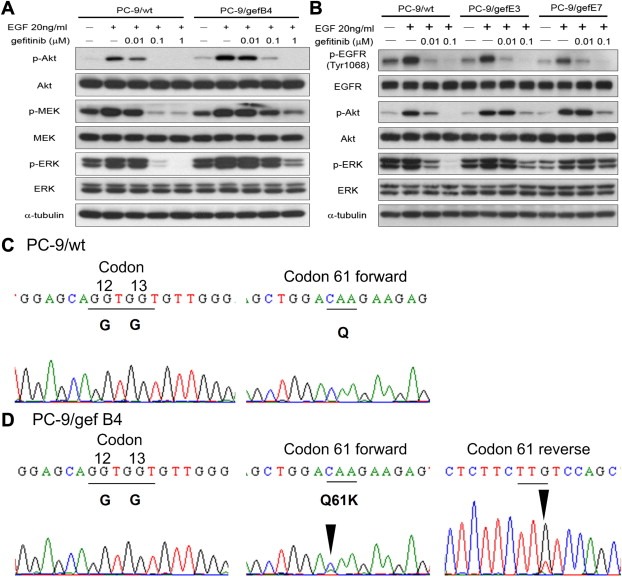
Persistent activation of the MAPK cascades in the resistant cells owing to mutant NRAS gene. (A) The effect of gefitinib on phosphorylation of AKT, MEK and ERK in PC‐9/wt and PC‐9/gefB4 cells. (B) The effect of gefitinib on phosphorylation of AKT and ERK in PC‐9/gefE3, and PC‐9/gefE7 cells. Results were repeated with three independent experiments. (C) NRAS codon 12, 13, and 61 in the PC‐9/wt cells. (D) NRAS codon 12, 13, and 61 in the PC‐9/gef B4 cells.
The persistent activation of MAPK cascades and residual phosphor‐AKT activity in the resistant cells upon gefitinib treatment suggest that alteration in the resistant cells may be at or upstream to the level of the RAS subfamily. As KRAS mutation was not observed in the resistant cells and NRAS mutation may be related to erlotinib resistance in the 11–18 lung adenocarcinoma cells, which carry EGFR L858R mutation (Ohashi et al., 2012), we sequenced the exon 2 and exon 3 of the NRAS gene in the PC‐9/wt and the resistant cells. We observed heterozygous NRAS Q61K mutations in PC‐9/gef B4, PC‐9/gefE3, and PC‐9/gefE7 cells but not in the PC‐9/wt cell (Figure 2C and D, and Figure S1).
3.3. MEK inhibitors on gefitinib‐induced cytotoxicity
To evaluate the significance of MPAK cascades in the gefitinib‐resistance, specific MEK inhibitors, including AZD6244 and CI‐1040, were employed. AZD6244 inhibited cell viability of PC‐9/wt cell and the resistant cells at micromolar level; administration of AZD6244 did not influence the sensitivity of PC‐9/wt cell to gefitinib (Figure 3A). CI‐1040 was modestly less cytotoxic to PC‐9/wt cell and the resistant cells than AZD6244; administration of CI‐1040 had no effect on the sensitivity of PC‐9/wt cell to gefitinib (Figure S2A). While 100 nM AZD6244 was not toxic to PC‐9/wt and resistant cells (Figure 3B), co‐administration of AZD6244 sensitized PC‐9/gef B4, PC‐9/gefE3, and PC‐9/gefE7 to gefitinib in a dose‐dependent manner, (Figure 3C); the same finding was observed in the PC‐9 and resistant cells administrated with CI‐1040 (Figure S2B and S2C).
Figure 3.
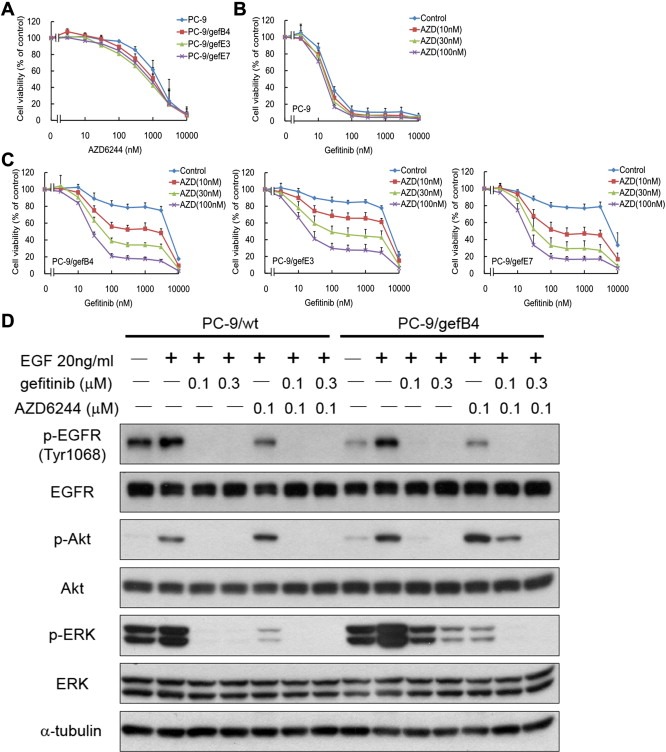
Cytotoxic effect and phosphorylation alterations by gefitinib and/or AZD6244 in PC‐9/wt and PC‐9/gef cells. (A) Effect of AZD6244 on cell viability of PC‐9/wt and PC‐9/gef cells. (B) Effect of AZD6244 on gefitinib sensitivity of PC‐9/wt cells. (C) Effect of AZD6244 on gefitinib sensitivity of PC‐9/gef B4, PC‐9/gefE3, and PC‐9/gefE7 cells. Values are the mean ± S.E.M. (n = 3). (D) Inhibition of AKT and ERK phosphorylation by gefitinib and/or AZD6244 in PC‐9/wt cells and PC‐9/gef B4 cells.
Using Western blot assay, we observed that AZD6244 (100 nM) and CI‐1040 (300 nM) inhibited EGF‐induced ERK phosphorylation in PC‐9/wt and PC‐9/gefB4 (Figure 3C and S2C). AZD6244 and CI‐1040 augmented phosphor‐AKT activities in the PC‐9/gefB4 cell but not in the PC‐9/wt cells, consistent with the idea that the AKT activity in the RAS mutant cells is partially negatively regulated by the MAPK cascades (Ebi et al., 2011). Furthermore, gefitinib plus AZD6244 or CI‐1040 completely abolished phosphorylation of ERK as well as AKT in PC‐9/gefB4 (Figure 3D and S2D). These data suggest that concomitant treatment of gefitinib and MEK inhibitors is required to block the EGFR and NRAS signals in the acquired‐resistant cells.
3.4. Gefitinib plus MEK inhibitors enhanced apoptosis in PC‐9/gefB4 cells
The involvement of apoptosis in cytotoxicity by gefitinib plus MEK inhibitors was studied by measuring apoptosis‐related proteins, including poly (ADP‐ribose) polymerase (PARP), caspase‐3, and caspase‐9 levels. Compared with PC‐9/wt cells which showed significant elevation in activated caspases 3 and 9 as well as PARP in the presence of gefitinib (100 nM), gefitinib was unable to induce profound apoptotic cell death in PC‐9/gefB4 cells (Figure 4A). Whereas AZD6244 (100 nM) or CI1040 (300 nM) alone did not induce cell death, gefitinib plus AZD6244 or CI‐1040 induced marked apoptosis in PC‐9/gefB4 cells (Figure 4A and B). The same augmentation of apoptosis by gefitinib plus AZD6244 or CI1040 were observed in PC‐9/gefE3 and PC‐9/gefE7 cells (Figure 4C and D).
Figure 4.
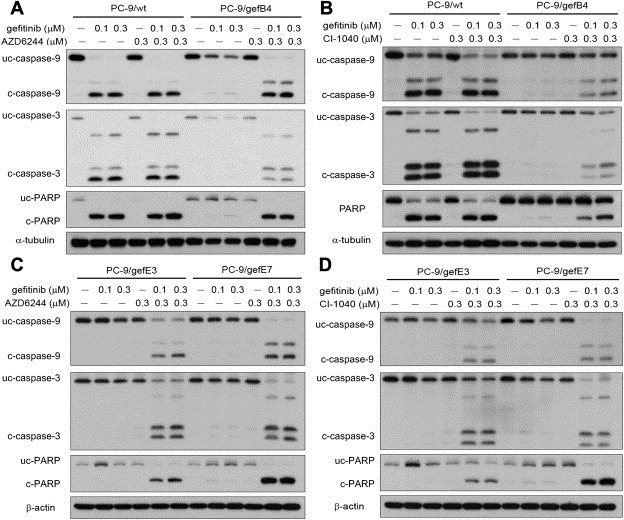
Apoptosis resulting from combined treatment with gefitinib and an MEK inhibitor. (A) apoptosis relating protein induced by gefitinib and/or AZD6244 in PC‐9/wt and PC‐9/gef B4 cells. (B) Apoptosis relating protein induced by gefitinib and/or CI‐1040 in PC‐9/wt and PC‐9/gef B4 cells. (C) apoptosis relating protein induced by gefitinib and/or AZD6244 in PC‐9/gefE3 and PC‐9/gefE7 cells. (D) apoptosis relating protein induced by gefitinib and/or CI‐1040 in PC‐9/gefE3 and PC‐9/gefE7 cells.
3.5. Gefitinib plus MEK inhibitors potentiated gefitinib‐induced anti‐tumor activity in PC‐9/gefB4 xenografts
In vivo studies were used to investigate the effect of AZD6244 or CI1040 on the anti‐tumor activity of gefitinib in Balb/c nude mice with PC‐9/wt and PC‐9/gefB4 xenografts (Figure 5). Gefitinib resulted in PC‐9/wt xenograft shrinkage and slightly slowed down PC‐9/gefB4 xenograft growth. AZD6244 alone slightly delayed the tumor growth of PC‐9/wt and PC‐9/gefB4 xenografts (Figure 5A and B). Whereas administration of AZD6244 had no effect on PC‐9/wt xenograft treated with gefitinib (Figure 5A), AZD6244 and gefitinib treatment completely abolished PC‐9/gefB4 xenograft growth (Figure 5B). Similarly, CI‐1040 did not affect anti‐xenograft effect of gefitinib in PC‐9/gef B4 xenograft (Figure 5C) and combination of both drugs completed inhibited PC‐9/gef B4 growth (Figure 5D).
Figure 5.
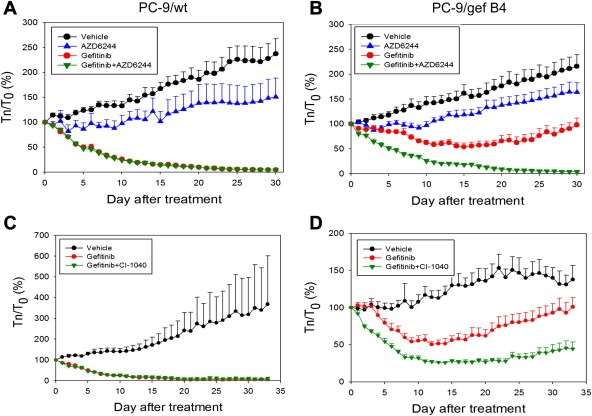
The anti‐tumor effect of gefitinib and AZD6244 or CI‐1040 in the mouse xenograft model. (A) PC‐9/Wt xenografts were treated with vehicle (●), AZD6244 (▵), gefitinib (○), or a combination of both (▾). (B) PC‐9/gefB4 xenografts were treated with vehicle (●), AZD6244 (▵), gefitinib (○), or a combination of both (▾). (C) PC‐9/Wt xenografts were treated with vehicle (●), gefitinib (○), or a combination of gefitinib and CI‐1040 (▾). (D) PC‐9/Gef B4 xenografts were reated with vehicle (●), gefitinib (○), or a combination of gefitinib and CI‐1040 (▾). Tn/T0: Tumor size on day n over tumor size on day 0 of drug treatment. Values are the mean ± S.E.M. (n = 4–9).
4. Discussion
Here we established a panel of acquired gefitinib‐resistant PC‐9 cell lines, which carry activating EGFR exon 19 deletion. We demonstrated that persistent activation of the MAPK cascades was the key mechanism underlying the resistance in these cell lines. We further demonstrated that MEK inhibitors sensitized the resistant cells to gefitinib in vitro and in vivo. In addition, NRAS mutation was found in the resistant PC‐9/gef cells but not in PC‐9/wt cells.
Several mechanisms, such as amplification of Met gene and secondary EGFR T790M mutation, are reportedly related with the acquired EGFR‐TKI resistance in vitro (Engelman and Janne, 2008); their clinical significances had been confirmed (Sequist et al., 2011). In an early stage clinical trial, combination of erlotinib and XL184, an MET inhibitor, results in partial response of a lung tumor harboring amplified MET gene (Wakelee et al., 2010). Afatinib (BIBW‐2992), an irreversible EGFR inhibitor, prolonged progression‐free survival of lung adenocarcinoma patients who had failed previous EGFR‐TKIs therapy in a randomized phase II/III trial (Miller et al., 2012). EGFR T790M specific TKIs are under development (Zhou et al., 2009). However, many tumors of lung adenocarcinoma patients who have failed EGFR‐TKIs therapy do not carry any known mechanisms. Novel strategies to overcome the acquired resistance in these tumors are warranted. In the present study, we showed that combination of an EGFR inhibitor and an MEK inhibitor overcome acquired EGFR‐TKI resistance in PC‐9 derived gefitinib resistant cells which does not harbor secondary EGFR mutation nor MET amplification. Several MEK inhibitors under development clinically do inhibit ERK phosphorylation in tumor specimen, and the therapeutic dose of these inhibitors can be reached in human (O'Neil et al., 2011; Rinehart et al., 2004). In a phase II trial, two NSCLC patients achieved partial response on AZD6244 monotherapy (Hainsworth et al., 2010). In a recent phase II trial focusing on KRAS mutant lung cancer, a combination of AZD6244 and docetaxel resulted in a response rate of 37% (Janne et al., 2012). Accordingly, a combined EGFR inhibitor and MEK inhibitor therapy appears to be therapeutically useful in such group of acquired EGFR‐TKI resistant lung adenocarcinoma, especially for those with aberrant RAS signaling.
NRAS mutation was observed in 1% of lung cancer specimens (Catalogue Of Somatic Mutation In Cancer, COSMIC database). Three NRAS mutation, all were Q61L mutation, were detected in 188 (1.6%) of lung adenocarcinomas (Ding et al., 2008). Little was known about the role of NRAS mutation in EGFR‐TKI resistance in lung adenocarcinoma harboring activating EGFR mutation. Furthermore, coexistence of EGFR mutation and NRAS mutation has never been reported in lung tumor specimens. Here we demonstrated the coexistence of the EGFR mutation and NRAS mutation in the acquired‐resistant cells. In all the gefitinib‐resistant PC9/gef subclones that we tested, NRAS mutation was present, suggesting that NRAS is an essential element for the resistant phenotype that we described in PC9/gef cells. Chang et al. previously demonstrated overexpression of Slug in the same PC9/gef cells indicating that NRAS Q61K and slug overexpression may occur in the same cell. As expression of Slug is under ERK regulation (Chen et al., 2009), it is possible that the mutant NRAS was responsive for the persistent activation of the MAPK cascades and subsequently affected slug expression. Acquired resistance to tyrosine kinase inhibitors due to secondary mutations are common (Choi et al., 2010; Gorre et al., 2001; Greger et al., 2012; Pao et al., 2005a). Continuous exposure of EGFR mutant cells to gefitinib may induce NRAS mutation in the PC‐9/wt cell we were studying. Despite of these, it is possible that, in the PC‐9 cells we were studying, the NRAS mutation may exist in a small portion of cancer cells at the beginning and is undetectable by using conventional sequencing technique. Turke et al. demonstrated that MET amplification pre‐exists in lung adenocarcinoma carrying mutant EGFR gene, and acquired resistance is from clonal selection on EGFR‐TKI treatment (Turke et al., 2010). In contrast to the direct sequencing which detected only one EGFR T790M mutation in 280 EGFR‐TKI treatment naive lung tumors (Inukai et al., 2006), Su et al. demonstrated that, using high sensitive mass spectrometry based assay, EGFR T790M mutation was observed in 25.2% TKI treatment‐naïve lung adenocarcinomas and was associated with lower response rate and shorter progression‐free survival to EGFR‐TKI treatment (Su et al., 2012). Because of different growth kinetics between EGFR‐TKI sensitive lung cancer cell lines and its isogenic acquired resistant cell lines (Chmielecki et al., 2011), pre‐existed EGFR T790M mutant cells may only be detected when the cells are exposed to EGFR inhibitors and the EGFR T790M mutant cells become dominant. Our study detected NRAS mutation only in the gefitinib resistant cells, indicating that NRAS mutation may be pre‐existed in the lung adenocarcinoma carrying mutant EGFR gene, and the proportion of the NRAS mutation in the tumor cells may be too low to be detected by traditional sequencing techniques. As no NRAS mutation was detected by high sensitive methods in two studies published recently focusing on acquired resistance to EGFR‐TKIs in lung adenocarcinoma carrying mutant EGFR gene (Ohashi et al., 2012; Sequist et al., 2011), the contribution of NRAS mutation in clinical EGFR TKI resistance in EGFR mutation positive cells is still not clear. We showed here that gefitinib plus MEK inhibition may completely reverse gefitinib resistance. However, mechanisms other than NRAS mutation may have contributed to MEK activation and gefitinib resistance as well.
In conclusion, persistent activation of the MAPK cascade possibly owing to NRAS mutation results in the acquired gefitinib resistance of lung adenocarcinoma harboring activation EGFR mutation. Combination of an EGFR inhibitor and an MEK inhibitor overcomes EGFR‐TKI resistance and warrants clinical evaluation.
Funding
This study was supported by National Science Council, Taiwan Grant No. NSC95‐2314‐B‐002‐227‐MY3, and Department of Health Grant No. DOH 94‐TD‐B‐111‐001.
Conflict of interest
James Chih‐Hsin Yang received honorarium from Astrazeneca and Pfizer for speech and advisory role.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2012.09.002.
Huang Ming-Hung, Lee Jih-Hsiang, Chang Ya-Ju, Tsai Hsin-Hui, Lin Yu-Lin, Lin Anya Maan-Yuh, Yang James Chih-Hsin, (2013), MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib, Molecular Oncology, 7, doi: 10.1002/mol2.2013.7.issue-1.
Contributor Information
Anya Maan-Yuh Lin, Email: myalin@ym.edu.tw.
James Chih-Hsin Yang, Email: chihyang@ntu.edu.tw.
References
- Bean, J. , Brennan, C. , Shih, J.Y. , Riely, G. , Viale, A. , Wang, L. , Chitale, D. , Motoi, N. , Szoke, J. , Broderick, S. , 2007. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. U. S. A.. 104, 20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, T.H. , Tsai, M.F. , Su, K.Y. , Wu, S.G. , Huang, C.P. , Yu, S.L. , Yu, Y.L. , Lan, C.C. , Yang, C.H. , Lin, S.B. , 2011. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am. J. Respir. Crit. Care Med.. 183, 1071–1079. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Zhu, G. , Li, Y. , Padia, R.N. , Dong, Z. , Pan, Z.K. , Liu, K. , Huang, S. , 2009. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res.. 69, 9228–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielecki, J. , Foo, J. , Oxnard, G.R. , Hutchinson, K. , Ohashi, K. , Somwar, R. , Wang, L. , Amato, K.R. , Arcila, M. , Sos, M.L. , 2011. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl Med.. 3, 90ra59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y.L. , Soda, M. , Yamashita, Y. , Ueno, T. , Takashima, J. , Nakajima, T. , Yatabe, Y. , Takeuchi, K. , Hamada, T. , Haruta, H. , 2010. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med.. 363, 1734–1739. [DOI] [PubMed] [Google Scholar]
- Ding, L. , Getz, G. , Wheeler, D.A. , Mardis, E.R. , McLellan, M.D. , Cibulskis, K. , Sougnez, C. , Greulich, H. , Muzny, D.M. , Morgan, M.B. , 2008. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donev, I.S. , Wang, W. , Yamada, T. , Li, Q. , Takeuchi, S. , Matsumoto, K. , Yamori, T. , Nishioka, Y. , Sone, S. , Yano, S. , 2011. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin. Cancer Res.. 17, 2260–2269. [DOI] [PubMed] [Google Scholar]
- Ebi, H. , Corcoran, R.B. , Singh, A. , Chen, Z. , Song, Y. , Lifshits, E. , Ryan, D.P. , Meyerhardt, J.A. , Benes, C. , Settleman, J. , 2011. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J. Clin. Invest.. 121, 4311–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman, J.A. , Janne, P.A. , 2008. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res.. 14, 2895–2899. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Gorre, M.E. , Mohammed, M. , Ellwood, K. , Hsu, N. , Paquette, R. , Rao, P.N. , Sawyers, C.L. , 2001. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Greger, J.G. , Eastman, S.D. , Zhang, V. , Bleam, M.R. , Hughes, A.M. , Smitheman, K.N. , Dickerson, S.H. , Laquerre, S.G. , Liu, L. , Gilmer, T.M. , 2012. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther.. 11, 909–920. [DOI] [PubMed] [Google Scholar]
- Guo, A. , Villen, J. , Kornhauser, J. , Lee, K.A. , Stokes, M.P. , Rikova, K. , Possemato, A. , Nardone, J. , Innocenti, G. , Wetzel, R. , 2008. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. U. S. A.. 105, 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth, J.D. , Cebotaru, C.L. , Kanarev, V. , Ciuleanu, T.E. , Damyanov, D. , Stella, P. , Ganchev, H. , Pover, G. , Morris, C. , Tzekova, V. , 2010. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J. Thorac. Oncol.. 5, 1630–1636. [DOI] [PubMed] [Google Scholar]
- Inukai, M. , Toyooka, S. , Ito, S. , Asano, H. , Ichihara, S. , Soh, J. , Suehisa, H. , Ouchida, M. , Aoe, K. , Aoe, M. , 2006. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res.. 66, 7854–7858. [DOI] [PubMed] [Google Scholar]
- Janne, P.A. , Shaw, A.T. , Pereira, J.R. , Jeannin, G. , Vansteenkiste, J. , Barrios, C.H. , Franke, F.A. , Grinsted, L. , Smith, P.D. , Zazulina, V. , 2012. Phase II double-blind, randomized study of selumetinib (SEL) plus docetaxel (DOC) versus DOC plus placebo as second-line treatment for advanced KRAS mutant non-small cell lung cancer (NSCLC). J. Clin. Oncol.. 30, 7503 [Google Scholar]
- Li, C. , Fang, R. , Sun, Y. , Han, X. , Li, F. , Gao, B. , Iafrate, A.J. , Liu, X.Y. , Pao, W. , Chen, H. , 2011. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS ONE. 6, e28204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, V.A. , Hirsh, V. , Cadranel, J. , Chen, Y.M. , Park, K. , Kim, S.W. , Zhou, C. , Su, W.C. , Wang, M. , Sun, Y. , 2012. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol.. 13, 528–538. [DOI] [PubMed] [Google Scholar]
- Mok, T.S. , Wu, Y.L. , Thongprasert, S. , Yang, C.H. , Chu, D.T. , Saijo, N. , Sunpaweravong, P. , Han, B. , Margono, B. , Ichinose, Y. , 2009. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med.. 361, 947–957. [DOI] [PubMed] [Google Scholar]
- O'Neil, B.H. , Goff, L.W. , Kauh, J.S. , Strosberg, J.R. , Bekaii-Saab, T.S. , Lee, R.M. , Kazi, A. , Moore, D.T. , Learoyd, M. , Lush, R.M. , 2011. Phase II study of the mitogen-activated protein kinase 1/2 inhibitor selumetinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol.. 29, 2350–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, K. , Sequist, L.V. , Arcila, M.E. , Moran, T. , Chmielecki, J. , Lin, Y.L. , Pan, Y. , Wang, L. , de Stanchina, E. , Shien, K. , 2012. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. U. S. A.. 109, E2127–E2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, M. , Kuwano, M. , 2006. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res.. 12, 7242–7251. [DOI] [PubMed] [Google Scholar]
- Pao, W. , Miller, V.A. , Politi, K.A. , Riely, G.J. , Somwar, R. , Zakowski, M.F. , Kris, M.G. , Varmus, H. , 2005. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med.. 2, e73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao, W. , Wang, T.Y. , Riely, G.J. , Miller, V.A. , Pan, Q. , Ladanyi, M. , Zakowski, M.F. , Heelan, R.T. , Kris, M.G. , Varmus, H.E. , 2005. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med.. 2, e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart, J. , Adjei, A.A. , Lorusso, P.M. , Waterhouse, D. , Hecht, J.R. , Natale, R.B. , Hamid, O. , Varterasian, M. , Asbury, P. , Kaldjian, E.P. , 2004. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol.. 22, 4456–4462. [DOI] [PubMed] [Google Scholar]
- Sequist, L.V. , Waltman, B.A. , Dias-Santagata, D. , Digumarthy, S. , Turke, A.B. , Fidias, P. , Bergethon, K. , Shaw, A.T. , Gettinger, S. , Cosper, A.K. , 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl Med.. 3, 75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos, M.L. , Rode, H.B. , Heynck, S. , Peifer, M. , Fischer, F. , Kluter, S. , Pawar, V.G. , Reuter, C. , Heuckmann, J.M. , Weiss, J. , 2010. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res.. 70, 868–874. [DOI] [PubMed] [Google Scholar]
- Su, K.Y. , Chen, H.Y. , Li, K.C. , Kuo, M.L. , Yang, J.C. , Chan, W.K. , Ho, B.C. , Chang, G.C. , Shih, J.Y. , Yu, S.L. , 2012. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J. Clin. Oncol.. 30, 433–440. [DOI] [PubMed] [Google Scholar]
- Tang, Z. , Du, R. , Jiang, S. , Wu, C. , Barkauskas, D.S. , Richey, J. , Molter, J. , Lam, M. , Flask, C. , Gerson, S. , 2008. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br. J. Cancer. 99, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turke, A.B. , Zejnullahu, K. , Wu, Y.L. , Song, Y. , Dias-Santagata, D. , Lifshits, E. , Toschi, L. , Rogers, A. , Mok, T. , Sequist, L. , 2010. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 17, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai, V. , Kirtikara, K. , 2006. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc.. 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wakelee, H.A. , Gettinger, S.N. , Engelman, J.A. , Janne, P.A. , West, H.J. , Subramaniam, D.S. , Leach, J.W. , Wax, M.B. , Yaron, Y. , Lara, P. , 2010. A phase Ib/II study of XL184 (BMS 907351) with and without erlotinib (E) in patients (pts) with non-small cell lung cancer (NSCLC). J. Clin. Oncol.. 28, (suppl) abstr 3017 [Google Scholar]
- Zhou, C. , Wu, Y.L. , Chen, G. , Feng, J. , Liu, X.Q. , Wang, C. , Zhang, S. , Wang, J. , Zhou, S. , Ren, S. , 2011. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol.. 12, 735–742. [DOI] [PubMed] [Google Scholar]
- Zhou, W. , Ercan, D. , Chen, L. , Yun, C.H. , Li, D. , Capelletti, M. , Cortot, A.B. , Chirieac, L. , Iacob, R.E. , Padera, R. , 2009. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 462, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data