

Abstract
The formation of new blood vessels (angiogenesis) is required for the growth of most tumors. The tumor microenvironment also induces lymphangiogenic factors that promote metastatic spread. Anti‐angiogenic therapy targets the mechanisms behind the growth of the tumor vasculature. During the past two decades, several strategies targeting blood and lymphatic vessels in tumors have been developed. The blocking of vascular endothelial growth factor (VEGF)/VEGF receptor‐2 (VEGFR‐2) signaling has proven effective for inhibition of tumor angiogenesis and growth, and inhibitors of VEGF‐C/VEGFR‐3 involved in lymphangiogenesis have recently entered clinical trials. However, thus far anti‐angiogenic treatments have been less effective in humans than predicted on the basis of pre‐clinical tests in mice. Intrinsic and induced resistance against anti‐angiogenesis occurs in patients, and thus far the clinical benefit of the treatments has been limited to modest improvements in overall survival in selected tumor types. Our current knowledge of tumor angiogenesis is based mainly on experiments performed in tumor‐transplanted mice, and it has become evident that these models are not representative of human cancer. For an improved understanding, angiogenesis research needs models that better recapitulate the multistep tumorigenesis of human cancers, from the initial genetic insults in single cells to malignant progression in a proper tissue environment. To improve anti‐angiogenic therapies in cancer patients, it is necessary to identify additional molecular targets important for tumor angiogenesis, and to get mechanistic insight into their interactions for eventual combinatorial targeting. The recent development of techniques for manipulating the mammalian genome in a precise and predictable manner has opened up new possibilities for the generation of more reliable models of human cancer that are essential for the testing of new therapeutic strategies. In addition, new imaging modalities that permit visualization of the entire mouse tumor vasculature down to the resolution of single capillaries have been developed in pre‐clinical models and will likely benefit clinical imaging.
Keywords: Tumor angiogenesis, Tumor lymphangiogenesis, VEGF / VEGFR, Endothelial cell, Anti-angiogenesis, Mouse models for cancer, Angiopoietin / Tie, Tumor imaging
1. Introduction
The formation of new blood vessels is required for tumor growth beyond a few millimeters in size, due to the diffusion limit of oxygen and nutrients. To grow beyond this limiting size, both tumor and stromal cells secrete various proangiogenic factors that stimulate the sprouting of new vessels from the surrounding tissues. Instead of targeting the tumor cells directly, anti‐angiogenic therapy aims to impair the nutritional and oxygen supply of the growing tumor by reducing the tumor blood vessel network. The concept is based on the early hypothesis promoted by Judah Folkman, that the inhibition of tumor angiogenesis factors should prevent tumor growth (Folkman, 1971).
Transgenic and gene targeting techniques have resulted in a large number of inbred and genetically defined mouse strains to study both developmental and tumor angiogenesis. The tumor models generated have provided pre‐clinical platforms to test the safety and efficacy of novel treatments targeted against the tumor vasculature. Studies in mice have revealed multiple growth factors that regulate vascular development and have also identified signaling pathways that provide potential targets for anti‐angiogenic treatment. Among these, the vascular endothelial growth factor family (VEGF‐A, ‐B, ‐C, ‐D, and placenta growth factor, PlGF) is the most extensively studied. VEGF (or VEGF‐A) is the key inducer of developmental angiogenesis via activation of VEGF receptor 2 (VEGFR‐2), and is probably also the most important signaling pathway for tumor vessel growth. Accordingly, blocking VEGF was found to efficiently inhibit tumor growth in various preclinical models (Chung and Ferrara, 2011; Saharinen et al., 2011), and this led to the development of the first FDA‐approved anti‐angiogenic antibody for clinical use (bevacizumab, a humanized monoclonal antibody directed against VEGF) (Hurwitz et al., 2004).
Similarly to angiogenesis, lymphangiogenesis does not occur in normal adult tissues, but is often activated in the tumor microenvironment. However, lymphatic vessels in solid tumors are often dysfunctional, leading to high interstitial fluid pressure inside the tumors that can reduce the effectiveness of anti‐cancer drugs (Alitalo and Detmar, 2012, Alitalo, 2011). Tumor‐induced alterations in the lymphatic vasculature promote lymph node metastasis, which is a major determinant for the staging and prognostic evaluation of common human cancers. The molecular mechanisms regulating lymphangiogenesis have been less studied than those regulating blood vessels; nevertheless, the major lymphangiogenic signaling pathway is known to be activated by VEGF‐C or VEGF‐D via their receptor VEGFR‐3. The first lymphangiogenesis inhibitors are currently being evaluated in clinical trials for patients with advanced solid tumors. These include VEGFR‐3 blocking monoclonal antibodies that have shown activity also as anti‐angiogenic and anti‐metastatic agents in mouse models (Tammela et al., 2008).
Although various anti‐angiogenic compounds have shown promising anti‐tumor activity in mice, either as single agents or in combination with chemotherapy, thus far the clinical benefit has been limited to modest improvements in overall survival of patients with certain cancer types. For example, the VEGF blocking antibody bevacizumab in combination with chemotherapy extends the survival of advanced colon cancer patients four to five months on average (Hayes, 2011; Hurwitz et al., 2004; Jain, 2008). So called ‘metronomic’ chemotherapy, where relatively low doses of the drug are administered frequently and continuously, seems to be effective when combined with specific anti‐angiogenic compounds (Kerbel and Kamen, 2004). Because of mainly unknown mechanisms, most cancer patients respond only transiently or are resistant to anti‐VEGF (Bergers and Hanahan, 2008; Crawford and Ferrara, 2009a; Shojaei, 2012). Although anti‐angiogenic inhibitors are generally well tolerated, they do have certain side effects, at least partly due to the blocking of molecules important for endothelial cell (EC) homeostasis (Baffert et al., 2006; Lee et al., 2007). Furthermore, in some recent reports, VEGF inhibitors appeared to promote tumor invasiveness in mice under certain conditions (Chung et al., 2012; Ebos et al., 2009; Maione et al., 2012; Paez‐Ribes et al., 2009; Sennino et al., 2012), although this could not be confirmed in a subsequent larger study (Singh et al., 2012a).
At the moment, the outstanding questions include why the anti‐angiogenic drugs are less effective in the treatment of human cancer than in experimental tumors in mice, and what would be the most successful strategy to control angiogenesis and lymphangiogenesis in cancer patients. It is important to note that anti‐angiogenic therapy does not significantly prolong survival in most patients, but we currently lack biomarkers for the identification of those patients who would benefit most from the therapy. Furthermore, it seems that no single anti‐angiogenic agent alone is sufficient to prevent tumor progression in human cancer patients, and the recent findings in mouse models linking angiogenesis inhibition and tumor cell invasiveness may limit the use of anti‐VEGF agents. In order to improve the efficacy and safety of anti‐angiogenic treatments, the identification of novel genes regulating tumor angiogenesis, inhibitors against their protein products, and new treatment modalities are required. The generation of experimental mouse models that better represent the clinical features of tumorigenesis and cancer treatment in patients are also necessary for a better understanding of angiogenesis in cancer biology and therapy. Such models are obligatory tools for the discovery of ways in which anti‐angiogenic drugs should be used to best inhibit tumor growth and metastasis.
In pre‐clinical models, the effects of anti‐angiogenic treatments are commonly evaluated from histological samples collected at the time when the mice are sacrificed. This does not allow gathering of dynamic data on the process of tumor angiogenesis, and high resolution imaging of lymphatic vessels in living mice has not been possible without injectable tracers. Advances in imaging instrumentation and data analysis have recently resolved many technical obstacles for high resolution and deeper imaging depth techniques to monitor tumor vasculature in vivo. These developments should soon impact also clinical imaging (Vakoc et al., 2012; Wang and Hu, 2012).
Here, we review the fundamental concepts learned and recent progress in the generation and use of mouse models to study angiogenesis and lymphangiogenesis in cancer. These models have been essential for the identification of genes and pathways important in tumor vasculature and have enabled the validation of the efficacy and safety of new anti‐angiogenic tumor therapies before their testing in clinical trials. Imaging modalities that permit visualization of the entire mouse tumor blood and lymphatic vasculature down to the resolution of single capillaries have recently been developed in pre‐clinical models, and these techniques will also be reviewed. We will furthermore discuss the advantages and disadvantages of the mouse models used, as well as future challenges and possible clinical implications.
2. Analysis of tumor vasculature in tumor transplantation models
2.1. Transplantation strategies
The most commonly used method to analyze angiogenesis or lymphangiogenesis in cancer has been transplantation of a tumor cell line into isogenic or immunodeficient mice (Figure 1A). The use of several well characterized tumor lines results in rapid tumor formation within a few weeks (Table 1). Tumor cells can be transplanted into different microenvironments: often subcutaneously to enable study of the formation of the primary tumor, intravenously for studies of metastatic organ seeding, or orthotopically to the organ of origin of the malignant cells so that the model mimics tumor growth within the most relevant tissue environment (Loi et al., 2011). Notably, the genetic background of the host mice has a great influence on their sensitivity to angiogenesis, as first shown by D'Amato et al. (Rohan et al., 2000). Considerable variation between the different tumor transplantation models also occurs in the growth pattern of the vessels, leukocyte recruitment, vessel pericytes and the periendothelial matrix. Furthermore, only a few transplanted tumors produce macrometastases before the host mouse needs to be terminated because of the large size of the primary tumor.
Figure 1.
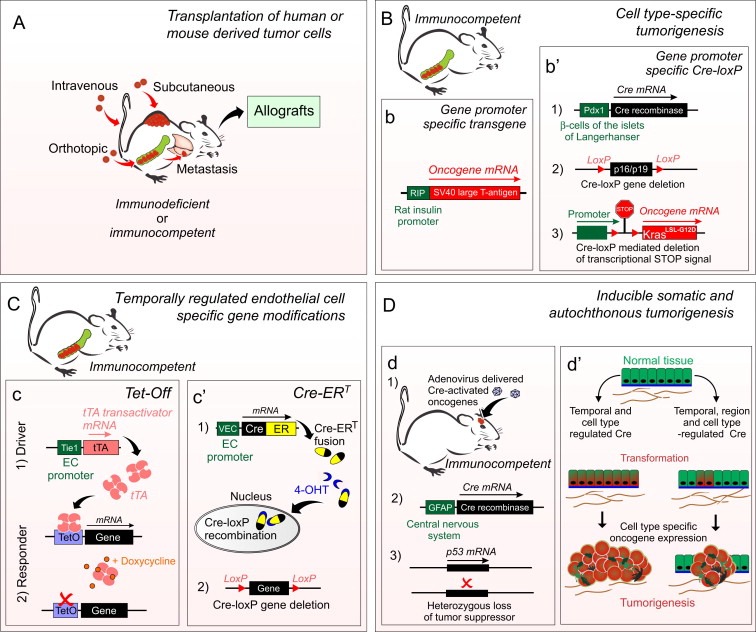
Mouse models to study tumor angiogenesis. A, Tumor cells, of either human or mouse origin are cultured on dishes and transplanted subcutaneously, intravenously, or orthotopically. Transplanted cells can be genetically modified to investigate the role of genes of interest or labeled for cell tracing and in vivo imaging. Transplantation can be combined with genetic models. The propagated tumor cells can be again transferred into new host mice (allografts). Disadvantages are that the tumor is commonly implanted to an ectopic tissue location and the tumor cell lines are often propagated in culture for numerous passages, where they may acquire additional abnormal properties and thus poorly mimic the original cancer. In a syngeneic model (where transplanted cells and host mice are genetically similar), the immune system is not compromised, and thus it may more closely mimic the tumor micro‐environment in patients. B–D, Genetic mouse models allow temporal and spatial control of tumorigenesis in immunocompetent mice. b, The RIP1‐Tag2 line (Hanahan, 1985) is presented in detail in the text. b', In a model for pancreatic ductal adenocarcinoma (Singh et al., 2012a) three modified alleles are combined; 1) Pdx1 promoter‐regulated Cre results in pancreas‐specific 2) deletion of p16Ink4a tumor suppressor gene product p19Arf and 3) the expression of an activated Kras oncogene. Cre deletes a DNA sequence between the two loxP sites oriented in the same direction. The DNA sequence between the loxP sites can contain a tumor suppressor gene (gene deletion) and/or transcription stop‐signals (transgene activation). C, Both spatial and temporal regulation can be achieved by employing tetracycline (Tet) dependent strategies. c, In the Tet‐Off system, transcriptional activator protein (tTA) binds to the tetracycline‐responsive promoter element (TetO) and activates transgene expression. In the presence of tetracycline (or semisynthetic doxycycline), tTA is released and the transgene is not expressed. In the Tet‐On system, reverse tetracycline‐controlled transactivator protein (rtTA) is not bound in the absence of doxycycline, resulting in an inactive transgene. tTA or rtTA expression can be driven by tissue‐specific promoters, thereby regulating target gene expression in a tissue‐specific manner, e.g. Ang2 or VEGF‐C in ECs under the Tie1 or VE‐Cadherin promoter (Holopainen et al., 2012; Lohela et al., 2008). Tet‐dependent strategies also allow repetitive switching to induce and suppress gene expression. c', A fusion protein comprising Cre and the mutant ligand‐binding domain of the estrogen receptor (Cre‐ER). Cre‐ERT produced in specific cells (e.g. in ECs under VE‐Cadherin promoter, (Wang et al., 2010b) remains in the cytoplasm in the absence of 4‐hydroxytamoxifen (4‐OHT, active metabolite of tamoxifen). When bound to 4‐OHT, CreERT is translocated to the nucleus where it catalyzes site‐specific Cre‐LoxP recombination. D. Human cancers are considered to arise from single transformed cell(s) that are propagated over time. d, This can be mimicked using a combination of inherited alleles and viral gene transfer to trigger cell transformation and local tumorigenesis starting from a selected set of somatic cells (Marumoto et al., 2009). d', In models based on a cell‐type specific gene promoter, a relative large number of cells within the organ are simultaneously transformed. Accompanying inherited alleles and viral gene transfer region‐specific expression of activated forms of the oncoproteins occur only in a few cells. Disadvantages of the most sophisticated genetically engineered mouse models include breeding schedules, resulting in high costs and a delay before achieving the “clinical” phenotypes. A–D, depending on the model, the extent of metastasis varies.
Table 1.
Examples of transplantation and genetic mouse models used in tumor angiogenesis and lymphangiogenesis studies.
| Category | Description of the model and characteristics of tumor blood and lymphatic vessels | References |
|---|---|---|
| Tumor transplantation (Mouse syngeneic) | ||
| Lewis lung carcinoma | Tumor cell line from spontaneous lung carcinoma of the C57BL mouse strain. Widely used in tumor angiogenesis and metastasis models. Induces tumor lymphangiogenesis. | (Tsai et al., 2011; O'Reilly et al., 1994) |
| B16 melanoma | Tumor cell line from spontaneous melanoma of the C57BL mouse strain. Several sublines with differing metastatic properties developed by Josh Fidler. Widely used as a model for pulmonary metastasis. Induces tumor lymphangiogenesis. | (Duong et al., 2012; Nasarre et al., 2009; Chung et al., 2012) |
| CT26 colon carcinoma | CT26 is a chemically induced, undifferentiated colon carcinoma cell line from the BALB/c mouse strain. Generates vascularized tumors and induces tumor lymphangiogenesis. | (Belur et al., 2011; Viana et al., 2013) |
| 66cl4 mammary carcinoma | Derived from a spontaneous mammary carcinoma in the BALB/c mouse strain. Metastasizes to the lungs primarily through the lymphatic vasculature. | (Karnezis et al., 2012; Wang et al., 2012) |
| AX osteosarcoma | c‐Myc‐overexpressing Ink4a/ARF−/− bone marrow derived stromal cell line. Produces high‐density tumor vascularization and peritumoral lymphatic vessels. | (Kubota et al., 2009) |
| Tumor transplantation (Human xenografts) | ||
| LNM35 lung carcinoma | A human lung cancer cell line. Allows orthotopic implantation into immunodeficient mice. Induces tumor angiogenesis, lymphangiogenesis and metastasis. | (Kozaki et al., 2000; He et al., 2005; Holopainen et al., 2012) |
| MDA‐MB‐231 breast adenocarcinoma | A human breast adenocarcinoma cell line derived from pleural effusion. Generates vascularized tumors, induces tumor lymphangiogenesis and metastasis. | (Price et al., 1990; Matsui et al., 2008) |
| CWR22Rv‐1 prostate carcinoma | A human prostate carcinoma cell line derived from a serially propagated xenograft in mice. Induces tumor lymphangiogenesis and metastasis. | (Burton et al., 2008; Holleran et al., 2000) |
| Genetic mouse models (Transgenic) | ||
| RIP1‐Tag2 | The mice express SV40 large T‐antigen under the rat insulin promoter, resulting in multistage carcinogenesis of pancreatic islets. Highly vascularized tumors, no tumor lymphangiogenesis. A subset of adenomas show an invasive phenotype. | (Hanahan, 1985; Mandriota et al., 2001) |
| MMTV‐PyMT | The polyoma middle T oncogene expressed under the mouse mammary tumor virus (MMTV) promoter and enhancer. Results in rapid formation of multifocal mammary adenocarcinomas. High metastasis incidence in the lungs and lymph nodes. Is not very lymphangiogenic. | (Guy et al., 1994; Gao et al., 2008) |
| K14‐HPV16 | Human papilloma virus oncogenes driven by the K14 promoter in basal squamous epithelium. Develops epidermal hyperplasias by one month of age. Induces tumor angiogenesis and lymphangiogenesis. Develops an invasive phenotype and metastasizes in ∼20% of mice to regional lymph nodes. | (Coussens et al., 1996; Joyce et al., 2004) |
| Genetic mouse models (Gene targeted) | ||
| Men1 | Targeted gene deletion of the menin tumor suppressor gene. Develops first pancreatic islet cell hyperplasia at ∼nine months of age and later multiple endocrine tumors, especially in the pituitary and parathyroid glands. | (Crabtree et al., 2001; Korsisaari et al., 2008) |
| Genetic mouse models (Mutagenesis induced) | ||
| Apc‐Min | ENU mutagenesis‐induced point mutation in the Apc‐tumor suppressor gene. Heterozygous mice are highly susceptible to spontaneous intestinal adenomas. Only few Apc‐Min tumors show an invasive phenotype, and they rarely metastasize. | (Moser et al., 1993; Korsisaari et al., 2007) |
| Spatially controlled tumorigenesis via cell type specific Cre‐recombinase | ||
| PDA, pancreatic ductal adeno carcinoma | Model of pancreatic ductal adenocarcinoma (PDA, KPC‐mice). Conditional expression of oncogenic Kras (G12D) and inactive p53 alleles (R172H) under pancreatic‐specific Cre recombinase (Pdx1‐Cre). Develops refractory and poorly vascularized pancreatic tumors. Metastases are observed in 80% of mice, most often in the liver and lungs. | (Olive et al., 2009; Cook et al., 2012; Hingorani et al., 2005) |
| Local induction of tumorigenesis in somatic cells using a combination of viral transfection and inducible alleles | ||
| GBM, glioblastoma multiforme | Cre‐loxP controlled lentiviral vectors encoding oncogenic H‐RasV12 and activated form of AKT injected into specific brain regions of tumor suppressor p53 heterozygous mice. Transformation occurs in GFAP‐Cre expressing central nervous system cells. Somatic transformation of only few cells occurs in the specific brain region that mimics natural development of glioblastoma multiforme. | (Marumoto et al., 2009) |
| NSCLC, non‐small cell lung cancer | Mice carry a conditionally activated oncogene (KrasLSL‐G12D) and simultaneously deleted tumor suppressor (p53) allele. Transformation of epithelial cells in the lungs is locally induced via replication‐deficient Cre expressing adenoviral vector administered intranasally. | (Jackson et al., 2005; Singh et al., 2010, 2012a) |
In xenograft transplantation, human derived tumor cells or a piece of solid tumor tissue is transplanted into an immunodeficient mouse. One advantage of xenografting is that a tumor of human origin can be used; however, due to a lack of immune cell contribution, xenograft models often fail to mimic the natural tumor microenvironment. Instead, immunodeficient mice have shown altered tumor growth properties due to a loss of protective immune responses and tumor‐promoting inflammation (Dranoff, 2011). Most often athymic nude mice or mice with severe combined immunodeficiency (SCID, lacking T and B lymphocytes) are used. Recent studies have shown that the non‐obese diabetic (NOD)‐SCID mice, which also lack the gamma chain of the IL‐2 receptor and thus are deficient of mature T cells, B cells, and natural killer (NK) cells, provide the most immunodeficient host for the scoring of clonogenic tumor “stem” cells (Quintana et al., 2012).
In a syngeneic model, the recipient mice and transplanted tumor cells share a common inbred genetic background; therefore tissues transplanted are not rejected by the recipient host's immune system. As the host immune system is not compromised, this model better represents the natural tumor environment. Syngeneic models also allow combination studies using genetically modified mice over‐expressing or lacking gene(s) of interest. It should be noted, however, that continuously growing tumor cell lines in culture undergo genetic drift and selection that may deviate strongly from the evolution of tumor cells in vivo. In addition, both the xeno‐ and syngrafts in general involve the implantation of a large number of tumor cells, thus lacking the stepwise progression of cancer from a single cell to the malignant disease in the tumor's natural microenvironment, making the models artificial in many ways.
The development of humanized host mice has provided some improvements over xenografting strategies, and such mice may better represent tumor heterogeneity in patients. Humanized mice are immunodeficient, complemented with the components of a human immune system, including dendritic, B and T cells, which allows implantation of human tumor material. Alternatively, specific target proteins in the mice can be genetically changed into their human counterparts. For example, a knock‐in‐mutation has been generated to convert the mouse VEGF gene product to its human homolog, permitting the testing of human VEGF‐specific agents in mice (Gerber et al., 2007). In addition, carcinogenic agents can induce specific tumor types in mice. Most commonly, a two‐stage chemical carcinogenesis model has been used in the skin and liver (Abel et al., 2009). There are also certain mouse lines that spontaneously develop cancer, but these are seldom used.
2.2. Anti‐angiogenic therapy in tumor transplant models
In a pioneering experiment, monoclonal antibodies blocking VEGF bioavailability reduced tumor blood vessel density and suppressed the growth of human tumor cell lines (rhabdomyosarcoma, glioblastoma multiforme, leiomyosarcoma) implanted into immunodeficient nude mice (Kim et al., 1993). Later, VEGF blockage has been intensively studied using dozens of human tumor cells lines and various tumor types as xenografts in immunodeficient mice (Gerber and Ferrara, 2005). These experiments have convincingly demonstrated a significant anti‐angiogenic activity and tumor growth inhibition ranging from 25% to 95% (Crawford and Ferrara, 2009b), indicating that VEGF blocking alone is sufficient to reduce human tumor growth in immunodeficient mice. Furthermore, after the pruning of the immature tumor vasculature by anti‐angiogenic treatments, the remaining vessels are often structurally and functionally “normalized” (Carmeliet and Jain, 2011). This may permit better delivery and increased efficiency of cytotoxic drugs against malignant cells and impede metastatic cell dissemination into the circulation.
In addition to the studies of VEGF signaling, tumor transplantation models have been used to investigate the importance of other signaling pathways involved in tumor vasculature and metastasis formation. Among them, the context dependent Tie2 agonist/antagonist Ang2 is expressed in remodeling vessels but not in the resting vasculature, making it an attractive candidate for anti‐angiogenic therapies (Maisonpierre et al., 1997; Thurston and Daly, 2012). The importance of Ang2 in tumor progression and metastasis has been tested using a wide variety of tumor transplantation models. Ang2 has been overexpressed in an endothelium‐specific and inducible manner in transgenic mice implanted with syngeneic tumors (Holopainen et al., 2012), and also in tumor cells including human colon adenocarcinoma and orthotopic pancreatic ductal adenocarcinoma xenografts (Cao et al., 2007; Schulz et al., 2011). The importance of endogenous Ang2 has been evaluated in gene‐targeted Ang2 deficient mice implanted with syngeneic tumors (Nasarre et al., 2009), using Ang2‐selective inhibitors (Oliner et al., 2004) and antibodies (Daly et al., 2013; Holopainen et al., 2012) in tumor‐transplanted immunodeficient mice, as well as human monoclonal Ang2 antibodies in subcutaneous and orthotopic xenograft models (Brown et al., 2010). The results of the above transplantation experiments suggest that Ang2 promotes the early stages of tumor growth, and that Ang2 can increase the plasticity of tumor vasculature via pericyte loss, by decreasing the integrity of EC cell–cell junctions, and by improving the survival of ECs (Daly et al., 2013). Currently, Ang2‐blocking agents are under evaluation in clinical trials (Daly et al., 2013; Saharinen et al., 2011), http://www.clinicaltrials.gov).
In summary, due to its practicality, the transplantation of tumor material into mice has been the most common in vivo method in experimental oncology, and tumor transplantation models have been used to study the pre‐clinical efficiency and safety of virtually all medical interventions aimed against tumor angiogenesis. Transplant tumor models have, however, severe limitations when one tries to mimic the natural multistep development of spontaneous and autochthonous tumorigenesis (Figure 2A). In fact, most anti‐angiogenic agents that have shown significant single‐agent anti‐tumor activities in transplanted mouse models have failed to do so in clinical trials of patients suffering from advanced malignancies. To address the shortcomings of tumor transplantation models, several genetically engineered mouse models have been developed that may better mimic the progression of human cancer and a proper environment.
Figure 2.
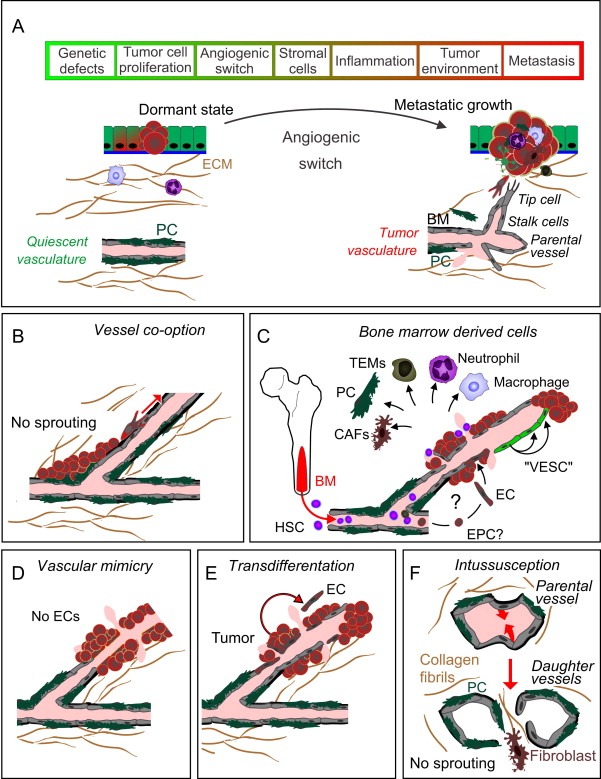
Multistep tumor progression of human cancer and the various vascularization mechanisms. A, Multiple sequential steps are required from the initial genetic defect(s) to the development of metastatic disease. Tumors can be present for years in patients before diagnosis, and metastatic disease usually requires a longer period, which differs from the rapid growth of most transplanted tumors in mice. In the avascular phase, a tumor may remain in a dormant state. An angiogenic switch is induced by tumor environmental factors, prominently by hypoxia, and stimulated also by inflammation or hypoglycemia (Niland and Eble, 2012) via pro‐angiogenic factors, resulting in sprouting angiogenesis (Potente et al., 2011). B–F, Non‐sprouting modes of angiogenesis are less dependent on the signals and cellular interactions that regulate sprouting angiogenesis, and can be refractory to e.g. VEGFR2/VEGF‐targeted anti‐angiogenic therapies. However, the importance of non‐canonical mechanisms for tumor angiogenesis is still under debate. B, In vascular co‐option, tumor cells migrate along pre‐existing vessels. In a mouse model of glioma, vascular co‐option may represent the initial mechanism, which precedes the angiogenic switch (Holash et al., 1999). Growing tumors can also co‐opt lymphatic vessels (Sleeman and Thiele, 2009). C, Bone marrow (BM) ‐residing stem and progenitor cells can stimulate tumor angiogenesis via paracrine mechanisms, but their differentiation to ECs appears to occur only very rarely and probably is not a significant mechanism for angiogenesis in most tumors. “Vascular endothelial stem cells” (VESC, green) reside in the vessel wall and show a long‐term self‐renewal capacity and capability to form ECs in adult mice. D, In solid tumors, rare blood‐filled channels have been found aligned by tumor cells (Maniotis et al., 1999). E, Some recent evidence suggests that in gliomas, tumor vasculature could be composed of a significant amount of ECs that have transdifferentiated from the tumor cells (Marumoto et al., 2009; Soda et al., 2011). F, Intussusceptive angiogenesis refers to splitting of the original vessel via protrusions formed by the ECs on opposite sides of the vessel, resulting in the formation of a transluminal connective tissue pillar, which may contribute to rapid vascular remodeling in some tumors (Ribatti and Djonov, 2012).
3. Analysis of the tumor vasculature in genetically engineered mouse models
3.1. Transgenic and gene‐targeted mice
The identification of tumor‐specific driver mutations and the simultaneous development of genetic engineering techniques in mice have allowed the generation of mouse lines that carry the exact genetic defects implicated in the development of human cancer. Two basic techniques are used to create genetically modified mouse lines: injection of an exogenous transgene to fertilized mouse oocytes, resulting in random chromosomal integration of the transgene, and homologous recombination of introduced DNA in cultured mouse embryonic stem cells (ES cells), allowing locus‐specific targeted mutation of an endogenous gene. In these categories, the mouse lines over‐express an oncogene, are deficient of a tumor suppressor gene, or carry target mutations in specific gene(s) that can be expressed in a temporally and spatially controlled manner (Figure 1).
The development of transgenic techniques during the eighties/nineties led to the generation of several cancer models that are based on forced expression of oncogenes or mutations in tumor suppressors found in human malignancies (Hanahan et al., 2007). Some of the most commonly used transgenic mice in tumor angiogenesis studies are listed in Table 1. In some cases, the transgenic mice better mimic the stepwise pathogenesis of human cancer than transplanted tumors. Their additional genetic modification is possible, and they provide more reliable models to study the interplay between tumor and stromal cells in their natural tissue microenvironments. A possible shortcoming is that the models are usually based on mutations in a single gene, and therefore they do not fully mimic the genetic complexity and heterogeneity of human tumors. This may affect the results when comparing therapy responses between pre‐clinical mouse models and cancer patients. Thus, mice that carry combinations of oncogenic mutations are becoming increasingly important for such studies, as the stepwise progression of human cancer involves successive genetic insults in numerous loci (Farago et al., 2012; Singh et al., 2012b).
Expression of a transgene can be regulated to some extent, but this is affected by random integration of DNA into the genome. Site‐specific homologous recombination in ES cells has been widely used to generate gene deletion alleles; however, global deficiency of key players in angiogenesis (e.g. VEGF) results in early lethality in conventional “knock‐out” models, preventing their use in tumor models in adult animals. The generation of conditional knockout mice and temporal regulation of gene modifications via Cre‐lox, FLP‐FRT, and Tet‐On/Tet‐Off – technologies has allowed a more detailed analysis of gene functions in tumor vasculature in adult mice (Figure 1 and Table 1). Identification of EC‐specific promoter sequences to drive gene expression in an EC‐restricted manner has enabled gene modifications in a spatially controlled manner in the vasculature (Table 2).
Table 2.
Mouse lines expressing blood EC and lymphatic EC specific Cre or a fluorescent protein reporter.
| Mouse line | Description of mouse line | References |
|---|---|---|
| EC‐specific constitutive Cre‐deleter mouse lines | ||
| Tie1‐Cre | The Tie1 gene promoter drives Cre expression in transgenic mice in most embryonic ECs from E8‐9 onwards, but not in all ECs in adult vasculature. There may be expression in some hematopoietic cells. | (Gustafsson et al., 2001) |
| Tie2‐Cre | Tie2 gene promoter and enhancer sequences linked to Cre drive expression in endothelium through embryogenesis and adulthood. May be expressed also in certain mesenchymal cells. | (Kisanuki et al., 2001) |
| Flk1‐Cre | Vegfr‐2 gene promoter and enhancer sequences driving Cre expression in transgenic mice. Cre is expressed in ECs in embryos from E11.5 or E13.5 onwards, depending on the mouse line, except in the lung microvasculature. | (Licht et al., 2004) |
| VE Cadherin‐Cre | VE‐cadherin gene promoter‐Cre. Early expression in EC of yolk sac at E7.5, more continuous EC expression by E14.5 and onwards. Shows ubiquitous EC expression in the adult, including LECs and some hematopoietic cells. | (Alva et al., 2006) |
| LEC‐specific constitutive Cre‐deleter mouse line | ||
| Lyve1‐Cre | GFP‐Cre knock in construct in mouse Lyve‐1 gene locus. Cre is expressed in LECs of the lymph nodes, but also in a varying fraction of other ECs and in some hematopoietic cells. | (Pham et al., 2010) |
| EC‐specific inducible Cre‐deleter mouse lines | ||
| VEC‐Cre‐ERT2 | Ve‐cadherin (Cdh5) promoter‐Cre‐ERT2. Administration of tamoxifen (4‐OHT) induces translocation of Cre‐ERT2 to the nucleus where it mediates Cre‐loxP site‐specific combination. Allows EC specific Cre‐recombination in a temporally and spatially controlled manner. | (Sorensen et al., 2009) |
| Pdgfb‐CreERT2 | Platelet‐derived growth factor B promoter‐Cre. Allows EC specific Cre‐recombination in most vascular beds of newborn mice, but not in all organs in adults (e.g. in central nervous system). | (Claxton et al., 2008) |
| LEC‐specific Cre‐deleter mouse line | ||
| Prox1‐CreERT2 | Prox1 gene promoter driven Cre‐ERT2 deletes in the lymphatic vasculature and in other Prox1‐expressing cells, for example in the heart and liver, but not in ECs, except in venous valves. | (Bazigou et al., 2011) |
| Genetic fluorescent‐protein reporter mouse lines for visualization of lymphatic vessels in living mice | ||
| Vegfr3‐EGFPLuc | GFP‐luciferase fusion protein knock in construct in mouse Vegfr3‐gene, expressed in LECs. Allows fluorescent and luminescent imaging of lymphatic vasculature in embryos and during the first weeks after birth, but expression is strongly down‐regulated during postnatal development. | (Martinez‐Corral et al., 2012) |
| Prox1‐GFP | Fluorescent GFP protein is expressed under the Prox1 promoter in LECs. Recapitulates faithfully the endogenous Prox1 expression. Allows fluorescent imaging of the lymphatic vasculature in embryos and adults. | (Choi et al., 2011) |
| Prox1‐mOrange2 | Fluorescent mOrange2 protein expressed under the Prox1 promoter in LECs. Similar expression pattern as in Prox1‐GFP mice (Choi et al., 2011). Allows fluorescent imaging of lymphatic vasculature in embryos and adults. | (Hagerling et al., 2011) |
| Prox1‐tdTomato | Fluorescent tdTomato protein imaging of lymphatic vasculature. | (Truman et al., 2012) |
| Genetic fluorescent‐protein reporter mouse lines for visualization of blood vessels in living mice | ||
| Tie2‐GFP | Tie2 gene promoter and enhancer sequences control GFP expression in transgenic mice. Allows fluorescent imaging of blood vessels during embryogenesis and in adults. | (Motoike et al., 2000) |
| Tie1‐GFP | Tie1 promoter fragment drives GFP expression in transgenic mice. GFP is expressed in the ECs in embryos, but is down‐regulated in the adult vasculature. | (Iljin et al., 2002) |
3.2. The Apc+/min – intestinal adenoma model
Transgenic Apc+/min mice carry a heterozygous truncating mutation in the adenomatous polyposis coli (Apc) tumor suppressor gene. The wild‐type Apc gene regulates the Wnt signaling pathway via β‐catenin (Nathke, 2004). Somatic and rare inherited APC mutations cause colorectal cancer and familial adenomatous polyposis, respectively, in humans. The Apc+/min mice are highly susceptible to spontaneous intestinal adenomas that appear at approximately six weeks of age (Moser et al., 1993). The adenomas arise from somatic stem cells located in the intestinal crypt base (Barker et al., 2009), thus having a natural tumor microenvironment for stepwise cancer progression. The adenomas in the Apc+/min mice are also highly vascularized, providing a useful model for tumor angiogenesis studies during early intestinal tumorigenesis. However, few Apcþ/min tumors have an invasive phenotype (Boivin et al., 2003) and unlike in humans, in mice the tumors appear first in the small intestine where they soon cause intestinal obstruction and death. Therefore, tumor progression is limited in this model, and metastasis does not occur. Genetic studies have revealed several Min modifying alleles, and a variety of different Apc mutant mice have been generated that more accurately model human polyposis and colon cancer (McCart et al., 2008).
Apc+/min mice have been used to test the efficiency of VEGF blocking monoclonal antibodies (Korsisaari et al., 2007), an inhibitor against VEGFR tyrosine kinases (AZD2171) (Goodlad et al., 2006), and a dual kinase inhibitor against VEGFR2 and epidermal growth factor (EGF) receptors (Vandetanib), that targets both the VEGFR2‐dependent vasculature and the EGFR‐dependent tumor cells (Alferez et al., 2008). In addition, Apc+/min mice have been used in genetic combination experiments, where they are crossed with transgenic mice carrying mutations in other genes of interest, for example to generate genetic deletion of VEGF in the intestinal epithelium of the Apc+/min mice using Cre‐LoxP technology (Korsisaari et al., 2007). These genetic studies have validated the anti‐tumor results obtained with tumor xenografts in immunodeficient mice, and have provided more compelling evidence that VEGFR‐2 signaling plays an important role in the development and progression of certain cancers in mice. They have also shown the efficacy of anti‐VEGF therapy especially in the early benign tumors before the major steps of tumor progression. The Apc+/min transgene in a combination of other gene alleles has also been used to investigate the roles of the cyclin‐dependent kinase Cdk4 (Abedin et al., 2010) and thrombospondin‐1 TSP‐1 (Gutierrez et al., 2003) in tumor angiogenesis.
3.3. RIP1‐Tag2 – pancreatic islet tumors
RIP1‐Tag2 refers to a transgenic mouse line that expresses SV40 large T‐antigen under the insulin promoter, resulting in highly vascularized pancreatic islet tumors (Hanahan, 1985). The RIP1‐Tag2 mice show a natural, stepwise progression of cancer; initial formation of hyperplastic/dysplastic islets occurs at approximately five weeks of age, followed by an angiogenic switch and the development of solid adenomas. RIP1‐Tag2 mice have probably been the most widely used genetic mouse model to investigate the causal role of tumor angiogenesis in cancer progression, as well as the various components involved, such as inflammatory cells, specific growth factors, the extracellular matrix, perivascular cells, and endogenous as well as designed angiogenesis inhibitors.
The importance of VEGF signaling has been studied in RIP1‐Tag2 mice using specific inhibitors and genetic experiments with double transgenic mice. The Cre‐LoxP mediated deletion of VEGF in pancreatic β cells of the RIP1‐Tag2 mice prior to the development of hyperplasia inhibited the angiogenic switch and subsequent tumor growth (Inoue et al., 2002). In line with this genetic experiment, specific function‐blocking antibodies against VEGFR2 (DC101) inhibited the angiogenic switch (Casanovas et al., 2005). Interestingly, it was also found that at later stages, the tumors can become resistant to anti‐VEGF blockage, possibly due to the upregulation of FGF‐signaling, which may rescue the tumor vasculature (Casanovas et al., 2005). The RIP1‐Tag2 mice have also been used to demonstrate a rapid regrowth of the tumor vasculature through the vascular basement membrane sleeves following withdrawal of VEGF signaling inhibitors (Mancuso et al., 2006). The RIP1‐Tag2 tumors metastasize rarely, but their lymphatic metastasis can be enhanced by transgenic overexpression of VEGF‐C under the RIP1 promoter (Mandriota et al., 2001). RIP1‐Tag2 mice have also been used to demonstrate a role for perivascular cells, including PDGFR‐β expressing pericytes, in the maintenance of tumor blood vessels (Bergers et al., 2003) and in inhibition of metastasis (Xian et al., 2006).
To investigate the role of Ang1 and Ang2 in the RIP1‐Tag2 model, Christofori et al. expressed their cDNAs in the pancreatic β cells under the RIP1 promoter. Ang1 expression in tumor cells had only minor effects on angiogenesis or tumor growth. In contrast, Ang2 increased the numbers of infiltrating leukocytes and resulted in poorly perfused and highly disorganized vessels lacking pericyte coverage, suggesting opposite roles for Ang1 and Ang2 in the regulation of EC integrity and plasticity in tumor blood vessels (Fagiani et al., 2011). Interestingly, the Ang2‐specific blocking antibodies inhibited angiogenesis and growth of late‐stage RIP1‐Tag2 tumors (Lewis and Ferrara, 2011), which were resistant to VEGF/VEGFR2 blockage (Casanovas et al., 2005). Other important mediators shown to have a role in tumor angiogenesis in the RIP1‐Tag2 mice include matrix metalloproteinase 9 (MMP‐9) (Bergers et al., 2000), which is produced by tumor infiltrating neutrophils (Nozawa et al., 2006), heparanase (Joyce et al., 2005), and cathepsins (Joyce et al., 2004). These enzymes are upregulated in angiogenic lesions and can modulate the extracellular matrix in the stroma and increase the mobilization of matrix and heparan sulfate‐associated growth factors such as VEGF. Other genes investigated in the RIP1‐Tag2 model include semaphorin 3A, which may function as an endogenous angiogenesis inhibitor that is able to normalize the tumor vasculature (Maione et al., 2009), and the Shb signaling adaptor protein, whose deletion restricts tumor expansion possibly by suppressing VEGF‐dependent tumor angiogenesis (Akerblom et al., 2012).
The RIP1‐Tag2 model has also been successfully used to test the efficiency of anti‐angiogenic molecules including endostatin, angiostatin, TNP470 and small‐molecule receptor tyrosine kinase inhibitors in different stages of pancreatic cancer (Bergers et al., 1999; You et al., 2011). A very significant clinical advance was finally obtained with this model when Hanahan et al. showed that sunitinib inhibits the growth of the RIP1‐Tag2 tumors, which provided a paradigm for the treatment of human pancreatic neuroendocrine tumors (PNET) (Olson et al., 2011).
3.4. MEN1 – multiple endocrine neoplasia
Heterozygous loss of the tumor suppressor gene menin results in endocrine tumors in mice (Crabtree et al., 2001). These closely resemble the human form of cancer characterized by multiple endocrine neoplasias that are due to mutations in the human homolog, the MEN1 gene. The Men1 mouse line was generated using targeted gene deletion in ES cells (Greenberg et al., 1995). In this model, VEGF blocking (with the anti‐VEGF monoclonal antibody, G6‐31) reduced the growth of the endocrine tumors in the pancreas and pituitary gland (Korsisaari et al., 2008).
4. Analysis of tumor vasculature in inducible genetic mouse models that mimic spontaneous and autochthonous cancer development
The advantage of inducible genetic models is that the expression of genes of interest can be temporally regulated in selected somatic cells, thus better mimicking the multistage tumor progression in human patients that most often initiates from single somatic cells. The timing and cell type specificity of gene expression or deletions has been most often controlled using tetracycline (tet) regulated alleles or Cre DNA recombinase selectively expressed under cell type‐specific promoters, or locally introduced using replication‐incompetent viruses encoding Cre (Figure 1). In a similar manner, the Flp recombinase may also be used (Cheon and Orsulic, 2011). The tumorigenesis occurs in immunocompetent mice, and the resulting histopathology is often similar to the human tumors. Some of the disadvantages include that the development of novel genetic mouse models takes years before they can be utilized for studies, and tumorigenesis takes a relatively long time when compared to transplantation experiments, making these sophisticated genetic models more costly and time‐consuming.
4.1. Tet‐On/Tet‐Off – regulated models
In the Tet‐On/Tet‐Off strategy, gene expression is regulated in transgenic mice by tetracycline analogs (often doxycycline) that can be administrated in drinking water or chow (Gossen and Bujard, 1992). A transgene can be either induced (Tet‐On) or repressed (Tet‐Off) in the presence of doxycycline. In the Tet‐Off system, double‐transgenic mice are generated by a combination of two genes in the same mouse: a cell type specific promoter of choice controls the expression of the transactivator (tTA), which induces the expression of the responder gene by binding to its engineered tetracycline operator (tetO) sequence doxycycline‐dependently (Figure 1C).
The Tet‐regulated system has been widely used in combination with tumor transplantation in order to investigate the role of an excess of angiogenic factors (such as VEGF, FGF, Ang2, HIF‐1), which are either induced in the transplanted tumor cells (Giavazzi et al., 2001; Yoshiji et al., 1997) or in ECs (Holopainen et al., 2012). Hypoxia inducible factor‐1 (HIF‐1) is a key transcription factor for the hypoxia response of ischemic tissue, and it induces the expression of many genes that control tumor angiogenesis, including VEGF and Ang2. In the conditional TetON‐HIF‐1 transgenic mice (Oladipupo et al., 2011b), skin neovascularization (controlled with a skin keratin promoter) was induced via a doxycycline‐regulated constitutively active form of HIF‐1. The blocking of VEGFR2 with monoclonal antibodies (DC101) prevented angiogenesis in this model. However, in the late stages, the efficacy was dramatically reduced, suggesting that not all vessels maintain their dependence on VEGFR‐2 (Oladipupo et al., 2011b). The TetON‐HIF‐1 inducible system was also used to investigate the role of VEGF in HIF‐1 mediated neovascularization by combining TetON‐HIF‐1, K14‐Cre, and VEGFflox/flox alleles in a mouse (TetON‐HIF‐1:VEGFΔ). In these mice HIF‐1 is activated in basal keratinocytes (via the keratinocyte specific K5‐promoter) in the absence of VEGF, which was conditionally deleted (VEGFflox/flox allele in K14‐Cre expressing keratinocytes). In the resulting TetON‐HIF‐1:VEGFΔ mice, VEGF was found to be dispensable for initial endothelial sprouting. However, VEGF was essential for the subsequent phase of HIF‐1‐mediated neovascularization despite the robust expression of other angiogenic growth factors (such as PlGF, PAI‐1, MMPs) that were not sufficient to compensate for the lack of VEGF in this model (Oladipupo et al., 2011a).
4.2. Spatial control of tumorigenesis via Cre‐recombinase
In an interesting study, tumor vasculature in pancreatic ductal adenocarcinoma (PDA) was analyzed using tumor transplant and inducible genetic models in parallel (Olive et al., 2009). PDAs are characterized by an extensive connective tissue stroma and when compared to many other solid tumors, they are poorly vascularized and show high resistance to chemotherapy. The poor perfusion probably contributes to inefficient drug delivery, resulting in high patient lethality. Among the somatic mutations that cause PDA, activating mutations in the KRAS proto‐oncogene are found in a majority of cases. PDA tumor transplantation models poorly mimic the human disease, as the characteristic fibrotic stroma is not developed in xenografts and in contrast to the human PDA, the transplanted PDAs in mice are sensitive to chemotherapeutics (Olive et al., 2009). To overcome these shortcomings in transplantation experiments, a genetic mouse model (KPC mice) was generated by activation of the expression of mutated Kras (KrasLSL‐G12D/+) and Trp53 (p53LSL‐R172H/+ or p53LSL‐R270H/+) in pancreatic cells using Cre‐recombinase under the Pdx1‐promoter (Hingorani et al., 2005). This produced PDA with fibrotic stroma and poorly developed vasculature, thus resembling well the characteristics of the human tumors, and this model was used to study the effect of Hedgehog signaling inhibitors on tumor growth. Normally, Hedgehog signaling regulates embryonic patterning, and in human cancers, aberrant Hedgehog signaling has been reported (McMillan and Matsui, 2012). Interestingly, in KPC mice a combination of chemotherapy (gemcitabine) with a Hedgehog signaling inhibitor (Smoothened inhibitor, IPI‐926) efficiently eliminated tumor associated fibroblasts, thus reducing the fibrous stroma, but the treatment also increased tumor vascular density (Olive et al., 2009). The increase in tumor perfusion enhanced chemotherapeutic drug delivery, but resulted in only a transient stabilization of disease, suggesting an adaptive resistance mechanism.
This same KPC mouse model for PDA has been recently used to test the effect of a Notch pathway antagonist (gamma secretase inhibitor MRK003) on tumor vasculature (Cook et al., 2012). Dysregulated Notch signaling has been implicated in human cancers (Kalaitzidis and Armstrong, 2011). In this signaling system, ligand (Delta‐like or Jagged) binding to the transmembrane Notch receptor results in the γ‐secretase‐dependent release of the Notch intracellular domain (NICD) and its translocation to the nucleus, where it functions as a transcriptional activator. In the KPC mice, the γ‐secretase inhibitor MRK003 inhibited intratumoral Notch signaling, and in combination with chemotherapy (gemcitabine), it prolonged the survival of the tumor‐bearing mice. The combination of γ‐secretase inhibition and chemotherapy efficiently depleted ECs in the established intratumoral vasculature, resulting in widespread hypoxia and tumor necrosis (Cook et al., 2012). Collectively, these results using the genetic KPC model suggest that the dysfunctional vasculature, particularly in PDA, may represent an attractive target for vascular therapies, and that at least in some tumor types, genetic mouse models may mimic human cancer better than tumor transplantation models.
4.3. Spatiotemporal control of tumorigenesis via Cre‐ERT
The tamoxifen‐inducible Cre‐ERT system is based on a designed recombinant fusion protein in which the ligand‐binding domain of the mutant estrogen receptor (ER) is fused to the Cre recombinase (Cre‐ERT and its more effective and less leaky form, Cre‐ERT2) (Feil et al., 1996; Indra et al., 1999). Nuclear translocation of Cre‐ERT is induced by an active metabolite of tamoxifen, 4‐hydroxytamoxifen (4‐OHT). To obtain cell‐type restricted activation of Cre‐ERT, specific promoters or local 4‐OHT administration can be used. Inducible alleles are constructed with loxP sites flanking the transcriptional stop sequence (LSL, Lox‐Stop‐Lox, Figure 1B). In the same mice, simultaneous deletion of tumor suppressor genes (commonly p53 or Rb) to increase tumorigenity can be obtained. Another interesting tamoxifen‐inducible model is a switchable beta‐cell specific c‐Myc‐ERT oncoprotein that has been used to analyze the robust induction of angiogenesis by the Myc oncoprotein (Shchors et al., 2006; Shchors and Evan, 2007).
4.4. Local induction of tumorigenesis in few somatic cells using a combination of virally transferred genes and cell‐type specific Cre
Human cancer is thought to arise from a single cell that has suffered somatic mutations, resulting in the activation of oncogenes and/or the deletion of tumor suppressor genes. The cellular microenvironment varies between tumor types depending on where the tumor cells originate, and this profoundly influences the characteristics of the tumors, including their blood and lymphatic vasculature. A shortcoming in the more generic genetic models is that tumor cell transformation is simultaneously induced in all cells of a given cell type where the promoter used is active (Figure 1D). In order to develop mouse models in which only a few cells are initially transfected, strategies that are based on replication‐deficient viral vectors carrying inductive genes and mice with loxP flanked germ‐line genes have been developed. For example, tumor angiogenesis in glioblastoma has been studied in a mouse model in which tumorigenesis is induced in only a few dozen adult glial cells in a region‐ and cell type‐specific manner (Marumoto et al., 2009). In this model, Cre‐loxP‐controlled lentiviral vectors encoding H‐Ras oncogene or activated Akt serine kinase were injected into specific brain regions of tumor suppressor p53 heterozygous mice, which also expressed Cre in the central nervous system cells under the control of the GFAP promoter. In these mice, Cre induced activation of mutated H‐Ras and Akt in virally transfected GFAP‐expressing cells in the hippocampus, resulting in local cell transformation (Marumoto et al., 2009). The model has been used to study the differentiation of glioma stem cell‐like cells into vascular ECs in the brain, but the strategy appears versatile enough to be used in combination with other Cre lines for somatic induction of tumorigenesis also in other organs.
4.5. Can the clinical activity of anti‐angiogenic drugs be better predicted in genetic than in conventional mouse tumor models?
Because inducible genetic mouse models are expensive and take a long time to generate, an important question is how much better they can actually predict the efficacy of anti‐angiogenic drugs than the more commonly used tumor transplantation models. In an important study (Singh et al., 2010), the clinical activities of EGF and VEGF inhibitors were tested in different combinations with chemotherapies using inducible genetic mouse lines for non‐small‐cell lung cancer (NSCLC, KrasLSL‐G12D; P53frt/frt) and pancreatic carcinoma (KrasLSL‐G12D; P16/p19fl/fl; Pdx1‐Cre) (Aguirre et al., 2003; Jackson et al., 2005; Singh et al., 2010). The authors then retrospectively compared anti‐angiogenic and anti‐tumor outcomes in these mice with the results of clinical trials to evaluate how well mouse models reflected the human therapeutic outcomes. As in the clinical NSCLC trial, a VEGF blocking antibody (B20‐4.1.1.) improved the overall survival of mice when used in combination with chemotherapy (carboplatin) (Sandler et al., 2006). A similar result was obtained with xenografts of the KRAS mutant NSCLC cell line. On the other hand, in the genetic mouse model of pancreatic carcinoma, the combination treatment showed improved survival in half of the mice, but no beneficial response in the other half. In the corresponding human trials, bevacizumab in combination with gemcitabine failed to show any clinical benefit (Van Cutsem et al., 2009).
Thus, based on current data it is not yet clear whether the most sophisticated genetic models are significantly better in predicting clinical activities of anti‐angiogenic drugs than the more conventional models (Francia and Kerbel, 2010; Singh and Ferrara, 2012). Currently, most anti‐angiogenic agents are pre‐clinically tested in tumor transplantation models or in the more generic genetic models (such as RIP1‐Tag2), but such studies in inducible genetic models have been limited. In both types of models, predicting the clinical efficacy of anti‐angiogenic agents has been difficult. This has been problematic not only with anti‐VEGF therapies, but also with other promising anti‐angiogenic drugs, such as the fragment of collagen XVIII that was initially reported to provide anti‐angiogenic activity in several primary tumors growing in syngeneic mice (O'Reilly et al., 1997) and in the RIP‐Tag2 model (Bergers et al., 1999), but which failed in clinical trials in the U.S. Intriguingly, a modified endostatin has now been approved by the Chinese State Food and Drug Administration as a treatment for non‐small‐cell lung carcinoma (Fu et al., 2009). It is also important to note that the poor predictive value of the pre‐clinical models and the resulting high failure rate in clinical trials occurs in oncology in general; more than 90% of phase three clinical trials fail to show any benefit (Singh and Ferrara, 2012).
Some controversial results have also been obtained regarding the role of placenta growth factor (PlGF) in tumor angiogenesis in pre‐clinical mouse models. PlGF is a member of the VEGF family, that binds to VEGFR1 but not VEGFR2. Fischer et al. (2007) originally reported that an antibody against PlGF can inhibit tumor growth, metastasis, angiogenesis and lymphangiogenesis in mouse syngeneic tumors. This result was not, however, confirmed in a subsequent larger study using four different PlGF blocking antibodies in tumor transplantation experiments in immunodeficient and immunocompetent mice (Bais et al., 2010). The anti‐tumor effects of PlGF blocking antibodies were further studied in carcinogenesis and transgenic tumor models (Van de Veire et al., 2010), providing evidence that only some tumors responded to the treatment. In a recent study, Yao et al. (2011), found that among twelve human tumor cell lines, the efficacy of anti‐PlGF treatment correlated with VEGFR1 expression and activity in tumor cells, suggesting that anti‐PlGF treatment may primarily target VEGFR1‐positive tumor cells, rather than ECs (Yao et al., 2011).
5. Diverse mechanisms of tumor vascularization and resistance to anti‐angiogenic therapy
In human cancer patients as well as in mouse models, the tumor vasculature can become resistant to anti‐VEGF treatments, resulting in tumor re‐growth. Such refractoriness is likely one reason for the poor responses to angiogenic therapies in human patients, and studies in mouse models have pointed to several potential mechanisms involved. Those suggested in various publications include the activation of alternative signaling pathways that are insensitive to VEGF inhibition, recruitment of bone marrow and pro‐angiogenic stromal cells, increase in cell invasion and metastasis due to VEGF blockage, activation of non‐canonical mechanisms for angiogenesis, regrowth of the vasculature involving drug resistant ECs that may be generated by tumor cell transdifferentation, and heterogeneity of tumor ECs (Bergers and Hanahan, 2008; Crawford and Ferrara, 2009a; Shojaei, 2012).
5.1. Heterogeneity of ECs in the tumor vasculature
It has been considered that tumor ECs represent a genetically more stable cell population than tumor cells, thus providing a more homogenous and less drug‐resistant target to prevent tumor growth than the genetically highly variable malignant cells. However, in one study, tumor‐associated ECs isolated from human renal cell carcinomas were reported to contain frequent chromosomal abnormalities (Akino et al., 2009). In line with this observation, phage‐display profiling of ECs from transgenic tumor models (HPV16‐induced epidermal carcinogenesis and RIP1‐Tag2 pancreatic islet cell carcinoma) indicated differences between normal and tumor vasculature (Seaman et al., 2007) between premalignant and malignant stages of squamous cell carcinoma (Hoffman et al., 2003) and between tumor types and anatomical locations (Joyce et al., 2003). Furthermore, at least in gliomas, some vascular ECs may be derived from tumor cells (Marumoto et al., 2009; Soda et al., 2011). Such genetic heterogeneity could contribute to the resistance to single‐agent therapy and may increase variation in the responses to anti‐angiogenic agents. When compared to the normal vasculature, tumor blood vessels are also structurally and functionally abnormal, and they show a high degree of heterogeneity that poses a considerable challenge for anti‐angiogenic therapies. Morphologically, the tumor vessels are dilated and tortuous. At the cellular level, tumor ECs show a high variation in shape, junctions between ECs are discontinuous, the perivascular matrix and cell support are aberrant, and inflammatory cells can be present in high numbers. This correlates with abnormal function: tumor vessels are poorly perfused and leaky, which increases the interstitial pressure, impairing drug delivery to the tumor. The loss of endothelial integrity may also facilitate metastatic cell intravasation and thus enhance tumor cell dissemination via the bloodstream. Lastly, EC phenotypes can be significantly different between various tumor types, and they also vary within a single tumor (Nagy et al., 2010).
5.2. Non‐canonical mechanisms of tumor vasculature growth
In sprouting angiogenesis, angiogenic growth factors secreted from tumor and stromal cells activate ECs in pre‐existing blood vessels located near the tumor cells. The distinct steps of resulting sprouting angiogenesis are relatively well characterized, and specific inhibitors against the key molecules have been developed (Potente et al., 2011). Certain tumor types can also employ non‐sprouting modes of angiogenesis that may operate in parallel with sprouting angiogenesis. These non‐canonical mechanisms of angiogenesis may provide blood supply before induction of the angiogenic switch, or occur as an adaptive response to changes in the tumor environment. The importance of non‐sprouting angiogenic modes in tumor biology is still under investigation, and these may account in part for the resistance toward anti‐angiogenic therapies. The non‐sprouting mechanisms include vessel co‐option (Holash et al., 1999), intussusceptive angiogenesis (Caduff et al., 1986), vasculogenic mimicry (Maniotis et al., 1999), and differentiation of glioma stem cell‐like cells into vascular ECs (Marumoto et al., 2009; Soda et al., 2011) (Figure 2).
5.2.1. Intussusceptive angiogenesis
In contrast to sprouting angiogenesis, EC proliferation is not required in intussusceptive growth. Instead, new vascular channels are generated via the formation of connective tissue pillars that can form rapidly. VEGF, Ang‐1, PDGF, Epo, and laminar shear stress have been implicated in this process; however, no intussusceptive‐specific molecules have so far been identified (Ribatti and Djonov, 2012). Intussusceptive tumor angiogenesis in mice has been studied using human colon adenocarcinoma xenografts in the dorsal skinfold chamber (Patan et al., 1996) and in colorectal carcinoma transplants in syngeneic C57Bl/6 mice (Paku et al., 2011; Patan et al., 1996). In mammary carcinoma allografts, a switch from sprouting to intussusceptive angiogenesis appeared responsible for the development of resistance to anti‐VEGF treatment and tumor recovery after termination of anti‐VEGF therapy (Hlushchuk et al., 2008).
5.2.2. Vessel co‐option
Instead of inducing active angiogenesis, malignant cells can grow along pre‐existing host vessels in well vascularized tissues, such as the brain, and are thus refractory to anti‐angiogenic therapy (Holash et al., 1999). Vessel co‐option was studied in transplantation experiments with a human melanoma tumor cell line (Kusters et al., 2002), a murine glioma cell line in the brain of immunodeficient mice (Winkler et al., 2009), and in a xenografting experiment using human primary glioblastomas in immunodeficient nude rats (Sakariassen et al., 2006). Sakariassen et al. found that transplanted human glioblastomas co‐opted the host vasculature, resulting in an invasive phenotype concurrent with the expression of neural stem cell markers, but inducing no angiogenesis in the host brain, suggesting that malignant tumor growth is not necessarily angiogenesis‐dependent. When compared to the first‐generation tumors, a less stem cell‐like gene expression pattern, a less invasive, and an angiogenesis‐dependent phenotype was observed when tumors were re‐grafted and passaged in new animals. The change did not occur parallel with progressive genetic derangements or clonal selection, suggesting that tumor cells can alter their phenotypes via transcriptional regulation (Sakariassen et al., 2006). Microscopic imaging of RFP‐labeled gliomas and GFP‐labeled ECs in living mice has revealed that even a single glioma cell can alter vessel diameter, creating a more favorable microenvironment for co‐opted glioma growth, without the necessity of sprouting angiogenesis. Interestingly, in this same model it was found that the perivascular surface of host capillaries was the primary trail for the invading glioma cells (Winkler et al., 2009).
5.2.3. Vascular mimicry and glioblastoma stem cell differentiation into tumor ECs
In the process of vascular mimicry (Maniotis et al., 1999), tumor cells form vessel‐like tubular structures that conduct blood flow, but are devoid of an EC lining. In addition, mosaic vessels containing both ECs and tumor cells have been reported (di Tomaso et al., 2005; Shaifer et al., 2010). Vascular mimicry can be advantageous in the areas of tumors that are poorly perfused via blood vessels, and these additional channels may also contribute to the draining of interstitial fluid, which is often impaired in solid tumors. However, the importance of vascular mimicry is still under debate and a puzzling question concerning vascular mimicry is why blood does not coagulate in such channels. Vascular mimicry was observed in athymic nude mice orthotopically injected with human glioma stem cell derived tumors (Dong et al., 2010) and with glioma cell lines (Shaifer et al., 2010). Additional studies (Ricci‐Vitiani et al., 2010; Soda et al., 2011; Wang et al., 2010a) have suggested that glioblastoma tumor‐derived stem cell‐like cells are capable of giving rise to a significant portion of the blood vessel lining ECs in the tumors. In the glioblastoma tumor angiogenesis model of Marumoto et al. (2009), a significant part of the tumor vasculature consisted of tumor‐derived cells, suggesting that glioblastoma cells can transdifferentiate into ECs and contribute to the formation of tumor blood vessels. Interestingly, in in vitro experiments, hypoxia promoted the transdifferentiation, which may be stimulated by lower oxygen levels in the tumor environment (Soda et al., 2011). However, the glioblastoma‐derived ECs lacked VEGFR2 and were refractory to VEGFR inhibition (Soda et al., 2011). Similar results were obtained in orthotopic and subcutaneous xenograft transplantation experiments in immunocompromised mice (Ricci‐Vitiani et al., 2010). In this model a relatively high number (ranging from 20 to 90%) of blood vessel lining cells carried the same genomic alteration as the implanted tumor cells, indicating that a significant portion of the tumor ECs may have a neoplastic origin. Importantly, selective targeting of stem cell‐like tumor‐derived ECs using transfection by the thymidine kinase gene followed by ganciclovir treatment resulted in diminished capillary density and tumor growth retardation (Ricci‐Vitiani et al., 2010). In another study, Notch signaling was found to contribute to the initial tumor stem cell differentiation to EC progenitors, while VEGF‐VEGFR2 signaling contributed to the maturation of the tumor derived cells into ECs (Francescone et al., 2012; Wang et al., 2010a). The glioblastoma stem cell‐like cells may also transdifferentiate into vascular mural‐like cells that may contribute to the functionality of tumor vessels (Scully et al., 2012).
6. Bone marrow‐derived cells in tumor angiogenesis
6.1. Progenitor cell differentiation into ECs
It is well documented that tumor angiogenesis is promoted by the homing of a variety of bone marrow (BM)‐derived cells into tumors. Both a paracrine contribution (secretion of pro‐angiogenic molecules) and direct endothelial progenitor cell (EPC) incorporation into the growing blood vessels and their differentiation into functional ECs have been reported (Figure 2C). However, the latter concept is still very controversial (Fang and Salven, 2011; Rafii and Lyden, 2008). EPCs from wild‐type BM were originally reported to rescue tumor angiogenesis in angiogenesis‐defective Id‐mutant mice (Lyden et al., 1999, 2001). Similar results appeared later in a syngeneic lung tumor transplantation model and in the transgenic MMTV‐PyMT breast cancer (polyoma virus middle T oncoprotein expressed in mammary epithelial cells) (Gao et al., 2008). The integration of BM‐derived cells into the tumor vasculature was also described in the transgenic RIP1‐Tag5 pancreatic insulinoma (Ganss and Hanahan, 1998) and AlbTag hepatocellular carcinoma (Spring et al., 2005). However, the role of EPCs in tumor vasculature could not be confirmed by others (Fang and Salven, 2011). Instead, genetic fate mapping studies indicated that tissue‐resident endothelial stem/progenitor cells, rather than hematopoietic circulating cells, contribute to angiogenesis at least in the healing of mouse digits after amputation (Rinkevich et al., 2011). Recently, Fang et al. (2012) identified a small population of blood vessel wall resident c‐kit (CD117+) positive ECs that the authors called “vascular endothelial stem cells” (VESC). The authors further showed that these cells have a long‐term self‐renewal capacity and are capable of forming functional blood vessels in adult mice. Interestingly, VESCs may also participate in tumor angiogenesis, as c‐kit deficiency in these cells resulted in impaired angiogenesis and retardation of subcutaneously injected syngeneic B16 melanomas (Fang et al., 2012).
6.2. The paracrine role of BM‐derived cells in the tumor vasculature
In many mouse models, BM‐derived cells are closely associated, but not directly incorporated into the endothelium of tumor vessels. Instead, the BM‐derived cells promote the expansion of the existing vasculature in a paracrine manner. Such BM‐derived cells include tumor associated macrophages, Tie2‐expressing mononuclear cells, mast cells, eosinophils, and neutrophils (Fang and Salven, 2011; Sica et al., 2008).
The depletion of tumor‐associated myeloid cells in various experimental mouse models significantly decreased vascular density and tumor progression (De Palma et al., 2005; Kubota et al., 2009; Lin et al., 2007). De Palma et al. (2005) showed that Tie2‐receptor expressing BM‐derived proangiogenic monocytes (TEM) are selectively recruited to the RIP1‐Tag2 tumors and to orthotopic human glioblastoma xenografts. In conditional transgenic mice, the expression of the herpes virus thymidine kinase under the control of the Tie2‐promoter and simultaneous administration of ganciclovir selectively depleted TEMs and inhibited angiogenesis in human glioma xenografts, resulting in tumor regression.
Neutrophils that produce the proangiogenic matrix metalloprotease type 9 (MMP‐9) are also recruited to sites of angiogenesis. MMP‐9 degrades the perivascular matrix and increases the bioavailability of matrix‐bound VEGF (Bergers et al., 2000). On the other hand, VEGF, induced by hypoxia, plays a role in the recruitment of a subset of proangiogenic neutrophils that express high levels of MMP‐9 and the chemokine receptor CXCR4 (Christoffersson et al., 2012). Transient depletion of neutrophils from dysplastic cell islets and pancreatic tumors of RIP1‐Tag2 mice using specific antibodies inhibited the angiogenic switch, suggesting an important role for neutrophils in the early stages of tumorigenesis in the RIP1‐Tag2 mice (Nozawa et al., 2006). Accordingly, inhibition of neutrophil infiltration by interleukin‐8 neutralizing antibodies reduced tumor angiogenesis in an orthotopic xenotransplantation model (Bekes et al., 2011). The importance of tumor infiltrating macrophages in mammary gland tumors was investigated in the MMTV‐PyMT mouse breast cancer model (Lin et al., 2006, 2007). Inhibition of VEGF‐producing macrophage infiltration by depleting the macrophage colony‐stimulating factor CSF‐1 resulted in a delayed angiogenic switch and malignant transition (Lin et al., 2006). To confirm the role of tumor‐associated macrophages in the angiogenic switch, Lin et al. conditionally induced VEGF expression in the mammary epithelium in a bi‐transgenic mouse line (Lin et al., 2007), in which the expression of the VEGF transgene was temporally controlled via the Tet‐On system specifically in the mammary epithelium using the MMTV promoter. This resulted in leukocyte, mostly macrophage, infiltration, stimulating tumor angiogenesis. In a mouse model for osteosarcoma metastasis, C57Bl6 mice were subcutaneously transplanted with c‐Myc‐overexpressing Ink4a/ARF −/− bone marrow‐derived stromal cells. The tumors were characterized by high‐density tumor vasculature and peritumoral lymphatic vessels, while CSF‐1 inhibition suppressed the tumor angiogenesis and lymphangiogenesis, and decreased tumor regrowth following interruption of the treatment (Kubota et al., 2009).
7. Adverse side effects of anti‐VEGF therapy
Similar signaling pathways operate in tumor angiogenesis and developmental angiogenesis, and some of these may also be required for the homeostatic functions of the normal vasculature. Therefore, an important question is how different the mechanisms in pathological angiogenesis vs. physiological angiogenesis are, and which anti‐angiogenic molecular targets are important in the maintenance of the quiescent vasculature. Genetic deletion experiments in mice are valuable tools to investigate the efficiency and safety of treatments targeting the key players in angiogenesis.
VEGF inhibition is generally well tolerated; however, recent studies in mice have revealed some unexpected results that could limit the use of anti‐VEGF agents in cancer therapy. VEGF gene expression in the ECs seems to be required for the steady state homeostasis of the ECs in the mature vasculature (Lazarus and Keshet, 2011). On the other hand, in certain conditions, VEGF blockage was reported to promote tumor cell invasion and metastasis (Ebos et al., 2009; Ebos and Kerbel, 2011; Loges et al., 2009; Paez‐Ribes et al., 2009). However, the evidence from various mouse models is controversial, and so far there is no evidence of increased invasiveness or metastasis associated with anti‐VEGF therapy in human patients (Singh et al., 2012a).
In a study by Singh et al. (2012a), four different genetic mouse models were used to investigate tumor cell metastasis after anti‐VEGF antibody treatment: pancreatic ductal adenocarcinoma (KrasLSL‐G12D; p16/p19fl/fl; Pdx1‐Cre, resulting in pancreas‐specific, simultaneous deletion of p16INK4A tumor suppressor gene and activation of Kras oncogene), non‐small cell lung cancer (KrasLSL‐G12D; p53frt/frt), SV40 large T‐antigen‐driven transgenic pancreatic neuroendocrine tumors (PNET, phenocopies the RIP1‐Tag2 model), and a small cell lung carcinoma model (conditional inactivation of tumor suppressors Rb1 and Trp53 in mouse lung epithelial cells via Cre expressing adenoviruses). In all of these four models, the anti‐VEGF antibodies decreased the tumor burden and increased overall survival, either as a single agent or in combination with chemotherapy, and without increased incidence of metastasis.
Chung et al. have compared the metastatic effects of various inhibitors of VEGF and VEGFRs using either intravenous injection of B16F10 melanoma or orthotopic implantation of a mouse mammary tumor cell line (Chung et al., 2012). Blocking antibodies against VEGF (B20‐4.1.1 and G6.31), VEGFR2 (DC101) or VEGF‐Trap that neutralizes VEGF, VEGF‐B and PlGF, did not promote metastasis. In contrast, metastasis was increased with high doses of the small molecular weight tyrosine kinase inhibitors sunitinib, sorafenib and imatinib that block VEGFRs but also target other tyrosine kinases. Sunitinib, which most strongly increased metastasis in these mouse models, also increased vascular permeability (Chung et al., 2012). These results suggested that the increase in metastasis may be facilitated by off‐target inhibition of homeostatic EC functions by this class of less specific pharmacological inhibitors. However, the explanation for the controversial results might not be this straightforward, as an increase in tumor invasion and metastasis was observed in two recent studies using the RIP1‐Tag2 mice and treatment with VEGF blocking antibodies (Maione et al., 2012; Sennino et al., 2012). In these studies the anti‐VEGF treatment reduced the tumor burden, but also led to increased tumor hypoxia, c‐Met activation, and increased invasiveness (Sennino et al., 2012). Interestingly, tumor invasion and metastasis were suppressed by concurrent inhibition of c‐Met and VEGF.
8. Mouse models of tumor lymphangiogenesis and metastasis
Similarly to the blood vessels, the growth of lymphatic vessels (lymphangiogenesis) does not occur in normal adult tissues but is activated in solid tumors. Metastatic cells use peritumoral lymphatic vessels as a route for their dissemination to the lymph nodes. In many clinical studies, a correlation between the extent of tumor lymphatic vasculature and lymph node metastasis has been reported, and additional mechanistic evidence for a causal connection is accumulating (Sleeman and Thiele, 2009). In the clinic, lymph node metastasis is a major determinant of the staging and prognostic evaluation of various common human cancers (Alitalo and Detmar, 2012, Tammela and Alitalo, 2010). On the other hand, intratumoral lymphatic vessels are considered to be poorly functional and are not necessarily required for lymphatic metastasis (Padera et al., 2002). The dysfunctional lymphatic drainage within tumors may lead to high interstitial fluid pressure, which likely reduces the effectiveness of anti‐cancer drugs.
Development of the lymphatic vasculature has been studied during normal mouse development in genetic models. These experiments have pointed to molecules that are either critical for lymphangiogenesis (such as Prox‐1, COUP‐TFII, TBX1, ITG9; and PDPN) as well as growth factor families that have overlapping functions in both lymphatic and blood vessels (such as VEGF‐C, VEGF‐D, VEGFR3, VEGFR2; Tie1, Tie2, Ang1, Ang2; NRP2, ephrinB2 and EphB4, Dll4, and MMP2) (Alitalo and Detmar, 2012, Alitalo, 2011; Detry et al., 2012; Niessen et al., 2011). Most of the current knowledge of tumor lymphangiogenesis and related molecular mechanisms is gained from xenograft transplantation models performed in immunodeficient mice. In these models, several human tumor cell lines induce lymphangiogenesis, including lung LNM35 (He et al., 2005), breast MDA‐MB‐231 (Price et al., 1990), prostate CWR22Rv‐1 (Burton et al., 2008; Holleran et al., 2000), and urinary bladder cancer cell line MBT‐2 (Horiguchi et al., 2000; Yang et al., 2011). Molecules found to be important for lymphatic vessel development in embryos have been the primary targets for the inhibition of tumor lymphangiogenesis and metastasis in pre‐clinical mouse models. VEGF‐C expression under the rat insulin promoter (RipVEGF‐C) in β‐cells of the pancreas resulted in an extensive network of lymphatic vessels around the Langerhans islets. To investigate the effects of this on tumor progression, these mice were crossed with the RIP1‐Tag2 mice. The resulting double‐transgenic mice formed tumors surrounded by an extensive lymphatic network, and they developed frequent peripancreatic lymph node metastases in contrast to the tumors of mice lacking the VEGF‐C transgene, which did not metastasize (Mandriota et al., 2001). Overexpression of VEGF‐C in various human tumor cell lines transplanted into mice also promoted tumor lymphangiogenesis and metastasis (Burton et al., 2008; Karpanen et al., 2001; Mattila et al., 2002; Skobe et al., 2001a, 2001b). On the other hand, anti‐VEGFR3 antibodies and a soluble form of VEGFR3 (VEGF‐C/D trap) reduced tumor lymphangiogenesis and metastasis in transplantation experiments (Burton et al., 2008; He et al., 2005; Lin et al., 2005; Roberts et al., 2006; Yang et al., 2011). Besides regulating the number of lymphatic vessels, VEGFR3 signaling has been shown to promote dilation of peritumoral lymphatic vessels that may facilitate the transfer of clusters of metastatic tumor cells into the lymph nodes (He et al., 2005; Karpanen et al., 2001; Tammela et al., 2011). In addition to VEGF‐C, also VEGF‐D has been implicated in the metastatic spread of tumor cells via lymphatic vessels (Stacker et al., 2001). In a mammary adenocarcinoma model in immunocompetent mice, VEGF‐D induced the enlargement of collecting lymphatic vessels draining the solid tumor. The increase in lymphatic vessel diameter was found to be prostaglandin, VEGFR3 and VEGFR2 dependent. This study also confirmed that the enlargement of collecting lymphatic vessels could profoundly increase tumor metastasis to the draining lymph nodes (Karnezis et al., 2012). In a genetic mouse model, RipVEGF‐D/RIP1‐Tag2 mice exhibited peritumoral lymphangiogenesis and lymph node metastasis (Mandriota et al., 2001), but they also had lung metastases, inflammation, and reduced primary tumor growth (Kopfstein et al., 2007). Collectively, the transplantation and genetic studies have demonstrated that both VEGF‐C and VEGF‐D via VEGFR3 activation increase tumor lymphangiogenesis, lymphatic vessel diameter and metastasis to regional lymph nodes and distant organs.
Mouse models have also verified additional signaling pathways in tumor lymphangiogenesis. Antibodies blocking the EphB4 ligand ephrinB2 reduced the number of blood and lymphatic vessels as well as tumor growth in a xenograft transplantation model (Abengozar et al., 2012). In addition, genetic deletion of the Tie2 ligand Ang2 resulted in a lymphatic phenotype in gene targeted mice (Gale et al., 2002). Transgenic expression of Ang1 or Ang2 in pancreatic β cells led to the formation of lymphatic vessels, but not blood vessels, and when these mice were crossed with the tumorigenic RIP1‐Tag2 background, Ang2 increased peritumoral lymphangiogenesis, but not metastasis (Fagiani et al., 2011). However, in another model, Ang2 overexpression in pancreatic cancer cell lines did enhance the rate of lymphatic metastasis (Schulz et al., 2011). Furthermore, antibodies blocking neuropilin‐2 (Nrp2), which potentiates VEGF‐C‐induced lymphangiogenesis during embryonic and postnatal development, reduced tumoral lymphangiogenesis and metastasis to sentinel lymph nodes and distant organs (Caunt et al., 2008; Xu et al., 2010). Analogously to tumor angiogenesis, inflammatory cells such as tumor‐associated macrophages (TAM) produce VEGF‐C and stimulate lymphangiogenesis (Moussai et al., 2011). In an orthotopic urinary bladder cancer model, blocking of VEGF‐C and VEGF‐D as well as the depletion of TAMs with clodronate liposomes inhibited lymphangiogenesis and lymphatic metastasis, suggesting that TAMs promote tumor lymphangiogenesis in a paracrine manner (Yang et al., 2011). In addition, in the mouse osteosarcoma model (c‐Myc‐overexpressing Ink4a/ARF−/− BM‐derived stromal cell line AX), CSF‐1 inhibition suppressed tumor lymphangiogenesis (Kubota et al., 2009). On the other hand, lymphatic endothelial cells (LECs) in turn may secrete chemokines, such as CCL21 and CXCL12, which stimulate the invasion of tumor cells into the lymphatic system (Kim et al., 2010; Shields et al., 2007).
9. Imaging the tumor vasculature in mouse models
9.1. Light microscopy of vascular parameters
In pre‐clinical studies, microvessel densities are counted from histological sections of tumor biopsies after immunohistochemical staining. This does not permit the longitudinal monitoring of the vasculature of the selected tumor. Furthermore, fixed samples do not allow evaluation of vessel function, and the quantification of the vasculature depends on the selection of the antibodies and staining techniques used. The method is unreliable in some tumors, for example when the tumor mass decreases in parallel with the number of blood vessels, and the thin tissue sections used in the analyses do not allow 3D visualization of vascular networks (McDonald and Choyke, 2003; Wang et al., 2008). Fluorescence‐based methods employ appropriate light sources and selective optics for fluorophore excitation and detection of emitted light from the specimen. They allow simultaneous imaging of different cell populations in the tumor, and several commercial antibodies can be used to label tumor and stromal cells, including ECs and LECs. Fluorescence light microscopy provides sub‐cellular resolution, and confocal microscopy, which reduces the out‐of‐focus signal through a pinhole aperture, allows 3D analysis of tumor vasculature. However, due to scattering and absorption of the light in thick sections or in whole‐mount preparations, the imaging depth of standard laser scanning confocal microscope is limited to the tumor surface (∼40–150 μm). The relatively slow imaging with the high energy lasers may also cause phototoxicity and photodamage in live samples. In addition, depending on the choice of objective, laser wavelength, and the probe, the imaging field and depth are reduced. To increase three‐dimensional imaging volumes at relative high spatial resolution, additional instruments have been developed (Table 3).
Table 3.
Imaging techniques to analyze three‐dimensional architecture and functionality of blood and lymphatic vessels at single capillary resolution.
| Technique | Description, limitations and advances | References |
|---|---|---|
| Optical tomography | ||
| OPT | Based on collection of a series of projections of bright‐field or fluorescence light images in multiple directions that are combined for 3D reconstructions. Allows visualization of vasculature at the single capillary resolution. Both fluorescence (also tissue autofluorescence) and colored contrasting (such as X‐gal) can be used. Antibody penetration for staining can be problematic in certain tissues and optical clearing of the tissue is often necessary. Label‐free in vivo applications are under development, including the visualization of zebrafish vasculature. | (Bassi et al., 2011; Quintana and Sharpe, 2011) |
| Light sheet‐based fluorescence microscopy | ||
| LSM | Imaging is based on illumination of a specimen within a thin light sheet and detection of emitted light from a perpendicular angle. The technique is used in various microscopy set‐ups and allows imaging of the vasculature at the single capillary resolution. Current in vivo applications are for small model organisms. Requires fluorescent contrast agents, optical clearing of tissues may be necessary, and antibody penetration for staining can be problematic in certain tissues. Advantages are high imaging speed, low fluorophore bleaching, and low phototoxicity. | (Tomer et al., 2011; Jahrling et al., 2009; Erturk et al., 2012) |
| Multiphoton microscopy | ||
| MPM | Imaging is based on localized excitation of a fluorophore by two relatively low energy photons. The multiphoton microcopy increases imaging depth (∼300–400 μm) over single‐photon laser scanning confocal microscopy and provides images at the single cell resolution. Because of longer excitation wavelengths, MPM results in minimal fluorophore bleaching, low phototoxicity and light scattering, from tissues enabling improved resolution and viability. Allows second‐harmonic visualization of non‐labeled fibrillar collagen. Imaging of the vasculature requires fluorescent contrast agents. Narrow field of image and limited imaging depth are not sufficient to image a whole mouse tumor. | (Brown et al., 2001; Wyckoff et al., 2011) |
| Optical coherence tomography | ||
| OFDI | Optical frequency domain imaging. This improved optical coherence tomography (OCT) technique provides wide‐field (whole mouse tumor scale) label‐free imaging of blood and lymph vessels. The image is based on analysis of light propagation and scattering in the tissue. Deep imaging depth (>1 mm) at the single capillary resolution. Allows simultaneous analysis of tumor blood‐ and lymphatic vessel based on intrinsic mechanisms of contrast. Because of optical scattering, OCT imaging depth is limited in tissues (<2 mm) can be used in window chambers or in endoscopic configurations. | (Vakoc et al., 2009; Vakoc et al., 2012; Jung et al., 2010) |
| Spectral imaging | ||
| Hyper‐spectral imaging | Allows label‐free quantification of hemoglobin oxygenation at the microvessel resolution. Is based on the differential light absorption of oxy‐ and deoxyhemoglobins. | (Moy et al., 2011; Sorg et al., 2005) |
| Photoacoustic tomography | ||
| OR‐PAM | Optical resolution photoacoustic tomography. The image is based on optical excitation of biomolecules by a pulsed laser light and detection of ultrasonic waves that are received by acoustic detectors. Allows label‐free structural and functional imaging of microcirculation. The imaging depth is proportional to the lateral resolution; at the imaging depth ∼1.2 mm the resolution is ∼2.5 μm. The all‐optical photoacoustic skanner provides label‐free imaging of whole mouse tumor vasculature at ∼10 mm imaging depth at 100 μm spatial resolution. | (Wang and Hu, 2012; Laufer et al., 2012) |
9.2. Development of non‐invasive high resolution imaging methods to analyze tumor vasculature
In order to evaluate the therapeutic potential of an anti‐angiogenic therapy it would be beneficial to monitor the tumor vasculature at single capillary resolution while retaining deep imaging depth into the tumor. However, longitudinal studies of tumor vasculature at this resolution in living mice have proven difficult. Current non‐invasive imaging modalities used in the clinic include positron emission tomography (PET), computed tomography (CT), and magnetic resonance imaging (MRI). The advantage of PET, CT and MRI is the deep imaging depth and large field of view, and advanced techniques such as dynamic contrast enhanced MRI and high resolution MRI and CT have been developed (Ullrich et al., 2011; Vakoc et al., 2012; Yang and Knopp, 2011). However, magnetic or ionizing contrasting agents have to be used, and these methods suffer from poor soft‐tissue contrast and relatively low resolution in vivo (>∼50 μm), which does not permit imaging of tumors at the cellular level or visualization of the smallest capillaries, which are 5–10 μm in diameter.
In intravital microcopy imaging of experimental tumors, fluorescence contrast agents are injected intravenously to label blood vessels. When compared to MRI and CT, the resolution is improved, but the imaging depth is limited. In certain tissues (such as the tail or ear), no invasive preparation is necessary. To improve resolution and penetration, tumors can be exposed by creating a microscopy window chamber or skin flap suitable for subcutaneous and certain orthotopic tumors (e.g. a cranial window for gliomas or mammary fat pad chamber for breast tumors). Using specific instruments, fluorescent dyes, reporter genes, and different tracers, intravital imaging allows quantitative measurements of vessel dimensions and densities, and the dynamics and functionality of the tumor microenvironment (such as red blood cell velocity, vessel permeability and tissue oxygenation). In parallel, the characteristics, such as cellular interactions, fluid pressure, and tissue pH of the tumor stroma can be studied (Lunt et al., 2010; Moy et al., 2011; Palmer et al., 2011). Intravital imaging has profited substantially from the development of multiphoton microscopic techniques that have several advantages over epi‐fluoresence and laser confocal microscopy. Longer wavelength and lower energy excitation light cause less bleaching and phototoxicity, permitting prolonged imaging periods (Fukumura et al., 2010). In addition, longer wavelength light is less prone to scattering in thick tissues, allowing greater imaging depths (200–∼400 μm), and it creates a so‐called second harmonic effect for the visualization of unlabeled collagen fibrils (Wyckoff et al., 2011). However, the field of view is limited (a few hundred micrometers, depending on the objective), allowing visualization of only a fraction of a mouse tumor.
Some of the shortcomings of fluorescent optic imaging of tumor microvasculature can be avoided by optical coherence tomography (OCT) (Vakoc et al., 2009, 2012) and photoacoustic tomography (PAM) (Wang and Hu, 2012) (Table 3). OCT is a label‐free optical imaging method in which the image is formed from differential light scattering from red blood cells, lymph, tumor stroma and other tissues. OCT utilizes long wavelength light (near‐infrared), providing deeper imaging depths at a relatively high resolution. However, due to optical scattering, the tissue penetration of OCT is limited (<2 mm). In PAM, the target tissue is excited by pulsed laser light, and the pressure waves from the tissue are recorded by an ultrasonic transducer. The advance is non‐invasive imaging and a higher imaging depth. PAM also provides a label‐free functional data on the vasculature, including blood oxygenation (Wang and Hu, 2012).
9.3. Intravital microscopy of lymphatic vessels
In lymphangiography, functional lymphatic vessels are most often visualized using a tracer dye, such as Evans blue or fluorescent dextran, that is injected into tumors or the nearby tissue, from where it is taken up by the lymphatic vessels. In intravital microscopy, the dye‐filled lymphatic vasculature is imaged close to the site of injection using a microscopy window chamber, permitting local functional analysis of lymph flow and structural characteristics such as lymphatic vessel diameter and density (Fukumura and Jain, 2008). However, this methodology results in local and often uneven lymphatic labeling, the dyes may interfere with lymphatic function, in longitudinal analysis repeated injections are necessary, and the tracer may not be able to enter lymphatic vessels in the parts of the tumor tissue or metastatic lymph node where the lymphatic vessels are functionally compromised.
Recently it has become possible to image lymphatic vessels in a non‐invasive manner using genetic reporter mice or OCT. In the genetic models fluorescent reporter constructs are expressed in LECs (Table 2) (Choi et al., 2011; Hagerling et al., 2011; Martinez‐Corral et al., 2012; Truman et al., 2012). Examples include targeted insertion of GFP‐Luc1 fusion cDNA into the Vegfr3‐gene locus (Martinez‐Corral et al., 2012) or a fluorescent reporter under the control of the Prox1 gene promoter (Choi et al., 2011; Hagerling et al., 2011; Truman et al., 2012). In addition, OCT has been used to image lymphatic vessels without contrast agents (Jung et al., 2010; Vakoc et al., 2009). In OCT, the lymph‐filled vessels appear as low‐scattering regions (transparent), contrasting with the higher scattering stromal tissue or blood vessels. Notably, high‐resolution OCT methods (OFDI) detect the smallest lymphatic capillaries and even individual lymphatic valves (Vakoc et al., 2009).
10. Future perspectives
Cancer is currently one of the leading causes of death worldwide, and the global cancer burden is expected to nearly double to 21.4 million cases in the next two decades (http://www.cancer.org). Therefore, there is a great need for improved technologies for diagnosis and treatment. Angiogenesis plays a rate‐limiting role in tumor growth, and tumor‐induced changes in lymphatic vessels increase metastatic spread, providing a rationale to treat cancer by targeting tumor blood and lymphatic vasculature. Unfortunately, the benefit of the thus far developed anti‐angiogenic treatments has been limited to only modest improvements in overall survival in select tumor types, while the first anti‐lymphangiogenic agents targeting VEGF‐C/VEGFR‐3 are being evaluating in clinical trials. The majority of our current knowledge of the mechanistic principles of tumor angiogenesis is based on mouse models, and this is likely to continue in the near future. The development of novel therapies that improve anti‐angiogenic therapy of human cancer patients requires pre‐clinical models that phenocopy the pathogenesis of human cancer. In addition to the models discussed here, new exciting models have emerged that may help to advance research in tumor angiogenesis and lymphangiogenesis (van Miltenburg and Jonkers, 2012). These include inducible genetic models (Farago et al., 2012; Perez‐Mancera et al., 2012), models for metastasis (Francia et al., 2011), as well as non‐germline chimeric mouse models (Huijbers et al., 2011) and TALEN‐mediated gene targeting (Sung et al., 2013), which can speed up the development of new in vivo platforms. New models should provide safer and more effective targets for the control of tumor angiogenesis and lymphangiogenesis. It can also be envisioned that both pre‐clinical models and the clinic would profit from high‐resolution and deep imaging techniques to monitor tumor blood and lymphatic vasculature, as well as possible cellular events that coincidence with treatment failure.
Acknowledgments
The authors thank Drs. Tuomas Tammela and Veli‐Pekka Ronkainen for critical comments, and M.Sc. Renata Prunskaite‐Hyyryläinen for OPT image. Research in the authors' laboratories has been supported by the Academy of Finland, the Sigrid Juselius Foundation and Finnish Cancer Research Organizations, and by the European Research Council. We apologize to the authors whose work could not be cited due to space limitations.
Eklund Lauri, Bry Maija, Alitalo Kari, (2013), Mouse models for studying angiogenesis and lymphangiogenesis in cancer, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.007.
References
- Abedin, Z.R. , Ma, Z. , Reddy, E.P. , 2010. Increased angiogenesis in Cdk4(R24C/R24C):Apc(+/Min) intestinal tumors. Cell Cycle 9, 2456–2463. [DOI] [PubMed] [Google Scholar]
- Abel, E.L. , Angel, J.M. , Kiguchi, K. , DiGiovanni, J. , 2009. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat. Protoc. 4, 1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abengozar, M.A. , de Frutos, S. , Ferreiro, S. , 2012. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood 119, 4565–4576. [DOI] [PubMed] [Google Scholar]
- Aguirre, A.J. , Bardeesy, N. , Sinha, M. , Lopez, L. , Tuveson, D.A. , Horner, J. , Redston, M.S. , DePinho, R.A. , 2003. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerblom, B. , Zang, G. , Zhuang, Z.W. , Calounova, G. , Simons, M. , Welsh, M. , 2012. Heterogeneity among RIP-Tag2 insulinomas allows vascular endothelial growth factor-A independent tumor expansion as revealed by studies in Shb mutant mice: implications for tumor angiogenesis. Mol. Oncol. 6, 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akino, T. , Hida, K. , Hida, Y. , 2009. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am. J. Pathol. 175, 2657–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alferez, D. , Wilkinson, R.W. , Watkins, J. , 2008. Dual inhibition of VEGFR and EGFR signaling reduces the incidence and size of intestinal adenomas in Apc(Min/+) mice. Mol. Cancer Ther. 7, 590–598. [DOI] [PubMed] [Google Scholar]
- Alitalo, A. , Detmar, M. , 2012. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 31, 4499–4508. [DOI] [PubMed] [Google Scholar]
- Alitalo, K. , 2011. The lymphatic vasculature in disease. Nat. Med. 17, 1371–1380. [DOI] [PubMed] [Google Scholar]
- Alva, J.A. , Zovein, A.C. , Monvoisin, A. , Murphy, T. , Salazar, A. , Harvey, N.L. , Carmeliet, P. , Iruela-Arispe, M.L. , 2006. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767. [DOI] [PubMed] [Google Scholar]
- Baffert, F. , Le, T. , Sennino, B. , Thurston, G. , Kuo, C.J. , Hu-Lowe, D. , McDonald, D.M. , 2006. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am. J. Physiol. Heart Circ. Physiol. 290, H547–H559. [DOI] [PubMed] [Google Scholar]
- Bais, C. , Wu, X. , Yao, J. , 2010. PlGF blockade does not inhibit angiogenesis during primary tumor growth. Cell 141, 166–177. [DOI] [PubMed] [Google Scholar]
- Barker, N. , Ridgway, R.A. , van Es, J.H. , van de Wetering, M. , Begthel, H. , van den Born, M. , Danenberg, E. , Clarke, A.R. , Sansom, O.J. , Clevers, H. , 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611. [DOI] [PubMed] [Google Scholar]
- Bassi, A. , Fieramonti, L. , D'Andrea, C. , Mione, M. , Valentini, G. , 2011. In vivo label-free three-dimensional imaging of zebrafish vasculature with optical projection tomography. J. Biomed. Opt. 16, 100502 [DOI] [PubMed] [Google Scholar]
- Bazigou, E. , Lyons, O.T. , Smith, A. , Venn, G.E. , Cope, C. , Brown, N.A. , Makinen, T. , 2011. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J. Clin. Invest. 121, 2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekes, E.M. , Schweighofer, B. , Kupriyanova, T.A. , Zajac, E. , Ardi, V.C. , Quigley, J.P. , Deryugina, E.I. , 2011. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 179, 1455–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belur, L.R. , Podetz-Pedersen, K.M. , Sorenson, B.S. , Hsu, A.H. , Parker, J.B. , Carlson, C.S. , Saltzman, D.A. , Ramakrishnan, S. , McIvor, R.S. , 2011. Inhibition of angiogenesis and suppression of colorectal cancer metastatic to the liver using the sleeping beauty transposon system. Mol. Cancer 10, 14-4598-10-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers, G. , Brekken, R. , McMahon, G. , 2000. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers, G. , Hanahan, D. , 2008. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers, G. , Javaherian, K. , Lo, K.M. , Folkman, J. , Hanahan, D. , 1999. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 284, 808–812. [DOI] [PubMed] [Google Scholar]
- Bergers, G. , Song, S. , Meyer-Morse, N. , Bergsland, E. , Hanahan, D. , 2003. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 111, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin, G.P. , Washington, K. , Yang, K. , 2003. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124, 762–777. [DOI] [PubMed] [Google Scholar]
- Brown, E.B. , Campbell, R.B. , Tsuzuki, Y. , Xu, L. , Carmeliet, P. , Fukumura, D. , Jain, R.K. , 2001. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat. Med. 7, 864–868. [DOI] [PubMed] [Google Scholar]
- Brown, J.L. , Cao, Z.A. , Pinzon-Ortiz, M. , 2010. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol. Cancer Ther. 9, 145–156. [DOI] [PubMed] [Google Scholar]
- Burton, J.B. , Priceman, S.J. , Sung, J.L. , Brakenhielm, E. , An, D.S. , Pytowski, B. , Alitalo, K. , Wu, L. , 2008. Suppression of prostate cancer nodal and systemic metastasis by blockade of the lymphangiogenic axis. Cancer Res. 68, 7828–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caduff, J.H. , Fischer, L.C. , Burri, P.H. , 1986. Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat. Rec. 216, 154–164. [DOI] [PubMed] [Google Scholar]
- Cao, Y. , Sonveaux, P. , Liu, S. , Zhao, Y. , Mi, J. , Clary, B.M. , Li, C.Y. , Kontos, C.D. , Dewhirst, M.W. , 2007. Systemic overexpression of angiopoietin-2 promotes tumor microvessel regression and inhibits angiogenesis and tumor growth. Cancer Res. 67, 3835–3844. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. , Jain, R.K. , 2011. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 10, 417–427. [DOI] [PubMed] [Google Scholar]
- Casanovas, O. , Hicklin, D.J. , Bergers, G. , Hanahan, D. , 2005. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8, 299–309. [DOI] [PubMed] [Google Scholar]
- Caunt, M. , Mak, J. , Liang, W.C. , 2008. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell 13, 331–342. [DOI] [PubMed] [Google Scholar]
- Cheon, D.J. , Orsulic, S. , 2011. Mouse models of cancer. Annu. Rev. Pathol. 6, 95–119. [DOI] [PubMed] [Google Scholar]
- Choi, I. , Chung, H.K. , Ramu, S. , 2011. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood 117, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersson, G. , Vagesjo, E. , Vandooren, J. , Liden, M. , Massena, S. , Reinert, R.B. , Brissova, M. , Powers, A.C. , Opdenakker, G. , Phillipson, M. , 2012. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 120, 4653–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, A.S. , Ferrara, N. , 2011. Developmental and pathological angiogenesis. Annu. Rev. Cell. Dev. Biol. 27, 563–584. [DOI] [PubMed] [Google Scholar]
- Chung, A.S. , Kowanetz, M. , Wu, X. , Zhuang, G. , Ngu, H. , Finkle, D. , Komuves, L. , Peale, F. , Ferrara, N. , 2012. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J. Pathol. 227, 404–416. [DOI] [PubMed] [Google Scholar]
- Claxton, S. , Kostourou, V. , Jadeja, S. , Chambon, P. , Hodivala-Dilke, K. , Fruttiger, M. , 2008. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis 46, 74–80. [DOI] [PubMed] [Google Scholar]
- Cook, N. , Frese, K.K. , Bapiro, T.E. , 2012. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J. Exp. Med. 209, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens, L.M. , Hanahan, D. , Arbeit, J.M. , 1996. Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am. J. Pathol. 149, 1899–1917. [PMC free article] [PubMed] [Google Scholar]
- Crabtree, J.S. , Scacheri, P.C. , Ward, J.M. , 2001. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. U S A 98, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, Y. , Ferrara, N. , 2009. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol. Sci. 30, 624–630. [DOI] [PubMed] [Google Scholar]
- Crawford, Y. , Ferrara, N. , 2009. VEGF inhibition: insights from preclinical and clinical studies. Cell Tissue Res. 335, 261–269. [DOI] [PubMed] [Google Scholar]
- Daly, C. , Eichten, A. , Castanaro, C. , 2013. Angiopoietin-2 functions as a tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 73, 108–118. [DOI] [PubMed] [Google Scholar]
- De Palma, M. , Venneri, M.A. , Galli, R. , Sergi Sergi, L. , Politi, L.S. , Sampaolesi, M. , Naldini, L. , 2005. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8, 211–226. [DOI] [PubMed] [Google Scholar]
- Detry, B. , Erpicum, C. , Paupert, J. , 2012. Matrix metalloproteinase-2 governs lymphatic vessel formation as an interstitial collagenase. Blood 119, 5048–5056. [DOI] [PubMed] [Google Scholar]
- di Tomaso, E. , Capen, D. , Haskell, A. , Hart, J. , Logie, J.J. , Jain, R.K. , McDonald, D.M. , Jones, R. , Munn, L.L. , 2005. Mosaic tumor vessels: cellular basis and ultrastructure of focal regions lacking endothelial cell markers. Cancer Res. 65, 5740–5749. [DOI] [PubMed] [Google Scholar]
- Dong, J. , Zhang, Q. , Huang, Q. , 2010. Glioma stem cells involved in tumor tissue remodeling in a xenograft model. J. Neurosurg. 113, 249–260. [DOI] [PubMed] [Google Scholar]
- Dranoff, G. , 2011. Experimental mouse tumour models: what can be learnt about human cancer immunology?. Nat. Rev. Immunol. 12, 61–66. [DOI] [PubMed] [Google Scholar]
- Duong, T. , Proulx, S.T. , Luciani, P. , Leroux, J.C. , Detmar, M. , Koopman, P. , Francois, M. , 2012. Genetic ablation of SOX18 function suppresses tumor lymphangiogenesis and metastasis of melanoma in mice. Cancer Res. 72, 3105–3114. [DOI] [PubMed] [Google Scholar]
- Ebos, J.M. , Kerbel, R.S. , 2011. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 8, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos, J.M. , Lee, C.R. , Cruz-Munoz, W. , Bjarnason, G.A. , Christensen, J.G. , Kerbel, R.S. , 2009. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erturk, A. , Becker, K. , Jahrling, N. , Mauch, C.P. , Hojer, C.D. , Egen, J.G. , Hellal, F. , Bradke, F. , Sheng, M. , Dodt, H.U. , 2012. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat. Protoc. 7, 1983–1995. [DOI] [PubMed] [Google Scholar]
- Fagiani, E. , Lorentz, P. , Kopfstein, L. , Christofori, G. , 2011. Angiopoietin-1 and -2 exert antagonistic functions in tumor angiogenesis, yet both induce lymphangiogenesis. Cancer Res. 71, 5717–5727. [DOI] [PubMed] [Google Scholar]
- Fang, S. , Salven, P. , 2011. Stem cells in tumor angiogenesis. J. Mol. Cell. Cardiol. 50, 290–295. [DOI] [PubMed] [Google Scholar]
- Fang, S. , Wei, J. , Pentinmikko, N. , Leinonen, H. , Salven, P. , 2012. Generation of functional blood vessels from a single c-kit+ adult vascular endothelial stem cell. PLoS Biol. 10, e1001407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago, A.F. , Snyder, E.L. , Jacks, T. , 2012. SnapShot: lung cancer models. Cell 149, 246–246.e1. [DOI] [PubMed] [Google Scholar]
- Feil, R. , Brocard, J. , Mascrez, B. , LeMeur, M. , Metzger, D. , Chambon, P. , 1996. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. U S A 93, 10887–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, C. , Jonckx, B. , Mazzone, M. , 2007. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131, 463–475. [DOI] [PubMed] [Google Scholar]
- Folkman, J. , 1971. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186. [DOI] [PubMed] [Google Scholar]
- Francescone, R. , Scully, S. , Bentley, B. , Yan, W. , Taylor, S.L. , Oh, D. , Moral, L. , Shao, R. , 2012. Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 protein activation. J. Biol. Chem. 287, 24821–24831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia, G. , Cruz-Munoz, W. , Man, S. , Xu, P. , Kerbel, R.S. , 2011. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat. Rev. Cancer 11, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia, G. , Kerbel, R.S. , 2010. Raising the bar for cancer therapy models. Nat. Biotechnol. 28, 561–562. [DOI] [PubMed] [Google Scholar]
- Fu, Y. , Tang, H. , Huang, Y. , Song, N. , Luo, Y. , 2009. Unraveling the mysteries of endostatin. IUBMB Life 61, 613–626. [DOI] [PubMed] [Google Scholar]
- Fukumura, D. , Duda, D.G. , Munn, L.L. , Jain, R.K. , 2010. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation 17, 206–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura, D. , Jain, R.K. , 2008. Imaging angiogenesis and the microenvironment. APMIS 116, 695–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale, N.W. , Thurston, G. , Hackett, S.F. , 2002. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by angiopoietin-1. Dev. Cell 3, 411–423. [DOI] [PubMed] [Google Scholar]
- Ganss, R. , Hanahan, D. , 1998. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 58, 4673–4681. [PubMed] [Google Scholar]
- Gao, D. , Nolan, D.J. , Mellick, A.S. , Bambino, K. , McDonnell, K. , Mittal, V. , 2008. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319, 195–198. [DOI] [PubMed] [Google Scholar]
- Gerber, H.P. , Ferrara, N. , 2005. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res. 65, 671–680. [PubMed] [Google Scholar]
- Gerber, H.P. , Wu, X. , Yu, L. , 2007. Mice expressing a humanized form of VEGF-A may provide insights into the safety and efficacy of anti-VEGF antibodies. Proc. Natl. Acad. Sci. U S A 104, 3478–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giavazzi, R. , Giuliani, R. , Coltrini, D. , Bani, M.R. , Ferri, C. , Sennino, B. , Tosatti, M.P. , Stoppacciaro, A. , Presta, M. , 2001. Modulation of tumor angiogenesis by conditional expression of fibroblast growth factor-2 affects early but not established tumors. Cancer Res. 61, 309–317. [PubMed] [Google Scholar]
- Goodlad, R.A. , Ryan, A.J. , Wedge, S.R. , Pyrah, I.T. , Alferez, Poulsom, R. , Smith, N.R. , Mandir, N. , Watkins, A.J. , Wilkinson, R.W. , 2006. Inhibiting vascular endothelial growth factor receptor-2 signaling reduces tumor burden in the ApcMin/+ mouse model of early intestinal cancer. Carcinogenesis 27, 2133–2139. [DOI] [PubMed] [Google Scholar]
- Gossen, M. , Bujard, H. , 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U S A 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, N.M. , DeMayo, F. , Finegold, M.J. , Medina, D. , Tilley, W.D. , Aspinall, J.O. , Cunha, G.R. , Donjacour, A.A. , Matusik, R.J. , Rosen, J.M. , 1995. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. U S A 92, 3439–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson, E. , Brakebusch, C. , Hietanen, K. , Fassler, R. , 2001. Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J. Cell Sci. 114, 671–676. [DOI] [PubMed] [Google Scholar]
- Gutierrez, L.S. , Suckow, M. , Lawler, J. , Ploplis, V.A. , Castellino, F.J. , 2003. Thrombospondin 1-a regulator of adenoma growth and carcinoma progression in the APC(Min/+) mouse model. Carcinogenesis 24, 199–207. [DOI] [PubMed] [Google Scholar]
- Guy, C.T. , Muthuswamy, S.K. , Cardiff, R.D. , Soriano, P. , Muller, W.J. , 1994. Activation of the c-Src tyrosine kinase is required for the induction of mammary tumors in transgenic mice. Genes Dev. 8, 23–32. [DOI] [PubMed] [Google Scholar]
- Hagerling, R. , Pollmann, C. , Kremer, L. , Andresen, V. , Kiefer, F. , 2011. Intravital two-photon microscopy of lymphatic vessel development and function using a transgenic Prox1 promoter-directed mOrange2 reporter mouse. Biochem. Soc. Trans. 39, 1674–1681. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , 1985. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315, 115–122. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Wagner, E.F. , Palmiter, R.D. , 2007. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 21, 2258–2270. [DOI] [PubMed] [Google Scholar]
- Hayes, D.F. , 2011. Bevacizumab treatment for solid tumors: boon or bust?. JAMA 305, 506–508. [DOI] [PubMed] [Google Scholar]
- He, Y. , Rajantie, I. , Pajusola, K. , Jeltsch, M. , Holopainen, T. , Yla-Herttuala, S. , Harding, T. , Jooss, K. , Takahashi, T. , Alitalo, K. , 2005. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer Res. 65, 4739–4746. [DOI] [PubMed] [Google Scholar]
- Hingorani, S.R. , Wang, L. , Multani, A.S. , Combs, C. , Deramaudt, T.B. , Hruban, R.H. , Rustgi, A.K. , Chang, S. , Tuveson, D.A. , 2005. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hlushchuk, R. , Riesterer, O. , Baum, O. , Wood, J. , Gruber, G. , Pruschy, M. , Djonov, V. , 2008. Tumor recovery by angiogenic switch from sprouting to intussusceptive angiogenesis after treatment with PTK787/ZK222584 or ionizing radiation. Am. J. Pathol. 173, 1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, J.A. , Giraudo, E. , Singh, M. , Zhang, L. , Inoue, M. , Porkka, K. , Hanahan, D. , Ruoslahti, E. , 2003. Progressive vascular changes in a transgenic mouse model of squamous cell carcinoma. Cancer Cell 4, 383–391. [DOI] [PubMed] [Google Scholar]
- Holash, J. , Maisonpierre, P.C. , Compton, D. , Boland, P. , Alexander, C.R. , Zagzag, D. , Yancopoulos, G.D. , Wiegand, S.J. , 1999. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284, 1994–1998. [DOI] [PubMed] [Google Scholar]
- Holleran, J.L. , Miller, C.J. , Culp, L.A. , 2000. Tracking micrometastasis to multiple organs with lacZ-tagged CWR22R prostate carcinoma cells. J. Histochem. Cytochem. 48, 643–651. [DOI] [PubMed] [Google Scholar]
- Holopainen, T. , Saharinen, P. , D'Amico, G. , 2012. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J. Natl. Cancer Inst. 104, 461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi, Y. , Larchian, W.A. , Kaplinsky, R. , Fair, W.R. , Heston, W.D. , 2000. Intravesical liposome-mediated interleukin-2 gene therapy in orthotopic murine bladder cancer model. Gene Ther. 7, 844–851. [DOI] [PubMed] [Google Scholar]
- Huijbers, I.J. , Krimpenfort, P. , Berns, A. , Jonkers, J. , 2011. Rapid validation of cancer genes in chimeras derived from established genetically engineered mouse models. Bioessays 33, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz, H. , Fehrenbacher, L. , Novotny, W. , 2004. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350, 2335–2342. [DOI] [PubMed] [Google Scholar]
- Iljin, K. , Petrova, T.V. , Veikkola, T. , Kumar, V. , Poutanen, M. , Alitalo, K. , 2002. A fluorescent Tie1 reporter allows monitoring of vascular development and endothelial cell isolation from transgenic mouse embryos. FASEB J. 16, 1764–1774. [DOI] [PubMed] [Google Scholar]
- Indra, A.K. , Warot, X. , Brocard, J. , Bornert, J.M. , Xiao, J.H. , Chambon, P. , Metzger, D. , 1999. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 27, 4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, M. , Hager, J.H. , Ferrara, N. , Gerber, H.P. , Hanahan, D. , 2002. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 1, 193–202. [DOI] [PubMed] [Google Scholar]
- Jackson, E.L. , Olive, K.P. , Tuveson, D.A. , Bronson, R. , Crowley, D. , Brown, M. , Jacks, T. , 2005. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 65, 10280–10288. [DOI] [PubMed] [Google Scholar]
- Jahrling, N. , Becker, K. , Dodt, H.U. , 2009. 3D-reconstruction of blood vessels by ultramicroscopy. Organogenesis 5, 145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, R.K. , 2008. Lessons from multidisciplinary translational trials on anti-angiogenic therapy of cancer. Nat. Rev. Cancer 8, 309–316. [DOI] [PubMed] [Google Scholar]
- Joyce, J.A. , Baruch, A. , Chehade, K. , Meyer-Morse, N. , Giraudo, E. , Tsai, F.Y. , Greenbaum, D.C. , Hager, J.H. , Bogyo, M. , Hanahan, D. , 2004. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 5, 443–453. [DOI] [PubMed] [Google Scholar]
- Joyce, J.A. , Freeman, C. , Meyer-Morse, N. , Parish, C.R. , Hanahan, D. , 2005. A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene 24, 4037–4051. [DOI] [PubMed] [Google Scholar]
- Joyce, J.A. , Laakkonen, P. , Bernasconi, M. , Bergers, G. , Ruoslahti, E. , Hanahan, D. , 2003. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 4, 393–403. [DOI] [PubMed] [Google Scholar]
- Jung, Y. , Zhi, Z. , Wang, R.K. , 2010. Three-dimensional optical imaging of microvascular networks within intact lymph node in vivo. J. Biomed. Opt. 15, 050501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzidis, D. , Armstrong, S.A. , 2011. Cancer: the flipside of Notch. Nature 473, 159–160. [DOI] [PubMed] [Google Scholar]
- Karnezis, T. , Shayan, R. , Caesar, C. , 2012. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 21, 181–195. [DOI] [PubMed] [Google Scholar]
- Karpanen, T. , Egeblad, M. , Karkkainen, M.J. , Kubo, H. , Yla-Herttuala, S. , Jaattela, M. , Alitalo, K. , 2001. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 61, 1786–1790. [PubMed] [Google Scholar]
- Kerbel, R.S. , Kamen, B.A. , 2004. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 4, 423–436. [DOI] [PubMed] [Google Scholar]
- Kim, K.J. , Li, B. , Winer, J. , Armanini, M. , Gillett, N. , Phillips, H.S. , Ferrara, N. , 1993. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362, 841–844. [DOI] [PubMed] [Google Scholar]
- Kim, M. , Koh, Y.J. , Kim, K.E. , Koh, B.I. , Nam, D.H. , Alitalo, K. , Kim, I. , Koh, G.Y. , 2010. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res. 70, 10411–10421. [DOI] [PubMed] [Google Scholar]
- Kisanuki, Y.Y. , Hammer, R.E. , Miyazaki, J. , Williams, S.C. , Richardson, J.A. , Yanagisawa, M. , 2001. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242. [DOI] [PubMed] [Google Scholar]
- Kopfstein, L. , Veikkola, T. , Djonov, V.G. , Baeriswyl, V. , Schomber, T. , Strittmatter, K. , Stacker, S.A. , Achen, M.G. , Alitalo, K. , Christofori, G. , 2007. Distinct roles of vascular endothelial growth factor-D in lymphangiogenesis and metastasis. Am. J. Pathol. 170, 1348–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsisaari, N. , Kasman, I.M. , Forrest, W.F. , Pal, N. , Bai, W. , Fuh, G. , Peale, F.V. , Smits, R. , Ferrara, N. , 2007. Inhibition of VEGF-A prevents the angiogenic switch and results in increased survival of Apc+/min mice. Proc. Natl. Acad. Sci. U S A 104, 10625–10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsisaari, N. , Ross, J. , Wu, X. , 2008. Blocking vascular endothelial growth factor-A inhibits the growth of pituitary adenomas and lowers serum prolactin level in a mouse model of multiple endocrine neoplasia type 1. Clin. Cancer Res. 14, 249–258. [DOI] [PubMed] [Google Scholar]
- Kozaki, K. , Miyaishi, O. , Tsukamoto, T. , Tatematsu, Y. , Hida, T. , Takahashi, T. , Takahashi, T. , 2000. Establishment and characterization of a human lung cancer cell line NCI-H460-LNM35 with consistent lymphogenous metastasis via both subcutaneous and orthotopic propagation. Cancer Res. 60, 2535–2540. [PubMed] [Google Scholar]
- Kubota, Y. , Takubo, K. , Shimizu, T. , Ohno, H. , Kishi, K. , Shibuya, M. , Saya, H. , Suda, T. , 2009. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 206, 1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters, B. , Leenders, W.P. , Wesseling, P. , Smits, D. , Verrijp, K. , Ruiter, D.J. , Peters, J.P. , van Der Kogel, A.J. , de Waal, R.M. , 2002. Vascular endothelial growth factor-A(165) induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res. 62, 341–345. [PubMed] [Google Scholar]
- Laufer, J. , Johnson, P. , Zhang, E. , Treeby, B. , Cox, B. , Pedley, B. , Beard, P. , 2012. In vivo preclinical photoacoustic imaging of tumor vasculature development and therapy. J. Biomed. Opt. 17, 056016 [DOI] [PubMed] [Google Scholar]
- Lazarus, A. , Keshet, E. , 2011. Vascular endothelial growth factor and vascular homeostasis. Proc. Am. Thorac. Soc. 8, 508–511. [DOI] [PubMed] [Google Scholar]
- Lee, S. , Chen, T.T. , Barber, C.L. , Jordan, M.C. , Murdock, J. , Desai, S. , Ferrara, N. , Nagy, A. , Roos, K.P. , Iruela-Arispe, M.L. , 2007. Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, C.E. , Ferrara, N. , 2011. Multiple effects of angiopoietin-2 blockade on tumors. Cancer Cell 19, 431–433. [DOI] [PubMed] [Google Scholar]
- Licht, A.H. , Raab, S. , Hofmann, U. , Breier, G. , 2004. Endothelium-specific Cre recombinase activity in flk-1-Cre transgenic mice. Dev. Dyn. 229, 312–318. [DOI] [PubMed] [Google Scholar]
- Lin, E.Y. , Li, J.F. , Bricard, G. , Wang, W. , Deng, Y. , Sellers, R. , Porcelli, S.A. , Pollard, J.W. , 2007. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol. 1, 288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, E.Y. , Li, J.F. , Gnatovskiy, L. , Deng, Y. , Zhu, L. , Grzesik, D.A. , Qian, H. , Xue, X.N. , Pollard, J.W. , 2006. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66, 11238–11246. [DOI] [PubMed] [Google Scholar]
- Lin, J. , Lalani, A.S. , Harding, T.C. , 2005. Inhibition of lymphogenous metastasis using adeno-associated virus-mediated gene transfer of a soluble VEGFR-3 decoy receptor. Cancer Res. 65, 6901–6909. [DOI] [PubMed] [Google Scholar]
- Loges, S. , Mazzone, M. , Hohensinner, P. , Carmeliet, P. , 2009. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell 15, 167–170. [DOI] [PubMed] [Google Scholar]
- Lohela, M. , Helotera, H. , Haiko, P. , Dumont, D.J. , Alitalo, K. , 2008. Transgenic induction of vascular endothelial growth factor-C is strongly angiogenic in mouse embryos but leads to persistent lymphatic hyperplasia in adult tissues. Am. J. Pathol. 173, 1891–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi, M. , Di Paolo, D. , Becherini, P. , 2011. The use of the orthotopic model to validate antivascular therapies for cancer. Int. J. Dev. Biol. 55, 547–555. [DOI] [PubMed] [Google Scholar]
- Lunt, S.J. , Gray, C. , Reyes-Aldasoro, C.C. , Matcher, S.J. , Tozer, G.M. , 2010. Application of intravital microscopy in studies of tumor microcirculation. J. Biomed. Opt. 15, 011113 [DOI] [PubMed] [Google Scholar]
- Lyden, D. , Hattori, K. , Dias, S. , 2001. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7, 1194–1201. [DOI] [PubMed] [Google Scholar]
- Lyden, D. , Young, A.Z. , Zagzag, D. , 1999. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 401, 670–677. [DOI] [PubMed] [Google Scholar]
- Maione, F. , Capano, S. , Regano, D. , Zentilin, L. , Giacca, M. , Casanovas, O. , Bussolino, F. , Serini, G. , Giraudo, E. , 2012. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Invest. 122, 1832–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione, F. , Molla, F. , Meda, C. , Latini, R. , Zentilin, L. , Giacca, M. , Seano, G. , Serini, G. , Bussolino, F. , Giraudo, E. , 2009. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J. Clin. Invest. 119, 3356–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre, P.C. , Suri, C. , Jones, P.F. , 1997. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60. [DOI] [PubMed] [Google Scholar]
- Mancuso, M.R. , Davis, R. , Norberg, S.M. , 2006. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Invest. 116, 2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota, S.J. , Jussila, L. , Jeltsch, M. , 2001. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 20, 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis, A.J. , Folberg, R. , Hess, A. , Seftor, E.A. , Gardner, L.M. , Pe'er, J. , Trent, J.M. , Meltzer, P.S. , Hendrix, M.J. , 1999. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am. J. Pathol. 155, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Corral, I. , Olmeda, D. , Dieguez-Hurtado, R. , Tammela, T. , Alitalo, K. , Ortega, S. , 2012. In vivo imaging of lymphatic vessels in development, wound healing, inflammation, and tumor metastasis. Proc. Natl. Acad. Sci. U S A 109, 6223–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto, T. , Tashiro, A. , Friedmann-Morvinski, D. , Scadeng, M. , Soda, Y. , Gage, F.H. , Verma, I.M. , 2009. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 15, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui, J. , Funahashi, Y. , Uenaka, T. , Watanabe, T. , Tsuruoka, A. , Asada, M. , 2008. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin. Cancer Res. 14, 5459–5465. [DOI] [PubMed] [Google Scholar]
- Mattila, M.M. , Ruohola, J.K. , Karpanen, T. , Jackson, D.G. , Alitalo, K. , Harkonen, P.L. , 2002. VEGF-C induced lymphangiogenesis is associated with lymph node metastasis in orthotopic MCF-7 tumors. Int. J. Cancer 98, 946–951. [DOI] [PubMed] [Google Scholar]
- McCart, A.E. , Vickaryous, N.K. , Silver, A. , 2008. Apc mice: models, modifiers and mutants. Pathol. Res. Pract. 204, 479–490. [DOI] [PubMed] [Google Scholar]
- McDonald, D.M. , Choyke, P.L. , 2003. Imaging of angiogenesis: from microscope to clinic. Nat. Med. 9, 713–725. [DOI] [PubMed] [Google Scholar]
- McMillan, R. , Matsui, W. , 2012. Molecular pathways: the hedgehog signaling pathway in cancer. Clin. Cancer Res. 18, 4883–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser, A.R. , Mattes, E.M. , Dove, W.F. , Lindstrom, M.J. , Haag, J.D. , Gould, M.N. , 1993. ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc. Natl. Acad. Sci. U S A 90, 8977–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike, T. , Loughna, S. , Perens, E. , 2000. Universal GFP reporter for the study of vascular development. Genesis 28, 75–81. [DOI] [PubMed] [Google Scholar]
- Moussai, D. , Mitsui, H. , Pettersen, J.S. , Pierson, K.C. , Shah, K.R. , Suarez-Farinas, M. , Cardinale, I.R. , Bluth, M.J. , Krueger, J.G. , Carucci, J.A. , 2011. The human cutaneous squamous cell carcinoma microenvironment is characterized by increased lymphatic density and enhanced expression of macrophage-derived VEGF-C. J. Invest. Dermatol. 131, 229–236. [DOI] [PubMed] [Google Scholar]
- Moy, A.J. , White, S.M. , Indrawan, E.S. , 2011. Wide-field functional imaging of blood flow and hemoglobin oxygen saturation in the rodent dorsal window chamber. Microvasc. Res. 82, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, J.A. , Chang, S.H. , Shih, S.C. , Dvorak, A.M. , Dvorak, H.F. , 2010. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 36, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasarre, P. , Thomas, M. , Kruse, K. , Helfrich, I. , Wolter, V. , Deppermann, C. , Schadendorf, D. , Thurston, G. , Fiedler, U. , Augustin, H.G. , 2009. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res. 69, 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke, I.S. , 2004. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu. Rev. Cell. Dev. Biol. 20, 337–366. [DOI] [PubMed] [Google Scholar]
- Niessen, K. , Zhang, G. , Ridgway, J.B. , Chen, H. , Kolumam, G. , Siebel, C.W. , Yan, M. , 2011. The Notch1-Dll4 signaling pathway regulates mouse postnatal lymphatic development. Blood 118, 1989–1997. [DOI] [PubMed] [Google Scholar]
- Niland, S. , Eble, J.A. , 2012. Integrin-mediated cell-matrix interaction in physiological and pathological blood vessel formation. J.Oncol. 2012, 125278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa, H. , Chiu, C. , Hanahan, D. , 2006. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. U S A 103, 12493–12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladipupo, S. , Hu, S. , Kovalski, J. , Yao, J. , Santeford, A. , Sohn, R.E. , Shohet, R. , Maslov, K. , Wang, L.V. , Arbeit, J.M. , 2011. VEGF is essential for hypoxia-inducible factor-mediated neovascularization but dispensable for endothelial sprouting. Proc. Natl. Acad. Sci. U S A 108, 13264–13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladipupo, S.S. , Hu, S. , Santeford, A.C. , Yao, J. , Kovalski, J.R. , Shohet, R.V. , Maslov, K. , Wang, L.V. , Arbeit, J.M. , 2011. Conditional HIF-1 induction produces multistage neovascularization with stage-specific sensitivity to VEGFR inhibitors and myeloid cell independence. Blood 117, 4142–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner, J. , Min, H. , Leal, J. , 2004. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 6, 507–516. [DOI] [PubMed] [Google Scholar]
- Olive, K.P. , Jacobetz, M.A. , Davidson, C.J. , 2009. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, P. , Chu, G.C. , Perry, S.R. , Nolan-Stevaux, O. , Hanahan, D. , 2011. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc. Natl. Acad. Sci. U S A 108, E1275–E1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, M.S. , Boehm, T. , Shing, Y. , Fukai, N. , Vasios, G. , Lane, W.S. , Flynn, E. , Birkhead, J.R. , Olsen, B.R. , Folkman, J. , 1997. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285. [DOI] [PubMed] [Google Scholar]
- O'Reilly, M.S. , Holmgren, L. , Shing, Y. , Chen, C. , Rosenthal, R.A. , Moses, M. , Lane, W.S. , Cao, Y. , Sage, E.H. , Folkman, J. , 1994. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79, 315–328. [DOI] [PubMed] [Google Scholar]
- Padera, T.P. , Kadambi, A. , di Tomaso, E. , 2002. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 296, 1883–1886. [DOI] [PubMed] [Google Scholar]
- Paez-Ribes, M. , Allen, E. , Hudock, J. , Takeda, T. , Okuyama, H. , Vinals, F. , Inoue, M. , Bergers, G. , Hanahan, D. , Casanovas, O. , 2009. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paku, S. , Dezso, K. , Bugyik, E. , Tovari, J. , Timar, J. , Nagy, P. , Laszlo, V. , Klepetko, W. , Dome, B. , 2011. A new mechanism for pillar formation during tumor-induced intussusceptive angiogenesis: inverse sprouting. Am. J. Pathol. 179, 1573–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, G.M. , Fontanella, A.N. , Shan, S. , Hanna, G. , Zhang, G. , Fraser, C.L. , Dewhirst, M.W. , 2011. In vivo optical molecular imaging and analysis in mice using dorsal window chamber models applied to hypoxia, vasculature and fluorescent reporters. Nat. Protoc. 6, 1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patan, S. , Munn, L.L. , Jain, R.K. , 1996. Intussusceptive microvascular growth in a human colon adenocarcinoma xenograft: a novel mechanism of tumor angiogenesis. Microvasc. Res. 51, 260–272. [DOI] [PubMed] [Google Scholar]
- Perez-Mancera, P.A. , Guerra, C. , Barbacid, M. , Tuveson, D.A. , 2012. What we have learned about pancreatic cancer from mouse models. Gastroenterology 142, 1079–1092. [DOI] [PubMed] [Google Scholar]
- Pham, T.H. , Baluk, P. , Xu, Y. , Grigorova, I. , Bankovich, A.J. , Pappu, R. , Coughlin, S.R. , McDonald, D.M. , Schwab, S.R. , Cyster, J.G. , 2010. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 207, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente, M. , Gerhardt, H. , Carmeliet, P. , 2011. Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887. [DOI] [PubMed] [Google Scholar]
- Price, J.E. , Polyzos, A. , Zhang, R.D. , Daniels, L.M. , 1990. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res. 50, 717–721. [PubMed] [Google Scholar]
- Quintana, E. , Piskounova, E. , Shackleton, M. , Weinberg, D. , Eskiocak, U. , Fullen, D.R. , Johnson, T.M. , Morrison, S.J. , 2012. Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci. Trans. Med. 4, 159ra149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana, L. , Sharpe, J. , 2011. Preparation of mouse embryos for optical projection tomography imaging. Cold Spring Harb Protoc. 2011, 664–669. [DOI] [PubMed] [Google Scholar]
- Rafii, S. , Lyden, D. , 2008. Cancer. A few to flip the angiogenic switch. Science 319, 163–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti, D. , Djonov, V. , 2012. Intussusceptive microvascular growth in tumors. Cancer Lett. 316, 126–131. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani, L. , Pallini, R. , Biffoni, M. , 2010. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468, 824–828. [DOI] [PubMed] [Google Scholar]
- Rinkevich, Y. , Lindau, P. , Ueno, H. , Longaker, M.T. , Weissman, I.L. , 2011. Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature 476, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, N. , Kloos, B. , Cassella, M. , Podgrabinska, S. , Persaud, K. , Wu, Y. , Pytowski, B. , Skobe, M. , 2006. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res. 66, 2650–2657. [DOI] [PubMed] [Google Scholar]
- Rohan, R.M. , Fernandez, A. , Udagawa, T. , Yuan, J. , D'Amato, R.J. , 2000. Genetic heterogeneity of angiogenesis in mice. FASEB J. 14, 871–876. [DOI] [PubMed] [Google Scholar]
- Saharinen, P. , Eklund, L. , Pulkki, K. , Bono, P. , Alitalo, K. , 2011. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 17, 347–362. [DOI] [PubMed] [Google Scholar]
- Sakariassen, P.O. , Prestegarden, L. , Wang, J. , 2006. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc. Natl. Acad. Sci. U S A 103, 16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler, A. , Gray, R. , Perry, M.C. , Brahmer, J. , Schiller, J.H. , Dowlati, A. , Lilenbaum, R. , Johnson, D.H. , 2006. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 355, 2542–2550. [DOI] [PubMed] [Google Scholar]
- Schulz, P. , Fischer, C. , Detjen, K.M. , 2011. Angiopoietin-2 drives lymphatic metastasis of pancreatic cancer. FASEB J. 25, 3325–3335. [DOI] [PubMed] [Google Scholar]
- Scully, S. , Francescone, R. , Faibish, M. , Bentley, B. , Taylor, S.L. , Oh, D. , Schapiro, R. , Moral, L. , Yan, W. , Shao, R. , 2012. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci. 32, 12950–12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman, S. , Stevens, J. , Yang, M.Y. , Logsdon, D. , Graff-Cherry, C. , St Croix, B. , 2007. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell 11, 539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennino, B. , Ishiguro-Oonuma, T. , Wei, Y. , 2012. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2, 270–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaifer, C.A. , Huang, J. , Lin, P.C. , 2010. Glioblastoma cells incorporate into tumor vasculature and contribute to vascular radioresistance. Int. J. Cancer 127, 2063–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors, K. , Evan, G. , 2007. Tumor angiogenesis: cause or consequence of cancer?. Cancer Res. 67, 7059–7061. [DOI] [PubMed] [Google Scholar]
- Shchors, K. , Shchors, E. , Rostker, F. , Lawlor, E.R. , Brown-Swigart, L. , Evan, G.I. , 2006. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 20, 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, J.D. , Emmett, M.S. , Dunn, D.B. , Joory, K.D. , Sage, L.M. , Rigby, H. , Mortimer, P.S. , Orlando, A. , Levick, J.R. , Bates, D.O. , 2007. Chemokine-mediated migration of melanoma cells towards lymphatics–a mechanism contributing to metastasis. Oncogene 26, 2997–3005. [DOI] [PubMed] [Google Scholar]
- Shojaei, F. , 2012. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 320, 130–137. [DOI] [PubMed] [Google Scholar]
- Sica, A. , Allavena, P. , Mantovani, A. , 2008. Cancer related inflammation: the macrophage connection. Cancer Lett. 267, 204–215. [DOI] [PubMed] [Google Scholar]
- Singh, M. , Couto, S.S. , Forrest, W.F. , 2012. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J. Pathol. 227, 417–430. [DOI] [PubMed] [Google Scholar]
- Singh, M. , Ferrara, N. , 2012. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat. Biotechnol. 30, 648–657. [DOI] [PubMed] [Google Scholar]
- Singh, M. , Lima, A. , Molina, R. , 2010. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat. Biotechnol. 28, 585–593. [DOI] [PubMed] [Google Scholar]
- Singh, M. , Murriel, C.L. , Johnson, L. , 2012. Genetically engineered mouse models: closing the gap between preclinical data and trial outcomes. Cancer Res. 72, 2695–2700. [DOI] [PubMed] [Google Scholar]
- Skobe, M. , Hamberg, L.M. , Hawighorst, T. , Schirner, M. , Wolf, G.L. , Alitalo, K. , Detmar, M. , 2001. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. Am. J. Pathol. 159, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe, M. , Hawighorst, T. , Jackson, D.G. , Prevo, R. , Janes, L. , Velasco, P. , Riccardi, L. , Alitalo, K. , Claffey, K. , Detmar, M. , 2001. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 7, 192–198. [DOI] [PubMed] [Google Scholar]
- Sleeman, J.P. , Thiele, W. , 2009. Tumor metastasis and the lymphatic vasculature. Int. J. Cancer 125, 2747–2756. [DOI] [PubMed] [Google Scholar]
- Soda, Y. , Marumoto, T. , Friedmann-Morvinski, D. , 2011. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. U S A 108, 4274–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, I. , Adams, R.H. , Gossler, A. , 2009. DLL1-mediated notch activation regulates endothelial identity in mouse fetal arteries. Blood 113, 5680–5688. [DOI] [PubMed] [Google Scholar]
- Sorg, B.S. , Moeller, B.J. , Donovan, O. , Cao, Y. , Dewhirst, M.W. , 2005. Hyperspectral imaging of hemoglobin saturation in tumor microvasculature and tumor hypoxia development. J. Biomed. Opt. 10, 44004 [DOI] [PubMed] [Google Scholar]
- Spring, H. , Schuler, T. , Arnold, B. , Hammerling, G.J. , Ganss, R. , 2005. Chemokines direct endothelial progenitors into tumor neovessels. Proc. Natl. Acad. Sci. U S A 102, 18111–18116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacker, S.A. , Caesar, C. , Baldwin, M.E. , Thornton, G.E. , Williams, R.A. , Prevo, R. , Jackson, D.G. , Nishikawa, S. , Kubo, H. , Achen, M.G. , 2001. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 7, 186–191. [DOI] [PubMed] [Google Scholar]
- Sung, Y.H. , Baek, I.J. , Kim, D.H. , Jeon, J. , Lee, J. , Lee, K. , Jeong, D. , Kim, J.S. , Lee, H.W. , 2013. Knockout mice created by TALEN-mediated gene targeting. Nat. Biotechnol. 31, 23–24. [DOI] [PubMed] [Google Scholar]
- Tammela, T. , Alitalo, K. , 2010. Lymphangiogenesis: molecular mechanisms and future promise. Cell 140, 460–476. [DOI] [PubMed] [Google Scholar]
- Tammela, T. , Saaristo, A. , Holopainen, T. , Yla-Herttuala, S. , Andersson, L.C. , Virolainen, S. , Immonen, I. , Alitalo, K. , 2011. Photodynamic ablation of lymphatic vessels and intralymphatic cancer cells prevents metastasis. Sci. Trans. Med. 3, 69ra11 [DOI] [PubMed] [Google Scholar]
- Tammela, T. , Zarkada, G. , Wallgard, E. , 2008. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454, 656–660. [DOI] [PubMed] [Google Scholar]
- Thurston, G. , Daly, C. , 2012. The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb Perspect. Med. 2, a006550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer, R. , Khairy, K. , Keller, P.J. , 2011. Shedding light on the system: studying embryonic development with light sheet microscopy. Curr. Opin. Genet. Dev. 21, 558–565. [DOI] [PubMed] [Google Scholar]
- Truman, L.A. , Bentley, K.L. , Smith, E.C. , 2012. ProxTom lymphatic vessel reporter mice reveal Prox1 expression in the adrenal medulla, megakaryocytes, and platelets. Am. J. Pathol. 180, 1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, M.S. , Chang, C.C. , Kuo, M.L. , Wu, Y.T. , 2011. Vascular endothelial growth factor-A and changes in a tumor-bearing mouse model with Lewis lung cancer. Oncol. Lett. 2, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich, R.T. , Jikeli, J.F. , Diedenhofen, M. , Bohm-Sturm, P. , Unruh, M. , Vollmar, S. , Hoehn, M. , 2011. In-vivo visualization of tumor microvessel density and response to anti-angiogenic treatment by high resolution MRI in mice. PLoS One 6, e19592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc, B.J. , Fukumura, D. , Jain, R.K. , Bouma, B.E. , 2012. Cancer imaging by optical coherence tomography: preclinical progress and clinical potential. Nat. Rev. Cancer 12, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc, B.J. , Lanning, R.M. , Tyrrell, J.A. , 2009. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat. Med. 15, 1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem, E. , Vervenne, W.L. , Bennouna, J. , 2009. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J. Clin. Oncol. 27, 2231–2237. [DOI] [PubMed] [Google Scholar]
- Van de Veire, S. , Stalmans, I. , Heindryckx, F. , 2010. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell 141, 178–190. [DOI] [PubMed] [Google Scholar]
- van Miltenburg, M.H. , Jonkers, J. , 2012. Using genetically engineered mouse models to validate candidate cancer genes and test new therapeutic approaches. Curr. Opin. Genet. Dev. 22, 21–27. [DOI] [PubMed] [Google Scholar]
- Viana, C.T. , Campos, P.P. , Carvalho, L.A. , Cenedezi, J.M. , Lavall, L. , Lopes, M.T. , Ferreira, M.A. , Andrade, S.P. , 2013. Distinct types of tumors exhibit differential grade of inflammation and angiogenesis in mice. Microvasc. Res 86, 44–51. [DOI] [PubMed] [Google Scholar]
- Wang, C.A. , Jedlicka, P. , Patrick, A.N. , Micalizzi, D.S. , Lemmer, K.C. , Deitsch, E. , Casas-Selves, M. , Harrell, J.C. , Ford, H.L. , 2012. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J. Clin. Invest. 122, 1895–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Stockard, C.R. , Harkins, L. , 2008. Immunohistochemistry in the evaluation of neovascularization in tumor xenografts. Biotech. Histochem. 83, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L.V. , Hu, S. , 2012. Photoacoustic tomography: in vivo imaging from organelles to organs. Science 335, 1458–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Chadalavada, K. , Wilshire, J. , Kowalik, U. , Hovinga, K.E. , Geber, A. , Fligelman, B. , Leversha, M. , Brennan, C. , Tabar, V. , 2010. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468, 829–833. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Nakayama, M. , Pitulescu, M.E. , 2010. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486. [DOI] [PubMed] [Google Scholar]
- Winkler, F. , Kienast, Y. , Fuhrmann, M. , Von Baumgarten, L. , Burgold, S. , Mitteregger, G. , Kretzschmar, H. , Herms, J. , 2009. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia 57, 1306–1315. [DOI] [PubMed] [Google Scholar]
- Wyckoff, J. , Gligorijevic, B. , Entenberg, D. , Segall, J. , Condeelis, J. , 2011. High-resolution multiphoton imaging of tumors in vivo. Cold Spring Harb Protoc. 2011, 1167–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian, X. , Hakansson, J. , Stahlberg, A. , Lindblom, P. , Betsholtz, C. , Gerhardt, H. , Semb, H. , 2006. Pericytes limit tumor cell metastasis. J. Clin. Invest. 116, 642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Yuan, L. , Mak, J. , 2010. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 188, 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Kim, C. , Kim, M.J. , Schwendener, R.A. , Alitalo, K. , Heston, W. , Kim, I. , Kim, W.J. , Koh, G.Y. , 2011. Soluble vascular endothelial growth factor receptor-3 suppresses lymphangiogenesis and lymphatic metastasis in bladder cancer. Mol. Cancer 10, 36-4598-10-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Knopp, M.V. , 2011. Quantifying tumor vascular heterogeneity with dynamic contrast-enhanced magnetic resonance imaging: a review. J. Biomed. Biotechnol. 2011, 732848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, J. , Wu, X. , Zhuang, G. , Kasman, I.M. , Vogt, T. , Phan, V. , Shibuya, M. , Ferrara, N. , Bais, C. , 2011. Expression of a functional VEGFR-1 in tumor cells is a major determinant of anti-PlGF antibodies efficacy. Proc. Natl. Acad. Sci. U S A 108, 11590–11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiji, H. , Harris, S.R. , Thorgeirsson, U.P. , 1997. Vascular endothelial growth factor is essential for initial but not continued in vivo growth of human breast carcinoma cells. Cancer Res. 57, 3924–3928. [PubMed] [Google Scholar]
- You, W.K. , Sennino, B. , Williamson, C.W. , Falcon, B. , Hashizume, H. , Yao, L.C. , Aftab, D.T. , McDonald, D.M. , 2011. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 71, 4758–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]