

Abstract
Breast cancer is the most common type of cancer in women. A substantial fraction of breast cancers have acquired mutations that lead to activation of the phosphoinositide 3‐kinase (PI3K) signaling pathway, which plays a central role in cellular processes that are essential in cancer, such as cell survival, growth, division and motility. Oncogenic mutations in the PI3K pathway generally involve either activating mutation of the gene encoding PI3K (PIK3CA) or AKT (AKT1), or loss or reduced expression of PTEN. Several kinases involved in PI3K signaling are being explored as a therapeutic targets for pharmacological inhibition. Despite the availability of a range of inhibitors, acquired resistance may limit the efficacy of single‐agent therapy. In this review we discuss the role of PI3K pathway mutations in human breast cancer and relevant genetically engineered mouse models (GEMMs), with special attention to the role of PI3K signaling in oncogenesis, in therapeutic response, and in resistance to therapy. Several sophisticated GEMMs have revealed the cause‐and‐effect relationships between PI3K pathway mutations and mammary oncogenesis. These GEMMs enable us to study the biology of tumors induced by activated PI3K signaling, as well as preclinical response and resistance to PI3K pathway inhibitors.
Keywords: Mouse models, PI3K signaling, GEMM-ESCs, Breast cancer, Resistance
1. Introduction
Breast cancer is by far the most common cancer and the most frequent cause of cancer‐related death in women (Ferlay et al., 2010). Breast cancer is a very heterogeneous disease, with marked variation in genetic mutations and protein expression. The epidermal growth factor receptor HER2 (or ERBB2) and estrogen receptor alpha (ERα) are widely used targets to select breast cancer patients for treatment with HER2 targeting therapeutics or endocrine agents. In addition to these two targets, many breast tumors are characterized by activation of the PI3K pathway (Stemke‐Hale et al., 2008). This signaling pathway drives cellular processes such as growth, cell cycling, survival and motility, which are associated with cancer. Indeed, increased PI3K signaling leads to oncogenesis in several experimental models. A diversity of pharmacological inhibitors has been developed to inactivate this signaling pathway in cancer cells (Courtney et al., 2010). Thus far, however, it seems that monotherapy with a single targeted inhibitor is not capable of eradicating cancer. Various mechanisms of resistance to PI3K pathway inhibitors have been identified in cancer cells, including the activation of feedback loops or upregulation of receptor tyrosine kinases (RTKs). To counteract the inevitable resistance against inhibitors, it will be necessary to employ combination therapies. Finding effective combination therapies is, however, a daunting task. Since tumors contain different combinations of mutations, different combinations of inhibitors may be required for effective treatment of individual patients. There is a need for realistic preclinical models to predict response to treatment. Using patient‐derived data on mutation status of PI3K signaling, advanced genetically engineered mouse models (GEMMs) have been developed to mimic human genetics of breast cancer. These models enable us to study the impact of each mutation on breast cancer development. In preclinical research, GEMMs can play an important role in a “mouse cancer clinic” setting. Mouse models can be treated with inhibitors that are in development, and the biology of response and resistance can be studied. Candidate resistance mechanisms may be identified and targeted in combination therapies. Here, we review the role of PI3K signaling in human breast cancer, the currently published mouse models, the clinical and preclinical studies with targeted therapy, and resistance to PI3K pathway inhibition.
2. PI3K signaling in human breast cancer
Cellular survival, growth and division normally support development and maintenance of tissues and organs. In cancer, cellular division has become uncoupled from physiological regulation, and as a result, a pathological increase in cell number takes place, disturbing organ function. Cancer cells may become invasive and, in the worst case, form metastases in other organs, which is the most life‐threatening consequence of cancer. The PI3K pathway is a major contributor to cellular proliferation and survival and aberrations in components of the PI3K pathways are frequently found in human cancers including breast cancer. Oncogenic mutations in the PI3K signaling pathway generally involve either activating mutations in the genes encoding the kinases PI3K (PIK3CA) or AKT (AKT1), or loss or reduced expression of the phosphatases PTEN, SHIP or INPP4B (Stemke‐Hale et al., 2008).
2.1. The PI3K signaling pathway
The PI3K pathway is an intracellular signaling pathway which signals from growth factor receptors at the cell membrane down to many effector proteins through many kinases, including PI3K, AKT and mTOR (Figure 1). In addition to its role in normal cell function, the PI3K pathway is also one of the most frequently activated signaling pathways in cancer. Phosphoinositide 3‐kinases (PI3Ks) act by phosphorylating phosphatidylinositol (PtdIns) lipid substrates, which activate multiple effector proteins, governing many cellular processes, including cell cycle progression, growth, survival and migration. There are three classes of PI3K isoforms. Class I PI3Ks are heterodimeric lipid kinases composed of two subunits: the p110 catalytic subunit and the p85 regulatory subunit. Three p110 isoforms have been described: p110α, p110β, and p110δ, encoded by PIK3CA, PIK3CB and PIK3CD, respectively. As reviewed recently (Vanhaesebroeck et al., 2010), the α and β isoforms are expressed ubiquitously. Expression of the p110δ isoform is largely restricted to the immune system. Signaling to PI3K may be provided by growth factor binding to tyrosine kinase receptors, or by Ras and other small GTPases, and G protein‐coupled receptors. Upon activation, PI3K converts phosphatidyl‐inositol‐4‐5‐bisphosphate (PIP2) to phosphatidyl‐inositol‐3,4,5‐trisphosphate (PIP3), which facilitates recruitment of PDK1 and AKT. Subsequently PDK1 phosphorylates AKT on T308, and full activation of AKT is established by phosphorylation on S473 by mTORC2, resulting in activation of growth, proliferation and survival signaling. Negative regulation of the PI3K signaling is provided by the phosphatases PTEN, SHIP and INPP4B, which dephosphorylate PIP3 and therefore function as tumor suppressors.
Figure 1.
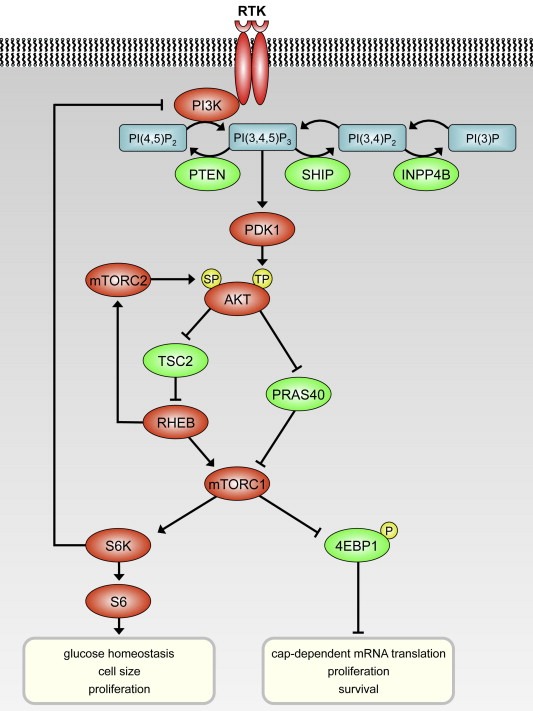
Simplified representation of the PI3K signaling pathway. Proteins that activate signaling are depicted in red; proteins that inhibit in green. Upon ligand binding to the receptor tyrosine kinase (RTK), PI3K is activated, leading to formation of PI(3,4,5)3. AKT is phosphorylated at threonine 308 (TP) by PDK1, and on serine 473 (SP) by mTORC2. Note that most kinases are activated by phosphorylation, whereas 4EBP1 is inactivated by phosphorylation.
2.2. PI3K pathway mutations in human breast cancer
The PI3K signaling pathway is frequently activated in breast cancer. PIK3CA is mutated in approximately 25–40% of all breast cancers (The Cancer Genome Atlas Network, 2012). It is the second most commonly mutated gene after TP53, and the most frequently mutated gene in the PI3K signaling network. Activating mutations in AKT are also reported in breast cancer, albeit at a lower frequency of around 8% (Carpten et al., 2007). PIK3CA and AKT mutations are more frequent in hormone receptor positive breast cancers (Gonzalez‐Angulo et al., 2011; Stemke‐Hale et al., 2008). Especially PIK3CA is often mutated in ER‐positive breast cancer, with a reported incidence of up to 41.3% (Campbell et al., 2004; Ellis et al., 2012; Loi et al., 2010; Santarpia et al., 2012; Stephens et al., 2012).
High‐throughput RNA sequencing has pointed to PI3K signaling as one of the overrepresented pathways in hormone receptor‐ and HER2‐negative (‘triple‐negative’) breast cancer (Shah et al., 2012). In triple‐negative breast cancers, the top three genes most frequently affected by somatic mutations are TP53, PIK3CA and PTEN, with PTEN loss occurring more frequently in this breast cancer subtype than in hormone receptor positive tumors. PTEN loss is also common in breast cancers in BRCA1 mutation carriers, underlining the relevance of this tumor suppressor in BRCA1‐associated hereditary breast cancer (Martins et al., 2012; Saal et al., 2008).
The prognostic role of PIK3CA mutations in breast cancer has been investigated in several retrospective studies, which yielded conflicting results. Recently, a large meta‐analysis of 2587 breast cancer patients from 12 independent studies showed that patients with tumors harboring a PIK3CA mutation have a better clinical outcome than those with a wildtype PIK3CA gene (Dumont et al., 2012), but this correlation with improved prognosis appears to be restricted to patients with mutations in the kinase domain of p110α and to postmenopausal women with ER‐positive breast cancer. Loss of PTEN and phosphorylation of AKT, as determined by immunohistochemistry, do not appear to be prognostic markers in breast cancer (Panigrahi et al., 2004).
PIK3CA or AKT mutations appear to occur early in breast cancer development. This is suggested by the complete concordance in mutation status between matched invasive and in situ tumor tissue in invasive breast carcinomas with an accompanying in situ component. (Dunlap et al., 2010). Analysis of PIK3CA mutations and PTEN loss in matched primary and metastatic breast cancer tissues revealed a high level of discordance in PTEN levels and PIK3CA mutations between primary tumors and metastases, which may confound patient selection based on analysis of primary tumor tissue and concurrently influence response to PI3K‐targeted therapies (Dupont Jensen et al., 2011; Gonzalez‐Angulo et al., 2011).
Since activating PIK3CA mutations and PTEN loss both lead to increased levels of phosphatidylinositol 3,4,5‐triphosphate, one might not expect them to co‐occur in tumors. Several studies have indeed reported statistically significant mutual exclusivity among PIK3CA, PTEN and AKT1 mutations (Saal et al., 2005; The Cancer Genome Atlas Network, 2012). However, other studies have shown that activating PIK3CA mutations and PTEN loss may co‐occur in breast cancer (Pérez‐Tenorio et al., 2007; Stemke‐Hale et al., 2008), which may point to functional differences between these genetic lesions.
2.3. PIK3CA mutations
In breast cancer, activating PIK3CA mutations occur mostly in exons 9 and 20, encoding the helical domain and the kinase domain of p110α, respectively. Helical domain mutations are generally glutamic acid (E) to lysine (K) substitutions at one of two favored positions: E542K or E545K. Exon 9 mutations relieve the inhibitory interaction between the N‐terminal SH‐domain of p85 and the helical domain of p110α, resulting in constitutive activation of the PI3K pathway. Both of these mutations correlate with poor prognosis (Barbareschi et al., 2007). The majority of the kinase domain mutations in exon 20 are H1047R (histidine to arginine) substitutions, which allow easier access of substrates to the catalytic sites of p110α by inducing an allosteric change, leading to constitutive PI3K pathway activation. The H1047R genotype has been associated with enhanced survival as compared to either wildtype or helical domain mutant PIK3CA (Barbareschi et al., 2007).
The different hotspot mutations in PIK3CA are not equally distributed between breast cancer subtypes. Helical domain mutations occur more frequently in the molecular luminal A subtype and in lobular carcinoma than in other molecular subtypes or ductal carcinoma, respectively (Barbareschi et al., 2007; The Cancer Genome Atlas Network, 2012). Indeed, mutations in the different domains of PIK3CA are not functionally identical. Helical domain mutant cells show increased directionality in chemotaxis assays in vitro, and increased intra‐ and extravasation in vivo, compared to both wildtype p110α and kinase domain mutant cells (Pang et al., 2009). Furthermore, the mechanisms underlying PI3K pathway activation seem to be different for both classes of p110α mutants. In case of the helical domain mutants, pathway activation requires interaction with RAS‐GTP, while kinase domain mutants depend on interaction with p85. These different gain‐of‐function mechanisms have been associated with different conformations of the molecule (Zhao and Vogt, 2010).
2.4. AKT mutation
AKT, also known as protein kinase B (PKB), was discovered as an oncogene in the mouse leukemia virus AKT8 (Staal, 1987). AKT can be phosphorylated by PDK1 at T308, and by mTOR complex 2 (mTORC2) at S473. AKT activates mTOR complex 1 (mTORC1), leading to activation of downstream targets such as 4E‐BP1 and S6 kinases (reviewed by Manning and Cantley, 2007). A small subset of breast cancers harbors an AKT point mutation causing substitution of glutamic acid by lysine at amino acid 17 (E17K). The E17K mutation promotes pathological localization of AKT to the plasma membrane, resulting in increased downstream signaling and decreased sensitivity of AKT to allosteric kinase inhibitors (Carpten et al., 2007).
2.5. PTEN loss
The PTEN tumor suppressor, identified in 1997, is a phosphatase that negatively regulates intracellular levels of phosphatidylinositol (3,4,5) triphosphate through dephosphorylation (Li et al., 1997; Stambolic et al., 1998). Reduced expression of PTEN in cancer can be caused by several mechanisms, including loss of heterozygosity at the gene locus, germline and somatic gene mutations, epigenetic silencing by methylation of the PTEN promoter, protein interactions, and PTEN protein degradation (Sadeq et al., 2011; Shetty et al., 2011).
During recent years, additional functions of PTEN have been discovered, as reviewed by Song (Song et al., 2012). PTEN possesses both lipid phosphatase and protein phosphatase activity, and plays tumor suppressive roles in both the cytoplasm and the nucleus. Several studies have reported multiple roles of both nuclear and cytoplasmic PTEN in the maintenance of genome stability. Nuclear PTEN was shown to promote association of APC/C with its activator CDH1, enhancing its E3 ligase activity, leading to degradation of oncoproteins such as polo‐like kinase 1 (PLK1) and Aurora kinases (AURKs) (Gao et al., 2009; Song et al., 2012, 2011). Nuclear PTEN also upregulates RAD51 to positively regulate DNA repair (Shen et al., 2007). Loss of PTEN leads to AKT‐mediated phosphorylation and cytoplasmic sequestration of the cell cycle regulator checkpoint kinase 1 (CHEK1), disrupting the G2‐M cell cycle checkpoint and leading to DNA double‐strand breaks (Puc et al., 2005). In support of a role of PTEN in the maintenance of genome stability, PTEN loss was shown to cause homologous recombination defects in human tumor cells, sensitizing them to inhibitors of poly(ADP‐ribose) polymerase (PARP) (Mendes‐Pereira et al., 2009).
2.6. INPP4B loss
PI3K activates intracellular signaling by the formation of phosphatidylinositol‐(3,4,5)trisphosphate (PI(3,4,5)P3 or PIP3; Figure 1). Control of signaling is maintained by PTEN, which dephosphorylates the 3‐position of PIP3. The 5‐position can be dephosphorylated by SHIP phosphatases to produce phosphatidylinositol‐3,4‐bisphosphate (PI(3,4)P2) (Damen et al., 1996). This phospholipid can be further dephosphorylated by inositol polyphosphate 4‐phosphatase type II (INPP4B). Thus, similarly to PTEN, INPP4B suppresses PI3K signaling. Indeed, knockdown of INPP4B leads to increased AKT activation, thereby promoting anchorage‐independent proliferation, migration and invasion, and in vivo tumor growth (Fedele et al., 2010; Gewinner et al., 2009). Although loss of SHIP family genes has not been correlated with solid tumor formation, frequent loss of INPP4B has been observed in basal‐like breast cancers (The Cancer Genome Atlas Network, 2012).
3. Pharmacological inhibition of PI3K signaling in breast cancer
Several pharmacological inhibitors of the PI3K signaling pathway have been developed and tested in preclinical models as well as in clinical trials (Table 1). The first available agents were rapamycin analogs (rapalogs), which inhibit PI3K signaling through allosteric inhibition of mTORC1. Despite their activity in tumor cell lines, rapalogs have shown only modest clinical activity, most likely due to the fact that mTORC1 inhibition results in abrogation of an S6K‐IRS1‐PI3K negative feedback loop, resulting in activation of AKT (Carracedo et al., 2008; O'Reilly et al., 2006). More recently, several small molecule inhibitors have been developed that function as an ATP‐competitive inhibitor of mTOR in both the mTORC1 and the mTORC2 complex (Chresta et al., 2010; Feldman et al., 2009; García‐Martínez et al., 2009; Thoreen et al., 2009; Yu et al., 2009). These inhibitors can completely block phosphorylation of 4E‐BP1 and have been reported to be more effective than rapamycin in inhibiting tumor growth. Also dual inhibitors of mTOR (TORC1/2) and PI3K, such as NVP‐BEZ235 and GDC‐0980, have been shown to inhibit signal transduction and tumor growth in vitro and in xenograft models (Maira et al., 2008; Wallin et al., 2011).
Table 1.
PI3K pathway inhibitors in clinical development.
| Inhibitor | Target | Company | Clinical trials | Reference |
|---|---|---|---|---|
| Everolimus (RAD‐001) | mTORC1 | Novartis | Phase II/III | Beuvink et al., 2001 |
| Temsirolimus | mTORC1 | Wyeth | Phase II/III | Dudkin et al., 2001 |
| AZD8055 | mTORC1/2 | Astrazeneca | Phase I | Chresta et al., 2010 |
| Ku‐0063794 | mTORC1/2 | Astrazeneca | – | García‐Martínez et al., 2009 |
| GDC‐0980 | Pan‐PI3K + mTORC1/2 | Genentech | Phase II | Sutherlin et al., 2011; Wallin et al., 2011 |
| NVP‐BEZ235 | Pan‐PI3K + mTORC1/2 | Novartis | Phase I/II | Maira et al., 2008 |
| GDC‐0941 | Pan‐PI3K | Genentech | Phase II | Folkes et al., 2008; O'Brien et al., 2010 |
| NVP‐BKM120 | Pan‐PI3K | Novartis | Phase II/III | Maira et al., 2012 |
| GDC‐0032 | PI3Kα | Genentech | Phase I | Friedman et al., 2012 |
| NVP‐BYL719 | PI3Kα | Novartis | Phase I/II | Fritsch et al., 2012 |
| AZD5363 | Pan‐AKT | AstraZeneca | Phase I | Davies et al., 2012 |
| GDC‐0068 | Pan‐AKT | Genentech | Phase I | Blake et al., 2012; Lin et al., 2013 |
| MK‐2206 | Pan‐AKT | Merck & Co | Phase II | Hirai et al., 2010 |
Since activation of the PI3K pathway in human cancers occurs mostly through activating mutations in PIK3CA, several inhibitors have been developed against this kinase. The pan‐class I PI3K inhibitors NVP‐BKM120 and GDC‐0941 show high activity in human cancer cell lines with mutations in PIK3CA (Maira et al., 2012; O'Brien et al., 2010). Also for AKT several inhibitors have been developed, including AZD5363 and MK‐2206 (Davies et al., 2012; Hirai et al., 2010).
3.1. Clinical intervention studies in breast cancer patients
The clinical use of mTOR inhibitors in breast cancer has been reviewed recently (Zagouri et al., 2012). The rapalog everolimus is one of the best‐described mTOR inhibitors in clinical trials, and is approved for various cancers. The drug forms a complex with FKBP12, which binds to mTOR and prevents downstream signaling. Everolimus has been developed both as an immunosuppressant to treat organ transplant rejection and as an anticancer agent. Results from clinical phase I, II and III trials in breast cancer patients have been published. In the neoadjuvant setting, an improved clinical response rate has been shown for everolimus with the aromatase inhibitor letrozole compared with letrozole alone (Baselga et al., 2009). Clinical studies evaluating everolimus in combination with the HER2 targeting monoclonal antibody trastuzumab and chemotherapeutics have yielded promising results with an overall response rate (ORR) of up to 44% and the combinations were well tolerated (Andre et al., 2010; Jerusalem et al., 2011; Morrow et al., 2011). In hormone receptor‐positive breast cancer, everolimus has been used in combination with aromatase inhibitors and tamoxifen. In the TAMRAD study, time to progression (TTP) and overall survival (OS) were increased in postmenopausal breast cancer patients who were treated with tamoxifen plus everolimus compared with tamoxifen alone. The combination is well tolerated, and the main toxicities are fatigue, stomatitis, rash, anorexia and diarrhea (Bachelot et al., 2012, 2011). The randomized phase III BOLERO‐2 trial compared everolimus and the aromatase inhibitor exemestane vs. exemestane and placebo in patients with hormone receptor‐positive advanced breast cancer who relapsed or progressed during previous adjuvant treatment with aromatase inhibitors (Baselga et al., 2012). The addition of everolimus extended median progression‐free survival from 3.2 to 7.8 months. The most common adverse events were stomatitis, anemia, hyperglycemia (which in fact has been used as a pharmacodynamic marker), fatigue and pneumonitis. This combination was recently approved by the U.S. Food and Drug Administration. Another phase III study (BOLERO‐3) is being performed to determine efficacy and safety of everolimus combined with vinorelbine and trastuzumab.
The rapalog temsirolimus inhibits mTOR kinase activity by forming a complex with the immunophilin FK506/rapamycin binding protein, which binds mTOR and inhibits its kinase activity. Temsirolimus has shown modest activity in clinical trials. In a phase III trial however, a combination with letrozole did not show a convincing benefit compared with letrozole alone. According to various trials, temsirolimus may prove to have clinical value in specific groups of breast cancer patients, but more work is needed to define the target population (reviewed by Zagouri et al., 2012).
Several other inhibitors of PI3K signaling in various stages of clinical development (Zardavas et al., 2012). BKM120 is a pan‐class I PI3K inhibitor that will be studied in two randomized phase III trials (BELLE‐2 and BELLE‐3), focusing on fulvestrant vs. fulvestrant and BKM120 therapy in postmenopausal women with hormone receptor‐positive breast cancer refractory to AI. Another pan‐PI3K inhibitor, GDC‐0941, is currently evaluated in a phase II trial with fulvestrant in patients with ER‐positive locally advanced or metastatic breast cancer. BEZ235 is a dual inhibitor of PI3K and mTOR, which is currently studied in phase I/II as monotherapy and in combination with trastuzumab in patients with HER2‐positive breast cancer after failure of trastuzumab treatment. Another dual PIK3/mTOR inhibitor, GDC‐0980, is currently evaluated in combination with fulvestrant in the same phase II trial as GDC‐0941. Of the different AKT inhibitors that are currently in clinical development, MK‐2206 is the most advanced, being studied in a phase II trial in breast cancer patients (Zardavas et al., 2012).
4. Resistance to PI3K pathway inhibitors
Despite the central role PI3K signaling in breast cancer, it is becoming clear that, even in tumors with PI3K pathway mutations, inhibitors of PI3K signaling do not cause tumor eradication or durable remission. Some tumors respond initially but become resistant after prolonged treatment, whereas other tumors do not respond at all. An explanation for the limited antitumor effects of PI3K pathway inhibition is that a number of negative feedback loops exist within this signaling network. When signaling is inhibited, negative feedback is also relieved, which may limit the therapeutic efficacy of inhibitors by re‐activation of signaling. As mentioned above, mTORC1 inhibition by rapalogs leads to AKT activation through abrogation of a negative feedback loop that controls RTK signaling. However, also combined mTORC1/2 inhibition can lead to activation of AKT through increased RTK‐PI3K‐PDK1 activity (Rodrik‐Outmezguine et al., 2011). Through a similar relief of feedback, induction of RTK expression and phosphorylation has been observed after inhibition of AKT (Chandarlapaty et al., 2011). Together, these studies support combined inhibition of mTOR and RTKs (Zhu et al., 2012). Similar effects have been observed for the PI3K inhibitor XL147, which leads to upregulation and phosphorylation of multiple RTKs in HER2‐overexpressing human breast cancer cells, attenuating its therapeutic efficacy. This may however be overcome by the addition of HER2/HER3 antagonists (Chakrabarty et al., 2012).
Another interesting mechanism of adaptive resistance to PI3K inhibition has been described in three‐dimensional (3D) cell cultures, in which breast cancer cells are grown as so‐called spheroids. The dual‐specificity PI3K/mTOR inhibitor BEZ235 has been shown to induce apoptosis exclusively in the inner region of tumor cell spheroids, whereas the matrix‐attached cells are resistant. This resistance has been associated with the upregulation and/or activation of multiple proteins, including several RTKs, anti‐apoptotic proteins, and transcription factors. Although these observations were made in ovarian cancer cells, subsequent experiments with 3D cultures of breast cancer cells resulted in a similar adaptive response to BEZ235, which could be overcome by combined inhibition of PI3K/mTOR and BCL2 (Muranen et al., 2012).
Activation of MEK‐ERK signaling is another relevant mechanism mediating resistance to PI3K signaling inhibitors. If both the PI3K pathway and the ERK pathway are activated in tumor cells, these cells are resistant to inhibition of either one of these pathways. In this setting, a combination of inhibitors for both pathways may be beneficial. 4E‐BP1 seems to be a key integrating downstream effector of both these pathways, and combined inhibition of both the PI3K and ERK pathways is effective in inhibiting phosphorylation of 4E‐BP1 and tumor growth (She et al., 2010).
5. The role of PI3K signaling in therapy resistance
The PI3K signaling pathway can also play a role in resistance to other targeted therapies. In breast cancer, PI3K signaling may play a role in resistance to both endocrine therapy and RTK inhibition. Understanding the biology of therapeutic response and mechanisms of resistance will be crucial for deciding which patient to treat with which therapeutics.
5.1. Resistance to endocrine therapy
For ER‐positive breast cancer, treatment with selective ER modulators (SERMs) or aromatase inhibitors (AI) is often chosen to inhibit the proliferative effects of estrogen on the tumor. However, not all patients benefit from such therapy, and there is a need for biomarkers to predict response. It seems that an increased activity of the PI3K signaling pathway correlates with poor response to endocrine therapy, and that this may be overcome by pharmacological inhibition of PI3K signaling. In support of this notion, the TAMRAD study has shown a clinical benefit of combining everolimus with tamoxifen in metastatic breast cancer, compared with tamoxifen alone (Bachelot et al., 2012). Unfortunately, assessment of PI3K pathway activity via measurement of single phosphorylated proteins in patient material can be difficult. In formalin‐fixed, paraffin‐embedded tissue, proteins can be rapidly dephosphorylated, and variation in fixation can complicate immunohistochemical detection and, a fortiori, semi‐quantitative measurement of phosphorylated proteins. These and other methodological issues need to be addressed before markers of PI3K pathway activity can be reliably validated as predictors of response to endocrine therapy (Beelen et al., 2012).
5.2. Resistance to RTK inhibitors
Trastuzumab (Herceptin) is a HER2/ERBB2‐targeting antibody used in patients with HER2‐overexpressing breast tumors. It has been found that rapid activation of PTEN contributes to the antitumor activity of trastuzumab, and that PTEN loss is associated with trastuzumab resistance in breast cancer (Nagata et al., 2004). A functional genetic screen in HER2‐overexpressing breast cancer cells identified low PTEN expression as a critical determinant of trastuzumab resistance, which can also be conferred by PIK3CA mutants. By using oncogenic PIK3CA mutations and low PTEN expression as determinants of trastuzumab resistance, patients at increased risk for progression can be identified (Berns et al., 2007). Several groups have confirmed the correlation of activated PI3K signaling with poor clinical response of HER2‐amplified breast cancer patients to trastuzumab treatment (Esteva et al., 2010; Gallardo et al., 2012; Razis et al., 2011). Interestingly, PTEN loss or activating PI3KCA mutations mark resistance to trastuzumab but not to lapatinib, a dual HER2/ERBB2 and EGFR inhibitor (O'Brien et al., 2010).
6. Mouse models of human cancer
Many studies have identified recurrent (epi)genetic alterations in human cancer. It is, however, difficult to identify the exact cause‐and‐effect relationships and the mechanisms by which these alterations promote malignant transformation, using observational data from patient material. While in vitro studies with cultured human cell lines aid in the empirical testing of hypotheses related to cellular transformation, they do not take into account some of the main hallmarks of cancer, as there is no immune system or vasculature. Human tumor cells can be xenografted into mice to study their in vivo behavior, with the caveat that cell lines do not fully represent the primary tumor as it grows in a patient, but rather a subpopulation from a tumor or effusion, selected by and adapted to growth in a laboratory environment in artificial media. Hence, tumor cell line xenograft models may be regarded as “animal culture systems” (Frese and Tuveson, 2007). More informative and clinically predictive in vivo models of human breast cancer can be produced using orthotopic transplantation of fresh human tumor tissue or introduction of cancer driver mutations in mice using genetic engineering.
6.1. PDX models of human cancer
By engrafting actual tumor tissue into immune‐deficient mice, in vivo models can be directly derived from patients. These models are termed patient‐derived tumor xenograft (PDX or PDTX) models (Tentler et al., 2012). PDX tumors may be serially passaged, with preservation of biological aspects such as gene expression patterns, mutational status, metastatic potential, drug responsiveness and tumor architecture (DeRose et al., 2011). Although PDX models permit in vivo propagation of human tumors that retain several characteristics of the patient's tumor, they also have their limitations. In practice, not all xenografted human breast cancers grow out in mice, although co‐engraftment of mesenchymal stem cells has been reported to promote growth and stability of the grafts (DeRose et al., 2011). Also metastatic potential is not always fully recapitulated in the PDX models, and the recipient mice lack important components of the immune system, which plays a major and complex role in tumor biology.
6.2. GEMMs of human cancer
Genetically engineered mouse models (GEMMs) are useful in vivo model systems to study the impact of specific genetic alterations on tumor onset, progression and/or therapy response. GEMMs of human cancer can be created through various approaches (Figure 2). By transferring a foreign gene into the genome of a mouse, a transgenic mouse model can be generated. In the strict sense, “transgenic” implies the insertion of genetic material from another species, but it is often applied to mouse models with insertion of a gene from the same species as well. The term “knock‐in” is used if the insertion is targeted into a specific locus, for example to replace an endogenous allele with a corresponding mutant allele. The Rosa26 (R26) and Col1a1 loci are frequently used as target sites for knock‐in strategies to achieve widespread expression of (candidate) cancer genes or shRNAs targeting tumor suppressors (Beard et al., 2006; Masui et al., 2005).
Figure 2.
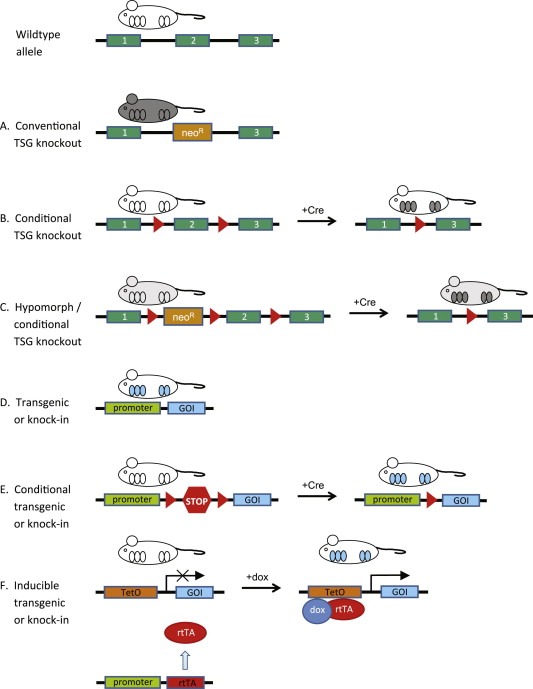
Types of GEMMs of PI3K‐driven breast cancer. Wildtype tissues are represented in white, while knockout tissues are depicted in gray. Tissues with expression of a transgene or knock in gene are depicted in blue. (A) Conventional knockout mice show tumor suppressor gene (TSG) inactivation in all tissues. (B) A conditional tumor suppressor gene (TSG) knockout can be engineered by flanking one or more critical exons with loxP sequences, which are substrates for the Cre site‐specific recombinase. Tissue specificity for a conditional knockout may be achieved by placing the Cre gene under control of a tissue‐specific promoter. (C) A hypomorphic mouse model can be engineered by intronic insertion of a neomycin cassette, which leads to transcriptional interference and lower expression of the targeted TSG. (D) A constitutive transgene can be inserted into the host genome in a construct containing a tissue‐specific promoter (TSP) to achieve tissue specific expression of the gene of interest (GOI). (E) In a conditional transgenic system, loxP sites (red triangles) flank a transcriptional termination sequence (represented as a STOP sign). Recombination by Cre leads to removal of the STOP sequence and tissue‐specific expression of the GOI. (F) Doxycycline‐inducible ‘tet‐on’ systems are controlled by tissue‐specific expression of a transactivator (rtTA), which can only bind the Tet operator (TetO) and drive expression of the GOI in the presence of doxycycline (dox).
To study loss‐of‐function mutations, endogenous genes can be inactivated or modified with great precision via homologous recombination in mouse embryonic stem cells, which can be used to generate mutant mice. In conventional transgenic mice and germline mutants, the mutation is present in all cells of an organ or even the whole animal. Genetically, this does not reflect sporadic cancer, which is initiated by somatic mutations in single cells within a non‐mutated (micro‐)environment. To inactivate genes in a cell‐type specific fashion, different site‐specific recombination systems have been employed in mice (Lewandoski, 2001). In case of the commonly used Cre/loxP system, the Cre recombinase specifically targets loxP sequences, which may be introduced into the mouse genome to flank critical exons of cancer genes or transcriptional terminators that prevent expression of (candidate) oncogenes (Figure 2). Tissue‐specific excision of the “floxed” alleles or transcriptional terminators may be achieved via transgenic Cre expression or via somatic gene transfer involving e.g. Cre encoding adenoviruses.
Mutations can be temporally controlled by making use of inducible systems, such as the doxycycline (dox) inducible system in which transcriptional repression or activation of target genes is controlled by binding of dox to the tet‐regulatable transcriptional activators tTA or rtTA, respectively (Lewandoski, 2001). Temporal control can also be achieved by fusing oncoproteins or Cre to a tamoxifen‐responsive mutant version of the estrogen receptor (ER) hormone‐binding domain. In this case, the oncoprotein or Cre recombinase is activated after administration of tamoxifen.
Several promoters can be used for (over)expression of transgenes in mouse mammary glands. The whey acidic protein (WAP), beta‐lactoglobulin (BLG) and mouse mammary tumor virus (MMTV)‐long terminal repeat (MMTV‐LTR) promoters are more or less selectively active in the mammary epithelial cells (Pittius et al., 1988; Stewart et al., 1984; Webster et al., 1995). The cytokeratin 14 (K14) gene promoter has also been used to drive Cre recombinase expression in mammary gland epithelium, but it is also active in the epidermis, meaning that tumorigenesis may also be induced in the skin (Jonkers et al., 2001).
6.3. Utility of GEMMs of human cancer
There are several ways in which GEMMs can contribute to fundamental and preclinical cancer research. Firstly, GEMMs allow for in vivo assessment of the causal role of genes that are found to be mutated or otherwise modified in human cancer patients. By selectively modifying a gene in a mouse and with the use of the proper control cohorts, cause and effect relationships in cancer can be identified with much stronger evidence than with use of correlative data that can be collected from patients.
Secondly, GEMMs permit in vivo analysis of all stages of tumor development. After induction of the genetically engineered mutation(s), early histologic lesions are formed, which can progress into more invasive and malignant tumors, and eventually may lead to metastatic disease in some models. In conventional transgenic mice, the disease‐initiating oncogenic event is simultaneously induced in many cells. Cre/loxP‐based conditional GEMMs, in which the initiating mutation occurs more stochastically, in a limited cell population, allow for development of tumors in a normal tissue environment (Frese and Tuveson, 2007; Jonkers and Berns, 2002). The developing tumors may accumulate additional genetic and epigenetic events resulting in an increased level of genetic complexity, which is characteristic for human cancers. These secondary events may play a role in tumor progression and metastasis. By comparing primary and metastatic tissues, metastasis‐specific traits may be identified, which can generate hypotheses for further studies on the metastatic process.
Thirdly, GEMM tumors arise in their natural tissue microenvironment in an immune competent host. Tumor cell extrinsic factors, such as angiogenesis, extracellular matrix and immunologic interactions can therefore be effectively studied in GEMMs (Walrath et al., 2010). For example, studies in sarcoma models have shown that immune cells influence tumor progression via immunoediting, leading to a selective advantage of tumor cells that escape T lymphocyte attack by losing tumor antigen expression or presentation of major histocompatibility complex I (DuPage et al., 2012). Also the impact of the immune system on chemotherapy response can be studied in GEMMs, for example by inactivating Rag genes, which renders mice T and B cell‐deficient (Ciampricotti et al., 2012; DeNardo et al., 2011).
Finally, GEMMs of human cancer may be useful for preclinical evaluation of novel chemoprevention and tumor intervention strategies. Chemopreventive agents can be tested in cancer‐prone GEMMs that have not yet developed tumors, and mice with primary tumors or metastatic disease can be treated with (combinations of) chemotherapy agents or targeted therapeutics. Therapeutic responses as well as intrinsic or acquired resistance mechanisms may be serially monitored during and after treatment. Therapeutic targets may also be validated in GEMMs by monitoring the effect of genetic target inactivation on tumor regression (Blasco et al., 2011).
GEMMs may improve the quality of preclinical intervention studies. Compared to tumor cell line xenografts, several studies with relevant GEMMs have shown a better prediction of therapy benefit in human patients. This has for example been described for Kras‐driven models for non‐small‐cell lung carcinoma and pancreatic adenocarcinoma (Singh et al., 2010). In an interesting co‐clinical trial, GEMMs of Kras‐mutant lung cancers were enrolled in ‘co‐clinical’ trial that mirrored an ongoing human clinical trial with the MEK inhibitor selumetinib in combination with docetaxel. In the lung epithelium, Kras, p53 and Lkb1 were mutated in different combinations. The genotypes correlated with different responses to single‐agent or combination therapy. The results from the preclinical study not only mimicked those of the human trial but also predicted resistance in patients with mutations in LKB1, which could be confirmed by retrospective analysis of LKB1 mutation status in the clinical trial. This work elegantly illustrates that co‐clinical trials may be useful to generate important hypotheses for clinical trials in human cancer patients (Chen et al., 2012).
Although there are important biological differences between mice and humans as well as legal and patent‐related barriers for the use of GEMMs, an increased use of these models in cancer research is expected to make a significant contribution to the oncology field (Sharpless and Depinho, 2006). The establishment of a growing number of “mouse cancer clinics” in the US and Europe illustrates the ambition of the research community to engineer and utilize mouse models for a range of cancer types, which may help us to better understand the biology of cancer, to predict therapeutic response and resistance of cancer treatments, and to guide clinical trials.
7. GEMMs of PI3K pathway‐driven breast cancer
7.1. PIK3CA models
Several GEMMs with tissue‐specific mutation of Pik3ca have been published (Table 2). Using the ROSA26 knock‐in system, R26‐Pik3ca H1047R;MMTV‐Cre mice have been generated (Adams et al., 2011). These mice develop mammary tumors of either adenosquamous carcinoma or adenomyoepithelioma phenotype. In combination with a heterozygous Trp53 knockout allele, tumorigenesis is accelerated and tumor phenotype is shifted to adenosquamous carcinoma or spindle cell/EMT type (Adams et al., 2011). Several groups have shown that expression of the H1047R Pik3ca mutant in the luminal mammary epithelium results in the formation of mammary tumors of several phenotypes, some of which express ER (Meyer et al., 2011; Tikoo et al., 2012; Yuan et al., 2013). In these tumors, spontaneous additional mutations were found in several genes, including Trp53. Indeed, co‐occurrence of PIK3CA and TP53 mutations has also been found in human breast cancer (Boyault et al., 2012).
Table 2.
GEMMs of PI3K‐driven breast cancer.
| GEMM | Type | Comments | Reference |
|---|---|---|---|
| Pten+/− | Knockout | Targeted disruption of Pten | (Podsypanina et al., 1999; Stambolic et al., 2000; Suzuki et al., 1998) |
| MMTV‐Cre; Ptenloxp/loxp | Conditional knockout | (Li et al., 2002) | |
| Ptenhy/+ | Hypomorphic allele | Transcriptional interference of Pten by insertion of CMV‐neoR in Pten intron 3 | (Alimonti et al., 2010) |
| MMTV‐myrAkt1 | Transgenic | Constitutive activation | (Blanco‐Aparicio et al., 2006; Schwertfeger et al., 2001) |
| R26‐Pik3caH1047R; MMTV‐Cre | Conditional transgenic | Rosa26‐lox‐stop‐lox‐Pik3caH1047R | (Adams et al., 2011) |
| WapiCre; Pik3caH1047RMMTV‐Cre; Pik3caH1047R | Conditional transgenic | CAGS‐lox‐stop‐lox‐Pik3caH1047R | (Meyer et al., 2011) |
| MMTV‐Cre; Pik3caH1047R | Conditional knock‐in | lox‐stop‐lox‐Pik3caH1047R in endogenous Pik3ca locus | (Tikoo et al., 2012) |
| MMTV‐Cre; Pik3caH1047R | Conditional knock‐in | lox‐stop‐lox‐Pik3caH1047R in endogenous Pik3ca locus | (Yuan et al., 2013) |
| MMTV‐rtTA; TetO‐PIK3CAH1047R | Inducible transgenic | Transgenic MMTV‐rtTATransgenic TetO‐PIK3CAH1047R | (Liu et al., 2011) |
GEMMs in which tumorigenesis is driven by mutant PI3K expression are suitable for preclinical intervention studies aimed at determining the response to pharmacologic inhibition of PI3K signaling. Tetracycline‐inducible expression of human PIKCAH1047R in the mammary gland has also been shown to induce tumors of heterogeneous pathological types, including adenocarcinomas and adenosquamous carcinomas (Liu et al., 2011). After doxycycline removal, two‐thirds of the tumors resumed growth after PIKCAH1047R inactivation. This could partially be explained by Met amplifications, leading to PI3K‐dependent tumor survival. Other tumors had Myc amplifications and were independent on PI3K signaling (Liu et al., 2011).
GEMMs have also been instrumental in elucidating isoform‐specific roles of p110 in tumorigenesis. Ablation of p110α impairs normal development of the mammary gland. Ablation of p110β in the mammary gland, however, results in increased ductal branching as well as tumor formation. It has been suggested that the less active p110β competes with the more active p110α for receptor binding sites, thereby downregulating signaling activity (Utermark et al., 2012).
7.2. AKT models
GEMMs have been instrumental in unraveling the in vivo functions of AKT in the mammary gland. AKT1 deficiency results in mammary gland developmental defects in mice (LaRocca et al., 2011). During pregnancy and lactation, prolactin generates signaling via AKT, leading to phosphorylation of STAT5. AKT1 ablation interferes with STAT5 phosphorylation and delays the differentiation of mammary epithelium, whereas ablation of AKT2 has the opposite effect (Maroulakou et al., 2008).
Overexpression or activation of AKT contributes to tumor development and malignancy in the mammary gland. Expression of human AKT1 under the control of the MMTV promoter causes delayed postlactational involution mammary gland, but no neoplastic transformation (Ackler et al., 2002). Ablation of Akt1 inhibits the development of mammary tumors in MMTV‐Neu and MMTV‐polyoma middle T (MMTV‐PyMT) transgenic mice, which is associated with reduced cell proliferation and survival. Conversely, Akt2 ablation accelerates tumorigenesis in these models (Maroulakou et al., 2007).
Addition of a myristoylation signal to the murine Akt1 gene activates the protein by localizing it to the plasma membrane. Mammary‐specific expression of myrAKT1 in MMTV‐myrAkt1 transgenic mice delays post‐lactation involution (Schwertfeger et al., 2001) and increases susceptibility of mice to induction of mammary tumors by the carcinogen 9,10‐dimethyl‐1,2 benzanthracene (DMBA). DMBA‐treated wildtype mice develop mostly sarcomatous mammary tumors, whereas MMTV‐myrAkt1 mice develop mainly ER‐positive adenocarcinomas or adenosquamous tumors (Blanco‐Aparicio et al., 2006). MyrAKT1 also accelerates tumorigenesis in MMTV‐c‐ErbB2 transgenic mice with overexpression of wildtype ERBB2. Tumors in these mice show a reduced requirement for signaling through the EGF family and resistance to ErbB2‐targeted therapies (Young et al., 2008). Mammary‐specific expression of myrAKT2 in MMTV‐Neu and MMTV‐PyMT mice does not affect the latency of tumor development, but it does increase metastasis to the lungs (Dillon et al., 2009). We do need to keep in mind, however, that expression of myrAKT1 or myrAKT2 in transgenic mice does not precisely copy the naturally occurring AKT mutations found in human breast cancer. It will therefore be important to generate mouse models with mammary‐specific expression of the AKTE17K hotspot mutant.
7.3. PTEN models
Following the discovery of PTEN, several groups generated Pten knockout mice. The Pten null genotype is embryonic lethal in mice, resulting in death occurring between gestation day E6.5 and E9.5 (Podsypanina et al., 1999; Suzuki et al., 1998). Pten heterozygous knockout (Pten +/−) mice are viable and develop a range of tumors and hyperplasia in multiple organs after an age of 6 months, with mammary tumors occurring in half of the female mice. Loss of PTEN expression is associated with basal‐like breast cancer in humans and, correspondingly, heterozygous inactivation of Pten leads to formation of basal‐like mammary tumors in mice (Saal et al., 2008).
Loss of heterozygosity at the Pten locus is seen in most tumors in Pten +/− mice (Stambolic et al., 2000). PTEN loss‐of‐function abrogates inhibitory regulation of PI3K signaling, leaving cells vulnerable to activating stimuli and possible oncogenic transformation. In vitro studies indicate that human breast cancer cells with PTEN loss mainly depend on the p110β isoform of PI3K (Torbett et al., 2008; Wee et al., 2008). When the p110β isoform is genetically inactivated in Pten +/− mice, prostate tumorigenesis is inhibited, while p110α inactivation protects from glomerulonephritis, pheochromocytoma and thyroid cancer induced by Pten loss. The incidence of mammary tumors remains unchanged (Berenjeno et al., 2012). Tissue‐specific deletion of Pten in the mouse mammary gland leads to precocious lobulo‐alveolar development, excessive ductal branching, delayed involution and severely reduced apoptosis, and tumor formation (Li et al., 2002).
Collaboration between PTEN loss and other oncogenic mutations has been demonstrated, for example in MMTV‐Wnt‐1 transgenic mice heterozygous for Pten, which showed accelerated formation of ductal carcinomas. In most of these tumors, the Pten wildtype allele was lost, indicating a selective growth advantage of Pten −/− cells (Li et al., 2001). In MMTV‐Neu transgenic mice with mammary gland‐specific overexpression of activated ERBB2, concomitant tissue‐specific inactivation of PTEN leads to acceleration of mammary tumor onset and increased metastasis (Schade et al., 2009).
In knock‐down experiments, even a small reduction of PTEN to 80% of normal levels results in tumorigenesis in several organs, with highest penetrance in the mammary gland. From these experiments it followed that loss of heterozygosity is not necessary for tumorigenesis in Pten +/− mice (Alimonti et al., 2010). These studies have contributed to the “continuum model of tumor suppression” (Berger et al., 2011), succeeding the two‐hit model, in which two mutations are required (Knudson, 1971). Conversely, PTEN overexpression leads to reduced Akt phosphorylation and interferes with differentiation and proliferation of the mammary gland in young mice, resulting in a lactation defect (Dupont et al., 2002).
8. Targeting PI3K signaling in GEMMs of human breast cancer
8.1. Single‐agent therapies
The antitumor efficacy of PI3K pathway inhibitors has been evaluated in several GEMMs of human cancer, including breast cancer. Using the dual PI3K/mTOR inhibitor BEZ235 in an Apc conditional knockout model of colorectal cancer, Roper et al. observed significant regression of tumors that lacked activating Pik3ca mutations, suggesting that antitumor activity of this inhibitor is not restricted to PIK3CA mutant cancers (Roper et al., 2011). Yuan et al. tested the in vivo activity of the pan‐class I PI3K inhibitor GDC‐0941 in a conditional knock‐in mouse model for breast cancer, in which the endogenous Pik3ca allele was modified to allow mammary gland‐specific expression of Pik3ca H1047R in mammary epithelium (Yuan et al., 2013). Pik3ca H1047R expressing mammary tumor explants grown as allografts showed variable responses to treatment with GDC‐0941, but in no instance tumor stasis or regression was observed, suggesting that GDC‐0941 has limited antitumor activity as single agent.
Although rapalogs show limited activity as single agents in most cancers, they may be effective against MYC‐driven lymphomas. Whereas short‐term‐dosing with rapamycin showed no activity against established lymphomas from Eμ‐Myc transgenic mice (Wendel et al., 2006, 2004), longer‐term mTORC1 inhibition with everolimus resulted in tumor regression and a significantly improved survival (Wall et al., 2013). A possible explanation for this unexpected response is that MYC‐driven lymphoma cells require mTORC1 activity to avoid entering senescence. In support of this notion, the response of Eμ‐Myc lymphomas to everolimus was dependent on a functional p53 pathway (Wall et al., 2013).
8.2. Combination therapies
Most PI3K pathway inhibitors show modest antitumor activity as single agent but synergize effectively with other targeted inhibitors or chemotherapy drugs in combination therapy. Clear examples are the mTORC1 inhibitor everolimus, the pan‐class I PI3K inhibitors BKM120 and GDC‐0941, and the dual PI3K/mTOR inhibitors BEZ235 and GDC‐0980, which are currently tested in combination with endocrine agents or trastuzumab in various phase II/III clinical trials in breast cancer patients. Some of these trials show major synergism between e.g. everolimus and the aromatase inhibitor exemestane (Baselga et al., 2012), prompting for preclinical in vivo evaluation of other combinations in GEMMs of human cancer.
The limited antitumor activity of rapalogs is caused by the fact that mTORC1 inhibition leads to loss of PI3K‐mediated feedback inhibition and consequent hyperactivation of AKT and mitogen‐activated protein kinase (MAPK) signaling via RTKs (Carracedo et al., 2008; O'Reilly et al., 2006). Combination of rapalogs and RTK inhibitors might therefore provide an improved therapeutic benefit in the treatment of cancer patients. To test, this concept, rapamycin was tested alone or in combination with the epidermal growth factor receptor (EGFR) inhibitor erlotinib in a GEMM of pancreatic neuroendocrine tumors. In contrast to either monotherapy, the combination treatment conferred unprecedented survival benefit in mice with aggressive multifocal cancer (Chiu et al., 2010). Interestingly, the antiapoptotic protein survivin was identified as a potential predictive biomarker for this combination therapy.
Other studies have focused on combinations of dual mTOR/PI3K inhibitors and MAPK‐kinase (MEK) inhibitors. Engelman et al. investigated the response of lung adenocarcinomas induced by tissue‐specific expression of mutant Pik3ca H1047R or Kras G12D to the dual PI3K/mTOR inhibitor BEZ235 alone or in combination with the MEK inhibitor ARRY‐142886 (Engelman et al., 2008). Whereas mutant Pik3ca H1047R tumors regressed in response to BEZ235, Kras G12D‐driven tumors did not respond to single‐agent therapy. However, combination of BEZ235 with ARRY‐142886 showed a marked synergy in shrinking mutant Kras G12D tumors, highlighting the potential of this combination against KRAS mutant lung cancers (Engelman et al., 2008). BEZ235 has also been shown to synergize with the MEK inhibitor AZD6244 in GEMMs of mutant EGFR T790M‐L858R‐driven lung cancer (Faber et al., 2009), HRas G12V‐driven melanoma and different GEMMs of human breast cancer (Roberts et al., 2012). Together, these results predict that combinations of PI3K/mTOR and MEK inhibitors will show antitumor activity in a wide range of human cancers.
Also combinations of PI3K pathway inhibitors with inhibitors of DNA repair pathways show promising results in GEMMs. Following the discovery that inactivation of poly(ADP‐ribose)‐polymerase (PARP), a key enzyme in DNA single‐strand break repair, is selectively toxic to BRCA1/2‐deficient cells, several selective PARP inhibitors have been developed (Yap et al., 2011). Recently, synergistic antitumor activity of a combination of the pan‐class I PI3K inhibitor BKM120 and the PARP inhibitor AZD2281 (olaparib) has been observed in a GEMM of BRCA1‐related breast cancer (Juvekar et al., 2012). Whereas treatment with olaparib alone showed only attenuated tumor growth in the mouse model (in which tumors still express a BRCA1‐Δ11 protein with residual activity), the combination of BKM120 and olaparib prolonged tumor‐doubling time from 5 to more than 70 days, suggesting that combined PI3K and PARP inhibition might be an effective treatment of BRCA1‐related tumors. Importantly, the combination of BKM120 and olaparib did not result in measurable toxicity, even in mice that were treated for over 3 months (Juvekar et al., 2012).
9. Future perspectives
9.1. Accelerated production of novel GEMMs of PI3K‐driven breast cancer
Genetically engineered mice have contributed greatly to the field of cancer research. Production of novel GEMMs of human cancer is hampered, however, by the fact that generation of complex multi‐allele models using conventional breeding is time‐consuming and requires large number of animals. Recent advances in embryonic stem cell (ESC) technology now permit efficient derivation of ESC lines from validated GEMMs (dubbed GEMM‐ESCs) and generation of chimeric mice harboring all desired modifications (Figure 3). Additional mutant alleles can be rapidly and reproducibly introduced into GEMM‐ESC via recombinase‐mediated cassette exchange (RMCE) and the resulting GEMM‐ESCs can be used to produce chimeric mice via microinjection into pre‐implementation embryos. The resulting chimeras are composed of normal cells and genetically engineered cells from which spontaneous tumors may arise (Dow and Lowe, 2012; Huijbers et al., 2011). In contrast to germline GEMMs, generating chimeric mice is relatively fast and requires fewer animals, as all genetic modifications are introduced ex vivo. Moreover, the GEMM‐ESC approach effectively eliminates any potential confounding effects due to variation in genetic background, as no breeding is involved. The GEMM‐ESC strategy has already been successfully employed to study the in vivo effects of (reversible) RNAi‐mediated suppression of p19‐ARF in a GEMM of mutant KRAS‐driven lung cancer (Premsrirut et al., 2011). Since generation of chimeric mice from individual GEMM‐ESCs is relatively fast, The GEMM‐ESC technology is particularly useful for in vivo analysis of multiple individual mutations within the same gene or pathway. For example, different mutant alleles of Pik3ca (E542K, E545K and H1047R), Akt1 (E17K) and short‐hairpin (sh)RNA expression vectors targeting Pten can be introduced into GEMM‐ESCs from established GEMMs of human breast cancer to study the effects of different PI3K pathway perturbations on tumor development, progression and response to PI3K pathway inhibitors.
Figure 3.
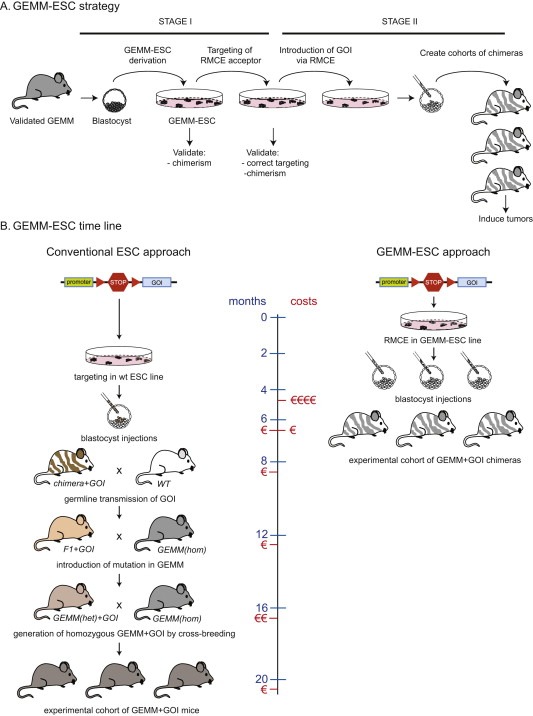
(A) Outline of the GEMM‐ESC strategy. Embryonic stem cell (ESC) lines are derived from genetically engineered mouse models (GEMMs) and equipped with vectors containing FRT sequences that permit rapid and reproducible introduction of GOI via FLP recombinase‐mediated cassette exchange (RMCE). (B) Time line and costs of introduction of a conditional GOI allele in a multi‐allele GEMM using conventional gene targeting vs. the GEMM‐ESC strategy. The conventional approach takes around 20 months and involves introduction of the allele via homologous recombination in wildtype ESCs, generation of chimeras, germline transmission of the mutant allele and subsequent breeding of the F1 mice to the GEMM model. One or more additional rounds of breeding are required to generate mice that are homozygous for all conditional TSG alleles present in the original GEMM. The GEMM‐ESC approach takes only 6 months and involves accelerated introduction of the conditional GOI allele into GEMM‐ESCs via recombinase‐mediated cassette exchange (RMCE) and direct production of an experimental cohort of chimeras via microinjection of the modified GEMM‐ESCs into blastocysts. Compared to the conventional approach, the GEMM‐ESC approach will deliver a considerable reduction in time and costs as well as full control over genetic background, as no breeding is required.
9.2. Elucidating mechanisms of response and resistance to PI3K pathway inhibition in GEMMs of human cancer
Although in vitro studies in cancer cell lines have identified a number or resistance mechanisms to PI3K pathway inhibitors, it can be expected that these mechanisms will not explain all forms of clinical resistance. In vivo tumor intervention studies in GEMMs of PI3K pathway‐driven cancer represent an alternative strategy that may uncover additional resistance mechanisms. A first example of such an alternative mechanism has been reported by Liu et al., who investigated genomic alterations in mammary tumors that grew back after doxycycline withdrawal in a GEMM with doxycycline‐inducible expression of PIKCA H1047R (Liu et al., 2011). A subset of recurrent tumors showed independence of PI3K signaling and acquired resistance to GDC‐0941. This resistance phenotype could be explained by Myc amplification, suggesting that c‐MYC elevation is a potential mechanism for resistance to PI3K‐targeted therapy in breast cancer (Liu et al., 2011).
Although comparative genomic analysis of sensitive and resistant tumors has already yielded novel mechanism of resistance to PI3K pathway inactivation, this approach can be cumbersome as many genomic alterations will be passenger mutations that do not contribute to the resistance phenotype. To accelerate the identification of genes that can confer resistance, retroviral or transposon‐based in vivo insertional mutagenesis strategies may be employed. These approaches are based on the fact that slow‐transforming retroviruses or engineered transposons may randomly integrate in the genome of mouse cells and activate or inactivate genes located at or near the insertion site. Cells with insertional mutations at or near genes that confer a selective advantage in their mutated state will expand clonally and create a pool of cells that are amenable to additional rounds of insertional mutagenesis and clonal expansion (Kool and Berns, 2009). Since this continuous process can contribute to every step in tumor development and progression, also acquired traits such as therapy resistance can be effectively studied using insertional mutagenesis. Retroviral and transposon insertion sites can be rapidly isolated from resistant tumors by ligation‐mediated PCR and mapped to the mouse genome after high‐throughput sequencing (Uren et al., 2009). This concept was elegantly demonstrated by Lauchle et al. who used a retroviral insertion mutagenesis screen in a GEMM of acute myeloid leukemia to identify several candidate genes mediating resistance to MEK inhibition (Lauchle et al., 2009).
Although slow‐transforming retroviruses can be employed to identify resistance genes, the utility of these viral agents is restricted to hematopoietic malignancies and mammary tumors due to their specific tissue tropism. In contrast, transposon‐based insertional mutagenesis systems can be engineered to function in virtually every mouse tissue. These mutagenesis systems are based on transposase enzymes that can mobilize defined genetic elements from a transgene array to new loci in the host genome via a ‘cut and paste’ mechanism. To date, the Sleeping Beauty (SB) and PiggyBac (PB) transposon systems have been successfully employed in genetically engineered mice. Inducible SB and PB transposase enzymes have been engineered to enable spatiotemporal control of the insertional mutagenesis process (Keng et al., 2009; March et al., 2011; Rad et al., 2010). The value of both transposon systems has been demonstrated in several in vivo insertional mutagenesis screens aimed at identifying novel cancer genes (reviewed in Copeland and Jenkins (2010)).
10. Concluding remarks
Since the original discovery of PTEN and the first (retroviral) oncogenic variant of PIK3CA in 1997 (Chang et al., 1997; Li et al., 1997; Steck et al., 1997), the PI3K network has been recognized as one of the most frequently mutated signaling pathways in human cancer. The PI3K pathway contains several critical and druggable kinases for which potent and selective inhibitors have been developed; however, the clinical development of these drugs has proven more difficult and less straightforward than initially anticipated. Several inhibitors show no or only modest antitumor activity as single agents, and in other cases tumors rapidly develop resistance after initial response. It is becoming clear that rational combinations of PI3K pathway inhibitors and other targeted therapeutics will be required to achieve durable remissions or – ultimately – complete tumor eradication. Analysis of mechanisms of resistance to single‐agent therapy may identify druggable targets that offer opportunities for rationally designed combination therapies.
GEMMs of human cancer offer unique opportunities to test antitumor activity of drug combinations in clinically relevant model organisms. A growing number of well‐characterized GEMMs of PI3K pathway driven cancer are available for preclinical drug testing and additional models can be relatively easily generated using the GEMM‐ESC approach. For breast cancer GEMMs, pieces of mammary tumor tissue can be effectively cryopreserved, resulting in a cumulative biobank of tumor fragments that can be used to generate allografts by orthotopic transplantation into syngeneic wildtype mice (Rottenberg et al., 2010). This approach allows for greater flexibility and permits evaluation of different (e.g. both single‐agent and combination) therapies on the same tumor. Genomic characterization of biobanked tumors may even permit pre‐selection of tumors with specific genomic features or mutations in defined cancer genes prior to allotransplantation and initiation of the intervention study.
GEMMs also offer opportunities to uncover relevant drug resistance mechanisms. In addition to pharmacological drug target inhibition, genetic approaches can be employed, permitting oncogene de‐induction in established tumors. Genes that confer therapy resistance or drive tumor recurrence after oncogene de‐induction can be identified using conventional genomic approaches (gene expression profiling, analysis of DNA copy number aberrations or mutational analysis through next‐generation sequencing) or by employing transposon‐based insertional mutagenesis strategies, which allow rapid mapping of loci that are mutated in multiple independent resistant tumors.
In conclusion, much progress has been made in modeling PI3K pathway‐driven breast cancer in GEMMs. Recapitulation of the most common genetic mutations in the PI3K pathway in GEMMs results in the formation of mammary tumors that closely resemble their human counterparts. The ensuing models have already been used in a number of preclinical tumor intervention studies with PI3K pathway inhibitors and other therapeutics, and we believe the best is yet to come. We therefore expect that the use of realistic and clinically relevant GEMMs will contribute significantly to furthering our understanding of PI3K‐driven breast cancer and play an increasingly important role in the development of smart combination therapies that maximize the response to PI3K pathway inhibition.
Acknowledgments
We thank the members of our laboratory for helpful discussions and comments on the manuscript. The work of the authors was supported by grants from the European Union, Top Institute (TI) Pharma, the TI Center for Translational Molecular Medicine (CTMM), the Netherlands Organization for Scientific Research (NWO), the Cancer Systems Biology Center and the Dutch Cancer Society (KWF).
Klarenbeek Sjoerd, van Miltenburg Martine H., Jonkers Jos, (2013), Genetically engineered mouse models of PI3K signaling in breast cancer, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.003.
References
- Ackler, S. , Ahmad, S. , Tobias, C. , Johnson, M.D. , Glazer, R.I. , 2002. Delayed mammary gland involution in MMTV-AKT1 transgenic mice. Oncogene 21, 198–206. [DOI] [PubMed] [Google Scholar]
- Adams, J.R. , Xu, K. , Liu, J.C. , Agamez, N.M.R. , Loch, A.J. , Wong, R.G. , Wang, W. , Wright, K.L. , Lane, T.F. , Zacksenhaus, E. , Egan, S.E. , 2011. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 71, 2706–2717. [DOI] [PubMed] [Google Scholar]
- Alimonti, A. , Carracedo, A. , Clohessy, J.G. , Trotman, L.C. , Nardella, C. , Egia, A. , Salmena, L. , Sampieri, K. , Haveman, W.J. , Brogi, E. , Richardson, A.L. , Zhang, J. , Pandolfi, P.P. , 2010. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 42, 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre, F. , Campone, M. , O'Regan, R. , Manlius, C. , Massacesi, C. , Sahmoud, T. , Mukhopadhyay, P. , Soria, J.-C. , Naughton, M. , Hurvitz, S.A. , 2010. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J. Clin. Oncol. 28, 5110–5115. [DOI] [PubMed] [Google Scholar]
- Bachelot, T. , Bourgier, C. , Cropet, C. , Guastalla, J.-P. , Ferrero, J.-M. , Leger-Falandry, C. , Soulie, P. , Eymard, J.-C. , Debled, M. , Spaeth, D. , Legouffe, E. , Delozier, T. , El Kouri, C. , Chidiac, J. , 2011. TAMRAD: a GINECO randomized phase II trial of everolimus in combination with tamoxifen versus tamoxifen alone in patients (pts) with hormone-receptor positive, HER2 negative metastatic breast cancer (MBC) with prior exposure to aromatase inhibitors (AI). Cancer Res. 70, Abstract S1–6 [Google Scholar]
- Bachelot, T. , Bourgier, C. , Cropet, C. , Ray-Coquard, I. , Ferrero, J.-M. , Freyer, G. , Abadie-Lacourtoisie, S. , Eymard, J.-C. , Debled, M. , Spaëth, D. , Legouffe, E. , Allouache, D. , El Kouri, C. , Pujade-Lauraine, E. , 2012. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J. Clin. Oncol. 30, 2718–2724. [DOI] [PubMed] [Google Scholar]
- Barbareschi, M. , Buttitta, F. , Felicioni, L. , Cotrupi, S. , Barassi, F. , Del Grammastro, M. , Ferro, A. , Dalla Palma, P. , Galligioni, E. , Marchetti, A. , 2007. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin. Cancer Res. 13, 6064–6069. [DOI] [PubMed] [Google Scholar]
- Baselga, J. , Campone, M. , Piccart, M. , Burris, H.A. , Rugo, H.S. , Sahmoud, T. , Noguchi, S. , Gnant, M. , Pritchard, K.I. , Lebrun, F. , Beck, J.T. , Ito, Y. , Yardley, D. , Deleu, I. , Perez, A. , Bachelot, T. , Vittori, L. , Xu, Z. , Mukhopadhyay, P. , Lebwohl, D. , Hortobagyi, G.N. , 2012. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 366, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga, J. , Semiglazov, V. , Van Dam, P. , Manikhas, A. , Bellet, M. , Mayordomo, J. , Campone, M. , Kubista, E. , Greil, R. , Bianchi, G. , Steinseifer, J. , Molloy, B. , Tokaji, E. , Gardner, H. , Phillips, P. , Stumm, M. , Lane, H.A. , Dixon, J.M. , Jonat, W. , Rugo, H.S. , 2009. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J. Clin. Oncol. 27, 2630–2637. [DOI] [PubMed] [Google Scholar]
- Beard, C. , Hochedlinger, K. , Plath, K. , Wutz, A. , Jaenisch, R. , 2006. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28. [DOI] [PubMed] [Google Scholar]
- Beelen, K. , Zwart, W. , Linn, S.C. , 2012. Can predictive biomarkers in breast cancer guide adjuvant endocrine therapy?. Nat. Rev. Clin. Oncol. 9, 529–541. [DOI] [PubMed] [Google Scholar]
- Berenjeno, I.M. , Guillermet-Guibert, J. , Pearce, W. , Gray, A. , Fleming, S. , Vanhaesebroeck, B. , 2012. Both p110α and p110β isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem. J. 442, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, A.H. , Knudson, A.G. , Pandolfi, P.P. , 2011. A continuum model for tumour suppression. Nature 476, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns, K. , Horlings, H.M. , Hennessy, B.T. , Madiredjo, M. , Hijmans, E.M. , Beelen, K. , Linn, S.C. , Gonzalez-Angulo, A.M. , Stemke-Hale, K. , Hauptmann, M. , Beijersbergen, R.L. , Mills, G.B. , Van de Vijver, M.J. , Bernards, R. , 2007. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12, 395–402. [DOI] [PubMed] [Google Scholar]
- Beuvink, I. , O'Reilly, T. , Zumstein, S. , Zilbermann, F. , Sedrani, R. , Kozma, S. , Thomas, G. , Lane, H.A. , 2001. Antitumor activity of RAD001, an orally active rapamycin derivative. Proc. Am. Assoc. Cancer Res. 42, 366 [Google Scholar]
- Blake, J.F. , Xu, R. , Bencsik, J.R. , Xiao, D. , Kallan, N.C. , Schlachter, S. , Mitchell, I.S. , Spencer, K.L. , Banka, A.L. , Wallace, E.M. , Gloor, S.L. , Martinson, M. , Woessner, R.D. , Vigers, G.P. , Brandhuber, B.J. , Liang, J. , Safina, B.S. , Li, J. , Zhang, B. , Chabot, C. , Do, S. , Lee, L. , Oeh, J. , Sampath, D. , Lee, B.B. , Lin, K. , Liederer, B.M. , Skelton, N.J. , 2012. Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J. Med. Chem. 55, 8110–8127. [DOI] [PubMed] [Google Scholar]
- Blanco-Aparicio, C. , Pérez-Gallego, L. , Pequeño, B. , Leal, J.F.M. , Renner, O. , Carnero, A. , 2006. Mice expressing myrAKT1 in the mammary gland develop carcinogen-induced ER-positive mammary tumors that mimic human breast cancer. Carcinogenesis 28, 584–594. [DOI] [PubMed] [Google Scholar]
- Blasco, R.B. , Francoz, S. , Santamaría, D. , Cañamero, M. , Dubus, P. , Charron, J. , Baccarini, M. , Barbacid, M. , 2011. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 19, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault, S. , Drouet, Y. , Navarro, C. , Bachelot, T. , Lasset, C. , Treilleux, I. , Tabone, E. , Puisieux, A. , Wang, Q. , 2012. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res. Treat. 132, 29–39. [DOI] [PubMed] [Google Scholar]
- Campbell, I.G. , Russell, S.E. , Choong, D.Y.H. , Montgomery, K.G. , Ciavarella, M.L. , Hooi, C.S.F. , Cristiano, B.E. , Pearson, R.B. , Phillips, W.A. , 2004. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 64, 7678–7681. [DOI] [PubMed] [Google Scholar]
- Carpten, J.D. , Faber, A.L. , Horn, C. , Donoho, G.P. , Briggs, S.L. , Robbins, C.M. , Hostetter, G. , Boguslawski, S. , Moses, T.Y. , Savage, S. , Uhlik, M. , Lin, A. , Du, J. , Qian, Y.-W. , Zeckner, D.J. , Tucker-Kellogg, G. , Touchman, J. , Patel, K. , Mousses, S. , Bittner, M. , Schevitz, R. , Lai, M.-H.T. , Blanchard, K.L. , Thomas, J.E. , 2007. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444. [DOI] [PubMed] [Google Scholar]
- Carracedo, A. , Ma, L. , Teruya-Feldstein, J. , Rojo, F. , Salmena, L. , Alimonti, A. , Egia, A. , Sasaki, A.T. , Thomas, G. , Kozma, S.C. , Papa, A. , Nardella, C. , Cantley, L.C. , Baselga, J. , Pandolfi, P.P. , 2008. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 118, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty, A. , Sánchez, V. , Kuba, M.G. , Rinehart, C. , Arteaga, C.L. , 2012. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. U. S. A. 109, 2718–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty, S. , Sawai, A. , Scaltriti, M. , Rodrik-Outmezguine, V. , Grbovic-Huezo, O. , Serra, V. , Majumder, P.K. , Baselga, J. , Rosen, N. , 2011. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H.W. , Aoki, M. , Fruman, D. , Auger, K.R. , Bellacosa, A. , Tsichlis, P.N. , Cantley, L.C. , Roberts, T.M. , Vogt, P.K. , 1997. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science 276, 1848–1850. [DOI] [PubMed] [Google Scholar]
- Chen, Z. , Cheng, K. , Walton, Z. , Wang, Y. , Ebi, H. , Shimamura, T. , Liu, Y. , Tupper, T. , Ouyang, J. , Li, J. , Gao, P. , Woo, M.S. , Xu, C. , Yanagita, M. , Altabef, A. , Wang, S. , Lee, C. , Nakada, Y. , Peña, C.G. , Sun, Y. , Franchetti, Y. , Yao, C. , Saur, A. , Cameron, M.D. , Nishino, M. , Hayes, D.N. , Wilkerson, M.D. , Roberts, P.J. , Lee, C.B. , Bardeesy, N. , Butaney, M. , Chirieac, L.R. , Costa, D.B. , Jackman, D. , Sharpless, N.E. , Castrillon, D.H. , Demetri, G.D. , Jänne, P.A. , Pandolfi, P.P. , Cantley, L.C. , Kung, A.L. , Engelman, J.A. , Wong, K.-K. , 2012. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, C.W. , Nozawa, H. , Hanahan, D. , 2010. Survival benefit with proapoptotic molecular and pathologic responses from dual targeting of mammalian target of rapamycin and epidermal growth factor receptor in a preclinical model of pancreatic neuroendocrine carcinogenesis. J. Clin. Oncol. 28, 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chresta, C.M. , Davies, B.R. , Hickson, I. , Harding, T. , Cosulich, S. , Critchlow, S.E. , Vincent, J.P. , Ellston, R. , Jones, D. , Sini, P. , James, D. , Howard, Z. , Dudley, P. , Hughes, G. , Smith, L. , Maguire, S. , Hummersone, M. , Malagu, K. , Menear, K. , Jenkins, R. , Jacobsen, M. , Smith, G.C.M. , Guichard, S. , Pass, M. , 2010. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70, 288–298. [DOI] [PubMed] [Google Scholar]
- Ciampricotti, M. , Hau, C.-S. , Doornebal, C.W. , Jonkers, J. , de Visser, K.E. , 2012. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat. Med. 18, 344–346. [DOI] [PubMed] [Google Scholar]
- Copeland, N.G. , Jenkins, N.A. , 2010. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer 10, 696–706. [DOI] [PubMed] [Google Scholar]
- Courtney, K.D. , Corcoran, R.B. , Engelman, J.A. , 2010. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 28, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen, J.E. , Liu, L. , Rosten, P. , Humphries, R.K. , Jefferson, A.B. , Majerus, P.W. , Krystal, G. , 1996. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc. Natl. Acad. Sci. U. S. A. 93, 1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, B.R. , Greenwood, H. , Dudley, P. , Crafter, C. , Yu, D.-H. , Zhang, J. , Li, J. , Gao, B. , Ji, Q. , Maynard, J. , Ricketts, S.-A. , Cross, D. , Cosulich, S. , Chresta, C.C. , Page, K. , Yates, J. , Lane, C. , Watson, R. , Luke, R. , Ogilvie, D. , Pass, M. , 2012. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 11, 873–887. [DOI] [PubMed] [Google Scholar]
- DeNardo, D.G. , Brennan, D.J. , Rexhepaj, E. , Ruffell, B. , Shiao, S.L. , Madden, S.F. , Gallagher, W.M. , Wadhwani, N. , Keil, S.D. , Junaid, S.A. , Rugo, H.S. , Hwang, E.S. , Jirström, K. , West, B.L. , Coussens, L.M. , 2011. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose, Y.S. , Wang, G. , Lin, Y.-C. , Bernard, P.S. , Buys, S.S. , Ebbert, M.T.W. , Factor, R. , Matsen, C. , Milash, B.A. , Nelson, E. , Neumayer, L. , Randall, R.L. , Stijleman, I.J. , Welm, B.E. , Welm, A.L. , 2011. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon, R.L. , Marcotte, R. , Hennessy, B.T. , Woodgett, J.R. , Mills, G.B. , Muller, W.J. , 2009. Akt1 and Akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 69, 5057–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow, L.E. , Lowe, S.W. , 2012. Life in the fast lane: mammalian disease models in the genomics era. Cell 148, 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudkin, L. , Dilling, M.B. , Cheshire, P.J. , Harwood, F.C. , Hollingshead, M. , Arbuck, S.G. , Travis, R. , Sausville, E.A. , Houghton, P.J. , 2001. Biochemical correlates of mTOR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin. Cancer Res. 7, 1758–1764. [PubMed] [Google Scholar]
- Dumont, A.G. , Dumont, S.N. , Trent, J.C. , 2012. The favorable impact of PIK3CA mutations on survival: an analysis of 2587 patients with breast cancer. Chin. J. Cancer 31, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap, J. , Le, C. , Shukla, A. , Patterson, J. , Presnell, A. , Heinrich, M.C. , Corless, C.L. , Troxell, M.L. , 2010. Phosphatidylinositol-3-kinase and AKT1 mutations occur early in breast carcinoma. Breast Cancer Res. Treat. 120, 409–418. [DOI] [PubMed] [Google Scholar]
- DuPage, M. , Mazumdar, C. , Schmidt, L.M. , Cheung, A.F. , Jacks, T. , 2012. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont, J. , Renou, J.P. , Shani, M. , Hennighausen, L. , LeRoith, D. , 2002. PTEN overexpression suppresses proliferation and differentiation and enhances apoptosis of the mouse mammary epithelium. J. Clin. Invest. 110, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont Jensen, J. , Laenkholm, A.-V. , Knoop, A. , Ewertz, M. , Bandaru, R. , Liu, W. , Hackl, W. , Barrett, J.C. , Gardner, H. , 2011. PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin. Cancer Res. 17, 667–677. [DOI] [PubMed] [Google Scholar]
- Ellis, M.J. , Ding, L. , Shen, D. , Luo, J. , Suman, V.J. , 2012. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman, J.A. , Chen, L. , Tan, X. , Crosby, K. , Guimaraes, A.R. , Upadhyay, R. , Maira, M. , McNamara, K. , Perera, S.A. , Song, Y. , Chirieac, L.R. , Kaur, R. , Lightbown, A. , Simendinger, J. , Li, T. , Padera, R.F. , García-Echeverría, C. , Weissleder, R. , Mahmood, U. , Cantley, L.C. , Wong, K.-K. , 2008. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 14, 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteva, F.J. , Guo, H. , Zhang, S. , Santa-Maria, C. , Stone, S. , Lanchbury, J.S. , Sahin, A.A. , Hortobagyi, G.N. , Yu, D. , 2010. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am. J. Pathol. 177, 1647–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber, A.C. , Li, D. , Song, Y. , Liang, M.-C. , Yeap, B.Y. , Bronson, R.T. , Lifshits, E. , Chen, Z. , Maira, S.-M. , García-Echeverría, C. , Wong, K.-K. , Engelman, J.A. , 2009. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc. Natl. Acad. Sci. U. S. A. 106, 19503–19508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele, C.G. , Ooms, L.M. , Ho, M. , Vieusseux, J. , O'Toole, S.A. , Millar, E.K. , Lopez-Knowles, E. , Sriratana, A. , Gurung, R. , Baglietto, L. , Giles, G.G. , Bailey, C.G. , Rasko, J.E.J. , Shields, B.J. , Price, J.T. , Majerus, P.W. , Sutherland, R.L. , Tiganis, T. , McLean, C.A. , Mitchell, C.A. , 2010. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl. Acad. Sci. U. S. A. 107, 22231–22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman, M.E. , Apsel, B. , Uotila, A. , Loewith, R. , Knight, Z.A. , Ruggero, D. , Shokat, K.M. , 2009. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7, e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay, J. , Shin, H.-R. , Bray, F. , Forman, D. , Mathers, C. , Parkin, D.M. , 2010. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917. [DOI] [PubMed] [Google Scholar]
- Folkes Tabladi, K. , Alderton, W.K. , Alix, S. , Baker, S.J. , Box, G. , Chuckowree, I.S. , Clarke, P.A. , Depledge, P. , Eccles, S.A. , Friedman, L.S. , Hayes, A. , Hancox, T.C. , Kugendradas, A. , Lensun, L. , Moore, P. , Olivero, A.G. , Pang, J. , Patel, S. , Pergl-Wilson, G.H. , Raynaud, F.I. , Robson, A. , Saghir, N. , Salphati, L. , Sohal, S. , Ultsch, M.H. , Valenti, M. , Wallweber, H.J. , Wan, N.C. , Wiesmann, C. , Workman, P. , Zhyvoloup, A. , Zvelebil, M.J. , Shuttleworth, S.J. , 2008. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Frese, K.K. , Tuveson, D.A. , 2007. Maximizing mouse cancer models. Nat. Rev. Cancer 7, 645–658. [DOI] [PubMed] [Google Scholar]
- Friedman, L. , Ross, L.B. , Wallin, J. , Guan, J. , Prior, W.W. , Wu, E. , Nannini, M. , Sampath, D. , 2012. Selective PI3K and dual PI3K/mTOR inhibitors enhance the efficacy of endocrine therapies in breast cancer models. Cancer Res. 72, (Suppl. 24) Abstract P5-19-02 [Google Scholar]
- Fritsch, C.M. , Schnell, C. , Chatenay-Rivauday, C. , Guthy, D.A. , De Pover, A. , Wartmann, M. , Brachmann, S. , Maira, S.M. , Huang, A. , Quadt, C. , Hofmann, F. , Caravatti, G. , 2012. NVP-BYL719, a novel PI3Kalpha selective inhibitor with all the characteristics required for clinical development as an anti-cancer agent. Cancer Res. 72, (Suppl. 8) Abstract 3748 [Google Scholar]
- Gallardo, A. , Lerma, E. , Escuin, D. , Tibau, A. , Muñoz, J. , Ojeda, B. , Barnadas, A. , Adrover, E. , Sánchez-Tejada, L. , Giner, D. , Ortiz-Martínez, F. , Peiró, G. , 2012. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer 106, 1367–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, D. , Inuzuka, H. , Tseng, A. , Chin, R.Y. , Toker, A. , Wei, W. , 2009. Phosphorylation by Akt1 promotes Skp2 cytoplasmic localization and impairs APC/Cdh1-mediated Skp2 destruction. Nat. Cell. Biol. 11, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez, J.M. , Moran, J. , Clarke, R.G. , Gray, A. , Cosulich, S.C. , Chresta, C.M. , Alessi, D.R. , 2009. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 421, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewinner, C. , Wang, Z.C. , Richardson, A. , Teruya-Feldstein, J. , Etemadmoghadam, D. , Bowtell, D. , Barretina, J. , Lin, W.M. , Rameh, L. , Salmena, L. , Pandolfi, P.P. , Cantley, L.C. , 2009. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 16, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo, A.M. , Ferrer-Lozano, J. , Stemke-Hale, K. , Sahin, A. , Liu, S. , Barrera, J.A. , Burgues, O. , Lluch, A.M. , Chen, H. , Hortobagyi, G.N. , Mills, G.B. , Meric-Bernstam, F. , 2011. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol. Cancer Ther. 10, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, H. , Sootome, H. , Nakatsuru, Y. , Miyama, K. , Taguchi, S. , Tsujioka, K. , Ueno, Y. , Hatch, H. , Majumder, P.K. , Pan, B.-S. , Kotani, H. , 2010. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- Huijbers, I.J. , Krimpenfort, P. , Berns, A. , Jonkers, J. , 2011. Rapid validation of cancer genes in chimeras derived from established genetically engineered mouse models. Bioessays 33, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerusalem, G. , Fasolo, A. , Dieras, V. , Cardoso, F. , Bergh, J. , Vittori, L. , Zhang, Y. , Massacesi, C. , Sahmoud, T. , Gianni, L. , 2011. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res. Treat. 125, 447–455. [DOI] [PubMed] [Google Scholar]
- Jonkers, J. , Berns, A. , 2002. Conditional mouse models of sporadic cancer. Nat. Rev. Cancer 2, 251–265. [DOI] [PubMed] [Google Scholar]
- Jonkers, J. , Meuwissen, R. , Van der Gulden, H. , Peterse, H. , Van der Valk, M. , Berns, A. , 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 29, 418–425. [DOI] [PubMed] [Google Scholar]
- Juvekar, A. , Burga, L.N. , Hu, H. , Lunsford, E.P. , Ibrahim, Y.H. , Balmañà, J. , Rajendran, A. , Papa, A. , Spencer, K. , Lyssiotis, C.A. , Nardella, C. , Pandolfi, P.P. , Baselga, J. , Scully, R. , Asara, J.M. , Cantley, L.C. , Wulf, G.M. , 2012. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2, 1048–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng, V.W. , Villanueva, A. , Chiang, D.Y. , Dupuy, A.J. , Ryan, B.J. , Matise, I. , Silverstein, K.A.T. , Sarver, A. , Starr, T.K. , Akagi, K. , Tessarollo, L. , Collier, L.S. , Powers, S. , Lowe, S.W. , Jenkins, N.A. , Copeland, N.G. , Llovet, J.M. , Largaespada, D.A. , 2009. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat. Biotechnol. 27, 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson, A.G. , 1971. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A. 68, 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool, J. , Berns, A. , 2009. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat. Rev. Cancer 9, 389–399. [DOI] [PubMed] [Google Scholar]
- LaRocca, J. , Pietruska, J. , Hixon, M. , 2011. Akt1 is essential for postnatal mammary gland development, function, and the expression of Btn1a1. PLoS One 6, e24432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauchle, J.O. , Kim, D. , Le, D.T. , Akagi, K. , Crone, M. , Krisman, K. , Warner, K. , Bonifas, J.M. , Li, Q. , Coakley, K.M. , Diaz-Flores, E. , Gorman, M. , Przybranowski, S. , Tran, M. , Kogan, S.C. , Roose, J.P. , Copeland, N.G. , Jenkins, N.A. , Parada, L. , Wolff, L. , Sebolt-Leopold, J. , Shannon, K. , 2009. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature 461, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski, M. , 2001. Conditional control of gene expression in the mouse. Nat. Rev. Genet. 2, 743–755. [DOI] [PubMed] [Google Scholar]
- Li, G. , Robinson, G.W. , Lesche, R. , Martinez-Diaz, H. , Jiang, Z. , Rozengurt, N. , Wagner, K.-U. , Wu, D.-C. , Lane, T.F. , Liu, X. , Hennighausen, L. , Wu, H. , 2002. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development 129, 4159–4170. [DOI] [PubMed] [Google Scholar]
- Li, J. , Yen, C. , Liaw, D. , Podsypanina, K. , Bose, S. , Wang, S.I. , Puc, J. , Miliaresis, C. , Rodgers, L. , McCombie, R. , Bigner, S.H. , Giovanella, B.C. , Ittmann, M. , Tycko, B. , Hibshoosh, H. , Wigler, M.H. , Parsons, R. , 1997. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Podsypanina, K. , Liu, X. , Crane, A. , Tan, L.K. , Parsons, R. , Varmus, H.E. , 2001. Deficiency of Pten accelerates mammary oncogenesis in MMTV-Wnt-1 transgenic mice. BMC Mol. Biol. 2, 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J. , Sampath, D. , Nannini, M.A. , Lee, B.B. , Degtyarev, M. , Oeh, J. , Savage, H. , Guan, Z. , Hong, R. , Kassees, R. , Lee, L.B. , Risom, T. , Gross, S.D. , Liederer, B. , Koeppen, H. , Skelton, N. , Wallin, J.J. , Belvin, M. , Punnoose, E.A. , Friedman, L.S. , Lin, K. , 2013. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. ([Epub ahead of print]) [DOI] [PubMed] [Google Scholar]
- Liu, P. , Cheng, H. , Santiago, S. , Raeder, M. , Zhang, F. , Isabella, A. , Yang, J. , Semaan, D.J. , Chen, C. , Fox, E.A. , Gray, N.S. , Monahan, J. , Schlegel, R. , Beroukhim, R. , Mills, G.B. , Zhao, J.J. , 2011. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat. Med. 17, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi, S. , Haibe-Kains, B. , Majjaj, S. , Lallemand, F. , Durbecq, V. , Larsimont, D. , Gonzalez-Angulo, A.M. , Pusztai, L. , Symmans, W.F. , Bardelli, A. , Ellis, P. , Tutt, A.N.J. , Gillett, C.E. , Hennessy, B.T. , Mills, G.B. , Phillips, W.A. , Piccart, M.J. , Speed, T.P. , McArthur, G.A. , Sotiriou, C. , 2010. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc. Natl. Acad. Sci. U. S. A. 107, 10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira, S.-M. , Pecchi, S. , Huang, A. , Burger, M. , Knapp, M. , Sterker, D. , Schnell, C. , Guthy, D. , Nagel, T. , Wiesmann, M. , Brachmann, S. , Fritsch, C. , Dorsch, M. , Chène, P. , Shoemaker, K. , De Pover, A. , Menezes, D. , Martiny-Baron, G. , Fabbro, D. , Wilson, C.J. , Schlegel, R. , Hofmann, F. , García-Echeverría, C. , Sellers, W.R. , Voliva, C.F. , 2012. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 11, 317–328. [DOI] [PubMed] [Google Scholar]
- Maira, S.-M. , Stauffer, F. , Brueggen, J. , Furet, P. , Schnell, C. , Fritsch, C. , Brachmann, S. , Chène, P. , De Pover, A. , Schoemaker, K. , Fabbro, D. , Gabriel, D. , Simonen, M. , Murphy, L. , Finan, P. , Sellers, W. , García-Echeverría, C. , 2008. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863. [DOI] [PubMed] [Google Scholar]
- Manning, B.D. , Cantley, L.C. , 2007. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March, H.N. , Rust, A.G. , Wright, N.A. , Ten Hoeve, J. , De Ridder, J. , Eldridge, M. , Van der Weyden, L. , Berns, A. , Gadiot, J. , Uren, A. , Kemp, R. , Arends, M.J. , Wessels, L.F.A. , Winton, D.J. , Adams, D.J. , 2011. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 43, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou, I.G. , Oemler, W. , Naber, S.P. , Klebba, I. , Kuperwasser, C. , Tsichlis, P.N. , 2008. Distinct roles of the three Akt isoforms in lactogenic differentiation and involution. J. Cell. Physiol. 217, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou, I.G. , Oemler, W. , Naber, S.P. , Tsichlis, P.N. , 2007. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 67, 167–177. [DOI] [PubMed] [Google Scholar]
- Martins, F.C. , De, S. , Almendro, V. , Gönen, M. , Park, S.Y. , Blum, J.L. , Herlihy, W. , Ethington, G. , Schnitt, S.J. , Tung, N. , Garber, J.E. , Fetten, K. , Michor, F. , Polyak, K. , 2012. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov. 2, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui, S. , Shimosato, D. , Toyooka, Y. , Yagi, R. , Takahashi, K. , Niwa, H. , 2005. An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res. 33, e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Pereira, A.M. , Martin, S.A. , Brough, R. , McCarthy, A. , Taylor, J.R. , Kim, J.-S. , Waldman, T. , Lord, C.J. , Ashworth, A. , 2009. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 1, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, D.S. , Brinkhaus, H. , Müller, U. , Müller, M. , Cardiff, R.D. , Bentires-Alj, M. , 2011. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. 71, 4344–4351. [DOI] [PubMed] [Google Scholar]
- Morrow, P.K. , Wulf, G.M. , Ensor, J. , Booser, D.J. , Moore, J.A. , Flores, P.R. , Xiong, Y. , Zhang, S. , Krop, I.E. , Winer, E.P. , Kindelberger, D.W. , Coviello, J. , Sahin, A.A. , Nuñez, R. , Hortobagyi, G.N. , Yu, D. , Esteva, F.J. , 2011. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J. Clin. Oncol. 29, 3126–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen, T. , Selfors, L.M. , Worster, D.T. , Iwanicki, M.P. , Song, L. , Morales, F.C. , Gao, S. , Mills, G.B. , Brugge, J.S. , 2012. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata, Y. , Lan, K.-H. , Zhou, X. , Tan, M. , Esteva, F.J. , Sahin, A.A. , Klos, K.S. , Li, P. , Monia, B.P. , Nguyen, N.T. , Hortobagyi, G.N. , Hung, M.-C. , Yu, D. , 2004. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127. [DOI] [PubMed] [Google Scholar]
- O'Brien, N.A. , Browne, B.C. , Chow, L. , Wang, Y. , Ginther, C. , Arboleda, J. , Duffy, M.J. , Crown, J. , O'Donovan, N. , Slamon, D.J. , 2010. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol. Cancer Ther. 9, 1489–1502. [DOI] [PubMed] [Google Scholar]
- O'Reilly, K.E. , Rojo, F. , She, Q.-B. , Solit, D. , Mills, G.B. , Smith, D. , Lane, H. , Hofmann, F. , Hicklin, D.J. , Ludwig, D.L. , Baselga, J. , Rosen, N. , 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, H. , Flinn, R. , Patsialou, A. , Wyckoff, J. , Roussos, E.T. , Wu, H. , Pozzuto, M. , Goswami, S. , Condeelis, J.S. , Bresnick, A.R. , Segall, J.E. , Backer, J.M. , 2009. Differential enhancement of breast cancer cell motility and metastasis by helical and kinase domain mutations of class IA phosphoinositide 3-kinase. Cancer Res. 69, 8868–8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi, A.R. , Pinder, S.E. , Chan, S.Y. , Paish, E.C. , Robertson, J.F.R. , Ellis, I.O. , 2004. The role of PTEN and its signalling pathways, including AKT, in breast cancer; an assessment of relationships with other prognostic factors and with outcome. J. Pathol. 204, 93–100. [DOI] [PubMed] [Google Scholar]
- Pérez-Tenorio, G. , Alkhori, L. , Olsson, B. , Waltersson, M.A. , Nordenskjöld, B. , Rutqvist, L.E. , Skoog, L. , Stål, O. , 2007. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin. Cancer Res. 13, 3577–3584. [DOI] [PubMed] [Google Scholar]
- Pittius, C.W. , Hennighausen, L. , Lee, E. , Westphal, H. , Nicols, E. , Vitale, J. , Gordon, K. , 1988. A milk protein gene promoter directs the expression of human tissue plasminogen activator cDNA to the mammary gland in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 85, 5874–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina, K. , Ellenson, L.H. , Nemes, A. , Gu, J. , Tamura, M. , Yamada, K.M. , Cordon-Cardo, C. , Catoretti, G. , Fisher, P.E. , Parsons, R. , 1999. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. U. S. A. 96, 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premsrirut, P.K. , Dow, L.E. , Kim, S.Y. , Camiolo, M. , Malone, C.D. , Miething, C. , Scuoppo, C. , Zuber, J. , Dickins, R.A. , Kogan, S.C. , Shroyer, K.R. , Sordella, R. , Hannon, G.J. , Lowe, S.W. , 2011. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145, 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puc, J. , Keniry, M. , Li, H.S. , Pandita, T.K. , Choudhury, A.D. , Memeo, L. , Mansukhani, M. , Murty, V.V.V.S. , Gaciong, Z. , Meek, S.E.M. , Piwnica-Worms, H. , Hibshoosh, H. , Parsons, R. , 2005. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 7, 193–204. [DOI] [PubMed] [Google Scholar]
- Rad, R. , Rad, L. , Wang, W. , Cadinanos, J. , Vassiliou, G. , Rice, S. , Campos, L.S. , Yusa, K. , Banerjee, R. , Li, M.A. , De la Rosa, J. , Strong, A. , Lu, D. , Ellis, P. , Conte, N. , Yang, F.T. , Liu, P. , Bradley, A. , 2010. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science 330, 1104–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razis, E. , Bobos, M. , Kotoula, V. , Eleftheraki, A.G. , Kalofonos, H.P. , Pavlakis, K. , Papakostas, P. , Aravantinos, G. , Rigakos, G. , Efstratiou, I. , Petraki, K. , Bafaloukos, D. , Kostopoulos, I. , Pectasides, D. , Kalogeras, K.T. , Skarlos, D. , Fountzilas, G. , 2011. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res. Treat. 128, 447–456. [DOI] [PubMed] [Google Scholar]
- Roberts, P.J. , Usary, J.E. , Darr, D.B. , Dillon, P.M. , Pfefferle, A.D. , Whittle, M.C. , Duncan, J.S. , Johnson, S.M. , Combest, A.J. , Jin, J. , Zamboni, W.C. , Johnson, G.L. , Perou, C.M. , Sharpless, N.E. , 2012. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin. Cancer Res. 18, 5290–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrik-Outmezguine, V.S. , Chandarlapaty, S. , Pagano, N.C. , Poulikakos, P.I. , Scaltriti, M. , Moskatel, E. , Baselga, J. , Guichard, S. , Rosen, N. , 2011. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 1, 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper, J. , Richardson, M.P. , Wang, W.V. , Richard, L.G. , Chen, W. , Coffee, E.M. , Sinnamon, M.J. , Lee, L. , Chen, P.-C. , Bronson, R.T. , Martin, E.S. , Hung, K.E. , 2011. The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PLoS One 6, e25132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg, S. , Pajic, M. , Jonkers, J. , 2010. Studying drug resistance using genetically engineered mouse models for breast cancer. Methods Mol. Biol. 596, 33–45. [DOI] [PubMed] [Google Scholar]
- Saal, L.H. , Gruvberger-Saal, S.K. , Persson, C. , Lövgren, K. , Jumppanen, M. , Staaf, J. , Jönsson, G. , Pires, M.M. , Maurer, M. , Holm, K. , Koujak, S. , Subramaniyam, S. , Vallon-Christersson, J. , Olsson, H. , Su, T. , Memeo, L. , Ludwig, T. , Ethier, S.P. , Krogh, M. , Szabolcs, M. , Murty, V.V.V.S. , Isola, J. , Hibshoosh, H. , Parsons, R. , Borg, A. , 2008. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 40, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal, L.H. , Holm, K. , Maurer, M. , Memeo, L. , Su, T. , Wang, X. , Yu, J.S. , Malmström, P.-O. , Mansukhani, M. , Enoksson, J. , Hibshoosh, H. , Borg, A. , Parsons, R. , 2005. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 65, 2554–2559. [DOI] [PubMed] [Google Scholar]
- Sadeq, V. , Isar, N. , Manoochehr, T. , 2011. Association of sporadic breast cancer with PTEN/MMAC1/TEP1 promoter hypermethylation. Med. Oncol. 28, 420–423. [DOI] [PubMed] [Google Scholar]
- Santarpia, L. , Qi, Y. , Stemke-Hale, K. , Wang, B. , Young, E.J. , Booser, D.J. , Holmes, F.A. , O'Shaughnessy, J. , Hellerstedt, B. , Pippen, J. , Vidaurre, T. , Gomez, H. , Valero, V. , Hortobagyi, G.N. , Symmans, W.F. , Bottai, G. , Di Leo, A. , Gonzalez-Angulo, A.M. , Pusztai, L. , 2012. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res. Treat. 134, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade, B. , Rao, T. , Dourdin, N. , Lesurf, R. , Hallett, M. , Cardiff, R.D. , Muller, W.J. , 2009. PTEN deficiency in a luminal ErbB-2 mouse model results in dramatic acceleration of mammary tumorigenesis and metastasis. J. Biol. Chem. 284, 19018–19026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertfeger, K.L. , Richert, M.M. , Anderson, S.M. , 2001. Mammary gland involution is delayed by activated akt in transgenic mice. Mol. Endocrinol. 15, 867–881. [DOI] [PubMed] [Google Scholar]
- Shah, S.P. , Roth, A. , Goya, R. , Oloumi, A. , Ha, G. , 2012. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless, N.E. , Depinho, R.A. , 2006. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov. 5, 741–754. [DOI] [PubMed] [Google Scholar]
- She, Q.-B. , Halilovic, E. , Ye, Q. , Zhen, W. , Shirasawa, S. , Sasazuki, T. , Solit, D.B. , Rosen, N. , 2010. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 18, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, W.H. , Balajee, A.S. , Wang, J. , Wu, H. , Eng, C. , Pandolfi, P.P. , Yin, Y. , 2007. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170. [DOI] [PubMed] [Google Scholar]
- Shetty, P.J. , Pasupuleti, N. , Chava, S. , Nasaruddin, K. , Hasan, Q. , 2011. Altered transcription and expression of PTEN in breast tumors: is it regulated by hypermethylation?. Breast Dis. 33, 27–33. [DOI] [PubMed] [Google Scholar]
- Singh, M. , Lima, A. , Molina, R. , Hamilton, P. , Clermont, A.C. , Devasthali, V. , Thompson, J.D. , Cheng, J.H. , Bou Reslan, H. , Ho, C.C.K. , Cao, T.C. , Lee, C.V. , Nannini, M.A. , Fuh, G. , Carano, R.A.D. , Koeppen, H. , Yu, R.X. , Forrest, W.F. , Plowman, G.D. , Johnson, L. , 2010. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat. Biotechnol. 28, 585–593. [DOI] [PubMed] [Google Scholar]
- Song, M.S. , Carracedo, A. , Salmena, L. , Song, S.J. , Egia, A. , Malumbres, M. , Pandolfi, P.P. , 2011. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, M.S. , Salmena, L. , Pandolfi, P.P. , 2012. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 13, 283–296. [DOI] [PubMed] [Google Scholar]
- Staal, S.P. , 1987. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A. 84, 5034–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic, V. , Suzuki, A. , De la Pompa, J.L. , Brothers, G.M. , Mirtsos, C. , Sasaki, T. , Ruland, J. , Penninger, J.M. , Siderovski, D.P. , Mak, T.W. , 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39. [DOI] [PubMed] [Google Scholar]
- Stambolic, V. , Tsao, M.S. , Macpherson, D. , Suzuki, A. , Chapman, W.B. , Mak, T.W. , 2000. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 60, 3605–3611. [PubMed] [Google Scholar]
- Steck, P.A. , Pershouse, M.A. , Jasser, S.A. , Yung, W.K. , Lin, H. , Ligon, A.H. , Langford, L.A. , Baumgard, M.L. , Hattier, T. , Davis, T. , Frye, C. , Hu, R. , Swedlund, B. , Teng, D.H. , Tavtigian, S.V. , 1997. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362. [DOI] [PubMed] [Google Scholar]
- Stemke-Hale, K. , Gonzalez-Angulo, A.M. , Lluch, A. , Neve, R.M. , Kuo, W.-L. , Davies, M. , Carey, M. , Hu, Z. , Guan, Y. , Sahin, A. , Symmans, W.F. , Pusztai, L. , Nolden, L.K. , Horlings, H. , Berns, K. , Hung, M.-C. , Van de Vijver, M.J. , Valero, V. , Gray, J.W. , Bernards, R. , Mills, G.B. , Hennessy, B.T. , 2008. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 68, 6084–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, P.J. , Tarpey, P.S. , Davies, H. , Van Loo, P. , Greenman, C. , 2012. The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, T.A. , Pattengale, P.K. , Leder, P. , 1984. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 38, 627–637. [DOI] [PubMed] [Google Scholar]
- Sutherlin, D.P. , Bao, L. , Berry, M. , Castanedo, G. , Chuckowree, I. , Dotson, J. , Folks, A. , Friedman, L. , Goldsmith, R. , Gunzner, J. , Heffron, T. , Lesnick, J. , Lewis, C. , Mathieu, S. , Murray, J. , Nonomiya, J. , Pang, J. , Pegg, N. , Prior, W.W. , Rouge, L. , Salphati, L. , Sampath, D. , Tian, Q. , Tsui, V. , Wan, N.C. , Wang, S. , Wei, B. , Wiesmann, C. , Wu, P. , Zhu, B.Y. , Olivero, A. , 2011. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J. Med. Chem. 54, 7579–7587. [DOI] [PubMed] [Google Scholar]
- Suzuki, A. , De la Pompa, J.L. , Stambolic, V. , Elia, A.J. , Sasaki, T. , Del Barco Barrantes, I. , Ho, A. , Wakeham, A. , Itie, A. , Khoo, W. , Fukumoto, M. , Mak, T.W. , 1998. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 8, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Tentler, J.J. , Tan, A.C. , Weekes, C.D. , Jimeno, A. , Leong, S. , Pitts, T.M. , Arcaroli, J.J. , Messersmith, W.A. , Eckhardt, S.G. , 2012. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network, 2012. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen, C.C. , Kang, S.A. , Chang, J.W. , Liu, Q. , Zhang, J. , Gao, Y. , Reichling, L.J. , Sim, T. , Sabatini, D.M. , Gray, N.S. , 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikoo, A. , Roh, V. , Montgomery, K.G. , Ivetac, I. , Waring, P. , Pelzer, R. , Hare, L. , Shackleton, M. , Humbert, P. , Phillips, W.A. , 2012. Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERα-positive tumors. PLoS One 7, e36924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbett, N.E. , Luna-Moran, A. , Knight, Z.A. , Houk, A. , Moasser, M. , Weiss, W. , Shokat, K.M. , Stokoe, D. , 2008. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem. J. 415, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren, A.G. , Mikkers, H. , Kool, J. , Van der Weyden, L. , Lund, A.H. , Wilson, C.H. , Rance, R. , Jonkers, J. , Van Lohuizen, M. , Berns, A. , Adams, D.J. , 2009. A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat. Protoc. 4, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utermark, T. , Rao, T. , Cheng, H. , Wang, Q. , Lee, S.H. , Wang, Z.C. , Iglehart, J.D. , Roberts, T.M. , Muller, W.J. , Zhao, J.J. , 2012. The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 26, 1573–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck, B. , Guillermet-Guibert, J. , Graupera, M. , Bilanges, B. , 2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell. Biol. 11, 329–341. [DOI] [PubMed] [Google Scholar]
- Wall, M. , Poortinga, G. , Stanley, K.L. , Lindemann, R.K. , Bots, M. , Chan, C.J. , Bywater, M.J. , Kinross, K.M. , Astle, M.V. , Waldeck, K. , Hannan, K.M. , Shortt, J. , Smyth, M.J. , Lowe, S.W. , Hannan, R.D. , Pearson, R.B. , Johnstone, R.W. , McArthur, G.A. , 2013. The mTORC1 inhibitor everolimus prevents and treats Eμ-myc lymphoma by Restoring oncogene-induced Senescence. Cancer Discov. 3, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin, J.J. , Edgar, K.A. , Guan, J. , Berry, M. , Prior, W.W. , Lee, L. , Lesnick, J.D. , Lewis, C. , Nonomiya, J. , Pang, J. , Salphati, L. , Olivero, A.G. , Sutherlin, D.P. , O'Brien, C. , Spoerke, J.M. , Patel, S. , Lensun, L. , Kassees, R. , Ross, L. , Lackner, M.R. , Sampath, D. , Belvin, M. , Friedman, L.S. , 2011. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol. Cancer Ther. 10, 2426–2436. [DOI] [PubMed] [Google Scholar]
- Walrath, J.C. , Hawes, J.J. , Van Dyke, T. , Reilly, K.M. , 2010. Genetically engineered mouse models in cancer research. Adv. Cancer Res. 106, 113–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, J. , Wallace, R.M. , Clark, A.J. , Whitelaw, C.B. , 1995. Tissue-specific, temporally regulated expression mediated by the proximal ovine beta-lactoglobulin promoter in transgenic mice. Cell. Mol. Biol. Res. 41, 11–15. [PubMed] [Google Scholar]
- Wee, S. , Wiederschain, D. , Maira, S.-M. , Loo, A. , Miller, C. , deBeaumont, R. , Stegmeier, F. , Yao, Y.-M. , Lengauer, C. , 2008. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U. S. A. 105, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, H.-G. , De Stanchina, E. , Fridman, J.S. , Malina, A. , Ray, S. , Kogan, S. , Cordon-Cardo, C. , Pelletier, J. , Lowe, S.W. , 2004. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428, 332–337. [DOI] [PubMed] [Google Scholar]
- Wendel, H.-G. , Malina, A. , Zhao, Z. , Zender, L. , Kogan, S.C. , Cordon-Cardo, C. , Pelletier, J. , Lowe, S.W. , 2006. Determinants of sensitivity and resistance to rapamycin-chemotherapy drug combinations in vivo. Cancer Res. 66, 7639–7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, T.A. , Sandhu, S.K. , Carden, C.P. , De Bono, J.S. , 2011. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 61, 31–49. [DOI] [PubMed] [Google Scholar]
- Young, C.D. , Nolte, E.C. , Lewis, A. , Serkova, N.J. , Anderson, S.M. , 2008. Activated Akt1 accelerates MMTV-c-ErbB2 mammary tumourigenesis in mice without activation of ErbB3. Breast Cancer Res. 10, R70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, K. , Toral-Barza, L. , Shi, C. , Zhang, W.-G. , Lucas, J. , Shor, B. , Kim, J. , Verheijen, J. , Curran, K. , Malwitz, D.J. , Cole, D.C. , Ellingboe, J. , Ayral-Kaloustian, S. , Mansour, T.S. , Gibbons, J.J. , Abraham, R.T. , Nowak, P. , Zask, A. , 2009. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 69, 6232–6240. [DOI] [PubMed] [Google Scholar]
- Yuan, W. , Stawiski, E. , Janakiraman, V. , Chan, E. , Durinck, S. , Edgar, K.A. , Kljavin, N.M. , Rivers, C.S. , Gnad, F. , Roose-Girma, M. , Haverty, P.M. , Fedorowicz, G. , Heldens, S. , Soriano, R.H. , Zhang, Z. , Wallin, J.J. , Johnson, L. , Merchant, M. , Modrusan, Z. , Stern, H.M. , Seshagiri, S. , 2013. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene 32, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagouri, F. , Sergentanis, T.N. , Chrysikos, D. , Filipits, M. , Bartsch, R. , 2012. mTOR inhibitors in breast cancer: a systematic review. Gynecol. Oncol 127, 662–672. [DOI] [PubMed] [Google Scholar]
- Zardavas, D. , Fumagalli, D. , Loi, S. , 2012. Phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway inhibition: a breakthrough in the management of luminal (ER+/HER2-) breast cancers?. Curr. Opin. Oncol. 24, 623–634. [DOI] [PubMed] [Google Scholar]
- Zhao, L. , Vogt, P.K. , 2010. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle 9, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Zhang, X. , Liu, Y. , Zhang, S. , Liu, J. , Ma, Y. , Zhang, J. , 2012. Antitumor effect of the mTOR inhibitor everolimus in combination with trastuzumab on human breast cancer stem cells in vitro and in vivo. Tumour Biol. 33, 1349–1362. [DOI] [PubMed] [Google Scholar]