

Abstract
Colorectal cancer (CRC) is the third most common cancer in the UK, with over 37,500 people being diagnosed every year. Survival rates for CRC have doubled in the last 30 years and it is now curable if diagnosed early, but still over half of all sufferers do not survive for longer than 5 years after diagnosis. The major complication to treating this disease is that of metastasis, specifically to the liver, which is associated with a 5 year survival of less than 5%. These statistics highlight the importance of the development of earlier detection techniques and more targeted therapeutics. The future of treating this disease therefore lies in increasing understanding of the mutations which cause tumourigenesis, and insight into the development and progression of this complex disease. This can only be achieved through the use of functional models which recapitulate all aspects of the human disease. There is a wide range of models of CRC available to researchers, but all have their own strengths and weaknesses. Here we review how CRC can be modelled and discuss the future of modelling this complex disease, with a particular focus on how genetically engineered mouse models have revolutionised this area of research.
Keywords: Colorectal cancer, Mouse models, Genetic modification
Highlights
-
►
The complexity of colorectal cancer requires a range of different models in order to develop potential therapeutics.
-
►
There are many models available, each with their own advantages and disadvantages.
-
►
Mouse models are a particularly potent model of colorectal cancer due to their genetic tractability.
-
►
A combination of approaches is necessary when modelling this disease.
1. Colorectal cancer: pathways and progression
The development of sporadic CRC is a multistep process in which an accumulation of genetic mutations leads to the progression from normal intestinal epithelium to dysplastic tissue to benign adenoma through to metastatic carcinoma. Studies comparing human tumour tissue to normal tissue have highlighted many key mutations which are commonly involved in colorectal tumourigenesis, discussed below, and this knowledge has been pivotal to the development of many of the available models of CRC.
Based on studies of the frequency of gene mutations at various stages of progression in human tumours, Fearon and Vogelstein (1990) proposed a model whereby loss of function of Adenomatous polyposis coli (APC) initiates the formation of a benign lesion, followed by an activating mutation in KRAS, allelic loss of the 18q locus and mutation of p53, which all contribute to the progression to malignant disease. This model has now been further explored in order to ascertain the importance of timing of mutations on tumour progression, and found that interestingly, it is surprisingly rare for KRAS and p53 mutations to co‐exist within a tumour, hinting at the complexity of the nature of colorectal tumourigenesis as they drive different pathways to tumour progression (Smith et al., 2002).
The technique of deep sequencing has enabled the identification of a range of drivers of colorectal tumourigenesis, leading to further refinement of the Fearon–Vogelstein model. In order to identify common mutations and drivers of CRC, deep sequencing has been used to uncover genes altered between normal and tumour tissue in a small number of colorectal cancers. Once a number of potential tumour driving mutations was identified, the genetic status of these genes could then be analysed in a much larger number of samples (Wood et al., 2007). Despite the clear identification of common loss of function mutations in APC, p53 and activating mutations in KRAS, the authors present the idea that a range of other less common mutations contributes to the tumour phenotype by providing a small fitness advantage to the tumour via disruption of a few key pathways.
APC is a tumour suppressor which functions by negatively regulating the Wnt‐signalling pathway. Mutations in APC result in constitutive activation of the canonical‐Wnt‐signalling pathway leading to a build up of nuclear β‐catenin and have been found in more than 80% of all sporadic human CRC tumours. Nuclear β‐catenin functions as a transcription factor for a range of Wnt‐target genes such as C‐myc and Lef1. Wnt‐target genes have a variety of functions, but most play some role in the control of essential homeostatic processes such as mitosis, apoptosis and migration, mis‐regulation of which can result in cancer.
More than 5% of all cases of CRC are due to inherited disorders, most commonly Familial Adenomatous Polyposis (FAP) and Hereditary Non‐Polyposis Colorectal Cancer (HNPCC). These familial forms of CRC can teach us a great deal about the genes involved in tumourigenesis and it is interesting to note that sufferers of Familial Adenomatous Polyposis (FAP) carry a heterozygous mutation for APC and a significant number of colorectal tumours from HNPCC patients display somatic mutations of APC highlighting the importance of APC as a tumour suppressor within the colon.
In order to understand the complex nature of CRC, including how it develops and how it should be treated, it is essential to use models which recapitulate the human disease as closely as possible. CRC patients present most frequently with few large tumours located in the large intestine, which are capable of invasion and metastasis. Modelling of CRC must therefore take these criteria into account.
2. Different approaches to modelling CRC
Patients present with CRC at a range of stages of the disease, and the combination of mutations which can promote the disease is huge, which make it a difficult disease to model accurately. In order to develop therapeutics for this complex disease it is necessary to understand which mutations are key to tumour progression, why some tumours respond better to certain therapies than others and the cellular processes which are necessary for disease progression which could be potential drug targets.
2.1. Human cell lines
Cell lines established from tumour tissue derived from patients with CRC are widely used to model the disease, and there are a number of established cell lines which are readily available for this purpose. As these cell lines are obtained from human patients, they are directly relevant to human disease and cell culture techniques enable high throughput experiments which are quick and relatively inexpensive. This makes cell culture an attractive model system for the disease, and are therefore widely used.
By mimicking tumour behaviour in culture, certain characteristics of the tumour cells can be identified, such as properties which represent the cells ability to aggregate, migrate and invade. These properties can be quantified and any changes assessed when the cells are exposed to a range of therapeutics. For example, a range of CRC cell lines (HCT‐116 p53 wild type, HT‐29, HCT‐116 p53−/− and SW‐620 cells) was used by Nautiyal et al. (2010) in order to assess the combined effects of curcumin and dasatinib on the ability of the cells to form colonies, invade through an extracellular matrix and the formation of tubules by endothelial cells. These readouts are commonly used as an indicator of metastatic capabilities, and in this study the enhanced inhibition of “metastasis” when the therapies were used in combination was confirmed in an in vivo model. This study not only highlights the utility of cell lines as a model for CRC, but also addresses the downfalls of the system by comparing a range of different cell lines and supporting the data with an in vivo model.
One major disadvantage of using cell lines is that the important interaction of the tumour cells with their environment is missing. The artificial environment in which these cells are maintained can not only radically alter the epigenetics of the tumour, but also many of the complex 3‐dimensional interactions between cells are lost. For example, by culturing cancer cells in a 2‐dimensional plane, the Oxygen levels between cells are kept relatively equal, which may be an unrealistic scenario within a growing tumour. The major limitation of cell culture is its inability to model the effects of organism response to a tumour, such as immune response and angiogenesis, two factors which are known to exert a large influence on the tumour development. Despite these drawbacks, culturing of a variety of CRC cell lines plays an important part in extending knowledge of genetic involved in CRC and the response of tumour cells to drugs and very frequently enables the elucidation of a mechanism of a therapy which is known to work in vivo. However, ideally, CRC needs to be modelled in situ in order to best represent the tumour environment and behaviour.
2.2. Xenografts
One attempt to overcome the limitations of cell culture is through the use of xenografts. For over thirty years, xenografts, whereby murine or human tumours are implanted into recipient mice, have been used to generate models of human CRC progression and metastasis, to great effect. Tumour cell lines or cell suspensions can be injected subcutaneously, directly into the colon or cecum, into the portal vein or via intraperitoneal injection. Cultured cells can be injected subcutaneously or surgically implanted into the site of interest. In order to prevent rejection of the tumour, recipient mice are either nude (which cannot generate T lymphocytes) or have severe combined immune deficiency (SCID). SCID mice have an altered adaptive immunity, but retain normal innate immunity. The tumour will develop within the mouse and enable the study of tumour progression in response to a specific therapeutic regime.
This method has been used to great effect when studying CRC. For example, Williams et al. (2002) used a model whereby LoVo human CRC cells were intradermally injected into nude mice in order to show an increase in tumour growth inhibition by radiotherapy treatment in combination with “Iressa”, a specific oral epidermal growth factor receptor‐tyrosine kinase inhibitor. This is a direct example of how xenograft models can be used to aid therapy development.
Orthotopic injection of cancer cells into a host mouse is another useful approach to model tumour invasion and metastasis. The rates of tumour development metastasis are highly variable and are dependent on a range of factors such as tumour cell line and site of implantation. Recently, Hackl et al. (2012) investigated the most clinically relevant method of performing xenografts from human cells lines and found that injection of the cancer cells into the caecal wall of SCID mice along with human chorionic gonadotropin had a high efficiency of tumour formation. Importantly, this method resulted in numbers of liver metastases which were comparable to the human disease.
The main disadvantages with this method are that although the tumours are growing in a more biologically relevant microenvironment than can be achieved in vitro, the mice are immunocompromised and so many of the complex interactions between tumour and host may be lost. Another, often overlooked, disadvantage of this method is the genetic and epigenetic changes which may occur in the tumour cells during resection, culture and implantation.
2.3. Primary xenografts
In order to overcome the issue of epigenetic and genetic changes synonymous with long term culture of cells, many researchers have been refining this technique to reduce the stages between tumour and xenograft. By performing primary xenografts whereby solid tumour or disassociated tumour cells are directly implanted into a host mouse without long periods of cell culture, many of these pitfalls can be avoided, this method however has its own limitations.
Previous attempts to develop a model of CRC which permits invasion and metastasis in a more reproducible way have been based on the artificial selection of the most aggressive tumour cells through serial xenografts. For example, it was found that serial orthotopic inoculation of cells derived from a human CRC liver metastasis into the colon of nude mice resulted in the production of an ultra‐metastatic tumour. Inoculation of these tumour cells consistently resulted in the production of liver metastasis, as well as metastasis into spleen and lymph (Sun et al., 1999). This is an important model in the study of highly metastatic human CRC and was the first effective method of reproducing the later stages of CRC, however the artificial selection of the most metastatic tumours within host mice resulted in a tumour very different to the primary tumour, both phenotypically and genotypically. The first 10 mice which were inoculated with the CRC liver metastasis cells did not show signs of disease metastasis themselves, but by the 10th passage, all mice presented clear metastasis and the tumour was deemed “ultra‐metastatic”. This change from the original tumour indicates that the tumour cells have accumulated features which make them less functionally relevant than freshly isolated tumour cells.
However, primary xenografts have been used to a great deal of success, and are exceptionally useful as a model for CRC. By subrenally implanting a capsule of disassociated human colon tumour cells into SCID mice, 17 out of 17 experimental mice developed tumours, with many showing liver metastasis (O'Brien et al., 2006). Importantly the histology and expression patterns of the xenografts accurately recapitulated that which was seen in the human tumour from which it originated, and so is a great advantage over the injection of cancer cell lines.
Over recent years there has been a drive to identify “cancer stem cells”, based on the idea that cancer cells are hierarchical, with only a small population of them capable of initiating tumour formation. The majority of this work is based on primary xenografts, whereby a human colorectal tumour or liver metastasis is disassociated and the cells separated into subpopulations of tumour cells according to their gene expression patterns. The different populations are orthotopically injected into immunocompromised mice and their tumour‐initiating efficiency analysed, with the idea that only populations of cells which can form tumours by xenograft contain “cancer stem cells”. This method has now identified populations of CRC initiating cells as expressing the gene CD44 (O'Brien et al., 2006), and has been used to increase our understanding of the expression profile of tumours which leads to metastasis, a subpopulation of CD44+ cells which express CD26, are associated with liver metastasis, and expression of CD26 in the primary tumour indicates an increased likelihood future metastasis (Pang et al., 2010).
Despite the accuracy of this model in mimicking the human disease there are still a number of drawbacks. For example, this method heavily relies on the availability of direct human samples, which is not feasible for many researchers and large scale studies requiring multiple samples can be difficult to achieve. By passaging the tumours through mice, this obstacle can be overcome, but as shown by Sun et al., the accumulation of further mutations in and between mice results in a less functionally relevant xenograft. As well as these logistical problems it is still important to note that it is still necessary for the host mice to be immunocompromised, so the model is not capable of reflecting the immunological component of the tumour/stromal interaction.
3. Genetically engineered mouse models
The advent of gene targeting has enabled a more controlled approach to producing models of colorectal cancer. Genetically engineered mice are an extremely advantageous model of CRC due to the availability of genetic and genomic information, the ease of genetic manipulation through mutagenesis techniques and the ability to monitor the effects on a whole organism. These advantages make mouse models indispensable not only when investigating the mechanisms of CRC and the genes involved in initiation and development, but also when testing therapeutics, enabling efficacy and toxicity of the treatments to be analysed but have their own limitations which will also be discussed here.
3.1. Models of HNPCC
Hereditary non‐polyposis colorectal cancer (HNPCC) is the most common cause of hereditary colorectal cancer. It is an autosomal dominant disorder which results in early‐onset tumours in the colon and rectum, with a small subset of patients also developing tumours in the stomach and small intestine. This disorder is due to mutations in the mismatch repair genes MLH1, MSH2 and MSH6, which causes microsatellite instability (MSI), characterised by increased rates of replication errors (Anwar et al., 2000). A mouse model was developed to help understand this disorder, which models the genetic scenario in humans by heterozygous deletions of these genes, but interestingly not develop early‐onset tumours. This is thought to be due to the short lifespan of mice. Homozygous knockout mice are cancer prone and develop tumours in multiple organs, and although they do develop gastrointestinal tumours, the cause of death is aggressive lymphoma (Prolla et al., 1998).
By combining homozygous mutations for the mismatch repair genes with various germ‐line APC mutations, much more accurate models of HNPCC have been developed which display a phenotype of multiple early‐onset intestinal tumours (Reitmair et al., 1996; Edelmann et al., 1999; Kuraguchi et al., 2001).
Biallelic germ‐line mutations in the MUTYH gene which expressed a DNA glycosylase responsible for base excision repair have been found in patients predisposed to a recessive form of hereditary multiple colorectal adenoma and carcinoma. These patients have an increased likelihood of G:C and T:A somatic mutations in the APC gene due to oxidative stress. In order to model this disease a Mutyh −/− mouse strain was developed and their response to oxidative stress in the form of exposure to dextran sulphate sodium (DSS) was assessed (Casorelli et al., 2010). It was found that loss of Mutyh results in higher susceptibility to chronic colitis due to oxidative stress, and that this appeared to be due to an inflammatory response. By providing a strong model for the disease this mouse model enabled the realisation of the importance of MUTYH in supporting intestinal maintenance by controlling the inflammatory response.
3.2. Models of FAP
The most widely used mouse models of CRC are derived from models of FAP. FAP is a dominantly inherited disorder which is characterised by hundreds of adenomas in the colon. These adenomas progress and by the age of 40 they have usually developed into CRC with a high risk of metastasis. A mouse termed the Min or Apc min/+ is widely used as model for FAP. This mouse model was generated by random mutagenesis using N‐ethyl‐N‐nitrosourea (ENU). Apc min/+ mice develop numerous benign adenomas in the small intestine, with the addition of a small number of polyps in the colon. The causative mutation of this model was found to be an autosomal dominant mutation in Apc; a transversion at codon 850, causing premature truncation (Moser et al., 1990, 1995). The Apc min/+ mouse model is heterozygous for this mutation and tumourigenesis requires loss of heterozygosity (LOH), a direct mimic of the human disorder.
The Apc min/+ mouse model has phenotypic and genetic similarities to FAP patients, making it an ideal model to investigate intestinal neoplasia and colon cancer. Unfortunately this model does not completely recapitulate the FAP phenotype, an example being that FAP patients develop adenomas predominately in the colon, whereas adenomas in Apc min/+ mice are predominantly localised to the small intestine. Another limitation of the Apc min/+ mouse model is that the adenomas are generally benign and do not progress to invasive colon cancer. This may be due to the short lifespan of the mice, meaning that the adenomas do not have sufficient time to accrue the necessary additional mutations required to progress any further.
The Apc min/+ mouse model has proved itself countless times as an indispensable tool in research into CRC, not only simply within the study of FAP but also within sporadic CRC in which loss of heterozygosity of APC is an early initiating stage. Using the Apc min/+ mouse it has been possible to test the effects of a range of therapeutics and environmental influences in a genotypically relevant model in which the response of the whole organism to both tumour growth and therapy can be assessed. The utility of this mouse model has resulted in direct patient benefit due to the wide variety of drug studies for which it has been used. For example, the Apc min/+ mouse model has been used to test the efficacy of a range of Non‐steroidal anti‐inflammatory drugs (NSAIDs) such as R‐flurbiprofen, Sulindac, Aspirin and Piroxicam (Jacoby et al., 1996; Wechter et al., 2000; Orner et al., 2003). These experiments showed that the utility of NSAIDs is based on their ability to inhibit COX activity. The efficacy of variety of dietary supplements has also been tested in the context of Apc min/+ mice with many, such as white tea, fish oil and caffeine showing an ability to reduce both tumour onset and burden (Corpet, 2012).
3.3. Refining the APC heterozygous model
Once the importance of APC inactivation in intestinal tumourigenesis had been established, further studies were conducted in order to probe the effect of different APC mutations and a summary of some of these mutations and their phenotypes can be found in Figure 1. This has been an important development as both FAP and sporadic patients present with a wide variety of different APC mutations, which can affect both the phenotype and the prognosis. Studies show that mouse models containing different mutations in the Apc gene differ by the number of polyps in the small intestine and levels of invasion. For example, a mouse model heterozygous for a truncation mutation in Apc at codon 716 (Apc Δ716/+) contain 10 times the number of polyps in the small intestine compared to the Apc min/+ mouse model (Oshima et al., 1995), whereas the insertion of a neomycin cassette into exon 15 (Apc 1638N) results in fewer polyps but with a marked increase in invasion (Fodde et al., 1994).
Figure 1.
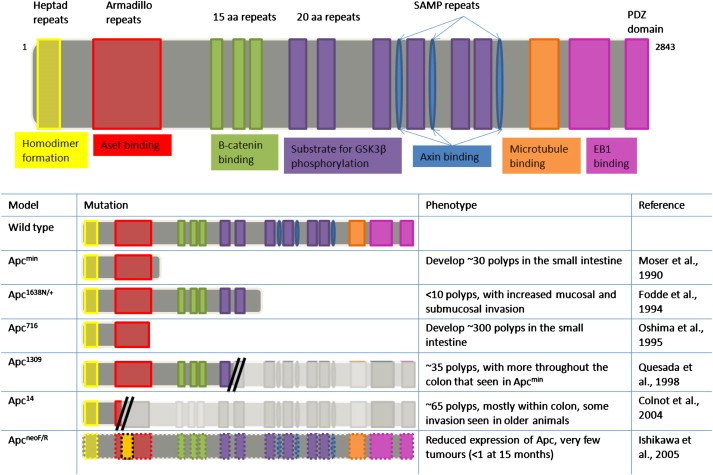
Diagrammatic representation of 6 of the most commonly used Apc mutations available, with a brief description of the phenotype.
Heterozygous Apc mutations have not only increased the range of research models available for study of the disease, but also each model has lead to insight into the relationship between APC mutation type, and patient prognosis. An example of this is the mouse model heterozygous for a truncation mutation in the Apc gene at codon 1309 (Apc 1309/+). This mouse model has a more severe phenotype than the Apc min/+ mouse model, including a shorter lifespan and an increase in polyps in the colon, and FAP patients with this mutation have an earlier onset of the disease and severe polyposis, making their phenotype more severe than patients with other APC mutations (Quesada et al., 1998).
As can be seen from Figure 1 there is a wide range of mutant models, and this range alone has provided insight into the nature of colorectal tumourigenesis. By comparing the phenotypes of the different models and the mutations which cause them, the “just right” hypothesis of Wnt‐signalling in CRC has been proposed. This theory is that levels of Wnt‐signalling between certain thresholds drive tumourigenesis. It has been supported by human data from FAP patients which show that the second “hit” to APC is dependent on the type of inherited mutation, with mutations which result in the retention of at least one β‐catenin down‐regulating domain being selected for as the most successful drivers of tumour clonality (Albuquerque et al., 2002).
3.4. Modifiers of Min
FAP patients with the same APC mutations present with a range of vastly different phenotypes, largely due to the combination of environment and genetic modifiers. Understanding the types of modifiers which interact with APC mutations and the effect that they cause on tumourigenesis can aid research in a number of significant ways. Insight into modifiers of the Min phenotype can improve our knowledge of tumourigenesis and help to increase the range of possible targets for therapy. In addition to these important factors, exploiting these modifiers enables the alteration of the Min phenotype towards a more patient relevant model. A number of these modifiers is shown in Table 1, and here we discuss some which have had the largest impact on understanding and treatment of CRC.
Table 1.
Selected examples of Modifiers of Min, with a brief description of the effect on tumour burden, location and invasion of loss of the modifiers on the Min phenotype.
| Modifier of Min | Alteration to phenotype | References |
|---|---|---|
| Mom‐1 | Mutations in Mom‐1 result in a longer lifespan and lower intestinal tumour burden in Min mice | (Moser et al., 1992) |
| Mom‐2 | Dominant mutations in Mom‐2 result in a reduction of polyp number, to a greater extent than Mom‐1 | (Silverman et al., 2002) |
| Mom‐3 | Mom‐3 mutations result in a broad range of polyp number and survival Pregnancy influences the phenotype | (Haines et al., 2005; Suraweera et al., 2006) |
| Mom‐5 | Mutations in Mom‐5 result in lower numbers of intestinal tumours | (Oikarinen et al., 2009) |
| Mom‐7 | Mutations in Mom7 cause an increase in polyps in the small intestine | (Kwong et al., 2007) |
| Foxl‐1 | Loss of Foxl‐1 modifies the Min phenotype by causing an increase in the number of colonic polyps as well as stomach tumours | (Perreault et al., 2005) |
| Ppar‐δ | Loss of Ppar‐δ in Min mice results in larger polyp size and reported to have a larger number of colonic polyps | (Harman et al., 2004; Reed et al., 2004) |
| Cox‐1/Cox‐2 | The additional loss of either Cox‐1 or Cox‐2 to an Apcmin/+ mouse results in a significantly reduced tumour burden | (Chulada et al., 2000) |
| Mbd2 | Loss of Mbd2 results in fewer, smaller adenomas and increases survival in Min mice | (Phesse et al., 2008) |
| Mbd4 | Loss of Mbd4 in Min mice results in a reduced survival due to an increased tumour burden | (Millar et al., 2002) |
| Prox‐1 | Loss of Prox‐1 results in a reduction in adenoma size and number in Min mice | (Petrova et al., 2008) |
| TGF‐β | Apc 1638N/wt mice that do not express TGF‐β results in an increase in more advanced and high grade lesions in the small intestine | (Muñoz et al., 2006) |
| Smad2 | Inactivating mutations in APC and Smad2 results in intestinal adenomas that invade into the stroma and vascular culture | (Hamamoto et al., 2002) |
| Smad3 | Loss of Smad3 in Min mice results in an increase in adenoma number, especially in the distal colon | (Sodir et al., 2006) |
| Smad | Mice containing inactivation of APC and Smad4 results in the presence of adenomas in the small intestine that invade into the stroma | (Takaku et al., 1998) |
| EphB2 | Loss of EphB2 activity in Min mice results in the presence of invasive intestinal adenomas but a reduction in the number of adenomas | (Batlle et al., 2005) |
| EphB3 | Loss of EphB3 in Min mice results in invasive intestinal adenomas and an increase in the size and number of colonic polyps | (Batlle et al., 2005) |
| EphB4 | Heterozygosity for EphB4 in Min mice results in larger intestinal adenomas and an increase in the number colonic polyps | (Dopeso et al., 2009) |
| p27 | Deficiency of p27 in Min mice results in an increase in adenoma number | (Philipp‐Staheli et al., 2002) |
| ERα | Heterozygous deficiency for Erα results in an increase in the number of colon tumours and the presence of invasive carcinomas | (Cho et al., 2007) |
Genes that play important roles in tumour initiation can be established using microarrays from Apc deficient mouse models. It was through such a study that the importance of the COX‐2 (PTGS2) gene was determined. COX‐2 was shown to be expressed from an early stage of polyp formation (Oshima et al., 1996). Cox‐2 knockout mice crossed with Apc Δ716/+ mice resulted in a drastic reduction in adenoma formation, and Apc Δ716/+ mice treated with chemical inhibitors of Cox‐2 had the same effect (Oshima et al., 1996). The Apc min/+ Cox‐2 −/− mouse model also caused a reduction in the number of intestinal polyps (Chulada et al., 2000). Celecoxib is a selective inhibitor of COX‐2. When administrated to Apc min/+ mice this drug caused a reduction in adenomas and polyps in the small intestine and the colon, and higher doses caused a reduction in adenoma size (Jacoby et al., 2000). The mice also had no apparent toxicity or loss in body weight. These promising results eventually lead to clinical trials where celecoxib was used to treat FAP patients. In one study celecoxib caused the reduction of colonic polyps in FAP patients, and the drug was well tolerated (Steinbach et al., 2000), however more recent studies have shown cardiovascular side effects (Solomon et al., 2005). This and other clinical trials have shown that the effects shown in the Apc min/+ mouse model are frequently also true for humans, highlighting the importance and effectiveness of mouse models.
One of the disadvantages of the Apc heterozygous mouse models is that the majority of adenomas is benign, as Wnt‐signalling deregulation is thought to be an important initiator of tumorigenesis but in these models is not sufficient to drive tumour progression. By artificially causing the mis‐regulation of other signalling pathways known to be involved in tumourigenesis it is possible to promote malignant transformation. The transforming growth factor‐β (TGF‐β) signalling pathway regulates cell processes such as proliferation, differentiation, motility and apoptosis, and therefore plays a central role in tissue homeostasis and development. The TGF‐β signalling pathway is commonly inactivated in colon cancer, with approximately 30% of colon cancers carrying mutations in the TGF‐β receptor II (a subunit of the TGF‐β receptor) (Bellam and Pasche, 2010). Interestingly, despite the wide acceptance of TGF‐β signalling as a tumour‐suppressing pathway in vitro studies has shown the role of TGF‐β in driving epithelial‐to‐mesenchymal transition, which is an important driver of invasion and metastasis (Miyazono, 2009). In order to fully elucidate the role of TGF‐β signalling within colorectal tumourigenesis, a number of mouse models targeting different stages of the pathway has been developed.
Muñoz et al., used a mouse model which do not express TGF‐β receptor II within the intestinal epithelium and found that these mice develop spontaneous tumours only very infrequently. However, when they crossed onto an Apc 1638N/wt background in order to study the effects of loss of TGF‐β signalling in the context of aberrant Wnt‐signalling the mouse presented with no significant increase in the number of neoplastic intestinal lesions than was seen in the Apc 1638N/wt mouse with functional TGF‐β receptor II, but with a drastically increased proportion of lesion categorised as advanced, high grade and adenocarcinomas (Muñoz et al., 2006). This mouse model has not only increased our understanding of factors which influence colorectal tumour progression, but also has provided researchers with means of studying tumourigenesis in a model which more closely mimics the human disease than was previously possible.
Loss of other components of the TGF‐β signalling pathway, such as Smad proteins, has been shown to have a similar effect in Apc heterozygous models. The loss of Smad proteins in Apc deficient mouse models also results in a higher level of tumour invasion, for example Smad4 mutations in the Apc Δ716/+ mouse model causes adenomas to become locally invasive (Takaku et al., 1998), and a study with the combined inactivation of Apc and Smad4 resulted in adenomas from the small intestine to invade into the stroma, although there was no change in the size or number of adenomas (Hamamoto et al., 2002). A mouse model containing Apc and Smad2 mutations contains adenomas that invade into the stroma and the vascular culture, although there was no difference in adenoma number, size or distribution in the intestine compared controls (Hamamoto et al., 2002). In addition to causing the adenomas to become invasive the Apc min+/− Smad3 −/− mouse model has an increase in adenoma number compared to the Apc min+/− mouse model. These adenomas were also predominantly located in the distal colon (Sodir et al., 2006). These mouse models support the importance of disrupted TGF‐β signalling in tumour invasion and progression, but not initiation.
Another disadvantage of the Apc min+/− mouse model is that the adenomas are predominantly located in the small intestine. Interestingly, a mouse model which more accurately recapitulates the human disease by presenting more invasive adenomas alongside a higher number of colonic polyps is the result of a deficiency for the Wnt‐target genes EphB receptors. The Apc min+/−, EphB4 +/− mouse model has larger adenomas in the small intestine as well as a higher number of polyps in the colon, compared to Apc min+/− mice (Dopeso et al., 2009). The loss of EphB3 in the Apc min+/− mouse model results in more invasive adenomas, as well as a higher number, and larger, polyps in the colon (Batlle et al., 2005). It is now thought that the loss of EphB3 activity in human colorectal cancer is a critical step in progression. In contrast the loss of EphB2 activity in the Apc min+/− mouse model results in a reduction of adenomas in the small intestine, though adenomas that were present were invasive (Batlle et al., 2005). The ability to genetically alter this family of receptors individually has enabled the sensitive dissection of the roles of members of this family within tumourigenesis.
As missense mutations of the important tumour suppressor p53 occur at a much higher frequency in more advanced human colorectal carcinomas (50–60%) than in colorectal adenomas (<10%) it has been proposed that p53 mutations are of great importance in colorectal tumour progression. It was thought that a model in which APCmin mice were crossed with p53−/− mice would result in no change to number of intestinal lesions but a higher level of invasion, however, the addition of p53 loss did not improve the utility of the Min model as was anticipated (Clarke et al., 1995).
3.5. Models of sporadic colorectal cancer
Sporadic CRC is the result of a build up of mutations all of which contribute to a tumourigenic phenotype with loss of heterozygosity (LOH) for APC being an early tumour‐initiating event (Gryfe et al., 1997). In order to understand the early stages of tumourigenesis, it is necessary to have temporal control over the introduction of initiating mutations. Modern genetic engineering techniques make both spatial and temporal control possible, and have provided an invaluable resource in the modelling of CRC.
4. Conditional genetic models of colorectal cancer
Cre‐lox technology is one of the most frequently used methods by which to conditionally inactivate genes of interest. This is achieved by producing genetically engineered mice which have essential exons of the gene of interest flanked by “LoxP” sites. Cre‐recombinase (Cre) will recombine between these two LoxP sites, cutting out the essential exons and thereby preventing expression of a functional gene product. The Cre can be linked to a promoter which is either tissue specific, or development dependent. This overcomes the issue of homozygous embryonic lethal mutations. Furthermore, by linking the Cre to an oestrogen‐receptor, expression of the Cre can be tissue specific, but unable to cause recombination between LoxP sites until induction with tamoxifen, which will remove the oestrogen‐receptor from the Cre enabling recombination to occur.
A number of different Cre‐recombinases and LoxP flanked genes has now been created and the wide variety of combinations of the two to produce an almost endless possibility of different DNA disruptions is proving invaluable in modelling CRC, as it enables researchers to have full spatial and temporal control over DNA mutations. Here a number of the commonly used methods has been reviewed, as well as specific examples of how this technology has been used to produce more accurate models of human disease.
4.1. APCflox/flox
One of the major impacts on patient prognosis is time of diagnosis, as most patients do not experience symptoms until the CRC is at a relatively late stage. In order to overcome this it is essential to model the very earliest stages of tumourigenesis. As loss of APC is well established as an early initiator of CRC, modelling the conditional loss of APC at a known timepoint enables the study of the early stage phenotype.
By flanking essential exons of the Apc gene with LoxP sites, a conditional mouse models for CRC was developed (Shibata et al., 1997), which was originally investigated by induction using adenovirus Cre to flush the rectum, which results in recombination within any exposed cells and resulted in the rapid development of colorectal adenomas. The APCflox/flox mouse has now been studied using a variety of different Cre‐recombinases resulting in a broad range of phenotypes. Using Ah‐Cre, in which expression of Cre‐recombinase is driven by the Cyp1a1 promoter and can be induced by β‐napthoflavone injection, enables the conditional loss of both alleles of Apc within the intestinal epithelium. This method was used to show that functional Apc is essential to the maintenance of normal intestinal homeostasis, and its loss results in immediately deregulated tissue morphology due to changes in migration, differentiation, proliferation and apoptosis (Sansom et al., 2004). These changes are due to the high levels of expression of Wnt‐target genes such as C‐myc.
This work was repeated using Villin‐Cre, which is induced by tamoxifen injection and also results in recombination within the intestinal epithelium (El Marjou et al., 2004). Using this Cre to cause homozygous deletion of APC resulted in a very similar phenotype as seen with Ah‐Cre, and the extreme tissue disruption resulted in the necessity of termination of the mice by day4 post induction (Andreu et al., 2005)
Interestingly, both of these models of homozygous deletion of APC resulted in an increased number of undifferentiated cells within the intestine, indicating the potential importance of the relationship between intestinal stem cells and cancer, and has been further explored through the use of an intestinal stem cell (ISC) specific Cre to delete APC within the ISC population.
ISCs are found at the base of the crypt where they are responsible for enabling the high rate of cell turnover required by the intestine. ISCs are capable of repopulating the entire crypt via production of all of the different intestinal epithelial lineages. The ISCs are identifiable by expression of the receptor protein Lgr5 (Barker et al., 2007). By driving expression of Cre using the Lgr5 floxed genes can be deleted specifically within the ISC population. Barker et al. (2008) used this method to conditionally delete APC within ISCs and showed them to be the cells of origin of CRC. Lgr5‐Cre driven deletion of APC in mice results in tumour formation at around 4–5 weeks, so that early stages of tumourigenesis can be studied more precisely than is possible using Villin or Ah‐Cre. It was through the use of Lgr5‐Cre and APCflox/flox that it was shown that ISCs are the cells of origin of CRC, which has altered the way researchers view tumourigenesis and tumour homeostasis.
Conditional mutagenesis has also enabled modelling the effects of loss of the Wnt‐target gene C‐myc on intestinal tumourigenesis. Loss of C‐myc suppresses virtually all phenotypes which are associated with Wnt‐activation due to loss of Apc (Sansom et al., 2007). This has increased the understanding of how loss of APC promotes tumourigenesis, as it shows that it is an entirely canonical‐Wnt‐dependent process.
4.2. APCflox/+
Homozygous deletion of APC under the control of Villin and Ah‐expressed Cre‐recombinases causes such a severe phenotype that although they are useful in understanding the tissue dysregulation that happens at the moment of LOH seen in humans at the earliest stages of tumourigenesis, the mice have a very short survival and so do no develop tumours. One solution to this is to use Cre‐recombinases which recombine in fewer cells, such as the Lgr5‐Cre, while another is to use Cre‐lox technology to heterozygously delete APC in order to study the effect of other genes which play a role in tumourigenesis in an APC deficient setting.
This method has been used to study the effects of the loss of the important tumour suppressor PTEN. Loss of PTEN results in increased levels of PIP3 which recruits AKT to the membrane, thereby increasing the levels of activated AKT. This effectively models the effects of constitutive PI3K pathway activation which is seen in 40% of human CRC tumours (Parsons et al., 2005). Studies have also linked loss of PTEN expression to poor clinical outcome of CRC patients (Li et al., 2009; Jang et al., 2010). Using Cre‐Lox technology Marsh et al. (2008) showed that Pten loss has no effect on the homeostasis of normal intestine, but does cause accelerated tumourigenesis when coupled with deficiency of Apc.
Another method of modelling Wnt‐dependent CRC is via the expression of a mutant form of β‐catenin which is far more stable in the cell and therefore resistant to proteasomal degradation. Mutations which result in a stabilised form of β‐catenin are found in a subset of human colon tumours which do not carry the APC mutation. When modelled in the mouse, conditional stabilisation of β‐catenin results in the formation of multiple tumours. Interestingly, the work was originally performed with expression of Cre‐recombinase being driven by the calbindin promoter (which is not expressed in the proliferative zone of the intestine) and resulted in few tumours (Romagnolo et al., 1999). This work was developed and Cre‐expression was then driven by a fatty acid binding protein gene promoter (which is expressed in the proliferative zone) and resulted in many thousands of adenoma (Harada et al., 1999). These mouse models of CRC lead to the idea that oncogenic mutations in cells at the base of the crypt play a more important role in CRC than the differentiated cells along the crypt‐villus axis.
4.3. Making conditional models of CRC more patient relevant
Mutations which result in the constitutive activation of the oncogene KRAS have been found in 40–50% of human CRCs (Bos et al., 1987) and the presence of these mutations is associated with poor patient prognosis (Esteller et al., 2001). Sporadic mutation of KRAS is thought to be an important stage in the progression of the tumour (Fearon and Vogelstein, 1990), and as yet, no mouse model recapitulates this aspect of the disease. In order to mimic this effect mouse models were created in which conditionally express an oncogenic K‐ras allele. Alone, this mutation did not disrupt intestinal homeostasis in the mouse, but in combination with Apc deficiency, it resulted in an increased rate of intestinal tumourigenesis as well as higher levels of invasion (Sansom et al., 2006). This improvement to the APCflox/+ model shows how knowledge of the human disease combined with advanced genetic engineering techniques can be used to produce a relevant and useful mouse model of CRC. The utility of this model is only hindered by the fact that the mice develop renal carcinomas which cause death before the intestinal tumours can fully invade and metastasis.
Location of tumours is also a fundamental issue in modelling CRC in the mouse. The mutations which cause tumourigenesis mainly in the large intestine in humans, result in a high number of tumours in the small intestine in mice. By developing a Cre which preferentially causes recombination within the colon and using it to delete one copy of APC within the mouse it has been possible to produce a more accurate model which results in a higher number of within the distal colon and rectum (Hinoi et al., 2007). This Cre, called CDX2‐NLS, does also cause recombination within the small intestine, but less efficiently than in the large intestine. The CDX2‐NLS APCflox/+ mice had a higher number of invasive carcinomas than seen in their Villin‐Cre APCflox/+ counterparts but no evidence of metastasis or spontaneous KRAS mutations was seen. Interestingly, the most invasive carcinoma seen was shown to have a p53 missense mutations, however this was only seen in 1 tumour in 1 mouse out of a cohort of 36, each with multiple tumours.
5. New approaches and technologies
5.1. Surgical Cre
Failure to produce advanced and metastatic tumours is a problem encountered by many mouse models of CRC due to the short lifespan of mice and models which produce too high a tumour burden for the mouse to survive long enough for the tumours to progress. The major problem with treating patients who present with CRC is addressing the issue of metastasis. The most common location for CRC metastases is the liver, and so the development of treatments requires the use of a model with the same common metastasis formation sites.
One method of accurately modelling sporadic and metastatic colon tumours has been developed by Hung et al. (2010) and involves the surgical application of adenovirus associated Cre specifically to the distal colon. This is achieved by opening the mouse under anaesthetic and clamping the colon 1 and 3 cm from the anus. Small abrasions were then caused in the clamped area using a small caliber brush and adenovirus Cre was injected into the blocked off colon and incubated for 10 min. The clamps were then removed and the animal closed. This procedure has been reported to be well tolerated by animals and so is a viable method for studying intestinal tumourigenesis.
This method enabled the conditional mutation of APC within the exposed tissue in the colon and resulted in isolated distal colon tumours, thereby recapitulating the disease. However, these tumours do not fully model the human disease however, as they do not present with sporadic KRAS mutations. Interestingly, when the authors used the method in mice with floxed APC as well as an inducible activation of KRAS, more advanced tumours were seen, as well as spontaneous gross liver metastases. This model presents a promising option for accurately modelling spontaneous CRC within the mouse.
Using this model of surgically introduced adeno‐Cre the efficacy of the mTOR pathway inhibiting drug rapamycin was able to be tested. Interestingly, colonic tumours as a result of APC mutations alone responded well to the drug with an 80% reduction in tumour size, whereas tumours with both APC and KRAS mutations did not respond. These data increase understanding of how therapy can be personalised depending on the mutations the individual presents with, and so displays the incredible utility of this model in accurately recapitulating the human disease. The only real drawback of this method is the difficulty of the surgical technique, making large cohorts of mice potentially unfeasible.
5.2. Sleeping beauty
Many of the genes currently being studied for their potential relationship with CRC were identified as having altered expression patterns in either human CRC tumours or tumours derived from mouse models. Simply looking at expression patterns within tumours does not enable the differentiation between expression changes/mutations which drive tumourigenesis and those which are simply passengers to the process. In order to fully understand the progressive series of mutations which result in tumourigenesis, it is necessary to start with the very earliest mutations and track which subsequent mutations are common to a high proportion of tumours. This has been done using insertional mutagenesis, such as the sleeping beauty transposon. This transposon inserts at random within the genome and thereby disrupts the function of gene into which it is inserted. By using the sleeping beauty transposon system in APCmin and APCfl/− mice, this model enabled the identification of many new drivers of CRC by comparing common insertion sites (March et al., 2011). The potential use of this model in CRC research is huge, as subsets of tumours with common insertion sites can be used to identify drivers of specific tumour types.
5.3. “Organoid culture”
In 2009 a technique was published by which single murine intestinal stem cells could be grown in culture to produce 3‐dimensional “intestinal organoids” with protruding crypt structures with all the cell types seen in the intestinal crypt in vivo (Sato et al., 2009 ). These cultures can be maintained long term and the organoids are self‐renewing at passage and their growth patterns can be analysed. It is easy to see how this technique will provide a useful model for CRC research, as organoid lines are established from the vast array of mouse models available, and their growth in response to a range of chemotherapeutics can be analysed. This will provide direct insight into the suitability of different therapies in relation to specific mutations in a high throughput and cost‐effective manner. Organoid culture is also now feasible from human tissue samples, from both normal and tumour tissue. This will enable the direct testing of drugs on patient samples in vitro, with potential to be used to design personalised therapy plans. Over the next few years the utility of this emerging method will be more fully established, but the work currently being undertaken to establish these cultures seems promising.
5.4. In silico
With the rapid advances in both computational technology and modelling techniques, it is perhaps unsurprising that mathematical modelling is beginning to play a role within research into colorectal tumourigenesis. Mathematical modelling has already played an important part in describing and understanding the dynamics in normal intestinal crypts (Van Leeuwen et al., 2006), but is now beginning to be taken more seriously as a method of modelling CRC. The idea that even the most simple of mathematical models can be useful indicates the potential of much more complex models to help understand an aspect of tumourigenesis that is so far undefined. More recently, computational models have been used to help identify a number of different drivers of tumourigenesis in a melanoma setting using previously collected data (Akavia et al., 2010), a method which could easily be applied to CRC in order to identify important potential therapeutic targets. Using computational techniques to model a biological process as complex as colorectal tumourigenesis will always have its drawbacks as the models will only ever be as robust as the data fed into them. This means that the predictions that they can produce are limited by the knowledge already available and relies heavily on data collected from in vivo models. However, the ability to take advantage of the plethora of information on CRC currently in the literature to establish previously unknown themes and predictions without a direct requirement for further experimentation is a huge advantage, and it is likely that the techniques currently being established will become a mainstay of CRC research in the future.
6. Conclusions
Despite the inability of a model of CRC to completely recapitulate all stages of the human disease, the use and impact of the available models has been far reaching. By careful selection of an appropriate model, most questions regarding the initiation, progression and development of CRC can now be asked. However, it is still vitally important that researchers carefully consider their choice of model for a specific research theme, as the selection of an inappropriate model can undermine otherwise valuable work. All of the models available have their own strengths and weaknesses, however it is the sheer range of mouse models available that make them the most versatile tool in the researchers' toolkit.
The versatility of genetically engineered mouse models has not only facilitated identification of a wide range of potential therapeutic targets, but also enabled the study of environmental factors, such as diet, on the occurrence and severity of CRC. Mouse models are unique in their ability to recapitulate the effects of subtle environmental factors on genetic predisposition, as has been shown by numerous studies of the effects of dietary supplements in Apc min/+ mice.
The use of both xenografts and APC+/− models is essential for the long term study of therapy efficacy continues to contribute to improved patient care, however, early diagnosis of the disease is still essential. Conditional mouse models of CRC are invaluable in this as they enable the study of the very early stages of tumourigenesis. Importantly, it is essential that researchers take advantage of the broad range of techniques available to them, as the imperfections of each individual model can often be accounted for by supporting data gained using a different model. The extensive utility of mouse models is clear, but researchers need to embrace the use of computational modelling and bioinformatics in order to increase the impact of the data that they produce.
The broad range of mutations capable of driving colorectal tumourigenesis makes CRC a very diverse disease, and so our biggest advantage is the variety of different models available. With this in mind, the development of targeted therapeutics for the treatment of CRC can only be possible through the use of a combination of the different model platforms discussed here.
Young Madeleine, Ordonez Liliana, Clarke Alan R., (2013), What are the best routes to effectively model human colorectal cancer?, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.006.
References
- Akavia, U.D. , Litvin, O. , 2010. An integrated approach to uncover drivers of cancer. Cell 143, (6) 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque, C. , Breukel, C. , 2002. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the β-catenin signaling cascade. Human Molecular Genetics 11, (13) 1549–1560. [DOI] [PubMed] [Google Scholar]
- Andreu, P. , Colnot, S. , 2005. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development 132, (6) 1443–1451. [DOI] [PubMed] [Google Scholar]
- Anwar, S. , Hall, C. , 2000. Hereditary non-polyposis colorectal cancer: an updated review. European Journal of Surgical Oncology 26, (7) 635–645. [DOI] [PubMed] [Google Scholar]
- Barker, N. , Van Es, J.H. , 2007. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, (7165) 1003–1007. [DOI] [PubMed] [Google Scholar]
- Barker, N. , Ridgway, R.A. , 2008. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, (7229) 608–611. [DOI] [PubMed] [Google Scholar]
- Batlle, E. , Bacani, J. , 2005. EphB receptor activity suppresses colorectal cancer progression. Nature 435, (7045) 1126–1130. [DOI] [PubMed] [Google Scholar]
- Bellam, N. , Pasche, B. , 2010. TGF-β signaling alterations and colon cancer. Cancer Genetics 155, 85–103. [DOI] [PubMed] [Google Scholar]
- Bos, J.L. , Fearon, E.R. , 1987. Prevalence of ras gene mutations in human colorectal cancers. Nature 327, (6120) 293–297. [DOI] [PubMed] [Google Scholar]
- Casorelli, I. , Pannellini, T. , 2010. The Mutyh base excision repair gene influences the inflammatory response in a mouse model of ulcerative colitis. PLoS One 5, (8) e12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, N.L. , Javid, S.H. , 2007. Estrogen receptors α and β are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+ mice. Cancer Research 67, (5) 2366–2372. [DOI] [PubMed] [Google Scholar]
- Chulada, P.C. , Thompson, M.B. , 2000. Genetic disruption of Ptgs-1, as well as of Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Research 60, (17) 4705–4708. [PubMed] [Google Scholar]
- Clarke, A.R. , Cummings, M.C. , 1995. Interaction between murine germline mutations in p53 and APC predisposes to pancreatic neoplasia but not to increased intestinal malignancy. Oncogene 11, (9) 1913–1920. [PubMed] [Google Scholar]
- Corpet, D. , 2012. Chemoprevention Database Colorectal Cancer Prevention. Retrieved 1/12/2012, 2012 [Google Scholar]
- Dopeso, H. , Mateo-Lozano, S. , 2009. The receptor tyrosine kinase EPHB4 has tumor suppressor activities in intestinal tumorigenesis. Cancer Research 69, (18) 7430–7438. [DOI] [PubMed] [Google Scholar]
- Edelmann, W. , Yang, K. , 1999. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Research 59, (6) 1301–1307. [PubMed] [Google Scholar]
- El Marjou, F. , Janssen, K.P. , 2004. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, (3) 186–193. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , Gonzalez, S. , 2001. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. Journal of Clinical Oncology 19, (2) 299–304. [DOI] [PubMed] [Google Scholar]
- Fearon, E.R. , Vogelstein, B. , 1990. A genetic model for colorectal tumorigenesis. Cell 61, (5) 759–767. [DOI] [PubMed] [Google Scholar]
- Fodde, R. , Edelmann, W. , 1994. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proceedings of the National Academy of Sciences of the United States of America 91, (19) 8969–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryfe, R. , Bapat, B. , 1997. Molecular biology of colorectal cancer. Current Problems in Cancer 21, (5) 233–299. [DOI] [PubMed] [Google Scholar]
- Hackl, C. , Man, S. , 2012. Metronomic oral topotecan prolongs survival and reduces liver metastasis in improved preclinical orthotopic and adjuvant therapy colon cancer models. Gut 65, (2) 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines, J. , Johnson, V. , 2005. Genetic basis of variation in adenoma multiplicity in Apc Min/+ Mom1 S mice. Proceedings of the National Academy of Sciences of the United States of America 102, (8) 2868–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto, T. , Beppu, H. , 2002. Compound disruption of Smad2 accelerates malignant progression of intestinal tumors in Apc knockout mice. Cancer Research 62, (20) 5955–5961. [PubMed] [Google Scholar]
- Harada, N. , Tamai, Y. , 1999. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. The EMBO Journal 18, (21) 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman, F.S. , Nicol, C.J. , 2004. Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nature Medicine 10, (5) 481–483. [DOI] [PubMed] [Google Scholar]
- Hinoi, T. , Akyol, A. , 2007. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Research 67, (20) 9721–9730. [DOI] [PubMed] [Google Scholar]
- Hung, K.E. , Maricevich, M.A. , 2010. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proceedings of the National Academy of Sciences of the United States of America 107, (4) 1565–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby, R.F. , Marshall, D.J. , 1996. Chemoprevention of spontaneous intestinal adenomas in the Apc Min mouse model by the nonsteroidal anti-inflammatory drug piroxicam. Cancer Research 56, (4) 710–714. [PubMed] [Google Scholar]
- Jacoby, R.F. , Seibert, K. , 2000. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Research 60, (18) 5040–5044. [PubMed] [Google Scholar]
- Jang, K.S. , Song, Y.S. , 2010. Clinicopathological significance of nuclear PTEN expression in colorectal adenocarcinoma. Histopathology 56, (2) 229–239. [DOI] [PubMed] [Google Scholar]
- Kuraguchi, M. , Yang, K. , 2001. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc 1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Research 61, (21) 7934–7942. [PubMed] [Google Scholar]
- Kwong, L.N. , Shedlovsky, A. , 2007. Identification of Mom7, a novel modifier of Apc Min/+ on mouse chromosome 18. Genetics 176, (2) 1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X.H. , Zheng, H.C. , 2009. PTEN expression and mutation in colorectal carcinomas. Oncology Reports 22, (4) 757–764. [DOI] [PubMed] [Google Scholar]
- March, H.N. , Rust, A.G. , 2011. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nature Genetics 43, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, V. , Winton, D.J. , 2008. Epithelial Pten is dispensable for intestinal homeostasis but suppresses adenoma development and progression after Apc mutation. Nature Genetics 40, (12) 1436–1444. [DOI] [PubMed] [Google Scholar]
- Millar, C.B. , Guy, J. , 2002. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 297, (5580) 403–405. [DOI] [PubMed] [Google Scholar]
- Miyazono, K. , 2009. Transforming growth factor-beta. signaling in epithelial-mesenchymal transition and progression of cancer. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences 85, (8) 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser, A. , Pitot, H. , 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science (New York, NY) 247, (4940) 322–324. [DOI] [PubMed] [Google Scholar]
- Moser, A.R. , Dove, W.F. , 1992. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. The Journal of Cell Biology 116, (6) 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser, A.R. , Luongo, C. , 1995. Apc Min: a mouse model for intestinal and mammary tumorigenesis. European Journal of Cancer 31, (7) 1061–1064. [DOI] [PubMed] [Google Scholar]
- Muñoz, N.M. , Upton, M. , 2006. Transforming growth factor β receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Research 66, (20) 9837–9844. [DOI] [PubMed] [Google Scholar]
- Nautiyal, J. , Banerjee, S. , 2010. Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. International Journal of Cancer 128, (4) 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, C.A. , Pollett, A. , 2006. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, (7123) 106–110. [DOI] [PubMed] [Google Scholar]
- Oikarinen, S.I. , Cleveland, A.G. , 2009. Genetic mapping of Mom5, a novel modifier of Apc Min-induced intestinal tumorigenesis. Carcinogenesis 30, (9) 1591–1596. [DOI] [PubMed] [Google Scholar]
- Orner, G.A. , Dashwood, W. , 2003. Suppression of tumorigenesis in the Apc Min mouse: down-regulation of β-catenin signaling by a combination of tea plus sulindac. Carcinogenesis 24, (2) 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima, M. , Oshima, H. , 1995. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proceedings of the National Academy of Sciences of the United States of America 92, (10) 4482–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima, M. , Dinchuk, J.E. , 1996. Suppression of intestinal polyposis in Apc(Delta 716) knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87, (5) 803–809. [DOI] [PubMed] [Google Scholar]
- Pang, R. , Law, W.L. , 2010. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 6, (6) 603–615. [DOI] [PubMed] [Google Scholar]
- Parsons, D. , Wang, T. , 2005. Colorectal cancer: mutations in a signalling pathway. Nature 436, (7052) 792 [DOI] [PubMed] [Google Scholar]
- Perreault, N. , Sackett, S.D. , 2005. Foxl1 is a mesenchymal Modifier of Min in carcinogenesis of stomach and colon. Science Signalling 19, (3) 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova, T.V. , Nykänen, A. , 2008. Transcription factor PROX1 induces colon cancer progression by promoting the transition from benign to highly dysplastic phenotype. Cancer Cell 13, (5) 407–419. [DOI] [PubMed] [Google Scholar]
- Phesse, T.J. , Parry, L. , 2008. Deficiency of Mbd2 attenuates Wnt signaling. Molecular and Cellular Biology 28, (19) 6094–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp-Staheli, J. , Kim, K.H. , 2002. Pathway-specific tumor suppression: reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell 1, (4) 355–368. [DOI] [PubMed] [Google Scholar]
- Prolla, T.A. , Baker, S.M. , 1998. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DMA mismatch repair. Nature Genetics 18, (3) 276–279. [DOI] [PubMed] [Google Scholar]
- Quesada, C.F. , Kimata, H. , 1998. Piroxicam and acarbose as chemopreventive agents for spontaneous intestinal adenomas in Apc gene 1309 knockout mice. Cancer Science 89, (4) 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, K.R. , Sansom, O.J. , 2004. PPARδ status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene 23, (55) 8992–8996. [DOI] [PubMed] [Google Scholar]
- Reitmair, A.H. , Cai, J.C. , 1996. MSH2 deficiency contributes to accelerated Apc-mediated intestinal tumorigenesis. Cancer Research 56, (13) 2922–2926. [PubMed] [Google Scholar]
- Romagnolo, B. , Berrebi, D. , 1999. Intestinal dysplasia and adenoma in transgenic mice after overexpression of an activated β-catenin. Cancer Research 59, (16) 3875–3879. [PubMed] [Google Scholar]
- Sansom, O. , Reed, K. , 2004. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes & Development 18, (12) 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom, O.J. , Meniel, V. , 2006. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proceedings of the National Academy of Sciences of the United States of America 103, (38) 14122–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom, O.J. , Meniel, V.S. , 2007. Myc deletion rescues Apc deficiency in the small intestine. Nature 446, (7136) 676–679. [DOI] [PubMed] [Google Scholar]
- Sato, T. , Vries, R.G. , 2009. Single Lgr5 stem cells build crypt villus structures in vitro without a mesenchymal niche. Nature 459, (7244) 262–265. [DOI] [PubMed] [Google Scholar]
- Shibata, H. , Toyama, K. , 1997. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278, (5335) 120–123. [DOI] [PubMed] [Google Scholar]
- Silverman, K.A. , Koratkar, R. , 2002. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Research 12, (1) 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, G. , Carey, F.A. , 2002. Mutations in APC, Kirsten-ras, and p53—alternative genetic pathways to colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America 99, (14) 9433–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodir, N.M. , Chen, X. , 2006. Smad3 deficiency promotes tumorigenesis in the distal colon of Apc Min/+ mice. Cancer Research 66, (17) 8430–8438. [DOI] [PubMed] [Google Scholar]
- Solomon, S.D. , McMurray, J.J.V. , 2005. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. New England Journal of Medicine 352, (11) 1071–1080. [DOI] [PubMed] [Google Scholar]
- Steinbach, G. , Lynch, P.M. , 2000. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. New England Journal of Medicine 342, (26) 1946–1952. [DOI] [PubMed] [Google Scholar]
- Sun, F.X. , Sasson, A.R. , 1999. An ultra-metastatic model of human colon cancer in nude mice. Clinical and Experimental Metastasis 17, (1) 51–58. [DOI] [PubMed] [Google Scholar]
- Suraweera, N. , Haines, J. , 2006. Genetic determinants modulate susceptibility to pregnancy-associated tumourigenesis in a recombinant line of Min mice. Human Molecular Genetics 15, (23) 3429–3435. [DOI] [PubMed] [Google Scholar]
- Takaku, K. , Oshima, M. , 1998. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 92, (5) 645–656. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen, I.M.M. , Byrne, H.M. , 2006. Crypt dynamics and colorectal cancer: advances in mathematical modelling. Cell Proliferation 39, (3) 157–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechter, W.J. , Murray, E.D. , 2000. Treatment and survival study in the C57BL/6J-APCMin/+(Min) mouse with R-flurbiprofen. Life Sciences 66, (8) 745–753. [DOI] [PubMed] [Google Scholar]
- Williams, K.J. , Telfer, B.A. , 2002. ZD1839 (‘Iressa’), a specific oral epidermal growth factor receptor-tyrosine kinase inhibitor, potentiates radiotherapy in a human colorectal cancer xenograft model. British Journal of Cancer 86, (7) 1157–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, L.D. , Parsons, D.W. , 2007. The genomic landscapes of human breast and colorectal cancers. Science Signalling 318, (5853) 1108–1113. [DOI] [PubMed] [Google Scholar]