

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal types of human cancer for which there are no effective therapies. Deep sequencing of PDAC tumors has revealed the presence of a high number of mutations (>50) that affect at least a dozen key signaling pathways. This scenario highlights the urgent need to develop experimental models that faithfully reproduce the natural history of these human tumors in order to understand their biology and to design therapeutic approaches that might effectively interfere with their multiple mutated pathways. Over the last decade, several models, primarily based on the genetic activation of resident KRas oncogenes knocked‐in within the endogenous KRas locus have been generated. These models faithfully reproduce the histological lesions that characterize human pancreatic tumors. Decoration of these models with additional mutations, primarily involving tumor suppressor loci known to be also mutated in human PDAC tumors, results in accelerated tumor progression and in the induction of invasive and metastatic malignancies. Mouse PDACs also display a desmoplastic stroma and inflammatory responses that closely resemble those observed in human patients. Interestingly, adult mice appear to be resistant to PDAC development unless the animals undergo pancreatic damage, mainly in the form of acute, chronic or even temporary pancreatitis. In this review, we describe the most representative models available to date and how their detailed characterization is allowing us to understand their cellular origin as well as the events involved in tumor progression. Moreover, their molecular dissection is starting to unveil novel therapeutic strategies that could be translated to the clinic in the very near future.
Keywords: Cancer models, Pancreatic ductal adenocarcinoma, Tumor microenvironment, Inflammation, Target validation, Therapeutic strategies
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal types of cancer, with an overall 5‐year survival below 4%. This is mostly due to the advance state of the disease at the time of diagnosis in most patients, which makes current therapeutics rather ineffective (Hidalgo, 2010; Vincent et al., 2011). Histological, genetic, and clinical studies have identified three different ductal preneoplastic lesions as potential precursors of PDAC. They include pancreatic intraepithelial neoplasias (PanINs) that originate within intralobular ducts, intraductal papillary mucinous neoplasms (IPMNs) that arise in the main pancreatic duct or its major branches, and mucinous cystic neoplasms (MCN) that are mucin‐producing epithelial neoplasms with a characteristic ovarian‐type stroma. MCNs are classified according to their degree of epithelial dysplasia including mucinous cystadenoma, borderline mucinous cystic neoplasms and mucinous cystic neoplasms with in situ carcinoma (Wilentz et al., 2000). All of these preneoplastic lesions are likely to represent progressive stages of the disease (Hruban et al., 2000; Maitra et al., 2005).
PanINs are the best characterized preneoplastic lesions at the anatomopathological and molecular levels (Hruban et al., 2005). PanINs can be classified in four grades, PanIN‐1A, PanIN‐1B, PanIN‐2, and PanIN‐3 or carcinoma‐in‐situ, based on the degree of dysplasia (Figure 1A). PanIN lesions are frequently associated with lobulocentric atrophy and acinar ductal metaplasia (ADM) structures that have been proposed to be precursors of PanIN lesions (Brune et al., 2006; Detlefsen et al., 2005). These histological alterations are genetically characterized by the cumulative acquisition of mutations in both oncogenes, mainly KRAS, and tumor suppressor genes. The latter are also frequently inactivated by epigenetic mechanisms. In addition, telomere shortening is considered one of the early events in pancreatic tumorigenesis and is thought to be responsible for inducing genetic instability (van Heek et al., 2002). The best characterized mutational event during early PDAC development is the activation of the KRAS oncogene, mainly via single point mutations in codon 12. Indeed as many as 74% of low‐grade PanIN lesions and 63% of ADM foci associated with PanINs in human patients contain such oncogenic mutations (Shi et al., 2009). This percentage increases with progression to invasive carcinoma up to more than 90% of the cases. The tumor suppressor most commonly mutated in PDAC is CDKN2A. This gene is found inactivated, deleted, or epigenetically silenced in 30–70% of PanIN lesions, a percentage that becomes as high as 95% in full‐blown tumors (Wilentz et al., 1998). Mutations in two additional tumor suppressor genes, TP53 and SMAD4, are often associated (50–75% of the cases) with progression of PanIN‐3 lesions to invasive PDAC tumors. Mutations in the RI and RII receptors for the transforming growth factor (TGF)‐β have been identified with low frequency (<5%). Other alterations frequently found in advanced PanINs and PDAC tumors include over expression of other growth factors and their receptors as well as activation of signaling pathways driven by NFkB, STAT3 and SRC (Goggins et al., 1998; Hahn et al., 1996; Huang et al., 2012; Korc, 1998; Redston et al., 1994; Sclabas et al., 2003; Shields et al., 2011). More recently, analysis of exomic sequences from PDAC patients has revealed that these tumors carry as many as 50 mutations involving at least 12 different core signaling pathways (Jones et al., 2008).
Figure 1.
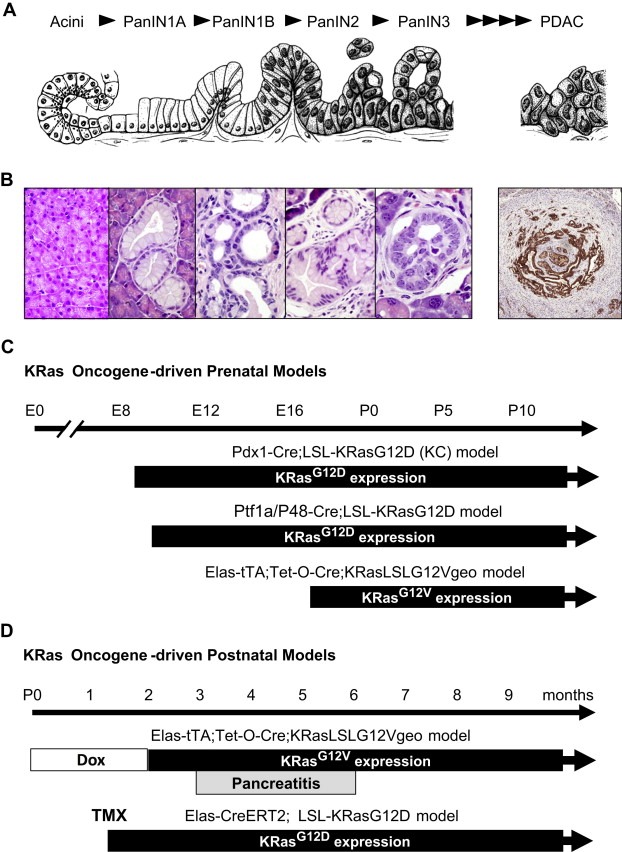
Genetically engineered mouse models of cancer faithfully reproduce the histological changes characteristic of human PDAC tumors. (A) Schematic diagram of the preneoplastic lesions that precede the development of PDAC (modified from the Hruban et al., 2005). (B) Representative PanIN lesions and PDAC observed in the Elas‐tTA; Tet‐O‐Cre; KRasLSLG12Vgeo strain. Similar lesions appear in the Pdx1‐Cre; LSL‐KRasG12D (KC) model and in the Ptf1a/P48‐Cre; LSL‐KRasG12D. (C) Schematic diagram of the timing at which the resident knocked in KRas oncogenes are expressed during embryonic development in the various models. (D) Schematic diagram of the timing at which the resident knocked‐in KRas oncogenes are expressed in adult mice in two independent models of PDAC. In the Elas‐tTA; Tet‐O‐Cre; KRasLSLG12Vgeo strain, the pregnant mothers and their offspring are exposed to doxycycline in the drinking water to prevent expression of the resident KRasG12V oncogene until the animals are at least 8 weeks old. These mice do not develop any lesions unless they undergo pancreatitis. In the Elas‐CreERT2; LSL‐KRasG12D model expression of the endogenous KRasG12D oncogene is mediated by exposure to tamoxifen, an estrogen analog that selectively activates the inducible CreERT2 recombinase.
IPMN and MCN are cystic lesions less well characterized at the molecular level. Recent studies have identified an average of 27 mutations in IPMNs and 16 in MCN lesions by exome sequencing (Wu et al., 2011a, 2011b). IPMN mutations included those in KRAS as well as in GNAS (a gene encoding for the guanine nucleotide binding, alpha stimulating protein) and RNF43 (RNF43 codes for a protein with intrinsic E3 ubiquitin ligase activity) genes. MCN mutations included, in addition of KRAS and RNF43 genes, inactivation of the TP53 tumor suppressor (Wu et al., 2011a, 2011b). Although KRAS appear in early lesions, their incidence increases with progressing dysplasia. In contrast, TP53 mutations appear late and are observed only in carcinomas, in combination with mutant KRAS genes (Jimenez et al., 1999).
In the case of IPMNs, KRAS and/or GNAS mutations are present in more than 95% of these lesions. Interestingly, most of the invasive adenocarcinomas in close association with IPMNs display the GNAS mutations present in these preneoplastic lesions (Wu et al., 2011b), indicating that IPMNs might be precursors of PDAC. An independent study using exome sequencing also revealed a high proportion of GNAS mutation (>40%) in IPMN lesions (Furukawa et al., 2011). The results indicate that GNAS mutations are selective for IPMNs suggesting that activation of G‐protein signaling may play a key role in the development of these lesions. Interestingly, IPMNs can be detected by computed tomography, allowing their surgical removal before they progress into malignant lesions.
2. Genetically engineered mouse (GEM) models of PDAC: prenatal models
Early attempts to express KRas oncogenes in ductal cells failed to induce PDAC (or any other type of tumor) in mice. The first model to faithfully reproduce the natural history of human PDAC in mice involved expression of a resident (knocked‐in) KRasG12D oncogene in all pancreatic lineages during early embryonic development (Hingorani et al., 2003) (Table 1). Briefly, a mouse strain carrying a conditional KRasG12D knocked‐in allele silenced by a floxed STOP transcriptional cassette (LSL‐KRasG12D) was crossed to transgenic strains that expressed the bacterial Cre recombinase under the control of either the Pdx1 or the Ptf1a/P48 promoters (Hingorani et al., 2003). The resulting strains, designated as Pdx1‐Cre; LSL‐KRasG12D (from now on KC, according to the nomenclature given in the original publication) and Ptf1a‐Cre; LSL‐KRasG12D, express the KRasG12D oncogene in all pancreatic lineages from early embryonic development (E8.5 in KC mice and E9.5 in Ptf1a‐Cre; LSL‐KRasG12D animals). These compound strains develop, with complete penetrance, the full spectrum of PanIN lesions histologically indistinguishable from those present in human patients. Indeed, these mouse PanINs also express mucins, cytokeratin‐19, and components of signaling pathways that include Cyclooxygenase‐2 (Cox‐2), EGFR, MMP‐7 and Hes1 (Hingorani et al., 2003). In addition, these mice display ADM, a benign lesion that precedes the appearance and possibly be a precursor of PanIN lesions (Zhu et al., 2007). PanIN lesions progress with long latencies to PDAC in a percentage of mice (Table 1). Addition of mutations in loci encoding tumor suppressors known to be mutated or inactivated in human PDAC such as INK4A, TP53, LKB1 or SMAD4 accelerate the progression of these PanIN lesions leading to the generation of invasive PDAC with complete penetrance (Table 2). A percentage of these compound mice also develop metastatic tumors. Some of the most important GEM PDAC models are summarized in recent reviews (Mazur and Siveke, 2012; Perez‐Mancera et al., 2012; Qiu and Su, 2012; Westphalen and Olive, 2012) and in Table 2.
Table 1.
List of basic GEMMs that develop PanIN lesions, organized by the prenatal or postnatal expression of the oncogenic initiating event. The targeted cell type is indicated.
| GEM model | Tumor initiation | Targeted cell type | Lesions (incidence %) | Observations | References | ||
|---|---|---|---|---|---|---|---|
| PanIN | PDAC | MET | |||||
| Elastase‐TGF‐α | Prenatal | Acinar | 100 | 10 | No | KrasG12D accelerates progression of mPanIN lesions to PDAC | Wagner et al., 2001 |
| Pdx1‐Cre; K‐Ras+/LSLG12D | Prenatal | Acinar, CAC, ductal, endocrine | 100 | 100 | Yes | Impossibility to asses cell of origin | Hingorani et al., 2003 |
| Ptf1a‐Cre; K‐Ras+/LSLG12D | Prenatal | Acinar, CAC, ductal, endocrine | 100 | 100 | Yes | Impossibility to asses cell of origin | Hingorani et al., 2003 |
| Elastase‐tTA; Tet‐O‐Cre; K‐Ras+/LSLG12Vgeo | Prenatal | Acinar | 90 | 10 | No | Pancreatitis accelerates PanIN development. | Guerra et al., 2007 |
| Nestin‐Cre; K‐Ras+/LSLG12D | Prenatal | Acinar, acinar precursors | 100 | No | No | Short survival due to CNS complications. PDAC in the context of pancreatitis. | Carriere et al., 2007 |
| Elastase‐tTA; Tet‐O‐Cre; K‐Ras+/LSLG12Vgeo plus caerulein | Postnatal (8 weeks) | Acinar | 100 | 20 | No | PanIN development requires pancreatitis. | Guerra et al., 2007 |
| Elastase‐CreER; K‐Ras+/LSLG12D | Postnatal (6 weeks) | Acinar | 36 | NA | No | Leaky system. Short term study. | De La O et al., 2008 |
| Elastase‐CreERT2; K‐Ras+/LSLG12D | Postnatal (6 weeks) | Acinar | 63 | No | No | PanIN development does not require pancreatitis. | Habbe et al., 2008 |
| Mist1‐CreERT2; K‐Ras+/LSLG12D | Postnatal (6 weeks) | Acinar | 30 | No | No | PanIN development does not require pancreatitis. | Habbe et al., 2008 |
| Pdx1‐CreER; K‐Ras+/LSLG12D | Postnatal (8 weeks) | Endocrine | 55 | Rare | No | The system may target acinar and precursor cells. | Gidekel‐Friedlander et al., 2009 |
| proCPA1CreERT2; K‐Ras+/LSLG12D | Postnatal (3 weeks) | Acinar | 10 | No | No | PanIN development require pancreatitis in older mice (5–8 weeks): PanIN in 33% mice. | Gidekel‐Friedlander et al., 2009 |
| RipCreER; K‐Ras+/LSLG12D | Postnatal (2–8 weeks) | Insulin expressing cells | 0 | No | No | Cooperation with pancreatitis | Gidekel‐Friedlander et al., 2009 |
MET (metastasis); NA: not analyzed.
Table 2.
List of current GEM models of PDAC.
| GEM model | Lesions | Metastasis | Survival | References |
|---|---|---|---|---|
| Pdx1‐Cre; K‐Ras+/LSLG12D; Ink4a/Arflox/lox | PanIN and PDAC | Yes | 2 m | Aguirre et al., 2003 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; p53R172H/+(KPC) | PanIN and PDAC | Yes | 5 m | Hingorani et al., 2005 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; Ink4a/Arf−/− | PanIN and PDAC | Yes | 5 m | Bardeesy et al., 2006 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; p53lox/lox | PanIN and PDAC | No | 3 m | Bardeesy et al., 2006 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; p53lox/lox; Ink4a/Arf−/− | PanIN and PDAC | Yes | 2 m | Bardeesy et al., 2006 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; Smad4lox/lox | IPMN and PDAC | Yes | 9 m | Bardeesy et al., 2006 |
| Ptf1a‐Cre; K‐Ras+/LSLG12D; TGFβIIRlox/lox | PanIN and PDAC | Yes | 2 m | Ijichi et al., 2006 |
| Elastase‐tTA; Tet‐O‐Cre; K‐Ras+/LSLG12Vgeo; p53−/− | PanIN and PDAC | Yes | 3–4 m | Guerra et al., 2007 |
| Ptf1a‐Cre; K‐Ras+/LSLG12D; Elastase‐TGF‐α | PanIN, IPMN, PDAC | Yes | 7 m | Siveke et al., 2007 |
| Ptf1a‐Cre; K‐Ras+/LSLG12D; Smad4lox/lox | MCN and PDAC | Yes | 8 m | Izeradjene et al., 2007 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; Brca2Tr/Δ11 | PanIN and PDAC | NA | NA | Skoulidis et al., 2010 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; p53R270H/+; Brca2Tr/+ | PanIN and PDAC | NA | <5 m | Skoulidis et al., 2010 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; p53R270H/+; Brca2Tr/Δ11 | PanIN and PDAC | Yes | 2.5 m | Skoulidis et al., 2010 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; Lkb1lox/lox | PanIN and PDAC | NA | 4.5 m | Morton et al., 2010 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; Notch1lox/lox | PanIN (high grade) | No | 3 m | Hanlon et al., 2010 |
| Ptf1a‐Cre; K‐Ras+/LSLG12D; Notch2lox/lox | MCN, anaplastic, PDAC | No | >12 m | Mazur et al., 2010 |
| Elastase‐tTA; Tet‐O‐Cre; K‐Ras+/LSLG12Vgeo; p53lox/lox | PanIN and PDAC | Yes | 3–4 m | Guerra et al., 2011 |
| Elastase‐tTA; Tet‐O‐Cre; K‐Ras+/LSLG12Vgeo; Ink4a/Arflox/lox | PanIN and PDAC | Yes | 3–4 m | Guerra et al., 2011 |
| Pdx1‐Cre; K‐Ras+/LSLG12D; Usp9x+/lox | PanIN and PDAC | NA | NA | Perez‐Mancera et al., 2012 |
m: Months; NA: Not analyzed.
Other important pathways involved in the progression of pancreatic cancer include the developmental Notch or Hedgehog signaling pathways. Notch signaling is a key regulator of pancreatic development (Apelqvist et al., 1999) and is upregulated in pancreatic cancer (Miyamoto et al., 2003). The role of Notch in PDAC development has been explored in GEM models. Expression of an active form of Notch1 (NIC) in KC and in Elas‐CreERT2; KRasG12D mice promoted PanIN formation (De La O et al., 2008). Interestingly, ablation of Notch1 accelerated PanIN development in KC mice (Hanlon et al., 2010). However, in a parallel study using Ptf1a‐Cre; LSL‐KRasG12D mice ablation of Notch1 had no effect on tumor development (Mazur et al., 2010).
PanINs display a Gli‐driven transcriptional program characterized by foregut developmental markers and elevated expression of canonical Gli target genes, suggesting an active role of the Hedgehog pathway in PanIN development (Thayer et al., 2003; Prasad et al., 2005). Indeed, tissue‐specific activation of a dominant active form of the GLI2 transcription factor induce tumor development in 30% of the mice, although these tumors do not appear to originate from PanIN lesions (Pasca di Magliano et al., 2006). However, if the Hedgehog pathway is activated at the same time as KRas oncogene expression in KC mice, animals develop extensive PanIN lesions that significantly reduce tumor latency and life span (Pasca di Magliano et al., 2006). Interestingly, deficiency of the G protein‐couple receptor Smoothened did not attenuate PanIN/PDAC development suggesting that Gli activation is decoupled from upstream Hedgehog signaling (Nolan‐Stevaux et al., 2009). Hedgehog inhibition with Smoothened inhibitors prolonged survival and slow down tumor development in GEM models (Feldmann et al., 2008; Olive et al., 2009). However, these effects are thought to be due to remodeling of tumor stroma (Olive et al., 2009) (see below).
3. Genetically engineered mouse (GEM) models of PDAC: postnatal models
In spite of the remarkable similarities between the PanIN and PDAC lesions observed in these GEM models with those of human patients, their etiology is distinct from that taking place in human patients. First of all, PDAC is not a pediatric disease. Hence, PDAC tumors are likely to arise due to sporadic mutations in adult individuals. In addition, PDAC patients are likely to suffer KRAS mutations in selected cell types not in the entire pancreas. A second generation of GEM tumor models in which a resident KRas oncogene is expressed in acinar cells has provided an alternative tool to address some of these issues (Guerra et al., 2007). This model was generated by crossing mice carrying a knocked‐in KRasLSLG12Vgeo allele with double transgenic mice (Elas‐tTA; Tet‐O‐Cre) that express a Cre recombinase under the control of the Elastase promoter following an inducible Tet‐Off strategy. These compound mice, if untreated, express the resident KRasG12V oncogene in a limited percentage (abound 20–30%) of acinar cells during late embryonic development (Guerra et al., 2007). Interestingly, they develop PanIN lesions with similar latencies and penetrance to mice expressing the KRasG12D oncogene in all pancreatic lineages. Moreover, a percentage of these mice develop PDAC by one year of age (Table 1; Figure 1B and C). Thus, suggesting that the cell origin in PDAC tumors is likely to be an acinar cell or an acinar precursor rather than cells of ductal lineages (see below). As indicated above, addition of mutations in Ink4a and TP53 tumor suppressors increase the penetrance of PDAC to 100% of the animals and significantly reduce tumor latency, inducing death of all animals by 6–8 month of age (Guerra et al., 2011) (Table 2).
The Elas‐tTA; Tet‐O‐Cre; KRasLSLG12Vgeo tumor model offers the possibility to turn on KRas oncogene expression in a controlled temporal manner by simply providing doxycycline in the drinking water. Unexpectedly, expression of the resident KRas oncogene in adult (≥60 day old) mice fails to induce pancreatic lesions including low‐grade PanINs (Guerra et al., 2007). Indeed, adult acinar cells are resistant to transformation by KRas even in the presence of inactivated TP53 or Ink4a/Arf tumor suppressors (Guerra et al., 2011). However, these mice readily develop PanIN lesions that progress to invasive PDAC tumors upon induction of pancreatitis (see below) (Table 1; Figure 1D). As expected, the presence of inactivated TP53 or Ink4a/Arf tumor suppressors increases the penetrance of tumor development to 100% and significantly shortened their latency (Guerra et al., 2007, 2011).
Mutational activation of either BRAF or PIK3CA is uncommon in PDAC (Jones et al., 2008). Yet, generation of GEM models of PDAC with mutations known to activate downstream effectors of KRas may provide relevant information regarding those signaling pathways critical for PDAC development. Interestingly, expression of the BRafV600E mutation in early pancreatic precursors results in embryonic lethality. However, expression of the same BRafV600E mutation expressed upon activation of the Pdx1‐CreERT2 transgene by exposure of P14 mice to tamoxifen led to widespread PanIN development (Collisson et al., 2012). However, these PanINs did not progress to PDAC tumors at least within one year. In contrast, activation of the PIK3CAH1047R oncogene under the control of the same Pdx1‐CreERT2 transgene failed to induce any PanIN lesion, suggesting that KRas oncogenes initiate PanIN lesions by activation of the Raf/Mek/Erk pathways rather than through the PI3K/Akt route (Collisson et al., 2012). These results indicate tumor initiation and maintenance is mediated by the RAF/MEK/ERK signaling pathway, suggesting that this pathway should be the focus of future therapeutic strategies.
4. Genetically engineered models of hereditary pancreatic cancer
Around 10% of patients with pancreatic cancer have a family history. These families have an increased risk of developing pancreatic cancer, estimated as a 2‐fold risk with one first‐degree relative. This risk is significantly higher in those families that carry germ line mutations in certain genes including BRCA2, CDKN2A/P16INK4a, STK11/LKB1, PRSS1 and PALB2 (Hruban et al., 2010). To date, two GEM models for hereditary pancreatic cancer have been generated by crossing the KC strain with mice carrying a truncated Brca2 locus. These mice develop PDAC tumors with higher penetrance and shorter latency than siblings carrying wild type Brca2 alleles. More importantly, these tumors retained the wild type Brca2 allele, indicating that LOH is not an essential requirement for tumor development (Skoulidis et al., 2010). In a similar study however, homozygous inactivation of Brca2 in a KRasG12D background led to the generation of acinar carcinomas, not PDAC (Rowley et al., 2011). Addition of a conditional Lkb1 floxed allele to the KC mouse model also resulted in increased numbers of PanINs leading to the development of PDAC tumors with full penetrance and reduced latency without detectable LOH (Morton et al., 2010). Interestingly, homozygous loss of Brca2 or Lkb1 in early pancreatic precursors in the absence of KRas oncogenes has distinct consequences. Whereas ablation of Brca2 does not induce histological alterations (Rowley et al., 2011), all Pdx1‐Cre; Lkb1lox/lox mice develop pancreatic mucinous cystadenomas with very short latencies (Morton et al., 2010). These observations illustrate that multiple pathways control malignant transformation of pancreatic cells.
5. Other mouse models of pancreatic cancer
GEM models of intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs) have also been developed. As indicated above, these pancreatic cystic preneoplasms have the potential to progress to invasive PDAC if they are not resected. Yet, these lesions are less defined than PanINs at the molecular level. Over the past few years, specific GEM models have been used to identify some of the genetic events involved in the development of these cystic neoplasms. For example, deletion of Smad4 in the pancreas compartment in the KC model induces the development of IPMNs (Bardeesy et al., 2006) and MCNs (Izeradjene et al., 2007). Interestingly, concomitant expression of Tgf‐α and KRasG12D leads to the development of cystic papillary neoplasms similar to IPMNs (Siveke et al., 2007). Furthermore, deletion of transcriptional intermediary factor (TIF)‐1γ, suggested to be involved in TGF‐β signaling in pancreas progenitor cells, cooperated with KRasG12D to induce IPMNs (Vincent et al., 2009). These GEM models illustrate that the alteration in the TGF‐α and TGF‐β pathways result in generation of cystic neoplasms and PanINs, which progress to PDAC.
Other pathways implicated in the development of these preneoplastic lesions are Pten and Notch. Pten deletion driven by the Pdx1‐Cre transgene induced the development of different histological lesions, such as metaplasia, rare PanINs, and carcinomas with papillary features that resemble IPMN (Stanger et al., 2005). Also, ablation of Notch2 in Ptf1a‐Cre; KRasG12D mice abrogates the development of the PanINs lesions characteristic of this strain, and induces MCN‐like lesions (Mazur et al., 2010). Finally, GEM models of endocrine pancreatic tumors, mainly the widely used RIP‐Tag model have been described (Hanahan, 1985).
6. PDAC cancer‐initiating cells
Tumor cells, both in PDAC tumors as well as in PanIN precursor lesions, display properties of ductal cells. Thus, it was initially assumed that PDAC might originate from any of the ductal cells types that populate pancreatic tissue. Early attempts to transform ductal cells by expressing a transgene encoding KRasG12D under the control of the cytokeratin‐19 promoter, failed to generate PDAC and even low‐grade PanIN lesions (Brembeck et al., 2003). However, expression of a resident KRasG12V oncogene in acinar cells under the control of the elastase promoter developed PanINs and PDAC lesions with high penetrance (Guerra et al., 2007). Indeed, the earliest histological change observed in GEM models of PDAC is ADM, a histological change also observed in human patients. These ductal‐like cells display both acinar and ductal markers suggesting that ADM might represent precursors of low‐grade PanINs (Guerra et al., 2007). Yet, it is possible that PanINs might be directly generated from acinar cells. In fact, some low‐grade PanINs contained cells expressing acinar (amylase) and ductal (cytokeratin 19) markers. These studies suggest that ADMs and PanIN lesions must originate either by transdifferentiation of elastase‐expressing acinar cells or by misdifferentiation of acinar precursors. Interestingly, induction of KRasG12V oncogene expression in adult acinar cells (8 week old mice) failed to induce ADM or PanIN lesions. These observations indicate that regardless of the mechanism by which PanIN and PDAC lesions arise, the cell of origin must be an embryonic‐like precursor, at least in a context in which there is no pancreatic injury (see below).
Yet, the precise cell of origin of these early preneoplastic lesions is still a matter of debate. Analyses of the number of PanIN lesions in GEM models of PDAC has revealed rather limited numbers of PanIN lesions in spite of the fact that in these mice the resident KRas oncogenes are expressed in either most embryonic pancreatic cells (KC and Ptf1a‐Cre; LSL‐KRasG12D models) or in as many as 30% of all embryonic acinar cells (Elas‐tTA; Tet‐O‐Cre; KRasLSLG12Vgeo model). Thus, only a small subset of embryonic cells must be susceptible to transformation by the resident KRas oncogenes. The cancer‐initiating cell, therefore, is unlikely to be a major cell type but a less abundant precursor. Alternatively, the low frequency of PanINs might be due to the need for additional mutational event(s) or epigenetic alteration(s) within the population of KRas‐expressing embryonic cells.
Other GEM models also point to the acinar cells as the cell of origin. For instance, mouse models that overexpress oncogenes in acinar cells, such as the Elastase‐TGF‐α transgenic strain (Wagner et al., 2001), or that target endogenous oncogenic KRas to the acinar cells with different Elastase‐Cre, Mist1‐Cre and Nestin‐Cre drivers (Carriere et al., 2007; De La O et al., 2008; Habbe et al., 2008), (Table 1).
Careful analysis of the expression pattern of the KRasG12V oncogene in Elas‐tTA; Tet‐O‐Cre; KRasLSLG12Vgeo mice using a beta‐Geo surrogate marker co‐expressed in a bicistronic fashion revealed the presence of KRas in few centroacinar, but not ductal cells (Guerra et al., 2007). These observations reinforce the concept that PanINs do not arise from normal ductal cells. Yet, they raise the possibility that the cell of origin of PanIN lesions might be centroacinar cells, which lie at the junction between acinar cells and the terminal end duct cells. Previous studies have shown that ALDH1‐expressing centroacinar and terminal duct cells express stem cell markers, expand in response to chronic epithelial injury, and contribute to endocrine and exocrine lineages following injection into cultured embryonic dorsal pancreatic buds (Rovira et al., 2010). More recently, Cre‐based lineage tracing experiments have shown that pancreatic acinar cells can originate under physiological conditions from Sox9‐expressing progenitors located in terminal ductal structures and centroacinar cells (Furuyama et al., 2011). However selective activation of the KRasG12D oncogene in Sox9 positive centroacinar and ductal cells failed to induce PanINs in young mice, with the possible exception of ductal cells present in large pancreatic ducts (Kopp et al., 2012). In contrast, loss of Pten expression in pancreatic cells during early embryonic development (Pdx1‐Cre; Ptenlox/lox strain) leads to the rapid expansion of centroacinar cells that overtake the resident acinar structures, and eventually generate PanIN lesions that occasionally progress to PDAC tumors. However, specific deletion of Pten in acinar cells does not lead to PanIN structures in Elastase‐CreER; Ptenlox/lox mice (Stanger et al., 2005). Thus, it is possible that centroacinar cells can generate PanIN lesions upon activation of their PI3Kinase pathway but not through the RAF/MEK/ERK pathway.
Interestingly, inactivation of a single Pten allele in KC mice resulted in metastatic PDAC tumors by a mechanism that involved silencing of the Ink4a/Arf locus and activation of the NFkB pathway (Ying et al., 2011). Whether loss of Pten can also generate metastatic tumors in KRas oncogene‐expressing acinar cells remains to be determined. Generation of knock‐in strains that express Cre recombinase from endogenous promoters that are completely selective for acinar and/or centroacinar cells should clarify the putative role of centroacinar and terminal duct cells in the initiation of PanIN lesions and PDAC.
7. Cancer‐initiating cells in the adult pancreas
Induction of KRasG12V oncogene expression in acinar cells during postnatal development results in the progressive reduction in the number of ADM and PanIN lesions, leading to complete disappearance when mice reach adulthood at 8–10 weeks of age (Guerra et al., 2007, 2011). Similar results were obtained using a related GEM model in which expression of a resident KRasG12D oncogene is mediated by a Cre recombinase driven by the procarboxypeptidase A1 promoter (Gidekel‐Friedlander et al., 2009) (Table 1). In general, tumor incidence gradually decreases, and tumor latency increases, as KRas expression is induced during late postnatal development. Differences in the age at which mice become resistant to KRas driven PanIN and PDAC formation have been reported. Maitra and coworkers have reported full induction of PanINs and PDAC tumors in adult (6 weeks of age) mice (Habbe et al., 2008) (Table 1; Figure 1D). These differences might be due to genetic background, pattern of expression of the Cre recombinase, conditional strategies, and/or leakiness of the inducible system. Yet, these observations, taken together suggest that PanIN lesions and PDAC are likely to result by transformation of progenitor cells that progressively loss their ability to respond to KRas oncogenic signaling when they reach a mature differentiated state.
Pdx1 is a marker for early pancreatic precursors (Guz et al., 1995). Its expression in adult mice is present at the highest levels in β‐cells of the islets and at much lower levels in other islet cell types and in acinar cells (Wu et al., 1997). Expression of KRasG12D in Pdx1 positive cells can lead to PanIN induction in adult mice under noninflammatory conditions (Gidekel‐Friedlander et al., 2009). Interestingly, the authors described a reduction in the number of lesions in older mice. Expression of KRasG12D in Pdx1 positive cells of young mice (2–4 weeks old) results in the efficient induction of low‐grade PanINs, some of which progress to high‐grade lesions. However, when KRasG12D expression in initiated in older, 8 week old mice, they only develop low‐grade PanINs in a small proportion of mice (Gidekel‐Friedlander et al., 2009). These results illustrate that the subpopulation of Pdx1‐expressing cells susceptible to transformation by the KRas oncogene also decreases as mice reach adulthood. Whether this population of Pdx1 expressing cells represents the cell of origin of PanIN and PDAC lesions is a possibility that needs to be further explored.
In a recent study, ablation of the transcription factor Sox9 from Ptf1a‐expressing acinar cells in young (3 week old) animals inhibited KRasG12D‐induced formation of ADM and PanIN lesions. Sox9 is not expressed in normal acinar cells. However, Sox9 is induced upon expression of the resident KRasG12D oncogene (Kopp et al., 2012). Likewise, forced expression of Sox9 in KRasG12D oncogene‐containing acinar cells facilitates the development of PanIN lesions. These results strongly suggest that Sox9 is a key player for the transformation of acinar cells into malignant ductal‐like structures such as PanINs and PDAC. Moreover, these results also suggest that PanIN lesions, at least during postnatal development, arise by Sox9 mediated reprogramming of otherwise normal acinar cells. These observations however, do not explain why acinar cells of adult mice are resistant to transformation by KRas oncogenes. The possibility that Sox9 may no longer be induced by oncogenic KRas in the adult pancreas should be examined.
8. The contribution of desmoplasia to PDAC development
A hallmark feature of PDAC is the big desmoplastic stroma that surrounds tumor cells. The stroma, which in most cases constitutes as much as 90% of the tumor volume, contains reactive “cancer‐associated” fibroblasts (CAFs), activated pancreatic stellate cells (PSCs), scattered inflammatory cells, partially collapsed microvasculature, nerve fibers, dense connective tissue, interstitial fluid, and numerous cytokines and growth factors that are embedded within the stromal matrix. In vitro studies involving co‐culturing tumoral cells with CAFs and PSCs or PSC conditioned media have suggested that the interaction between these cells play an important role in tumor progression. Indeed, it is possible that the stroma contributes to tumor development from the very early stages of the disease since both human and mouse low‐grade PanINs already display a layer of reactive stroma. Stroma remodeling is a process that mostly depends on oncogenic KRas signaling, since acute loss of mutant KRas in established PDAC tumors results in rapid quiescence and involution of the stroma cells (Collins et al., 2012).
Previous studies using mouse xenografts have illustrated that the presence of PSCs and/or CAFs enhance tumor incidence growth and metastasis of PDAC tumor cell lines (Apte and Wilson, 2012; Hwang et al., 2008; Kraman et al., 2010). These effects might be due, at least in part, to activation of the Hedgehog pathway in PSCs (Hwang et al., 2012), in response to secretion of Hedgehog ligands by the tumor cells (Yauch et al., 2008). Indeed, other pathways such as those driven by the PDGF receptor, TGF‐β, Notch and cyclooxygenase‐2 also seem to play a role (Apte and Wilson, 2012). The role of macrophages has also been explored. For example, CD40 agonists have been used to activate tumoricidal macrophages that infiltrate the tumor and cause degradation of surrounding fibrotic tumor‐stromal elements. This approach produced tumor regression in a subset of surgically incurable PDAC patients as well as in KPC mice, a GEM model derived from the KC strain, that expresses a mutated form of TP53 (TP53R172H) (Hingorani et al., 2005). This effect appears to be independent of T cells and cytotoxic agents (e.g. gemcitabine) (Beatty et al., 2011). In another study using a GEM tumor model in which the TGF‐β receptor II (Tgfbr2) is ablated in pancreatic epithelium in the context of endogenous KRasG12D expression (Ptf1a‐Cre; LSL‐KRasG12D; Tgfbr2lox/lox mice), Ijichi et al. (2011) reported high levels of expression of connective tissue growth factor (Ctgf), a profibrotic and tumor‐promoting factor, especially in the tumor‐stromal border area, suggesting an active tumor‐stromal interaction. Interestingly stromal production of Ctgf was induced by production of high levels of Cxc chemokines by the invasive PDAC tumor cells. Inhibition of the Cxcl–Cxcr2 pathway resulted in reduced levels of Ctgf in the stromal compartment, reduced angiogenesis and increased survival times (Ijichi et al., 2011). Interestingly, Cxcr2 inhibition did not synergize with gemcitabine, possibly because of toxic adverse effects or incomplete target inhibition (Ijichi et al., 2011).
The pro‐inflammatory cytokine GM‐CSF induces a paracrine circuit by engaging stromal myeloid cells to exert an immunosuppressive effect on local killer T cells (Bayne et al., 2012; Pylayeva‐Gupta et al., 2012). GM‐CSF has been found consistently upregulated in tumors of the KPC model as well as in human PanIN lesions and PDAC tumors. Lineage marking experiments illustrate that tumoral cells and not the stroma cells produce GM‐CSF (Bayne et al., 2012). Near‐complete abrogation of GM‐CSF mRNA by pharmacological inhibition using MEK or PI3K inhibitors in ductal cells expressing oncogenic KRas suggests that the Ras/RAF/MEK/ERK and PI3K effector pathways regulate GM‐CSF production in these cells at the level of transcription (Pylayeva‐Gupta et al., 2012). Cytotoxic CD8 T cells can recognize and clear incipient PDAC tumors, but oncogenic KRas signaling overcomes this effect (Bayne et al., 2012; Pylayeva‐Gupta et al., 2012). These results suggests that extensive secretion of GM‐CSF induced by the KRas oncogene is sufficient to drive the development of myeloid cells that suppressed antigen‐specific CD8 T cells. Abrogation of GM‐CSF secretion or neutralization of its activity inhibited tumor growth and maintenance. Conversely, depletion of CD8 rescues tumor growth. These findings suggest a therapeutic potential for disrupting the crosstalk between tumor cells and the immune system by targeting myeloid cells or the cytokines that regulate their differentiation (Bayne et al., 2012; Pylayeva‐Gupta et al., 2012).
9. Pancreatitis and PDAC
Chronic pancreatitis is one of the highest risk factors for the development of PDAC in humans (Lowenfels et al., 1993; Malka et al., 2002). The pooled risk estimate of seven studies for pancreatic cancer in chronic pancreatitis patients was reported to be 13.3 (Raimondi et al., 2010) and their cumulative risk after 20 years was reported to be 4%, at least tenfold higher than those who have not suffer from this chronic condition (Lowenfels et al., 1993). Interestingly, recent epidemiological studies have suggested a beneficial correlation between NSAIDs and pancreatic cancer risk (Bonifazi et al., 2010; Bradley et al., 2010; Rothwell et al., 2011; Tan et al., 2011). Moreover, patients who underwent surgery for the treatment of chronic pancreatitis had significantly lower incidences of pancreatic cancer (Ueda et al., 2012).
In GEM models of PDAC, pancreatitis appears to be an essential component for tumor development in adult mice. As illustrated by Guerra and co‐workers, adult acinar cells are resistant to transformation by some of the mutations most commonly found in human cancer, that is expression of KRas oncogenes and inactivation of the p16Ink4a/p19Arf and TP53 tumor suppressors (Guerra et al., 2011). However, adult mice expressing a resident KRasG12V oncogene in acinar cells develop PanINs and PDACs with high penetrance in the context of acute, chronic or even sporadic bouts of pancreatitis induced by exposure to the cholecystokinin analog, caerulein (Guerra et al., 2007, 2011).
Chronic pancreatitis cooperates more effectively with resident KRas oncogenes than acute pancreatitis. For instance, induction of acute pancreatitis in mice expressing an endogenous KRasG12D in all pancreatic cell lineages led to steatorrhea and formation of PanINs (Morris et al., 2010). However, these mice did not develop PDAC by 8 months of age. In contrast, chronic pancreatitis induced multifocal in situ as well as invasive PDAC even though these mice only expressed the resident KRas oncogene in a limited number of acinar cells (Guerra et al., 2007). It is possible that excessive tissue damage affects homeostatic recovery, limiting the ability of PanINs to progress to PDAC. Short periods of caerulein treatment such as one month with a daily dose are sufficient to induce PanIN in 50% of the mice. A small proportion of these mice progressed to develop PDAC within a year. Caerulein exposure for longer periods of time (3 months) results in PanIN development in all treated animals along with increased incidence of PDAC tumors within one year (20%) (Guerra et al., 2011). Interestingly, the effect of pancreatitis in inducing PanINs and PDAC lesions does not require concomitant expression of KRas oncogenes providing that some tissue damage, along with the subsequent inflammatory response remains within the pancreatic parenchyma at the time of KRasG12V expression (Guerra et al., 2011).
To date, it is not clear how pancreatitis causes adult mice to overcome their resistance to induction of PanIN lesions or PDAC by oncogenic KRas. Pancreatitis induces tissue damage that results in proliferation of acinar cells to repair the injured parenchyma. It is not clear whether this proliferation is mediated by the recruitment of putative progenitor cells or by dedifferentiation of mature acinar cells. Regardless of the mechanism, it is likely that these progenitor and/or differentiated acinar cells become susceptible to transformation by the resident KRas oncogene leading to the acquisition of ductal‐like properties, as observed in ADM and low‐grade PanIN lesions. Previous studies have illustrated that tissue repair induces expression of embryonic markers such as Pdx‐1, Sox9, Bmi1 and activates developmental signals, including Notch, Hedgehog and Wnt pathways (Fendrich et al., 2008; Jensen et al., 2005; Keefe et al., 2012; Morris et al., 2010; Siveke et al., 2008; Yoshida et al., 2008). Hebrok and co‐workers reported that oncogenic KRas induced misdifferentiation of the regenerating acinar cell compartment into ductal like structures by a mechanism that involved decreased expression of β‐catenin (Morris et al., 2010). More recently, they have shown that induction of Sox9 is a key element for the induction of ADM and PanIN lesions at least during postnatal development possibly by inducing expression of ductal genes (Kopp et al., 2012). Induction of acute pancreatitis in these young mice considerably accelerated the process of PanIN induction (Kopp et al., 2012).
Likewise, Bmi1 a gene expressed in a subpopulation of self‐renewing pancreatic acinar cells and in response to pancreatitis, contributes to regeneration of the exocrine pancreas through cell autonomous mechanisms, in part by regulating Cdkn2a expression, and non‐cell autonomous mechanisms (Fukuda et al., 2012). Cell lineage tracing with Bmi1‐CreER mice revealed a subpopulation of differentiated pancreatic acinar cells with proliferative capacity with self‐renewing capacity (Sangiorgi and Capecchi, 2009). Whether the main contribution of pancreatitis to the generation of PanIN lesions involves reprogramming of the refractory adult acinar cells remains to be determined. It is also possible that tissue damage mobilizes pools of adult stem or progenitor cells in an attempt to restore homeostasis. If so, these progenitor cells might be susceptible to KRas oncogene‐mediated transformation, leading to misdifferentiation into the ductal‐like lineage characteristic of early PanIN lesions. This process would be similar to the ability of KRas oncogenes to induce PDAC when expressed in pancreatic precursors during embryonic development. Finally, we have not excluded the possibility that caerulein may have a direct effect on its target cells, stimulating acinar cell proliferation (Douziech et al., 1998).
10. Inflammation and PDAC development
Pancreatitis‐induced tissue damage also results in an inflammatory response involving the innate and adaptive immune systems. Indeed, the inflammatory response observed during the early stages of tumor development in GEM models of PDAC appears to be primarily mediated by macrophages and T cells. The precise contribution of inflammation to PDAC development remains to be determined. Likewise, the mechanism by which inflammatory cells facilitate progression of PanIN lesions into invasive PDAC tumors, including generation of the desmoplastic stroma so characteristic of this tumor type, remains to be worked out.
Recent studies have illustrated that the inflammatory response might contribute to PanIN progression and PDAC development by inhibiting oncogene‐mediated senescence during the early stages of PanIN progression (Guerra et al., 2011). In PDAC development, the senescence barrier is lost when low‐grade PanIN‐1A and 1B lesions progress to high‐grade PanIN‐2/3s. However, in the presence of pancreatitis‐induced inflammation, low‐grade PanINs loose senescence markers. Abrogation of the senescence barrier is likely to facilitate progression of low‐grade PanINs into high‐grade lesions including invasive PDAC. Interestingly, inhibition of senescence by inflammatory cells is a reversible process since shortly after cessation of pancreatitis the expression of senescence markers is detected in the majority of low‐grade PanINs (Guerra et al., 2011). Senescence markers are also expressed in human low‐grade PanINs present in patients suffering from chronic pancreatitis. More importantly, only those patients with chronic pancreatitis who had received anti‐inflammatory therapy (either prednisolone or nonsteroidal anti‐inflammatory drugs) contained PanINs with senescent cells (Guerra et al., 2011). Further studies should establish if the presence of senescence markers in PanIN lesions, due to anti‐inflammatory therapy, slow the progression of PanINs into more malignant lesions thereby slowing tumor development in pancreatitis patients at risk of developing PDAC tumors. Indeed, some epidemiological studies have correlated nonsteroidal anti‐inflammatory drug therapy with decreased risk of developing pancreatic cancer (Bonifazi et al., 2010; Bradley et al., 2010; Rothwell et al., 2011). A more systematic use of GEM PDAC models should help to determine whether inhibiting the inflammation‐mediated loss of senescence in low‐grade PanINs prevents or delays PDAC development.
The inflammatory response is likely to contribute to PanIN progression by mechanisms beyond inhibition of senescence. In fact, the inflammatory response during PDAC development, regardless of whether animals undergo pancreatitis or not, is likely to be the result of multiple events contributed by a large variety of inflammatory cells. GEM models should help to dissect by genetic as well as pharmacological means the contribution of the main inflammatory pathways. For instance, treatment of Elas‐tTA; Tet‐O‐Cre; KRasLSLG12Vgeo mice exposed to caerulein for 3 months with sulindac, a nonsteroidal anti‐inflammatory drug and non specific Cox inhibitor, eliminated parenchyma atrophy and reduced the extent of the PanINs by as much as 90%. However, inhibition of the inflammatory response by sulindac treatment did not abrogate the process of tumorigenesis and some of the mice displayed PDAC. Indeed, preliminary results suggest that treated mice do not display a survival advantage. These observations are reminiscent of clinical results obtained with targeted drugs in which overall survival does not improve in spite of robust anti‐tumor responses.
Stat3 becomes activated during tumor development primarily as a response to interleukin‐6 trans‐signaling. Disruption of Stat3 in the developing exocrine pancreas significantly reduced PanIN development, especially those of high‐grade, and consequently, these mice had a lower incidence of PDAC tumors (Corcoran et al., 2011; Fukuda et al., 2011; Lesina et al., 2011). Yet, the reduction in tumor development observed in these mice can be due to non‐cell autonomous effects, since they also had fewer tumor‐associated macrophages and lost expression of Cox‐2, suggesting a role of Stat3 at the level of tumoral cells and in the microenvironment (Fukuda et al., 2011). In agreement with these studies, inactivation of Socs3, an inhibitor of the Stat3 signaling, promoted PanIN and PDAC development (Lesina et al., 2011). Interestingly, Socs3 is highly expressed in most PanINs, maybe acting as a negative feedback loop consequence of chronic Stat3 signaling. Collectively, these studies implicate Stat3 signaling in the inflammatory process that promotes PanIN and PDAC. Stat3 also has a role in the induction of PDAC during acute pancreatitis (Fukuda et al., 2011). Stat3 becomes immediately activated following acute pancreatitis and promotes the proliferation of acinar and ductal cells. Curiously, cell proliferation in PanINs did not seem to be significantly affected by the absence of Stat3. This may be explained by the predominant role of Stat3 in the pancreatic microenvironment during pancreatitis. Indeed, disruption of Stat3 in the exocrine pancreas decreased the levels of infiltrating inflammatory cells and altered their composition. Loss of Stat3 expression also insignificantly reduced the overall levels of cytokines. This appeared to be a consequence of decreased levels of inflammatory infiltrates and a reduction of cytokine production by acinar cells following exposure to caerulein (Fukuda et al., 2011). This role of Stat3 in promoting PanIN development in mice following induction of acute pancreatitis was also observed in older mice. Whereas 8‐month‐old Stat3‐expressing mice displayed weight loss, Stat3 expression in the exocrine pancreas did not acquire these phenotypes. However, these mice had low‐grade PanINs in the pancreata, indicating tumor initiation in the absence of Stat3 (Fukuda et al., 2011). Current efforts are beginning to decipher the molecular pathways responsible for the effects of Stat3 in pancreatic cancer. For example, the Stat3 target gene MMP7 appears to play an important role in advanced pancreatic cancer. Indeed, although MMP7 nullizygous mice develop low‐grade and high‐grade PanIN lesions with the same efficiency as control animals, the absence of MMP7 reduces progression of the lesions to PDACs (Fukuda et al., 2011). Moreover, in mice also harboring heterozygous disruptions in TP53, absence of MMP7 significantly reduced tumor size (Fukuda et al., 2011). Stat3 might therefore contribute to tumor initiation and progression by controlling various effector genes, such as MMP7, at different stages of PDAC development.
11. Genetic complexity of PDAC tumors
Most human PDAC tumors contain KRAS oncogenes and deletion of the P16INK4a tumor suppressor. Indeed, these mutations have been detected in a significant percentage of early lesions including low‐grade PanINs. In GEM models of PDAC, expression of KRas oncogenes appears to be essential for PanIN formation as well as progression to PDAC, providing that they are expressed in acinar cells or their precursors. Deletion of the p16Ink4a locus including the p19Arf tumor suppressor is not sufficient to induce ADM or PanIN lesions. Yet, loss of these tumor suppressors greatly synergizes with KRas oncogenes leading to the rapid induction of PDAC tumors as well as anaplastic sarcomatoid tumors (Aguirre et al., 2003; Guerra et al., 2011). Recently, an inducible GEM model has been used to illustrate that PDAC tumors are “addicted” to KRas oncogenic signaling, since both PanIN development as well as PDAC maintenance require KRas oncogene expression. Yet, PDAC tumors are known to accumulate a large number of mutations (Jones et al., 2008). Deep sequencing of exomic regions has illustrated not only a large variety of mutated genes, but the presence of independent clones harboring additional mutations even within the primary tumor (Campbell et al., 2010). These mutated genes have been loosely classified into twelve different oncogenic signaling pathways (Jones et al., 2008). Yet, tumors from different patients harbor a subset of completely different genes in most of these pathways. Preliminary results indicate that a similar situation may also take place in tumors derived from GEM models. Deep sequencing of exomic regions of eleven PDAC tumors harboring mutations in KRas and the TP53 tumor suppressor displayed an average of 14 additional mutations per tumor (our unpublished observations). Perhaps more importantly, none of the 150 mutated genes appeared in more than one tumor. Functional studies will be necessary to asses the real genetic complexity of PDAC tumors to determine how many of these oncogenic signaling pathways need to be blocked in order to obtain a significant and durable therapeutic response (see below).
Recent studies using the Sleeping Beauty transposon‐mediated insertional mutagenesis have illustrated that the genetic complexity of PDAC tumors could be even higher (Perez‐Mancera et al., 2012). In this study, Pérez‐Mancera and cols. identified 1150 significant candidate genes, whose mutations could contribute to PDAC development. These results contrast with the much lower number of somatic mutations (average of 63 per tumor) found in the exome of patient samples (Jones et al., 2008). Only 14% of the genes identify by Sleeping Beauty transposition are known to be mutated in human pancreatic cancer, suggesting that not all of these genes will be actually implicated in the development of PDAC tumors in human patients. Yet, this experimental approach might help to identify genes relevant for tumor development that have been missed by exomic sequencing or might be epigenetically targeted. For example, the gene more frequently identified by transposon‐mediated insertional mutagenesis was the X‐linked deubiquitinase Usp9x, which was found to be inactivated in over 50% of PDAC tumors (Perez‐Mancera et al., 2012). Although previous work has ascribed a pro‐survival role to USP9X in human neoplasia (Schwickart et al., 2010), its implication as a tumor suppressor in PDAC tumors was not predicted by deep sequencing technologies.
12. Targeting oncogenic signaling in PDAC tumors
One of the main advantages offered by GEM models of human cancer is the possibility to carry out validation studies of selective targets using genetic and/or pharmacological approaches at different stages of tumor development. Moreover, validated targets can be used in combination leading to the identification of therapeutic strategies unlikely to be uncovered in classical clinical trials.
Currently, the only treatment approved to treat PDAC tumors is gemcitabine, a nucleoside analog approved for patients with advanced or metastatic PDAC. Yet, the average increased survival offered by gemcitabine is minimal (4–5 months in a small percentage of responding patients) (Burris et al., 1997; Li et al., 2004). Interestingly a phase III clinical study reported that addition of Erlotinib, an EGFR inhibitor produced more responses than gemcitabine alone (Moore et al., 2007). Although the benefit of the combination was rather modest, it was quite surprising since the EGFR is supposed to signal upstream of the KRas oncogenes present in most human PDAC tumors. Indeed, these observations are at odds with extensive clinical data in human non small cell lung carcinomas (NSCLC), in which oncogenic mutations in the EGFR and KRAS loci are mutually exclusive (Shigematsu et al., 2005). Likewise, patients with colorectal tumors carrying KRAS oncogenes do not benefit from treatments involving inhibition of EGFR signaling (Karapetis et al., 2008). Yet, recent studies using GEM models of PDAC tumors driven by KRas oncogenes have illustrated that EGFR, unlike in lung and colorectal tumors, is essential for PDAC development. Animals carrying floxed alleles of the EGFR locus did not develop PanIN lesions or PDAC tumors even in the context of pancreatic injury (pancreatitis) or lacking the p16Ink4a/p19Arf tumor suppressors (Ardito et al., 2012; Navas et al., 2012). Unfortunately the observed requirement for EGFR signaling was bypassed in the absence of the TP53 tumor suppressor a mutation observed in about 50% of human PDAC tumors. Thus, it is likely that inhibition of EGFR in advanced pancreatic tumors may not be sufficient considering the genetic complexity of these tumors. Yet, inhibition of EGFR should cooperate with inhibitors affecting other signaling pathways in inducing more sustainable responses in PDAC patients.
Previous studies have also shown a strong synergy between the presence of activated Notch receptors and KRasG12D oncogene expression in PanIN development (De La O et al., 2008). Conditional ablation of different Notch receptors in KRasG12D‐driven pancreatic carcinogenesis illustrated that deficiency of Notch2 but not Notch1 reduces PanIN progression, prolongs survival, and leads to a phenotypical switch toward anaplastic pancreatic cancer with epithelial–mesenchymal transition (Mazur et al., 2010). Furthermore, loss of Notch1 simultaneously to the expression of oncogenic KRasG12D displayed increased tumor incidence and progression, implying that Notch1 can function as a tumor suppressor in PDAC development (Hanlon et al., 2010). These observations should stimulate the development of selective Notch inhibitors that block Notch 2 without affecting Notch1 receptors.
Notch inhibitors have also been tested in GEM models of PDAC. MRK‐003, a gamma‐secretase inhibitor has been shown to inhibit PanIN development in Pdx1‐Cre; LSL‐KRasG12D; P53lox/lox mice providing that the animals were tested during the very early stages of tumor development (Plentz et al., 2009). More recently this inhibitor has been used to treat advance tumors in the very same GEM model. Whereas treatment with MRK‐003 alone had no effect, this inhibitor was able to increase the survival of the tumored mice (9 days vs. 26 days) when used in combination with gemcitabine. This therapeutic combination killed tumor endothelial cells and promoted widespread hypoxic necrosis (Cook et al., 2012). These results suggest that the paucivascular nature of PDAC can be exploited as a therapeutic vulnerability, and the dual targeting of the tumor endothelium and neoplastic cells by gamma secretase inhibition may constitute a rationale for clinical translation.
Recent studies have also investigated the role of cathepsin B, a lysosomal protease upregulated in PDAC, in the initiation and progression of this tumor type in Pdx1‐Cre; LSL‐KRasG12D; P53lox/lox mice. Constitutive ablation of cathepsin B resulted in delayed progression of both PanIN and PDAC development as well as in a significant increased in overall survival (Gopinathan et al., 2012). These results suggest that cathepsin B might play an important role in the progression of PDAC and support further investigation of cathepsin B as a therapeutic target.
13. Targeting the stroma in PDAC
The stroma represents a critical barrier to the pharmacological treatment of pancreatic cancer. For instance, in the KPC model, gemcitabine cannot be delivered to the tumor cells. Limited depletion of stromal cells using a Hedgehog antagonist such as IPI‐926 (Infinity Pharmaceuticals), a smoothened inhibitor, allowed robust gemcitabine delivery into the tumor and increased therapeutic efficacy (Olive et al., 2009). Several clinical trials have been initiated to investigate the therapeutic effect of Hedgehog inhibitors in PDAC patients. Unfortunately, Infinity Pharmaceuticals announced a year ago that was halting its phase II trial (IPI‐926 plus gemcitabine, NCT01130142). This was a surprising decision in view of the encouraging results reported in a phase I trial in which the IPI‐926 plus gemcitabine arm led to a 31% partial response rates vs. 10% in the arm using only gemcitabine (ASCO 2011, abstract 4114).
Another potential target to deplete, or at least compromise the desmoplastic stroma of PDAC tumors, is SPARC (secreted protein acidic and rich in cysteine, also known as osteo), a protein found to be overexpressed by CAFs. SPARC is the target of albumin‐bound paclitaxel (nab‐paclitaxel also known as abraxane), a novel formulation for this cytotoxic drug that avoids the use of toxic solvents (Gradishar, 2006). A phase II clinical trial using nab‐paclitaxel along with gemcitabine has shown significant anti‐tumor activity with limited toxicity, a result that has led to more comprehensive phase III trial (Von Hoff et al., 2011). Preclinical studies in human‐derived pancreatic cancer xenografts suggested that the mechanism of action is through drug accumulation via binding of albumin to SPARC‐positive fibroblasts (Von Hoff et al., 2011). More recently, another laboratory confirmed the remarkable efficacy of nab‐paclitaxel in combination with gemcitabine as a therapeutic approach to treat PDAC tumors in another GEM tumor model (Frese et al., 2012). However, the mechanism of action for this drug is still a matter of debate, since they did not observed significant depletion of stroma in their KPC model. Instead, they observed that tumor cells exposed to nab‐paclitaxel were much more sensitive to gemcitabine, an effect attributed to increased stability of gemcitabine due to reactive oxygen species–mediated degradation of cytidine deaminase (Frese et al., 2012). Further investigations are required to establish the mechanism of action of his therapeutic strategy. Moreover, it will be interesting to develop novel therapeutic approaches to directly target SPARC or other proteins that might allow elimination of CAFs and other stromal components.
More recently, two groups have used the KPC model to target stromal tissue using a PEGylated hyaluronidase (Jacobetz et al., 2013; Provenzano et al., 2012). These studies identified prominent interstitial hydrostatic pressure within the stromal compartment that constricts blood vessels and prevents the passive efflux of delivered chemotherapeutics from the vasculature into the tumor. Elimination of the stromal hyaluronan diminishes this hydrostatic pressure, allowing efflux of the administered chemotherapeutic agents. These results suggest that a similar strategy could be use to improved chemotherapeutic delivery to patients with pancreatic cancer. GEM models of PDAC should play a key role in interrogating the contribution of the stroma to tumorigenesis. The combination of conditional strategies based on different recombinases (e.g. Cre and FLP) should make it possible to genetically manipulate the tumor microenvironment independently of targeting the tumor cells at different stages during disease progression.
14. Future GEM models: target validation and preclinical studies
GEM models available to date faithfully reproduce the histological changes that lead to the formation of PDAC tumors, specially those combining expression of resident KRas oncogenes with mutations in tumor suppressors known to be mutated in the corresponding human tumors, mainly P16INK4a, TP53 and SMAD4. Indeed, some of these GEM models also result in the generation of metastatic tumors whose behavior closely resembles that of their human counterparts.
However, available models are only partially suitable to carry out much needed target validation studies aimed at identifying those signaling pathways essential for tumor development, one of the main added value of GEM models. Today's PDAC models are almost exclusively based on the use of a single recombinase, the bacteriophage‐derived Cre recombinase. Hence, oncogene activation, tumor suppressor inactivation and target ablation all occur at the same time and in the same cells. These strategies prevent the analysis of target ablation in already formed tumors or preneoplastic lesions. Likewise, most of these models do not allow genetic evaluation of the role of stromal and immune cells in invasive or metastatic tumors. An effort needs to be done to combine the limited repertoire of recombinases available today to separate those events that lead to tumor development from those responsible for the formation of the desmoplastic stroma. Moreover, it is essential to separate tumor development from ablation of targets of predicted therapeutic value. Most human PDAC tumors display more than a dozen altered signaling pathways. Thus, identification of those signaling pathways, both in stromal as well as in tumor cells, essential for tumor development is an urgent need. Without this information, current and future clinical trials are likely to be doomed.
Acknowledgements
Work was supported by grants from the European Research Council (ERC‐AG/ 250297‐RAS AHEAD), the EU‐Framework Programme (HEALTH‐2010‐260791) and the Spanish Ministry of Economy and Competitiveness (SAF2011‐30173).
Guerra Carmen, Barbacid Mariano, (2013), Genetically engineered mouse models of pancreatic adenocarcinoma, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.002.
Contributor Information
Carmen Guerra, Email: mcguerra@cnio.es.
Mariano Barbacid, Email: mbarbacid@cnio.es.
References
- Aguirre, A.J. , Bardeesy, N. , Sinha, M. , Lopez, L. , Tuveson, D.A. , Horner, J. , Redston, M.S. , DePinho, R.A. , 2003. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelqvist, A. , Li, H. , Sommer, L. , Beatus, P. , Anderson, D.J. , Honjo, T. , Hrabe de Angelis, M. , Lendahl, U. , Edlund, H. , 1999. Notch signalling controls pancreatic cell differentiation. Nature 400, 877–881. [DOI] [PubMed] [Google Scholar]
- Apte, M.V. , Wilson, J.S. , 2012. Dangerous liaisons: pancreatic stellate cells and pancreatic cancer cells. J. Gastroenterol. Hepatol. 27, 69–74. [DOI] [PubMed] [Google Scholar]
- Ardito, C.M. , Gruner, B.M. , Takeuchi, K.K. , Lubeseder-Martellato, C. , Teichmann, N. , Mazur, P.K. , DelGiorno, K.E. , Carpenter, E.S. , Halbrook, C.J. , Hall, J.C. , Pal, D. , Briel, T. , Herner, A. , Trajkovic-Arsic, M. , Sipos, B. , Liou, G.-Y. , Storz, P. , Murray, N.R. , Threadgill, D.W. , Sibilia, M. , Washington, M.K. , Wilson, C.L. , Schmid, R.M. , Raines, E.W. , Crawford, H.C. , Siveke, J.T. , 2012. EGF Receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22, 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy, N. , Cheng, K.H. , Berger, J.H. , Chu, G.C. , Pahler, J. , Olson, P. , Hezel, A.F. , Horner, J. , Lauwers, G.Y. , Hanahan, D. , DePinho, R.A. , 2006. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 20, 3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne, L.J. , Beatty, G.L. , Jhala, N. , Clark, C.E. , Rhim, A.D. , Stanger, B.Z. , Vonderheide, R.H. , 2012. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty, G.L. , Chiorean, E.G. , Fishman, M.P. , Fishman, M.P. , Saboury, B. , Teitelbaum, U.R. , Sun, W. , Huhn, R.D. , Song, W. , Li, D. , Sharp, L.L. , Torigian, D.A. , O'Dwyer, P.J. , Vonderheide, R.H. , 2011. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifazi, M. , Gallus, S. , Bosetti, C. , Polesel, J. , Serraino, D. , Talamini, R. , Negri, E. , La Vecchia, C. , 2010. Aspirin use and pancreatic cancer risk. Eur. J. Cancer Prev. 19, 352–354. [DOI] [PubMed] [Google Scholar]
- Bradley, M.C. , Hughes, C.M. , Cantwell, M.M. , Napolitano, G. , Murray, L.J. , 2010. Non-steroidal anti-inflammatory drugs and pancreatic cancer risk: a nested casecontrol study. Br. J. Cancer 102, 1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brembeck, F.H. , Schreiber, F.S. , Deramaudt, T.B. , Deramaudt, T.B. , Craig, L. , Rhoades, B. , Swain, G. , Grippo, P. , Stoffers, D.A. , Silberg, D.G. , Rustgi, A.K. , 2003. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 63, 2005–2009. [PubMed] [Google Scholar]
- Brune, K. , Abe, T. , Canto, M. , O'Malley, L. , Klein, A.P. , Maitra, A. , Volkan, A.N. , Fishman, E.K. , Cameron, J.L. , Yeo, C.J. , Kern, S.E. , Goggins, M. , Hruban, R.H. , 2006. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am. J. Surg. Pathol. 30, 1067–1076. [PMC free article] [PubMed] [Google Scholar]
- Burris, H.A. , Moore, M.J. , Andersen, J. , Green, M.R. , Rothenberg, M.L. , Modiano, M.R. , Cripps, M.C. , Portenoy, R.K. , Storniolo, A.M. , Tarassoff, P. , Nelson, R. , Dorr, F.A. , Stephens, C.D. , Von Hoff, D.D. , 1997. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15, 2403–2413. [DOI] [PubMed] [Google Scholar]
- Campbell, P.J. , Yachida, S. , Mudie, L.J. , Stephens, P.J. , Pleasance, E.D. , Stebbings, L.A. , Morsberger, L.A. , Latimer, C. , McLaren, S. , Lin, M.L. , McBride, D.J. , Varela, I. , Nik-Zainal, S.A. , Leroy, C. , Jia, M. , Menzies, A. , Butler, A.P. , Teague, J.W. , Griffin, C.A. , Burton, J. , Swerdlow, H. , Quail, M.A. , Stratton, M.R. , Iacobuzio-Donahue, C. , Futreal, P.A. , 2010. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere, C. , Seeley, E.S. , Goetze, T. , Longnecker, D.S. , Korc, M. , 2007. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc. Natl. Acad. Sci. U.S.A. 104, 4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, M.A. , Bednar, F. , Zhang, Y. , Brisset, J.C. , Galbán, S. , Galbán, C.J. , Rakshit, S. , Flannagan, K.S. , Adsay, N.V. , Pasca di Magliano, M. , 2012. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 122, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson, E.A. , Trejo, C.L. , Silva, J.M. , Gu, S. , Korkola, J.E. , Heiser, L.M. , Charles, R.P. , Rabinovich, B.A. , Hann, B. , Dankort, D. , Spellman, P.T. , Phillips, W.A. , Gray, J.W. , McMahon, M. , 2012. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, N. , Frese, K.K. , Bapiro, T.E. , Jacobetz, M.A. , Gopinathan, A. , Miller, J.L. , Rao, S.S. , Demuth, T. , Howat, W.J. , Jodrell, D.I. , Tuveson, D.A. , 2012. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J. Exp. Med. 209, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran, R.B. , Contino, G. , Deshpande, V. , Tzatsos, A. , Conrad, C. , Benes, C.H. , Levy, D.E. , Settleman, J. , Engelman, J.A. , Bardeesy, N. , 2011. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 71, 5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La O, L.P. , Emerson, L.L. , Goodman, J.L. , Froebe, S.C. , Illum, B.E. , Curtis, A.B. , Murtaugh, L.C. , 2008. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. U.S.A. 105, 18907–18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detlefsen, S. , Sipos, B. , Feyerabend, B. , Klöppel, G. , 2005. Pancreatic fibrosis associated with age and ductal papillary hyperplasia. Virchows Arch. 447, 800–805. [DOI] [PubMed] [Google Scholar]
- Douziech, N. , Lajas, A. , Coulombe, Z. , Calvo, E. , Lainé, J. , Morisset, J. , 1998. Growth effects of regulatory peptides and intracellular signaling routes in human pancreatic cancer cell lines. Endocrine 9, 171–183. [DOI] [PubMed] [Google Scholar]
- Feldmann, G. , Habbe, N. , Dhara, S. , Bisht, S. , Alvarez, H. , Fendrich, V. , Beaty, R. , Mullendore, M. , Karikari, C. , Bardeesy, N. , Ouellette, M.M. , Yu, W. , Maitra, A. , 2008. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 57, 1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrich, V. , Esni, F. , Garay, M.V. , Feldmann, G. , Habbe, N. , Jensen, J.N. , Dor, Y. , Stoffers, D. , Jensen, J. , Leach, S.D. , Maitra, A. , 2008. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology 135, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese, K.K. , Neesse, A. , Cook, N. , Bapiro, T.E. , Lolkema, M.P. , Jodrell, D.I. , Tuveson, D.A. , 2012. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2, 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, A. , Wang, S.C. , Morris, J.P. , Folias, A.E. , Liou, A. , Kim, G.E. , Akira, S. , Boucher, K.M. , Firpo, M.A. , Mulvihill, S.J. , Hebrok, M. , 2011. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 19, 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, A. , Morris, J.P. , Hebrok, M. , 2012. Bmi1 is required for regeneration of the exocrine pancreas in mice. Gastroenterology 143, 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, T. , Kuboki, Y. , Tanji, E. , Yoshida, S. , Hatori, T. , Yamamoto, M. , Shibata, N. , Shimizu, K. , Kamatani, N. , Shiratori, K. , 2011. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci. Rep. 1, 161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama, K. , Kawaguchi, Y. , Akiyama, H. , Horiguchi, M. , Kodama, S. , Kuhara, T. , Hosokawa, S. , Elbahrawy, A. , Soeda, Koizumi, T.M. , Masui, T. , Kawaguchi, M. , Takaori, K. , Doi, R. , Nishi, E. , Kakinoki, R. , Deng, J.M. , Behringer, R.R. , Nakamura, T. , Uemoto, S. , 2011. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet. 43, 34–41. [DOI] [PubMed] [Google Scholar]
- Gidekel-Friedlander, S.Y. , Chu, G.C. , Snyder, E.L. , Girnius, N. , Dibelius, G. , Crowley, D. , Vasile, E. , DePinho, R.A. , Jacks, T. , 2009. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggins, M. , Shekher, M. , Turnacioglu, K. , Yeo, C.J. , Hruban, R.H. , Kern, S.E. , 1998. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 58, 5329–5332. [PubMed] [Google Scholar]
- Gopinathan, A. , Denicola, G.M. , Frese, K.K. , Cook, N. , Karreth, F.A. , Mayerle, J. , Lerch, M.M. , Reinheckel, T. , Tuveson, D.A. , 2012. Cathepsin B promotes the progression of pancreatic ductal adenocarcinoma in mice. Gut 61, 877–884. [DOI] [PubMed] [Google Scholar]
- Gradishar, W.J. , 2006. Albumin-bound paclitaxel: a next-generation taxane. Exp. Opin. Pharmacother. 7, 1041–1053. [DOI] [PubMed] [Google Scholar]
- Guerra, C. , Schuhmacher, A.J. , Cañamero, M. , Grippo, P.J. , Verdaguer, L. , Pérez-Gallego, L. , Dubus, P. , Sandgren, E.P. , Barbacid, M. , 2007. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-ras oncogenes in adult mice. Cancer Cell 11, 291–302. [DOI] [PubMed] [Google Scholar]
- Guerra, C. , Collado, M. , Navas, C. , Schuhmacher, A.J. , Hernández-Porras, I. , Cañamero, M. , Rodriguez-Justo, M. , Serrano, M. , Barbacid, M. , 2011. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene induced senescence. Cancer Cell 19, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guz, Y. , Montminy, M.R. , Stein, R. , Leonard, J. , Gamer, L.W. , Wright, C.V.E. , Teitelman, G. , 1995. Expression of murine STF-1, a putative insulin gene transcription factor, in β-cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 121, 11–18. [DOI] [PubMed] [Google Scholar]
- Habbe, N. , Shi, G. , Meguid, R.A. , Fendrich, V. , Esni, F. , Chen, H. , Feldmann, G. , Stoffers, D.A. , Konieczny, S.F. , Leach, S.D. , Maitra, A. , 2008. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. U.S.A. 105, 18913–18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, S.A. , Schutte, M. , Hoque, A.T. , Moskaluk, C.A. , da Costa, L.T. , Rozenblum, E. , Weinstein, C.L. , Fischer, A. , Yeo, C.J. , Hruban, R.H. , Kern, S.E. , 1996. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271, 350–353. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , 1985. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315, 115–122. [DOI] [PubMed] [Google Scholar]
- Hanlon, L. , Avila, J.L. , Demarest, R.M. , Troutman, S. , Allen, M. , Ratti, F. , Rustgi, A.K. , Stanger, B.Z. , Radtke, F. , Adsay, V. , Long, F. , Capobianco, A.J. , Kissil, J.L. , 2010. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 70, 4280–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo, M. , 2010. Pancreatic cancer. N. Engl. J. Med. 362, 1605–1617. [DOI] [PubMed] [Google Scholar]
- Hingorani, S.R. , Petricoin, E.F. , Maitra, A. , Rajapakse, V. , King, C. , Jacobetz, M.A. , Ross, S. , Conrads, T.P. , Veenstra, T.D. , Hitt, B.A. , Kawaguchi, Y. , Johann, D. , Liotta, L.A. , Crawford, H.C. , Putt, M.E. , Jacks, T. , Wright, Ch.V.E. , Hruban, R.A. , Lowy, A.M. , Tuveson, D.A. , 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. [DOI] [PubMed] [Google Scholar]
- Hingorani, S.R. , Wang, L. , Multani, A.S. , Combs, C. , Deramaudt, T.B. , Hruban, R.H. , Rustgi, A.K. , Chang, S. , Tuveson, D.A. , 2005. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hruban, R.H. , Wilentz, R.E. , Kern, S.E. , 2000. Genetic progresión in the pancreatic ducts. Am. J. Pathol. 156, 1821–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban, R.H. , Wilentz, R.E. , Maitra, A. , 2005. Identification and analysis of precursors to invasive pancreatic cancer. Methods Mol. Med. 103, 1–13. [DOI] [PubMed] [Google Scholar]
- Hruban, R.H. , Canto, M. , Goggins, M. , Schulick, R. , Klein, A.P. , 2010. Update on familial pancreatic cancer. Adv. Surg. 44, 293–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Huang, R. , Chang, W. , Jiang, T. , Huang, K. , Cao, J. , Sun, X. , Qiu, Z. , 2012. The expression and clinical significance of pSTAT3, VEGF and VEGF-C in pancreatic adenocarcinoma. Neoplasma 59, 52–61. [DOI] [PubMed] [Google Scholar]
- Hwang, R.F. , Moore, T. , Arumugam, T. , Ramachandran, V. , Amos, K.D. , Rivera, A. , Ji, B. , Evans, D.B. , Logsdon, C.D. , 2008. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, R.F. , Moore, T.T. , Hattersley, M. , Scarpitti, M. , Yang, B. , Devereaux, E. , Ramachandran, V. , Arumugam, T. , Ji, B. , Logsdon, C.D. , Brown, J.L. , Godin, R. , 2012. Inhibition of the Hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol. Cancer Res. 10, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi, H. , Chytil, A. , Gorska, A.E. , Aakre, M.E. , Fujitani, Y. , Fujitani, S. , Wright, C.V. , Moses, H.L. , 2006. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 20, 3147–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi, H. , Chytil, A. , Gorska, A.E. , Aakre, M.E. , Bierie, B. , Tada, M. , Mohri, D. , Miyabayashi, K. , Asaoka, Y. , Maeda, S. , Ikenoue, T. , Tateishi, K. , Wright, C.V. , Koike, K. , Omata, M. , Moses, H.L. , 2011. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Invest. 121, 4106–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeradjene, K. , Combs, C. , Best, M. , Gopinathan, A. , Wagner, A. , Grady, W.M. , Deng, C.X. , Hruban, R.H. , Adsay, N.V. , Tuveson, D.A. , Hingorani, S.R. , 2007. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 11, 229–243. [DOI] [PubMed] [Google Scholar]
- Jacobetz, M.A. , Chan, D.S. , Neesse, A. , Bapiro, T.E. , Cook, N. , Frese, K.K. , Feig, C. , Nakagawa, T. , Caldwell, M.E. , Zecchini, H.I. , Lolkema, M.P. , Jiang, P. , Kultti, A. , Thompson, C.B. , Maneval, D.C. , Jodrell, D.I. , Frost, G.I. , Shepard, H.M. , Skepper, J.N. , Tuveson, D.A. , 2013. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, J.N. , Cameron, E. , Garay, M.V. , Starkey, T.W. , Gianani, R. , Jensen, J. , 2005. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128, 728–741. [DOI] [PubMed] [Google Scholar]
- Jimenez, R.E. , Warshaw, A.L. , Z'graggen, K. , Hartwig, W. , Taylor, D.Z. , Compton, C.C. , Fernández-del Castillo, C. , 1999. Sequential accumulation of K-ras mutations and p53 overexpression in the progression of pancreatic mucinous cystic neoplasms to malignancy. Ann. Surg. 230, 501–509. discussion 509–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. , Zhang, X. , Parsons, D.W. , Lin, J.C. , Leary, R.J. , Angenendt, P. , Mankoo, P. , Carter, H. , Kamiyama, H. , Jimeno, A. , Hong, S.M. , Fu, B. , Lin, M.T. , Calhoun, E.S. , Kamiyama, M. , Walter, K. , Nikolskaya, T. , Nikolsky, Y. , Hartigan, J. , Smith, D.R. , Hidalgo, M. , Leach, S.D. , Klein, A.P. , Jaffee, E.M. , Goggins, M. , Maitra, A. , Iacobuzio-Donahue, C. , Eshleman, J.R. , Kern, S.E. , Hruban, R.H. , Karchin, R. , Papadopoulos, N. , Parmigiani, G. , Vogelstein, B. , Velculescu, V.E. , Kinzler, K.W. , 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapetis, C.S. , Khambata-Ford, S. , Jonker, D.J. , O'Callaghan, C.J. , Tu, D. , Tebbutt, N.C. , Simes, R.J. , Chalchal, H. , Shapiro, J.D. , Robitaille, S. , Price, T.J. , Shepherd, L. , Au, H.J. , Langer, C. , Moore, M.J. , Zalcberg, J.R. , 2008. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765. [DOI] [PubMed] [Google Scholar]
- Keefe, M.D. , Wang, H. , De La O, J.P. , Khan, A. , Firpo, M.A. , Murtaugh, L.C. , 2012. Beta-catenin is selectively required for the expansion and regeneration of mature pancreatic acinar cells in mice. Dis. Model. Mech. 5, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp, J.L. , von Figura, G. , Mayes, E. , Liu, F. , Dubois, C.L. , Morris, J.P. , Pan, F.C. , Akiyama, H. , Wright, Ch.V.E. , Jensen, K. , Hebrok, M. , Sander, M. , 2012. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korc, M. , 1998. Role of growth factors in pancreatic cancer. Surg. Oncol. Clin. N. Am. 7, 25–41. [PubMed] [Google Scholar]
- Kraman, M. , Bambrough, P.J. , Arnold, J.N. , Roberts, E.W. , Magiera, L. , Jones, J.O. , Gopinathan, A. , Tuveson, D.A. , Fearon, D.T. , 2010. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830. [DOI] [PubMed] [Google Scholar]
- Lesina, M. , Kurkowski, M.U. , Ludes, K. , Rose-John, S. , Treiber, M. , Klöppel, G. , Yoshimura, A. , Reindl, W. , Sipos, B. , Akira, S. , Schmid, R.M. , Algül, H. , 2011. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19, 456–469. [DOI] [PubMed] [Google Scholar]
- Li, D. , Xie, K. , Wolff, R. , Abbruzzese, J.L. , 2004. Pancreatic cancer. Lancet 363, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Lowenfels, A.B. , Maisonneuve, P. , Cavallini, G. , Ammann, R.W. , Lankisch, P.G. , Andersen, J.R. , Dimagno, E.P. , Andrén-Sandberg, A. , Domellof, L. , International Pancreatitis Study Group1993. Pancreatitis and the risk of pancreatic cancer. N. Engl. J. Med. 328, 1433–1437. [DOI] [PubMed] [Google Scholar]
- Maitra, A. , Fukushima, N. , Takaori, K. , Hruban, R.H. , 2005. Precursors to invasive pancreatic cancer. Adv. Anat. Pathol. 12, 81–91. [DOI] [PubMed] [Google Scholar]
- Malka, D. , Hammel, P. , Maire, F. , Rufat, P. , Madeira, I. , Pessione, F. , Lévy, P. , Ruszniewski, P. , 2002. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 51, 849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur, P.K. , Siveke, J.T. , 2012. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressin translational oncology. Gut 61, 1488–1500. [DOI] [PubMed] [Google Scholar]
- Mazur, P.K. , Einwachter, H. , Lee, M. , Sipos, B. , Nakhai, H. , Rad, R. , Zimber-Strobl, U. , Strobl, L.J. , Radtke, F. , Klöppel, G. , Schmid, R.M. , Siveke, J.T. , 2010. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A. 107, 13438–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, Y. , Maitra, A. , Ghosh, B. , Zechner, U. , Argani, P. , Iacobuzio-Donahue, C.A. , Sriuranpong, V. , Iso, T. , Meszoely, I.M. , Wolfe, M.S. , Hruban, R.H. , Ball, D.W. , Schmid, R.M. , Leach, S.D. , 2003. Notch mediates TGF alphainduced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3, 565–576. [DOI] [PubMed] [Google Scholar]
- Moore, M.J. , Goldstein, D. , Hamm, J. , Figer, A. , Hecht, J.R. , Gallinger, S. , Au, H.J. , Murawa, P. , Walde, D. , Wolff, R.A. , Campos, D. , Lim, R. , Ding, K. , Clark, G. , Voskoglou-Nomikos, T. , Ptasynski, M. , Parulekar, W. , National Cancer Institute of Canada Clinical Trials Group, 2007. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- Morris, J.P.I.V. , Cano, D.A. , Sekine, S. , Wang, S.C. , Hebrok, M. , 2010. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Invest. 120, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, J.P. , Jamieson, N.B. , Karim, S.A. , Athineos, D. , Ridgway, R.A. , Nixon, C. , McKay, C.J. , Carter, R. , Brunton, V.G. , Frame, M.C. , Ashworth, A. , Oien, K.A. , Evans, T.R. , Sansom, O.J. , 2010. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 39, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas, C. , Hernández-Porras, I. , Schumacher, A.J. , Sibilia, M. , Guerra, C. , Barbacid, M. , 2012. EGF receptor signaling is essential for K-Ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 22, 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan-Stevaux, O. , Lau, J. , Truitt, M.L. , Chu, G.C. , Hebrok, M. , Fernández-Zapico, M.E. , Hanahan, D. , 2009. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive, K.P. , Jacobetz, M.A. , Davidson, C.J. , Gopinathan, A. , McIntyre, D. , Honess, D. , Madhu, B. , Goldgraben, M.A. , Caldwell, M.E. , Allard, D. , Frese, K.K. , DeNicola, G. , Christine Feig, C. , Combs, C. , Winter, S.P. , Heather, I. , Reichelt, S. , Howat, W.J. , Chang, A. , Dhara, M. , Lifu Wang, L. , Ruckert, F. , Grutzmann, R. , Pilarsky, C. , Izeradjene, K. , Hingorani, S.R. , Huang, P. , Davies, S.E. , Plunkett, W. , Egorin, M. , Hruban, R.H. , Whitebread, N. , McGovern, K. , Adams, J. , Iacobuzio-Donahue, C. , Griffiths, J. , Tuveson, D.A. , 2009. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano, M. , Sekine, S. , Ermilov, A. , Ferris, J. , Dlugosz, A.A. , Hebrok, M. , 2006. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 20, 3161–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mancera, P.A. , Rust, A.G. , van der Weyden, L. , Kristiansen, G. , Li, A. , Sarver, A.L. , Silverstein, K.A.T. , Grutzmann, R. , Aust, D. , Rummele, P. , Knosel, T. , Herd, C. , Stemple, D.L. , Kettleborough, R. , Brosnan, J.A. , Li, A. , Morgan, R. , Knight, S. , Yu, J. , Stegeman, S. , Collier, L.S. , ten Hoeve, J.J. , Ridder, J. , Klein, A.P. , Goggins, M. , Hruban, R.H. , Chang, D.K. , Biankin, A.V. , Grimmond, S.M. , Australian Pancreatic Cancer Genome Initiative, Wessels, Lodewyk F.A. , Wood, S.A. , Iacobuzio-Donahue, Ch.A. , Pilarsky, Ch. , Largaespada, D.A. , Adams, D.J. , Tuveson, D.A. , 2012. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez–Mancera, P.A. , Guerra, C. , Barbacid, M. , Tuveson, D.A. , 2012. What we have learned about pancreatic cancer from mouse models. Gastroenterology 142, 1079–1092. [DOI] [PubMed] [Google Scholar]
- Plentz, R. , Park, J.S. , Rhim, A.D. , Abravanel, D. , Hezel, A.F. , Sharma, S.V. , Gurumurthy, S. , Deshpande, V. , Kenific, C. , Settleman, J. , Majumder, P.K. , Stanger, B.Z. , Bardeesy, N. , 2009. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 136, 1741–1749.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, N.B. , Biankin, A.V. , Fukushima, N. , Maitra, A. , Dhara, S. , Elkahloun, A.G. , Hruban, R.H. , Goggins, M. , Leach, S.D. , 2005. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 65, 1619–1626. [DOI] [PubMed] [Google Scholar]
- Provenzano, P.P. , Cuevas, C. , Chang, A.E. , Goel, V.K. , Daniel, D. , Von Hoff, D.D. , Hingorani, S.R. , 2012. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta, Y. , Lee, K.E. , Hajdu, C.H. , Miller, G. , Bar-Sagi, D. , 2012. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, W. , Su, G.H. , 2012. Challenges and advances in mouse modeling for human pancreatic tumorigenesis and metastasis. Cancer Metastasis Rev. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi, S. , Lowenfels, A.B. , Morselli-Labate, A.M. , Maisonneuve, P. , Pezzilli, R. , 2010. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract. Res. Clin. Gastroenterol. 24, 349–358. [DOI] [PubMed] [Google Scholar]
- Redston, M.S. , Caldas, C. , Seymour, A.B. , Hruban, R.H. , da Costa, L. , Yeo, C.J. , Kern, S.E. , 1994. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 54, 3025–3033. [PubMed] [Google Scholar]
- Rothwell, P.M. , Fowkes, F.G. , Belch, J.F. , Ogawa, H. , Warlow, C.P. , Meade, T.W. , 2011. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377, 31–41. [DOI] [PubMed] [Google Scholar]
- Rovira, M. , Scott, S.G. , Liss, A.S. , Jensen, J. , Thayer, S.P. , Leach, S.D. , 2010. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc. Natl. Acad. Sci. U.S.A. 107, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley, M. , Ohashi, A. , Mondal, G. , Mills, L. , Yang, L. , Zhang, L. , Sundsbak, R. , Shapiro, V. , Muders, M.H. , Smyrk, T. , Couch, F.J. , 2011. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology 140, 1303–1313.e1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangiorgi, E. , Capecchi, M.R. , 2009. Bmi1 lineage tracing identifies a selfrenewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc. Natl. Acad. Sci. U.S.A. 106, 7101–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickart, M. , Huang, X. , Lill, J.R. , Liu, J. , Ferrando, R. , French, D.M. , Maecker, H. , O'Rourke, K. , Bazan, F. , Eastham-Anderson, J. , Yue, P. , Dornan, D. , Huang, D.C. , Dixit, V.M. , 2010. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107. [DOI] [PubMed] [Google Scholar]
- Sclabas, G.M. , Fujioka, S. , Schmidt, C. , Evans, D.B. , Chiao, P.J. , 2003. NF-kB in pancreatic cancer. Int. J. Gastrointest. Cancer 33, 15–26. [DOI] [PubMed] [Google Scholar]
- Shi, C. , Hong, S.M. , Lim, P. , Kamiyama, H. , Khan, M. , Anders, R.A. , Goggins, M. , Hruban, R.H. , Eshleman, J.R. , 2009. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Mol. Cancer Res. 7, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, D.J. , Murphy, E.A. , Desgrosellier, J.S. , Mielgo, A. , Lau, S.K.M. , Barnes, L.A. , Lesperance, J. , Huang, M. , Schmedt, C. , Tarin, D. , Lowy, A.M. , Cheresh, D.A. , 2011. Oncogenic Ras/Src cooperativity in pancreatic neoplasia. Oncogene 30, 2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu, H. , Lin, L. , Takahashi, T. , Nomura, M. , Suzuki, M. , Wistuba, I.I. , Fong, K.M. , Lee, H. , Toyooka, S. , Shimizu, N. , Fujisawa, T. , Feng, Z. , Roth, J.A. , Herz, J. , Minna, J.D. , Gazdar, A.F. , 2005. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 97, 339–346. [DOI] [PubMed] [Google Scholar]
- Siveke, J.T. , Einwächter, H. , Sipos, B. , Lubeseder-Martellato, C. , Klöppel, G. , Schmid, R.M. , 2007. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell 12, 266–279. [DOI] [PubMed] [Google Scholar]
- Siveke, J.T. , Lubeseder-Martellato, C. , Lee, M. , Mazur, P.K. , Nakhai, H. , Radtke, F. , Schmid, R.M. , 2008. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology 134, 544–555. [DOI] [PubMed] [Google Scholar]
- Skoulidis, F. , Cassidy, L.D. , Pisupati, V. , Jonasson, J.G. , Bjarnason, H. , Eyfjord, J.E. , Karreth, F.A. , Lim, M. , Barber, L.M. , Clatworthy, S.A. , Davies, S.E. , Olive, K.P. , Tuveson, D.A. , Venkitaraman, A.R. , 2010. Germline Brca2 heterozygosity promotes Kras(G12D)-driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 18, 499–509. [DOI] [PubMed] [Google Scholar]
- Stanger, B.Z. , Stiles, B. , Lauwers, G.Y. , Bardeesy, N. , Mendoza, M. , Wang, Y. , Greenwood, A. , Cheng, K.-h. , McLaughlin, M. , Brown, D. , DePinho, R.A. , Wu, H. , Melton, D.A. , Dor, Y. , 2005. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 8, 185–195. [DOI] [PubMed] [Google Scholar]
- Tan, X.L. , Reid Lombardo, K.M. , Bamlet, W.R. , Oberg, A.L. , Robinson, D.P. , Anderson, K.E. , Petersen, G.M. , 2011. Aspirin, nonsteroidal anti-inflammatory drugs, acetaminophen, and pancreatic cancer risk: a clinic-based case-control study. Cancer Prev. Res. (Phila) 4, 1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer, S.P. , di Magliano, M.P. , Heiser, P.W. , Nielsen, C.M. , Roberts, D.J. , Lauwers, G.Y. , Qi, Y.P. , Gysin, S. , Fernández-del Castillo, C. , Yajnik, V. , Antoniu, B. , McMahon, M. , Warshaw, A.L. , Hebrok, M. , 2003. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, J. , Tanaka, M. , Ohtsuka, T. , Tokunaga, S. , Shimosegawa, T. , 2012. Research Committee of Intractable Diseases of the Pancreas. Surgery for chronic pancreatitis decreases the risk for pancreatic cancer: a multicenter retrospective analysis. Surgery 139, 3806–3816. [DOI] [PubMed] [Google Scholar]
- van Heek, N.T. , Meeker, A.K. , Kern, S.E. , Yeo, C.J. , Lillemoe, K.D. , Cameron, J.L. , Offerhaus, G.J. , Hicks, J.L. , Wilentz, R.E. , Goggins, M.G. , De Marzo, A.M. , Hruban, R.H. , Maitra, A. , 2002. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am. J. Pathol. 161, 1541–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, D.F. , Yan, K.P. , Treilleux, I. , Gay, F. , Arfi, V. , Kaniewski, B. , Marie, J.C. , Lepinasse, F. , Martel, S. , Goddard-Leon, S. , Iovanna, J.L. , Dubus, P. , Garcia, S. , Puisieux, A. , Rimokh, R. , Bardeesy, N. , Scoazec, J.Y. , Losson, R. , Bartholin, L. , 2009. Inactivation of TIF1gamma cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 5, e1000575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, A. , Herman, J. , Schulick, R. , Hruban, R.H. , Goggins, M. , 2011. Pancreatic cancer. Lancet 378, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff, D.D. , Ramanathan, R.K. , Borad, M.J. , Laheru, D.A. , Smith, L.S. , Wood, T.E. , Korn, R.L. , Desai, N. , Trieu, V. , Iglesias, J.L. , Zhang, H. , Soon-Shiong, P. , Shi, T. , Rajeshkumar, N.V. , Maitra, A. , Hidalgo, M. , 2011. Gemcitabine plus nabpaclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J. Clin. Oncol. 29, 4548–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, M. , Greten, F.R. , Weber, C.K. , Koschnick, S. , Mattfeldt, T. , Deppert, W. , Kern, H. , Adler, G. , Schmid, R.M. , 2001. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev. 15, 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen, C.B. , Olive, K.P. , 2012. Genetically engineered mouse models of pancreatic cancer. Cancer J. 18, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilentz, R.E. , Geradts, J. , Maynard, R. , Offerhaus, G.J. , Kang, M. , Goggins, M. , Yeo, C.J. , Kern, S.E. , Hruban, R.H. , 1998. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 58, 4740–4744. [PubMed] [Google Scholar]
- Wilentz, R.E. , Albores-Saavedra, J. , Hruban, R.H. , 2000. Mucinous cystic neoplasms of the pancreas. Semin. Diagn. Pathol. 17, 31–42. [PubMed] [Google Scholar]
- Wu, K.L. , Gannon, M. , Peshavaria, M. , Offield, M.F. , Henderson, E. , Ray, M. , Marks, A. , Gamer, L.W. , Wright, C.V.E. , Stein, R. , 1997. Hepatocyte nuclear factor 3β is involved in pancreatic β-cell-specific transcription of the pdx-1 gene. Mol. Cell. Biol. 17, 6002–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Jiao, Y. , Dal Molin, M. , Maitra, A. , de Wilde, R.F. , Wood, L.D. , Eshleman, J.R. , Goggins, M.G. , Wolfgang, C.L. , Canto, M.I. , Schulick, R.D. , Edil, B.H. , Choti, M.A. , Adsay, V. , Klimstra, D.S. , Offerhaus, G.J. , Klein, A.P. , Kopelovich, L. , Carter, H. , Karchin, R. , Allen, P.J. , Schmidt, C.M. , Naito, Y. , Diaz, L.A. , Kinzler, K.W. , Papadopoulos, N. , Hruban, R.H. , Vogelstein, B. , 2011. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. U.S.A. 108, 21188–21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Matthaei, H. , Maitra, A. , Dal Molin, M. , Wood, L.D. , Eshleman, J.R. , Goggins, M. , Canto, M.I. , Schulick, R.D. , Edil, B.H. , Wolfgang, C.L. , Klein, A.P. , Diaz, L.A. , Allen, P.J. , Schmidt, C.M. , Kinzler, K.W. , Papadopoulos, N. , Hruban, R.H. , Vogelstein, B. , 2011. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 3, 92ra66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch, R.L. , Gould, S.E. , Scales, S.J. , Tang, T. , Tian, H. , Ahn, C.P. , Marshall, D. , Fu, L. , Januario, T. , Kallop, D. , Nannini-Pepe, M. , Kotkow, K. , Marsters, J.C. , Rubin, L.L. , de Sauvage, F.J. , 2008. A paracrine requirement for hedgehog signalling in cancer. Nature 455, 406–410. [DOI] [PubMed] [Google Scholar]
- Ying, H. , Elpek, K.G. , Vinjamoori, A. , Zimmerman, S.M. , Chu, G.C. , Yan, H. , Fletcher-Sananikone, E. , Zhang, H. , Liu, Y. , Wang, W. , Ren, X. , Zheng, H. , Kimmelman, A.C. , Paik, J.H. , Lim, C. , Perry, S.R. , Jiang, S. , Malinn, B. , Protopopov, A. , Colla, S. , Xiao, Y. , Hezel, A.F. , Bardeesy, N. , Turley, S.J. , Wang, Y.A. , Chin, L. , Thayer, S.P. , DePinho, R.A. , 2011. Pten is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 1, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, T. , Shiraki, N. , Baba, H. , Goto, M. , Fujiwara, S. , Kume, K. , Kume, S. , 2008. Expression patterns of epiplakin in pancreas, pancreatic cancer and regenerating pancreas. Genes Cells 13, 667–678. [DOI] [PubMed] [Google Scholar]
- Zhu, L. , Shi, G. , Schmidt, C.M. , Hruban, R.H. , Konieczny, S.F. , 2007. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am. J. Pathol. 171, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]