

Abstract
The relative genetic simplicity of leukaemias, the development of which likely relies on a limited number of initiating events has made them ideal for disease modelling, particularly in the mouse. Animal models provide incomparable insights into the mechanisms of leukaemia development and allow exploration of the molecular pillars of disease maintenance, an aspect often biased in cell lines or ex vivo systems. Several of these models, which faithfully recapitulate the characteristics of the human disease, have been used for pre‐clinical purposes and have been instrumental in predicting therapy response in patients. We plea for a wider use of genetically defined animal models in the design of clinical trials, with a particular focus on reassessment of existing cancer or non‐cancer drugs, alone or in combination.
Keywords: Cancer, Leukaemia, Mouse models
1. Introduction
Leukaemias have been particularly striking to the non‐medical audience because they can hit at any age, including childhood and may have an extremely rapid unfavourable outcome. Leukaemias tremendously benefited from translational research initiated in the 1960's, which allowed their classification on morphologic and antigenic criteria. Soon after, the progress of cytogenetics led to the emergence of molecular classifications and to the identification of the main oncogenic drivers (Look, 1997). These advances were not only stimulated by the ability to sample the tumour easily, including during or after therapy, but also by the ability to flow sort leukaemic cells. Finally, even though leukaemias are rare (sometimes even orphan) and very diverse diseases, the relative simplicity of genetic alterations has permitted physio‐pathological studies, including the pioneering creation of animal models recapitulating the diseases and constituting precious tools for experimental therapeutics. These diseases are now much better understood in their physiopathology than most other cancers and some of them are even definitively cured. Clinically, the small numbers of leukaemia patients has facilitated tailored strategies and clinical protocols, allowing them to gain very significant survival benefits in the past 40 years. Haematology appears as a leading field in cancer biology at large, in particular to demonstrate that cancers constitute a juxtaposition of rare distinct diseases and that biology can pave the way to cures.
2. Leukaemias: a genetic metonymy of cancers?
2.1. Specific features of leukaemias
Cancers result from the accumulation of multiple genetic or epigenetic lesions in cells, progressively driving them from normal to fully malignant. This is well reflected by epidemiologic studies from the 1950's, demonstrating an exponential incidence of cancer rates as age progresses. More recently, genome sequencing has revealed the existence, in many solid tumours of 6–10 recurrent mutations likely to be the drivers of transformation and a 10‐fold larger number of random alterations considered as passenger ones (Jones et al., 2008).
In contrast to solid tumours, some leukaemias are not diseases of old age and many of them actually only occur in infants or young children. Others have a constant incidence with age. As expected, the genetics of these conditions vary and the earlier the disease occurs, the fewer genetic events are likely to be involved. The reason why haematopoietic cells are more susceptible to oncogenic transformation than epithelial or mesenchymal ones is not understood. In lymphocyte‐derived leukaemias, however, this likely reflects their ability to rearrange their genomes as part of their differentiation program (Haluska and Croce, 1987). It is also clear that some initiating events may occur very early in development, sometimes in utero (Ford et al., 1993; Hong et al., 2008).
For the purpose of our demonstration, we will focus on some well‐characterized diseases for which significant evidence points to a single dominant rate‐limiting genetic alteration. These are often the results from chromosomal translocations and represent highly attractive therapeutic targets, as demonstrated below.
2.2. Some leukaemias are quasi‐monogenic
The best‐studied disease to that respect is probably acute promyelocytic leukaemia (APL). APL incidence is poorly affected by age (Vickers et al., 2000). It is driven by a t(15,17) translocation present in over 98% of patients (Rowley et al., 1977), shown to yield the expression of the PML/RARA fusion protein (de Thé et al., 1990; de Thé et al., 1991). It was assumed that cooperating events were required for the full APL phenotype, because transgenic mice do not develop an immediate disease (see below). Yet, recent whole genome genetic exploration revealed the near absence of gene amplification, deletion, or mutations in addition to the main driving translocation (Akagi et al., 2009; Karnan et al., 2006; Wartman et al., 2011; Welch et al., 2012). The only rare additional alterations (Ras, UTX, ASLX1 or FLT3 mutations, Myc trisomy, KDM6A loss), shared with other myeloid leukaemias have been associated with disease progression and appear to accelerate the course of the disease, but may not be absolutely required for its onset. Some specific epigenetic changes were observed, but it is unclear whether those are acquired of just a marker of a specific differentiation stage (Figueroa et al., 2010). That APL genomes are so stable is likely an important explanation for the rare occurrence of therapy resistance. In fact, the delay observed in mice may reflect the time for PML/RARA to acquire the specific post‐translational modifications required for its full transforming activity (Zhu et al., 2005) or to tune down an inappropriately activated senescence program. Thus APL can be considered as an example of monogenic cancer.
Some other leukaemias share similar features with APL. Chronic myelogenous leukaemia is due to the Philadelphia chromosome, encoding the Bcr/Abl fusion. Careful epidemiological modelling has revealed that it can be explained by a single mutation model (Michor et al., 2006), despite the fact that its genome is quite unstable and that other genetic abnormalities are frequently observed, particularly when the disease progresses towards acute phase (Wada et al., 1994). This relative low number of driving abnormalities holds true for most translocation‐associated leukaemias. Thus, haematopoietic disorders are associated with fewer genetic alterations than most solid tumours, suggesting that their biologic modelling and reconstruction in model systems are likely to be much easier.
2.3. Darwinian selection and/or hierarchy?
The novel power of deep sequencing has allowed an unprecedented exploration of the Darwinian evolution of cancers, notably leukaemias. This has uncovered the existence of multiple parallel pathways of disease evolution after a common initiating event (Notta et al., 2011). Importantly, these clones evolve during the course of the disease and in response to therapy (Ding et al., 2012). Chemotherapy not only enhances genomic instability and favours new mutations, but also allows the clonal dominance of a pre‐existing minor tumour component resistant to therapy (Greaves and Maley, 2012). Similar findings have been made in other tumours, where deep sequencing has allowed the pioneering observation of clonal dynamics during spontaneous evolution and adaptation to therapy, providing one of the most striking example of rapid genetic drift under stress adaptation.
How can sense be made within this genetic catastrophe? In contrast to many solid tumours, where it is difficult to find recurrent alterations or construct pathways that would drive or initiate the disease, leukaemia are often characterised by well‐identified recurrent alterations, which are most likely to be the primary drivers of the disease. For example, elegant studies have demonstrated that the Tel/AML1 fusion is the initiating event of some B‐cell ALL (Anderson et al., 2011). Yet, molecular epidemiological studies have demonstrated that this fusion can be observed in a number of individuals that will never develop the disease (Brassesco, 2008). Its expression induces the formation of a small clone of stem cells which can persist for a long time, until other events occur or this clone is ultimately cleared (Hong et al., 2008). One of the important events of progression towards growth appears to be the loss of the remaining AML1 allele. Importantly, relapse often occurs through a reinitiation of leukaemia from the pre‐malignant clone, rather than from additional mutations conferring resistance to the fully malignant clone (van Delft et al., 2011). Thus these studies demonstrate that although multiple genes may be mutated in a haematologic malignancy, even sometimes in normal stem cells prior to actual transformation (Welch et al., 2012), there is a sharp hierarchy and a primary, if not single, driver alteration, at least in translocation‐associated diseases. The latter is obviously a prime candidate, not only for deconstructing the disease pathogenesis, but also for therapeutic purposes. Besides, genetic analyses can unravel the unexpected requirements of some specific pathways for tumour maintenance, but not for that of normal cells.
Several lines of evidence have suggested that cell‐lines only imperfectly reflect tumour biology. Not every leukaemia can be derived into a cell line, and for many of them significant concerns were raised as to the presence of additional genetic/epigenetic changes required to sustain growth ex vivo.
3. Mouse models of leukaemias
On the contrary, the creation of murine models of leukaemias has allowed significant advances we will review the different approaches in a historical order. The respective uses, advantages and limitations of each system are summarized in Figure and Table 1.
Table 1.
Comparison of the respective features of different technologies used to obtain mouse models of leukaemia.
| Advantages | Drawbacks | |
|---|---|---|
| Xenografts | • Human diseases • Ready to use | • Engraftment difficulties • Experimental reproducibility • Transplantability? • Cohorts size and homogeneity |
| Viral transformation‐derived models | • Fast • Allow large‐scale experiments | • Choice of oncogene(s) • Insertion site issues |
| Transgenics‐derived models | • Reproducible • Allow large‐scale experiments | • Choice of transgene(s) • Choice of promoter/cell type specificity issue • Time‐consuming |
3.1. Xenografts
Although growth of human leukaemia cell lines in rodent was described as early as in the 1960's, it is only recently that the use of profoundly immuno‐deficient mice has allowed the transplantation and propagation of primary human leukaemia cells (Imada, 2003). While these studies have been essential in analysing hierarchy in human myeloid malignancies and contributed to define human leukaemia stem cells (Greaves, 2010), they are often limited by the low efficiency of engraftment, precluding drug screening or pathway identification.
3.2. Retroviral transductions
Manipulation of the murine bone marrow, purification of haematopoietic progenitors, viral transduction of oncogenic proteins and reimplantation of these transformed cells in syngenic animals, a technically simple procedure was routinely performed to establish oncogene‐driven leukaemia models in mice, particularly with BCR/ABL, a very potent oncogene (Pear et al., 1998). The retroviral transduction/transplantation CML mouse model was shown to be a faithful representation of the human CML and was successfully used for genetic analyses of the driver's function, for the identification of critical downstream signalling pathways of BCR‐ABl and for testing the efficacy of therapeutic drugs in vivo. However issues related to the level of expression of the transduced oncogenes, as well as the nature of the precursor cell transduced have been raised. Moreover, for some unknown reason this approach is not suitable for all oncoproteins.
3.3. Transgenics
Because of the relative genetic simplicity of leukaemia, these were among the first diseases to be successfully modelled in transgenic experiments (Harris et al., 1988). Multiple issues were later raised, notably regarding the critical importance of the maturity of the cell population actually targeted by the promoter driving expression of the transgene. However, technological progress has permitted hundreds of models of haematopoietic malignancies to be generated that way. Multiple refinements have been obtained through the incredible power of mouse genome technology, so that most human haematopoietic malignancies now have several faithful equivalents in the mouse. These are allowing an extraordinary dissection of the biochemical and cellular mechanisms of disease development and maintenance.
The APL model has been an unprecedented example of molecular investigation of tumour pathogenesis. After some unsuccessful attempts using promoters that were only active in fully differentiated cells (Early et al., 1996), two promoters MRP8 and cathepsin G were found to yield APL‐like leukemias at an efficient rate (Brown et al., 1997; Grisolano et al., 1997; He et al., 1997). These model systems became invaluable to analyse APL pathogenesis. This first started with deciphering the role of the two fusion partners (Kogan et al., 2000), of partners proteins involved in the oncogenic complex (RXRA, HDAC) (Matsushita et al., 2006; Zhu et al., 2007), then with the identification of critical post‐translational modifications (Nasr et al., 2008; Zhu et al., 2005), culminating with the elucidation of the mechanisms of therapy response (de The and Chen, 2010; Kogan, 2009; Lallemand‐Breitenbach et al., 2012; Nasr et al., 2008)(see below). The pathogenesis of the variant form of the disease was also investigated (He et al., 2000). The almost perfect parallelism between mice and patient response was covered in many influential reviews (Kogan and Bishop, 1999; Lallemand‐Breitenbach et al., 2005; Piazza et al., 2001).
4. Mouse models as relevant pre‐clinical tools
These mouse models are particularly well‐suited to assess the effects of new drugs, but also to identify among existing drugs those that may have the greatest efficacy alone or in combination Figure 1. Leukaemic blasts are easily and rapidly transplantable, allowing the formation of large cohorts of mice with identical diseases that can be used for preclinical purposes (Lallemand‐Breitenbach et al., 2005; Nardella et al., 2011).
Figure 1.
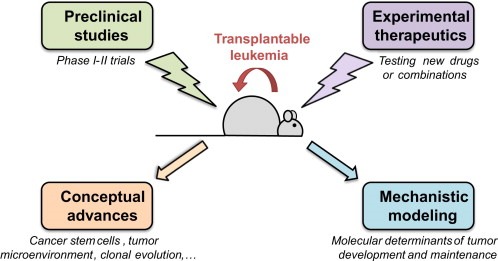
Schematic representation of the different possible uses of mouse models of leukemia.
4.1. Clinical utility: the APL paradigm
In some cases, these models have shown clinical utility, as curative regimens have been discovered in mice and later successfully transferred to patients (de The et al., 2012; Nasr and de The, 2010; Nasr et al., 2009). Perhaps the most demonstrative illustration of the combined power of mouse models and drug combinations is the cure of APL by the retinoic acid and arsenic association (Chen et al., 2011; Wang and Chen, 2008). Either drug alone was known to yield APL regression in patients and arsenic even allowed 70% cure rates (de The and Chen, 2010; Mathews et al., 2010). Studies of ex vivo differentiation demonstrated a clear antagonism between the two drugs, precluding their combination in patients (Shao et al., 1998). However subsequent studies in the mouse demonstrated that differentiation was not a valid end‐point reflecting APL eradication (Ablain and de The, 2011; Nasr et al., 2008). On the contrary, several different mouse models had predicted synergistic effects of the two drugs given frontline (Jing et al., 2001; Lallemand‐Breitenbach et al., 1999; Rego et al., 2000). Importantly, while a clear‐cut synergy for disease elimination and survival was found, an antagonism for differentiation was also observed, in line with the observations made on primary cells (Shao et al., 1998). It is remarkable that these different models, using different technologies and approaches all reached the same conclusion (Bernardi et al., 2002; Lallemand‐Breitenbach et al., 2005).
Ultimately, several clinical trials demonstrated the potency of the frontline combination and its superiority to the standard of care for overall survival, patient comfort and treatment cost (Estey et al., 2006; Hu et al., 2009; Shen et al., 2004; Tallman and Altman, 2009). Thus APL can be considered not only an exemplary clinical success of targeted therapies (de The and Chen, 2010), but also one of mouse modelling of human cancers (Lallemand‐Breitenbach et al., 2005).
4.2. Testing new drugs and associations?
It is becoming increasingly clear that cancer is a highly heterogeneous group of diseases. Each molecularly homogeneous group may end up being an orphan disease. It is now most unlikely that we will find universal strategies against tumours at large. As microbiologists learnt to adjust the antibiotics to the pathogenic bacteria strains, we will have to test thousands of drugs and/or combinations to find the one to which a specific cancer subset is exquisitely sensitive. In some cases, the number of available drugs, exceeds the actual number of patients available for phase I/II trials, not to mention combinations in phase III. Clearly, mice present an attractive alternative to demonstrate the potency of drugs in a defined genetic setting and even more to test associations between different drugs with different targets or even drugs targeting the same protein through non‐overlapping (and hence potentially synergistic) mechanisms.
In the APL example, mice were used to test new drugs, such as nucleotide derivatives (Guillemin et al., 2002; Nasr et al., 2008), arsenicals (Kroemer and de The, 1999; Lallemand‐Breitenbach et al., 1999) or rexinoids (Altucci et al., 2005). Actually, on the basis of the APL studies, several centres have gone forward to implement the systematic coupling of mouse modelling and patient trials, an approach known as the “co‐clinical trial project” (Nardella et al., 2011). Indeed, it is anticipated that patients could benefit from early drug testing to tailor treatment regimen not only on tumour genetics (targeted therapy), but also on the actual in vivo tumour sensitivity.
4.3. Exploring new therapeutic endpoints
Most drug screening processes use growth arrest or apoptosis as their major endpoint. However, in line with the cancer stem cell hypothesis, multiple studies have identified drugs that efficiently kill the tumour bulk, but unfortunately cannot prevent rapid regrowth of the tumour. These agents most likely target progression events, but fail to assess “stemness” or self‐renewal. Although feasible, testing “stemness” ex vivo is highly cumbersome experimentally.
Conversely, in several mouse models of cancer, some recent studies have identified drugs or combinations that have modest effect on short‐term tumour growth, but dramatically affect the ability of tumour cells to transplant into secondary hosts. The latter only, actually predicts disease eradication and long‐term survival. For example, the combination of arsenic and interferon alpha appears to be selectively toxic for HTLVI‐infected cells that express the viral transactivator Tax (Bazarbachi et al., 1999; El‐Sabban et al., 2000). This interferon/arsenic combination cures mice transplanted with murine adult T cell leukaemia (ATL) cells derived from Tax transgenics (El Hajj et al., 2010). However, this combination does not induce rapid growth arrest or apoptosis. Indeed, the tumours continue to grow on a therapy that is ultimately curative! But in vivo treated ATL cells are incapable of transplanting leukaemia into secondary hosts, demonstrating that even a short treatment abolished “stemness” (El Hajj et al., 2010). Importantly, as in APL, cure of the animals was translated into clear‐cut benefit in patients (Kchour et al., 2009). Similar findings were made on calcineurin inhibition by cyclosporine A in T‐ALL, which appears to have the same effect (Medyouf et al., 2007) (J. Ghysdael, unpublished).
4.4. Understanding and manipulating tumour biology
Mouse models are also precious to address a number of very basic issues about cancer biology. The first issue is tumour heterogeneity and clonal selection. As described above, it is becoming increasingly clear that tumours constitute prototypic models of clone selection and evolution (Greaves and Maley, 2012). The advantage of the mouse system is that it allows external manipulation of several types of selective pressure and the brutal increase in genomic changes through mutagenic interventions to accelerate the tumour evolution (irradiation, ENU mutagenesis, retroviral mutagenesis) (Joslin et al., 2007; Kool and Berns, 2009; Uren et al., 2008). Similarly, treatment‐related selective pressure can be applied to promote (…) and to highlight a rapid evolution of the leukemic clone, together with highlighting the basis for therapy resistance. These are key issues, with major implications for clinical management of the disease, that obviously cannot be efficiently tackled in patient‐derived materials.
Novel imaging strategies have allowed an unprecedented level of morphological analyses of cell interaction and in vivo migration. Cell labelling combined to two‐photon imaging has permitted the visualisation of multiple cellular events, including migration, invasion, interaction with the haematopoietic niche or the stroma, formation of immunological synapse, destruction by immune cells, phagocytosis (Bengtsson et al., 2011; Rauch et al., 2009). In that respect, mouse models have enabled a complete reassessment of the relationship between tumour and stroma, together with the realisation that the latter is not restricted to epithelial tumours. Indeed, cellular interactions between leukemic cells and the microenvironment have been discovered (Zhang et al., 2012). Thus, in many unrelated aspects, mouse models have driven major conceptual advances in cancer biology.
5. Perspective: using mice to promote more investigator‐driven trials
Science most often proceeds by chance and some of the most important breakthroughs came from unexpected, puzzling, findings that were subsequently explored in great details and often justified or explained a posteriori. These curative regimen were invariably first disregarded, progressively confirmed, ultimately becoming standard of care.
The last 20 years have seen an growing interest in the methodology of clinical trials. Very elaborate statistical methods, rigid procedures and quality controls, external data committees have been implemented in very large groups of patients. While this undoubtedly improved the intrinsic quality of trials aimed at comparing two predefined strategies, one could argue that this has paradoxically yielded a negative effect on the discovery process at large. With the current organisation of clinical research and the increasingly strict publications standards, it is probable that the princeps clinical observations of retinoic acid or arsenic in acute promyelocytic leukaemia (Chen et al., 1996; Huang et al., 1988; Sun et al., 1992), high dose cis‐platinum in testicular cancer, methotrexate in osteosarcoma (Jaffe, 1972), may experience difficulties to be published today. Yet, these represent some of the too rare examples of cancer cures by medical oncology.
Effective and innovative clinical trials should assess strategies, stratify cancer patients using genetics or other biological annotations, but also test combinations and/or dosage of existing drugs. Clearly, they should also be based on physio‐pathological disease models. In that sense, preclinical data from mouse is most likely going to emerge as one of the most important contributors to the cures to come.
Acknowledgements
We thank the Fondation des Treilles (France), where many of the ideas expressed in this article were matured. Research in the Paris Laboratory is supported by INSERM, CNRS, University Paris Diderot, Institut Universitaire de France, Ligue Nationale contre le Cancer, Institut National du Cancer, Prix Griffuel and the European Research Council (Senior grant 268729 – STEMAPL to HdT). J.A. was supported by a fellowship from Ecole Polytechnique and Association pour la Recherche contre le Cancer.
Ablain Julien, Nasr Rihab, Zhu Jun, Bazarbachi Ali, Lallemand-Breittenbach Valérie, de Thé Hugues, (2013), How animal models of leukaemias have already benefited patients, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.01.006.
References
- Ablain, J. , de The, H. , 2011. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 117, 5795–5802. [DOI] [PubMed] [Google Scholar]
- Akagi, T. , Shih, L.Y. , Kato, M. , Kawamata, N. , Yamamoto, G. , Sanada, M. , Okamoto, R. , Miller, C.W. , Liang, D.C. , Ogawa, S. , Koeffler, H.P. , 2009. Hidden abnormalities and novel classification of t(15;17) acute promyelocytic leukemia (APL) based on genomic alterations. Blood 113, 1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altucci, L. , Rossin, A. , Hirsch, O. , Nebbioso, A. , Vitoux, D. , Wilhelm, E. , Guidez, F. , Schiavone, E.M. , Grimwade, D. , Zelent, A. , De Thé, H. , Gronemeyer, H. , 2005. Rexinoid-triggered differentiation and tumours selective apoptosis of AML by protein kinase-A-mediated de-subordination of RXR. Cancer Res. 65, 8754–8765. [DOI] [PubMed] [Google Scholar]
- Anderson, K. , Lutz, C. , van Delft, F.W. , Bateman, C.M. , Guo, Y. , Colman, S.M. , Kempski, H. , Moorman, A.V. , Titley, I. , Swansbury, J. , Kearney, L. , Enver, T. , Greaves, M. , 2011. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469, 356–361. [DOI] [PubMed] [Google Scholar]
- Bazarbachi, A. , El-Sabban, M.E. , Nasr, R. , Quignon, F. , Awaraji, C. , Kersual, J. , Dianoux, L. , Zermati, Y. , Haidar, J.H. , Hermine, O. , de The, H. , 1999. Arsenic trioxide and interferon- alpha synergize to induce cell cycle arrest and apoptosis in HTLV-I transformed cells. Blood 93, 278–283. [PubMed] [Google Scholar]
- Bengtsson, N.E. , Kim, S. , Lin, L. , Walter, G.A. , Scott, E.W. , 2011. Ultra-high-field MRI real-time imaging of HSC engraftment of the bone marrow niche. Leuk. Off. J. Leuk. Soc. America, Leuk. Res. Fund U.K 25, 1223–1231. [DOI] [PubMed] [Google Scholar]
- Bernardi, R. , Grisendi, S. , Pandolfi, P.P. , 2002. Modelling haematopoietic malignancies in the mouse and therapeutical implications. Oncogene 21, 3445–3458. [DOI] [PubMed] [Google Scholar]
- Brassesco, M.S. , 2008. Leukemia/lymphoma-associated gene fusions in normal individuals. Genet. Mol. Res. 7, 782–790. [DOI] [PubMed] [Google Scholar]
- Brown, D. , Kogan, S. , Lagasse, E. , Weissman, I. , Alcalay, M. , Pelicci, P.G. , Atwater, S. , Bishop, J.M. , 1997. A PML RAR alpha transgene initiates murine acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U S A 94, 2551–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G.-Q. , Zhu, J. , Shi, X.-G. , Ni, J.-H. , Zhong, H.-J. , Si, G.-Y. , Jin, X.-L. , 1996. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia. As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood 88, 1052–1061. [PubMed] [Google Scholar]
- Chen, S.J. , Zhou, G.B. , Zhang, X.W. , Mao, J.H. , de The, H. , Chen, Z. , 2011. From an old remedy to a magic bullet: molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood 117, 6425–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de The, H. , Chen, Z. , 2010. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783. [DOI] [PubMed] [Google Scholar]
- de Thé, H. , Chomienne, C. , Lanotte, M. , Degos, L. , Dejean, A. , 1990. The t(15;17) translocation of acute promyelocytic leukemia fuses the retinoic acid receptor a gene to a novel transcribed locus. Nature 347, 558–561. [DOI] [PubMed] [Google Scholar]
- de Thé, H. , Lavau, C. , Marchio, A. , Chomienne, C. , Degos, L. , Dejean, A. , 1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 66, 675–684. [DOI] [PubMed] [Google Scholar]
- de The, H. , Le Bras, M. , Lallemand-Breitenbach, V. , 2012. The cell biology of disease: acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 198, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, L. , Ley, T.J. , Larson, D.E. , Miller, C.A. , Koboldt, D.C. , Welch, J.S. , Ritchey, J.K. , Young, M.A. , Lamprecht, T. , McLellan, M.D. , McMichael, J.F. , Wallis, J.W. , Lu, C. , Shen, D. , Harris, C.C. , Dooling, D.J. , Fulton, R.S. , Fulton, L.L. , Chen, K. , Schmidt, H. , Kalicki-Veizer, J. , Magrini, V.J. , Cook, L. , McGrath, S.D. , Vickery, T.L. , Wendl, M.C. , Heath, S. , Watson, M.A. , Link, D.C. , Tomasson, M.H. , Shannon, W.D. , Payton, J.E. , Kulkarni, S. , Westervelt, P. , Walter, M.J. , Graubert, T.A. , Mardis, E.R. , Wilson, R.K. , DiPersio, J.F. , 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early, E. , Moore, M.A. , Kakizuka, A. , Nason-Burchenal, K. , Martin, P. , Evans, R.M. , Dmitrovsky, E. , 1996. Transgenic expression of PML/RARalpha impairs myelopoiesis. Proc. Natl. Acad. Sci. U S A 93, 7900–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj, H. , El-Sabban, M. , Hasegawa, H. , Zaatari, G. , Ablain, J. , Saab, S.T. , Janin, A. , Mahfouz, R. , Nasr, R. , Kfoury, Y. , Nicot, C. , Hermine, O. , Hall, W. , de The, H. , Bazarbachi, A. , 2010. Therapy-induced selective loss of leukemia-initiating activity in murine adult T cell leukemia. J. Exp. Med. 207, 2785–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sabban, M.E. , Nasr, R. , Dbaibo, G. , Hermine, O. , Abboushi, N. , Quignon, F. , Ameisen, J.C. , Bex, F. , de The, H. , Bazarbachi, A. , 2000. Arsenic-interferon-alpha-triggered apoptosis in HTLV-I transformed cells is associated with tax down-regulation and reversal of NF-kappaB activation. Blood 96, 2849–2855. [PubMed] [Google Scholar]
- Estey, E. , Garcia-Manero, G. , Ferrajoli, A. , Faderl, S. , Verstovsek, S. , Jones, D. , Kantarjian, H. , 2006. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood 107, 3469–3473. [DOI] [PubMed] [Google Scholar]
- Figueroa, M.E. , Lugthart, S. , Li, Y. , Erpelinck-Verschueren, C. , Deng, X. , Christos, P.J. , Schifano, E. , Booth, J. , van Putten, W. , Skrabanek, L. , Campagne, F. , Mazumdar, M. , Greally, J.M. , Valk, P.J. , Lowenberg, B. , Delwel, R. , Melnick, A. , 2010. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, A.M. , Ridge, S.A. , Cabrera, M.E. , Mahmoud, H. , Steel, C.M. , Chan, L.C. , Greaves, M. , 1993. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature 363, 358–360. [DOI] [PubMed] [Google Scholar]
- Greaves, M. , 2010. Cancer stem cells: back to Darwin?. Semin. Cancer Biol. 20, 65–70. [DOI] [PubMed] [Google Scholar]
- Greaves, M. , Maley, C.C. , 2012. Clonal evolution in cancer. Nature 481, 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisolano, J.L. , Wesselschmidt, R.L. , Pelicci, P.G. , Ley, T.J. , 1997. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RARalpha under control of cathepsin G regulatory sequences. Blood 89, 376–387. [PubMed] [Google Scholar]
- Guillemin, M.C. , Raffoux, E. , Vitoux, D. , Kogan, S. , Soilihi, H. , Lallemand-Breitenbach, V. , Zhu, J. , Janin, A. , Daniel, M.T. , Gourmel, B. , Degos, L. , Dombret, H. , Lanotte, M. , De The, H. , 2002. In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J. Exp. Med. 196, 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haluska, F.G. , Croce, C.M. , 1987. Molecular mechanisms of chromosome translocation in human B- and T-cell neoplasia. Ann. N. Y. Acad. Sci. 511, 196–206. [DOI] [PubMed] [Google Scholar]
- Harris, A.W. , Pinkert, C.A. , Crawford, M. , Langdon, W.Y. , Brinster, R.L. , Adams, J.M. , 1988. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med. 167, 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, L. , Bhaumik, M. , Tribioli, C. , Rego, E.M. , Ivins, S. , Zelent, A. , Pandolfi, P.P. , 2000. Two critical hits for promyelocytic leukemia. Mol. Cell. 6, 1131–1141. [DOI] [PubMed] [Google Scholar]
- He, L.-Z. , Tribioli, C. , Rivi, R. , Peruzzi, D. , Pelicci, P.G. , Soares, V. , Cattoretti, G. , Pandolfi, P.P. , 1997. Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc. Natl. Acad. Sci. U S A 94, 5302–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, D. , Gupta, R. , Ancliff, P. , Atzberger, A. , Brown, J. , Soneji, S. , Green, J. , Colman, S. , Piacibello, W. , Buckle, V. , Tsuzuki, S. , Greaves, M. , Enver, T. , 2008. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science 319, 336–339. [DOI] [PubMed] [Google Scholar]
- Hu, J. , Liu, Y.F. , Wu, C.F. , Xu, F. , Shen, Z.X. , Zhu, Y.M. , Li, J.M. , Tang, W. , Zhao, W.L. , Wu, W. , Sun, H.P. , Chen, Q.S. , Chen, B. , Zhou, G.B. , Zelent, A. , Waxman, S. , Wang, Z.Y. , Chen, S.J. , Chen, Z. , 2009. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U S A 106, 3342–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. , Ye, Y. , Chen, R. , Chai, J. , Lu, J. , Zhoa, L. , Gu, L. , Wang, Z. , 1988. Use of all trans retinoic acid in the treatment of acute promyelocytic leukaemia. Blood 72, 567–572. [PubMed] [Google Scholar]
- Imada, K. , 2003. Immunodeficient mouse models of lymphoid tumors. Int. J. Hematol. 77, 336–341. [DOI] [PubMed] [Google Scholar]
- Jaffe, N. , 1972. Recent advances in the chemotherapy of metastatic osteogenic sarcoma. Cancer 30, 1627–1631. [DOI] [PubMed] [Google Scholar]
- Jing, Y. , Wang, L. , Xia, L. , Chen, G. , Chen, Z. , Miller, W.H. , Waxman, S. , 2001. Combined effect of all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia cells in vitro and in vivo. Blood 97, 264–269. [DOI] [PubMed] [Google Scholar]
- Jones, S. , Zhang, X. , Parsons, D.W. , Lin, J.C. , Leary, R.J. , Angenendt, P. , Mankoo, P. , Carter, H. , Kamiyama, H. , Jimeno, A. , Hong, S.M. , Fu, B. , Lin, M.T. , Calhoun, E.S. , Kamiyama, M. , Walter, K. , Nikolskaya, T. , Nikolsky, Y. , Hartigan, J. , Smith, D.R. , Hidalgo, M. , Leach, S.D. , Klein, A.P. , Jaffee, E.M. , Goggins, M. , Maitra, A. , Iacobuzio-Donahue, C. , Eshleman, J.R. , Kern, S.E. , Hruban, R.H. , Karchin, R. , Papadopoulos, N. , Parmigiani, G. , Vogelstein, B. , Velculescu, V.E. , Kinzler, K.W. , 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joslin, J.M. , Fernald, A.A. , Tennant, T.R. , Davis, E.M. , Kogan, S.C. , Anastasi, J. , Crispino, J.D. , Le Beau, M.M. , 2007. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood 110, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnan, S. , Tsuzuki, S. , Kiyoi, H. , Tagawa, H. , Ueda, R. , Seto, M. , Naoe, T. , 2006. Genomewide array-based comparative genomic hybridization analysis of acute promyelocytic leukemia. Genes Chromosomes Cancer 45, 420–425. [DOI] [PubMed] [Google Scholar]
- Kchour, G. , Tarhini, M. , Kooshyar, M.M. , El Hajj, H. , Wattel, E. , Mahmoudi, M. , Hatoum, H. , Rahimi, H. , Maleki, M. , Rafatpanah, H. , Rezaee, S.A. , Yazdi, M.T. , Shirdel, A. , de The, H. , Hermine, O. , Farid, R. , Bazarbachi, A. , 2009. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood 113, 6528–6532. [DOI] [PubMed] [Google Scholar]
- Kogan, S.C. , 2009. Curing APL: differentiation or destruction?. Cancer Cell 15, 7–8. [DOI] [PubMed] [Google Scholar]
- Kogan, S.C. , Bishop, J.M. , 1999. Acute promyelocytic leukemia: from treatment to genetics and back. Oncogene 18, 5261–5267. [DOI] [PubMed] [Google Scholar]
- Kogan, S.C. , Hong, S.H. , Shultz, D.B. , Privalsky, M.L. , Bishop, J.M. , 2000. Leukemia initiated by PMLRARalpha: the PML domain plays a critical role while retinoic acid-mediated transactivation is dispensable. Blood 95, 1541–1550. [PubMed] [Google Scholar]
- Kool, J. , Berns, A. , 2009. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat. Rev. Cancer 9, 389–399. [DOI] [PubMed] [Google Scholar]
- Kroemer, G. , de The, H. , 1999. Arsenic trioxide, a novel mitochondriotoxic anti-cancer agent?. J. Natl. Cancer Inst. 91, 743–745. [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach, V. , Guillemin, M.-C. , Janin, A. , Daniel, M.-T. , Degos, L. , Kogan, S.C. , Bishop, J.M. , de The, H. , 1999. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J. Exp. Med. 189, 1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach, V. , Zhu, J. , Chen, Z. , de The, H. , 2012. Mechanisms of APL cure through PML/RARA degradation by As2O3 . Trends Mol. Med. 18, 36–42. [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach, V. , Zhu, J. , Kogan, S. , Chen, Z. , de The, H. , 2005. Opinion: how patients have benefited from mouse models of acute promyelocytic leukaemia. Nat. Rev. Cancer 5, 821–827. [DOI] [PubMed] [Google Scholar]
- Look, A.T. , 1997. Oncogenic transcription factors in the human acute leukemias. Science 278, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Mathews, V. , George, B. , Chendamarai, E. , Lakshmi, K.M. , Desire, S. , Balasubramanian, P. , Viswabandya, A. , Thirugnanam, R. , Abraham, A. , Shaji, R.V. , Srivastava, A. , Chandy, M. , 2010. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: long-term follow-up data. J. Clin. Oncol. 28, 3866–3871. [DOI] [PubMed] [Google Scholar]
- Matsushita, H. , Scaglioni, P.P. , Bhaumik, M. , Rego, E.M. , Cai, L.F. , Majid, S.M. , Miyachi, H. , Kakizuka, A. , Miller, W.H. , Pandolfi, P.P. , 2006. In vivo analysis of the role of aberrant histone deacetylase recruitment and RAR alpha blockade in the pathogenesis of acute promyelocytic leukemia. J. Exp. Med. 203, 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medyouf, H. , Alcalde, H. , Berthier, C. , Guillemin, M.C. , dos Santos, N.R. , Janin, A. , Decaudin, D. , de The, H. , Ghysdael, J. , 2007. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 13, 736–741. [DOI] [PubMed] [Google Scholar]
- Michor, F. , Iwasa, Y. , Nowak, M.A. , 2006. The age incidence of chronic myeloid leukemia can be explained by a one-mutation model. Proc. Natl. Acad. Sci. U S A 103, 14931–14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella, C. , Lunardi, A. , Patnaik, A. , Cantley, L.C. , 2011. The APL paradigm and the co-clinical trial project. Cancer Discov. 1, 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr, R. , de The, H. , 2010. Eradication of acute promyelocytic leukemia-initiating cells by PML/RARA-targeting. Int. J. Hematol. 91, 742–747. [DOI] [PubMed] [Google Scholar]
- Nasr, R. , Guillemin, M.C. , Ferhi, O. , Soilihi, H. , Peres, L. , Berthier, C. , Rousselot, P. , Robledo-Sarmiento, M. , Lallemand-Breitenbach, V. , Gourmel, B. , Vitoux, D. , Pandolfi, P.P. , Rochette-Egly, C. , Zhu, J. , de The, H. , 2008. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 14, 1333–1342. [DOI] [PubMed] [Google Scholar]
- Nasr, R. , Lallemand-Breitenbach, V. , Zhu, J. , Guillemin, M.C. , de The, H. , 2009. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin. Cancer Res. 15, 6321–6326. [DOI] [PubMed] [Google Scholar]
- Notta, F. , Mullighan, C.G. , Wang, J.C. , Poeppl, A. , Doulatov, S. , Phillips, L.A. , Ma, J. , Minden, M.D. , Downing, J.R. , Dick, J.E. , 2011. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469, 362–367. [DOI] [PubMed] [Google Scholar]
- Pear, W.S. , Miller, J.P. , Xu, L. , Pui, J.C. , Soffer, B. , Quackenbush, R.C. , Pendergast, A.M. , Bronson, R. , Aster, J.C. , Scott, M.L. , Baltimore, D. , 1998. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 92, 3780–3792. [PubMed] [Google Scholar]
- Piazza, F. , Gurrieri, C. , Pandolfi, P.P. , 2001. The theory of APL. Oncogene 20, 7216–7222. [DOI] [PubMed] [Google Scholar]
- Rauch, D. , Gross, S. , Harding, J. , Niewiesk, S. , Lairmore, M. , Piwnica-Worms, D. , Ratner, L. , 2009. Imaging spontaneous tumorigenesis: inflammation precedes development of peripheral NK tumors. Blood 113, 1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego, E.M. , He, L.Z. , Warrell, R.P. , Wang, Z.G. , Pandolfi, P.P. , 2000. Retinoic acid (RA) and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL) unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc. Natl. Acad. Sci. U S A 97, 10173–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley, J.D. , Golomb, H.M. , Dougherty, C. , 1977. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1, 549–550. [DOI] [PubMed] [Google Scholar]
- Shao, W. , Fanelli, M. , Ferrara, F.F. , Riccioni, R. , Rosenauer, A. , Davison, K. , Lamph, W.W. , Waxman, S. , Pelicci, P.G. , Lo Coco, F. , Avvisati, G. , Testa, U. , Peschle, C. , Gambacorti-Passerini, C. , Nervi, C. , Miller, W.H.J. , 1998. Arsenic trioxide as an inducer of apoptosis and loss of PML/RARalpha protein in acute promyelocytic leukemia cells. J. Natl. Cancer Inst. 90, 124–133. [DOI] [PubMed] [Google Scholar]
- Shen, Z.X. , Shi, Z.Z. , Fang, J. , Gu, B.W. , Li, J.M. , Zhu, Y.M. , Shi, J.Y. , Zheng, P.Z. , Yan, H. , Liu, Y.F. , 2004. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U S A 101, 5328–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H.D. , Ma, L. , Hu, H.X. , Zhang, T.D. , 1992. Use of Ai-Ling n.1 injection, combined with pattern identification theory of chinese traditional medicine, in the treatment of acute promyelocytic leukemia: report from 32 patients. Chin. J. Integrat. Chin. West. Med. 12, 170–171. [Google Scholar]
- Tallman, M.S. , Altman, J.K. , 2009. How I treat acute promyelocytic leukemia. Blood 114, 5126–5135. [DOI] [PubMed] [Google Scholar]
- Uren, A.G. , Kool, J. , Matentzoglu, K. , de Ridder, J. , Mattison, J. , van Uitert, M. , Lagcher, W. , Sie, D. , Tanger, E. , Cox, T. , Reinders, M. , Hubbard, T.J. , Rogers, J. , Jonkers, J. , Wessels, L. , Adams, D.J. , van Lohuizen, M. , Berns, A. , 2008. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 133, 727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft, F.W. , Horsley, S. , Colman, S. , Anderson, K. , Bateman, C. , Kempski, H. , Zuna, J. , Eckert, C. , Saha, V. , Kearney, L. , Ford, A. , Greaves, M. , 2011. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117, 6247–6254. [DOI] [PubMed] [Google Scholar]
- Vickers, M. , Jackson, G. , Taylor, P. , 2000. The incidence of acute promyelocytic leukemia appears constant over most of a human lifespan, implying only one rate limiting mutation. Leukemia 14, 722–726. [DOI] [PubMed] [Google Scholar]
- Wada, C. , Shionoya, S. , Fujino, Y. , Tokuhiro, H. , Akahoshi, T. , Uchida, T. , Ohtani, H. , 1994. Genomic instability of microsatellite repeats and its association with the evolution of chronic myelogenous leukemia. Blood 83, 3449–3456. [PubMed] [Google Scholar]
- Wang, Z.Y. , Chen, Z. , 2008. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 111, 2505–2515. [DOI] [PubMed] [Google Scholar]
- Wartman, L.D. , Larson, D.E. , Xiang, Z. , Ding, L. , Chen, K. , Lin, L. , Cahan, P. , Klco, J.M. , Welch, J.S. , Li, C. , Payton, J.E. , Uy, G.L. , Varghese, N. , Ries, R.E. , Hoock, M. , Koboldt, D.C. , McLellan, M.D. , Schmidt, H. , Fulton, R.S. , Abbott, R.M. , Cook, L. , McGrath, S.D. , Fan, X. , Dukes, A.F. , Vickery, T. , Kalicki, J. , Lamprecht, T.L. , Graubert, T.A. , Tomasson, M.H. , Mardis, E.R. , Wilson, R.K. , Ley, T.J. , 2011. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J. Clin. Invest. 121, 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, J.S. , Ley, T.J. , Link, D.C. , Miller, C.A. , Larson, D.E. , Koboldt, D.C. , Wartman, L.D. , Lamprecht, T.L. , Liu, F. , Xia, J. , Kandoth, C. , Fulton, R.S. , McLellan, M.D. , Dooling, D.J. , Wallis, J.W. , Chen, K. , Harris, C.C. , Schmidt, H.K. , Kalicki-Veizer, J.M. , Lu, C. , Zhang, Q. , Lin, L. , O'Laughlin, M.D. , McMichael, J.F. , Delehaunty, K.D. , Fulton, L.A. , Magrini, V.J. , McGrath, S.D. , Demeter, R.T. , Vickery, T.L. , Hundal, J. , Cook, L.L. , Swift, G.W. , Reed, J.P. , Alldredge, P.A. , Wylie, T.N. , Walker, J.R. , Watson, M.A. , Heath, S.E. , Shannon, W.D. , Varghese, N. , Nagarajan, R. , Payton, J.E. , Baty, J.D. , Kulkarni, S. , Klco, J.M. , Tomasson, M.H. , Westervelt, P. , Walter, M.J. , Graubert, T.A. , Dipersio, J.F. , Ding, L. , Mardis, E.R. , Wilson, R.K. , 2012. The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Ho, Y.W. , Huang, Q. , Maeda, T. , Lin, A. , Lee, S.U. , Hair, A. , Holyoake, T.L. , Huettner, C. , Bhatia, R. , 2012. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 21, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J. , Nasr, R. , Peres, L. , Riaucoux-Lormiere, F. , Honore, N. , Berthier, C. , Kamashev, D. , Zhou, J. , Vitoux, D. , Lavau, C. , de The, H.s , 2007. RXR is an essential component of the oncogenic PML/RARA complex in vivo. Cancer Cell 12, 23–35. [DOI] [PubMed] [Google Scholar]
- Zhu, J. , Zhou, J. , Peres, L. , Riaucoux, F. , Honore, N. , Kogan, S. , de The, H. , 2005. A sumoylation site in PML/RARA is essential for leukemic transformation. Cancer Cell 7, 143–153. [DOI] [PubMed] [Google Scholar]