

Abstract
Long Interspersed Nuclear Element‐1 (LINE‐1 or L1) is an autonomous, mobile element within the human genome that transposes via a “copy and paste” mechanism and relies upon L1‐encoded endonuclease and reverse transcriptase (RT) activities to compromise genome integrity. L1 has been implicated in various forms of cancer, but its role in the regulation of the oncogenic phenotype is not understood. The present studies were conducted to evaluate mechanisms of genetic regulatory control in HepG2 cells by human L1, or a D702Y mutant deficient in RT activity, and their influence on cellular phenotype. Forced expression of synthetic L1 ORF1p and ORF2p was associated with formation of cytoplasmic foci and minor association with the nuclear compartment. While de novo L1 mobilizations were only identified in cells expressing wild type L1, and were absent in the D702Y mutant, changes in gene expression profiles involved RT dependent as well as RT independent mechanisms. Synthetic L1 altered the expression of 24 in silico predicted genetic targets; ten of which showed RT‐dependence, ten RT‐independence, and four reciprocal regulatory control by both wild type and RT mutant. Of five targets examined, only VCAM1 and PTPRB colocalized with newly retrotransposed wild type L1. Biological discretization to partition patterns of gene expression into unique frequencies identified adhesion, inflammation, and cellular metabolism as key processes targeted for molecular interference with disruption of epithelial‐to‐mesenchymal programming seen irrespective of the RT phenotype. These findings establish L1 as a key regulator of genome plasticity and EMT via mechanisms independent of RT activity.
Keywords: Epithelial-to-mesenchymal transition, Long interspersed nuclear element-1, Genetic reprogramming, Retrotransposons, Reverse transcriptase
Highlights
L1 reprograms the HepG2 genome via mechanisms that are independent of reverse transcriptase activity.
Biological discretization identified adhesion, inflammation, and metabolism as processes targeted for molecular interference.
The findings established L1 as a regulator of genome plasticity via mechanisms not directly coupled to retrotransposition.
1. Introduction
A prototypical human Long Interspersed Nuclear Element‐1 (LINE‐1 or L1) is ∼6 kb long and consists of an internal promoter located in the 5′ untranslated region (5′ UTR), two open reading frames (ORFs) which encode two proteins (ORF1p and ORF2p), both required for retrotransposition, and a short 3′ UTR with a poly (A) tail and signal (Swergold, 1990; Dombroski et al., 1991). ORF1p is a 40‐kDa protein with RNA binding and nucleic acid chaperone activities (Hohjoh and Singer, 1996). ORF2p is a 150‐kDa protein with endonuclease (EN) and reverse transcriptase (RT) activities, and a C‐terminal cysteine‐rich motif containing a zinc knuckle that mediates ORF2–DNA interactions, and/or facilitates the extended polymerization of RT domain (Martin, 2010; Mathias et al., 1991; Feng et al., 1996). The entire L1 sequence is flanked by so called target duplication sites. In a diploid human genome, an estimated 80–100 full‐length retrotransposition competent L1s (RC‐L1s) are present, with a small number termed “hot L1s” because of their high retrotransposition frequencies in cultured cells (Piskareva and Schmatchenko, 2006; Moran et al., 1996). L1s are mobilized through target‐primed reverse transcription (TPRT), also known as a “copy and paste” mechanism.
RC‐L1s are tightly regulated in somatic tissues (except in embryonic cells) where expression is repressed via DNA methylation and covalent protein modifications (Montoya‐Durango and Ramos, 2010). When activated, RC‐L1s mobilize throughout the genome to induce insertional mutations that cause genomic instability. Organisms have developed exquisite defense mechanisms against L1 mobilization, including APOBEC proteins, TREX1 exonuclease, Piwi proteins and Piwi‐interacting RNAs (Takaori‐Kondo, 2006; Brennecke et al., 2007; Houwing et al., 2007; Carmell et al., 2007). This laboratory and others have shown that environmental injury reactivates L1 in somatic tissues (Schiff et al., 1991; Lu and Ramos, 2003; Stribinskis and Ramos, 2006). L1 reactivation involves transcriptional activation by proteins that bind to the L1 regulatory region, as well as epigenetic modulation of DNA methyltransferases, histone acetyltransferases and deacetyltransferases (Teneng et al., 2011). Unscheduled reactivation has been proposed by McClintock to reshuffle the genome to afford survival advantage to the organism (McClintock, 1984). In accord with this view, L1 retrotransposition is an integral component of the global genomic response to environmental stress (Stribinskis and Ramos, 2006).
L1 is believed to modulate genetic programs via mechanisms that require formation of ribonucleoprotein particles in cis that shuttle L1 proteins back into the nucleus, followed by reverse transcription of L1 at sites of single strand breakage for reintegration of L1 (Kolosha and Martin, 1997). Integration of new L1s has been described as interspersed, and most insertions non‐targeted (Lander, 1996; Levin and Moran, 2011). De novo L1 mobilizations occur near or within intronic regions at a loosely defined sequence of 5‐TTTT/A‐3 (Beck et al., 2011). Cytogenetic studies, however, have revealed that older L1s specifically mobilized into gene poor and AT rich regions of the genome (Korenberg and Rykowski, 1988; Lander et al., 2001). Irrespective of the mechanism, modulation of genetic programs in mammalian cells is believed to be largely dependent upon insertion of L1s at new locations to disrupt chromatin architecture, afford new transcriptional regulatory sites and/or mediate alternative splicing. Specific molecular targets for regulatory control by L1 in mammalian cells have not been exhaustively identified, but gene mutations have been described (Beck et al., 2011; Goodier and Kazazian, 2008). Limited information is also available concerning the degree to which L1 modulates genes not directly impacted by insertional mutation.
Earlier we described an in silico regulatory genetic network for L1 derived from Boolean inferences using a coefficient of determination (COD) algorithm (Ramos et al., 2007). A total of 34 targets were identified as putative genetic targets and it was hypothesized that their co‐expression with L1 revealed the architecture of a biological regulatory network for L1 in mammalian cells. A limited number of studies were conducted at that time to evaluate the biological connectivity of elements within the network, with evidence presented that PAS proteins were key nodes for regulatory control of L1 in mammalian cells (Ramos et al., 2007).
Details of the mechanisms by which L1s influence the genome landscape are lacking. The present study was conducted to evaluate the impact of an RC‐synthetic L1 or its D702Y mutant deficient in RT activity on genetic expression in HepG2 cells focusing on a discretized biological regulatory network for L1 (Ramos et al., 2007). HepG2 cells were chosen as a model to evaluate the influence of L1 on genetic programming because this line retains highly functional activity of PAS proteins involved in the regulation of L1 functions (Teneng et al., 2007; Jennen et al., 2010). Evidence is presented here that 1) wild type synthetic L1 undergoes complete cycles of retrotransposition associated with de novo genome mobilizations; 2) reprogramming of the HepG2 genome by L1 involves RT‐independent mechanisms that are not directly coupled to retrotransposition; and 3) in silico prediction of the L1 genetic network coupled to biological validation identified adhesion, inflammation and cellular metabolism as key pathways controlling plasticity of the HepG2 genome.
2. Material and methods
2.1. Cloning
All cloning was completed using Infusion Cloning System (Catalog number 639691, Clontech, Mountain View, CA). Briefly, luciferase was removed from the pGL4.15Luc2P (Hygro) vector by double digestion with BglII and FseI to generate pB001CTR. pB001CTR was linearized with BglII and a double FlagMyc tag cloned into the pB001CTR to generate pB002. pB002 was linearized with BglII followed by cloning of the L1 ORF2 PCR fragment, or its mutant carrying a single point mutation (D702Y) in ORF2 that destroys RT activity. pB003WT and pB004Mut were linearized with XhoI for cloning of the double HaStrep tag. pB005WT and pB006Mut vectors were linearized with XhoI and a concatener of L1‐ORF1, L1‐UTR and CMV promoter cloned to generate pB011WT and pB012Mut which were in turn linearized with FseI. A concatener of a neomycin cassette, a globin intron inserted in neomycin gene and a CMV promoter, were cloned into the Fse1 site in opposite orientation to generate pB015WT and pB016Mut.
2.2. Generation of stable cell lines
Cells were transfected with pB015WT, pB016MUT and pB001CTR using lipofectamine. Transfection medium was removed and replaced with 2 ml of complete RPM1‐1654 medium containing 10% FBS plus 0.1 nM non‐essential amino acids. Cells were incubated under standard conditions for 3 days before daily selection with hygromycin until the appearance of clones (∼12–14 days). Single foci were trypsinized, diluted and expanded into 10 cm2 plates for 7 days and retrypsinized and expanded in 100 cm2 plates until cells reached 80–90% confluence. Cells were frozen in complete medium using 5% DMSO or processed immediately for RNA or protein. Total protein was extracted using the m‐PER reagent (Thermo Scientific, Rockford, IL) and L1 ORF1 and ORF2 proteins detected using antibodies against HA (HA.C5: sc‐57595, Santa Cruz, CA) or Flag (Anti‐Flag (F1804): Sigma, St. Louis, MO), respectively.
2.3. Indirect immunofluorescence
Cells stably expressing L1 proteins were grown on glass coverslips to 80% confluence, rinsed with PBS (phosphate‐buffered saline, pH 7.4) twice, fixed in 3% paraformaldehyde for 15 min at 4 °C and rinsed twice with PBS. Fixed cells were permeabilized with 0.1% Triton‐X100 for 5 min, blocked in 2% dry milk for 15 min, rinsed with PBS twice and incubated with conjugated primary antibodies (HA tag (6E2) Mouse mAB‐Alexa‐488 conjugate or Anti‐flag M2‐Alexa Fluor‐594 conjugate (1/1000), (Cell Signaling Technology Inc., Danvers, MA)) in PBS containing 1% dry milk and 0.1% Triton‐X100 overnight. Cells were washed three times with PBS and incubated in 1 μg/ml of Hoescht nuclear dye for 20 min and washed 2×. Cells were visualized in CARL ZEISS AXIOVERT 200 inverted microscope at 63× magnification.
2.4. Quantitative PCR (qPCR)
RNA was extracted using RNeasy Mini kit (Qiagen, Maryland, cat# 74104) following manufacturers' instructions. Five hundred ng of total RNA was combined with 50 ng of random hexamers and 10 mM dNTP in a final volume of 12 μl. This mixture was incubated at 65 °C for 5 min before cooling on ice for 2 min. SuperScript Reverse Transcriptase (1 μl), RNaseOUT™ ribonuclease inhibitor (1 μl), 5× First strand buffer (4 μl), and DTT (2 μl) were added to each tube (Invitrogen Corporation, Carlsbad, CA). Samples were incubated at 25 °C for 5 min, followed by incubation at 50 °C for 1 h. Enzymes in the reaction mix were inactivated by incubation at 70 °C for 15 min. Primers were designed against coding regions (Table 1) to yield template sizes of ∼120 bp for each gene using Primer3plus software (http://www.bioinformatics.nl/cgi‐bin/primer3plus/primer3plus.cgi). Special care was taken to design primers that spanned introns at the exon–exon junction to avoid amplification of genomic DNA. Quantitative real time PCR was carried out using 10 μl containing: 2.0 μl of cDNA, 0.5 μl gene‐specific primer pairs, and 5.0 μl SyberGreen dye (Molecular Probes, Eugene, Oregon). Real Time PCR amplification was completed using 7900HT Fast Real‐Time PCR system in accordance with manufacturer's instructions. Data was analyzed using the standard curve method and melting curves were used to ensure no primer dimer formation. To evaluate the efficiency of each primer set, the slope and R 2 for each standard curve was determined. A different primer set was used if the slope was greater than 4 or the R 2 was below 0.9. A quality primer set was characterized by a slope of ∼−3.34 and R 2 ≥ 0.9. Statistical analysis was completed using ANOVA followed by Tukey's post hoc analysis.
Table Table 1.
Primers used for RT‐PCR analysis of genes within the L1 regulatory network. Primer sets not capitalized were used to amplify probes for FISH analysis.
| Primer name | Primer sequence | Primer name | Primer sequence |
|---|---|---|---|
| AGL‐SybG‐1f | 5′‐GCATTGCCTTCAAACACAGGT‐3′ | PKIA‐SybG‐1f | 5′‐TACATGATATCCTGGTTTCCTCTG‐3′ |
| AGL‐SybG‐1r | 5′‐CACCTGAATAAAACCCTTGG‐3′ | PKIA‐SybG‐1r | 5′‐GCATCTTCTTCACCTTCTGTCTT‐3′ |
| CCL2‐sybG‐lf | 5′‐TGGTCTTGAAGATCACAGC‐3′ | POSTN‐SybG‐1f | 5′‐CAGCGCTATTCTGACGCCTC‐3′ |
| CCL2‐sybG‐lr | 5′‐CACCAATAGGAAGATCTCAGTGC‐3′ | POSTN‐SybG‐1r | 5′‐CCAAGTTGTCCCAAGCCTCATTAC‐3′ |
| CDO1‐SybG‐lf | 5′‐GCCAAGTTCGACCAGTACA‐3′ | PREI4‐SybG‐lf | 5′‐TCTTTGCAATATGTGGAAGCTG‐3′ |
| CDO1‐SybG‐lr | 5′‐TGAATACTGCTGCCATGTCC‐3′ | PREI4‐SybG‐lr | 5′‐CCACAACACACTGTCTCCTGTC‐3′ |
| CHI3L3‐SybG‐lf | 5′‐GACCTGCCCCGTTCAGTG‐3′ | PTPRB‐SybG‐1f | 5′‐CGTGTCAAACATGCCAATG‐3′ |
| CHI3L3‐SybG‐lr | 5′‐GTCCAGGTTGAGGCCATCAAAG‐3′ | PTPRB‐SybG‐1r | 5′‐AGAGGTCTCTGTCAGCTAGGG‐3′ |
| CLIC3‐SybG‐lf | 5′‐TGTTTGTCAAGGCGAGTGAG‐3′ | RBM39‐SybG‐1f | 5′‐GCAAGGACAGTCTTCTGTATGC‐3′ |
| CLIC3‐SybG‐1r | 5′‐CGTGGTGAGGGTGAAAGGTA‐3′ | RBM39‐SybG‐1r | 5′‐TCATCCTCACATCTCGAACCT‐3′ |
| COL3A1‐SybG‐1f | 5′‐CCTCCAACTGCTCCTACTCG‐3 | RGS‐SybG‐1f | 5′‐GCAAATATGGTCTTGCTGCAT‐3′ |
| COL3A1‐SybG‐1r | 5′‐ACCCTGGTTGTCCTGGAATA‐3′ | RGS‐SybG‐1r | 5′‐ACAGCTTTTGGGGTGATTTG‐3′ |
| CXCL1‐SybG‐1f | 5′‐CTGCCAGTGCTTGCAGAC‐3′ | SDCBP‐SybG‐1f | 5′‐TTCTGCTCCTATCCCTCACG‐3′ |
| CXCL1‐SybG‐lr | 5′‐CTGCCAGTGCTTGCAGAC‐3′ | SDCBP‐SybG‐1r | 5′‐GCCACATTTGCACGTATTTCT‐3′ |
| CYP2A4‐SybG‐1f | 5′‐GAGGCCAAGGTCCATGAG‐3′ | SGPLl‐SybG‐1f | 5′‐TGTAAATGGACATTGCACC‐3′ |
| CYP2A4‐SybG‐1r | 5′‐GTGGATCACTGCCTCCATGT‐3′ | SGPLl‐SybG‐1r | 5′‐CTGGCTGGAAGACAAACTC‐3′ |
| DPB‐SybG‐1f | 5′‐CTGACCTCTCGGGACACAC‐3′ | SLC32A2‐SybG‐1f | 5′‐CGTCCTACTCCACGGCTAC‐3′ |
| DPB‐SybG‐1r | 5′‐GGAATGCTTGATAGGGCAAG‐3′ | SLC32A2‐SybG‐1r | 5′‐TCTCTCTGACCACTTGATCC‐3′ |
| DNAJB9‐SybG‐1f | 5′‐CAGAGCGCCAAATCAAGAAG‐3′ | TEK‐SybG‐1f | 5′‐GCTCCATCCAAAAGACTTTAAC‐3′ |
| DNAJB9‐SybG‐1r | 5′‐TGCTTCTGCAATCTCTCTGAA‐3′ | TEK‐SybG‐1r | 5′‐CTGCAGACCCAAACTCCTGA‐3′ |
| ELD‐SybG‐1f | 5′‐GTACCCAGGGCAGCCATAC‐3′ | VAMP3‐SybG‐2f | 5′‐TGAGTTAGACGACCGTGCAG‐3′ |
| ELD‐SybG‐1r | 5′‐TCCATAAGGAGGAATCTGCTG‐3′ | VAMP3‐SybG‐2r | 5′‐GCCCACATCTTGCAATTCTT‐3′ |
| EVI2A‐SybG‐1f | 5′‐CACGGACATGGAACACACA‐3′ | VCAM1‐SybG‐1f | 5′‐CCTGAGCCCTGTGAGTTTTG‐3′ |
| EVI2A‐SybG‐1r | 5′‐CCACAGACGGGTATAGTTTGC‐3′ | VCAM1‐SybG‐1r | 5′‐GAGTAGAGCTCCACCTGGAT‐3′ |
| GJA‐SybG‐1f | 5′‐GTCAGCCTGGGGAGATGAG‐3′ | PKIA‐Probe‐1f | 5′‐ctgcgtccatccataatgag‐3′ |
| GJA‐SybG‐1r | 5′‐AGCGCACATGAGAGATTGG‐3′ | PKIA‐Probe‐1r | 5′‐taggccagagagtgcctgtt‐3′ |
| GNA12‐SybG‐1f | 5′‐CAGCTGAATTACTTTCCTAGTAAGC‐3′ | POSTN‐Probe‐1f | 5′‐cattttggcaatgtccatgt‐3′ |
| GNA12‐SybG‐1r | 5′‐CCACCATCTTAAAGGGGATCT‐3′ | POSTN‐Probe‐1r | 5′‐ttctgcaatcctttccagagt‐3′ |
| ICAM1‐SybG‐1f | 5′‐GAGCTTCGTGTCCTGTATGG‐3′ | PTPRB‐Probe‐1f | 5′‐ccattttcgaggctccttat‐3′ |
| ICAM1‐SybG‐lr | 5′‐GCCTGGCACATTGGAGTC‐3′ | PTPRB‐Probe‐1r | 5′‐ttggcatgtgactgcttctt‐3′ |
| MGST1‐SybG‐1f | 5′‐AGAGCCCACCTGAATGACCT‐3′ | VCAM1‐Probe‐1f | 5′‐tgggtttgcggttaaatctc‐3′ |
| MGST1‐SybG‐1r | 5′‐TGAAGTGCAGGATGGCTGTA‐3′ | VCAM1‐Probe‐1 | 5′‐caggcttttcctcatcttcg‐3′ |
| PAH‐SybG‐1f | 5′‐TGACCACCCTGGTTTTAAAGAT‐3′ | CCL2‐Probe‐1f | 5′‐acacgtttccttccagcagt‐3′ |
| PAH‐SybG‐1r | 5′‐GTATTCCACTCGAGGGATGG‐3′ | CCL2‐Probe‐1r | 5′‐ttaccttcaggccacattcc‐3′ |
2.5. Degenerate oligonucleotide primed polymerase chain reaction (DOP‐PCR)
Two L1 degenerate primers (L1‐retro‐1f, 5′‐GGATAGCATTGGGAGATATACCT‐3′; and L1‐retro‐1r, 5′‐ATTGAACAAGATGGATTGCACGC‐3′) were used to amplify the spliced 1 kb neomycin gene from HepG2 genomic DNA. The 1 kb neomycin gene was gel purified and 10 ng of the product used for Degenerate Oligonucleotide Primed PCR (DOP‐PCR). One unit of Go Taq DNA Polymerase (Cat # M3178, Promega, USA) was incubated with 10 ng of neomycin gene template DNA, 1× of 5× Go Tag Colorless Flexi buffer, 300 nM of each primer, 2 mM each of dATP, dCTP, dGTP, 1.5 mM dTTP and 0.5 mM dTTP‐Biotin. Cycling parameters were 95 °C for 2 min, 35 cycles of 95 °C for 30 s; 62 °C for 30 s; 72 °C for 1 min, followed by a 2 min denaturation at 94 °C. Products were visualized on 1% agarose gels, band exercised, purified and quantified using a nanodrop. For the synthesis of gene specific probes, each probe was first amplified from genomic DNA, then labeled with Fluorescein‐dTTP (FITC‐dTTP) using MIRUS FISH labeling kit (Cat # MIR 6510) at 37 °C for 1 h. Primers for the initial PCR of gene specific probes are presented in Table 1.
2.6. Fluorescence in situ hybridization
Stably HepG2 transfected cells were incubated with colcemid (0.1 μg/ml) for 1.5 h to arrest cells at metaphase. Cells were trypsinized, centrifuged at 126g for 5 min and incubated in hypotonic solution (75 mM KCl) for 20 min at 37 °C. Three drops of the Carnoy fixative solution (methanol/glacial acetic acid 3:1) was added before centrifugation at 126g for 5 min. This step was repeated 4× or until the pellet became visibly white. Cell suspensions of 10–20 μl were dropped onto clean dry slides and exposed to hot steam for 30 s. Metaphase spread chromosomes were treated with 200 μl of RNase A (100 μg/ml) for 1 h at 37 °C, washed twice in 2× SSC buffer, rinsed in 10 mM HCL and treated with 1% pepsin solution for 10 min at 37 °C. Cells were rewashed twice in 2× SSC buffer, dehydrated in an ethanol series (70%, 80%, 90% and 100%) and allowed to air dry. Chromosomes were aged at 65 °C for 1 h, denatured in 70% formamide in 2× SSC for 60 s and dehydrated using ethanol series. Biotin or FITC labeled probes (150 ng) were dissolved in hybridization buffer (50% formamide, 10% dextran sulfate, 0.1% SDS, 300 ng/ml Salmon Sperm DNA in 2× SSC), heated at 72 °C for 10 min, and cooled on ice for 2 min. The probe solution (30 μl) was added to each slide, covered with 22 × 22 mm glass coverslip, and the edges sealed with rubber cement. Slides were heated at 72 °C for 5 min and the temperature gradually dropped to 37 °C on a heat block before incubation overnight in a humid chamber at 37 °C. Slides were immersed in 2× SSC buffer to remove the coverslip, washed twice at 45 °C in wash buffer (20% formamide in 0.1× SSC), 0.1 SSC buffer and 2× SSC buffer. Slides were equilibrated in detection buffer (0.2% Tween 20 in 4× SSC) for 10 min, blocked for 30 min in Blocking buffer (5% bovine albumin, 0.2% Tween 20 in 4× SSC) and incubated with secondary antibody (Streptavidin‐FITC or Streptavidin‐CY3 (for double FISH) (5 μg/ml)) for 1 h. Slides were washed twice in 2× SSC buffer, stained with DAPI for 10 min, mounted with a coverslide, the edges sealed with nail polish and images taken using the Axiovert inverted microscope at 63× magnification.
3. Results
3.1. Synthetic L1 undergoes complete cycles of retrotransposition and de novo insertions into the HepG2 genome
To gain insight into the functionality of genetic targets within an in silico predicted L1 biological regulatory network, unique tags were introduced into the L1 sequence for tracking of ectopic L1 in HepG2 cells. ORF1 was tagged with HA and STREP, while ORF2 was tagged with FLAG and MYC, and both tags were placed at the C‐terminal end of each ORF without disruption of the 63bp located between the two ORFs (Figure 1A). For constitutive expression of L1 proteins, a CMV promoter was placed upstream of the L1 UTR (Figure 1A). A retrotransposition incompetent L1 with aspartate to tyrosine substitution (D702Y) in the RT domain of ORF2 (Moran et al., 1996; Mathias et al., 1991) was also constructed (Figure 1A). The vector design and fidelity of sequences were confirmed by restriction enzyme digestion and nucleotide sequencing. The backbone of the vector control was engineered to remove known mammalian transcription factor binding sites, and its use in stable transfection experiments corrected for non‐specific contributions of endogenous L1 to changes in gene expression. Synthetic vectors (Figure 1A) were used to generate HepG2 cell lines that stably express tagged L1 ORF1 and ORF2 proteins.
Figure 1.
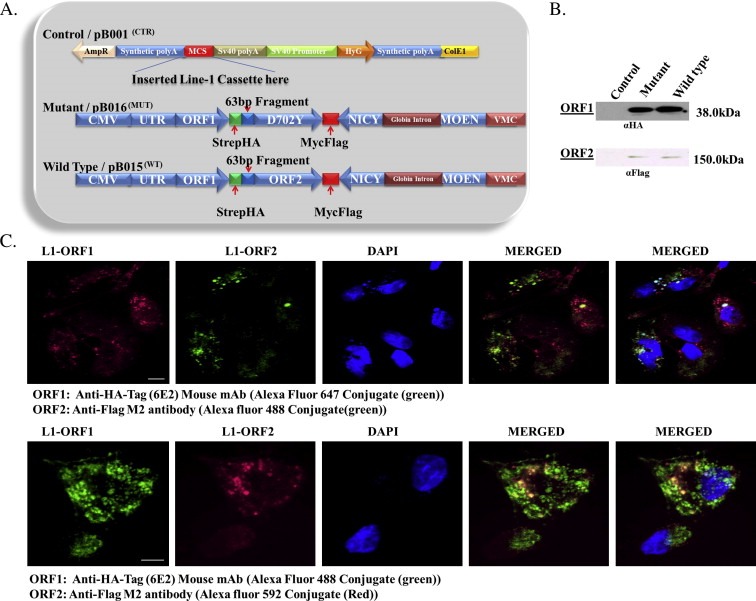
Expression of synthetic L1 in HepG2 cells. (A) Schematic diagram of L1 vectors constructed to determine the influence of L1 on genetic targets in HepG2 cells. Control (pB001CTR) is the vector backbone, the L1 cassette was cloned into the multiple cloning site (MCS). Wild type (pB015WT) represents wild type L1 and mutant (pB016MUT) represents mutant L1. L1 ORF1 was tagged with Strep and HA tags (green), while ORF2 was tagged with Myc and Flag (red). Wild type and mutant both contain a neomycin cassette (blue) in the antisense orientation to assay for retrotransposition. (B) Western blot of ORF1 and ORF2 proteins with antibodies directed against HA and Flag, respectively. The 41 kDA and 151 kDA bands which are absent in control were detected by HA and Flag antibodies, respectively, indicating the expression of ORF1 and ORF2 proteins. All signals were obtained from the same membrane. (C) Indirect immunofluorescence of ORF1 (green, top panel; red, bottom panel) and ORF2 (red top panel; green, bottom panel) protein using conjugated HA (αHA tag (6E2) mouse‐Alexa‐488) and Flag tag (αFlag M2‐Alexa‐594) antibodies. Hoescht (blue) stains the nucleus with colocalization of the two proteins shown in yellow. Scale bar is 20 μm.
Western blotting of HepG2 cells expressing synthetic wild type L1, mutant L1 or empty vector was completed using antibodies against HA or Flag for ORF1p and ORF2p, respectively. In keeping with published work (Goodier et al., 2004, 2007), the expression of ORF1p was greater than that of ORF2 protein in both wild type and mutant clones (Figure 1B and C). ORF1 and ORF2 proteins were expressed at comparable levels in wild type and mutant cells, but no synthetic L1 proteins was detected in cells expressing the empty vector (Figure 1B). To establish functional linkage between ORF1 and ORF2 proteins, the co‐localization of proteins was examined (Figure 1C). Punctate staining of ORF1 (red, top panel) and ORF2 (green, top panel), or ORF1 (green, bottom panel) and ORF2 (red, bottom panel) co‐localized as cytoplasmic foci, with lesser staining detected in the nucleus (Figure 1C). Despite evidence that the CMV promoter can be silenced by DNA methylation within 6 h when delivered as a transgene (Brooks et al., 2004); expression of the synthetic construct was detected for up to 2 weeks after selection without changes in expression. Next, experiments were designed to monitor the integration and splicing of the neomycin cassette (Figure 2A). Evaluation of L1 retrotransposition was completed by direct PCR or by fluorescence in situ hybridization (FISH) using primers or probes directed against the neomycin cassette (Figure 2A). While integration into the genome was readily detected in wild type and RT‐mutant cells, spliced neomycin cDNA was only detected in cells expressing wild type L1, indicating that only wild type L1 undergoes complete cycles of retrotransposition (i.e. mobilization) (Figure 2B). The L1 RT mutant while expressed is not able to support retrotransposition because of a defective RT. Further, neomycin probe was labeled with biotin‐dTTP using Degenerate Oligonucleotide Primed PCR (DOP‐PCR) which generates a labeled neomycin probe of increased size due to biotin‐dTTP incorporation (Figure 2C). Using this probe, FISH analysis was completed on wild type, mutant and control cells. No staining was seen for neomycin in mutant and control cells, while unique spots were detected in wild type cells (Figure 2D). Given that the reconstitution and insertion of neomycin is strictly dependent upon complete cycles of retrotransposition, these results confirm our earlier finding (Figure 2B) that only cells expressing wild type L1 undergo complete cycles of retrotransposition.
Figure 2.
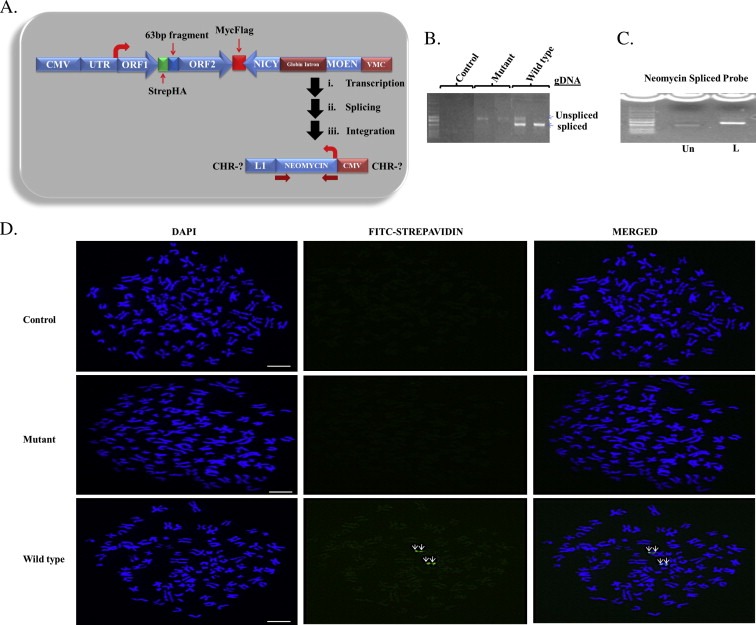
Retrotransposition of stably expressed synthetic L1 in HepG2 cells. (A) Schematic diagram of the L1 vector containing the neomycin cassette. The thick arrows indicate splicing and integration of neomycin gene into the genome after ectopic expression of L1. (B) Spliced neomycin and un‐spliced neomycin gene from wild type cells and un‐spliced neomycin genes from mutant cells indicate the retrotransposition competency and incompetency respectively in these cells. No band is seen for cells expressing the control plasmid. (C) Degenerate Oligonucleotide Primed PCR (DOP‐PCR) product of neomycin gene from HepG2 genomic DNA. Result shows an increase in size of the biotin‐dTTP labeled (L) probe compared to the unlabeled probe (Un). (C) Fluorescence in situ hybridization (FISH) of synthetic L1 in HepG2 cell lines. The first column from left to right in C shows the 4′,6‐diamidino‐2‐phenylindole (DAPI) staining of chromosome spreads, the second column shows Streptavidin‐Fluorescein (FITC) staining of the neomycin probe and the third column shows the merged image of the two stains. Results show that only wild type L1 undergoes complete cycle of retrotransposition. Scale bar is 10 μm.
3.2. Reprogramming of genetic expression by L1 involves RT‐dependent and RT‐Independent mechanisms
The unique roles of L1 in the regulation of genome integrity are poorly understood. To address this question, computational algorithms have been used to identify genes within a putative L1 regulatory network (Ramos et al., 2007). Although the nature of predicted interactions was not fully resolved, coordinate expression of genetic elements within the network was hypothesized to be critical to the regulation of the adaptive response of mammalian cells to L1 activation.
RT‐PCR analysis of targets within the predicted L1 regulatory network showed that 24 targets were differentially regulated by expression of synthetic L1. Chemokine (C–C motif) ligand 2 (CCL2), Cytochrome P450, family 2, subfamily a, polypeptide 4 (CYP2A4), Microsomal glutathione S‐transferase 1 (MGST1), Phenylalanine hydroxylase (PAH), Periostin, Osteoblast specific factor (POSTN), Protein tyrosine phosphatase, receptor type, B (PTPRB), and Vascular cell adhesion molecule 1 (VCAM1), showed RT‐dependent increases in expression as evidenced by specific increases in only RC‐L1 transfected cells (Figure 3A). Extracellular link domain‐containing 1 (ELD), Syndecan binding protein (SDCBP), and Mesicle‐associated membrane protein 3 (VAMP3) showed RT‐dependent decreases in expression (Figure 3B). The remainder of the targets including Chemokine (C–X–C motif) ligand 1 (CXCL1), Ectopic viral integration site 2a (EVI2A), Gap junction membrane channel protein alpha 1 (GJA1), RNA binding motif protein 39 (RBM39), Chloride intracellular channel 3 (CLIC3), Protein kinase inhibitor alpha (PKIA), Regulator of G‐protein Signaling (RGS), Cysteine dioxygenase 1 (CDO1), D site albumin promoter binding protein (DBP), DnaJ (Hsp40) homolog, subfamily B member 9 (DNAJB9), Myosin light polypeptide 7 (MYL7), Nemo‐like kinase (NLK), Preimplantation protein 4 (PREI4) were regulated irrespective of RT status (Figure 3C–E). Four of the above referenced targets, CXCL1, EVI2A, GJA1, and RBM39 showed reciprocal profiles such that expression was increased by wild type L1, but decreased by mutant L1 compared to control (Figure 3C). CLIC3, PKIA, and RGS transcripts were increased in cells expressing both wild type and mutant L1 (Figure 3D). All other targets including CDO1, DBP, DNAJB9, MYL7, NLK and PREI4 were downregulated in both wild type and mutant (Figure 3E). Only one gene, Intercellular Adhesion Molecule 1 (ICAM1), was not regulated by RC‐L1, but show marked downregulation by mutant (Figure 3E). Four targets, Amylo‐1,6‐glucosidase (AGL), Guanine nucleotide binding protein, alpha 12 (GNA12), Sphingosine phosphate lyase 1 (SGPL1), and Solute carrier family 34 (sodium phosphate), member 2 (SLC34A2), showed no change in expression levels compared to control (Figure 3G), and the transcripts of three other genes (Endothelial‐specific receptor tyrosine kinase (TEK), Chitinase 3‐like 3 (CHI3L3), and Procollagen, type III (COL3A1)) were undetectable in HepG2 cells, implicating tissue‐specific expression. These findings established greater than 90% predictive accuracy for the previously predicted regulatory network and demonstrate that regulation of genes within the L1 network occurs via mechanisms that are largely RT‐independent.
Figure 3.
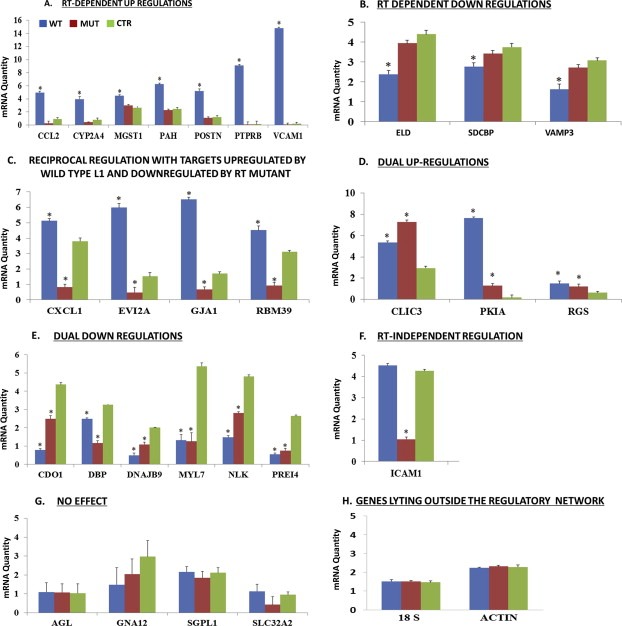
Real time PCR of COD predicted genetic regulatory elements within the L1 network. Total RNA was extracted from cells stably expressing L1 wild type (pB015WT or WT (blue)), mutant (pB016MUT or MUT (brown)) or control vector (pB001CTR or CTR (green)) and transcript levels for each gene determined using standard curve methodology. (A) RT‐dependent upregulation. (B) RT‐dependent downregulation. (C) Reciprocal regulation with targets upregulated by wild type L1 and downregulated by RT mutant. (D) Dual upregulation. (E) Dual downregulation. (F) RT‐independent. (G) No effect. (H) Genes lying outside the regulatory network. Statistical analysis was completed using ANOVA followed by Tukey's post hoc analysis.
3.3. De novo insertion‐dependent and ‐independent modulation of selected targets
Next, we addressed the question of whether de novo insertion (i.e. retrotransposition/mobilization) of L1 near or within coding regions of genes within the network mediates differential regulation. FISH analysis was completed on VCAM1, CCL2, POSTN and PTPRB because these targets showed L1 RT dependent modulation, while PKIA was tested as a target regulated irrespective of RT status. Each probe was located within the 5′‐untranslated region (UTR) of the target (Figure 4A) and labeled with fluorescein (FITC)‐dTTP using the MIRUS FISH labeling kit. As shown in Figure 4B, a faint band is seen for labeled gene specific probes which are indications that UV emission at ∼300 nm is not optimal for fluorescent label DNA (Figure 4B). The FITC labeled gene‐specific probe and biotin‐labeled neomycin probe were used in dual fish analysis for each target (Figure 4C). The strong colocalization of signal for VCAM1 and PTPRB suggests that differential regulation of these targets may be linked to retrotransposition of L1. However, the neomycin signal did not colocalize for CCL2, POSTN, and PKIA at any time indicating that the regulation of genes within the L1 network is not coupled to retrotransposition or insertional dysregulation of genetic targets.
Figure 4.
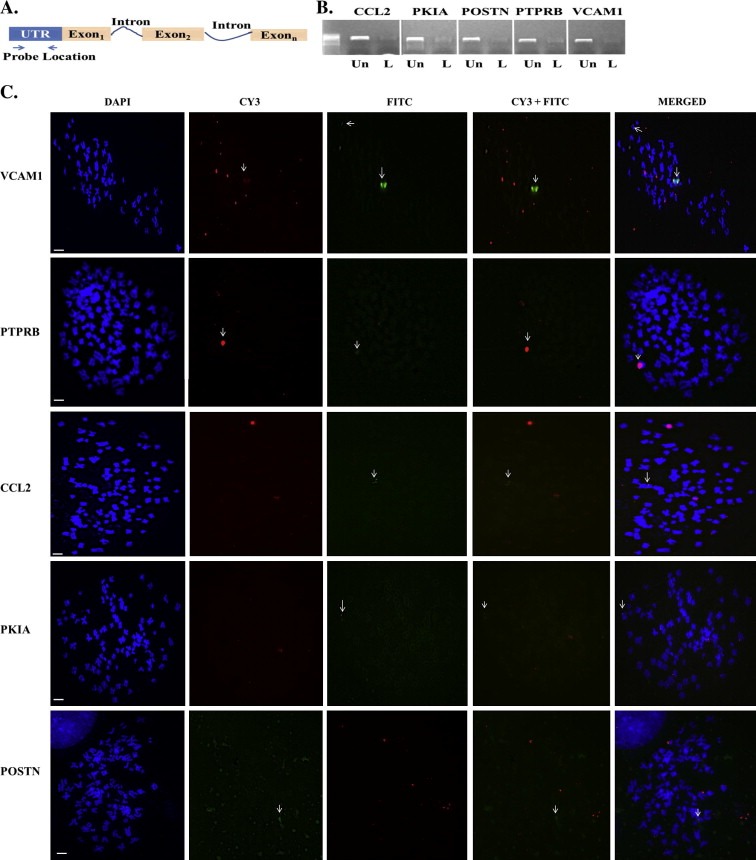
Differential regulation of VCAM1 and PTPRB, but not CCL2, PKIA and POSTN involves de novo insertion of L1. (A) Schematic diagram showing location of probes for each target. Each probe is ∼500 bp from the 5′‐untranslated region (UTR). (B) FITC‐dTTP labeled (L) and unlabeled (Un) probes for each target. Again size differences between labeled and unlabeled probes indicate incorporation of FITC‐dTTP. (C) Dual fish analysis of L1 and targets (CCL2, VCAM2, PKIA, POSTN and PTPRB) within the L1 genetic network. Column 1 shows DAPI staining (Blue) of chromosomes, column 2 shows CY3 staining of each the gene‐specific probe (red), column 3 shows FITC staining of the neomycin probe (green), column 4 shows matched CY3 and FITC staining and column 5 shows the merged staining for all dyes. Notice that the neomycin signal colocalized with VCAM1 and PTPRB gene specific probes, but not with CCL2, PKIA and POSTN gene specific probes. Scale bar is 50 μm.
3.4. Further discretization of the L1 genetic regulatory network identifies adhesion, inflammation and cellular metabolism as key pathways controlling plasticity of the HepG2 genome
Ingenuity pathway analysis of targets modulated by L1 identified cellular adhesion/extracellular matrix, inflammation, and metabolism as major pathways targeted by L1 in HepG2 cells. To confirm this assertion, experiments were conducted to examine the expression of several markers of hepatocyte differentiation in HepG2 cells expressing wild type and mutant L1. HepG2 cells undergo epithelial to mesenchymal transition (EMT) under conditions of cellular stress, with vast recapitulation of developmental programming, loss of epithelial polarity, reorganization of the cytoskeleton and acquisition of migratory potential (Lee et al., 2012). To determine if reprogramming of the HepG2 genome by L1is associated with EMT, the expression of liver‐specific albumin (ALB) and α‐fetoprotein (αFP), and N‐ and E‐cadherins (CDH1 and N‐CDH2) and zona occludins‐1 (ZO‐1) adhesion molecules was examined by indirect immunofluorescence (Figure 5). Vimentin (VIM) was examined as a marker of mesenchymal conversion. The expression of ALB was decreased (Figure 5A) while that of αFP increased in both wild type and mutant cell lines compared to control (Figure 5B). This phenotypic switch was also associated with loss of N‐CDH2, CDH1 and ZO‐1 expression (Figure 5C–E), and marked increases in VIM expression (Figure 5F). These data show that reprogramming of the HepG2 genome by L1 culminates in EMT and this response is independent of L1‐encoded RT activity or retrotransposition. A revised version of the discretized L1 gene–gene interaction network (Ramos et al., 2007) is shown in Figure 6. A total of 24 targets were differently regulated by synthetic L1, with ten (blue) regulated in an RT‐dependent manner, one (green) RT independent, eleven (Red) both RT dependent and RT independent regulation, four (Black) unchanged, and six undetected in HepG2 cells.
Figure 5.
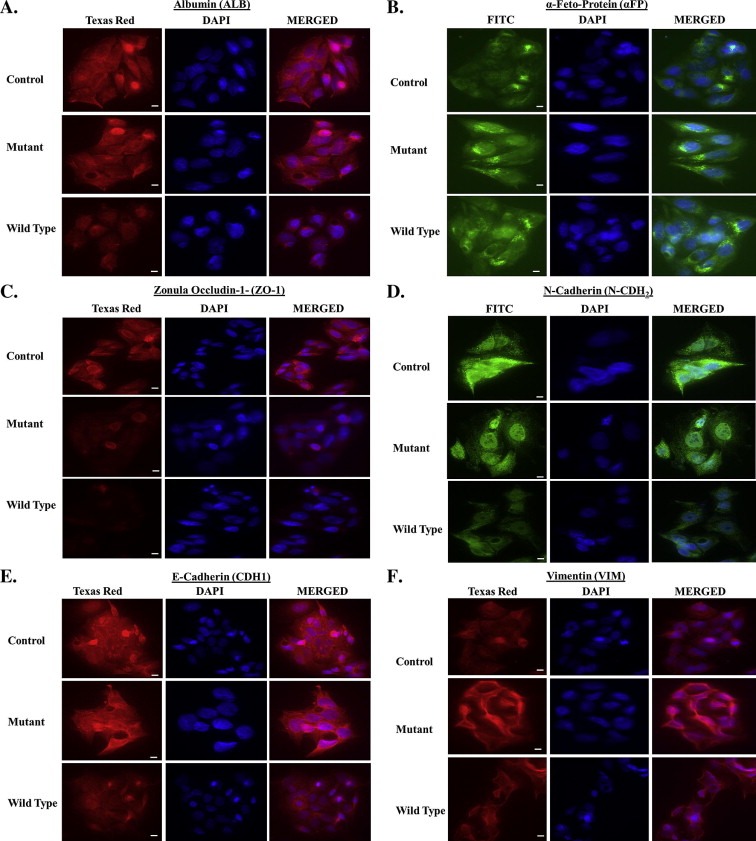
Epithelial‐to‐mesenchymal transition in L1 expressing HepG2 cells. (A) Expression of liver‐specific albumin (ALB) is reduced in wild type and mutant compared to controls. (B) Expression of a‐fetoprotein (a‐FP), a marker of the embryonic or neoplastic liver phenotype, was increased in wild type and mutant cells. (C–E) Expression of epithelial markers including Zona Occlidins‐1 (ZO‐1), N‐Cadherin (N‐CHD2), and E‐Cadherin (CDH1) was decreased in wild type and mutant compared to controls. (F) Vimentin (VIM), a marker of the mesenchymal phenotype, was overexpressed in wild type and mutant cells compared to controls. Scale bar is 50 μm.
Figure 6.
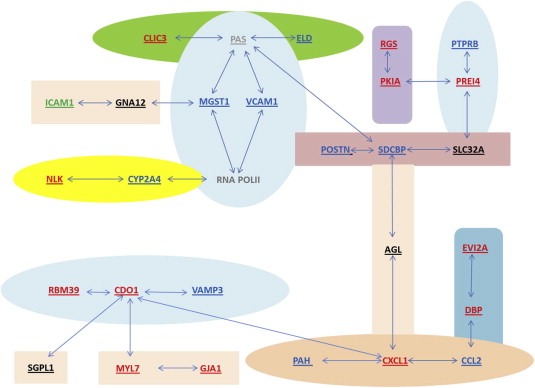
L1 gene regulatory network after biological evaluation in HepG2 cells. A revised network depicting genes within the predicted network that were differentially regulated by forced expression of L1. Eleven targets [Chemokine (C–C motif) ligand 2 (CCL2); Cytochrome P450, family 2, subfamily a, polypeptide 4 (CYP2A4); Microsomal glutathione S‐transferase 1 (MGST1); Phenylalanine hydroxylase (PAH); Periostin, Osteoblast specific factor (POSTN); Protein tyrosine phosphatase, receptor type, B (PTPRB); Vascular cell adhesion molecule 1 (VCAM1); Extracellular link domain‐containing 1 (ELD); Syndecan binding protein (SDCBP); and Mesicle‐associated membrane protein 3 (VAMP3)] were regulated in RT‐dependent manner (Figure 3A and B), thirteen targets [Cysteine dioxygenase 1 (CDO1); Chloride intracellular channel 3 (CLIC3); Chemokine (C–X–C motif) ligand 1 (CXCL1); D site albumin promoter binding protein (DBP); DNAJ (Hsp40) homolog, subfamily B member 9 (DNAJB9); Gap junction membrane channel protein alpha 1 (GJA1); Ectopic viral integration site 2a (EVI2A); Myosin light polypeptide 7 (MYL7); Nemo‐like kinase (NLK); Protein kinase inhibitor alpha (PKIA); Preimplantation protein 4 (PREI4) and RNA binding motif protein 39 (RBM39)] in both RT‐dependent and RT‐independent manner (Figure 3C–E), four of the genes [Amylo‐1,6‐glucosidase (AGL), Sphingosine phosphate lyase 1 (SGPL1), Solute carrier family 34 (sodium phosphate), member 2 (SLC34A2), Guanine nucleotide binding protein, alpha 12 (GNA12)] (Figure 3F) showed no change in expression compared to control and the transcripts of three other targets [Endothelial‐specific receptor tyrosine kinase (TEK), Chitinase 3‐like 3 (CHI3L3), and Procollagen, type III (COL3A1)] were undetectable in all samples, suggesting tissue specific expression for these targets.
4. Discussion
The defining attribute of L1s is their ability to remodel the genome landscape via retrotransposition. Retrotransposons afford germ cells and somatic cells a considerable degree of genome plasticity during evolution, as well as during adaptation to toxic assaults. L1s are highly active during genetic sorting in early embryogenesis, but undergo epigenetic silencing via DNA methylation and heterochromatin formation of pericentromeric and telomeric regions within the genome. With the exception of mutational inactivation of selected loci (Goodier and Kazazian, 2008; Montoya‐Durango et al., 2009), or the silencing of neighboring regions via chromatin remodeling (Ramos et al., 2011), little is understood about the complexity of biological control exerted by L1s. In the present study, we establish for the first time key elements of the functionality of L1 within the mammalian genome, and elucidate the biological connectivity of genetic elements within a regulatory network that reprograms the HepG2 genome. Evidence is presented that 1) L1 inserts into the HepG2 genome; 2) reprogramming of genetic expression by L1 involves RT‐dependent as well as RT‐independent mechanisms not coupled to retrotransposition; and 3) genetic reprogramming targets adhesion, inflammation and cellular metabolism pathways that modulate HepG2 plasticity and mediate EMT. A major discovery of the present investigation was the finding that reprogramming of the mammalian genome by L1 is not solely dependent upon retrotransposition, but instead, can involve selective deregulation of genes within a specified genetic regulatory network via RT‐independent mechanisms.
4.1. Expression profiles of synthetic L1 in HepG2 cells
Forced expression of wild type and mutant L1s was associated with stable integration into the HepG2 genome, and strong expression of synthetic proteins that colocalize as cytoplasmic foci, and to access the nuclear compartment (Figures 1 and 2). Synthetic ORF1 and ORF2 proteins mediate active retrotransposition events only when RT function is preserved and is lost in synthetic constructs with deficient RT activity. An important question in understanding the role of L1 is whether L1 insertions are random or guided (Levin and Moran, 2011). Interestingly, L1 insertions have been shown to preferentially target AT rich regions of the genome, with insertions often found within the most frequently mutated genes and 50–200 kb away from housekeeping genes and actively transcribing genes (Lee et al., 2012). Expression of L1 proteins was primarily cytoplasmic, suggesting that transfected cells have in place the machinery for complete cycles of transposition. Although we do not know if the neoplastic phenotype is permissive of retrotransposition, we have established previously that retrotransposition occurs efficiently in different cell types (Stribinskis and Ramos, 2006; Tomilin, 1999).
4.2. Reprogramming of the HepG2 genome
The L1 network was determined using a coefficient of determination (COD) algorithm that identified genes predictive of L1 expression on the basis of Boolean ideology (Kim et al., 2000). Biological connectivity was established for AHR/PAS proteins (Ramos et al., 2007), but connectivity of other genetic elements, or their relevance in regulatory control is unknown. Thus, we set out to query the fidelity of the predicted L1 regulatory network. The multiplicity of L1 insertion sites within the HepG2 genome suggests that a master switch of differentiation programming is likely not targeted by L1 and that instead genetic regulatory control by L1 involves genes that participate in disparate cellular processes. An important finding was the fidelity of computational prediction of the COD algorithm, with greater than 90% accuracy in predicting unknown biological relationships. Analyses of genes within the L1 regulatory network identified multiple clusters discretized on the basis of RT phenotype, with less than half of the genetic targets exhibiting strict RT‐dependence. In fact, most genetic targets within the L1 network exhibited patterns of dual regulatory control by both wild type and mutant RT.
The simplest explanation for the modulation of genetic targets within the network is that L1 inserts in the vicinity of targets to regulate expression. This implies multiplicity of insertions and/or cross regulatory functions dependent upon the L1 sequence. In this scenario, L1 regulation would involve integration into regions that afford secondary regulatory control. For example, L1 sequences can act in cis as transcriptional regulatory units for general transcription factors, or as binding sites for hormone receptor‐dependent transcriptional enhancers (Islam et al., 1993). In accord with this view, an enhancer of apolipoprotein (a) has been shown to reside within an L1 element (Ashworth et al., 1990). At the posttranscriptional level, L1 sequences can provide novel splicing sites within coding genes, thereby generating new transcripts, or giving rise to aberrant mRNA transcripts. Insertion of L1 sequences into specific introns considerably reduces transcriptional elongation of the targeted gene (Ferrigno et al., 2001), while the bidirectional L1 promoter can provide new promoters in both sense and antisense orientation, thus triggering activation of existing genes, or modulation of tissue‐specific expression (Speek, 2001). In fact, 13% of developmentally‐regulated genes in mouse preimplantation embryos contain retrotransposon‐derived sequences, suggesting that retroelements provide alternative promoters to regulate developmental programs (Ting et al., 1992). The loss of L1 expression arrests the development of murine pre‐implantation embryos, and this developmental arrest is associated with substantial reprogramming of gene expression (Hata and Sakaki, 1997). While most of what is presently understood about L1 is based on retrotransposition mechanisms of genetic control, compelling evidence is presented here that the RT‐incompetent mutant incapable of retrotransposition modulated a large number of elements within the network (Figure 3). As such, co‐expression of L1 with other elements within the network likely serves biological functions distinct from insertional dysregulation secondary to retrotransposition. This conclusion is also consistent with the finding that several genetic targets regulated in an RT‐dependent manner did not show colocalization with de novo inserted L1 sequences (Figure 4). Silencing of L1 using specific siRNAs and epigenetic reactivation of L1 by DNA damaging agents are currently been tested to characterize the reversibility of genetic change within the regulatory network (Ramos et al., unpublished data).
Epigenetic regulation of L1 sequences through DNA methylation and covalent modification represents another important mechanism for regulation of gene expression. The L1 promoter contains 34 CpG sites, and is typically heavily methylated (Teneng et al., 2011). Thus, if L1 sequences retrotranspose near genes, this may create methylation centers within the vicinity of the insertion leading to formation of heterochromatin and repression of transcription. Epigenetic silencing of genes within the L1 network in HepG2 cells is unlikely since as noted earlier several of the genetic elements modulated by L1 expression were not direct targets for insertional mutation (Figure 4). FISH experiments showed that de novo L1 insertions co‐localized with VCAM1 and PTPRB genes. This finding is intriguing in light of evidence that expression of L1 and VCAM1 strongly correlates with atherosclerosis, where the presence of L1 sequences in VCAM1 lead to co‐expression of these genes (Baccarelli et al., 2010). Collectively, these findings show that contrary to previously accepted views, the functionality of L1 within the mammalian genome is not strictly dependent upon retrotransposition.
The question remains as to how L1 reproducibly modulates the cellular phenotype of HepG2 cells. The expression of L1 generates RNAs known to function as self‐regulating silencing RNAs, or as downstream regulators of other cellular RNAs. The bidirectional promoter (5′UTR) of L1 has been shown to produce minus strand double stranded L1 RNA (Swergold, 1990) which can be processed by Dicer to siRNA that target L1 mRNA or mRNAs of other genes for degradation. Loss of Dicer has been shown to increase the expression of ectopic L1 with minimal effect on the expression of endogenous L1. Thus, L1 RNAs may serve as bridge between the coding and non‐coding domains of the genome. The regulation of some targets may result from secondary effects exerted by genes directly regulated by L1. This is plausible since modulation of most targets was independent of physical proximity or retrotransposition.
4.3. Biological implications for plasticity of the HepG2 genome
Pathway analyses identified adhesion, inflammation, and cellular metabolism as key pathways targeted by L1 in HepG2 cells. These pathways are closely linked to malignant transformation and tumorigenesis. These findings are in keeping with previous reports showing that siRNA knockdown of ORF2 reduces the proliferation of human hepatoma cell lines in a dose dependent manner (Yang et al., 2003), while ORF1p is abundantly expressed in invasive malignant breast carcinoma compared with nonmalignant carcinoma, or normal tissues (Kuo et al., 1998), and ectopic expression of L1 modulates differentiation programs of embryonic kidney cells (Ramos et al., 2011). An emerging theme is the use of RT inhibitors for inhibition of tumor growth in vivo (Sciamanna et al., 2005), with tumors totally arrested or considerably reduced when tumorigenic cell lines (A‐375 melanoma, PC3 prostate carcinoma, H69 small lung carcinoma, and HT29 colon carcinoma) treated with efavirenz, an RT inhibitor (Sciamanna et al., 2005). Many of the genetic elements within the L1 network including, PTPRB, CDO1, EVI2A, NLK, PKIA, PREI4, and CLIC3 are involved in tumorigenesis, so it became important to determine if one or more of these targets participates in the regulation of differentiation programs by L1.
Evidence was also obtained that HepG2 cells expressing wild type and mutant L1 exhibit the EMT phenotype. L1s are transcriptionally silenced in somatic cells, but highly active in embryonic, transformed and undifferentiated cells. The inverse relationship between L1 and phenotypic changes implicate L1 in the regulation of differentiation programs. EMT plays a key role in embryonic development as well as malignant transformation which ultimately confers increased migratory activity and features that sustain the malignant phenotype (Lee et al., 2012). The finding that most of the genes dysregulated in the genetic network are involved in cellular adhesion and inflammation reaffirms the involvement of L1 in EMT. Lastly, viral nucleic acids including retrotransposon DNA/RNA are detected by pathogen recognition receptors leading to production of type I interferons (IFNs) through activation of NF‐κB and interferon regulatory factors (IRFs) (Akira et al., 2006). This pathway readily connects to several of the genetic targets present in the L1 network (data not shown).
Analysis of genomic architecture often relies on a linear process to decipher key determinants of genetic control. The advent of functional genomic technologies has challenged this notion suggesting that gene function is not as rigid as once thought and that genes support varied cellular functions. L1 has been shown to be involved in genomic plasticity, reprograming during early development and under stressful conditions. The findings presented here established biological connectivity of an in silico‐predicted genetic regulatory network of L1 and show that L1 regulation of genes is not strictly dependent upon retrotransposition. The degree to which L1 mediates human oncogenesis may be dependent upon disruption of genetic elements within the L1 regulatory network, but our understanding of the complex interactions of L1 retrotransposon with the host genome is in its infancy.
Conflict of interest
The authors declare no potential competing interests.
Funding
This work was supported in part by grants from the National Institute of Environmental Health Sciences (ES014443 and ES017274) and Astra Zeneca to KSR.
Acknowledgments
The helpful commentary provided by Dr. Diego Montoya‐Durango is gratefully acknowledged.
Bojang Pasano, Roberts Ruth A., Anderton Mark J., Ramos Kenneth S., (2013), Reprogramming of the HepG2 genome by long interspersed nuclear element‐1, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.04.003.
References
- Akira, S. , Uematsu, S. , Takeuchi, O. , 2006. Pathogen recognition and innate immunity. Cell 124, 783–801. [DOI] [PubMed] [Google Scholar]
- Ashworth, A. , Skene, B. , Swift, S. , Lovell-Badge, R. , 1990. Zfa is an expressed retroposon derived from an alternative transcript of the Zfx gene. Embo J. 9, 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli, A. , Wright, R. , Bollati, V. , Litonjua, A. , Zanobetti, A. , Tarantini, L. , Sparrow, D. , Vokonas, P. , Schwartz, J. , 2010. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 21, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, C.R. , Garcia-Perez, J.L. , Badge, R.M. , Moran, J.V. , 2011. LINE-1 elements in structural variation and disease. Annu. Rev. Genom. Hum. Genet. 12, 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke, J. , Aravin, A.A. , Stark, A. , Dus, M. , Kellis, M. , Sachidanandam, R. , Hannon, G.J. , 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila . Cell 128, 1089–1103. [DOI] [PubMed] [Google Scholar]
- Brooks, A.R. , Harkins, R.N. , Wang, P. , Qian, H.S. , Liu, P. , Rubanyi, G.M. , 2004. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 6, 395–404. [DOI] [PubMed] [Google Scholar]
- Carmell, M.A. , Girard, A. , van de Kant, H.J. , Bourc'his, D. , Bestor, T.H. , de Rooij, D.G. , Hannon, G.J. , 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev. Cell 12, 503–514. [DOI] [PubMed] [Google Scholar]
- Dombroski, B.A. , Mathias, S.L. , Nanthakumar, E. , Scott, A.F. , Kazazian, H.H. , 1991. Isolation of an active human transposable element. Science 254, 1805–1808. [DOI] [PubMed] [Google Scholar]
- Feng, Q. , Moran, J.V. , Kazazian, H.H. , Boeke, J.D. , 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87, 905–916. [DOI] [PubMed] [Google Scholar]
- Ferrigno, O. , Virolle, T. , Djabari, Z. , Ortonne, J.P. , White, R.J. , Aberdam, D. , 2001. Transposable B2 SINE elements can provide mobile RNA polymerase II promoters. Nat. Genet. 28, 77–81. [DOI] [PubMed] [Google Scholar]
- Goodier, J.L. , Kazazian, H.H. , 2008. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135, 23–35. [DOI] [PubMed] [Google Scholar]
- Goodier, J.L. , Ostertag, E.M. , Engleka, K.A. , Seleme, M.C. , Kazazian, H.H. , 2004. A potential role for the nucleolus in L1 retrotransposition. Hum. Mol. Genet. 13, 1041–1048. [DOI] [PubMed] [Google Scholar]
- Goodier, J.L. , Zhang, L. , Vetter, M.R. , Kazazian, H.H. , 2007. LINE-1 ORF1 protein localizes in stress granules with other RNA-binding proteins, including components of RNA interference RNA-induced silencing complex. Mol. Cell. Biol. 27, 6469–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata, K. , Sakaki, Y. , 1997. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene 189, 227–234. [DOI] [PubMed] [Google Scholar]
- Hohjoh, H. , Singer, M.F. , 1996. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. Embo J. 15, 630–639. [PMC free article] [PubMed] [Google Scholar]
- Houwing, S. , Kamminga, L.M. , Berezikov, E. , Cronembold, D. , Girard, A. , van den Elst, H. , Filippov, D.V. , Blaser, H. , Raz, E. , Moens, C.B. , Plasterk, R.H. , Hannon, G.J. , Draper, B.W. , Ketting, R.F. , 2007. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 129, 69–82. [DOI] [PubMed] [Google Scholar]
- Islam, T.C. , Bugge, T.H. , Bohm, S. , 1993. The long terminal repeat of VL30 retrotransposons contains sequences that determine retinoic acid-induced transcription in cultured keratinocytes. J. Biol. Chem. 268, 3251–3259. [PubMed] [Google Scholar]
- Jennen, D.G. , Magkoufopoulou, C. , Ketelslegers, H.B. , van Herwijnen, M.H. , Kleinjans, J.C. , van Delft, J.H. , 2010. Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol. Sci. 115, 66–79. [DOI] [PubMed] [Google Scholar]
- Kim, S. , Dougherty, E.R. , Chen, Y. , Sivakumar, K. , Meltzer, P. , Trent, J.M. , Bittner, M. , 2000. Multivariate measurement of gene expression relationships. Genomics 67, 201–209. [DOI] [PubMed] [Google Scholar]
- Kolosha, V.O. , Martin, S.L. , 1997. In vitro properties of the first ORF protein from mouse LINE-1 support its role in ribonucleoprotein particle formation during retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 94, 10155–10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenberg, J.R. , Rykowski, M.C. , 1988. Human genome organization: Alu, lines, and the molecular structure of metaphase chromosome bands. Cell 53, 391–400. [DOI] [PubMed] [Google Scholar]
- Kuo, K.W. , Sheu, H.M. , Huang, Y.S. , Leung, W.C. , 1998. Expression of transposon LINE-1 is relatively human-specific and function of the transcripts may be proliferation-essential. Biochem. Biophys. Res. Commun. 253, 566–570. [DOI] [PubMed] [Google Scholar]
- Lander, E.S. , 1996. The new genomics: global views of biology. Science 274, 536–539. [DOI] [PubMed] [Google Scholar]
- Lander, E.S. , Linton, L.M. , Birren, B. , Nusbaum, C. , Zody, M.C. , Baldwin, J. , Devon, K. , Dewar, K. , Doyle, M. , FitzHugh, W. , 2001. Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lee, E. , Iskow, R. , Yang, L. , Gokcumen, O. , Haseley, P. , Luquette, L.J. , Lohr, J.G. , Harris, C.C. , Ding, L. , Wilson, R.K. , Wheeler, D.A. , Gibbs, R.A. , Kucherlapati, R. , Lee, C. , Kharchenko, P.V. , Park, P.J. , 2012. Landscape of somatic retrotransposition in human cancers. Science 337, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, H.L. , Moran, J.V. , 2011. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K.P. , Ramos, K.S. , 2003. Redox regulation of a novel L1Md-A2 retrotransposon in vascular smooth muscle cells. J. Biol. Chem. 278, 28201–28209. [DOI] [PubMed] [Google Scholar]
- Martin, S.L. , 2010. Nucleic acid chaperone properties of ORF1p from the non-LTR retrotransposon, LINE-1. RNA Biol. 7, 706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias, S.L. , Scott, A.F. , Kazazian, H.H. , Boeke, J.D. , Gabriel, A. , 1991. Reverse transcriptase encoded by a human transposable element. Science 254, 1808–1810. [DOI] [PubMed] [Google Scholar]
- McClintock, B. , 1984. The significance of responses of the genome to challenge. Science 226, 792–801. [DOI] [PubMed] [Google Scholar]
- Montoya-Durango, D.E. , Ramos, K.S. , 2010. L1 retrotransposon and retinoblastoma: molecular linkages between epigenetics and cancer. Curr. Mol. Med. 10, 511–521. [DOI] [PubMed] [Google Scholar]
- Montoya-Durango, D.E. , Liu, Y. , Teneng, I. , Kalbfleisch, T. , Lacy, M.E. , Steffen, M.C. , Ramos, K.S. , 2009. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat. Res. 665, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, J.V. , Holmes, S.E. , Naas, T.P. , DeBerardinis, R.J. , Boeke, J.D. , Kazazian, H.H. , 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87, 917–927. [DOI] [PubMed] [Google Scholar]
- Piskareva, O. , Schmatchenko, V. , 2006. DNA polymerization by the reverse transcriptase of the human L1 retrotransposon on its own template in vitro. FEBS Lett. 580, 661–668. [DOI] [PubMed] [Google Scholar]
- Ramos, K.S. , He, Q. , Kalbfleisch, T. , Montoya-Durango, D.E. , Teneng, I. , Stribinskis, V. , Brun, M. , 2007. Computational and biological inference of gene regulatory networks of the LINE-1 retrotransposon. Genomics 90, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, K.S. , Montoya-Durango, D.E. , Teneng, I. , Nanez, A. , Stribinskis, V. , 2011. Epigenetic control of embryonic renal cell differentiation by L1 retrotransposon. Birth Defects Res. A, Clin. Mol. Teratol. 91, 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff, R. , Itin, A. , Keshet, E. , 1991. Transcriptional activation of mouse retrotransposons in vivo: specific expression in steroidogenic cells in response to trophic hormones. Genes Dev. 5, 521–532. [DOI] [PubMed] [Google Scholar]
- Sciamanna, I. , Landriscina, M. , Pittoggi, C. , Quirino, M. , Mearelli, C. , Beraldi, R. , Mattei, E. , Serafino, A. , Cassano, A. , Sinibaldi-Vallebona, P. , Garaci, E. , Barone, C. , Spadafora, C. , 2005. Inhibition of endogenous reverse transcriptase antagonizes human tumor growth. Oncogene 24, 3923–3931. [DOI] [PubMed] [Google Scholar]
- Speek, M. , 2001. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol. Cell. Biol. 21, 1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stribinskis, V. , Ramos, K.S. , 2006. Activation of human long interspersed nuclear element 1 retrotransposition by benzo(a)pyrene, an ubiquitous environmental carcinogen. Cancer Res. 66, 2616–2620. [DOI] [PubMed] [Google Scholar]
- Swergold, G.D. , 1990. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol. Cell. Biol. 10, 6718–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaori-Kondo, A. , 2006. APOBEC family proteins: novel antiviral innate immunity. Int. J. Hematol. 83, 213–216. [DOI] [PubMed] [Google Scholar]
- Teneng, I. , Stribinskis, V. , Ramos, K.S. , 2007. Context-specific regulation of LINE-1. Genes Cells 12, 1101–1110. [DOI] [PubMed] [Google Scholar]
- Teneng, I. , Montoya-Durango, D.E. , Quertermous, J.L. , Lacy, M.E. , Ramos, K.S. , 2011. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics 6, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting, C.N. , Rosenberg, M.P. , Snow, C.M. , Samuelson, L.C. , Meisler, M.H. , 1992. Endogenous retroviral sequences are required for tissue-specific expression of a human salivary amylase gene. Genes Dev. 6, 1457–1465. [DOI] [PubMed] [Google Scholar]
- Tomilin, N.V. , 1999. Control of genes by mammalian retroposons. Int. Rev. Cytol. 186, 1–48. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Zhang, L. , Zhang, Y. , Kazazian, H.H. , 2003. An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res. 31, 4929–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]