

Abstract
The cancer invasion front (CIF), a spatially‐recognized area due to the frequent presence of peritumoral desmoplastic reaction, represents a cancer site where many hallmarks of cancer metastasis occur. It is now strongly suggested that the desmoplastic microenvironment holds crucial information for determining tumor development and progression. Despite extensive research on tumor‐host cell interactions at CIFs, the exact paracrine molecular network that is hardwired into the proteome of the stromal and cancer subpopulations remains partially understood. Here, we interrogated the signaling pathways and the molecular functional signatures across the proteome of a desmoplastic coculture model system of colorectal cancer progression. We discovered a group of bone morphogenetic protein (BMP) antagonists that coordinates major biological programs in CIFs, including cell proliferation, invasion, migration and differentiation processes. Using a mathematical model of cancer cell progression, coupled to in vitro cell migration assays, we demonstrated that the prominent BMP antagonist gremlin‐1 (GREM1) may trigger motility of cancer cell cohorts. Our data collectively demonstrate that the desmoplastic CIFs deploy a microenvironmental signature, based on BMP antagonism, in order to regulate the motogenic fates of cancer cell cohorts invading the adjacent stroma.
Keywords: Colorectal cancer, Cancer-associated fibroblasts, Desmoplasia, Bone morphogenetic protein, Gremlin-1, Migration
Highlights
We perform enrichment map analysis in a desmoplastic protein dataset.
We demonstrate the overrepresentation of gremlin‐1 in the cancer invasion front.
Our mathematical model predicts that gremlin‐1 increases migration in cancer cells.
Our experimental assays verify such predictions.
Abbreviations
- ATCC
American type culture collection
- BMP(I)
bone morphogenetic protein (inhibitor)
- CAF
cancer-associated fibroblast
- CIF
cancer invasion front
- COL12A1
collagen type XII
- CRC
colorectal cancer
- DMEM
Dulbecco's modified Eagle's medium
- DPD
desmoplastic protein dataset
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- FBS
fetal bovine serum
- FST
follistatin
- FSTL3
follistatin-like 3
- GO
gene ontology
- GREM1
gremlin-1
- HGF
hepatocyte growth factor
- HTRA3
high temperature requirement A3
- IPA
ingenuity pathway analysis
- IPKB
ingenuity pathway knowledgebase
- LOX
lysine-6-oxidase
- uPA
urokinase-type plasminogen activator
- PDGF
platelet-derived growth factor
- α-SMA
alpha-smooth muscle actin
- TGF-β
transforming growth factor-beta
- VEGF(R2)
vascular endothelial growth factor (receptor-2)
1. Introduction
Carcinoma growth and metastatic dissemination have been associated with the formation of a permissive tumor microenvironment, especially supported by cancer‐associated fibroblasts (CAFs), endothelial cells and immune cells, among others (Hanahan and Weinberg, 2011). An area of particular interest for the progression of most carcinomas is the invasive tumor margins, also known as cancer invasion front (CIF). In CIFs, cancer cell cohorts from the primary tumor migrate in the underlying stroma, excavating a metastatic pathway towards blood and/or lymphatic vessels and come in direct contact with CAFs and other recruited stromal cells. CIFs represent the most evident sites where tumor‐host cell interactions occur in a paracrine, juxtacrine and mechanical fashion (Karagiannis et al., 2012b). Quite interestingly, specific metastatic or other phenotypic [(i.e. epithelial‐to‐mesenchymal transition (EMT)] signatures have been associated with these interactions (De Wever and Mareel, 2003; De Wever et al., 2008b).
Heterotypic signaling between cancer cells and recruited fibroblasts is perhaps the most prominent interaction occurring at CIF sites, usually resulting in the formation of desmoplastic lesions, characterized by abundant extracellular matrix (ECM) deposition and accumulative proliferation of peritumoral CAFs (Elenbaas and Weinberg, 2001; Kalluri and Zeisberg, 2006; Kunz‐Schughart and Knuechel, 2002a; Xing et al., 2010). Several lines of evidence suggest that CAFs are recruited by cancer cell‐secreted factors, such as transforming growth factor‐beta (TGF‐β) and platelet‐derived growth factor (PDGF) (Bierie and Moses, 2006; Kalluri and Zeisberg, 2006). Upon their recruitment, CAFs initiate the expression of a smooth muscle‐like gene and protein machinery, reminiscent of the one observed by myofibrolasts during wound healing (Hanahan and Weinberg, 2011). Such CAFs contribute a wide spectrum of secreted factors, including chemokines and cytokines in desmoplastic CIFs, promoting various metastatic phenomena such as cancer cell proliferation, invasion, migration and EMT (Kunz‐Schughart and Knuechel, 2002b).
Desmoplasia seems to present with unique biological behavior in different carcinomas. For instance, pancreatic ductal adenocarcinomas are extremely desmoplastic (90% of the tumor mass is covered by CAFs) and remarkably, this microenvironment prevents the support of tumor‐associated angiogenesis (Olive et al., 2009; Pandol et al., 2009). In contrast, desmoplastic reaction is not often found in prostate cancers as the prostatic stroma is more muscular, but it may promote angiogenesis (Ayala et al., 2003; Yang et al., 2005). The diverse biological behavior of CAFs in different types of cancer may arise from the fact that CAFs can be recruited from a wide variety of progenitor cells, including resident fibroblasts, bone marrow‐derived mesenchymal stem cells, endothelial cells, or even cancer cells themselves (Xing et al., 2010). Therefore, depending on the recruiter cytokines that each type of cancer secretes, there might be a differential response of CAF recruitment from all these progenitors, and subsequently the generated CAF subpopulations may present with different biological repertoires. Thus, it is of particular importance to examine tissue‐specific profiles of desmoplastic CIFs and delineate mechanisms of neoplastic progression for cancers that may exert signals through CAFs.
Previously, we have underscored the potential of mass spectrometry‐based proteomic approaches in providing mechanistic insights on tumor cell dissemination and metastasis (Karagiannis et al., 2010). In one such study, we successfully characterized the protein profile of the CRC desmoplastic CIF (Karagiannis et al., 2012a). Our analysis defined a desmoplastic signature, named desmoplastic protein dataset (DPD), which showed particular enrichment in myofibroblastic proteins and fetoproteins. To our knowledge, the DPD is one of the most complete proteomic signatures characterizing the invasion front of any type of cancer. Within DPD, many typical CAF markers were identified and/or verified, including hepatic growth factor (HGF) and urokinase‐type plasminogen activator (uPA). In addition, DPD served as a platform for dissecting novel markers for characterization of CAF populations. For instance, we demonstrated that a‐SMA‐positive CAFs in CRC patients are characterized by aberrant co‐expression of collagen type XII (COL12A1), a typical collagen‐organizer molecule, participating in collagen cross‐linking and further regulation and stabilization of the desmoplastic lesions (Karagiannis et al., 2012a). In the present study, we wished to examine whether DPD can provide information, particularly involved in the paracrine regulation of CRC development and progression. To achieve this, we utilized pathway enrichment analysis on DPD to gain a global overview of the molecular networks associated with the regulation of CRC metastasis by CAFs and subsequently undertook a bioinformatics approach to further explore the most promising candidate pathways.
2. Materials and methods
2.1. Bioinformatics
2.1.1. Proteomic datasets
The DPD was retrieved from our previous study (Karagiannis et al., 2012a).
2.1.2. Performance and visualization of enrichment analysis
Enrichment map profiling was performed as follows: BINGO (v.2.44) was utilized to calculate over‐representation of Gene Ontology (GO) “biological process” terms among the input gene list (DPD; Supplementary Table 1) (Maere et al., 2005). The hypergeometric test was performed to assess the significance of the enrichment and resulting p‐values were FDR corrected using the Benjamini & Hochberg method (p < 0.05). Data from the enriched GO annotations was exported. Functional enrichment visualization was constructed by the enrichment map plugin in Cytoscape (Merico et al., 2011, 2010) using the exported BINGO results as the input. The Jaccard coefficient was used at a cutoff of 0.5 to connect related GO biological process terms and create an enrichment network. Parameters for the selection of the enrichment results, which appear on the network, were set to a p‐value cutoff of 0.005 and FDR < 10%. The generated clusters within the enrichment map were named after a commercially‐available word cloud algorithm for word frequencies (Wordle®) (http://www.wordle.net/).
2.1.3. Protein–protein interaction and pathway analyses
Protein–protein interaction analysis was performed via STRING tool (http://string‐db.org/) (Jensen et al., 2009). Pathway analysis was performed with Ingenuity Pathway analysis (Ingenuity® Systems; IPA) software (http://www.ingenuity.com/), as previously described (Karagiannis et al., 2013; Prassas et al., 2011). For details, refer to Supplementary Materials and Methods.
2.1.4. Statistical analysis
Enrichment in statistical binary tests was examined by Fisher's exact test; p < 0.05 was considered significant. The statistical software SPSS (version 20) was used.
2.2. Simulations of cancer development using mathematical modeling
We simulated the transverse view mode of an individual cancer cell cohort migrating in the cancer‐associated stroma (Figure 3B), using a hybrid of continuous and cellular automata models, developed using the R language for statistical computing (R Foundation for Statistical Computing, 2008). The mathematical model implements principles from the simulation of avascular tumour growth models, and well‐established mathematical characterizations of cell chemotaxis and enzyme diffusion as described elsewhere (Chaplain and Anderson, 1996; Sherratt and Chaplain, 2001; Sun et al., 2005; Ward and King, 1997, 1999) with certain modifications. The cells were allowed to move on a continuous‐scale in space, and enzyme concentrations were updated discretely, according to the discretized partial differential equations used to model them. This approach, thus, allowed for detailed observation of changes in the morphology of the cancer cell cohort over time. The model captures regulatory factors of tumor invasion and progression, by focusing on the interaction between chemotactic gradients and cell–cell adhesion, and in parallel by omitting the role of fibronectin as an agent in tumor morphogenesis. The inclusion of dynamic cell states was also omitted, since the cells can evenly receive nutrients (i.e. oxygen) through diffusion, apart from the inversely proportional decrease in Gremlin‐1 concentration to a cell's distance from the invasion front. The latter concept is further supported by the fact that our testing hypothesis states that the expression of gremlin‐1 in the tumor microenvironment is context‐dependent, thus its secretion will be limited in the desmoplastic stroma and subsequently be diffused from that area. For parameterization, values were estimated using previously described settings (Alarcon et al., 2003; Anderson, 2005; Perumpanani et al., 1996; Silva et al., 2009; Sun et al., 2005). A thorough description is presented in Supplementary Materials and Methods.
Figure 3.
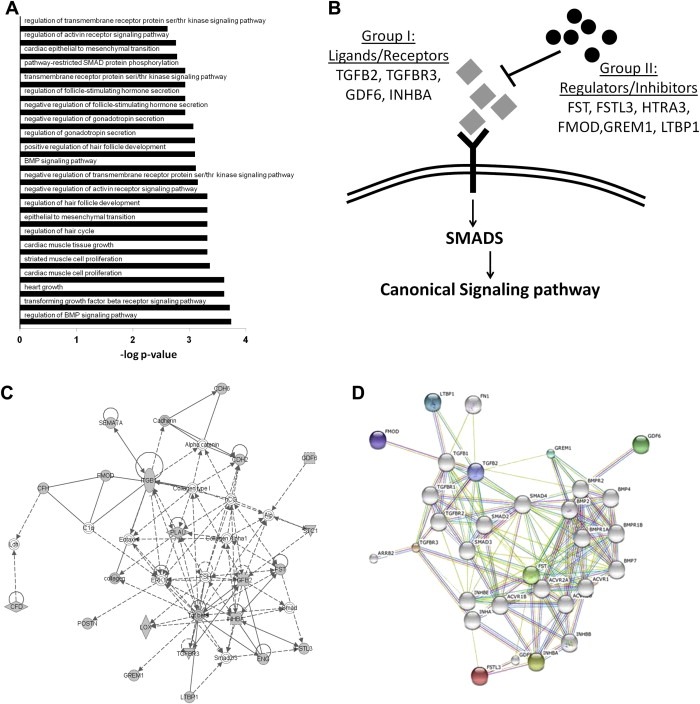
(A) Enriched GO terms for the genes found in the three TGF‐β‐superfamily‐associated clusters. (B) Categorization of proteins belonging to the TGF‐β superfamily of proteins into group I (Ligands/Receptors) and group II (Regulators/Inhibitors). (C) IPA network demonstrating that members of the TGF‐β‐superfamily of proteins are grouped together. (D) STRING network demonstrating that members of the TGF‐β‐superfamily of proteins are grouped together. Colored nodes correspond to members of the TGF‐β‐superfamily of proteins found in DPD, while white nodes represent other members of the TGF‐β pathway.
2.3. In‐vitro experiments
2.3.1. Reagents
The recombinant human proteins gremlin‐1 (rhGREM1) and bone morphogenetic protein‐7 (rhBMP7) were purchased from Sigma–Aldrich.
2.3.2. Cell lines
The human SW480 and SW620 colon cancer cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD) and were maintained in their favorable media, according to the manufacturer's instructions. All experiments were performed within the first 3 passages from the initiation of all cultures.
2.3.3. Under‐the‐agarose cell migration assays
The under‐the‐agarose cell migration assays were performed in Petri dishes, as previously described (Heit and Kubes, 2003). Experimental conditions were applied to the four surrounding punched wells, depending on the question asked. SW480/SW620 cells stained with crystal violet in the central cavity were allowed to migrate towards the surrounding gradients. To determine preferential cell migration towards specific gradients, we measured the percentage of crystal violet intensity that corresponded to the surface of each individual gradient after the termination of all experiments. At least two independent assays in triplicates were repeated for each experiment. An extended description is presented in Supplementary Materials and Methods.
3. Results
3.1. Overview of enrichment map profiling of DPD
We have previously generated in vitro cocultures of colon cancer cells and normal colonic fibroblasts, to mimic the desmoplastic tumor‐host cell interface in CRC. By performing proteomic analysis of coculture CM, we proposed the secreted 152‐protein DPD signature (Karagiannis et al., 2012a) (Supplementary Table 1). Since desmoplasia affects cancer development and progression (Kunz‐Schughart and Knuechel, 2002b), we reasoned that DPD might hold key factors, regulating malignancy at CRC CIFs. To test this, we unraveled overrepresented themes in DPD through enrichment analysis in Gene Ontology (GO) annotations for biological process. This analysis resulted in the significant (p < 0.05) overrepresentation of 155 GO terms (Supplementary Table 2). Since several annotations are branched together, we visualized the analysis as an enrichment network, which algorithmically clustered GO terms with highly similar content, using the enrichment map plug‐in in cytoscape environment (Supplementary Figure 1). To better define their nature, we used a word‐frequency detection algorithm to automatically generate prevailing keywords in these clusters. The resulting keywords corresponded to well‐established hallmarks of cancer, namely “cell proliferation”, “cell migration”, “cell motility”, “cell adhesion”, “neoangiogenesis”, “neovascularization”, “development and morphogenesis”, “inflammation” among others (Figure 1). Representative GO terms corresponding to “hubs” of these clusters are shown in Figure 2A.
Figure 1.
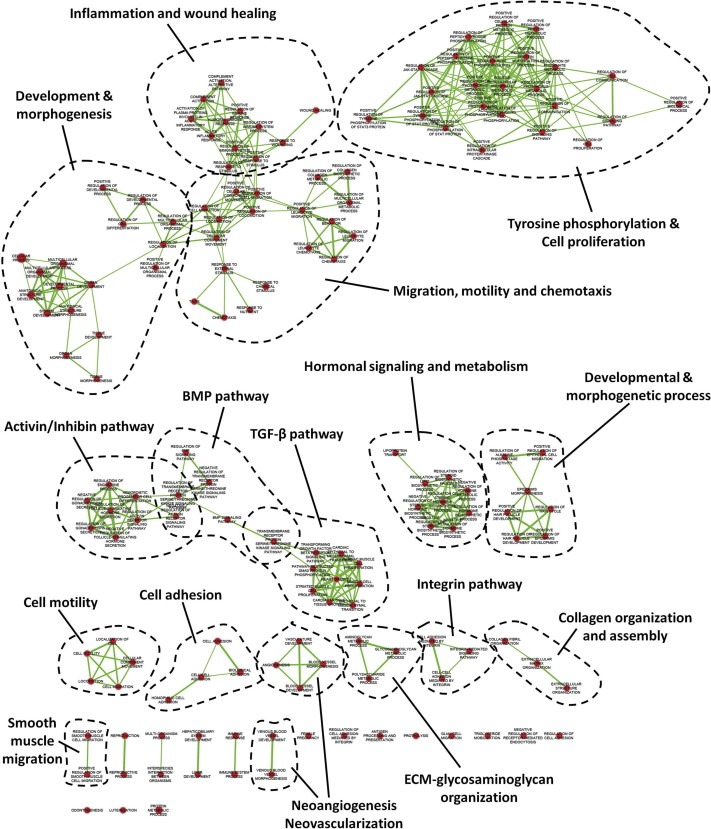
Enrichment map profiling of DPD reveals overrepresentation of functional clusters associated with cancer development and progression. Nodes represent GO terms and lines demonstrate their connectivity. Dashed lines encircle groups of relevant GO terms into functional clusters, named from word cloud algorithm.
Figure 2.
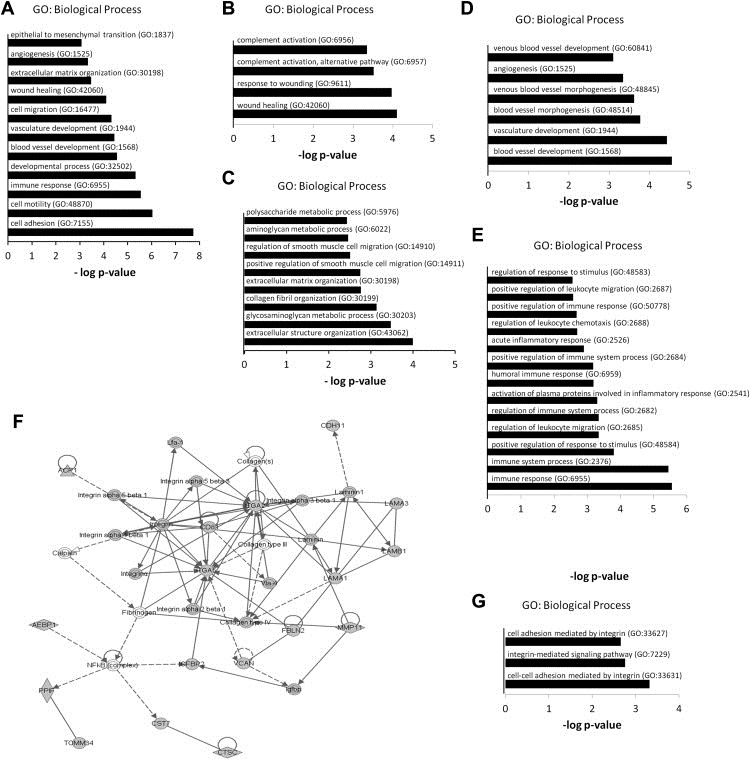
Bioinformatic characterization of the enrichment map verifies the desmoplastic nature of DPD. (A) Enriched GO terms found as hubs of the functional clusters of the enrichment map. (B) Enriched GO terms for the genes found in the “wound healing” cluster. (C) Enriched GO terms for the genes found in the combined “smooth muscle migration”, “collagen organization and assembly” and “ECM‐glycosaminoglycan organization” clusters. (D) Enriched GO terms for the genes found in the combined “neoangiogenesis” and “neovascularization clusters. (E) Enriched GO terms for the genes found in the “inflammation” cluster. (F) IPA network demonstrating the integrin/laminin axis as regulatory hub within the network. (G) Enriched GO terms for the genes found in the “integrin pathway” cluster.
3.2. Enrichment map profiling verifies the desmoplastic nature of DPD
The enrichment map analysis indicated four distinct molecular signatures, collectively verifying the desmoplastic nature of DPD: (I) the presence of the “wound healing” cluster, (II) the presence of clusters associated with fibroblast‐to‐myofibroblast phenotype switching, (III) the presence of clusters associated with regulation of other stromal responses (i.e. angiogenesis and inflammation) and, (IV) the presence of clusters regulating matrix‐to‐cell adhesions (i.e. integrin signaling), reminiscent of those in desmoplastic CIFs.
-
(I)
The recruitment of CAFs by cancer cells has been paralleled with the transdifferentiation of resident/quiescent fibroblasts into myofibroblasts in tissues undergoing wound healing (Dvorak, 1986). Consistent with this, a “wound healing” cluster was determined in the enrichment map profiling of DPD (Figure 1). This cluster was comprised of two corresponding GO terms (i.e. “response to wounding” and “wound healing”) in close proximity to annotations associated with immune response and inflammation. It was also linked to the function of proteins, such as members of the complement system (e.g. C4A, C4B, CFD and CFH) (Figure 2B), which have been previously linked to both wound healing and cancer progression (Glaros et al., 2009; Stover, 2010).
-
(II)
The myofibroblastic phenotype of CAFs is characterized by increased proliferation, migration, contractility, ECM‐remodeling and collagen organization/assembly, all of which are regulated from a de novo‐deployed smooth muscle protein expression machinery (De Wever et al., 2008a; Kalluri and Zeisberg, 2006). Associated clusters, such as the “smooth muscle migration”, “collagen organization and assembly” and “ECM‐glycosaminoglycan organization” were found in the enrichment map (Figure 1). Of note, the corresponding GO terms (Figure 2C) demarcate various ECM‐regulatory proteins, including lysine‐6‐oxidase (LOX) and COL12A1, which have both been associated with regulation of matrix stiffness and mechanotransduction of the peritumoral desmoplastic stroma (Karagiannis et al., 2012b). We have previously demonstrated COL12A1 expression in α‐SMA positive CAFs, as well as in cancer cells lining the desmoplastic CIFs and tumor budding cells in CRC patients (Supplementary Figure 2) (Karagiannis et al., 2012a).
-
(III)
Desmoplasia has been considered as a positive regulator of the angiogenic switch, with the exception of a few cancer types, such as pancreatic ductal adenocarcinoma (Olive et al., 2009). In CRC, colonic CAFs may locally promote angiogenesis (Hewitt et al., 1991; Huang et al., 2012), an observation supported by the enrichment map profiling of DPD, whereby two relevant clusters, “neoangiogenesis” and “neovascularization”, were found (Figures 1 and 2D). Moreover, an “inflammation” cluster was found in the enrichment map (Figures 1 and 2E) verifying accumulative literature that desmoplasia participates in reciprocal signaling with the inflammatory microenvironment (Mueller et al., 2007; Quante et al., 2011).
-
(IV)
An independent pathway analysis of the DPD using IPA demonstrated that members of the integrin family and the focal adhesion machinery (i.e. ITGA2 and ITGA5) serve as major regulatory hubs in the dataset (Figure 2F). In addition, an “integrin pathway” cluster with representative GO terms related to the integrin signaling pathway was also found in the enrichment map (Figures 1 and 2G). These data verify previous literature (Karagiannis et al., 2012b; Navab et al., 2011) that an altered cell‐to‐matrix program and focal adhesion machinery is supported at desmoplastic CIFs.
3.3. The TGF‐β superfamily of proteins harbors the major regulatory network in DPD
After verifying the desmoplastic nature of DPD, we sought to characterize specific pathways that could potentially harbor causative regulation of neoplastic progression in desmoplastic CIFs. Interestingly, three clusters corresponding to the three canonical pathways of the TGF‐β‐superfamily of proteins (“TGF‐β pathway”, “activin/inhibin pathway”, “BMP pathway”) were all overrepresented in the enrichment map profiling of DPD (Figure 1). The corresponding GO terms were associated with the regulation of signal transduction of these pathways, as well as with various developmental processes regulated by them, in particular EMT and cell differentiation (Figure 3A).
The TGF‐β‐superfamily cluster comprised of ligands/receptors (i.e. TGFB2, TGFBR3, GDF6, INHBA) that normally participate in signal propagation (group I), as well as of regulatory proteins (i.e. FST, FSTL3, HTRA3, GREM1, LTBP1) that antagonize signal transduction (group II) (Figure 3B). As suggested by two independent pathway analyses (IPA and STRING), which use diverse algorithms for generating protein–protein interaction networks, both group I and group II members of the TGF‐β‐superfamily cluster were found under a common network, nucleated around representative components/hubs of the TGF‐β‐pathway (Figure 3C and D). Specifically, all three signaling pathways of the superfamily [i.e. TGF‐beta pathway, activin/inhibin pathway and bone morphogenetic protein (BMP) pathway] were represented in the DPD (Figures 1, 3C & 3D). Of note though, group I proteins exclusively belonged to the TGF‐β and activin/inhibin pathways and no ligands/receptors of the BMP pathway were found in this group. On the contrary, group II proteins belonged to the BMP pathway, comprising of the BMP antagonists gremlin‐1 (GREM1), follistatin (FST), follistatin‐like 3 (FSTL3) and high‐temperature requirement A3 (HTRA3). Consequently, cancer cell‐CAF interactions seem to preferentially disrupt the BMP signaling pathway propagation at desmoplastic CIFs through secretion of multiple BMPIs.
3.4. BMPIs may be involved in cancer/metastasis hallmarks
We next sought to investigate whether the secreted BMPIs identified in DPD could participate in the regulation of malignant traits of metastasis. To interrogate this, we performed enrichment analysis to test whether group I or II genes are significantly overrepresented in any of the defined clusters of the enrichment map. Interestingly, several clusters corresponding to specific cancer and metastasis hallmarks (i.e. cell proliferation, migration, motility, adhesion, various developmental processes, angiogenesis, neovascularization, and inflammation) were enriched for both receptors/ligands of the TGF‐beta superfamily of proteins (group I) and BMPIs (group II) (Figure 4A). Thus, our current working model suggests that BMPIs may be expressed at desmoplastic CIFs by either cancer cells or CAFs, to regulate cancer‐specific biological programs (e.g. invasion/migration of cancer cells) as well as stromatogenic responses (e.g. angiogenesis/neovascularization) (Figure 4B).
Figure 4.
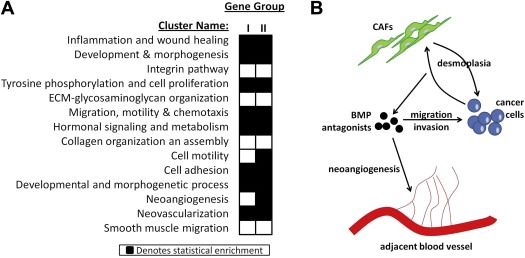
BMPIs are overrepresented in various clusters of the enrichment map. (A) Heatmap demonstrating statistical enrichment of a specific cluster identified in the enrichment map in either group I or group II genes, with the latter group corresponding to the BMPIs. (B) A working model that demonstrates the potential regulation of various biological programs and stromatogenic responses by the BMPIs in desmoplastic CIFs.
3.5. Simulation of cancer cell migration using mathematical modeling predicts microenvironmental regulation by GREM1 gradient
Data from the previous section formulated the working hypothesis that BMPIs may regulate localized invasion of various tumor populations into the adjacent cancer‐associated stroma. We wished to predict whether BMPI expression in desmoplastic CIFS would indeed have such an impact on the migratory behavior of cancer cells.
To proceed to the generation of a mathematical model capable of such prediction, we first defined the biological interface to be modeled: According to the traditional concept of the metastatic cascade, most carcinomas initially develop in‐situ whereby they are separated from the adjacent normal stroma with a basement membrane. At some point, the basement membrane is breached and cancer populations are migrating in collective configuration through the adjacent stroma, excavating a pathway towards the lymphatic or blood vessels (Figure 5A; left illustration) (Hanahan and Weinberg, 2011). A transverse section of these migratory collectives in the submucosa gives the appearance of “tumor islets” surrounded by cancer associated stroma. In this study, we define this collective as “cancer cell cohort” (Figure 5A; middle illustration). Three distinct cancer subpopulations present with a diverse repertoire of interactions with the surrounding stroma: the “tumor core” cells do not contact the desmoplastic stroma, the “tumor invasion front” cells come in direct contact with the desmoplastic stroma and the “tumor budding” cells are small groups of cells that have detached from the invasion front and further migrate in the desmoplastic stroma away from the rest of the collective (Figure 5A; right illustration) (Mitrovic et al., 2012).
Figure 5.
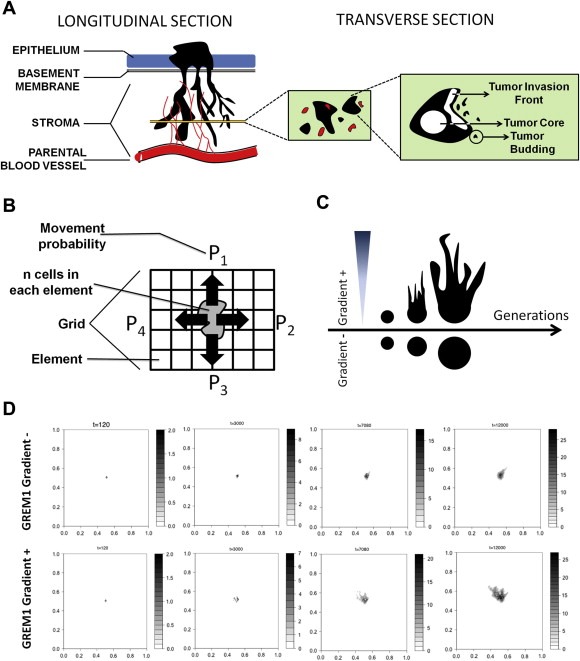
Mathematical modeling of cancer cell migration predicts microenvironmental regulation by GREM1 gradients. (A) The biological concept illustrated in the mathematical model: Cancer cells are migrating in collective configuration through the adjacent stroma, forming octopus‐like projections (left illustration; longitudinal section). A transverse section of these invasive collectives will have the appearance of tumor islets surrounded by cancer associated stroma (middle illustration). Each one of these collectives is termed as cancer cell cohort and has three distinct cancer subpopulations, the “tumor core” cells, the “tumor invasion front” cells and the “tumor budding” cells (right illustration). (B) Visualization of the mathematical model. The diverse tumor subpopulations (n number of cells in each element) respond to microenvironmental stimuli, thus depicting a movement probability (P1‐4) towards the various elements of the grid. (C) Hypothesis of the mathematical model: The generation of a microenvironmental GREM1 gradient from an element of the grid will affect cell migration towards the source of the gradient. Absence of such gradient will not generate a preferential migratory behavior towards specific elements of the grid. (D) Representative time snapshots of the mathematical modeling demonstrating the developing cancer cell cohort in either the absence of GREM1 gradient (first row) or the presence of it (second row). Note, that the cancer cell cohorts growing in the absence of GREM1 gradient is phenotypically reminiscent with the avascular tumor growth model described by Ward (1997) (Ward and King, 1997), whereas the cancer cell cohorts growing in the presence of GREM1 gradient actually develop branching formation, supporting the generated hypothesis of panel (C).
The developed mathematical model simulates one single cancer cell cohort surrounded by its desmoplastic stroma (Figure 5B). The cancer cell cohort grows on a matrix (i.e. grid) following previous models of tumor growth (Anderson, 2005; Chaplain and Anderson, 1996; Ward and King, 1999). In this study, we particularly focused on the migratory behavior of the cancer cell cohort over a considerable number of simulation generations. The migratory behavior of each cell depends on the microenvironmental stimuli (i.e. BMPI expression) present in the grid, where the expected direction of cell taxis is determined by the differential environmental states of neighboring grid cells (Figure 5B). The details in constructing the motogenic dynamics are thoroughly explained in Supplementary Materials & Methods.
The biological working hypothesis, explained in the previous section and illustrated in Figure 4B could be translated in this mathematical model as follows. The application of gradients of stromatogenic motogens, such as GREM1, around cancer cell cohorts, will affect the migratory response of the tumor subpopulations, causing a branching morphology in the cohort spreading (Figure 5C). On the contrary, the increased migratory behavior of a cancer cell cohort in the absence of a GREM1 microenvironmental gradient will induce a random spreading of the cancer cells without evident direction (Figure 5C).
The mathematical model allows for the observation of a vast plethora of generations to draw conclusions on the morphology of the cohorts. Indeed, the model suggests that after a considerable number of generations, a branching morphology is induced in the cancer cell cohort, which always sprouts towards the randomly‐appearing microenvironmental GREM1 gradient in the grid (Figure 5D, Supplementary Video 1). In the absence of such gradient, migration occurs against no specific direction on the grid, thus giving a more circular appearance in the evolving cohorts (Figure 5D, Supplementary Video 2). Overall, microenvironmental gradients of GREM1 are predicted to cause branching formation of cancer cell cohorts invading the stroma.
3.6. GREM1 mediates the direction of migration of CRC cells in vitro and in silico
To test the accuracy of this prediction in a simplistic in vitro setting, we performed under‐the‐agarose cell migration assays. Such assays depend on the generation of chemotactic gradients of motogens that are diffused in agarose gels. In a control experiment where no motogen gradients were applied on the setting, we were able to observe an equal distribution of migratory cancer cell populations (SW480) in each quadrant, which measured approximately 25% of the total crystal violet staining intensity observed in the entire Petri dish (Supplementary Figure 3). Following the optimization of the assay, we proceeded to investigating the influence of chemotactic gradients of GREM1 and/or BMP7 on the migration of both SW480 and SW620 cells. The tested motogen, in this case rhGREM1, was initially diluted in wells positioned in the periphery of Petri dishes and over time the developing gradients affected the migratory behavior of cancer cell populations residing in the central cavity. To monitor migration of the cancer cell cohort under the influence of the GREM1 motogen, we fixed and stained the cancer cells with crystal violet after the termination of all experiments, and subsequently used imaging software to calculate the percentage of cancer cells positioned on each quadrant corresponding to one of the four individual gradients.
Our initial observations on this setup revealed that more than 70% of SW480/SW620 cells migrated towards a rhGREM1 gradient (2 μg/mL) (quadrant B), while lower populations (<10%) migrated towards the absence of such gradient (quadrant A), or towards a gradient of rhBMP7 (500 ng/mL) (quadrant C) (Figure 6A). Especially the latter is an interesting observation, because it demonstrates that BMPs not only decrease cancer cell migration as also suggested by the literature (Alarmo and Kallioniemi, 2010), but may also act as repellents of cancer cell motility. Finally, only a small SW480/SW620 population (∼10%) migrated towards a gradient consisting of admixed rhBMP7 and rhGREM1 (quadrant D) (Figure 6A). This suggests that when rhGREM1 gradient is disrupted by addition of rhBMP7, then under‐the‐agarose cancer cell migration is abrogated in this specific quadrant, possibly through reduction of rhGREM1 bioavailability from its covalent binding with rhBMP7. Moreover, this observation indicates that the observed high migratory behavior of SW480/SW620 cells in quadrant B is indeed GREM1‐specific.
Figure 6.
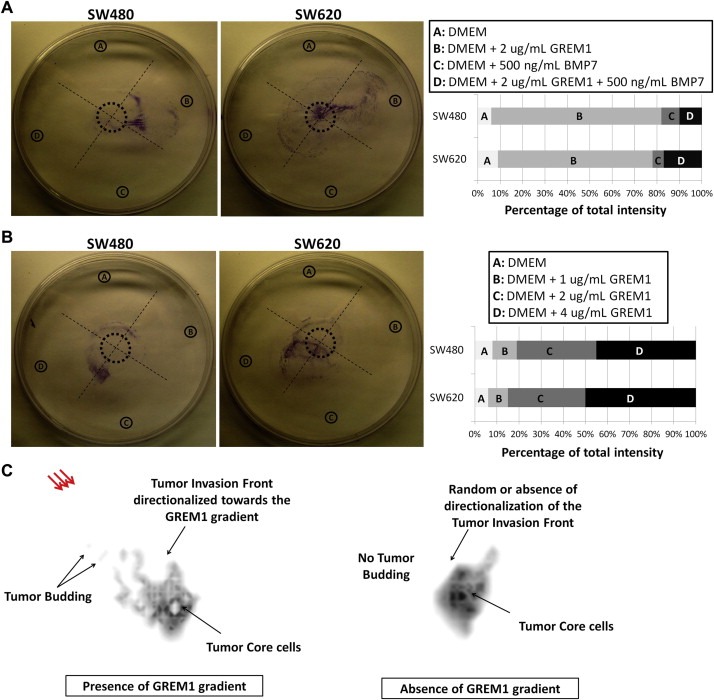
GREM1 mediates the direction of migration of CRC cells in vitro and in silico. (A–B) under‐the‐agarose cell migration assays. Panels on the right describe in detail the gradients induced in each of the quadrants A–D. The corresponding graphs below describe the crystal violet intensity found in each individual quadrant, as percentage of the total fluorescence that was detected in the entire plate. The values are mean percentages of intensities from three independent replicates. The figures show one representative replicate from each cell line. (C) Magnified snapshot (generation #7080) of the mathematical modeling demonstrating the developing cancer cell cohort in either the absence of GREM1 gradient (right schematic) or the presence of it (red arrows; left schematic). The black arrows with labels and descriptions compare the behavior of the three prominent cancer cell subpopulations: Tumor core, tumor invasion front, and tumor budding cells.
Subsequent experiments on this setup revealed that the SW480/SW620 cancer cells migrated towards GREM1 gradients in a dose‐dependent fashion. Specifically, approximately 50% of the cells migrated towards quadrant D, which was the strongest gradient (4 μg/mL of rhGREM1), approximately 30% towards quadrant C (2 μg/mL of rhGREM1), while the remaining 20% of cells were collectively found on quadrants A and B, which held no or very weak (1 μg/mL of rhGREM1) gradients, respectively (Figure 6B).
In all of the in vitro experiments presented in this section, an increased cancer cell population was always found in the quadrants with the highest rhGREM1 concentrations (i.e. quadrant B of Figure 6A and quadrant D of Figure 6B). In all of the in silico predictions performed in the previous section, increased cancer cell motility, coupled to branching morphology and tumor budding formation (all of which are hallmarks of increased cancer cell migration) were exclusively observed in the presence of GREM1 gradients (Figure 6C). Therefore, when all in vitro and in silico findings were qualitatively co‐assessed, we found that they corresponded to each other with an optimal overlap, thus rendering the mathematical predictions (Figure 5) more concrete.
4. Discussion
Currently, pathway analysis software seems to be a major strategy for conceptualizing molecular pathways of interest in large proteomic datasets, especially those derived from mass spectrometry‐based experiments (Tiger et al., 2012). Due to the high load of information in such datasets, here, we performed an unbiased integrative pathway dissection of our previously described DPD (Karagiannis et al., 2012a), using three independent pathway analysis tools: enrichment map profiling, IPA and STRING. The enrichment map profiling pointed to the TGF‐β‐superfamily pathway, as an enriched canonical pathway in our dataset (Figure 1), whereas the remaining two pathway analyses, Ingenuity and STRING, specified the BMP antagonists as molecular hubs of interest in our dataset (Figure 3C–D). Therefore, these bioinformatic tools have been utilized in a complementary fashion and successfully narrowed the high candidate number down to the BMP‐antagonist signature, as potential marker of cancer progression in CRC desmoplastic CIFs. Our group advocates the utilization of more than one pathway analysis algorithm in such type of studies for ensuring data reproducibility, reduction of database‐specific and operator‐specific biases, as well as network robustness.
The confirmation of the desmoplastic nature of the DPD (Figure 2), from where GREM1 was selected and further investigated, should not be disregarded. Indeed it has been documented that BMP antagonists, and especially GREM1, are being produced in desmoplastic microenvironments of various types of cancer (Karagiannis et al., 2012b; Sneddon et al., 2006), as well as in other fibrotic conditions of non‐neoplastic diseases (Carvajal et al., 2008). Also, it has been shown that GREM1 is expressed by stromal cells of myofibroblastic phenotype (Kosinski et al., 2007), a cell type that is highly abundant in cancer desmoplasia (Kunz‐Schughart and Knuechel, 2002a). However, it should be mentioned that expression of GREM1 by cancer cells has also been documented (Namkoong et al., 2006), although interaction of these cancer cells with normal stroma seems to always be necessary for GREM1 overexpression (Karagiannis et al., 2012b). In the current study, the functional association of GREM1 gradients with increased migratory behavior of cancer cells constitutes a proof‐of‐principle notion that DPD could be potentially enriched in myofibroblastic/desmoplastic factors associated with the regulation of neoplastic development and progression. Thus, we believe that other proteins identified in DPD should be further explored in the future for potential impact on CRC progression.
In recent years, it has become clear that carcinogenesis is a rather complicated process and the understanding of the origins, growth and spread of cancer required an integrated or system‐wide approach (Materi and Wishart, 2007). In this work, we performed mathematical simulation to predict the role of GREM1 in the tumor microenvironment. We used a hybrid model, as it has been suggested by multiple researchers that it combines the advantages of both discrete (cellular automata) and continuous approaches (Coveney and Fowler, 2005; Ridgway et al., 2006), to address this issue. Our model has successfully driven our rationale towards a possible motogenic effect of GREM1 in desmoplastic tumor microenvironments and allowed us to successfully test this hypothesis in vitro. We anticipate that computational systems biology will continue to grow as a distinct sub‐discipline in systems biology and allow the assembling of computer simulations for intra‐ and intercellular processes (Materi and Wishart, 2007).
BMPs, especially BMP2, are produced by normal colonic epithelium to sustain epithelial phenotype at the level of the colonic crypt (Hardwick et al., 2004). Interestingly, BMP2 and BMP7 signaling have been documented to have tumor‐suppressive properties in colorectal and other types of cancer, particularly by reducing cancer cell proliferation, invasion, motility as well as blocking EMT (Beck et al., 2007, 2006; Flier et al., 2010; Wu et al., 2008; Zeisberg et al., 2005). On the other side, it has been hypothesized that BMP inhibitors, especially GREM1, could potentially antagonize BMP signaling at the cancer invasion front, thus supporting the maintenance of a mesenchymal niche (He et al., 2004; Sneddon and Werb, 2007; Sneddon et al., 2006). GREM1 is an already established mediator of EMT in non‐cancerous pathologies, as shown for instance in chronic allograft nephropathy (Carvajal et al., 2008). It therefore seems that BMP signaling is essential for normal epithelial integrity of the colon and is disrupted during CRC development and progression. For these reasons, we focused our interest on the group II genes belonging to the BMP cluster of the enrichment map, consisting of the BMP inhibitors FST, FSTL3 and HTRA3 on top of the more thoroughly investigated GREM1.
The BMP inhibitors identified were either highly‐selective against certain BMP ligands (e.g. GREM1 covalently binds to and heterodimerizes with BMP2, BMP4 and BMP7) (Kosinski et al., 2007) or less‐selective (e.g. FST and FSTL3 bind to BMPs with less affinity) (Beites et al., 2009) or even non‐selective (e.g. HTRA3 is an extracellular protease that remodels ECM in a manner that sequesters members of the TGF‐β superfamily and reduces their bioavailability) (Tocharus et al., 2004). However, they all clustered up together in our pathway analyses (Figure 3C & D) further demonstrating that they retain pivotal regulatory roles in desmoplastic CIFs. In this study, we decided to focus on GREM1, since it is the most highly‐selective among all these BMP inhibitors and provided its causative link with tumor progression as a proof‐of‐concept. However, by targeting only one member of the BMPI signature (i.e. GREM1) and given that the desmoplastic CIF is a source of multiple BMPIs, it would be reasonable to assume that FST, FSTL3 and HTRA3 could partially compensate for the GREM1 targeting. Thus, we believe that a holistic approach should be taken in investigating this pathway, and, more precisely, the entire BMPI signature should be examined in future studies.
The BMP pathway, a developmental pathway which promotes the sustenance of epithelial phenotype in various cell types and cell lineages, may modulate tumor‐suppressive effects in CRC and other cancers (Alarmo and Kallioniemi, 2010; Beck et al., 2006; Hsu et al., 2005; Wu et al., 2008; Ye et al., 2007). Recent data suggest that sporadic CRC may disrupt the tumor‐suppressive effects of BMP signaling by genomic mutations of SMAD4 and BMP receptor type II (BMPR2) (Kotzsch et al., 2008). Other lines of evidence suggest the deployment of epigenetic mechanisms for the disruption of BMP signaling in tumors of the gastrointestinal tract (Wen et al., 2006). In the current study, as well as in others (Sneddon et al., 2006), the paracrine production of BMP antagonists, especially GREM1, served as a documented mechanism for BMP2/7 impairment and promotion of tumor cell motility and proliferation. Reasonably, certain questions could be raised from all these observations. First, why does cancer develop a wide diversity of mechanisms to block BMP signaling? Second, are all of them or only a few of these mechanisms essential for successfully blocking BMP signaling in cancer cells?
To partially answer these questions, we speculate that the tumor microenvironment in CRC deploys a plethora of strategies, most of the times serving as “safety valves”, to ensure that BMP signaling will be either totally inhibited or effectively antagonized. Here, we refer to the totality of these strategies as “the multilayered BMP‐inhibition hypothesis”. This working hypothesis states that CRC cells of the advanced invasion front can only flourish in a microenvironment devoid of the tumor‐suppressive properties of the BMP signaling. To guarantee its absolute disruption, the cancer cells deploy multiple “layers” of BMP antagonism, including the genetic level (i.e. SMAD4/BMPR2 functional mutations), the epigenetic level (i.e. methylation of BMP2 gene promoter), as well as the proteomic level (i.e. antagonistic regulation of BMP ligands by paracrine production of BMP antagonists). The latter was also the focus of the current study.
Despite the fact that the under‐the‐agarose cell migration assays do not take into account the real complexity of the tumor microenvironment found in vivo, they are highly indicative on demonstrating the ability of microenvironmental stimuli to affect cancer cells cohorts. Evidently, our findings suggest that GREM1 is capable of inducing cancer cell migration in vitro, with profound chemotactic tendency of SW480/SW620 cells towards the source of GREM1 gradients. As expected, the addition of BMP7 together with GREM1 in one of the wells in this assay (Figure 6A) caused an antagonizing effect, reducing the bioavailability of GREM1 and slowing SW480/SW620 motility. This could suggest that GREM1 mediates its tumor‐promoting effects in a BMP7‐dependent manner.
Besides its implication in cancer cell motility, literature evidence suggests that GREM1 may stimulate endothelial cell intracellular signaling and migration in vitro, leading to a potent angiogenic response in vivo (Mitola et al., 2010; Stabile et al., 2007). This occurs due to the ability of GREM1 to bind to and activate phosphorylation of vascular endothelial growth factor receptor‐2 (VEGFR2) in a BMP‐independent fashion (Mitola et al., 2010), leading to the formation of VEGFR2/ανβ3 integrin complexes and angiogenesis promotion (Ravelli et al., 2012). Thus, GREM1 may act as non‐canonical VEGFR2 agonist, distinct from VEGF family members, that may play autocrine/paracrine functions in tumor neovascularization. In a complementary fashion, the BMPI group (i.e. group II genes) was found to be statistically enriched in most molecular clusters of the enrichment map, including the angiogenesis and cell motility clusters (Figure 4A). Conclusively, the tumorigenic properties of GREM1 as exerted by its documented motogenic and angiogenic responses may highlight the potential for therapeutic targeting of this molecule in neoplastic disease.
In conclusion, we have defined the functional potential of a robust and extensive proteomic signature of the desmoplastic CIF during CRC development and progression. Our results show that reciprocal interactions between cancer and host cells may result in the expression of BMPIs, such as GREM1, as an effective regulatory mechanism for dictating motogenic cancer cell fates.
5. Author contributions
Conceptualized study: G.S.K.; Designed, performed and interpreted bioinformatics analyses: G.S.K. and A.D.; Designed, simulated and interpreted the mathematical model: G.S.K., A.B. and A.D.; Designed, performed and interpreted in‐vitro experiments: G.S.K..; Wrote manuscript: G.S.K., A.B. and E.P.D.; Revised manuscript: G.S.K., A.B., A.D. and E.P.D.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary Table 1 List of 152 proteins with their respective gene names, corresponding to the desmoplastic protein dataset (DPD).
Supplementary Table 2 The output file of BINGO analysis, listing the significantly enriched GO annotations for biological process found in the DPD.
Video S1 Simulation of migratory behavior of a single cancer cell cohort invading the peritumoral stroma, under the influence of GREM1 microenvironmental gradient.
Video S2 Simulation of migratory behavior of a single cancer cell cohort invading the peritumoral stroma, without the influence of GREM1 microenvironmental gradient.
Acknowledgements
We would like to thank Dr. Costantina Petraki for Supplementary Figure 2; the staining of these tissues is reported previously (Karagiannis et al., 2012a). We would like to thank Antoninus Soosaipillai, Ihor Batruch, and Chris Smith for technical support. We would like to also thank Dr. Ioannis Prassas, Dr. Jane Bayani, Dr. Geeth Gunawardana and Dr. Uros Kuzmanov for suggestions on result interpretation, and Maria Pavlou for reading this article and providing valuable feedback.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.04.002.
Karagiannis George S., Berk Aaron, Dimitromanolakis Apostolos, Diamandis Eleftherios P., (2013), Enrichment map profiling of the cancer invasion front suggests regulation of colorectal cancer progression by the bone morphogenetic protein antagonist, gremlin‐1, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.04.002.
References
- Alarcon, T. , Byrne, H.M. , Maini, P.K. , 2003. A cellular automaton model for tumour growth in inhomogeneous environment. J. Theor. Biol. 225, 257–274. [DOI] [PubMed] [Google Scholar]
- Alarmo, E.L. , Kallioniemi, A. , 2010. Bone morphogenetic proteins in breast cancer: dual role in tumourigenesis?. Endocr. Relat. Cancer 17, R123–R139. [DOI] [PubMed] [Google Scholar]
- Anderson, A.R. , 2005. A hybrid mathematical model of solid tumour invasion: the importance of cell adhesion. Math. Med. Biol. 22, 163–186. [DOI] [PubMed] [Google Scholar]
- Ayala, G. , Tuxhorn, J.A. , Wheeler, T.M. , Frolov, A. , Scardino, P.T. , Ohori, M. , 2003. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin. Cancer Res. 9, 4792–4801. [PubMed] [Google Scholar]
- Beck, S.E. , Jung, B.H. , Del Rosario, E. , Gomez, J. , Carethers, J.M. , 2007. BMP-induced growth suppression in colon cancer cells is mediated by p21WAF1 stabilization and modulated by RAS/ERK. Cell. Signal. 19, 1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, S.E. , Jung, B.H. , Fiorino, A. , Gomez, J. , Rosario, E.D. , Cabrera, B.L. , 2006. Bone morphogenetic protein signaling and growth suppression in colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G135–G145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beites, C.L. , Hollenbeck, P.L. , Kim, J. , Lovell-Badge, R. , Lander, A.D. , Calof, A.L. , 2009. Follistatin modulates a BMP autoregulatory loop to control the size and patterning of sensory domains in the developing tongue. Development 136, 2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie, B. , Moses, H.L. , 2006. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520. [DOI] [PubMed] [Google Scholar]
- Carvajal, G. , Droguett, A. , Burgos, M.E. , Aros, C. , Ardiles, L. , Flores, C. , 2008. Gremlin: a novel mediator of epithelial mesenchymal transition and fibrosis in chronic allograft nephropathy. Transplant. Proc. 40, 734–739. [DOI] [PubMed] [Google Scholar]
- Chaplain, M.A. , Anderson, A.R. , 1996. Mathematical modelling, simulation and prediction of tumour-induced angiogenesis. Invasion Metastas. 16, 222–234. [PubMed] [Google Scholar]
- Coveney, P.V. , Fowler, P.W. , 2005. Modelling biological complexity: a physical scientist's perspective. J. R. Soc. Interface 2, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wever, O. , Demetter, P. , Mareel, M. , Bracke, M. , 2008. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 123, 2229–2238. [DOI] [PubMed] [Google Scholar]
- De Wever, O. , Mareel, M. , 2003. Role of tissue stroma in cancer cell invasion. J. Pathol. 200, 429–447. [DOI] [PubMed] [Google Scholar]
- De Wever, O. , Pauwels, P. , De Craene, B. , Sabbah, M. , Emami, S. , Redeuilh, G. , 2008. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell. Biol. 130, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak, H.F. , 1986. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Elenbaas, B. , Weinberg, R.A. , 2001. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell. Res. 264, 169–184. [DOI] [PubMed] [Google Scholar]
- Flier, S.N. , Tanjore, H. , Kokkotou, E.G. , Sugimoto, H. , Zeisberg, M. , Kalluri, R. , 2010. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 285, 20202–20212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaros, T. , Larsen, M. , Li, L. , 2009. Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front. Biosci. 14, 3988–3993. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hardwick, J.C. , Van Den Brink, G.R. , Bleuming, S.A. , Ballester, I. , Van Den Brande, J.M. , Keller, J.J. , 2004. Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology 126, 111–121. [DOI] [PubMed] [Google Scholar]
- He, X.C. , Zhang, J. , Tong, W.G. , Tawfik, O. , Ross, J. , Scoville, D.H. , 2004. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet. 36, 1117–1121. [DOI] [PubMed] [Google Scholar]
- Heit, B. , Kubes, P. , 2003. Measuring chemotaxis and chemokinesis: the under-agarose cell migration assay. Sci. STKE 2003, PL5 [DOI] [PubMed] [Google Scholar]
- Hewitt, R.E. , Leach, I.H. , Powe, D.G. , Clark, I.M. , Cawston, T.E. , Turner, D.R. , 1991. Distribution of collagenase and tissue inhibitor of metalloproteinases (TIMP) in colorectal tumours. Int. J. Cancer 49, 666–672. [DOI] [PubMed] [Google Scholar]
- Hsu, M.Y. , Rovinsky, S. , Penmatcha, S. , Herlyn, M. , Muirhead, D. , 2005. Bone morphogenetic proteins in melanoma: angel or devil?. Cancer Metastasis Rev. 24, 251–263. [DOI] [PubMed] [Google Scholar]
- Huang, W.H. , Chang, M.C. , Tsai, K.S. , Hung, M.C. , Chen, H.L. , Hung, S.C. , 2012. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 10.1038/onc.2012.458 Oct 22 [DOI] [PubMed] [Google Scholar]
- Jensen, L.J. , Kuhn, M. , Stark, M. , Chaffron, S. , Creevey, C. , Muller, J. , 2009. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , Zeisberg, M. , 2006. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401. [DOI] [PubMed] [Google Scholar]
- Karagiannis, G.S. , Pavlou, M.P. , Diamandis, E.P. , 2010. Cancer secretomics reveal pathophysiological pathways in cancer molecular oncology. Mol. Oncol. 4, 496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis, G.S. , Petraki, C. , Prassas, I. , Saraon, P. , Musrap, N. , Dimitromanolakis, A. , 2012. Proteomic signatures of the desmoplastic invasion front reveal collagen type XII as a marker of myofibroblastic differentiation during colorectal cancer metastasis. Oncotarget 3, 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis, G.S. , Poutahidis, T. , Erdman, S.E. , Kirsch, R. , Riddell, R.H. , Diamandis, E.P. , 2012. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical Pressure on cancer tissue. Mol. Cancer Res. 10, 1403–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis, G.S. , Weile, J. , Bader, G.D. , Minta, J. , 2013. Integrative pathway dissection of molecular mechanisms of moxLDL-induced vascular smooth muscle phenotype transformation. BMC Cardiovasc. Disord. 13, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski, C. , Li, V.S. , Chan, A.S. , Zhang, J. , Ho, C. , Tsui, W.Y. , 2007. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. U S A 104, 15418–15423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzsch, A. , Nickel, J. , Seher, A. , Heinecke, K. , van Geersdaele, L. , Herrmann, T. , 2008. Structure analysis of bone morphogenetic protein-2 type I receptor complexes reveals a mechanism of receptor inactivation in juvenile polyposis syndrome. J. Biol. Chem. 283, 5876–5887. [DOI] [PubMed] [Google Scholar]
- Kunz-Schughart, L.A. , Knuechel, R. , 2002. Tumor-associated fibroblasts (part I): active stromal participants in tumor development and progression?. Histol. Histopathol. 17, 599–621. [DOI] [PubMed] [Google Scholar]
- Kunz-Schughart, L.A. , Knuechel, R. , 2002. Tumor-associated fibroblasts (part II): functional impact on tumor tissue. Histol. Histopathol. 17, 623–637. [DOI] [PubMed] [Google Scholar]
- Maere, S. , Heymans, K. , Kuiper, M. , 2005. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. [DOI] [PubMed] [Google Scholar]
- Materi, W. , Wishart, D.S. , 2007. Computational systems biology in cancer: modeling methods and applications. Gene Regul. Syst. Bio. 1, 91–110. [PMC free article] [PubMed] [Google Scholar]
- Merico, D. , Isserlin, R. , Bader, G.D. , 2011. Visualizing gene-set enrichment results using the cytoscape plug-in enrichment map. Methods Mol. Biol. 781, 257–277. [DOI] [PubMed] [Google Scholar]
- Merico, D. , Isserlin, R. , Stueker, O. , Emili, A. , Bader, G.D. , 2010. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitola, S. , Ravelli, C. , Moroni, E. , Salvi, V. , Leali, D. , Ballmer-Hofer, K. , 2010. Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 116, 3677–3680. [DOI] [PubMed] [Google Scholar]
- Mitrovic, B. , Schaeffer, D.F. , Riddell, R.H. , Kirsch, R. , 2012. Tumor budding in colorectal carcinoma: time to take notice. Mod. Pathol. 25, 1315–1325. [DOI] [PubMed] [Google Scholar]
- Mueller, L. , Goumas, F.A. , Affeldt, M. , Sandtner, S. , Gehling, U.M. , Brilloff, S. , 2007. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 171, 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkoong, H. , Shin, S.M. , Kim, H.K. , Ha, S.A. , Cho, G.W. , Hur, S.Y. , 2006. The bone morphogenetic protein antagonist gremlin 1 is overexpressed in human cancers and interacts with YWHAH protein. BMC Cancer 6, 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navab, R. , Strumpf, D. , Bandarchi, B. , Zhu, C.Q. , Pintilie, M. , Ramnarine, V.R. , 2011. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. U S A 108, 7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive, K.P. , Jacobetz, M.A. , Davidson, C.J. , Gopinathan, A. , McIntyre, D. , Honess, D. , 2009. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandol, S. , Edderkaoui, M. , Gukovsky, I. , Lugea, A. , Gukovskaya, A. , 2009. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 7, S44–S47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perumpanani, A.J. , Sherratt, J.A. , Norbury, J. , Byrne, H.M. , 1996. Biological inferences from a mathematical model for malignant invasion. Invasion Metastas. 16, 209–221. [PubMed] [Google Scholar]
- Prassas, I. , Karagiannis, G.S. , Batruch, I. , Dimitromanolakis, A. , Datti, A. , Diamandis, E.P. , 2011. Digitoxin-induced cytotoxicity in cancer cells is mediated through distinct kinase and interferon signaling networks. Mol. Cancer Ther. 10, 2083–2093. [DOI] [PubMed] [Google Scholar]
- Quante, M. , Tu, S.P. , Tomita, H. , Gonda, T. , Wang, S.S. , Takashi, S. , 2011. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2008. R: A Language and Environment for Statistical Computing R Foundation for Statistical Computing; Vienna, Austria: 3-900051-07-0 <http://www.R-project.org> [Google Scholar]
- Ravelli, C. , Mitola, S. , Corsini, M. , Presta, M. , 2012. Involvement of alpha(v)beta (3) integrin in gremlin-induced angiogenesis. Angiogenesis 16, 235–243. [DOI] [PubMed] [Google Scholar]
- Ridgway, D. , Broderick, G. , Ellison, M.J. , 2006. Accommodating space, time and randomness in network simulation. Curr. Opin. Biotechnol. 17, 493–498. [DOI] [PubMed] [Google Scholar]
- Sherratt, J.A. , Chaplain, M.A. , 2001. A new mathematical model for avascular tumour growth. J. Math. Biol. 43, 291–312. [DOI] [PubMed] [Google Scholar]
- Silva, A.S. , Gatenby, R.A. , Gillies, R.J. , Yunes, J.A. , 2009. A quantitative theoretical model for the development of malignancy in ductal carcinoma in situ. J. Theor. Biol. 262, 601–613. [DOI] [PubMed] [Google Scholar]
- Sneddon, J.B. , Werb, Z. , 2007. Location, location, location: the cancer stem cell niche. Cell Stem Cell 1, 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon, J.B. , Zhen, H.H. , Montgomery, K. , van de Rijn, M. , Tward, A.D. , West, R. , 2006. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc. Natl. Acad. Sci. U S A 103, 14842–14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabile, H. , Mitola, S. , Moroni, E. , Belleri, M. , Nicoli, S. , Coltrini, D. , 2007. Bone morphogenic protein antagonist Drm/gremlin is a novel proangiogenic factor. Blood 109, 1834–1840. [DOI] [PubMed] [Google Scholar]
- Stover, C. , 2010. Dual role of complement in tumour growth and metastasis (Review). Int. J. Mol. Med. 25, 307–313. [DOI] [PubMed] [Google Scholar]
- Sun, S. , Wheeler, M.F. , Obeyesekere, M. , Patrick, C.W. , 2005. A deterministic model of growth factor-induced angiogenesis. Bull. Math. Biol. 67, 313–337. [DOI] [PubMed] [Google Scholar]
- Tiger, C.F. , Krause, F. , Cedersund, G. , Palmer, R. , Klipp, E. , Hohmann, S. , 2012. A framework for mapping, visualisation and automatic model creation of signal-transduction networks. Mol. Syst. Biol. 8, 578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocharus, J. , Tsuchiya, A. , Kajikawa, M. , Ueta, Y. , Oka, C. , Kawaichi, M. , 2004. Developmentally regulated expression of mouse HtrA3 and its role as an inhibitor of TGF-beta signaling. Dev. Growth Differ. 46, 257–274. [DOI] [PubMed] [Google Scholar]
- Ward, J.P. , King, J.R. , 1997. Mathematical modelling of avascular-tumour growth. IMA J. Math. Appl. Med. Biol. 14, 39–69. [PubMed] [Google Scholar]
- Ward, J.P. , King, J.R. , 1999. Mathematical modelling of avascular-tumour growth. II: modelling growth saturation. IMA J. Math. Appl. Med. Biol. 16, 171–211. [PubMed] [Google Scholar]
- Wen, X.Z. , Akiyama, Y. , Baylin, S.B. , Yuasa, Y. , 2006. Frequent epigenetic silencing of the bone morphogenetic protein 2 gene through methylation in gastric carcinomas. Oncogene 25, 2666–2673. [DOI] [PubMed] [Google Scholar]
- Wu, W.K. , Sung, J.J. , Wu, Y.C. , Li, Z.J. , Yu, L. , Cho, C.H. , 2008. Bone morphogenetic protein signalling is required for the anti-mitogenic effect of the proteasome inhibitor MG-132 on colon cancer cells. Br. J. Pharmacol. 154, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, F. , Saidou, J. , Watabe, K. , 2010. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 15, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. , Tuxhorn, J.A. , Ressler, S.J. , McAlhany, S.J. , Dang, T.D. , Rowley, D.R. , 2005. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 65, 8887–8895. [DOI] [PubMed] [Google Scholar]
- Ye, L. , Lewis-Russell, J.M. , Kyanaston, H.G. , Jiang, W.G. , 2007. Bone morphogenetic proteins and their receptor signaling in prostate cancer. Histol. Histopathol. 22, 1129–1147. [DOI] [PubMed] [Google Scholar]
- Zeisberg, M. , Shah, A.A. , Kalluri, R. , 2005. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 280, 8094–8100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary Table 1 List of 152 proteins with their respective gene names, corresponding to the desmoplastic protein dataset (DPD).
Supplementary Table 2 The output file of BINGO analysis, listing the significantly enriched GO annotations for biological process found in the DPD.
Video S1 Simulation of migratory behavior of a single cancer cell cohort invading the peritumoral stroma, under the influence of GREM1 microenvironmental gradient.
Video S2 Simulation of migratory behavior of a single cancer cell cohort invading the peritumoral stroma, without the influence of GREM1 microenvironmental gradient.