

Abstract
Histone deacetylase inhibitors such as Vorinostat display anti‐neoplastic activity against a variety of solid tumors. Here, we have investigated the anti‐tumoral activity of Vorinostat on endometrial cancer cells. We have found that Vorinostat caused cell growth arrest, loss of clonogenic growth and apoptosis of endometrial cancer cells. Vorinostat‐induced the activation of caspase‐8 and ‐9, the initiators caspases of the extrinsic and the intrinsic apoptotic pathways, respectively. Next, we investigated the role of the extrinsic pathway in apoptosis triggered by Vorinostat. We found that Vorinostat caused a dramatic decrease of FLIP mRNA and protein levels. However, overexpression of the long from of FLIP did not block Vorinostat‐induced apoptosis. To further investigate the role of extrinsic apoptotic pathway in Vorinostat‐induced apoptosis, we performed an shRNA‐mediated knock‐down of caspase‐8. Surprisingly, downregulation of caspase‐8 alone caused a marked decrease in clonogenic ability and reduced the growth of endometrial cancer xenografts in vivo, revealing that targeting caspase‐8 may be an attractive target for anticancer therapy on endometrial tumors. Furthermore, combination of caspase‐8 inhibition and Vorinostat treatment caused an enhancement of apoptotic cell death and a further decrease of clonogenic growth of endometrial cancer cells. More importantly, combination of Vorinostat and caspase‐8 inhibition caused a nearly complete inhibition of tumor xenograft growth. Finally, we demonstrate that cell death triggered by Vorinostat alone or in combination with caspase‐8 shRNAs was inhibited by the anti‐apoptotic protein Bcl‐XL. Our results suggest that combinatory therapies using Vorinostat treatment and caspase‐8 inhibition can be an effective treatment for endometrial carcinomas.
Keywords: Endometrial cancer, Vorinostat, Histone deacetylase inhibitors, Caspase-8
Highlights
Vorinostat display a promising anti‐neoplastic activity in different tumor types.
Inhibition of caspase‐8 has been associated to resistance to apoptosis.
Inhibition of caspase‐8 combined with Vorinostat has high anti‐tumor activity.
Such combination may be an attractive therapy for endometrial carcinomas.
1. Introduction
Endometrial carcinoma (EC) is the most commonly diagnosed gynecologic malignancy in the western world. The majority of these cancers are curable, but a subset about 15–20% of endometrial carcinomas exhibits an aggressive phenotype (Yeramian et al., 2012). EC can be divided into two main clinicopathological variants. Type I ECs are endometrioid EC are estrogen‐related tumors, frequently well differentiated that appear in pre‐ and perimenopausal women. Type I ECs are associated frequently with four main molecular alterations: microsatellite instability and mutations of K‐RAS, PTEN, and β‐catenin. Type II ECs are nonendometrioid EC (papillary serous and clear cell carcinomas) that tend to occur in older women, are estrogen‐unrelated tumors, frequently aneuploid, associated with p53 mutations, and clinically more aggressive tumors (Matias‐Guiu et al., 2001).
Traditionally, cancer has been regarded to originate from genetic alterations such as mutations, deletions, rearrangements as well as gene amplifications, leading to abnormal expression of tumor suppressor genes and oncogenes. An increasing body of evidence indicates that in addition to changes in DNA sequence, epigenetic alterations contribute to cancer initiation and progression. In contrast to genetic mutations, epigenetic changes are reversible and, therefore, they represent an attractive target for cancer therapy (Hagelkruys et al., 2011). Histone acetylation is a reversible process whereby histone and non‐histone protein acetyltransferases transfer the acetyl motif from acetyl co‐enzyme A to lysines. HDACs remove the acetyl groups re‐establishing the positive charge of the proteins (Marks and Xu, 2009), resulting in chromatin condensation and transcriptional repression, including a decrease in the expression of tumor suppressor genes (gene silencing) (Petruccelli et al., 2011). For that reason histone deacetylases (HDACs) play a central role in the epigenetic regulation of gene expression (Petrella et al., 2011). Histone deacetylase inhibitors (HDACi), such as Vorinostat, are a relatively new class of epi‐drugs that play important roles in epigenetic or non‐epigenetic regulation, inducing cell death, apoptosis, and cell cycle arrest in cancer cells (Kim and Bae, 2011). Current investigations suggest that HDACi, such as Vorinostat, show anti‐tumor activity against solid tumors, including the non‐small cell lung cancer (NSCLC) (Nagji et al., 2010), breast cancer (Palmieri et al., 2009), colon cancer (Walker et al., 2009), ovarian carcinoma (Yang et al., 2009), thyroid carcinomas (Borbone et al., 2010), gastric cancer (Shin et al., 2012) and others. Moreover, preliminary clinical results show a promising anti‐neoplastic activity in most hematologic malignancies, including, multiple myeloma (MM) (a clonal B‐cell neoplasm that accounts for 10% of all malignant hematologic neoplasms) (Pei et al., 2004). Its use in hematology led to Vorinostat approval by the US Food and Drug Administration (FDA) in October 2006 for the treatment of cutaneous manifestations in patients with cutaneous T‐cell lymphomas (CTCL) that have progressive, persistent or recurrent disease (Stimson et al., 2009). It is worth mentioning that normal cells have been reported to be significantly less sensitive than tumor cells to all anti‐neoplastic effects of Vorinostat (Gaymes et al., 2006).
Apoptotic cell death induced by chemotherapeutic drugs such as HDACi can be transduced through two distinct pathways: the intrinsic pathway, which emerges from mitochondria or the extrinsic pathway, which is activated by the engagement of death receptors (Carew et al., 2008). In the extrinsic pathway, activation of death receptors receptor leads to the formation of the Death‐Inducing Signaling Complex (DISC). The intracellular Death Domain (DD) of these receptors recruits Fas Associated DD‐containing protein (FADD) which, in turn, binds pro‐caspase‐8. After recruitment to the DISC, pro‐caspase‐8 is activated by autoproteolytic cleavage resulting in the initiation of apoptotic signaling (Bodmer et al., 2000; Kischkel et al., 2000; Sprick et al., 2000). One of the critical regulators of apoptosis triggered by death receptors is the FLICE‐Inhibitory Protein (FLIP) (Thome et al., 1997). High levels of FLIP are found in many tumor tissues including endometrial carcinoma. We have demonstrated that the long form of FLIP (FLIP‐L) plays a key role in the regulation of sensitivity of endometrial carcinoma cells (ECC) to death receptor‐induced apoptosis (Dolcet et al., 2005). Paradoxically, increasing evidences demonstrate non‐apoptotic functions for caspases and other apoptosis‐related proteins (Lamkanfi et al., 2007; Schwerk and Schulze‐Osthoff, 2003). Such alternative functions are particularly important for DISC components such as FADD or caspase‐8. Although caspase‐8 and FADD are well‐known apoptotic inducers, it has been demonstrated that they can have pro‐apoptotic, anti‐apoptotic or other apoptosis‐unrelated functions (Dillon et al., 2012; van Raam and Salvesen, 2012).
The present study was designed to define the biological and therapeutic effects of Vorinostat on endometrial cancer cells in vitro and in vivo. We have also investigated that role of FLIP‐L and caspase‐8 in mediating the anti‐tumoral effects of Vorinostat. We have found that combination of Vorinostat with caspase‐8 inhibition dramatically reduces tumorogenic growth of endometrial cancer cells, indicating that such combination may be an effective therapy for endometrial carcinomas.
2. Materials and methods
2.1. Cell lines, culture conditions and transfection
The Ishikawa 3‐H‐12 (IK) and AN3CA cell lines were obtained from the American Type Culture Collection (Manassas, VA), RL‐95/2 and HEC‐1‐A (HEC) cells were a gift from Dr Reventos (Hospital Vall d'Hebron, Barcelona). EC cells were grown in Dulbecco's modified Eagle's medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (Invitrogen, Inc., Carlsbad, CA), 1 mmol/L HEPES (Sigma), 1 mmol/L sodium pyruvate (Sigma), 2 mmol/L l‐glutamine (Sigma), and 1% of penicillin/streptomycin (Sigma) at 37 °C with saturating humidity and 5% CO2.
2.2. Chemical reagents
Vorinostat (suberoylanilide hydroxamic acid) was obtained from Merck. A 50 mM stock was prepared in dimethylsulphoxide (DMSO) and stored at −80 °C in 5 μl aliquots until further use. 2,5‐diphenyl tetrazolium bromide assay (MTT), DMSO, 5‐bromodeoxyuridine (BrdU), bis‐benzimide fluorescent dye (Hoechst 33258), and monoclonal antibody to tubulin were from Sigma (St Louis, MO, USA). Z‐LEHD‐fmk and z‐IETD‐fmk were from Calbiochem (La Jolla, CA, USA).
2.3. Cell viability assays and assessment of apoptosis
The general mitochondrial activity of EC cell lines was determined by assaying reduction of MTT (3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5 diphenyltetrazolium bromide) to formazan. EC cells were plated on M96‐well plates at 15 × 103 cells per well. After the indicated treatments, cells were incubated for 30 min with 0.5 mg/ml of MTT reagent and lysed with dimethylsulphoxide (DMSO) to dissolve the blue formazan crystals produced by the mitochondrial succinate dehydrogenase of the living cells. Cell viability proportionate to optical density was measured using a colorimetric assay of mitochondria activity. Drug resistance was represented as the percentage of live cells surviving after drug treatment relative to control cells. Absorbances were measured using a spectrophotometer (Bio‐Rad, Richmond, CA, USA.) at a dual wavelength of 595 nm and 620 nm.
Apoptotic cells were identified by nuclear staining with bis‐benzimide fluorescent dye (Hoechst 33258), after the indicated treatments, to a final concentration of 0.5 mg/ml to each M24 well. Cells were counted under epifluorescence microscope (Leica Microsystems, Wetzlar, Germany).
2.4. Xenografting and Vorinostat administration
Immunodeficient female SCID hr/hr mice (age, 12 weeks; weight 20–25 g) were maintained in Specific Pathogen Free (SPF) conditions. Animals were subcutaneously injected with HEC‐1A cells (1.5 × 106) suspended in 100 μl PBS + Matrigel (1:1). Tumors were allowed to growth for 38 days. Xenografted mice were treated with an intraperitoneal injection of Vorinostat at 50 mg/kg/day for 5 days/week during two consecutive weeks. Tumors were measured weekly with calipers. Tumor size was calculated using the formula: Tumor weight (TW, mg) = (D × d2)/2 = mm3.
2.5. Histology and immunehistochemical analysis
Mice were euthanized by cervical dislocation after 2 weeks of Vorinostat injections. Tumor samples were collected and formaline‐fixed overnight at 4 °C. Tumors were paraffin embedded for further histologic analysis. Paraffin blocks were sectioned at 3 μm, dried for 1 h at 65 °C before pre‐treatment procedure of deparaffinization, rehydration and epitope retrieval in the Pre‐Treatment Module, PT‐LINK (DAKO) at 95 °C for 20 min in 50× Tris/EDTA buffer, pH 9. Before staining the sections, endogenous peroxidase was blocked. The antibodies used were anti‐Acetyl‐Histone H4 (Lys12) (Cell Signallig Technology). Appropriate negative controls were also tested. Representative images were taken with Leica DMD108 microscope.
2.6. 5‐Bromodeoxyuridine incorporation
For the determination of DNA and after the indicated treatments, cells were incubated with 3 ng/ml of 5‐bromodeoxyuridine (BrdU) (Sigma) for 20 min and then fixed with 4% paraformaldehyde. After DNA denaturing with 2 mol/L HCl for 30 min and neutralization with 0.1 mol/L Na2B4O7 (pH 8.5) for 2 min, cells were blocked in phosphate‐buffered saline (PBS) solution containing 5% horse serum, 5% fetal bovine serum, 0.2% glycine, and 0.1% Triton X‐100 for 1 h. Subsequently, cells were subjected to indirect immunofluorescence with a mouse anti‐BrdU monoclonal antibody (Dako, Glostrup, Denmark), and fluorescein isothiocyanate‐conjugated anti‐mouse secondary antibody (Molecular Probes, Eugene, OR, USA.). Nuclei were counterstained with 5 mg/ml Hoechst 33258 and cells were visualized under an epifluorescence microscope (Leica Microsystems, Wetzlar, Germany).
2.7. Western Blot analysis
EC cell lines were washed with cold phosphate‐buffered saline (PBS) and lysed with lysis buffer (2% sodium dodecyl sulfate, 125 mmol/L Tris–HCl, pH 6.8). Protein concentrations were determined with a protein assay kit (Bio‐Rad, Hercules, CA). Equal amounts of proteins were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore). Nonspecific binding was blocked by incubation with TBST (20 mmol/L Tris–HCl, pH 7.4, 150 mmol/L NaCl, 0.1% Tween‐20) plus 5% of non‐fat milk. Membranes were incubated with the primary ab overnight at 4 °C. Signal was detected with ECL Advance (Amersham‐Pharmacia, Buckinghamshire, UK). The ab used were: FLIP (monoclonal, NF6; Alexis Biochemicals, Lausen, Switzerland), Cyclin D1 (monoclonal, DCS‐6; Santa Cruz Biotechnology), anti‐human‐FADD (monoclonal, BD Pharmingen). Ab to active Caspase‐3 and caspase‐9 were obtained from Cell‐Signal (Beverly, MA), active caspase‐8 (monoclonal, Calbiochem), anti‐Flag (monoclonal, Sigma), anti‐Bcl‐XL (polyclonal, BD Pharmingen), α‐tubulin (monoclonal, Sigma) and anti‐Acetyl‐Histone H4 (Lys12) (polyclonal, Cell‐Signal) were used as a Vorinostat control.
Membranes were then incubated with peroxidase‐coupled anti‐mouse or anti‐rabbit secondary ab (Amersham‐Pharmacia, Uppsala, Sweden) for 1 h, followed by chemiluminescent detection with ECL Advance (Amersham‐Pharmacia, Buckinghamshire, UK).
2.8. Real‐time quantitative polymerase chain reaction
For RNA preparation, we used RNeasy Mini kit® with DNase I (Qiagen) to keep the same level of quality among all the RNA samples. Expression of mRNA was measured with the use of TaqMan Gene Expression Assays on demand (Applied Biosystems, Foster City, CA, USA). The following assays were used: Flip/CFLAR (Hs01116280_m1), and GAPDH (Hs99999905_m1) as a control.
Quantitative reverse transcriptase polymerase chain reaction (RT‐PCR) analysis was performed in triplicate with the ABI Prism 7900 Sequence Detector System (Applied Biosystems). The PCR cycling conditions were the standard 95 °C for 10 min for one cycle, 95 °C for 15 s, and 60 °C for 1 min for 40 cycles.
2.9. Lentiviral production and infection
Lentiviral‐based vectors for RNA interference‐mediated gene silencing (FSV) consisted on an U6 promoter for expression of short‐hairpin RNAs (shRNAs) and the Venus variant of YFP under the control of an SV40 promoter for monitoring transduction efficiency. Oligonucleotides to produce plasmid‐based shRNA were cloned into the FSVsi vector using AgeI–BamHI restriction sites. Lentiviral particles were produced in 293T (human embryonic kidney cells) cotransfected by the calcium phosphate method with the above plasmid plus plasmids coding for the envelope and the packaging systems (VSV‐G and D8.9, respectively).
The 293T cells were allowed to produce lentiviral particles during 3–4 days in the same culture medium of the cultured cells. Culture medium was collected, centrifuged for 10 min at 2500 r.p.m., filtered through a 0.45 mm filter (Millipore, Bedford, MA) and then concentrated by centrifugation through a filter column of 100 kDa (VWR International LLC, West Chester, PE, USA) for 1 h at 4000 r.p.m. Cells were incubated overnight in the presence of medium containing lentiviral particles. After this period, medium was replaced by fresh medium and cells were incubated for two additional days to allow endogenous protein knock‐down or protein overexpression. The sequence of shRNA: targeting caspase‐8 was: 5′‐ GAATCACAGACTTTGGACAA ‐3′.
3. Results
3.1. Vorinostat reduces growth of endometrial cancer cells in vitro and in vivo
First, we assessed the effects of Vorinostat on cell viability, clonogenic growth and proliferation on three different endometrial cancer cells lines (IK, RL‐95 and HEC‐1‐A). To ensure that Vorinostat treatment was effective, we analyzed the levels histone H4 acetylation by Western Blot. Increasing the dose of Vorinostat resulted in an increased acetylation of histone H4, suggesting that it was effectively blocking HDACi activity (Suppl Figure 1A). Next, we analyzed the effects of Vorinostat on cell viability by MTT assay, on autonomous growth ability by clonogenic assay and on cell proliferation by BrdU incorporation. Vorinostat caused a dose‐ and time‐dependent decrease of cell viability of the three cell lines analyzed (Figure 1A). Similarly, Vorinostat inhibited clonogenic growth of the three endometrial cancer cells lines (Figure 1B) and reduced its BrdU incorporation (Figure 1C), suggesting that Vorinostat treatment caused a decrease in cell proliferation. Consistently, Vorinostat caused a progressive reduction of cyclin D1 expression (Suppl Figure 1B). To assess anti‐tumoral activity of Vorinostat in vivo, we injected HEC‐1A cells subcutaneously to SCID mice and tumors were allowed to grow for 38 days. At this point, engrafted mice were intraperitoneally injected with 0.5 mg/kg 5 days/week for two weeks. Vorinostat treatment caused a marked reduction of tumoral size, indicating that Vorinostat reduced tumoral growth of xenografted endometrial cancer cells in vivo (Figure 1D).
Figure 1.
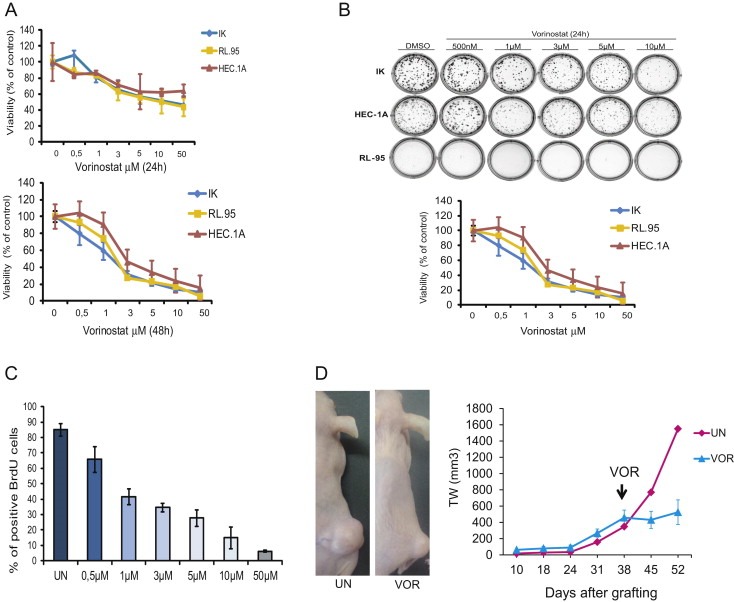
Vorinostat treatment reduces cell viability, clonogenicity and proliferation in endometrial carcinoma cell lines. A, IK, RL and HEC cells were treated for 24 (top) or 48 h (bottom) with the indicated doses of Vorinostat and cell viability was assessed by MTT. Results are expressed as percentage of survival over control values. B, IK, RL and HEC cells were seeded as a single‐cell suspension with a specified number of cells. After allowing cells time to attach (6 h), Vorinostat or the vehicle control was added at specified concentrations and treated for 24 h. Ten to fourteen days after seeding, survival curves (from counting the number of colonies) were generated. C, Endometrial cancer cells were incubated for 24 h with indicated doses of Vorinostat, then labeled with BrdU for 1 h. The cells were fixed and stained using an anti‐BrdU antibody. DNA was stained with Hoechst dye. The percentage of cells positive for BrdU staining was quantified in at least 300 cells (Hoechst dye‐positive cells). Each value represents the average and standard deviation. D, Representative images and tumor weight measurements of HEC‐1A xenografts from mice injected with Vorinostat (VOR) or with vehicle alone (UN).
3.2. Vorinostat activates intrinsic and extrinsic apoptotic pathways and triggers apoptotic cell death of endometrial cancer cells
Having ascertained that Vorinostat causes a reduction in cell viability, we further investigated whether Vorinostat was able to induce apoptotic cell death of endometrial cancer cell lines. First, to evidence apoptotic cell death, we performed a Hoechst staining of IK, RL and HEC cells treated with increasing doses of Vorinostat. Vorinostat caused a dose‐dependent increase of the number of cells displaying apoptotic nuclear morphology (Figure 2A). Next, we investigated the molecular mechanisms involved in transduction of apoptotic cell death. For this purpose, we analyzed the activation of the initiator caspases of the extrinsic/death receptor (caspase‐8) and intrinsic/mitochondrial (caspase‐9) apoptotic pathways by Western Blot. Treatment of IK cells with Vorinostat 5 μM resulted in processing of both caspase‐8 and caspase‐9 (Figure 2B), suggesting that both extrinsic and intrinsic pathways are activated by Vorinostat. Similar results were obtained with HEC‐1A (Figure 2C) and RL‐95 cells (Figure 2D).
Figure 2.
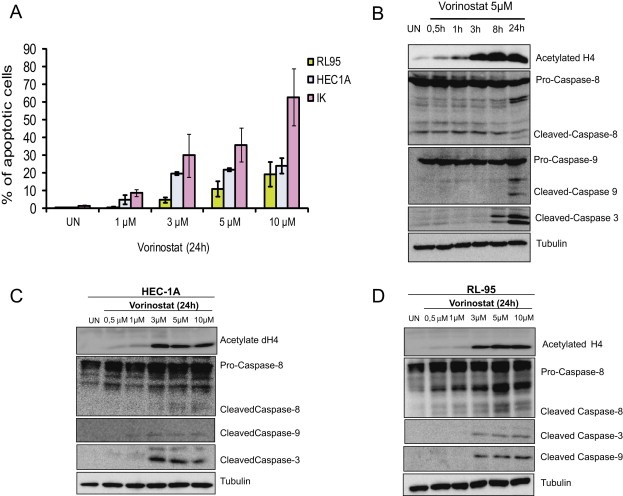
Vorinostat induces apoptotic cell death on endometrial cancer cell lines. A, Quantification of Hoechst‐stained apoptotic nuclei of IK, HEC and RL cells treated for 24 h with increasing doses of Vorinostat. Results are expressed as percentage of viability over control values. B, IK cells were treated with Vorinostat 5 μM Vorinostat for the indicated periods of time or left untreated (UN). Cell lysates were prepared, and 20 μg protein was electrophoresed, blotted, and probed with the indicated antibodies. C, HEC‐1‐A and D, RL‐95 cells were treated with increasing doses of Vorinostat for 24 h. Cell lysates were prepared and 20 μg of protein were electrophoresed, blotted, and probed with the indicated antibodies. Anti‐tubulin was used to ensure equal protein loading and anti‐acetylated‐H4 was used to ensure that Vorinostat treatment was effective.
3.3. FLIP is not involved in regulation of Vorinostat‐induced apoptosis
Once demonstrated that Vorinostat was able to induce caspase‐8 cleavage, we sought to investigate the role of the extrinsic signaling pathway in the transduction of apoptotic cell death. In our laboratory, we have previously demonstrated that high levels of FLIP‐L inhibit death receptor‐induced apoptotic cell death in endometrial cancer cells (Dolcet et al., 2005; Llobet et al., 2011, 2008, 2010). Since FLIP‐L competes with caspase‐8 to bind FADD at the DISC, we analyzed whether Vorinostat caused a decrease of FLIP‐L levels, leading to the activation of caspase‐8. Western Blot analysis of FLIP‐L protein revealed that Vorinostat caused a dose‐dependent decrease of FLIP‐L protein levels in IK, HEC‐1A and RL‐95 cells (Figure 3A). Such reduction in protein levels correlated with a reduction of FLIP mRNA expression (Figure 3B). On the light of these results, we wondered whether such a dramatic reduction in FLIP‐L levels was involved in anti‐tumoral effects of Vorinostat. For this purpose, we infected three different endometrial cancer cell lines (IK, HEC‐1‐A and AN3CA) with lentiviruses carrying FLIP‐L cDNA (pEIGW‐FLIP) or the empty vector (pEIGW), and we assessed the clonogenic ability and the induction of apoptosis after Vorinostat treatment. Surprisingly, FLIP‐L overexpression did not inhibit Caspase‐3 activation of IK cells treated with Vorinostat (Figure 3C). To demonstrate that FLIP‐L was correctly overexpressed, we performed a Western Blot to detect flag‐tag (Figure 3C). Furthermore, FLIP‐L overexpression was unable to restore clonogenic ability of three different endometrial cancer cell lines (IK, HEC‐1‐A and AN3CA) treated with Vorinostat (Figure 3D). These results suggest that the reduction of FLIP‐L is not related with anti‐tumoral activity of Vorinostat.
Figure 3.
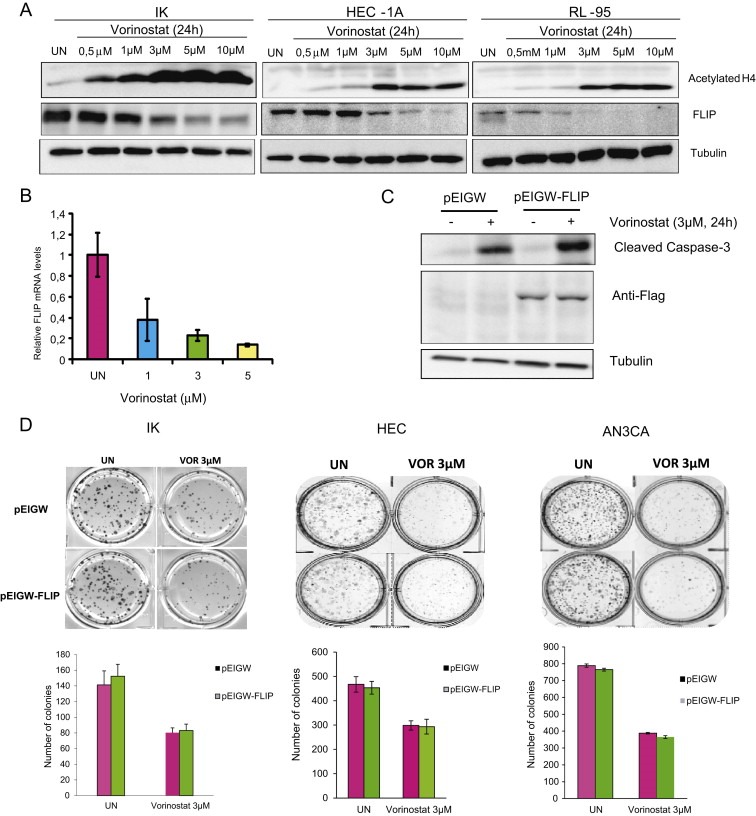
Downregulation of FLIP is not involved in anti‐tumoral activity of Vorinostat. A, IK, HEC‐1A and RL‐95 cells were treated with increasing doses of Vorinostat for 24 h. Cell lysates were subjected to Western Blot with Acetylate H4, anti‐FLIP antibody and tubulin antibody. B, IK cells were treated with indicated doses of Vorinostat and cell lysates were analyzed by quantitative RT‐PCR to determine the levels of FLIP mRNA. C, IK cells transfected with either control (pEIGW) or a plasmid expressing FLIP (pEIGW‐FLIP) and treated with 3 μM Vorinostat and after indicated times protein cell lysates were subjected to Western Blot with antibodies against Caspase‐3, anti‐Flag to ensure specific overexpression of FLIP and anti‐tubulin to ensure equal protein loading. D, Representative images and quantification of clonogenic ability of IK, HEC‐1‐A and AN3CA cells infected with lentiviruses carrying either control (pEIGW) or overexpression of FLIP (pEIGW‐FLIP) plasmid and left untreated (UN) or treated with 3 μM Vorinostat (VOR).
3.4. Downregulation of caspase‐8 reduces tumoral growth and enhances anti‐tumoral activity of Vorinostat
As mentioned above, the cleavage of caspase‐8 suggests that Vorinostat activates death receptor pathway. Previous studies from our laboratory demonstrated that Caspase‐8 knock‐down blocks apoptosis triggered by death receptor‐induced apoptosis (Llobet et al., 2011, 2008). Therefore, we decided to investigate whether inhibition of caspase‐8 expression results in impairment of apoptosis induced by Vorinostat. To address this point, we infected IK and HEC cells with lentiviruses carrying caspase‐8 shRNA and we analyzed induction of apoptosis in presence or absence of Vorinostat. Surprisingly, caspase‐8 knock‐down resulted in a further increase of Caspase‐3 activation caused by Vorinostat (Figure 4A). We further investigated the effects of caspase‐8 knock‐down on congenic ability of IK, HEC and AN3CA endometrial cancer cell lines in presence or absence of Vorinostat. Treatment of endometrial cancer cells with Caspase‐8 shRNA plus Vorinostat caused a nearly complete loss clonogenic growth (Figure 4B). Noteworthy, caspase‐8 shRNAs caused a significant reduction of clonogenic ability of endometrial cancer cell lines even in absence of Vorinostat (Figure 4B). These results suggest an important role for caspase‐8 in the regulation of growth of endometrial cancer cells.
Figure 4.
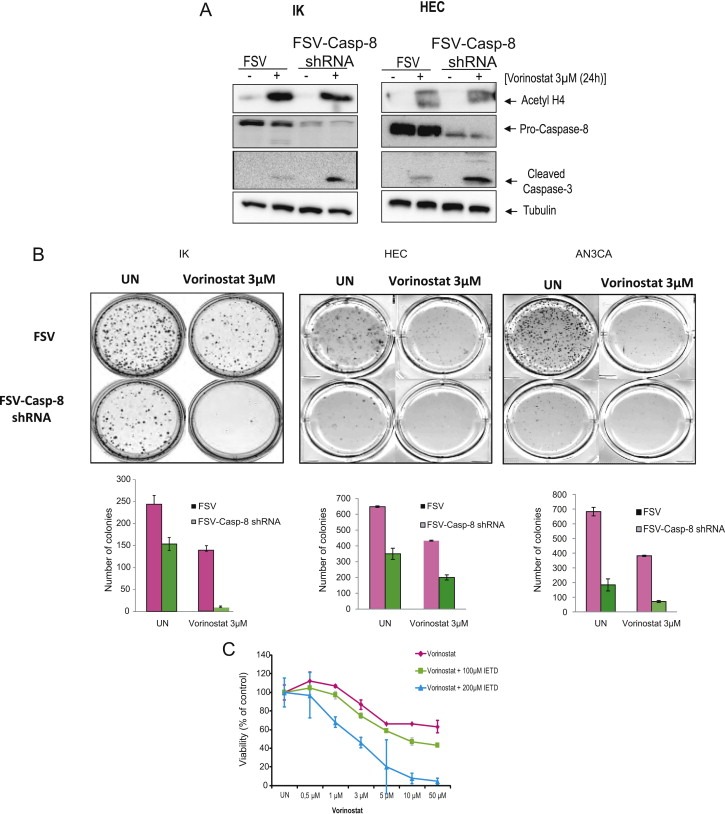
Downregulation of Caspase‐8 by specific shRNA enhances sensitivity of IK cells to Vorinostat‐induced apoptosis. A, IK and HEC cells were infected with lentiviruses carrying Caspase‐8 shRNA, and treated with 3 μM Vorinostat or left untreated (UN), protein cell lysates were subjected to Western Blot with antibodies against Caspase‐3, anti‐Caspase‐8 and anti‐Acetylated‐H4. Membranes were reprobed with anti‐tubulin to ensure equal protein loading. B, IK, HEC and AN3CA cells were infected with lentiviruses carrying Caspase‐8 shRNA, seeded as a single‐cell suspension and treated with 3 μM Vorinostat for 24 h. Ten to fourteen days after seeding, survival curves (from counting the number of colonies) were generated. C, IK cells were pretreated with (100 and 200 μM) of the inhibitor of Caspase‐8 activity z‐IETD‐fmk (IETD), after 2 h, IK cells were treated with increasing doses of Vorinostat for 24 h and cell viability was assessed by MTT.
Finally, to investigate whether the effects of caspase‐8 on Vorinostat‐induced apoptosis were caused by the expression of caspase‐8 or were caused by its protease activity, we treated IK cells with Vorinostat plus the inhibitor of caspase‐8 activity z‐IETD‐fmk (IETD). Addition of IETD caused a marked decrease on cell viability of Vorinostat‐treated cells (Figure 4C), suggesting that proteolytic activity of caspase‐8 induced by Vorinostat is protecting and has an anti‐apoptotic role.
3.5. Combination of caspase‐8 knock‐down and Vorinostat treatment supress growth of endometrial cancer cells in vivo
The results obtained in vitro enabled us to analyze the effects of caspase‐8 shRNA alone or in combination with Vorinostat on endometrial cancer cells in vivo. To address this point, HEC‐1A cells were infected with lentivitures carrying caspase‐8 shRNA (FSV‐Casp‐8 shRNA) or the control vector (FSV) and subcutaneously xenotransplanted to SCID mice. Tumor size was measured weekly. Tumors with caspase‐8 donwregulation displayed a substantial reduction in growth compared to those infected with control vector (Figure 5A). We further analyzed the effects of Vorinostat on growth of tumors depleted of caspase‐8. For this purpose, HEC‐1A cells infected with FSV or FSV‐Caspase‐8 were xenotranplanted and tumors were allowed to growth for 38 days. At this point, mice were injected with Vorinostat for two weeks (see material and methods for protocol of administration), and tumor size was measured weekly. Combination of caspase‐8 shRNA plus Vorinostat produced an impressive reduction of tumor weight (Figures 5B and 5C). In fact, those tumors showed a nearly complete inhibition of growth over time (Figure 5C). To rule out the possibility that caspase‐8 was affecting inhibition of acetylation caused by Vorinostat, at the end of the protocol, tumors were dissected and the levels of acetylated histone H4 were assessed by immunohistochemistry. Tumors from Vorinostat‐treated mice showed an increased staining for acetylated histone H4, regardless of Caspase‐8 status (Figure 5D).
Figure 5.
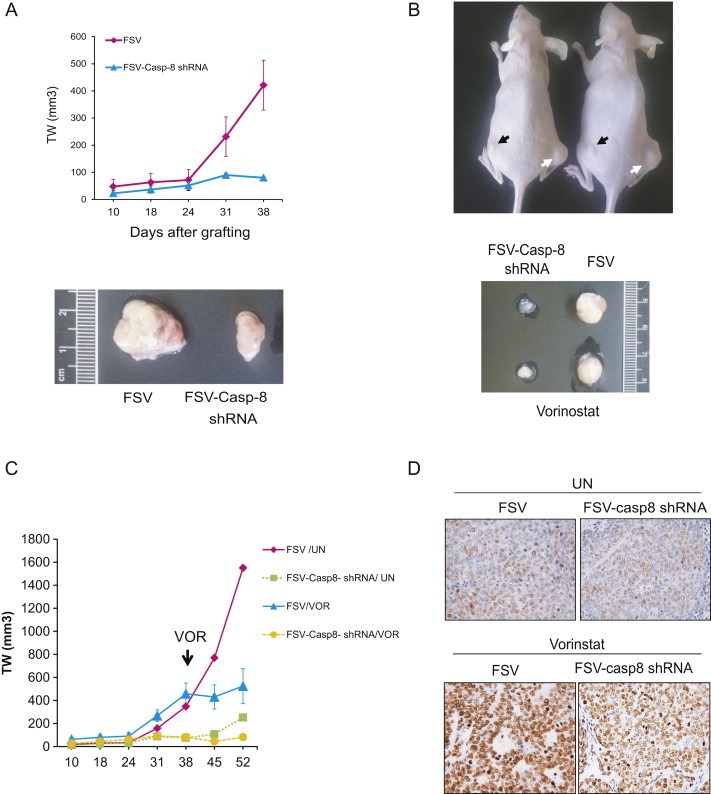
Combination of Caspase‐8 dowregulation with Vorinostat treatment inhibits tumoral growth of endometrial cancer cells in vivo. A, Tumor weight measurement and representative images of tumors developed from HEC‐1A cells infected with lentiviruses carrying capase‐8 shRNA (FSV‐Casp‐8 shRNA) or the control vector (FSV) and subcutaneously grafted in SCID mice. B, Top, representative images of two mice xenografted with HEC‐1A cells infected with Caspase‐8 shRNA (black arrows) or the control vector (white arrows) and treated with Vorinostat. Bottom, images corresponding to the dissected tumors. Note the difference in tumor size. C, Tumor weight measurements of HEC‐1A tumors carrying Caspase‐8 shRNA (FSV‐Casp‐8 shRNA) or the control vector (FSV). Tumor xenografts were grown subcutaneously and, at day 38, mice were injected with Vorinostat (VOR) or with vehicle alone (UN). D, Representative micrograph of acetylated H4 immunostaining of tumors with Caspase‐8 shRNA (FSV‐Casp‐8 shRNA) or the control vector (FSV) dissected from mice treated with Vorinostat (VOR) or left untreated (UN).
3.6. Bcl‐XL overexpression suppresses apoptosis induced by combination of Vorinostat and caspase‐8 shRNA
Having demonstrated that blockade of caspase‐8 increases Vorinostat‐induced cell death; we sought to investigate whether such enhancement was caused by increased signaling through the intrinsic pathway. As we show in Figure 2, Vorinostat triggered activation of caspase‐9, suggesting activation of intrinsic pathway. We first analyzed the contribution of intrinsic pathway in driving apoptotic signals of Vorinostat itself. For this purpose, we infected IK cells with lentiviruses carrying the anti‐apoptotic Bcl‐XL or we treated IK cells with the caspase‐9 inhibitor LEHD‐fmk. Overexpression of Bcl‐XL caused a dramatic reduction of apoptotic cells (Figure 6A) and consistently, abolished the activation of Caspase‐3 (Figure 6B). Similarly, treatment of IK cells with Vorinostat plus LEHD‐fmk inhibited apoptotic cell death caused by Vorinostat (Figure 6C). These results suggest that activation of mitochondrial apoptotic pathway is necessary to transduce apoptosis signals of Vorinostat. Finally, we investigated the role of mitochondrial pathway in the transduction of the apoptotic signals caused by co‐treatment of Vorinostat and caspase‐8 shRNA. For this purpose IK cells were co‐infected with lentiviruses carrying Bcl‐XL cDNA and lentiviruses caspase‐8 shRNA. Bcl‐XL overexpression decreased the number of apoptotic cell death caused by co‐treatment of caspase‐8 shRNA plus Vorinostat (Figure 6D) and, consistently, caused an inhibition of Caspase‐3 activation (Figure 6E).
Figure 6.
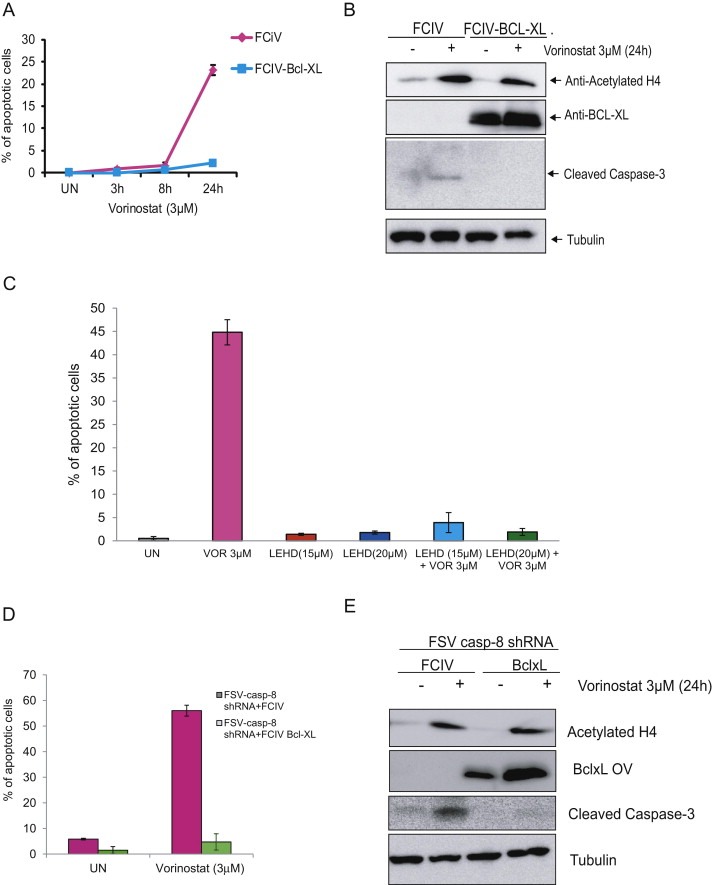
Bcl‐XL overexpression reduces apoptosis induced by combination of Vorinostat and Caspase‐8 shRNA. A, IK cells were infected with lentiviruses carrying Bcl‐XL CDNA (FCIV‐Bcl‐XL) or the empty vector (FCIV) and treated with 3 μM Vorinostat at indicated times. The graph represents a quantification of nuclei displaying nuclear apoptotic morphology. B, Western Blot analysis of whole lysates from IK cells infected with lentiviruses carrying FCIV‐Bcl‐XL or the empty vector (FCIV). The membranes were reprobed with Acetyl H4, Caspase‐3, Bcl‐XL and tubulin to ensure equal protein loading. C, Quantification of Hoechst‐stained apoptotic nuclei of IK cells treated with Vorinostat (VOR) plus the caspase‐9 inhibitor LEHD‐fmk D, Quantification of Hoechst‐stained apoptotic nuclei of IK cells co‐infected with lentiviruses carrying overexpression of Bcl‐XL and lentiviruses Caspase‐8 shRNA and treated with 3 μM Vorinostat for 24 h. E, IK cells were infected with lentiviruses carrying FCIV‐Bcl‐XL and either the empty vector (FCIV), and after 3 days, IK cells were treated for 24 h with 3 μM Vorinostat and lysed. Membrane was reprobed using anti cleaved Caspase‐3, anti‐Acetyl H4, anti‐Bcl‐XL (to demonstrate specific overexpression of Bcl‐XL) and anti‐tubulin to ensure equal protein loading.
4. Discussion
In the present work, we have studied the effects of Vorinostat on endometrial cancer cells. Vorinostat has been shown to be an effective anti‐neoplastic agent for different types of tumors. However, in case of endometrial cancer cells, treatment with Vorinostat or other HDCAi has been reported to result in different outcomes, including cell growth arrest, apoptosis (Kerr et al., 2012; Sarfstein et al., 2011; Takai et al., 2004), differentiation (Uchida et al., 2005) or induction of migration and invasion (Uchida et al., 2007). Here, we have demonstrated that Vorinostat‐induced proliferative arrest, inhibition of clonogenic growth, apoptotic cell death and more importantly, a marked reduction of tumoral xenografts in vivo. These results support an anti‐tumoral effect of Vorinostat on endometrial cancer cells.
Next, we concentrated our investigations in the study of the molecular mechanisms involved in the transduction of cell death triggered by Vorinostat. We found that Vorinostat caused the activation of both intrinsic and extrinsic initiator caspases (caspase‐9 and caspase‐8, respectively). The activation of the intrinsic pathway was expected, because Vorinostat induces DNA damage, a well‐known inducer of apoptosis by the intrinsic pathway. In contrast, the activation of the extrinsic apoptotic pathway and its role in transduction of apoptotic cell death triggered by Vorinostat is more controversial. To this regard, we have found that Vorinostat caused a dramatic decrease of FLIP mRNA and protein levels. Previous works demonstrated that Vorinostat‐induced apoptosis of malignant mesothelioma or colon cancer cells was requires FLIP downregulation and activation of caspase‐8 (Hurwitz et al., 2012; Kerr et al., 2012). Moreover, it has been shown that reduction of FLIP protein levels caused by Vorinostat alone or in combination with other chemotherapeutic drugs sensitizes cancer cells to TRAIL‐induced apoptosis (Carlisi et al., 2009; Frew et al., 2008; Sung et al., 2010; Yerbes and Lopez‐Rivas, 2012; Zhang et al., 2008). Surprisingly, we have found that, despite FLIP‐L was markedly downregulated, overexpression of exogenous FLIP‐L failed to block apoptosis triggered by Vorinostat. These results suggested that extrinsic pathway might not be important in mediating apoptotic effects of Vorinostat. To further substantiate that activation of extrinsic pathway was dispensable to transduce apoptotic signals, we decided to inhibit expression of caspase‐8. We have previously demonstrated that inhibition of either caspase‐8 was able to block apoptosis triggered by death receptors in endometrial cancer cells (Llobet et al., 2011, 2008). Therefore, if apoptosis triggered by Vorinostat was mediated by the extracellular pathway, knock‐down of these proteins should impair Vorinostat‐induced apoptosis. However, caspase‐8 downregulation enhanced apoptosis triggered by Vorinostat. These results suggested a protective effect of caspase‐8 in Vorinostat‐induced apoptosis. Classically, alterations leading to loss of caspase‐8 activity have been considered as mechanisms of apoptosis resistance to chemotherapeutic drugs (Kim et al., 2001; McKee and Thiele, 2006). However, our investigations demonstrate that Caspase‐8 can be a double‐edged sword. On the one hand, inhibition of caspase‐8 impairs apoptotic signaling by the extrinsic pathway. Such blockade results in resistance to apoptosis, a hallmark of malignancy. On the other hand, inhibition of caspase‐8 enhances Vorinostat‐induced apoptosis, indicating that inhibition of caspase‐8 may be effective in combination with Vorinostat.
Our results support a protective role of caspase‐8 in apoptosis triggered by Vorinostat. Such a protective function of caspase‐8 has been supported by in vivo and in vitro evidences (Dillon et al., 2012; van Raam and Salvesen, 2012). Mice lacking proteins involved in apoptosis such as caspase‐9 or Fas knock‐outs lead to defects related to an overabundance of cells (Kuida et al., 1998; Watanabe‐Fukunaga et al., 1992). Conversely, caspase‐8 knock‐out mice is embryonic lethal by a failure of yolk sac vascularization and hematopoiesis (Varfolomeev et al., 1998). Such phenotype led to the hypothesis that these proteins have roles in cellular functions beyond apoptosis. Many studies have demonstrated other cellular functions for caspase‐8, including regulation of cell cycle (Kennedy et al., 1999), NF‐kB activation (Lemmers et al., 2007), cell migration and metastasis (Barbero et al., 2008, 2009; Torres et al., 2010). To this regard, we found that knock‐down of caspase‐8 caused a marked decrease of clonogenic ability in vitro and growth of xenografts in vivo, even in absence of Vorinostat. These results support the hypothesis that caspase‐8 has pro‐survival functions in endometrial cancer cells. We have also demonstrated that apoptosis induced by Vorinostat or Vorinostat caspase‐8 shRNA was impaired by Bcl‐XL overexpression. This result indicates that the increased apoptotic cell death observed is ultimately mediated by the mitochondrial pathway. It is worth to mention that, in other cell types such as lymphocytes, one of the signaling pathways regulated by caspase‐8 is the NF‐kB pathway. Because of the importance of NF‐kB signaling pathway in regulation of cell proliferation and survival, we assessed that NF‐kB activity on endometrial cancer cell lines depleted of caspase‐8. However, inhibition of caspase‐8 did not affect NF‐kB activity. Moreover, overexpression of the p65 subunit of NF‐kB failed to restore the clonogenic ability of endometrial cancer cell lines (Suppl Figure 3). These results suggest that the decrease of cell viability observed in cells depleted of caspase‐8 is independent of NF‐kB activity.
Finally, the combination of Vorinostat with caspase‐8 shRNA caused a dramatic reduction of tumor growth of endometrial cancer cells. It is worth to mention that combination of these two treatments caused a nearly complete inhibition of tumoral growth in vivo. Therefore, we believe that combination of Vorinostat and caspase‐8 inhibition may be an attractive therapeutic strategy for endometrial cancer treatment.
5. Conclusions
Based on our results, we conclude that Vorinostat and caspase‐8 inhibition are effective to reduce growth of endometrial cancer cells and, combination of these two treatments displays highly efficient anti‐neoplastic effects in vivo and in vitro. Therefore, such combinatory therapies using targeting caspase‐8 and histone deacetylation may be an effective treatment for endometrial cancer.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
Supported by grants FIS PI10/00604, PI10/00922, RD06/0020/1034, 2009SGR794, and 2004XT00090, Grupos estables AECC, Catalunya contra el cáncer and programa de intensificación de la investigación, Instituto Carlos III. L.B. holds an Alicia Cuello de Merigó Fellowship, N.E. and C.M hold a fellowship from FIS. We want to thank Lidia Mónica Domingo and Lidia Parra for their technical support.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.03.003.
Bergadà Laura, Sorolla Annabel, Yeramian Andree, Eritja Nuria, Mirantes Cristina, Matias-Guiu Xavier, Dolcet Xavier, (2013), Combination of Vorinostat and caspase‐8 inhibition exhibits high anti‐tumoral activity on endometrial cancer cells, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.03.003.
References
- Barbero, S. , Barila, D. , Mielgo, A. , Stagni, V. , Clair, K. , Stupack, D. , 2008. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. The Journal of Biological Chemistry 283, 13031–13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero, S. , Mielgo, A. , Torres, V. , Teitz, T. , Shields, D.J. , Mikolon, D. , Bogyo, M. , Barila, D. , Lahti, J.M. , Schlaepfer, D. , Stupack, D.G. , 2009. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Research 69, 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer, J.L. , Holler, N. , Reynard, S. , Vinciguerra, P. , Schneider, P. , Juo, P. , Blenis, J. , Tschopp, J. , 2000. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nature Cell Biology 2, 241–243. [DOI] [PubMed] [Google Scholar]
- Borbone, E. , Berlingieri, M.T. , De Bellis, F. , Nebbioso, A. , Chiappetta, G. , Mai, A. , Altucci, L. , Fusco, A. , 2010. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene 29, 105–116. [DOI] [PubMed] [Google Scholar]
- Carew, J.S. , Giles, F.J. , Nawrocki, S.T. , 2008. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Letters 269, 7–17. [DOI] [PubMed] [Google Scholar]
- Carlisi, D. , Lauricella, M. , D'Anneo, A. , Emanuele, S. , Angileri, L. , Di Fazio, P. , Santulli, A. , Vento, R. , Tesoriere, G. , 2009. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. European Journal of Cancer (Oxford, England: 1990) 45, 2425–2438. [DOI] [PubMed] [Google Scholar]
- Dillon, C.P. , Oberst, A. , Weinlich, R. , Janke, L.J. , Kang, T.B. , Ben-Moshe, T. , Mak, T.W. , Wallach, D. , Green, D.R. , 2012. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Reports 1, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcet, X. , Llobet, D. , Pallares, J. , Rue, M. , Comella, J.X. , Matias-Guiu, X. , 2005. FLIP is frequently expressed in endometrial carcinoma and has a role in resistance to TRAIL-induced apoptosis. Lab Invest 85, 885–894. [DOI] [PubMed] [Google Scholar]
- Frew, A.J. , Lindemann, R.K. , Martin, B.P. , Clarke, C.J. , Sharkey, J. , Anthony, D.A. , Banks, K.M. , Haynes, N.M. , Gangatirkar, P. , Stanley, K. , Bolden, J.E. , Takeda, K. , Yagita, H. , Secrist, J.P. , Smyth, M.J. , Johnstone, R.W. , 2008. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proceedings of the National Academy of Sciences of the United States of America 105, 11317–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaymes, T.J. , Padua, R.A. , Pla, M. , Orr, S. , Omidvar, N. , Chomienne, C. , Mufti, G.J. , Rassool, F.V. , 2006. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis?. Molecular Cancer Research: MCR 4, 563–573. [DOI] [PubMed] [Google Scholar]
- Hagelkruys, A. , Sawicka, A. , Rennmayr, M. , Seiser, C. , 2011. The biology of HDAC in cancer: the nuclear and epigenetic components. Handbook of Experimental Pharmacology 206, 13–37. [DOI] [PubMed] [Google Scholar]
- Hurwitz, J.L. , Stasik, I. , Kerr, E.M. , Holohan, C. , Redmond, K.M. , McLaughlin, K.M. , Busacca, S. , Barbone, D. , Broaddus, V.C. , Gray, S.G. , O'Byrne, K.J. , Johnston, P.G. , Fennell, D.A. , Longley, D.B. , 2012. Vorinostat/SAHA-induced apoptosis in malignant mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. European Journal of Cancer (Oxford, England: 1990) 48, 1096–1107. [DOI] [PubMed] [Google Scholar]
- Kennedy, N.J. , Kataoka, T. , Tschopp, J. , Budd, R.C. , 1999. Caspase activation is required for T cell proliferation. The Journal of Experimental Medicine 190, 1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, E. , Holohan, C. , McLaughlin, K.M. , Majkut, J. , Dolan, S. , Redmond, K. , Riley, J. , McLaughlin, K. , Stasik, I. , Crudden, M. , Van Schaeybroeck, S. , Fenning, C. , O'Connor, R. , Kiely, P. , Sgobba, M. , Haigh, D. , Johnston, P.G. , Longley, D.B. , 2012. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death and Differentiation 19, 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.J. , Bae, S.C. , 2011. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. American Journal of Translational Research 3, 166–179. [PMC free article] [PubMed] [Google Scholar]
- Kim, P.K. , Mahidhara, R. , Seol, D.W. , 2001. The role of caspase-8 in resistance to cancer chemotherapy. Drug Resistance Updates: Reviews and Commentaries in Antimicrobial and Anticancer Chemotherapy 4, 293–296. [DOI] [PubMed] [Google Scholar]
- Kischkel, F.C. , Lawrence, D.A. , Chuntharapai, A. , Schow, P. , Kim, K.J. , Ashkenazi, A. , 2000. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12, 611–620. [DOI] [PubMed] [Google Scholar]
- Kuida, K. , Haydar, T.F. , Kuan, C.Y. , Gu, Y. , Taya, C. , Karasuyama, H. , Su, M.S. , Rakic, P. , Flavell, R.A. , 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325–337. [DOI] [PubMed] [Google Scholar]
- Lamkanfi, M. , Festjens, N. , Declercq, W. , Vanden Berghe, T. , Vandenabeele, P. , 2007. Caspases in cell survival, proliferation and differentiation. Cell Death and Differentiation 14, 44–55. [DOI] [PubMed] [Google Scholar]
- Lemmers, B. , Salmena, L. , Bidere, N. , Su, H. , Matysiak-Zablocki, E. , Murakami, K. , Ohashi, P.S. , Jurisicova, A. , Lenardo, M. , Hakem, R. , Hakem, A. , 2007. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. The Journal of Biological Chemistry 282, 7416–7423. [DOI] [PubMed] [Google Scholar]
- Llobet, D. , Eritja, N. , Domingo, M. , Bergada, L. , Mirantes, C. , Santacana, M. , Pallares, J. , Macia, A. , Yeramian, A. , Encinas, M. , Moreno-Bueno, G. , Palacios, J. , Lewis, R.E. , Matias-Guiu, X. , Dolcet, X. , 2011. KSR1 is overexpressed in endometrial carcinoma and regulates proliferation and TRAIL-induced apoptosis by modulating FLIP levels. The American Journal of Pathology 178, 1529–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llobet, D. , Eritja, N. , Encinas, M. , Llecha, N. , Yeramian, A. , Pallares, J. , Sorolla, A. , Gonzalez-Tallada, F.J. , Matias-Guiu, X. , Dolcet, X. , 2008. CK2 controls TRAIL and Fas sensitivity by regulating FLIP levels in endometrial carcinoma cells. Oncogene 27, 2513–2524. [DOI] [PubMed] [Google Scholar]
- Llobet, D. , Eritja, N. , Yeramian, A. , Pallares, J. , Sorolla, A. , Domingo, M. , Santacana, M. , Gonzalez-Tallada, F.J. , Matias-Guiu, X. , Dolcet, X. , 2010. The multikinase inhibitor Sorafenib induces apoptosis and sensitises endometrial cancer cells to TRAIL by different mechanisms. European Journal of Cancer (Oxford, England: 1990) 46, 836–850. [DOI] [PubMed] [Google Scholar]
- Marks, P.A. , Xu, W.S. , 2009. Histone deacetylase inhibitors: potential in cancer therapy. Journal of Cellular Biochemistry 107, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias-Guiu, X. , Catasus, L. , Bussaglia, E. , Lagarda, H. , Garcia, A. , Pons, C. , Munoz, J. , Arguelles, R. , Machin, P. , Prat, J. , 2001. Molecular pathology of endometrial hyperplasia and carcinoma. Human Pathology 32, 569–577. [DOI] [PubMed] [Google Scholar]
- McKee, A.E. , Thiele, C.J. , 2006. Targeting caspase 8 to reduce the formation of metastases in neuroblastoma. Expert Opinion on Therapeutic Targets 10, 703–708. [DOI] [PubMed] [Google Scholar]
- Nagji, A.S. , Cho, S.H. , Liu, Y. , Lee, J.K. , Jones, D.R. , 2010. Multigene expression-based predictors for sensitivity to Vorinostat and Velcade in non-small cell lung cancer. Molecular Cancer Therapeutics 9, 2834–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri, D. , Lockman, P.R. , Thomas, F.C. , Hua, E. , Herring, J. , Hargrave, E. , Johnson, M. , Flores, N. , Qian, Y. , Vega-Valle, E. , Taskar, K.S. , Rudraraju, V. , Mittapalli, R.K. , Gaasch, J.A. , Bohn, K.A. , Thorsheim, H.R. , Liewehr, D.J. , Davis, S. , Reilly, J.F. , Walker, R. , Bronder, J.L. , Feigenbaum, L. , Steinberg, S.M. , Camphausen, K. , Meltzer, P.S. , Richon, V.M. , Smith, Q.R. , Steeg, P.S. , 2009. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 15, 6148–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, X.Y. , Dai, Y. , Grant, S. , 2004. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 10, 3839–3852. [DOI] [PubMed] [Google Scholar]
- Petrella, A. , Fontanella, B. , Carratu, A. , Bizzarro, V. , Rodriquez, M. , Parente, L. , 2011. Histone deacetylase inhibitors in the treatment of hematological malignancies. Mini Reviews in Medicinal Chemistry 11, 519–527. [DOI] [PubMed] [Google Scholar]
- Petruccelli, L.A. , Dupere-Richer, D. , Pettersson, F. , Retrouvey, H. , Skoulikas, S. , Miller, W.H. , 2011. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One 6, e20987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfstein, R. , Bruchim, I. , Fishman, A. , Werner, H. , 2011. The mechanism of action of the histone deacetylase inhibitor vorinostat involves interaction with the insulin-like growth factor signaling pathway. PLoS One 6, e24468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerk, C. , Schulze-Osthoff, K. , 2003. Non-apoptotic functions of caspases in cellular proliferation and differentiation. Biochemical Pharmacology 66, 1453–1458. [DOI] [PubMed] [Google Scholar]
- Shin, H. , Lee, Y.S. , Lee, Y.C. , 2012. Sodium butyrate-induced DAPK-mediated apoptosis in human gastric cancer cells. Oncology Reports 27, 1111–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprick, M.R. , Weigand, M.A. , Rieser, E. , Rauch, C.T. , Juo, P. , Blenis, J. , Krammer, P.H. , Walczak, H. , 2000. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 12, 599–609. [DOI] [PubMed] [Google Scholar]
- Stimson, L. , Wood, V. , Khan, O. , Fotheringham, S. , La Thangue, N.B. , 2009. HDAC inhibitor-based therapies and haematological malignancy. Annals of Oncology: Official Journal of the European Society for Medical Oncology/ESMO 20, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Sung, E.S. , Kim, A. , Park, J.S. , Chung, J. , Kwon, M.H. , Kim, Y.S. , 2010. Histone deacetylase inhibitors synergistically potentiate death receptor 4-mediated apoptotic cell death of human T-cell acute lymphoblastic leukemia cells. Apoptosis: An International Journal on Programmed Cell Death 15, 1256–1269. [DOI] [PubMed] [Google Scholar]
- Takai, N. , Desmond, J.C. , Kumagai, T. , Gui, D. , Said, J.W. , Whittaker, S. , Miyakawa, I. , Koeffler, H.P. , 2004. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 10, 1141–1149. [DOI] [PubMed] [Google Scholar]
- Thome, M. , Schneider, P. , Hofmann, K. , Fickenscher, H. , Meinl, E. , Neipel, F. , Mattmann, C. , Burns, K. , Bodmer, J.L. , Schroter, M. , Scaffidi, C. , Krammer, P.H. , Peter, M.E. , Tschopp, J. , 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386, 517–521. [DOI] [PubMed] [Google Scholar]
- Torres, V.A. , Mielgo, A. , Barbero, S. , Hsiao, R. , Wilkins, J.A. , Stupack, D.G. , 2010. Rab5 mediates caspase-8-promoted cell motility and metastasis. Molecular Biology of the Cell 21, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, H. , Maruyama, T. , Nagashima, T. , Asada, H. , Yoshimura, Y. , 2005. Histone deacetylase inhibitors induce differentiation of human endometrial adenocarcinoma cells through up-regulation of glycodelin. Endocrinology 146, 5365–5373. [DOI] [PubMed] [Google Scholar]
- Uchida, H. , Maruyama, T. , Ono, M. , Ohta, K. , Kajitani, T. , Masuda, H. , Nagashima, T. , Arase, T. , Asada, H. , Yoshimura, Y. , 2007. Histone deacetylase inhibitors stimulate cell migration in human endometrial adenocarcinoma cells through up-regulation of glycodelin. Endocrinology 148, 896–902. [DOI] [PubMed] [Google Scholar]
- van Raam, B.J. , Salvesen, G.S. , 2012. Proliferative versus apoptotic functions of caspase-8 hetero or homo: the caspase-8 dimer controls cell fate. Biochimica et biophysica acta 1824, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev, E.E. , Schuchmann, M. , Luria, V. , Chiannilkulchai, N. , Beckmann, J.S. , Mett, I.L. , Rebrikov, D. , Brodianski, V.M. , Kemper, O.C. , Kollet, O. , Lapidot, T. , Soffer, D. , Sobe, T. , Avraham, K.B. , Goncharov, T. , Holtmann, H. , Lonai, P. , Wallach, D. , 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276. [DOI] [PubMed] [Google Scholar]
- Walker, T. , Mitchell, C. , Park, M.A. , Yacoub, A. , Graf, M. , Rahmani, M. , Houghton, P.J. , Voelkel-Johnson, C. , Grant, S. , Dent, P. , 2009. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Molecular Pharmacology 76, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Fukunaga, R. , Brannan, C.I. , Copeland, N.G. , Jenkins, N.A. , Nagata, S. , 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356, 314–317. [DOI] [PubMed] [Google Scholar]
- Yang, Y.T. , Balch, C. , Kulp, S.K. , Mand, M.R. , Nephew, K.P. , Chen, C.S. , 2009. A rationally designed histone deacetylase inhibitor with distinct antitumor activity against ovarian cancer. Neoplasia (New York, N.Y.) 11, 552–563. 553 pp. following 563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeramian, A. , Moreno-Bueno, G. , Dolcet, X. , Catasus, L. , Abal, M. , Colas, E. , Reventos, J. , Palacios, J. , Prat, J. , Matias-Guiu, X. , 2012. Endometrial carcinoma: molecular alterations involved in tumor development and progression. Oncogene 32, 403–413. [DOI] [PubMed] [Google Scholar]
- Yerbes, R. , Lopez-Rivas, A. , 2012. Itch/AIP4-independent proteasomal degradation of cFLIP induced by the histone deacetylase inhibitor SAHA sensitizes breast tumor cells to TRAIL. Investigational New Drugs 30, 541–547. [DOI] [PubMed] [Google Scholar]
- Zhang, G. , Park, M.A. , Mitchell, C. , Hamed, H. , Rahmani, M. , Martin, A.P. , Curiel, D.T. , Yacoub, A. , Graf, M. , Lee, R. , Roberts, J.D. , Fisher, P.B. , Grant, S. , Dent, P. , 2008. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 14, 5385–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data