

Abstract
Chronic myeloid leukaemia (CML) is driven by the fusion protein Bcr‐Abl, a constitutively active tyrosine kinase playing a crucial role in initiation and maintenance of CML phenotype. Despite the great efficacy of the Bcr‐Abl‐specific inhibitor imatinib, resistance to this drug is recognized as a major problem in CML treatment. We found that in LAMA84 cells, characterized by imatinib‐resistance caused by BCR‐ABL1 gene amplification, the pro‐survival protein kinase CK2 is up‐regulated as compared to the sensitive cells. CK2 exhibits a higher protein‐level and a parallel enhancement of catalytic activity. Consistently, CK2‐catalysed phosphorylation of Akt‐Ser129 is increased. CK2 co‐localizes with Bcr‐Abl in the cytoplasmic fraction as judged by subcellular fractionation and fluorescence immunolocalization. CK2 and Bcr‐Abl are members of the same multi‐protein complex(es) in imatinib‐resistant cells as demonstrated by co‐immunoprecipitation and co‐sedimentation in glycerol gradients. Cell treatment with CX‐4945, a CK2 inhibitor currently in clinical trials, counteracts CK2/Bcr‐Abl interaction and causes cell death by apoptosis. Interestingly, combination of CX‐4945 with imatinib displays a synergistic effect in reducing cell viability. Consistently, knockdown of CK2α expression by siRNA restores the sensitivity of resistant LAMA84 cells to low imatinib concentrations. Remarkably, the CK2/Bcr‐Abl interaction and the sensitization towards imatinib obtained by CK2‐inhibition in LAMA84 is observable also in other imatinib‐resistant CML cell lines.
These results demonstrate that CK2 contributes to strengthen the imatinib‐resistance phenotype of CML cells conferring survival advantage against imatinib. We suggest that CK2 inhibition might be a promising tool for combined strategies in CML therapy.
Keywords: Chronic myeloid leukaemia, CK2, Bcr‐Abl, Imatinib‐resistance, Inhibitor
Highlights
CK2 signalling is up‐regulated in imatinib‐resistant as compared to sensitive chronic myeloid leukaemia cells.
CK2 and Bcr‐Abl co‐localise and are interacting proteins.
Inhibition of CK2 causes cell death by apoptosis of chronic myeloid leukaemia cells.
Down‐regulation of CK2 signalling can overcome the imatinib‐resistance.
1. Introduction
The cytogenetic hallmark of the chronic myeloid leukaemia (CML) is the chromosomal translocation t(9; 22)(q34; q11), yielding the Philadelphia chromosome and generating a fusion gene that encodes Bcr‐Abl, a constitutively active protein tyrosine kinase. Signal transduction pathways activated by Bcr‐Abl kinase activity promote cell survival and proliferation while protecting cells from apoptosis (Goldman and Melo, 2003). Since Bcr‐Abl plays a critical role in the initiation and maintenance of the CML phenotype, targeting its tyrosine kinase activity is the therapeutic strategy of choice. Imatinib mesylate is a potent inhibitor selective for Bcr‐Abl that has become the frontline therapy for CML patients. However, despite the high effectiveness of this therapeutic approach, up to one third of CML patients develop either resistance or intolerance to imatinib and require alternative therapy (Bixby and Talpaz, 2009; Roychowdhury and Talpaz, 2011). The various mechanisms of imatinib‐resistance described up to now, either Bcr‐Abl‐dependent (gene amplification or mutation) or Bcr‐Abl‐independent (decreased imatinib bioavailability or activation of alternative signalling pathway) (Bixby and Talpaz, 2009; Roychowdhury and Talpaz, 2011) have provided the opportunity for second‐generation or combination therapies aimed at preventing resistance or restoring response to the drug (Santos et al., 2011; Stegmeier et al., 2010; Roychowdhury and Talpaz, 2011).
Protein kinase CK2 is a ubiquitous, highly conserved and pleiotropic Ser/Thr kinase, endowed with constitutive activity, independent of any known second messenger or phosphorylation events. CK2 is usually present as a tetrameric holoenzyme composed of two catalytic subunits (α and/or α′) and a dimer of regulatory (β) subunits. It phosphorylates a huge number of protein substrates, implicated in fundamental cell processes and is essential for cell life (Meggio and Pinna, 2003; Ruzzene and Pinna, 2010; Salvi et al., 2009). CK2 is abnormally elevated in a wide variety of tumours, where it plays a global role as an anti‐apoptotic and pro‐survival agent (Ahmad et al., 2008; St‐Denis and Litchfield, 2009) and there is strong evidence that it operates as a cancer driver by creating a cellular environment favourable to neoplasia (Ruzzene and Pinna, 2010). Different data suggest that CK2 may also have a significant role in the pathogenesis of haematopoietic tumours, including CML (Piazza et al., 2012), where a relationship between CK2 and Bcr‐Abl has been suggested (Hériché and Chambaz, 1998, 2003, 2007). The role of CK2 in imatinib‐resistance, however, has never been explored.
In this study we compare the properties of the protein kinase CK2 in imatinib‐sensitive and resistant LAMA84 cell lines. We also analyse the potential cross‐talk between CK2 and Bcr‐Abl and the possibility of using CK2‐specific inhibitors for combined therapy to overcome the imatinib‐resistance of CML cells.
2. Materials and methods
2.1. Materials and antibodies
[γ33P]ATP was purchased from Perkin–Elmer (Waltham, MA). Protease inhibitor cocktail was from Calbiochem (Darmstadt, Germany), while phosphatase inhibitor cocktails and β‐casein from Sigma–Aldrich (Dorset, U.K.). Imatinib mesylate was from Cayman Chemical (Ann‐Arbor, MI), while CX‐4945 was provided by Cylene‐Pharmaceuticals (S. Diego, CA). Inhibitors GNF‐2 and staurosporine, and other chemicals were from Sigma–Aldrich. RRRADDSDDDDD peptide (Ruzzene et al., 2010) and recombinant CK2 (α2β2) (Lolli et al., 2012) were kindly provided by Dr. Oriano Marin and Dr. Andrea Venerando (University of Padova, Italy), respectively. Anti‐CK2α (Sarno et al., 1996) and anti‐phospho‐Akt(Ser129) (Di Maira et al., 2005) antibodies were raised in rabbit. Anti‐c‐Abl was from Calbiochem, anti‐CK2β, CrkL and phospho‐CrkL(Tyr207) from Epitomics (Burlingame, CA), anti‐CK2α′, Akt, phospho‐Akt(Ser473), Lyn, lamin B, LDH and rpS6 from Santa Cruz Biotechnology (Santa Cruz, CA), anti‐phospho‐tyrosine from Millipore Corporation (Billerica, MA), anti‐PARP from Roche (Basel, Switzerland) and anti‐tubulin from Sigma–Aldrich.
2.2. Cell culture
KCL‐22, K562 and LAMA84 cell lines, either sensitive or resistant to imatinib, were kindly supplied by Dr. C. Gambacorti‐Passerini (le Coutre et al., 2000; Redaelli et al., 2010). Cells were maintained in RPMI 1640 supplemented with 10% foetal calf serum, 2 mM l‐glutamine, 100U/ml penicillin and 100 mg/ml streptomycin in the absence (sensitive) or presence (resistant) of imatinib (3 μM, 0.6 μM and 1.5 μM for KCL‐22, K562 and LAMA84 cells, respectively).
2.3. Cell lysis and western blot analysis
Cells were lysed as previously described (Di Maira et al., 2007). Protein concentration was determined by Bradford method. Proteins were subjected to 9% or 11% SDS‐PAGE, blotted on Immobilon‐P membranes (Sigma–Aldrich), processed in western‐blot with the indicated antibodies and developed using an enhanced chemiluminescent detection system (ECL). Immunostained bands were quantified by means of a Kodak‐Image‐Station 4000 MM‐PRO and analysis with Carestream Molecular Imaging software (New‐Haven, CT).
2.4. Immunoprecipitation experiments
Indicated lysate proteins were immunoprecipitated overnight with the specific antibody, followed by addition of protein A‐Sepharose. The immunocomplexes, washed three times with 50 mM Tris–HCl, pH 7.5, were analysed by western‐blot.
2.5. RNA extraction and real‐time quantitative PCR
Total RNA from S‐ and R‐LAMA84 cells was extracted using TRIzol reagent (Life‐Technologies, Carlbad, CA) and 1 μg RNA was reverse transcribed with TaqMan® Reverse Transcription Reagents (Life‐Technologies) according to the manufacturer's instructions and subsequently used for real‐time quantitative PCR. Amplification and quantification was performed with Power SYBR® Green PCR Master Mix (Life‐Technologies) and a Rotor‐Gene 3000 system (Corbett Life Science, Concorde, NSW). The oligonucleotide primers (Sigma–Aldrich) used for CK2α were: 5′‐GAGAGGAGGTCCCAACATCA‐3′ (sense) and 5′‐TGACATTATGGGGCTTGACA‐3′ (antisense), and for β‐actin: 5′‐GGACTTCGAGCAAGAGATGG‐3′ (sense) and 5′‐AGCACTGTGTTGGCGTACAG‐3′ (antisense). Expression levels were normalized to β‐actin.
2.6. CK2 kinase activity assay
Lysate proteins were incubated for 10 min at 30 °C in 25 μl of a phosphorylation medium containing 50 mM Tris–HCl (pH 7.5), 100 mM NaCl, 12 mM MgCl2, 400 μM synthetic peptide‐substrate RRRADDSDDDDD and 20 μM [γ33P]ATP (1000 cpm/pmol). Assays were stopped by absorption onto phosphocellulose filters. Filters were washed four times in 75 mM phosphoric acid (Ruzzene et al., 2010) and analysed by a Scintillation Counter (PerkinElmer).
2.7. In‐gel kinase assay of CK2α
The activity displayed by CK2α subunit alone was determined by running cell lysates on an 11% SDS‐PAGE containing the CK2‐substrate β‐casein (0.5 mg/ml). After electrophoresis, the activity of CK2α toward the co‐localized β‐casein was detected by incubating the gel with the above described phosphorylation medium containing 1 mM [γ33P]ATP (Ruzzene et al., 2010). Radioactive 33P‐β‐casein was evidenced by analysing the dried gel with a Cyclone Plus Storage PhosphorSystem (PerkinElmer).
2.8. Subcellular fractionation by differential centrifugation
Cells (8 × 106) were re‐suspended in a hypotonic buffer (10 mM Tris/acetate, pH 7.4, containing protease and phosphatase inhibitor cocktails), incubated for 5 min on ice and broken by Dounce homogenization. The solution was immediately adjusted to 0.25 M saccharose, 1 mM MgCl2 and subjected to differential centrifugation to separate nuclei, mitochondria, microsomes and cytosol (Kang and Welch, 1991). Pellets were re‐suspended in a volume of lysis buffer corresponding to that of cytosol. Same volumes of different fractions were analysed by western‐blot.
2.9. Immunolocalization of CK2 and Bcr‐Abl by fluorescence microscopy
Cells (5 × 105) were seeded on polylysine‐coated glass coverslips, allowed to adhere overnight, fixed with 4% para‐formaldehyde in PBS for 20 min at room temperature and permeabilised with 0.1% Triton X–100 in PBS for 10 min at 4 °C. For dual labelling, cells were first incubated with mouse anti‐Abl antibody (1:10) overnight at 4 °C, followed by 1 h incubation with anti‐mouse IgG/FITC conjugated antibody (1:50) at 37 °C. Cells were then incubated with rabbit anti‐CK2α antibody (1:50) for 1 h at 37 °C followed by goat anti‐rabbit Alexa‐Fluor 633 conjugated antibody (1:500) for 1 h at 37 °C. Nuclei were stained with Hoechst 33342. Fluorescence images were captured using a LEICA‐TCS SP5 confocal microscopy (Wetzlar, Germany), equipped with HCX PL APO lambda blue 63 × 1.4 oil immersion objective. Images were processed with the LAS‐AF software.
2.10. Glycerol gradient sedimentation
Cells (20 × 106) were lysed with the above‐described lysis buffer containing 10 mM KCl and 0.2% triton X‐100. 400 μg of lysates were layered on the top of a 3.6 ml of a glycerol linear gradient (10%–40%) in 50 mM Hepes, pH 8, 1 mM EDTA, 1 mM DTT, protease and phosphatase inhibitors. The tubes were centrifuged at 100 000× g for 18 h at 4 °C and fractionated from the bottom into 20 fractions.
2.11. RNA interference
Cells (1.5 × 106) were transfected with 30 nM CK2α specific siGENOME SMARTpool siRNAs (Dharmacon, Lafayette, CO, USA) or aspecific siRNA siCONTROL riscfree#1 (Dharmacon), as control, using the transfecting reagent INTERFERin (Polyplus‐transfection SA, Illkirch, France), according to the manufacturer's recommendations.
2.12. Cell viability assay
Cell viability was detected by the method of MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐3,5‐diphenyltetrazolium bromide), incubating 15 × 103 cells/100 μl in a 96‐well plate under different conditions. 1 h before the incubation end, 10 μl of MTT solution (5 mg/ml in PBS) was added to each well. Incubations were stopped by addition of 20 μl of a pH 4.7 solution containing 20% (w:v) SDS, 50% (v:v) N,N‐dimethylformamide, 2% (v:v) acetic acid and 25 mM HCl. Plates were read at λ540 nm absorbance, in a Titertek Multiskan Plus plate reader (Flow Laboratories, Sutton, U.K.).
2.13. Combined treatments
The combination index (CI) (Chou, 2006) for the combined treatment with imatinib and CX‐4945 was calculated with the software Calcusyn (Biosoft, Cambridge, U.K.).
2.14. Statistical analysis
Data are presented as means ± SD and mean differences were analysed using t‐test. A p < 0.05 was considered as statistically significant.
3. Results
3.1. Protein‐level and activity of protein kinase CK2 in imatinib‐sensitive and ‐resistant CML cells
The CML cell lines KCL‐22, K562 and LAMA84, either sensitive (S) or resistant (R) to imatinib, were characterized with specific antibodies. In these cell lines the imatinib‐resistance is neither due to Bcr‐Abl mutations (le Coutre et al., 2000; Redaelli et al., 2010) nor to a multidrug resistance phenotype (le Coutre et al., 2000; Zanin et al., 2012). Western blot analysis of equal amounts of cell lysates shows that the protein‐level of Bcr‐Abl, while similar in parental and imatinib‐resistant KCL‐22 and K562 cell lines, is about four‐fold higher in imatinib‐resistant as compared to imatinib‐sensitive LAMA84 cells (Figure 1A). This finding is consistent with the notion that in this cell line imatinib‐resistance is associated with an overexpression of the oncokinase mediated by gene amplification (le Coutre et al., 2000). As expected, the parallel analysis with anti‐phospho‐tyrosine antibody shows that overexpressed Bcr‐Abl is constitutively active as judged from its autophosphorylation and the phosphorylation of the key Bcr‐Abl substrate CrkL (de Jong et al., 1997), which are higher in R‐LAMA84 than in sensitive cells (Figure 1A).
Figure 1.

Expression analysis of Bcr‐Abl, Lyn and CK2 in different CML cell lines. (A,B) 30 μg (A) or 10 μg (B) of lysate proteins were analysed by Western blot. Anti‐p‐Tyr immunostaining was superimposed on Bcr‐Abl band, which was detected by anti‐Abl antibody. Anti‐α‐tubulin Western blot is shown as a loading control. Figure is representative of at least five separate experiments. (C) Real‐time quantitative PCR analysis of CK2α gene expression was performed using cDNA obtained from reverse transcribed total RNA from S‐LAMA84 and R‐LAMA84 cells. The CK2α mRNA level was assessed by quantitative PCR analysis as described in Materials and methods. Results were normalized to β‐actin mRNA used as internal standard and the expression level of CK2α gene in R‐LAMA84 is normalized to that of S‐LAMA84 cells. Reported values are means ± SD of four separate experiments performed in triplicate.
Since up‐regulation of the Src‐kinase Lyn has been described to be associated with imatinib‐resistance (Ptasznik et al., 2004), the protein‐level of this Src family tyrosine kinase was also analysed in the leukaemic cells. In all CML cell lines Lyn is similarly expressed in imatinib‐resistant and sensitive cells (Figure 1A).
The expression of the protein kinase CK2 was next examined using antibodies toward the kinase catalytic (α and α′) and regulatory (β) subunits (Figure 1B). While CK2 subunits are similarly expressed in both imatinib‐sensitive and ‐resistant KCL‐22 and K562 cell lines, the amount of CK2α and β subunits is approximately two‐fold higher in R‐LAMA84 cell line as compared to the sensitive counterpart. At variance, the expression of CK2α′ is similar in sensitive and resistant cells. The outcome that CK2 is more abundant in R‐LAMA84 cells prompted us to further characterizes the two LAMA84 cell variants. To assess whether the high CK2α amount detected in resistant cells might be due to altered regulation at transcription level, CK2α mRNA amount was examined by means of relative semi‐quantitative RT‐PCR (not shown) and real‐time quantitative PCR (Figure 1C). Both methods showed that comparable levels of mRNA are present in S‐LAMA84 and R‐LAMA84 cells.
The activity of cellular CK2 was then tested using increasing amounts of cell lysates by in vitro kinase assay toward a CK2‐specific peptide‐substrate. Consistent with the protein level, the cellular kinase activity is about two‐fold higher in R‐LAMA84 cells than in parental cell line (Figure 2A). To further characterize the detected kinase activity, a parallel analysis was performed using the lysates of cells treated with imatinib or CX‐4945, a potent and selective CK2‐inhibitor currently in clinical trials for the treatment of different tumours (Siddiqui‐Jain et al., 2010). As expected, CX‐4945 strongly reduces CK2 activity in the two cell lines, while imatinib treatment does not affect it (Figure 2B).
Figure 2.

Analysis of CK2 activity in S‐LAMA84 and R‐LAMA84 cells. (A) The kinase activity of cellular CK2 was tested, as detailed in Materials and methods, toward the peptide‐substrate RRRADDSDDDDD in a phosphorylation medium containing the indicated micrograms of lysate proteins. Reported values are means ± SD of four separate experiments. (B) Cells, incubated for 24h with vehicle DMSO (Ctrl), CX‐4945 (3 μM) or imatinib (0.5 μM in S‐LAMA84 and 2 μM in R‐LAMA84), were lysed and CK2 activity was tested in 1 μg of lysate proteins as described in (A). Reported values are means ± SD of four separate experiments. (C) The kinase activity of monomeric CK2α was analysed by an in‐gel kinase assay. The indicated micrograms of lysate proteins were loaded on a polyacrylamide gel containing the CK2‐substrate β‐casein and CK2α activity was detected as detailed in Materials and methods. The 33P‐phosphorylation of β‐casein, evidenced by a Cyclone Plus Storage PhosphorSystem is expressed in Digital Light Units (DLU). Reported values are means ± SD of four separate experiments.
Cellular CK2 was also studied by analysing the activity displayed by its catalytic subunit α. To this purpose CK2α was separated on a polyacrylamide gel containing the CK2‐substrate β‐casein and the activity of the α‐subunit toward the co‐localized substrate was determined by a radioactive in‐gel kinase assay. Equal amounts of cellular lysates show a higher 33P‐phosphorylation of β‐casein in R‐LAMA84 cells as compared to the parental counterpart (Figure 2C).
3.2. Protein quantification and subcellular distribution of CK2 in LAMA84 cells
CK2 level was found markedly increased in highly proliferating myeloblastic cells from CML patients in comparison with normal granulocytes (Phan‐Dinh‐Tuy et al., 1985). This finding prompted us to perform a relative quantification of cellular CK2α and β subunits in LAMA84 cells by comparative analysis with recombinant CK2 holoenzyme (α2β2) containing equimolar amounts of the two subunits. The comparison suggests that the two CK2 subunits are expressed at very high levels in both CML cell lines and confirms that imatinib‐resistant cells contain about twice as much CK2α and β subunits (Figure 3A). In particular, the densitometric analysis suggests that in imatinib‐resistant cells the amount of CK2α represents about 0.3% of total proteins.
Figure 3.

Quantification and subcellular distribution of CK2 in S‐LAMA84 and R‐LAMA84 cells. (A) The indicated ng of recombinant CK2 holoform (α2β2) and μg of lysate proteins from S‐LAMA84 and R‐LAMA84 cells were analysed by Western blot with anti‐CK2α (left panel) and anti‐CK2β (right panel) antibodies. Anti‐α‐tubulin Western blot is shown as a loading control. Means of densitometric values ± SD, expressed in arbitrary units (a.u.), are reported above the relative subunit bands. Cellular CK2 subunit amounts were calculated by densitometric analysis and extrapolation from the calibration curve built on the signal of recombinant CK2. (B) (Left panel) Cells were disrupted by Dounce homogenization and subcellular fractionation was performed by differential centrifugation as detailed in Materials and methods. Subcellular fractions (N, nuclei; Mt, mitochondria; C, cytosol and Mc, microsomes) were resuspended in an equal volume and the same volume of resulting fractions was immunoblotted with the indicated antibodies including the organelle‐specific antibodies against lamin B (nuclei), lactate dehydrogenase (LDH) (cytosol) and S6 ribosomal protein (microsomes and nucleoli). The Figure is representative of five separate experiments. (Right panel) Bars report the mean values ± SD of the densitometric analysis of the CK2‐subunit bands obtained as in left panel. Densitometric values are expressed in arbitrary units. *p < 0.05. (C) Confocal microscopy of double immunofluorescence staining of R‐LAMA84 cells with anti‐CK2α (red) and anti‐Abl (green) antibodies. Nuclei were stained with Hoechst 33342. Co‐localization of red and green fluorescences is visualized by the yellow fluorescence appearing after merging of both signals. Violet appears from the merging of nuclear staining and red fluorescence.
There is ample evidence that CK2 is distributed in nearly every subcellular compartment, where it plays different functions, and that the subcellular localization of CK2 is tightly regulated (Filhol and Cochet, 2009). Therefore studying the distribution of this kinase may be a key to understand its function. The comparison of CK2 subcellular localization in the two cell variants (Figure 3B) reveals that the amount of CK2α′ is similar in the different subcellular compartments of S‐LAMA84 and R‐LAMA84 cells. In contrast, while CK2α level is comparable in nuclei and almost undetectable in mitochondria, it is overexpressed in the cytosolic and microsomal fractions of R‐LAMA84 cells. Likewise, the protein‐level of CK2β is consistently higher in cytosol and microsomes.
It is noteworthy that, in resistant cells, CK2 is overexpressed in the cytoplasm (cytosol and microsomes), where Bcr‐Abl is also mainly retained in CML cells and where it interacts with most proteins involved in the oncogenic pathway (Cilloni and Saglio, 2012). This prompted us to perform a confocal microscopy immunofluorescence analysis looking for a possible co‐localization of the two protein kinases, which are both overexpressed in R‐LAMA84 cells. CK2 fluorescence is observable in the nucleus but is mostly localized in the cytoplasm, where Bcr‐Abl is exclusively visible and appears to co‐localize with CK2 (Figure 3C). Immunolocalization performed in parallel with S‐LAMA84 cells showed a similar distribution of CK2 fluorescence, which is more evident in the cytoplasm (results not shown), while Bcr‐Abl localization was unfeasible because the oncokinase fluorescence was not detectable (see also Figure 1A).
3.3. CK2 and Bcr‐Abl interact in CML cells
CK2 and Bcr‐Abl co‐localization prompted us to check if the two protein kinases are interacting proteins. To this purpose, Bcr‐Abl‐immunoprecipitates obtained from LAMA84 cellular lysates were probed with anti‐CK2 antibodies (Figure 4Aa). Interestingly, while CK2 does not co‐immunoprecipitate with Bcr‐Abl in S‐LAMA84 cells, a substantial amount of both CK2α and β subunits is detectable in R‐LAMA84 cells. Consistently, Bcr‐Abl is present in CK2α immunocomplexes only in imatinib‐resistant cells (Figure 4Ab). A parallel analysis was also performed to compare LAMA84 with K562 and KCL‐22 cell lines, where Bcr‐Abl and CK2 are similarly expressed in S and R variants (Figure 4Ac). Differently from LAMA84, in K562 and KCL‐22 the interaction between Bcr‐Abl and CK2 is detectable also in imatinib‐sensitive cells. Interestingly, in K562 the association observed is higher in imatinib‐resistant than in sensitive cells as in the case of LAMA84 cells (Figure 4Ac).
Figure 4.

Analysis of CK2 and Bcr‐Abl interaction in S‐LAMA84 and R‐LAMA84 cells. (Aa, Ab) S‐ and R‐LAMA84 cells were lysed and 300 μg of lysate proteins were immunoprecipitated with a control antibody from the same class (Ctrl) and anti‐Abl antibody (Aa), or pre‐immune serum (Ctrl) and anti‐CK2α antibody (Ab). The immunocomplexes were then analysed by Western blot with the indicated antibodies. (Ac) LAMA84, K562 and KCL‐22 cells were immunoprecipitated with anti‐Abl antibody and immunocomplexes were analysed by anti‐CK2α immunostaining. (Ba) LAMA84, K562 and KCL‐22 cells were lysed and lysate proteins (300 μg) were immunoprecipitated with anti‐CK2α antibody. The immunocomplexes were then analysed by Western blot with anti‐phospho‐tyrosine (anti‐p‐Tyr) followed by anti‐CK2α antibodies. (Bb,Bc) R‐LAMA84 cells were treated with vehicle, CX‐4945 (5 μM) or imatinib (3 μM) for 24h and then lysed. (Bb) Cellular lysates were analysed by Western blot with the indicated antibodies. (Bc) 300 μg of cellular lysates were immunoprecipitated by anti‐CK2α antibody and immunocomplexes were immunostained with anti‐p‐Tyr followed by anti‐CK2α antibodies. Figure is representative of at least four separate experiments.
Since it has been shown that Abl tyrosine kinase phosphorylates CK2α in vitro (Hériché and Chambaz, 1998), the lysates of the CML cell lines were immunoprecipitated with anti‐CK2α antibody and analysed for the presence of phospho‐tyrosine (p‐Tyr) (Figure 4B). In LAMA84 cells CK2α is Tyr‐phosphorylated only in the imatinib‐resistant counterpart, while in K562 and KCL‐22 the subunit is Tyr‐phosphorylated in both cell variants (Figure 4Ba). Interestingly, in the case of K562 the extent of Tyr‐phosphorylation is higher in the imatinib‐resistant than in the sensitive cells, as observed in LAMA84 cells suggesting a relationship between higher CK2 association to Bcr‐Abl (Figure 4Bc) and higher CK2α Tyr‐phosphorylation. Parallel experiments demonstrated that Tyr‐phosphorylation is undetectable in CK2β immunoprecipitates (data not shown).
To assess whether CK2 itself, or Bcr‐Abl (or eventually both) might be responsible for this Tyr‐phosphorylation, imatinib‐resistant LAMA84 cells were treated for 24 h with vehicle, CX‐4945 or imatinib. CX‐4945, which neither affects the protein‐level nor the activity of Bcr‐Abl (Figure 4Bb), does not reduce the Tyr‐phosphorylation extent of immunoprecipitated CK2α (Figure 4Bc), ruling out the possibility that CK2 catalytic subunit might undergo Tyr‐autophosphorylation in R‐LAMA84 cell line, as found in other mammalian cells (Vilk et al., 2008). On the contrary, imatinib greatly decreases both the Bcr‐Abl activation state (Figure 4Bb) and the extent of CK2α Tyr‐phosphorylation (Figure 4Bc), consistent with the concept that Bcr‐Abl is the kinase responsible for this phosphorylation. Experiments aimed at highlighting the effect of this Tyr‐phosphorylation on CK2 catalytic activity failed to show any significant difference between CK2α immunoprecipitated in comparable amounts from control or imatinib‐treated R‐LAMA84 cells (not shown).
To evaluate the role played by the activity of each kinase on the reciprocal binding, CK2/Bcr‐Abl interaction was analysed in R‐LAMA84 cells treated with different kinase inhibitors (Figure 5A). Intriguingly, the treatment with CX‐4945 almost abrogates the interaction occurring between CK2 and Bcr‐Abl, while imatinib and GNF‐2, an allosteric non‐ATP competitive inhibitor of Bcr‐Abl (Adrián et al., 2006), do not affect this binding. The additional finding that staurosporine, added at a concentration ineffective toward CK2 but able to inhibit most protein kinases (Meggio et al., 1995) including Bcr‐Abl (not shown), does not counteract the interaction between CK2 and Bcr‐Abl (Figure 5A), corroborates the hypothesis that CK2 kinase activity plays a specific role in the binding. Consistently, a highly reduced amount of Bcr‐Abl is detectable in CK2α immunoprecipitates from R‐LAMA84 cells treated with CX‐4945 (Figure 5B). This finding prompted us to assess whether CK2‐catalysed phosphorylation of Bcr‐Abl might be a prerequisite for the interaction of the two kinases. However, phosphorylation assays performed in vitro by adding recombinant CK2 holoenzyme to Bcr‐Abl immunoprecipitated from R‐LAMA84 lysates, do not support the hypothesis that Bcr‐Abl might be a target–substrate of CK2 (not shown).
Figure 5.

Effect of CX‐4945 on CK2/Bcr‐Abl interaction. (A) R‐LAMA84 cells were treated with vehicle, CX‐4945 (5 μM), imatinib (3 μM), GNF‐2 (10 μM) or staurosporine (1 μM) for 24 h. Lysate proteins (300 μg) were immunoprecipitated by anti‐Abl antibody and immunocomplexes were analysed by Western‐blot. (B) R‐LAMA84 cells were treated with the indicated inhibitors as in (A). Lysate proteins (300 μg) were immunoprecipitated with anti‐CK2α antibody and then probed with the indicated antibodies. (C) R‐LAMA84 cells, treated for 24 h with vehicle (Ctrl) or 5 μM CX‐4945, were lysed and lysate proteins were separated on glycerol gradient as detailed in Materials and methods. Molecular weight standards were run on separated tubes: bovine serum albumin (66 kDa), alcohol dehydrogenase (150 kDa), apoferritin (443 kDa) and thyroglobulin (669 kDa). 40 μl of the resulting fractions were analysed by Western blot. The densitometric analysis of CK2α and Bcr‐Abl bands is reported above the relative gradient. (Right panel) Fractions 8–12 of each gradient were pooled and immunoprecipitated with anti‐CK2α antibody. The immunocomplexes were analysed by Western blot. Figure is representative of four separate experiments.
To further analyse the CK2/Bcr‐Abl interaction, R‐LAMA84 cells treated with vehicle or CX‐4945 were lysed under mild conditions and subjected to glycerol gradient sedimentation (Figure 5C). In control cells CK2α subunit co‐migrates with most Bcr‐Abl (fractions 8–12), suggesting that they are partners of the same complex(es) as confirmed by their co‐immunoprecipitation observed using the pooled fractions 8–12 of the gradient (Figure 5C, right panel). Interestingly, CX‐4945‐treatment, which does not significantly change the sedimentation profile of Bcr‐Abl, makes CK2α to shift towards fractions containing complexes displaying lower molecular weights (fractions 10–14), implying that CK2 dissociates from Bcr‐Abl as corroborated by the reduced co‐immunoprecipitation of the two oncokinases (Figure 5C, right panel).
3.4. Effect of CK2 down‐regulation on CML cell viability
The effect of imatinib on LAMA84 cell viability was compared with that of CX‐4945. As expected, the DC50 values (concentration inducing the 50% of cell death) calculated for imatinib are about 0.3 and 2.1 μM, in sensitive and resistant cell lines, respectively (Figure 6A). Treatment with CX‐4945 reduces the viability of both S‐LAMA84 and R‐LAMA84 cells with DC50 values of about 8 and 5 μM, respectively (Figure 6B).
Figure 6.

Cell death induction by imatinib and CX‐4945 in LAMA84 cells. (A–D) S‐LAMA84 and R‐LAMA84 were treated with the indicated concentration of imatinib (A,C) or CX‐4945 (B,D) for 48 h. (A,B) Cell viability was assessed by MTT method and expressed as percentage of controls. *p < 0.01, **p < 0.05 vs S‐LAMA84 cells. (C,D) Cellular lysates (30 μg) were analysed by Western blot. Anti‐PARP antibody recognizes the full length protein and its p85 fragment. Figure is representative of five separate experiments.
Apoptosis occurrence was then analysed by comparing the cleavage of the caspase substrate PARP in the two cell variants. As expected, PARP is almost completely cleaved by treatment with 0.5 μM imatinib in sensitive LAMA84 cells, an event parallelled by the proteolysis of Bcr‐Abl, Akt and α‐tubulin (Figure 6C). On the contrary, treatment with up to 1 μM imatinib does not induce any appreciable effect in resistant cells (Figure 6C). The opposite is observable with CX‐4945, which is not effective up to 5 μM concentration in S‐LAMA84 cells, while the same concentration of inhibitor induces an almost complete cleavage of PARP and of the other analysed proteins in R‐LAMA84 cells (Figure 6D). This outcome supports the hypothesis that imatinib‐resistant cells are more dependent on CK2 activity for their survival than sensitive cells.
Since CK2 phosphorylates Akt at Ser129 inducing an increased activity of this pro‐survival kinase (Di Maira et al., 2005), the phosphorylation state of this residue was evaluated upon treatment with the two inhibitors. Consistent with the higher CK2 activity, the extent of Akt Ser129 phosphorylation is higher in imatinib‐resistant than in sensitive cells under basal conditions (Figure 6C and D) suggesting an up‐regulation of Akt signalling in R‐LAMA84 cells. Moreover, while imatinib treatment does not affect Ser129 phosphorylation (Figure 6C), CX‐4945 strongly reduces the phosphorylation of this Akt residue, which, in both cell variants, is almost abrogated by 2 μM CX‐4945, a concentration not affecting the total Akt amount (Figure 6D).
We have recently found that inhibition of CK2 by CX‐4945 reduces also the viability of the CML cell lines K562 and KCL‐22 (Zanin et al., 2012). In all the tested CML lines, CK2 inhibition induces cell death also in the imatinib‐resistant variants (see Figure 6B and Zanin et al., 2012), independently of the CK2 expression level (Figure 1B); this prompted us to investigate whether CX‐4945 might sensitize resistant cells to imatinib. To this purpose, cells were treated with CX‐4945 and imatinib either separately or in combination. We then examined if the combined treatment induced a higher degree of cell death compared to the separate treatments. Interestingly, low concentrations of CX‐4945 are able to significantly increase the effect of imatinib on all the resistant CML cell lines analysed (Figure 7). The values of the combination index (which denotes synergism if <1) (Chou, 2006) are 0.57 for R‐LAMA84, 0.75 for R‐K562, and 0.87 for R‐KCL‐22, demonstrating that the combined treatment promotes a synergistic reduction of cell viability more pronounced in R‐LAMA84 and R‐K562 cells.
Figure 7.

Synergistic effect of CX‐4945 and imatinib treatment on CML cell viability. (A) R‐LAMA84, (B) R‐K562 or (C) R‐KCL‐22 were treated for 48 h with the indicated concentration of imatinib, CX‐4945 or with the two drugs in combination by increasing simultaneously the concentration of both compounds added at 1:3 (A), 1:4 (B), and 1:1 (C) imatinib:CX‐4945 ratio. Viability, assessed by MTT method and expressed as percentage of controls, was plotted as function of imatinib concentration (left panel), or CX‐4945 concentration (right panel). *p < 0.01, **p < 0.05 vs cells treated with a single inhibitor.
To further support a specific role of CK2 in imatinib‐resistance, we knocked down the expression of CK2α in LAMA84 cells by performing RNA‐interference experiments. A decrease of CK2α protein‐level of about 52% and 80% was obtained in S‐ and R‐LAMA84 cells, respectively (Figure 8A). Also CK2 activity was reduced, although to a lesser extent (about 30% and 56% in S‐ and R‐LAMA84 cells, respectively) (Figure 8B), due to the contribution of the other catalytic subunit (α′) not affected by the silencing procedure. Moreover, CK2α down‐regulation by siRNA greatly reduces the interaction occurring between CK2 and Bcr‐Abl (Figure 8C) as previously shown in LAMA84 cells treated with the CK2‐inhibitor CX‐4945 (Figure 5A,B). When we treated control and CK2α down‐regulated cells with increasing concentration of imatinib, we found that no significant effect on cell viability was induced in sensitive cells by CK2α silencing (Figure 8D). On the contrary, in imatinib‐resistant cells, CK2α down‐regulation promotes a higher sensitivity to low imatinib concentrations (Figure 8E), confirming the data obtained with the pharmacological blockade of CK2 (Figure 7A).
Figure 8.
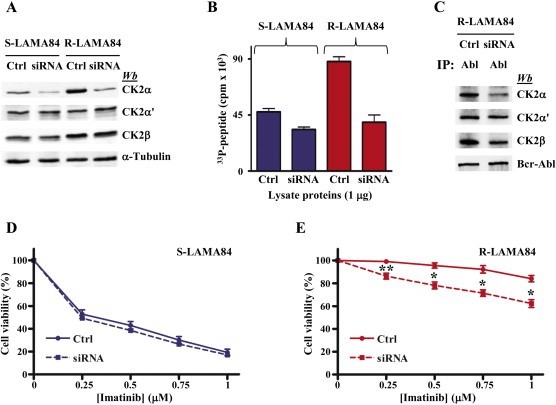
Effect of CK2α knocking down by siRNA on LAMA84 sensitivity to imatinib. S‐LAMA84 and R‐LAMA84 cells were transfected with aspecific siRNA (Ctrl) or CK2α specific siRNA. (A, B and C) After 96 h, cells were lysed, and (A) 10 μg of lysate proteins were analysed by Western blot, (B) 1 μg of lysate proteins was tested for CK2 activity toward the specific peptide RRRADDSDDDDD, and (C) 300 μg of R‐LAMA84 cell proteins were immunoprecipitated with anti‐Abl antibody and then analysed by Western blot with the indicated antibodies. Panels A–C are representative of four separate experiments (D, E) 48 h after transfection, S‐LAMA84 (D) or R‐LAMA84 (E) cells were treated for 48 h with the indicated imatinib concentrations and cell viability was analysed by MTT method. *p < 0.01, **p < 0.05 vs control cells. Panels D and E are representative of five separate experiments performed in triplicate.
4. Discussion
In this study, we provide the first evidence that, among different adaptations described to be associated with imatinib‐resistance, CK2‐dependent signalling represents an additional mechanism that can be exploited to ensure survival to CML cells. In particular, we found that in imatinib‐resistant LAMA84 cell line, characterized by a BCR‐ABL1 gene amplification, CK2 is upregulated in comparison with the parental cell line. While CK2α′ is equally expressed, the level of CK2α and β subunits is about two‐fold higher in R‐LAMA84 than in sensitive cells (Figure 1A and B). CK2 protein increase, which is accompanied by a parallel increase of cellular CK2 catalytic activity (Figure 2), appears related to an altered regulation at protein level since the mRNA amount of CK2α is very similar in the two cell variants (Figure 1C). These results are in agreement with studies where abnormally high level of CK2 protein and activity in cancer cells is not accompanied by a parallel mRNA increase (Di Maira et al., 2007; Trembley et al., 2009).
It is noteworthy that CK2 has been found overrepresented in highly proliferating myeloblastic cells from CML patients in blast crisis (Phan‐Dinh‐Tuy et al., 1985), a phase in which Bcr‐Abl overexpression has been associated to imatinib‐resistance (Barnes et al., 2005; Gorre et al., 2001; Keeshan et al., 2001; Virgili and Nacheva, 2010). Considering the pro‐survival function of CK2, it is conceivable that its increased level represents a device to escape apoptosis. Although CK2 up‐regulation is not an absolute requirement for the resistant phenotype (Figure 1B and Di Maira et al., 2008), overexpression of CK2α, either alone or in combination with the β subunit has been already associated in other cancer cell lines with resistance mechanisms, either related to a multidrug resistance phenotype or induced by specific drugs (Di Maira et al., 2008; Matsumoto et al., 2001).
CK2 nuclear concentration has been reported to be particularly high in cancer cells (Trembley et al., 2009). In contrast, in S‐LAMA84 and R‐LAMA84 cells CK2 is mainly present in the cytoplasm (Figure 3B,C). We also show that CK2 and Bcr‐Abl co‐localize in the cytoplasm, where the CK2‐targets related to imatinib‐resistance are presumably placed. Phospho‐proteomic analyses are in progress to identify the proteins whose phosphorylation, sensitive to CX‐4945‐inhibition, is evoked/increased in R‐LAMA84 as compared to sensitive cells.
CK2 and Bcr‐Abl co‐localization reflects the finding that, in resistant LAMA84 cells, these two oncokinases are members of the same multi‐protein complex(es) as demonstrated by their co‐immunoprecipitation and co‐sedimentation in glycerol‐gradients (Figures 4A, 5C). CK2α and Bcr‐Abl interaction is also detectable in the other CML cell lines analysed, K562 and KCL‐22 (Figure 4Ac). Our results demonstrate that in the case of LAMA84 and K562 cells the two oncokinases interact more in imatinib‐resistant than in sensitive cells (Figure 4Ac). The occurrence of an interaction between CK2 and Bcr‐Abl has been previously described in cells overexpressing the two protein kinases and in lymphoblastic cells obtained from Bcr‐Abl transgenic mouse (Hériché and Chambaz, 1998; Mishra et al., 2003). The region responsible for CK2 interaction was localized to residues 242–413 of the Bcr moiety of Bcr‐Abl (Mishra et al., 2003).
In the attempt to detect reciprocal phosphorylation of the two protein kinases in CML cell lines we disclosed a Bcr‐Abl‐dependent Tyr‐phosphorylation of CK2α, which is more evident in R‐LAMA84 and R‐K562 cells (Figure 4Ba), where also Bcr‐Abl/CK2 interaction is higher (Figure 4Ac). The Tyr‐phosphorylation of CK2α, however, is not required for the interaction occurring between the two kinases, which is not affected by imatinib. On the contrary, inhibition of CK2 almost abrogates the binding between the two enzymes (Figure 5).
A significant contribution of CK2 to chronic myeloid leukaemia is supported by data obtained from cell treatments with the highly selective CK2 inhibitor CX‐4945, which affects neither the amount nor the activity of Bcr‐Abl (Figure 4Bb). Indeed the viability of both imatinib‐sensitive and ‐resistant CML cells is significantly reduced whenever CK2 activity is inhibited by CX‐4945, consistent with the general anti‐apoptotic and pro‐survival role played by CK2 in cancer cells (Figure 6B and Zanin et al., 2012). Interestingly, CX‐4945 added in combination with imatinib promotes a synergistic effect on the cell viability of imatinib‐resistant CML variants, partially rescuing the response to imatinib. The synergism is especially evident in R‐LAMA84 and R‐K562 cells (Figure 7), where Bcr‐Abl/CK2 interaction is also higher (Figure 4Ac). In this respect, we can hypothesize that the interaction occurring between CK2 and Bcr‐Abl might be one of the molecular mechanisms reinforcing the imatinib‐resistance but also offering the possibility to sensitize cells to imatinib by CK2 down‐regulation and consequent binding disruption (Figure 5 for CK2 inhibition by CX‐4945, and Figure 8C for CK2 knock‐down by siRNA).
The hypothesis that imatinib‐resistant cells become partially dependent on CK2 for their survival was confirmed by the observation that the CX‐4945 concentrations required to induce apoptosis in R‐LAMA84 cells are lower than those effective in S‐LAMA84 cells (Figure 6B,D).
It has been proposed that the high CK2 level observed in cancer cells may generate an environment, which favours cancer progression by promoting/fostering multiple oncogenic pathways (Ruzzene and Pinna, 2010). Since some of these deregulated pathways are also under the control of Bcr‐Abl (Perrotti et al., 2010; Quintás‐Cardama and Cortes, 2009), CK2 might, on one hand, potentiate the Bcr‐Abl oncogenic signalling and, on the other, strengthen the imatinib‐resistant phenotype by activating key molecular events able to circumvent the drug inhibitory effects on Bcr‐Abl pathways. Pertinent to this, we have found that Akt‐signalling is reinforced in R‐LAMA84 cells by the increased phosphorylation of the CK2 target‐residue Ser129 (Figure 6C,D).
Imatinib is the first‐line therapy for chronic myeloid leukaemia, but resistance to this drug frequently occurs and causes therapy failure. This study identifies the protein kinase CK2 as a player in CML imatinib resistance, where it supports the Bcr‐Abl oncogenic potential conferring survival advantage against imatinib. Down‐regulation of CK2 rescues the response to imatinib. We suggest that CK2 inhibitors, with special reference to CX‐4945, a compound already in clinical trials for the treatment of different tumours, might represent promising drugs for combined strategies in CML therapy.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by grants from AIRC (Italian Association for Cancer Research), Project IG 10312 to LAP and from University of Padova (Progetto Ateneo 2011) to MR. The authors would like to thank Cylene‐Pharmaceuticals (Dr. Sean O'Brien) for kindly providing CX‐4945.
Borgo Christian, Cesaro Luca, Salizzato Valentina, Ruzzene Maria, Massimino Maria Lina, Pinna Lorenzo A., Donella‐Deana Arianna, (2013), Aberrant signalling by protein kinase CK2 in imatinib‐resistant chronic myeloid leukaemia cells: Biochemical evidence and therapeutic perspectives, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.08.006.
References
- Adrián, F.J. , Ding, Q. , Sim, T. , Velentza, A. , Sloan, C. , Liu, Y. , Zhang, G. , Hur, W. , Ding, S. , Manley, P. , Mestan, J. , Fabbro, D. , Gray, N.S. , 2006. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol.. 2, 95–102. [DOI] [PubMed] [Google Scholar]
- Ahmad, K.A. , Wang, G. , Unger, G. , Slaton, J. , Ahmed, K. , 2008. Protein kinase CK2–a key suppressor of apoptosis. Adv. Enzym. Regul.. 48, 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, D.J. , Palaiologou, D. , Panousopoulou, E. , Schultheis, B. , Yong, A.S.M. , Wong, A. , Pattacini, L. , Goldman, J.M. , Melo, J.V. , 2005. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res.. 65, 8912–8919. [DOI] [PubMed] [Google Scholar]
- Bixby, D. , Talpaz, M. , 2009. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematol. Am. Soc. Hematol. Educ. Prog.. 461–476. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev.. 58, 621–681. [DOI] [PubMed] [Google Scholar]
- Cilloni, D. , Saglio, G. , 2012. Molecular pathways: BCR-ABL. Clin. Cancer Res.. 18, 930–937. [DOI] [PubMed] [Google Scholar]
- de Jong, R. , ten Hoeve, J. , Heisterkamp, N. , Groffen, J. , 1997. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 14, 507–513. [DOI] [PubMed] [Google Scholar]
- Di Maira, G. , Salvi, M. , Arrigoni, G. , Marin, O. , Sarno, S. , Brustolon, F. , Pinna, L.A. , Ruzzene, M. , 2005. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ.. 12, 668–677. [DOI] [PubMed] [Google Scholar]
- Di Maira, G. , Brustolon, F. , Bertacchini, J. , Tosoni, K. , Marmiroli, S. , Pinna, L.A. , Ruzzene, M. , 2007. Pharmacological inhibition of protein kinase CK2 reverts the multidrug resistance phenotype of a CEM cell line characterized by high CK2 level. Oncogene. 26, 6915–6926. [DOI] [PubMed] [Google Scholar]
- Di Maira, G. , Brustolon, F. , Tosoni, K. , Belli, S. , Krämer, S.D. , Pinna, L.A. , Ruzzene, M. , 2008. Comparative analysis of CK2 expression and function in tumor cell lines displaying sensitivity vs. resistance to chemical induced apoptosis. Mol. Cell. Biochem.. 316, 155–161. [DOI] [PubMed] [Google Scholar]
- Filhol, O. , Cochet, C. , 2009. Protein kinase CK2 in health and disease: cellular functions of protein kinase CK2: a dynamic affair. Cell. Mol. Life Sci.. 66, 1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, J.M. , Melo, J.V. , 2003. Chronic myeloid leukemia–advances in biology and new approaches to treatment. N. Engl. J. Med.. 349, 1451–1464. [DOI] [PubMed] [Google Scholar]
- Gorre, M.E. , Mohammed, M. , Ellwood, K. , Hsu, N. , Paquette, R. , Rao, P.N. , Sawyers, C.L. , 2001. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Hériché, J.K. , Chambaz, E.M. , 1998. Protein kinase CK2alpha is a target for the Abl and Bcr-Abl tyrosine kinases. Oncogene. 17, 13–18. [DOI] [PubMed] [Google Scholar]
- Kang, H.S. , Welch, W.J. , 1991. Characterization and purification of the 94-kDa glucose-regulated protein. J. Biol. Chem.. 266, 5643–5649. [PubMed] [Google Scholar]
- Keeshan, K. , Mills, K.I. , Cotter, T.G. , McKenna, S.L. , 2001. Elevated Bcr-Abl expression levels are sufficient for a haematopoietic cell line to acquire a drug-resistant phenotype. Leukemia. 15, 1823–1833. [DOI] [PubMed] [Google Scholar]
- le Coutre, P. , Tassi, E. , Varella-Garcia, M. , Barni, R. , Mologni, L. , Cabrita, G. , Marchesi, E. , Supino, R. , Gambacorti-Passerini, C. , 2000. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 95, 1758–1766. [PubMed] [Google Scholar]
- Lolli, G. , Pinna, L.A. , Battistutta, R. , 2012. Structural determinants of protein kinase CK2 regulation by autoinhibitory polymerization. ACS Chem. Biol.. 7, 1158–1163. [DOI] [PubMed] [Google Scholar]
- Matsumoto, Y. , Takano, H. , Kunishio, K. , Nagao, S. , Fojo, T. , 2001. Expression of drug resistance genes in VP-16 and mAMSA-selected human carcinoma cells. Jpn. J. Cancer Res.. 92, 778–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio, F. , Donella-Deana, A. , Ruzzene, M. , Brunati, A.M. , Cesaro, L. , Guerra, B. , Meyer, T. , Mett, H. , Fabbro, D. , Furet, P. , Dobrowolska, G. , Pinna, L.A. , 1995. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur. J. Biochem.. 234, 317–322. [DOI] [PubMed] [Google Scholar]
- Meggio, F. , Pinna, L.A. , 2003. One-thousand-and-one substrates of protein kinase CK2?. FASEB J.. 17, 349–368. [DOI] [PubMed] [Google Scholar]
- Mishra, S. , Reichert, A. , Cunnick, J. , Senadheera, D. , Hemmeryckx, B. , Heisterkamp, N. , Groffen, J. , 2003. Protein kinase CKIIalpha interacts with the Bcr moiety of Bcr/Abl and mediates proliferation of Bcr/Abl-expressing cells. Oncogene. 22, 8255–8262. [DOI] [PubMed] [Google Scholar]
- Mishra, S. , Pertz, V. , Zhang, B. , Kaur, P. , Shimada, H. , Groffen, J. , Kazimierczuk, Z. , Pinna, L.A. , Heisterkamp, N. , 2007. Treatment of P190 Bcr/Abl lymphoblastic leukemia cells with inhibitors of the serine/threonine kinase CK2. Leukemia. 21, 178–180. [DOI] [PubMed] [Google Scholar]
- Perrotti, D. , Jamieson, C. , Goldman, J. , Skorski, T. , 2010. Chronic myeloid leukemia: mechanisms of blastic transformation. J. Clin. Invest.. 120, 2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan-Dinh-Tuy, F. , Henry, J. , Boucheix, C. , Perrot, J.Y. , Rosenfeld, C. , Kahn, A. , 1985. Protein kinases in human leukemic cells. Am. J. Hematol.. 19, 209–218. [DOI] [PubMed] [Google Scholar]
- Piazza, F. , Manni, S. , Ruzzene, M. , Pinna, L.A. , Gurrieri, C. , Semenzato, G. , 2012. Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia. 26, 1174–1179. [DOI] [PubMed] [Google Scholar]
- Ptasznik, A. , Nakata, Y. , Kalota, A. , Emerson, S.G. , Gewirtz, A.M. , 2004. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat. Med.. 10, 1187–1189. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama, A. , Cortes, J. , 2009. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 113, 1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redaelli, S. , Boschelli, F. , Perini, P. , Pirola, A. , Viltadi, M. , Gambacorti-Passerini, C. , 2010. Synergistic activity of the Src/Abl inhibitor bosutinib in combination with imatinib. Leukemia. 24, 1223–1227. [DOI] [PubMed] [Google Scholar]
- Roychowdhury, S. , Talpaz, M. , 2011. Managing resistance in chronic myeloid leukemia. Blood Rev.. 25, 279–290. [DOI] [PubMed] [Google Scholar]
- Ruzzene, M. , Di Maira, G. , Tosoni, K. , Pinna, L.A. , 2010. Assessment of CK2 constitutive activity in cancer cells. Methods Enzymol.. 484, 495–514. [DOI] [PubMed] [Google Scholar]
- Ruzzene, M. , Pinna, L.A. , 2010. Addiction to protein kinase CK2: a common denominator of diverse cancer cells?. Biochim. Biophys. Acta. 1804, 499–504. [DOI] [PubMed] [Google Scholar]
- Salvi, M. , Sarno, S. , Cesaro, L. , Nakamura, H. , Pinna, L.A. , 2009. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta. 1793, 847–859. [DOI] [PubMed] [Google Scholar]
- Santos, F.P.S. , Kantarjian, H. , Quintás-Cardama, A. , Cortes, J. , 2011. Evolution of therapies for chronic myelogenous leukemia. Cancer J.. 17, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno, S. , Vaglio, P. , Meggio, F. , Issinger, O.G. , Pinna, L.A. , 1996. Protein kinase CK2 mutants defective in substrate recognition. Purification and kinetic analysis. J. Biol. Chem.. 271, 10595–10601. [DOI] [PubMed] [Google Scholar]
- Siddiqui-Jain, A. , Drygin, D. , Streiner, N. , Chua, P. , Pierre, F. , O'Brien, S.E. , Bliesath, J. , Omori, M. , Huser, N. , Ho, C. , Proffitt, C. , Schwaebe, M.K. , Ryckman, D.M. , Rice, W.G. , Anderes, K. , 2010. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits pro-survival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res.. 70, 10288–10298. [DOI] [PubMed] [Google Scholar]
- St-Denis, N.A. , Litchfield, D.W. , 2009. Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell. Mol. Life Sci.. 66, 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier, F. , Warmuth, M. , Sellers, W.R. , Dorsch, M. , 2010. Targeted cancer therapies in the twenty-first century: lessons from imatinib. Clin. Pharmacol. Ther.. 87, 543–552. [DOI] [PubMed] [Google Scholar]
- Trembley, J.H. , Wang, G. , Unger, G. , Slaton, J. , Ahmed, K. , 2009. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell. Mol. Life Sci.. 66, 1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilk, G. , Weber, J.E. , Turowec, J.P. , Duncan, J.S. , Wu, C. , Derksen, D.R. , Zien, P. , Sarno, S. , Donella-Deana, A. , Lajoie, G. , Pinna, L.A. , Li, S.S. , Litchfield, D.W. , 2008. Protein kinase CK2 catalyzes tyrosine phosphorylation in mammalian cells. Cell Signal. 20, 1942–1951. [DOI] [PubMed] [Google Scholar]
- Virgili, A. , Nacheva, E.P. , 2010. Genomic amplification of BCR/ABL1 and a region downstream of ABL1 in chronic myeloid leukaemia: a FISH mapping study of CML patients and cell lines. Mol. Cytogenet.. 3, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanin, S. , Borgo, C. , Girardi, C. , O'Brien, S.E. , Miyata, Y. , Pinna, L.A. , Donella-Deana, A. , Ruzzene, M. , 2012. Effects of the CK2 inhibitors CX-4945 and CX-5011 on drug-resistant cells. PLoS One. 7, e49193 [DOI] [PMC free article] [PubMed] [Google Scholar]