

Abstract
Epithelial‐mesenchymal transition (EMT) is associated with reduced sensitivity to many chemotherapeutic drugs, including EGFR tyrosine kinase inhibitors. Here, we investigated if this reduced sensitivity also contributes to resistance to crizotinib, an ALK inhibitor of lung cancer that exhibits the EML4‐ALK translocation. We established a crizotinib‐resistant subline (H2228/CR), which was derived from the parental H2228 cell line by long‐term exposure to increasing concentrations of crizotinib. Characteristics associated with EMT, including morphology, EMT marker proteins, and cellular mobility, were analyzed. Compared with H2228 cells, the growth of H2228/CR cells was independent of EML4‐ALK, and H2228/CR cells showed cross‐resistance to TAE‐684 (a second‐generation ALK inhibitor). Phenotypic changes to the spindle‐cell shape were noted in H2228/CR cells, which were accompanied by a decrease in E‐cadherin and increase in vimentin and AXL. In addition, H2228/CR cells showed increased secretion and expression of TGF‐β1. Invasion and migration capabilities were dramatically increased in H2228/CR cells. Applying TGF‐β1 treatment to parental H2228 cells for 72 h induced reversible EMT, leading to crizotinib resistance, but this was reversed by the removal of TGF‐β1. Suppression of vimentin in H2228/CR cells by siRNA treatment restored sensitivity to crizotinib. Furthermore, these resistant cells remained highly sensitive to the Hsp90 inhibitors, similar to the parental H2228 cells. In conclusion, we suggest EMT is possibly involved in acquired resistance to crizotinib, and that HSP90 inhibitors could be a promising option for the treatment of EMT.
Keywords: Lung cancer, ALK, Resistance, Epithelial–mesenchymal transition
Highlights
Two crizotinib‐resistant models in H2228 lung cancer cells were established by treatment with crizotinib and TGF‐β1.
Epithelial‐to‐mesenchymal transition (EMT) with increased TGF‐β1 signaling was found in resistant cells.
Resistant cells escaped from dependence to ALK signaling while they were still sensitive to HSP90 inhibitors.
Down‐regulation of TGF‐β type II receptor was induced by HSP90 inhibitors.
EMT may be involved in acquired resistance to crizotinib and HSP90 inhibitors could be a promising treatment option.
1. Introduction
ALK was originally identified in anaplastic large‐cell lymphoma cells, in which translocation between the ALK gene on chromosome 2 and the nucleophosmin (NPM) gene on chromosome 5 leads to the expression of the NPM‐ALK fusion oncogene (Morris et al., 1994). Similarly, expression of the echinoderm microtubule‐associated protein‐like 4 (EML4)‐ALK fusion oncogene, which is caused by a small inversion within chromosome 2p, was found in non‐small cell lung cancer (NSCLC) (Soda et al., 2007). Subsequently, other fusion partners comprised of ALK and TFG, KIF5B, and KLC1 were identified (Rikova et al., 2007; Takeuchi et al., 2009; Togashi et al., 2012), although their proportions are not large in NSCLC. The incidence of NSCLC with ALK rearrangements is reportedly 3–5%. This tends to present in young and non‐ or light smokers who possess wild‐type EGFR and KRAS genes (Shaw et al., 2009; Solomon et al., 2009; Wong et al., 2009). The oncogenic potential that drives transformation and maintains tumor cell growth in cells expressing ALK fusion proteins has been reported in several studies (Chen et al., 2010, 2007, 2008).
Crizotinib is an orally administered drug that was initially developed as a c‐Met kinase inhibitor. However, since crizotinib was found to demonstrate cross‐reactivity with other receptor tyrosine kinases, including ALK and ROS1 (Bergethon et al., 2012; Christensen et al., 2007), it has been actively evaluated in ALK‐positive lung cancer patients (Kwak et al., 2010), thereby leading to accelerated approval as an ALK inhibitor by the US Food and Drug Administration (FDA). According to results of a global phase II study that was presented at the 48th Annual Meeting of the American Society of Clinical Oncology (ASCO), the overall response rate to crizotinib is 59.8% and median progression‐free survival is 8.1 months (Kim et al., 2012).
Despite the impressive efficacy of crizotinib for the treatment of ALK‐positive lung cancer, acquired resistance eventually develops in most patients. As secondary T790M mutation on exon 20 of the EGFR gene is the most common mechanism of EGFR tyrosine kinase inhibitors (EGFR‐TKIs) (Bell et al., 2005; Vikis et al., 2007), gatekeeper mutations such as L1196M, L1152R, C1156Y, and F1174L hinder drug binding and are frequently detected in crizotinib‐resistant samples (Choi et al., 2010; Katayama et al., 2011). Other mutations such as G1269A, G1202R, and S1206Y may be involved in the conformational changes that affect the affinity of crizotinib (Doebele et al., 2012; Katayama et al., 2012). There are several other possible mechanisms, including ALK amplification (Katayama et al., 2011), EGFR or c‐KIT activation (Katayama et al., 2012; Sasaki et al., 2011), and the acquisition of KRAS (Doebele et al., 2012), although their exact roles remain to be determined. However, our understanding of resistance mechanisms and frequency is very limited at the present because of the small number of experimental results and included patients, which indicates that more preclinical and clinical data should be accumulated to provide improved diagnostic and therapeutic strategies.
Epithelial‐mesenchymal transition (EMT) is the cellular process of morphological changing from the epithelial polarized shape to the mesenchymal fibroblastoid shape, in addition to accompanying behavioral changes such as enhanced mobility (Lee et al., 2006; Savagner, 2010). Recently several studies have reported that this phenomenon is strongly associated with cancer cell stemness (Chiou et al., 2010; Scheel and Weinberg, 2012; Dave et al., 2012) and resistance to drugs such as EGFR‐TKIs (Rho et al., 2009; Sequist et al., 2011; Yauch et al., 2005; Thomson et al., 2005). In our current study, we investigated how EMT contributes to crizotinib resistance and other therapeutic options that could be used to overcome EMT‐mediated resistance.
2. Materials and methods
2.1. Cell culture and reagents
The H2228 cell line was purchased from the American Type Culture Collection (Rockville, MD). The H3122 cell line was a gift from Adi F. Gazdar (UT Southwestern, Dallas, TX). Cells were cultured in 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA) at 37 °C in an atmosphere with 5% CO2. Crizotinib, TAE‐684, 17‐DMAG, and AUY‐922 were obtained from Selleck Chemicals Co. Ltd (Houston, TX). The 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) solution and TGF‐β1 were purchased from Sigma (St. Louis, MO) and R&D Systems (Minneapolis, MN), respectively.
2.2. Establishment of crizotinib resistance in H2228 cells (H2228/CR)
H2228/CR cells were developed by chronic, repeated exposure to crizotinib. Over a period of 8 months, H2228 cells were continuously exposed to increasing concentrations of the drug in culture and the surviving cells were cloned. These cells could survive exposure >100 nM of crizotinib. In all studies, resistant cells were cultured in drug‐free medium for >1 week to eliminate the effects of crizotinib.
2.3. MTT assay
Cells were seeded onto 96‐well plates and incubated overnight, then treated with their respective agents for an additional 3 days. Cell viability was determined using the previously described MTT‐based method (Carmichael et al., 1987). Each assay consisted of 8 replicate wells and was repeated at least three times. Data are expressed as the percentage survival of the control, which was calculated using absorbance after correcting for background noise.
2.4. Western blot analysis
Whole cell lysates were prepared using EBC lysis buffer (50 mM Tris–HCl [pH 8.0], 120 mM NaCl, 1% Triton X‐100, 1 mM EDTA, 1 mM EGTA, 0.3 mM phenylmethylsulfonyl fluoride, 0.2 mM sodium orthovanadate, 0.5% NP40, and 5 U/mL aprotinin) and centrifuged. Proteins were separated using SDS‐PAGE and transferred to PVDF membranes (Invitrogen) for Western blot analysis. Membranes were probed with antibodies against p‐ALK (Tyr1604), ALK, p‐Akt (Ser473), cytokeratin‐18, snail, p‐MET (Tyr1234/1235), vimentin, p‐AXL (Tyr702) (all from Cell Signaling Technology, Beverly, MA), Akt, p‐Erk (Thr202/Tyr204), Erk, E‐cadherin, TGF‐β1, TβRI, TβRII, p‐EGFR (Tyr1173), EGFR, MET, β‐actin, and AXL (all from Santa Cruz Biotechnology, Santa Cruz, CA) as the first antibody, and then membranes were treated with horseradish peroxidase‐conjugated secondary antibody. All membranes were developed using an enhanced chemiluminescence system (Thermo Scientific, Rockford, IL). Densitometric analysis was performed with ImageJ software offered by the NIH (http://rsb.info.nih.gov/nih‐image/).
2.5. Invasion and migration assays
The cell migration and invasion assays were performed using Transwell (6.5‐mm diameter, 8‐μm pore polycarbonate membrane), which was obtained from Corning (Cambridge, MA). Cells (105) in 200 μL medium were placed in the upper chamber, and the lower chamber was filled with 1 mL serum‐free media supplemented with 0.1% bovine serum albumin. After incubation for 24 h, non‐migrating cells were removed using cotton swabs, and the cells that migrated to the lower surface of the filter were stained using the Diff‐Quick kit (Fisher Scientific, Pittsburgh, PA). Cells were counted using a microscope. The migration assay was performed using the same procedure with filters that had been coated with extracellular matrix on the upper surface (BD Biosciences, Bedford, MA). Triplicate results are expressed as the mean (standard deviation).
2.6. Transfection of small interfering RNA
Small interfering RNA (siRNA) oligonucleotides specific to ALK, E‐cadherin, vimentin, and the siRNA control were obtained from Santa Cruz Biotechnology. Introduction of siRNA was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instructions. Target gene expression was measured 24 h later using western blot analysis. For the MTT assay, cells were seeded onto 96‐well plates after siRNA transfection, then treated with the indicated drugs for 72 h.
3. Results
3.1. The EML4‐ALK dependency is absent in H2228/CR cells
To investigate the mechanisms of acquired resistance to crizotinib, we selected H2228 (variant 3) and H3122 (variant 1) cell lines with EML4‐ALK rearrangement. Both cell lines were shown to be highly sensitive to ALK inhibitor, crizotinib. Therefore, these cell lines have clinical relevance for determining the mechanisms of acquired resistance to crizotinib. Crizotinib‐resistant cells were established by stepwise selection using increasing concentrations of crizotinib over a period of 8 months, as described in the Materials and Methods section. These resistant cells acquired approximately 10‐fold higher 50% inhibitory concentrations (IC50) to crizotinib compared with H2228 cells (crizotinib IC50 = 0.1 μM and >1 μM in H2228 cells and H2228/CR cells, respectively; Figure 1A). Furthermore, resistant cells showed cross‐resistance to TAE‐684, a more potent second‐generation ALK inhibitor (Figure 1B). Crizotinib and TAE‐684 could inhibit the activation of ALK and its downstream signals, such as Akt and Erk, in both H2228 and H2228/CR cells, although TAE‐684 was considerably more effective than crizotinib (Figure 1C). To further determine if H2228/CR cells depend on ALK signaling for growth, we suppressed the ALK gene by siRNA treatment. The growth of parental H2228 cells was significantly inhibited, while that of H2228/CR cells was unaffected (Figure 1D and E). This suggests that H2228/CR cells are independent of ALK signaling. H2228/CR cells did not have any secondary mutations that caused resistance to direct sequencing (data not shown).
Figure 1.
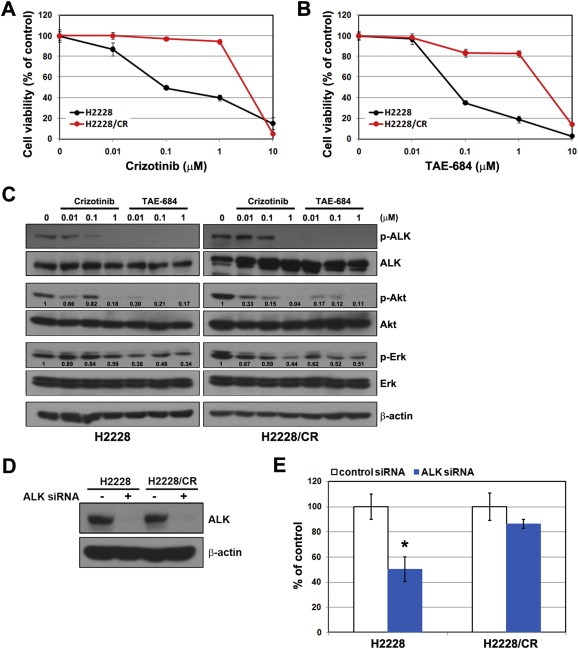
Establishment of acquired resistance to crizotinib in the H2228 cell line. (A, B) Cells were treated with the indicated doses of crizotinib or TAE‐684 for 72 h. Cell viability was determined using the MTT assay. (C) Cells were treated with or without the indicated doses of crizotinib or TAE‐684 for 3 h. Changes in ALK‐related signals were assessed using Western blot analysis. The densitometry values were determined relative to control after normalization to non‐phospho form of each protein. (D, E) Control and ALK siRNA (100 nM) were introduced into parental or resistant cells, and ALK silencing was confirmed by Western blot analysis. Cell viability was measured using the MTT assay 72 h later. Bars represent the standard deviations. *P < 0.001 compared with control siRNA.
3.2. EMT is induced in H2228/CR cells
We observed morphologic differences between the H2228 and H2228/CR cell lines using a light microscope. The round shape of the H2228/CR cells changed to a spindle form, suggesting that EMT‐like changes might have occurred (Figure 2A). To confirm the induction of EMT in H2228/CR cells, we analyzed the expression of epithelial and mesenchymal marker proteins using western blots (Figure 2B). Compared with the H2228 cells, E‐cadherin and cytokeratin‐18 expression were drastically reduced and vimentin expression was increased in H2228/CR cells. In addition, the levels of several factors that induce EMT were increased, such as snail, TGF‐β1, and AXL. TGF‐β1 was also elevated in the culture medium, indicating enhanced secretion. EGFR activation was not found to be associated with acquired resistance to crizotinib because the activity and expression levels of EGFR were reduced in H2228/CR cells. Next, we examined migratory and invasive potential, which are considered functional hallmarks of EMT. We found that the ability to migrate and invade was enormously increased in H2228/CR cells (Figure 2C). Taken together, these findings indicate that the acquisition of resistance to crizotinib in H2228/CR cells is accompanied by morphologic and functional changes that are consistent with EMT.
Figure 2.
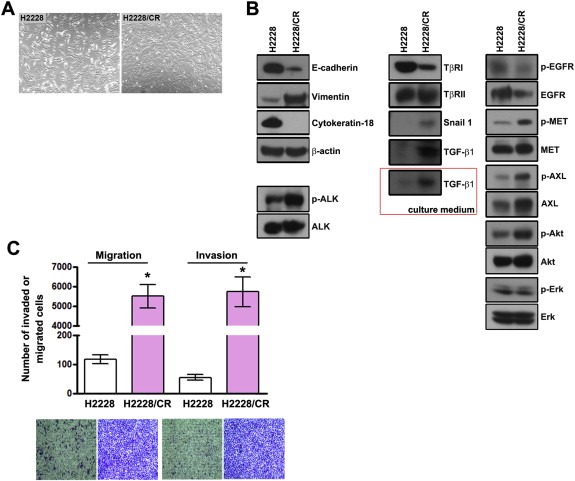
Induction of EMT in H2228/CR cells. (A) H2228 parental cells and crizotinib‐resistant cells (H2228/CR) were evaluated to determine any morphologic changes consistent with EMT using a light microscope. H2228/CR cells were composed mainly of spindle‐shaped cells compared with H2228 cells. (B) Cell lysates from H2228 and H2228/CR cells were subjected to Western blot analysis. The indicated antibodies were used to evaluate the levels of EMT‐related marker proteins, MET, AXL, and EGFR signals. The secretion of TGF‐β1 was confirmed in the culture media. (C) The migration and invasion assays were performed using Transwell. Cells were seeded onto either collagen‐ or Matrigel‐coated polycarbonate filters to determine their migratory and invasive potentials, respectively. Cells were incubated for 24 h in modified Boyden chambers, and the cells that penetrated through the filter were stained and counted using a light microscope. Experiments were repeated in triplicate. Bars represent the standard deviations. *P < 0.001 compared with H2228 cells.
3.3. TGF‐β1‐induced EMT leads to ALK inhibitor resistance
We previously reported the induction of reversible EMT by TGF‐β1 treatment in A549 lung cancer cells (Rho et al., 2009). To further determine if EMT leads to resistance against ALK inhibitors, we measured the response to ALK inhibitors under the conditions of TGF‐β1‐induced EMT. When treated with TGF‐β1 (10 ng/mL) for 72 h, H2228 cells were scattered and elongated and found the loss of epithelial marker proteins (E‐cadherin and cytokeratin‐18) and an increase in vimentin (designated H2228/TGF‐β1), which were reversed by the removal of TGF‐β1 (designated H2228/TGF‐β1/R; Figure 3A and B). However, TGF‐β1‐induced EMT was not observed in H3122 cells, indicating cell line specificity. H2228/TGF‐β1 cells were more resistant to ALK inhibitors than their parental cells, whereas sensitivity to ALK inhibitors was partially restored in H2228/TGF‐β1/R cells (Figure 3C), suggesting an association between EMT and response to ALK inhibitors.
Figure 3.
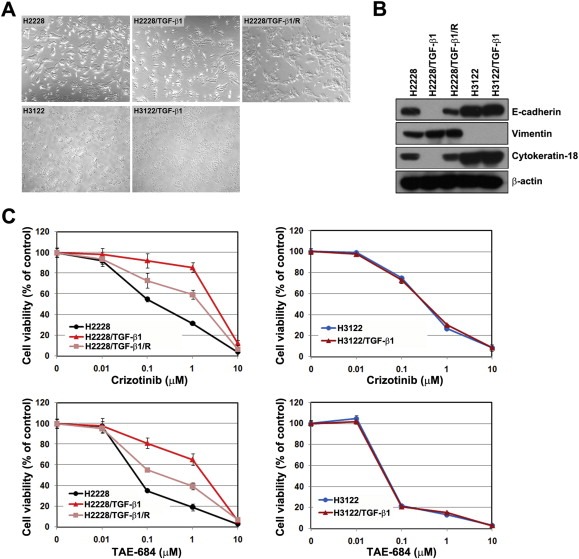
TGF‐β1 stimulated EMT, resulting in resistance to ALK inhibitors. H2228 and H3122 cells were treated with TGF‐β1 (10 ng/mL) for 72 h, and these cells are referred to as H2228/TGF‐β1 and H3122/TGF‐β1, respectively. H2228/TGF‐β1 cells were incubated for 24 h in the complete medium without TGF‐β1, and these cells are referred to as H2228/TGF‐β1/R. (A) Cells were observed under a light microscope. (B) Cell lysates from each cell line were subjected to Western blot analysis. The indicated antibodies were used to evaluate EMT‐related marker proteins. (C) Cells were treated with the indicated doses of crizotinib or TAE‐684 in the presence or absence of TGF‐β1. Cell viability was measured using the MTT assay 72 h later. Bars represent the standard deviations.
3.4. Inhibition of vimentin by siRNA treatment restores sensitivity to ALK inhibitors
E‐cadherin and vimentin exert significant effects on EMT. E‐cadherin maintains cell–cell contacts and the architecture of epithelial tissue, whereas vimentin is an intermediate filament protein that is present in most mesenchymal cells and is necessary for cellular movement. To determine if modulation of E‐cadherin and vimentin affects sensitivity to ALK inhibitors, drug responses to ALK inhibitors were evaluated after transfection with E‐cadherin or vimentin siRNA. Treatment with siRNA resulted in the decreased expression of both proteins, but expression was not completely abolished (Figure 4A). Reduced expression of E‐cadherin did not affect sensitivity to ALK inhibitors. However, decreased vimentin restored responsiveness to ALK inhibitors in H2228/CR cells (Figure 4B).
Figure 4.
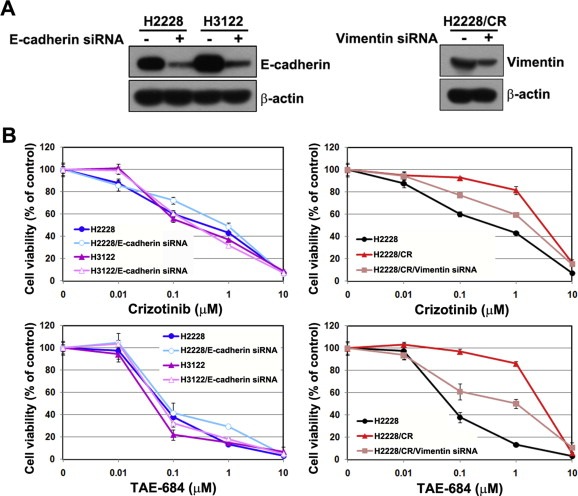
Suppression of vimentin enhanced sensitivity to ALK inhibitors in H2228/CR cells. (A) Control, E‐cadherin, and vimentin siRNA (100 nM) were introduced to the parental or resistant cells, and the suppression of each protein was confirmed by Western blot analysis. (B) After the transfection of each siRNA, cells were treated with the indicated doses of crizotinib or TAE‐684, and cell viability was measured 72 h later using the MTT assay. Bars represent the standard deviations.
3.5. Crizotinib‐resistant cells demonstrate no cross‐resistance to Hsp90 inhibitors
Some studies report that Hsp90 inhibitors overcome acquired resistance to ALK inhibitors as well as EGFR‐TKIs (Katayama et al., 2011; Sang et al., 2013; Sawai et al., 2008; Shimamura et al., 2008). Hsp90 is an abundant cellular chaperone protein and its inhibition degrades client proteins, including mutant EGFR and ALK. Thus, Hsp90 inhibitors affect mutant EGFR and ALK‐positive NSCLC cells regardless of the presence of any second mutations. Next, we examined if HSP90 inhibitors are still active in cells with EMT changes. 17‐DMAG and AUY‐922 effectively suppressed cell growth in parental H2228, H2228/CR, and H2228 cells with TGF‐β1‐induced EMT (Figure 5A). Treatment with these drugs led to the suppression of ALK activity, Akt, and Erk in both parental H2228 and H2228/CR cells (Figure 5B). Furthermore, treatment degraded TGF‐β type II receptor (TβRII), but even at low concentrations. TGF‐β type I receptor (TβRI) was not significantly affected. When cells were treated with Hsp90 inhibitors in a time‐dependent manner, the induction of PARP cleavage and Bim were similarly observed in both parental H2228 and H2228/CR cells. Interestingly, the restoration of E‐cadherin expression was found in H2228/CR cells after incubation for 48 h (Figure 5C).
Figure 5.
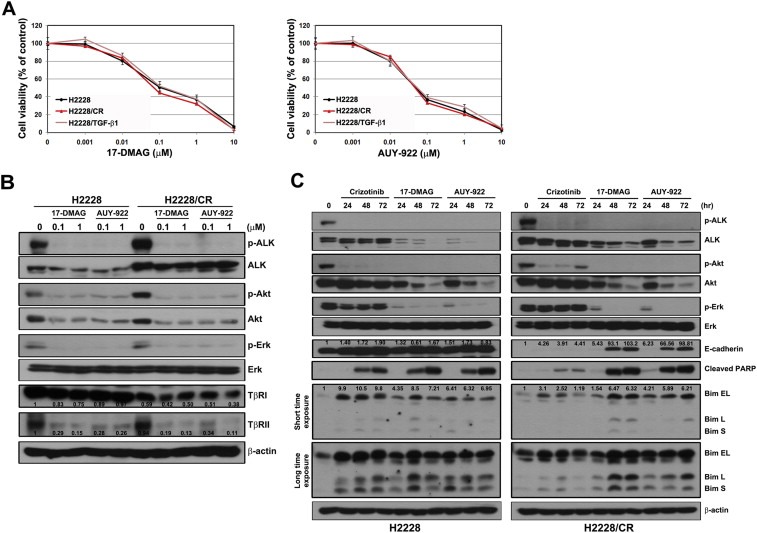
Hsp90 inhibitors can overcome crizotinib resistance caused by EMT. (A) Cells were treated with the indicated doses of 17‐DMAG or AUY‐2922. Cell viability was measured 72 h later using the MTT assay. Bars represent the standard deviations. (B) Cells were treated with or without the indicated doses of 17‐DMAG or AUY‐922 for 3 h. Changes in ALK‐related signaling and TGF‐β receptors were analyzed using Western blot analysis. The densitometry values were determined relative to control of H2228 after normalization to β‐actin protein. (C) Cells were treated with crizotinib (1 μM), 17‐DMAG (1 μM), or AUY‐922 (1 μM) for the indicated times. Cell lysates were subjected to Western blot analysis using the indicated antibodies. The densitometry values were determined relative to control after normalization to β‐actin protein.
4. Discussion
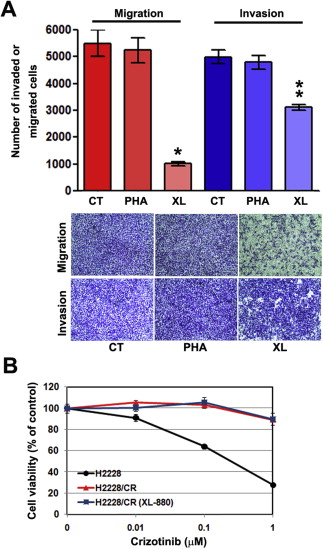
In our present study, we demonstrate that EMT could contribute to resistance against ALK inhibitors, including crizotinib and TAE‐684, whilst HSP90 inhibitors remain active in H2228 cells regardless of EMT. EMT is associated with the acquisition of stem cell‐like features (Singh and Settleman, 2010), cancer progression (Kalluri and Weinberg, 2009), and resistance to chemotherapy (Iwatsuki et al., 2010). We previously reported that acquired resistance to gefitinib leads to EMT in NSCLC cells and patient tissues (Chung et al., 2011; Rho et al., 2009). Furthermore, we found that the increased activation of AXL and EMT is associated with in vitro and in vivo acquired resistance to erlotinib in EGFR‐mutant lung cancer models (Zhang et al., 2012). Similarly, H2228/CR cells demonstrated increased AXL expression and activity. Although the inhibition of AXL could not overcome resistance to crizotinib, cellular migration and invasion both decreased (Supplementary Figure S1). This finding suggests that AXL signaling may not be directly involved in crizotinib resistance in these cells.
Although H2228 cells resistant to crizotinib due to EMT can be induced by short‐term treatment with TGF‐β1, our H2228/CR model established by chronic exposure to crizotinib closely resembles the clinical situation. However, we found that TGF‐β1 signaling is also involved in this model, indicating that the driving force for the induction of EMT does not differ between these two models. TGF‐β1 is a cytokine involved in multiple functions, including the regulation of cell growth, extracellular matrix remodeling, and the development of EMT, which leads to enhanced invasion, metastasis, and therapeutic resistance (Lee et al., 2006; Massague et al., 2000; Rho et al., 2009). Although the signaling pathways of TGF‐β1 are also diverse (Derynck and Zhang, 2003; Massague and Chen, 2000), interactions with TβRII are associated with the Smad group of intracellular signaling proteins and seems to be important for the development of EMT in lung cancer cells (Kasai et al., 2005; Zavadil and Bottinger, 2005). TβRII, in addition to TβRI, has been identified as an HSP90‐interacting protein. Wrighton et al. reported that the inhibition of HSP90 blocks TGF‐β1‐induced downstream signaling and transcriptional response and increases TβR ubiquitination and degradation, suggesting that HSP90 inhibitors could be used for the treatment of diseases with aberrantly activated TGF‐β1 signaling (Wrighton et al., 2008). Accordingly, the expression of TβRII was suppressed by HSP90 inhibitors in this study, which can explain the equivalent activities of these drugs in both H2228 and H2228/CR cells.
More diverse secondary mutations that decrease drug binding affinity have been found in association with crizotinib resistance than EGFR‐TKI. That is the reason why some investigators insist that it is closely related to imatinib resistance in chronic myeloid leukemia (O'Hare et al., 2007; Sang et al., 2013). Because these secondary mutations affect the binding affinity of the drug, the resistant cells obtained using this mechanism are still dependent on ALK signaling. Therefore, more potent second‐generation ALK inhibitors, such as TAE‐684 and AP26113, could demonstrate anticancer effects (Katayama et al., 2011). In contrast, we found in our present analyses that our H2228/CR cells are far less responsive to the complete blocking of ALK expression by siRNA treatment compared with parental H2228 cells. In addition, although TAE‐684 completely blocked ALK activation, it could not inhibit the proliferation of H2228/CR cells, indicating that they escape from addiction to ALK signaling.
In support of our current results, H3122 cells resistant to crizotinib via EMT were generated previously by Sang et al. (2013). In that study, epithelial markers decreased while the expression of vimentin, snail, Notch 1, caveolin, and Src were upregulated. Morphologic changes were also compatible with EMT. These cells were still sensitive to ganetespib, another HSP90 inhibitor developed by Synta Pharmaceuticals Corp (Lexington, MA), while several other ALK inhibitors such as CH5424802, ASP3026, and TAE684 were ineffective. These results are almost equivalent to our current observations. Sang et al. did not explain the effectiveness of HSP90 inhibitors against EMT cells, though they did report that the ALK fusion protein remained sensitive to ganetespib‐induced destabilization. It is sometimes misunderstood that overcoming the effects of HSP90 inhibitors in EMT is caused by ALK fusion proteins. Because they are fully dependent on ALK signaling, as demonstrated by this study, the suppression of TGF‐β1 signaling by HSP90 inhibitors could explain the drug effects. Interestingly, we also found that the reduced expression of E‐cadherin is recovered by treating H2228/CR cells with HSP90 inhibitors. This might be caused by the ability of the HSP90 inhibitors to reverse the driving forces of EMT.
In summary, EMT should be considered as a possible acquired resistance mechanism associated with crizotinib, and the use of HSP90 inhibitors may be a promising option for treating EMT. Further investigations that include clinical samples are needed to validate our results.
Supporting information
The following is the supplementary data related to this article:
Figure S1. Inhibition of AXL reduced cellular mobility in H2228/CR cells, but was not associated with sensitivity to crizotinib. (A) H2228/CR cells were seeded onto either collagen‐ or matrigel‐coated polycarbonate filters to determine their migratory and invasive potentials, respectively. Cells were treated with or without 1 μM PHA‐665752 (MET inhibitor) or 1 μM XL‐880 (dual MET and AXL inhibitor) for 24 h in modified Boyden chambers. Cells that penetrated through the filter were stained and counted using a light microscope. Experiments were repeated in triplicate. Bars represent the standard deviations. *p < 0.001 compared with control cells; **P < 0.005 compared with control cells. (B) Cells were treated with the indicated doses of crizotinib alone or a combination of crizotinib and XL‐880 (1 μM). CT, control; PHA, PHA‐665752; XL, XL‐880.
{kind=link}
Acknowledgments
This study was supported by a research grant (2012‐01) from Korean Association for the Study of Lung Cancer and a grant (2013‐563) from the Asan Institute for Life Sciences, Seoul, Korea.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.08.001.
Kim Hyeong Ryul, Kim Woo Sung, Choi Yun Jung, Choi Chang Min, Rho Jin Kyung, Lee Jae Cheol, (2013), Epithelial‐mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4‐ALK translocation, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.08.001.
Contributor Information
Jin Kyung Rho, Email: jkrho@amc.seoul.kr.
Jae Cheol Lee, Email: jclee@amc.seoul.kr.
References
- Bell, D.W. , Gore, I. , Okimoto, R.A. , Godin-Heymann, N. , Sordella, R. , Mulloy, R. , Sharma, S.V. , Brannigan, B.W. , Mohapatra, G. , Settleman, J. , Haber, D.A. , 2005. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet.. 37, 1315–1316. [DOI] [PubMed] [Google Scholar]
- Bergethon, K. , Shaw, A.T. , Ou, S.H. , Katayama, R. , Lovly, C.M. , McDonald, N.T. , Massion, P.P. , Siwak-Tapp, C. , Gonzalez, A. , Fang, R. , Mark, E.J. , Batten, J.M. , Chen, H. , Wilner, K.D. , Kwak, E.L. , Clark, J.W. , Carbone, D.P. , Ji, H. , Engelman, J.A. , Mino-Kenudson, M. , Pao, W. , Iafrate, A.J. , 2012. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol.. 30, 863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael, J. , DeGraff, W.G. , Gazdar, A.F. , Minna, J.D. , Mitchell, J.B. , 1987. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res.. 47, 936–942. [PubMed] [Google Scholar]
- Chen, Z. , Sasaki, T. , Tan, X. , Carretero, J. , Shimamura, T. , Li, D. , Xu, C. , Wang, Y. , Adelmant, G.O. , Capelletti, M. , Lee, H.J. , Rodig, S.J. , Borgman, C. , Park, S.I. , Kim, H.R. , Padera, R. , Marto, J.A. , Gray, N.S. , Kung, A.L. , Shapiro, G.I. , Janne, P.A. , Wong, K.K. , 2010. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res.. 70, 9827–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou, S.H. , Wang, M.L. , Chou, Y.T. , Chen, C.J. , Hong, C.F. , Hsieh, W.J. , Chang, H.T. , Chen, Y.S. , Lin, T.W. , Hsu, H.S. , Wu, C.W. , 2010. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res.. 70, 10433–10444. [DOI] [PubMed] [Google Scholar]
- Choi, Y.L. , Soda, M. , Yamashita, Y. , Ueno, T. , Takashima, J. , Nakajima, T. , Yatabe, Y. , Takeuchi, K. , Hamada, T. , Haruta, H. , Ishikawa, Y. , Kimura, H. , Mitsudomi, T. , Tanio, Y. , Mano, H. , Group, A.L.K.L.C.S. , 2010. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med.. 363, 1734–1739. [DOI] [PubMed] [Google Scholar]
- Christensen, J.G. , Zou, H.Y. , Arango, M.E. , Li, Q. , Lee, J.H. , McDonnell, S.R. , Yamazaki, S. , Alton, G.R. , Mroczkowski, B. , Los, G. , 2007. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther.. 6, 3314–3322. [DOI] [PubMed] [Google Scholar]
- Chung, J.H. , Rho, J.K. , Xu, X. , Lee, J.S. , Yoon, H.I. , Lee, C.T. , Choi, Y.J. , Kim, H.R. , Kim, C.H. , Lee, J.C. , 2011. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer. 73, 176–182. [DOI] [PubMed] [Google Scholar]
- Dave, B. , Mittal, V. , Tan, N.M. , Chang, J.C. , 2012. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res.. 14, 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck, R. , Zhang, Y.E. , 2003. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 425, 577–584. [DOI] [PubMed] [Google Scholar]
- Doebele, R.C. , Pilling, A.B. , Aisner, D.L. , Kutateladze, T.G. , Le, A.T. , Weickhardt, A.J. , Kondo, K.L. , Linderman, D.J. , Heasley, L.E. , Franklin, W.A. , Varella-Garcia, M. , Camidge, D.R. , 2012. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res.. 18, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsuki, M. , Mimori, K. , Yokobori, T. , Ishi, H. , Beppu, T. , Nakamori, S. , Baba, H. , Mori, M. , 2010. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci.. 101, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , Weinberg, R.A. , 2009. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai, H. , Allen, J.T. , Mason, R.M. , Kamimura, T. , Zhang, Z. , 2005. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res.. 6, 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama, R. , Khan, T.M. , Benes, C. , Lifshits, E. , Ebi, H. , Rivera, V.M. , Shakespeare, W.C. , Iafrate, A.J. , Engelman, J.A. , Shaw, A.T. , 2011. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. U S A. 108, 7535–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama, R. , Shaw, A.T. , Khan, T.M. , Mino-Kenudson, M. , Solomon, B.J. , Halmos, B. , Jessop, N.A. , Wain, J.C. , Yeo, A.T. , Benes, C. , Drew, L. , Saeh, J.C. , Crosby, K. , Sequist, L.V. , Iafrate, A.J. , Engelman, J.A. , 2012. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med.. 4, 120ra117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.W. , Ahn, M.J. , Shi, Y.K. , De Pas, T.M. , Yang, P.C. , Riely, G.J. , Crino, L. , Evans, T.L. , Liu, X.Q. , Han, J.Y. , Salgia, R. , Moro-Sibilot, D. , Ou, S.H.I. , Gettinger, S.N. , Wu, Y.L. , Lanzalone, S. , Polli, A. , Iyer, S. , Shaw, A.T. , 2012. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J. Clin. Oncol.. 30, [Google Scholar]
- Kwak, E.L. , Bang, Y.J. , Camidge, D.R. , Shaw, A.T. , Solomon, B. , Maki, R.G. , Ou, S.H. , Dezube, B.J. , Janne, P.A. , Costa, D.B. , Varella-Garcia, M. , Kim, W.H. , Lynch, T.J. , Fidias, P. , Stubbs, H. , Engelman, J.A. , Sequist, L.V. , Tan, W. , Gandhi, L. , Mino-Kenudson, M. , Wei, G.C. , Shreeve, S.M. , Ratain, M.J. , Settleman, J. , Christensen, J.G. , Haber, D.A. , Wilner, K. , Salgia, R. , Shapiro, G.I. , Clark, J.W. , Iafrate, A.J. , 2010. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med.. 363, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.M. , Dedhar, S. , Kalluri, R. , Thompson, E.W. , 2006. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol.. 172, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. , Blain, S.W. , Lo, R.S. , 2000. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 103, 295–309. [DOI] [PubMed] [Google Scholar]
- Massague, J. , Chen, Y.G. , 2000. Controlling TGF-beta signaling. Genes Dev.. 14, 627–644. [PubMed] [Google Scholar]
- Morris, S.W. , Kirstein, M.N. , Valentine, M.B. , Dittmer, K.G. , Shapiro, D.N. , Saltman, D.L. , Look, A.T. , 1994. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 263, 1281–1284. [DOI] [PubMed] [Google Scholar]
- O'Hare, T. , Eide, C.A. , Deininger, M.W. , 2007. Bcr-Abl kinase domain mutations and the unsettled problem of Bcr-AblT315I: looking into the future of controlling drug resistance in chronic myeloid leukemia. Clin. Lymphoma. Myeloma. 7, (Suppl. 3) S120–S130. [DOI] [PubMed] [Google Scholar]
- Rho, J.K. , Choi, Y.J. , Lee, J.K. , Ryoo, B.Y. , Na, I.I. , Yang, S.H. , Kim, C.H. , Lee, J.C. , 2009. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 63, 219–226. [DOI] [PubMed] [Google Scholar]
- Rikova, K. , Guo, A. , Zeng, Q. , Possemato, A. , Yu, J. , Haack, H. , Nardone, J. , Lee, K. , Reeves, C. , Li, Y. , Hu, Y. , Tan, Z. , Stokes, M. , Sullivan, L. , Mitchell, J. , Wetzel, R. , Macneill, J. , Ren, J.M. , Yuan, J. , Bakalarski, C.E. , Villen, J. , Kornhauser, J.M. , Smith, B. , Li, D. , Zhou, X. , Gygi, S.P. , Gu, T.L. , Polakiewicz, R.D. , Rush, J. , Comb, M.J. , 2007. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 131, 1190–1203. [DOI] [PubMed] [Google Scholar]
- Sang, J. , Acquaviva, J. , Friedland, J.C. , Smith, D.L. , Sequeira, M. , Zhang, C. , Jiang, Q. , Xue, L. , Lovly, C.M. , Jimenez, J.P. , Shaw, A.T. , Doebele, R.C. , He, S. , Bates, R.C. , Camidge, D.R. , Morris, S.W. , El-Hariry, I. , Proia, D.A. , 2013. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov.. 3, 430–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, T. , Koivunen, J. , Ogino, A. , Yanagita, M. , Nikiforow, S. , Zheng, W. , Lathan, C. , Marcoux, J.P. , Du, J. , Okuda, K. , Capelletti, M. , Shimamura, T. , Ercan, D. , Stumpfova, M. , Xiao, Y. , Weremowicz, S. , Butaney, M. , Heon, S. , Wilner, K. , Christensen, J.G. , Eck, M.J. , Wong, K.K. , Lindeman, N. , Gray, N.S. , Rodig, S.J. , Janne, P.A. , 2011. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res.. 71, 6051–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner, P. , 2010. The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol.. 21, (Suppl. 7) vii89–vii92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai, A. , Chandarlapaty, S. , Greulich, H. , Gonen, M. , Ye, Q. , Arteaga, C.L. , Sellers, W. , Rosen, N. , Solit, D.B. , 2008. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res.. 68, 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel, C. , Weinberg, R.A. , 2012. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin. Cancer Biol.. 22, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist, L.V. , Waltman, B.A. , Dias-Santagata, D. , Digumarthy, S. , Turke, A.B. , Fidias, P. , Bergethon, K. , Shaw, A.T. , Gettinger, S. , Cosper, A.K. , Akhavanfard, S. , Heist, R.S. , Temel, J. , Christensen, J.G. , Wain, J.C. , Lynch, T.J. , Vernovsky, K. , Mark, E.J. , Lanuti, M. , Iafrate, A.J. , Mino-Kenudson, M. , Engelman, J.A. , 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med.. 3, 75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, A.T. , Yeap, B.Y. , Mino-Kenudson, M. , Digumarthy, S.R. , Costa, D.B. , Heist, R.S. , Solomon, B. , Stubbs, H. , Admane, S. , McDermott, U. , Settleman, J. , Kobayashi, S. , Mark, E.J. , Rodig, S.J. , Chirieac, L.R. , Kwak, E.L. , Lynch, T.J. , Iafrate, A.J. , 2009. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol.. 27, 4247–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura, T. , Li, D. , Ji, H. , Haringsma, H.J. , Liniker, E. , Borgman, C.L. , Lowell, A.M. , Minami, Y. , McNamara, K. , Perera, S.A. , Zaghlul, S. , Thomas, R.K. , Greulich, H. , Kobayashi, S. , Chirieac, L.R. , Padera, R.F. , Kubo, S. , Takahashi, M. , Tenen, D.G. , Meyerson, M. , Wong, K.K. , Shapiro, G.I. , 2008. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res.. 68, 5827–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A. , Settleman, J. , 2010. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 29, 4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda, M. , Choi, Y.L. , Enomoto, M. , Takada, S. , Yamashita, Y. , Ishikawa, S. , Fujiwara, S. , Watanabe, H. , Kurashina, K. , Hatanaka, H. , Bando, M. , Ohno, S. , Ishikawa, Y. , Aburatani, H. , Niki, T. , Sohara, Y. , Sugiyama, Y. , Mano, H. , 2007. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 448, 561–566. [DOI] [PubMed] [Google Scholar]
- Soda, M. , Takada, S. , Takeuchi, K. , Choi, Y.L. , Enomoto, M. , Ueno, T. , Haruta, H. , Hamada, T. , Yamashita, Y. , Ishikawa, Y. , Sugiyama, Y. , Mano, H. , 2008. A mouse model for EML4-ALK-positive lung cancer. Proc. Natl. Acad. Sci. U S A. 105, 19893–19897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon, B. , Varella-Garcia, M. , Camidge, D.R. , 2009. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J. Clin. Oncol.. 4, 1450–1454. [DOI] [PubMed] [Google Scholar]
- Takeuchi, K. , Choi, Y.L. , Togashi, Y. , Soda, M. , Hatano, S. , Inamura, K. , Takada, S. , Ueno, T. , Yamashita, Y. , Satoh, Y. , Okumura, S. , Nakagawa, K. , Ishikawa, Y. , Mano, H. , 2009. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res.. 15, 3143–3149. [DOI] [PubMed] [Google Scholar]
- Thomson, S. , Buck, E. , Petti, F. , Griffin, G. , Brown, E. , Ramnarine, N. , Iwata, K.K. , Gibson, N. , Haley, J.D. , 2005. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res.. 65, 9455–9462. [DOI] [PubMed] [Google Scholar]
- Togashi, Y. , Soda, M. , Sakata, S. , Sugawara, E. , Hatano, S. , Asaka, R. , Nakajima, T. , Mano, H. , Takeuchi, K. , 2012. KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 7, e31323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikis, H. , Sato, M. , James, M. , Wang, D. , Wang, Y. , Wang, M. , Jia, D. , Liu, Y. , Bailey-Wilson, J.E. , Amos, C.I. , Pinney, S.M. , Petersen, G.M. , de Andrade, M. , Yang, P. , Wiest, J.S. , Fain, P.R. , Schwartz, A.G. , Gazdar, A. , Gaba, C. , Rothschild, H. , Mandal, D. , Kupert, E. , Seminara, D. , Viswanathan, A. , Govindan, R. , Minna, J. , Anderson, M.W. , You, M. , 2007. EGFR-T790M is a rare lung cancer susceptibility allele with enhanced kinase activity. Cancer Res.. 67, 4665–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, D.W. , Leung, E.L. , So, K.K. , Tam, I.Y. , Sihoe, A.D. , Cheng, L.C. , Ho, K.K. , Au, J.S. , Chung, L.P. , Pik Wong, M. , University of Hong Kong Lung Cancer Study, G., 2009. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 115, 1723–1733. [DOI] [PubMed] [Google Scholar]
- Wrighton, K.H. , Lin, X. , Feng, X.H. , 2008. Critical regulation of TGFbeta signaling by Hsp90. Proc. Natl. Acad. Sci. United State. America. 105, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch, R.L. , Januario, T. , Eberhard, D.A. , Cavet, G. , Zhu, W. , Fu, L. , Pham, T.Q. , Soriano, R. , Stinson, J. , Seshagiri, S. , Modrusan, Z. , Lin, C.Y. , O'Neill, V. , Amler, L.C. , 2005. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res.. 11, 8686–8698. [DOI] [PubMed] [Google Scholar]
- Zavadil, J. , Bottinger, E.P. , 2005. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 24, 5764–5774. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Lee, J.C. , Lin, L. , Olivas, V. , Au, V. , LaFramboise, T. , Abdel-Rahman, M. , Wang, X. , Levine, A.D. , Rho, J.K. , Choi, Y.J. , Choi, C.M. , Kim, S.W. , Jang, S.J. , Park, Y.S. , Kim, W.S. , Lee, D.H. , Lee, J.S. , Miller, V.A. , Arcila, M. , Ladanyi, M. , Moonsamy, P. , Sawyers, C. , Boggon, T.J. , Ma, P.C. , Costa, C. , Taron, M. , Rosell, R. , Halmos, B. , Bivona, T.G. , 2012. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet.. 44, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Figure S1. Inhibition of AXL reduced cellular mobility in H2228/CR cells, but was not associated with sensitivity to crizotinib. (A) H2228/CR cells were seeded onto either collagen‐ or matrigel‐coated polycarbonate filters to determine their migratory and invasive potentials, respectively. Cells were treated with or without 1 μM PHA‐665752 (MET inhibitor) or 1 μM XL‐880 (dual MET and AXL inhibitor) for 24 h in modified Boyden chambers. Cells that penetrated through the filter were stained and counted using a light microscope. Experiments were repeated in triplicate. Bars represent the standard deviations. *p < 0.001 compared with control cells; **P < 0.005 compared with control cells. (B) Cells were treated with the indicated doses of crizotinib alone or a combination of crizotinib and XL‐880 (1 μM). CT, control; PHA, PHA‐665752; XL, XL‐880.