

Abstract
CUB‐domain‐containing‐protein‐1 (CDCP1) is an integral membrane protein whose expression is up‐regulated in various cancer types. Although high CDCP1 expression has been correlated with poor prognosis in lung, breast, pancreas, and renal cancer, its functional role in tumor formation or progression is incompletely understood. So far it has remained unclear, whether CDCP1 is a useful target for antibody therapy of cancer and what could be a desired mode of action for a therapeutically useful antibody. To shed light on these questions, we have investigated the cellular effects of a therapeutic antibody candidate (RG7287). In focus formation assays, prolonged RG7287 treatment prevented the loss of contact inhibition caused by co‐transformation of NIH3T3 cells with CDCP1 and Src. In a xenograft study, MCF7 cells stably overexpressing CDCP1 reached the predefined tumor volume faster than the parental MCF7 cells lacking endogenous CDCP1. This tumor growth advantage was abolished by RG7287 treatment. In vitro, RG7287 induced rapid tyrosine phosphorylation of CDCP1 by Src, which was accompanied by translocation of CDCP1 to a Triton X‐100 insoluble fraction of the plasma membrane. Triggering these effects required bivalency of the antibody suggesting that it involves CDCP1 dimerization or clustering. However, this initial activation of CDCP1 was only transient and prolonged RG7287 treatment induced internalization and down‐regulation of CDCP1 in different cancer cell lines. Antibody stimulated CDCP1 degradation required Src activity and was proteasome dependent. Also in three different xenograft models with endogenous CDCP1 expression RG7287 treatment resulted in significant tumor growth inhibition concomitant with substantially reduced CDCP1 levels as judged by immunohistochemistry and Western blotting. Thus, despite transiently activating CDCP1 signaling, the RG7287 antibody has a therapeutically useful mode of action.
Keywords: CDCP1, Src, Antibody therapy, Contact inhibition
Highlights
Antibody RG7287 induces CDCP1 clustering, tyrosine phosphorylation and downregulation.
RG7287 blocks transformation of NIH3T3 cells induced by Src and CDCP1 co‐transduction.
Stable overexpression of CDCP1 in MCF7 cells leads to faster growing xenograft tumors.
RG7287 treatment inhibits tumor growth in three different xenograft models.
1. Introduction
CUB‐domain‐containing‐protein‐1 (CDCP1) is a highly glycosylated, single pass type I transmembrane protein of 140 kD originally found due to its up‐regulation and tyrosine phosphorylation in breast, colorectal, and lung cancer (Hooper et al., 2003, 2009, 2006, 2001). In an analysis of 200 lung adenocarcinoma patients, high expression of CDCP1 correlated with higher relapse rate, poor prognosis, and lymph node metastasis (Ikeda et al., 2009). A study on pancreatic cancer also found that patients with high CDCP1 expression levels had a lower overall survival rate (Miyazawa et al., 2010). For renal clear cell carcinoma, CDCP1 up‐regulation was reported to occur in about 33% of 230 cases analyzed by immunohistochemistry (Awakura et al., 2008). Moreover, high CDCP1 levels correlated with indicators of advancing disease and predicted a poor prognosis during a median follow‐up time of 45 months. The molecular basis of such correlations is presently unclear, but could be related to the fact that CDCP1 has been found to promote cell migration, invasion, and tumor cell dissemination (Deryugina et al., 2009; Uekita et al., 2008). In agreement with this, a functional role of CDCP1 in mediating anoikis‐resistance (Uekita et al., 2007) and degradation of extracellular matrix (Miyazawa et al., 2010) has been reported. However, the poor prognosis of cancer patients with high CDCP1 expression levels could also be related to a higher proliferative capacity of their tumors. In a small study with 25 breast cancer patients, Ikeda et al. (2006) found that the CDCP1‐high cases also had higher levels of the proliferation‐associated Ki67 antigen than the CDCP1‐low cases.
CDCP1 consists of a large extracellular domain (ECD), which contains three CUB (Complement C1r/C1s, Uegf, Bmp1) domains, and a short intracellular portion (Scherl‐Mostageer et al., 2001). Little is known about the functional role of the ECD of CDCP1. The intracellular domain of CDCP1 has five potential tyrosine phosphorylation sites. Tyrosine734 (Y) of CDCP1 is the main phosphorylation site for Src family kinases (SFKs) (Brown et al., 2004). Phosphorylation of this site enables SH2 domain mediated SFK binding to CDCP1, which promotes further phosphorylation at other tyrosine residues (e.g. Y743 and Y762) creating docking sites for other interacting proteins like PKCδ (Benes et al., 2005). Moreover, tyrosine phosphorylation of Y734 provides a scaffold for Src‐mediated phosphorylation of PKCδ and this is required for the migration and invasion effects of CDCP1 (Miyazawa et al., 2010). Overall our understanding of downstream signaling events of CDCP1 is incomplete.
The physiological and pathophysiological stimuli that trigger tyrosine phosphorylation of CDCP1 in normal and cancer cells, respectively, are also poorly understood. CDCP1 seems to be part of an outside‐in signaling mechanism triggered by detachment and this involves clustering of CDCP1 in special membrane subdomains. Alvares et al. (2008) found that a certain activating anti‐CDCP1 antibody (ActGp140 mAb) can mimic this process. Addition of this antibody resulted in translocation of CDCP1 to the detergent resistant membrane fraction (DRM), and association with lipid raft components. The resulting close proximity to SFKs within lipid rafts appears to promote CDCP1 phosphorylation. However, besides antibody ligation and translocation of CDCP1 to DRMs, a parallel signal from cell adhesion is necessary, since ActGp140 mAb treatment of cells in suspension failed to induce CDCP1 phosphorylation. Moreover, not all anti‐CDCP1 antibodies induce its phosphorylation (Alvares et al., 2008). The fact that there are activating and non‐activating anti‐CDCP1 antibodies indicates that clustering induced phosphorylation has certain spatial or conformational requirements.
Whether CDCP1 is a useful target for antibody therapy of cancer has remained unclear due to the incomplete understanding of its functional role in tumorigenesis and progression as well as the unclear molecular details of its activation and downstream signaling. Moreover, some anti‐CDCP1 antibodies can promote its phosphorylation, and thus could potentially promote tumor growth and survival.
In this study, we investigated the mode of action of RG7287, an activating anti‐CDCP1 antibody, and show that it is a therapeutically useful antibody. In focus formation assays, RG7287 prevents loss of contact inhibition induced by the coexpression of Src and CDCP1. Additionally, in a time to event study mice that were inoculated with MCF7 cells overexpressing CDCP1 reached a predefined tumor volume faster than parental MCF7 cells, and more importantly mice bearing tumors of the CDCP1 overexpressing MCF7 cells that were treated with RG7287 reached the predefined tumor volume concurrently with the tumors of the parental MCF7 cells. In vitro, addition of RG7287 triggered a rapid increase in CDCP1 associated Src activity, translocation of CDCP1 to lipid rafts, and concomitantly CDCP1 phosphorylation. We show here that Src activity is required not only for CDCP1 phosphorylation, but also for its translocation to DRMs. Prolonged RG7287 treatment resulted in Src‐ and proteasome‐dependent down modulation of CDCP1. Moreover, RG7287 inhibited tumor growth in three mouse xenograft models in vivo. We suggest that this down‐regulation of CDCP1 is the underlying mode of action by which the RG7287 antibody showed efficacy.
2. Materials and methods
2.1. Cell culture, expression vectors, and antibodies
NCI‐H322M (NCI) and MCF7 (NCI) cells were grown in RPMI1640 medium, NIH‐3T3 and GP + E86 in DMEM, containing 2 mM Glutamine and 10% FCS (Invitrogen) at 37 °C with 5% CO2.
CDCP1 and Src cDNAs were cloned into the pLXSN retroviral expression vector (Clontech). CDCP1 cloned into pcDNA3.1 was used to stably express CDCP1 in MCF7 cells. Focus formation assays were performed as described before (Kapp et al., 2007).
Generation of the original mouse CDCP1 antibody, RG7287, has been previously described (Buhring et al., 2004). This antibody and its humanized version bind huCDCP1 with single digit nanomolar affinity (1.2 × 10−9) as measured by surface plasmon resonance. The Fab fragment of RG7287 was prepared by papain cleavage. Phospho‐CDCP1 was detected with a phospho‐specific rabbit monoclonal anti‐CDCP1 antibody raised against a peptide containing phosphorylated Y734. CDCP1, Src, phospho‐Src family kinases (Y416), and flotillin antibodies were obtained from Cell Signaling. LAMP1 and tubulin antibodies were purchased from Abcam and anti‐CD71 antibody was from Santa Cruz. For immunohistochemistry, anti‐CDCP1 antibody (MAB2666) from R&D Systems was used.
2.2. Inhibitors
Epoxomicin, PP2 and P3 were from Calbiochem and the Src Inhibitor No.5 (Sino5) was from Biaffin.
2.3. Western blotting
If cell fractionation was not required, RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 5 mM EDTA, and 0.1% SDS) was used for lysis. Proteins were separated and blotted using the NuPage and iBlot systems from Invitrogen and detected with enhanced chemiluminescence (Roche).
2.4. Preparation of DRM fractions
All steps were carried out on ice. Cells were washed with ice‐cold TBS (50 mM Tris–HCL, 150 mM NaCl pH 7.5) and lysed in TBS containing 1% Triton X‐100, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, and Halt phosphatase inhibitor for 30 min. Lysates were collected with a rubber policeman and homogenized with a Potter‐Elvehjem PTFE homogenizer. Cell debris was cleared by centrifugation at 1000 × g for 10 min. DRMs were precipitated at 20,800 × g for 30 min. The pellet was washed twice with lysis buffer, denatured with reducing NuPage sample buffer, and subjected to three cycles of boiling and snap freezing in liquid nitrogen.
2.5. Antibody internalization
H322M cells were incubated with RG7287 or an isotype control antibody for the indicated time periods at 37 °C or 4 °C for 30 min for the non‐internalizing control. After incubation, the cells were detached from the plates with Accutase (SIGMA) and counted. 1 × 106 cells were incubated with phycoerythrin (PE) labeled anti‐CDCP1 antibody (Biozol) for 30 min on ice. Cells were washed, resuspended in FACS buffer, and analyzed with FacsCanto. The percent internalization was calculated as follows: (geo meannon‐internalizing − geo meantreated)/geo meannon‐internalizing.
2.6. Immunocytochemistry
H322M cells were seeded onto sterile glass coverslips pre‐coated with FCS. Detection antibodies were diluted in blocking buffer (PBS with 5% FCS) and all washes were done 3× with PBS. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X‐100 in PBS for 5 min and blocked for 1 h. The coverslips were incubated with primary and secondary antibodies overnight and 1 h, respectively. The coverslips were mounted with ProLong® Gold reagent (Invitrogen). Fluorescence images were taken with an Olympus BX60 Microscope using an UPlanF1 100×/1.30OilPh3 objective.
2.7. Xenograft studies
In all studies, tumors cells were subcutaneously inoculated into the right flank of female SCID beige mice (Charles River Germany). MCF7 tumors were established with 1 × 107 cells (n = 9–10). When 200 mm3 was reached, treatment of vehicle or RG7287 (10 mg/kg) began and once weekly dosing continued until the animals were sacrificed upon reaching a pre‐defined volume of 1000 mm3.
In the other xenograft studies, animals were randomized into treatment and vehicle groups (n = 10), when the median tumor volume in each group was ≈175 mm3. Animals were treated once weekly for the indicated number of weeks with intraperitoneal injections of either vehicle or RG7287 antibody. Six days after the last treatment, animals were sacrificed. Tumors were excised, weighed, and analyzed.
2.8. Immunohistochemistry
Excised xenograft tumors were stained on Ventana Benchmark‐XT using anti‐CDCP1 (MAB2666) antibody (0.8 μg/ml).
2.9. Tumor lysates
Tumors were excised and dissociated with a scalpel in a petri dish on ice. 2 ml of Triton X‐100 lysis buffer were added per gram of tumor tissue and the tumor was further processed until completely homogenized. After incubation on ice for another 30 min, cell debris was removed by centrifugation for 30 min at 20,800 × g.
3. Results
3.1. RG7287 treatment blocks co‐transformation of NIH3T3 cells by CDCP1 and Src
We had previously found that co‐infecting NIH3T3 cells with retroviral constructs for CDCP1 and Src induced frequent loss of contact inhibition in focus formation assays (Kollmorgen et al., 2012). As shown in Figure 1A, infection of NIH3T3 cells with retroviruses encoding either only Src or only CDCP1 resulted in the formation of 15 foci or no foci at all, respectively. In contrast, co‐infection with both CDCP1 and Src encoding viruses resulted in formation of more than 300 foci clearly demonstrating the co‐transforming potential of these two gene products. Since CDCP1 is highly expressed in many tumors and has co‐transforming potential, we hypothesized that targeting CDCP1 could be therapeutically beneficial. To this end, we generated an antibody against the extracellular domain of CDCP1 (RG7287). In focus formation assays of NIH3T3 cells co‐infected with CDCP1 and Src where 5 μg/ml of RG7287 added to the medium at all times, the formation of foci was completely inhibited. One of the concerns when using a bivalent receptor targeted antibody is potential activation due to dimerization of the receptor. To elucidate the extent to which the bivalency plays a role in the prevention of focus formation, we generated a Fab fragment of RG7287. Treating the Src and CDCP1 infected NIH3T3 cells with equivalent amount of the monomeric Fab only inhibited the foci formation by 40% (Figure 1B), demonstrating that the bivalency of RG7287 is an essential part of its mode of action. In summary, co‐infection of NIH3T3 cells with CDCP1 and Src led to loss of contact inhibition, but addition of RG7287 to the medium throughout the duration of the assay prevented cell transformation and focus formation.
Figure 1.
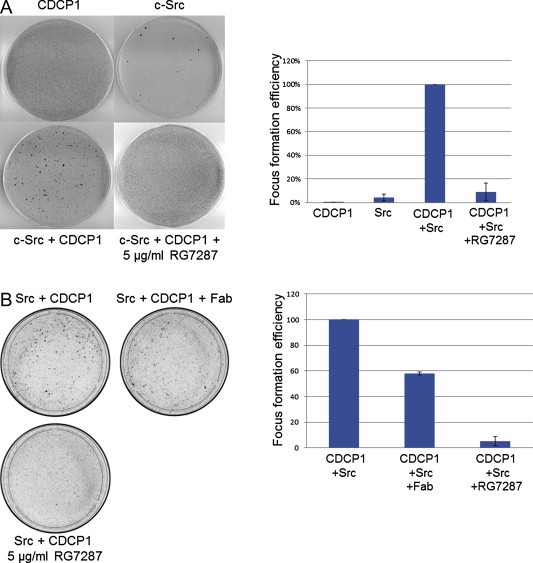
CDCP1‐Src co‐transformation is prevented by RG7287 treatment. 5 × 104 NIH3T3 cells were either infected singly with retroviruses (105 virus particles) containing CDCP1 and Src or with both together. After 48 h the cells were expanded to a 10 cm plate and kept in 4% FCS containing medium and either left untreated or incubated with 5 μg/ml of RG7287 antibody (A) and with equivalent amounts of the Fab from RG7287 (B). After 3 weeks, cells were stained with crystal violet and the number of foci was counted. The foci that formed in three separate experiments were counted. The focus formation efficiency in each experiment was calculated by normalizing to the untreated Src and CDCP1 co‐infected plates as 100%. The error bars indicate standard deviation.
3.2. MCF7 cells overexpressing CDCP1 reach predefined tumor volume faster than parental MCF7 cells
In order to investigate the direct effect of CDCP1 expression, MCF7, a cell line lacking endogenous CDCP1, was stably transfected with CDCP1. The parental and the CDCP1 overexpressing cells were compared in subcutaneous xenograft models. As seen in Figure 2, tumor volume of 1000 mm3 was achieved much faster in the MCF7 cells overexpressing CDCP1 compared to the parental MCF7 cells. Moreover, the animals with established tumors (200 mm3) from the MCF7‐CDCP1 cells that were treated with RG7287 showed tumor growth comparable to the parental cell line (RG7287 had no effect on wild‐type MCF7 xenograft tumors, data not shown), indicating that CDCP1 overexpression impacts tumor cell aggressiveness. And finally, supports the hypothesis that targeting CDCP1 with a therapeutic antibody will lead to decreased tumor growth.
Figure 2.

CDCP1 overexpressing MCF7 cells reach a predefined tumor volume faster than wild type MCF7 cells. Subcutaneous tumors were generated by injecting 1 × 107 cells into the flank of SCID beige mice, systemic estrogen was supplemented. The Kaplan–Meier curves compare the times to reach the pre‐defined tumor volume of 1000 mm3 between animals with established CDCP1 overexpressing and parental MCF7 tumors (n = 9–10). The time to reach 1000 mm3, is significantly shorter in CDCP1‐overexpressing MCF7 tumors (p = 0.0065) compared to wild type MCF7 tumors (33 days versus 42 days). RG7287 treatment of CDCP1‐overexpressing MCF7 tumors reversed this feature back to that of the wild type (41 days; p = 0.034). Time to event was analyzed using the pair‐wise log‐rank test.
3.3. RG7287 treatment triggers an initial phosphorylation of CDCP1
In order to further understand how RG7287 inhibits focus formation and slows the tumor growth in the MCF7 model overexpressing CDCP1, we investigated the effect of RG7287 antibody treatment in vitro. Initial experiments indicated that short incubation of cells with RG7287 triggered phosphorylation of CDCP1 on residue Y734 and caused CDCP1 to move to the Triton‐X100 insoluble fraction. To investigate the kinetics of these events, H322M cells were incubated with either RG7287 or an isotype control antibody and levels of total and phospho‐CDCP1 were analyzed by Western blot at various time points. As shown in Figure 3A, CDCP1 became phosphorylated within 1 min and concomitantly translocated to the DRM fraction. Peak levels of total and phospho‐CDCP1 in the insoluble fraction were reached at 20 min post stimulation and remained high for at least 1 h. To test whether bivalency of RG7287 was required for these effects, the same time course was performed using only the antigen‐binding fragment (Fab) of the same antibody (Figure 3B). This monovalent binder neither stimulated CDCP1 phosphorylation nor induced translocation of CDCP1 to a detergent insoluble membrane compartment indicating that these responses are triggered by antibody mediated dimerization or clustering of CDCP1. On the contrary, the Fab treatment reduced basal levels of phospho‐CDCP1 and in a time‐dependent fashion diminished total CDCP1 amount in the insoluble fraction (Figure 3B).
Figure 3.

Ligation with RG7287 antibody translocates CDCP1 to detergent insoluble fraction. H322M cells were incubated with either 10 μg/ml of control IgG or (A) RG7287 antibody or (B) the Fab fragment of RG7287 for the indicated times. After lysis in 1% Triton X‐100 buffer, the insoluble fraction was prepared and analyzed by Western blotting. The insoluble fraction was isolated and analyzed by Western blotting with antibodies against phospho‐ and total CDCP1, phospho‐ and total Src, the lipid raft marker protein flotillin.
3.4. Src inhibition prevents CDCP1 translocation to the insoluble fraction
CDCP1 is a known Src substrate and its partitioning into DRMs upon RG7287 binding could trigger phosphorylation by raft‐associated SFKs. The time course did not allow us to discriminate whether CDCP1 phosphorylation triggered its translocation to the insoluble fraction or vice versa. Therefore, we determined the effect of Src inhibitors on CDCP1 translocation. Pre‐incubation of H322M cells with the Src inhibitors PP2 and Sino5 for 1 h abrogated RG7287 stimulated CDCP1 tyrosine phosphorylation as expected, while the control PP3 had no effect (Figure 4). Blocking CDCP1 phosphorylation with Src inhibitors also prevented its translocation to the insoluble fraction as shown by the absence of total and phospho‐CDCP1 bands in insoluble fractions from PP2 and Sino5 pre‐treated cells, but not from the control samples. In contrast to CDCP1, the presence of Src in the insoluble fraction was unaffected by treatment with Src inhibitors. Thus, Src activity is not only required for CDCP1 phosphorylation but also for its translocation to the insoluble fraction. RG7287 mediated dimerization/clustering of CDCP1 triggers its phosphorylation by Src, which in turn mediates CDCP1 translocation to lipid rafts.
Figure 4.
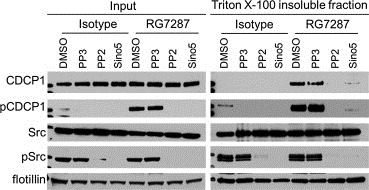
CDCP1 translocation to a detergent insoluble fraction is Src dependent. Prior to a 15 min incubation with 10 μg/ml RG7287 antibody, H322M cells were pre‐treated for 1 h with vehicle or the following compounds: Sino5 (4 μM), PP2, or PP3 (20 μM). The insoluble fraction was prepared and analyzed by Western blotting.
3.5. Prolonged RG7287 treatment down‐regulates CDCP1 protein levels
Since RG7287 seems to act like a CDCP1 agonist and activation of transmembrane signal transduction often leads to subsequent down‐regulation of the signal conveying receptors, we investigated the effect of prolonged RG7287 treatment on CDCP1 protein levels. H322M, MDA‐MB‐231, Kpl‐4, HCT116, HT29, and SW620 cells were treated with RG7287 for 5 h, and cell lysates were blotted for total and phospho‐CDCP1 levels. Total CDCP1 level was strongly reduced after prolonged treatment with RG7287 versus the isotype control (Figure 5A). Thus, RG7287 induced CDCP1 phosphorylation is only a transient response that is followed within 5 h by receptor degradation. In order to determine which cellular process is mainly responsible for the observed down‐regulation of CDCP1, we treated H322M cells with either the proteasome inhibitor epoxomicin or the lysosome inhibitor chloroquine or with both prior to incubation with RG7287 antibody for 5 h. Epoxomicin pre‐treatment completely prevented RG7287 induced degradation of CDCP1, while chloroquine only partially blocked it (Figure 5B). Even when cells were not stimulated with RG7287, total CDCP1 levels were highest in lysates of cells treated with epoxomicin and chloroquine suggesting that both degradation pathways also play some role for steady‐state CDCP1 levels in non‐stimulated cells. While after 5 h stimulation with RG7287, phospho‐CDCP1 levels were barely detectable, chloroquine pre‐treatment resulted in slight and epoxomicin or combination treatment in massive accumulation of phospho‐CDCP1 (compare lanes 5, 7, and 9 with lane 3 in Figure 5B).
Figure 5.

RG7287 treatment downregulates CDCP1 in many different tumor cell lines. (A) H322M, MDA‐MB231, Kpl‐4, HCT116, HT29, and SW620 cells were incubated with isotype control or RG7287 (10 μg/ml) for 5 h. The cells were lysed with RIPA buffer and analyzed by Western blotting. (B) H322M cells were treated for 1 h with either epoxomicin (1 μM) or chloroquine (1 μM), then 10 μg/ml of RG7287 or the isotype control was added and further incubated for 6 h before lysis in RIPA buffer and analysis by immunoblotting. (C) H322M cells were incubated with 20 μg human RG7287 or isotype control antibodies for the indicated times at 37 °C. Using Accutase the cells were removed from the plate. For each time point 1 × 106 cells were incubated with phycoerythrin (PE) labeled anti‐CDCP1 antibody for 30 min on ice. The median fluorescence intensity (MFI) for the non‐internalized control sample that was incubated only with the PE labeled antibody on ice was set to 100%. Histograms of each treatment time point are shown in addition. (D) 2 × 105 H322M cells were seeded on FCS coated glass coverslips. 10 μg/ml of either human isotype control or RG7287 antibody was added for 5 h. The cells were fixed with 4% PFA, permeabilized and stained for CDCP1 and LAMP1.
FACS analysis confirmed that CDCP1 internalized significantly within 1 h of RG7287 treatment of H322M cells. Maximal internalization (60–70%) was achieved within 5 h and sustained for at least 24 h (Figure 5C). We also examined by immunofluorescence microscopy the subcellular localization of internalized CDCP1 after prolonged RG7287 stimulation of H322M cells, by staining for CDCP1 and the lysosome marker LAMP1. Control antibody treated cells showed predominantly plasma membrane staining for CDCP1, which was especially prominent at cell–cell junctions. There was no significant overlap with the intracellular, vesicular staining pattern of an anti‐LAMP1 antibody (Figure 5D). In contrast to this, cells treated with RG7287 antibody for 5 h showed no prominent cell surface staining for CDCP1 anymore, but rather an accumulation of CDCP1 in intracellular vesicles. A small fraction of these co‐stained for LAMP1. In summary, we found that after RG7287 stimulation activated CDCP1 is degraded primarily via the proteasomal pathway and to a lesser degree also via lysosomes.
3.6. Src activation is necessary for RG7287 mediated down‐regulation of CDCP1
To determine whether internalization and degradation of CDCP1 require Src kinase activity, we tested the effects of Src inhibitors. When cells were pre‐incubated with DMSO or PP3, total CDCP1 was slightly down‐regulated within 1 h of RG7287 treatment and substantially reduced after 5 h (compare lane 4 with 3 and 12 with 11 in Figure 6). Pre‐treatment with two different Src inhibitors completely blocked the effect of RG7287 on CDCP1 degradation (compare lane 8 with 7 and 16 with 15). Thus, transient activation of CDCP1 by RG7287 and Src mediated phosphorylation events are both required for RG7287 stimulated degradation of CDCP1.
Figure 6.

Src activation is required for RG7287 induced CDCP1 downregulation. Src inhibitors Sino5 (4 μM) and PP2 and the control compound PP3 (20 μM) were added to H322M cells for 1 h prior to the addition of RG7287 antibody (10 μg/ml) for either 1 h or 5 h. The cells were lysed in RIPA buffer and analyzed by immunoblotting.
3.7. RG7287 treatment impedes tumor growth in subcutaneous xenograft models
To prove that prolonged RG7287 treatment represents a therapeutically useful mode of action for cancer targeted therapy we investigated whether RG7287 treatment was able to suppress tumor growth in vivo in cancer cell line xenografts. We tested RG7287 antibody therapy in one lung and two breast carcinoma xenograft models: H322M, Kpl4, and MDA‐MB231. Treatment of established xenograft tumors with RG7287 decreased tumor volume compared to the control group in the H322M, Kpl4 and MDA‐MB231 xenografts by 82%, 77% and 71%, respectively, (Figure 7A) clearly demonstrating the therapeutic potential of this antibody. After study termination, tumors were excised and a substantial reduction of CDCP1 protein levels in RG7287‐treated tumors was demonstrated by immunohistochemistry. As shown in Figure 7B, membrane staining for CDCP1 was almost completely absent in RG7287 treated tumors. Western blot analysis confirmed that total CDCP1 levels were readily detectable in lysates of vehicle, but not RG7287 treated tumors (Figure 7C). Thus, antibody induced down‐regulation of CDCP1 appears to cause substantial tumor growth inhibition of different xenografts suggesting that this is a therapeutically useful approach.
Figure 7.
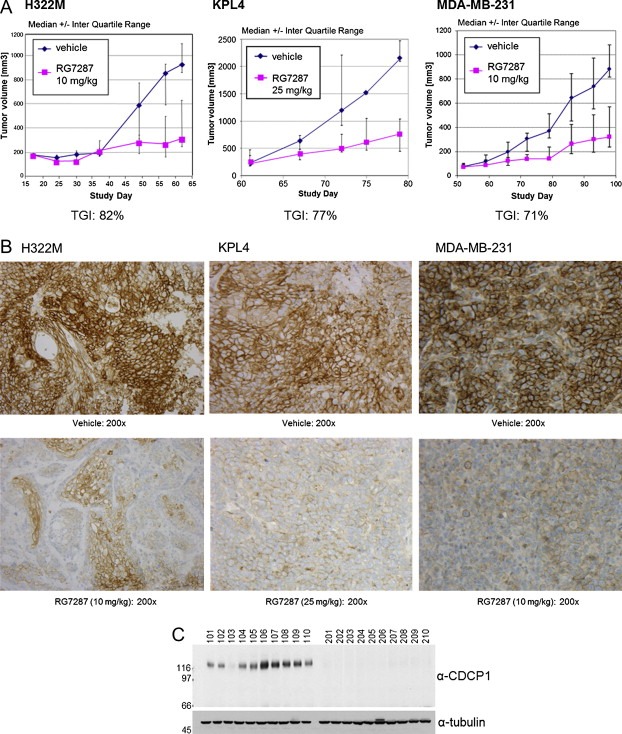
RG7287 inhibits tumor growth in subcutaneous xenograft models. Established subcutaneous H322M (innoculated with matrigel), Kpl4, and MDA‐MB231 (both innoculated without matrigel) tumors in SCID beige mice were treated once weekly (for 6, 3 and 7 weeks, respectively) with vehicle or 10 mg/kg of RG7287 antibody (25 mg/kg in case of Kpl4) and tumor volumes were measured with calipers. (A) The average tumor volume of vehicle and RG7287 treated H322M, Kpl4, and MDA‐MB231 xenografts models (n = 10) is plotted over time with standard deviation. (B) Tumors were explanted, fixed in 3.8% buffered formaldehyde solution, and embedded in Paraplast. CDCP1 was detected by immunohistochemistry using the mouse monoclonal antibody MAB2666. (C) Explanted tumors from H322M were lysed in Triton X‐100 lysis buffer. The tumor lysates were analyzed for CDCP1 levels and the loading control tubulin by Western blotting.
4. Discussion
Although up‐regulation of CDCP1 expression and tyrosine phosphorylation occurs in various cancers, its role in tumor development and progression is poorly understood. Early on CDCP1 was identified as a protein functionally involved in metastasis (Hooper et al., 2003) and was recently shown to promote invasion and peritoneal dissemination of gastric scirrhous carcinoma (Uekita et al., 2008). Also in highly metastatic pancreatic cells lines phospho‐CDCP1 levels are increased and regulate invasion, metastasis, and extracellular matrix degradation (Miyazawa et al., 2010).
Despite mounting evidence that CDCP1 represents a promising target for cancer targeted therapy, development of therapeutically useful antibodies is hampered by several issues: Firstly, CDCP1 is to varying extents also expressed on normal tissues where its physiological role is poorly understood. Secondly, although some interaction partners have been identified, understanding CDCP1 signaling still represents a significant challenge. Not even the physiological and pathophysiological activation mechanisms of CDCP1 signaling are fully characterized. Thirdly, this makes it difficult to define the desired mode of action for a therapeutically useful antibody. Finally, some antibodies against CDCP1 have been shown to stimulate CDCP1 phosphorylation (Alvares et al., 2008) raising concern that such agonistic antibodies could enhance tumor progression and metastasis. These difficulties have led Siva et al. (2008) to evaluate an immunotoxin approach with a saporin‐conjugated anti‐CDCP1 antibody in PC‐3 xenografts (Siva et al., 2008). While this antibody‐drug‐conjugate blocked metastasis irrespective of the route of administration, it only blocked primary tumor growth when given intravenously. Severe body weight loss limited the maximally applicable dose indicating acute, albeit reversible, systemic toxicity of the drug. Dose‐limiting toxicity is unlikely to occur with an unconjugated CDCP1 antibody that affects CDCP1 signaling by downregulating it from the cell surface.
In this study, we show that RG7287 is an agonistic antibody that, when continuously present, can strongly reduce the CDCP1 levels on the surface of cancer cells in vitro and in vivo. Its bivalency and ability to crosslink CDCP1 molecules is required for inducing Src‐dependent CDCP1 phosphorylation and translocation to the insoluble fraction. Translocation of CDCP1 into DRMs has also been observed for another agonistic anti‐CDCP1 antibody (Alvares et al., 2008). Although we cannot exclude the possibility that both antibodies displace an inhibitory molecule from the extracellular domain of CDCP1 or induce a conformational change that stabilizes CDCP1 complexes in lipid rafts, the lack of effect of a Fab fragment of RG7287 strongly suggests that the molecular basis of this agonistic behavior is antibody‐induced cross‐linking of CDCP1. Recently, Cooper and Qian (2008) proposed that dimerization provides a robust activation mechanism for SFK‐dependent transmembrane signaling by receptors that lack intrinsic catalytic activity, if the following conditions are met: i) the receptor has SFK phosphorylation sites, to which SFKs bind in a phosphorylation dependent manner ii) an SFK bound to one receptor can phosphorylate the second receptor or its associated SFK in a dimer, and iii) dephosphorylation occurs via an unregulated protein tyrosine phosphatase (PTP) (Kapp et al., 2007). Although presently nothing is known about the PTP involved, CDCP1 fulfills the first two conditions and we hypothesize that any bivalent anti‐CDCP1 antibody that does not sterically interfere with receptor dimerization will induce CDCP1 activation.
Interestingly, CDCP1 phosphorylation has been reported to occur in response to detachment of MCF10A cells (Spassov et al., 2009). Besides agonistic antibodies and cell detachment, protease cleavage in the extracellular region of CDCP1 has been found to induce CDCP1 phosphorylation (He et al., 2010). Since we primarily observed full length phospho‐p140 after RG7287 treatment of H322M cells, with little induction of the cleaved phospho‐p70 form, CDCP1 activation by RG7287 is not a secondary event to proteolytic cleavage of CDCP1.
Agonistic activity can be a viable mode of action for a therapeutic antibody, if a transient activation is followed by sustained and pronounced target down‐regulation. Prolonged in vitro exposure of H322M cells to RG7287 antibody resulted in massive reduction of total as well as cell surface CDCP1 levels. CDCP1 degradation is preceded by phosphorylation, since proteasome inhibition led to massive accumulation of RG7287‐induced tyrosine phosphorylation of CDCP1. As Src inhibitors also block CDCP1 degradation, phosphorylation of CDCP1 or another SFK substrate must be necessary. Internalization of CDCP1 may require SFK activity as previously shown for other cell surface proteins (Cao et al., 2010; Wilde et al., 1999). Potential therapeutic usefulness of RG7287 treatment is indicated by focus formation assays, in which CDCP1 and Src cooperate in overcoming contact inhibition of NIH3T3 cells. Despite its agonistic property, the net effect of prolonged RG7287 antibody treatment was prevention of focus formation. Furthermore, overexpression of CDCP1 in the MCF7 xenograft model decreased the predetermined time to event compared to the parental model, moreover treating the overexpressing model with RG7287 prevented this acceleration of tumor growth. Additionally, we analyzed the effect of RG7287 on 3 different mouse xenograft models. Tumor growth was reduced more than 70% in H322M, MDA‐MB‐231, and Kpl4 models, clearly demonstrating the therapeutic usefulness of this particular antibody and of CDCP1 as a target for antibody therapy in these tumors. We also observed significant, but less pronounced tumor growth inhibition in other xenograft models (e.g. ∼30% in BxPC3) suggesting that, not surprisingly, the dependence of tumors on CDCP1 protein levels and function varies. Successful targeting of CDCP1 for cancer therapy will profit from further improvements in our knowledge about its interaction with other proteins in normal and tumor biology. Such progress will greatly facilitate clinical development by hopefully allowing identification of those patients whose primary tumor growth depends most on CDCP1 signaling.
Kollmorgen Gwendlyn, Niederfellner Gerhard, Lifke Alexander, Spohn Gloria J., Rieder Natascha, Vega Harring Suzana, Bauss Frieder, Burtscher Helmut, Lammers Reiner, Bossenmaier Birgit, (2013), Antibody mediated CDCP1 degradation as mode of action for cancer targeted therapy, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.08.009.
References
- Alvares, S.M. , Dunn, C.A. , Brown, T.A. , Wayner, E.E. , Carter, W.G. , 2008. The role of membrane microdomains in transmembrane signaling through the epithelial glycoprotein Gp140/CDCP1. Biochim. Biophys. Acta. 1780, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awakura, Y. , Nakamura, E. , Takahashi, T. , Kotani, H. , Mikami, Y. , Kadowaki, T. , Myoumoto, A. , Akiyama, H. , Ito, N. , Kamoto, T. , Manabe, T. , Nobumasa, H. , Tsujimoto, G. , Ogawa, O. , 2008. Microarray-based identification of CUB-domain containing protein 1 as a potential prognostic marker in conventional renal cell carcinoma. J. Cancer Res. Clin. Oncol.. 134, 1363–1369. [DOI] [PubMed] [Google Scholar]
- Benes, C.H. , Wu, N. , Elia, A.E. , Dharia, T. , Cantley, L.C. , Soltoff, S.P. , 2005. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. 121, 271–280. [DOI] [PubMed] [Google Scholar]
- Brown, T.A. , Yang, T.M. , Zaitsevskaia, T. , Xia, Y. , Dunn, C.A. , Sigle, R.O. , Knudsen, B. , Carter, W.G. , 2004. Adhesion or plasmin regulates tyrosine phosphorylation of a novel membrane glycoprotein p80/gp140/CUB domain-containing protein 1 in epithelia. J. Biol. Chem.. 279, 14772–14783. [DOI] [PubMed] [Google Scholar]
- Buhring, H.J. , Kuci, S. , Conze, T. , G.Rathke, Bartolovic, K. , Grunebach, F. , Scherl-Mostageer, M. , Brummendorf, T.H. , Schweifer, N. , Lammers, R. , 2004. CDCP1 identifies a broad spectrum of normal and malignant stem/progenitor cell subsets of hematopoietic and nonhematopoietic origin. Stem Cells. 22, 334–343. [DOI] [PubMed] [Google Scholar]
- Cao, H. , Chen, J. , Krueger, E.W. , McNiven, M.A. , 2010. SRC-mediated phosphorylation of dynamin and cortactin regulates the “constitutive” endocytosis of transferrin. Mol. Cell Biol.. 30, 781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, J.A. , Qian, H. , 2008. A mechanism for SRC kinase-dependent signaling by noncatalytic receptors. Biochemistry. 47, 5681–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina, E.I. , Conn, E.M. , Wortmann, A. , Partridge, J.J. , Kupriyanova, T.A. , Ardi, V.C. , Hooper, J.D. , Quigley, J.P. , 2009. Functional role of cell surface CUB domain-containing protein 1 in tumor cell dissemination. Mol. Cancer Res.. 7, 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y. , Wortmann, A. , Burke, L.J. , Reid, J.C. , Adams, M.N. , bdul-Jabbar, I. , Quigley, J.P. , Leduc, R. , Kirchhofer, D. , Hooper, J.D. , 2010. Proteolysis-induced N-terminal ectodomain shedding of the integral membrane glycoprotein CUB domain-containing protein 1 (CDCP1) is accompanied by tyrosine phosphorylation of its C-terminal domain and recruitment of Src and PKCdelta. J. Biol. Chem.. 285, 26162–26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper, J.D. , Zijlstra, A. , Aimes, R.T. , Liang, H. , Claassen, G.F. , Tarin, D. , Testa, J.E. , Quigley, J.P. , 2003. Subtractive immunization using highly metastatic human tumor cells identifies SIMA135/CDCP1, a 135kDa cell surface phosphorylated glycoprotein antigen. Oncogene. 22, 1783–1794. [DOI] [PubMed] [Google Scholar]
- Ikeda, J. , Oda, T. , Inoue, M. , Uekita, T. , Sakai, R. , Okumura, M. , Aozasa, K. , Morii, E. , 2009. Expression of CUB domain containing protein (CDCP1) is correlated with prognosis and survival of patients with adenocarcinoma of lung. Cancer Sci.. 100, 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, J.I. , Morii, E. , Kimura, H. , Tomita, Y. , Takakuwa, T. , Hasegawa, J.I. , Kim, Y.K. , Miyoshi, Y. , Noguchi, S. , Nishida, T. , Aozasa, K. , 2006. Epigenetic regulation of the expression of the novel stem cell marker CDCP1 in cancer cells. J. Pathol.. 210, 75–84. [DOI] [PubMed] [Google Scholar]
- Kapp, K. , Siemens, J. , Weyrich, P. , Schulz, J.B. , Haring, H.U. , Lammers, R. , 2007. Extracellular domain splice variants of a transforming protein tyrosine phosphatase alpha mutant differentially activate Src-kinase dependent focus formation. Genes Cells. 12, 63–73. [DOI] [PubMed] [Google Scholar]
- Kollmorgen, G. , Bossenmaier, B. , Niederfellner, G. , Haring, H.U. , Lammers, R. , 2012. Structural requirements for cub domain containing protein 1 (CDCP1) and Src dependent cell transformation. PLoS. One. 7, e53050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa, Y. , Uekita, T. , Hiraoka, N. , Fujii, S. , Kosuge, T. , Kanai, Y. , Nojima, Y. , Sakai, R. , 2010. CUB domain-containing protein 1, a prognostic factor for human pancreatic cancers, promotes cell migration and extracellular matrix degradation. Cancer Res.. 70, 5136–5146. [DOI] [PubMed] [Google Scholar]
- Scherl-Mostageer, M. , Sommergruber, W. , Abseher, R. , Hauptmann, R. , Ambros, P. , Schweifer, N. , 2001. Identification of a novel gene, CDCP1, overexpressed in human colorectal cancer. Oncogene. 20, 4402–4408. [DOI] [PubMed] [Google Scholar]
- Siva, A.C. , Wild, M.A. , Kirkland, R.E. , Nolan, M.J. , Lin, B. , Maruyama, T. , Yantiri-Wernimont, F. , Frederickson, S. , Bowdish, K.S. , Xin, H. , 2008. Targeting CUB domain-containing protein 1 with a monoclonal antibody inhibits metastasis in a prostate cancer model. Cancer Res.. 68, 3759–3766. [DOI] [PubMed] [Google Scholar]
- Spassov, D.S. , Baehner, F.L. , Wong, C.H. , McDonough, S. , Moasser, M.M. , 2009. The transmembrane src substrate Trask is an epithelial protein that signals during anchorage deprivation. Am. J. Pathol.. 174, 1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uekita, T. , Jia, L. , Narisawa-Saito, M. , Yokota, J. , Kiyono, T. , Sakai, R. , 2007. CUB domain-containing protein 1 is a novel regulator of anoikis resistance in lung adenocarcinoma. Mol. Cell Biol.. 27, 7649–7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uekita, T. , Tanaka, M. , Takigahira, M. , Miyazawa, Y. , Nakanishi, Y. , Kanai, Y. , Yanagihara, K. , Sakai, R. , 2008. CUB-domain-containing protein 1 regulates peritoneal dissemination of gastric scirrhous carcinoma. Am. J. Pathol.. 172, 1729–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde, A. , Beattie, E.C. , Lem, L. , Riethof, D.A. , Liu, S.H. , Mobley, W.C. , Soriano, P. , Brodsky, F.M. , 1999. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 96, 677–687. [DOI] [PubMed] [Google Scholar]