

Abstract
Introduction
Breast cancer risk of recurrence is known to span 20 years, yet existing prognostic signatures are best at predicting early recurrences (≤5 years). There is a critical need to identify those patients at risk of late‐relapse (>5 years), in order to select potential candidates for further treatment and to identify molecular targets for such treatment.
Methods
A total of 252 breast primary tumors were selected at the Netherlands Cancer Institute from a retrospective series of ER+, HER2− breast cancer patients with a follow‐up of at least 10 years. Gene expression analysis was performed using Agilent 4x44K microarrays. Patients were classified in 3 groups: no relapse (M0); relapse before 5 years (M0‐5) or after 5 years (M5‐15). We assessed the correlation of clinico‐pathological variables with late Distant Metastases (DM). We divided the patient series into a training set of untreated patients (n = 140) and a test set of treated patients (n = 112), to investigate whether a gene‐signature or single genes could be identified for predicting late DM. Pathway level late DM correlates were identified using PARADIGM and DAVID.
Results
Of the clinico‐pathologic variables tested, only lymph node status associated with late DM. A 241‐gene signature developed on the NKI training set was able to classify M5‐15 patients in the test set with a sensitivity of 77% and a specificity of 33% (AUC 0.654). This signature showed enrichment in genes involved in immune response and extracellular matrix. An alternative analysis of individual genes identified CH25H as an independent predictor of distant metastasis in our patient series.
Conclusions
We identified a gene signature for late metastasis in breast cancer. Our data are consistent with a model in which suppressed anti‐tumoral immunity enables dormant tumor cells to re‐enter the cell cycle to form metastases in response to extrinsic events in the microenvironment.
Keywords: Breast cancer, Late distant metastasis, Prognostic, Gene expression profiling, Microenvironment, CH25H gene, MammaPrint, PARADIGM
Highlights
Breast cancer prognostic classifications are based on clinical and molecular profiles.
But they do not allow to select patients for adjuvant treatment after 5 years.
44 K Genomic profiling allowed Identification of a 241‐late metastasis signature.
Specific molecular pathways related to late metastases are expressed.
CH25H regulates the metabolism of lipids and steroids and is correlated to late events.
1. Introduction
Breast cancer is a highly heterogeneous disease with respect to its molecular and clinico‐pathological characteristics. Estimates of the number of distinct breast cancer subtypes range from the five intrinsic subtypes (Luminal A, Luminal B, HER2, Basal, and Normal) to ten or more as proposed recently by Curtis et al. using an integrated genomic and transcriptomic approach (Curtis et al., 2012) For breast cancer patients, this can translate into very different prognoses as well as diverse sensitivities to treatment. Moreover, the risk of recurrence for breast cancer patients is known to span 20 years, with estrogen positive (ER+) and Luminal A patients especially prone to late onset metastases. The 2012 report of the Early Breast Cancer Trialist's Collaborative group (EBCTSG) provides a comprehensive view of the natural history of breast cancer and the effects of treatment on outcome for up to 15 years after adjuvant treatment. While confirming the long‐term benefit of radiotherapy, chemotherapy and adjuvant tamoxifen, which persist for up to 10 years after diagnosis, this overview also shows that even optimally treated patients continue to relapse after 5 years. For ER+ patients who received chemotherapy and 5 years of tamoxifen, the absolute risk of relapse is 16.4% over the first 5 years, a risk level that persists at 16.6% between years 5 and 10 rather than decreasing with time as occurs in ER negative disease (Chia and Wolff, 2011; Darby et al., 2011; Davies et al., 2011; Peto et al., 2012). These findings have been recently confirmed by long‐term follow up of major adjuvant trials of aromatase inhibitors with an annual recurrence risk after 5 years of approximately 2% per year, resulting in a similar overall absolute rate of recurrence in the first and second 5‐year period after diagnosis (Burstein and Griggs, 2012). These findings imply that patient management should cover both short‐term and long‐term risks of mortality and morbidity, ideally taking into account the evolving features of the disease after initial treatment.
Various prognostic classifiers are currently used as decision‐making tools for adjuvant treatment. Classical clinico‐pathological factors such as patient age, tumor size, number of positive nodes, tumor grade, and hormone receptor and HER2 status are integrated in tools like Adjuvant Online!® and routinely used for chemotherapy or hormonal therapy indications; more recently, molecular tools such as the 70‐gene profile Mammaprint® (Glas et al., 2006) or the Recurrence Score derived by Oncotype DX® (Paik et al., 2004) inform the utility of adjuvant systemic therapy. These tools are most powerful to identify patients who will develop distant relapse within the first 5 years after diagnosis, with decreasing predictive power over time (Buyse et al., 2006; Esserman et al., 2011). These results suggest that different mechanisms may be responsible for the development of early and late distant metastases.
Studies of large cohorts as well as more specific research on the timing of recurrence have demonstrated that the tumors at highest risk for relapse after 5 years are ER+, HER2− (Esserman et al., 2011). The use of anti‐estrogen therapy for longer than 5 years has been studied in several trials (Harbeck, 2008). These studies suggest that longer‐term hormonal therapy might improve disease‐free survival in some subgroups, but the clinical significance and magnitude of this benefit remain unclear. There is a critical need to identify those patients at risk of late‐relapse after 5 years of adequate hormonal treatment, in order to select potential candidates for further treatment and identify molecular targets for such treatment.
With the hypothesis that the ‘intrinsic’ molecular features present in a tumor at diagnosis may predict early or distant metastases (Weigelt et al., 2005), we initiated the present study aimed at exploring the differential molecular expression profiles of ER+, HER− breast cancer tumors based on outcome: we compared tumors of patients that did not develop relapse at 10 years, patients with early metastatic relapse before 5 years, and patients with late metastatic relapse after 5 years. We assessed the correlation of clinico‐pathological variables with late Distant Metastases (DM) and investigated whether a gene‐signature could be identified for the late metastasis group. In addition, we investigated single genes associated to the late metastatic process, and identified biological pathways revealed by comparative analysis of the expression profiles of the tumors.
2. Methods
2.1. Patient selection
A set of 252 patients was selected retrospectively at the Netherlands Cancer Institute (NKI‐AVL) from six different consecutive series: (2009, 2009, 2010, 2002; Saghatchian et al. (N = 9, submitted), Bedard et al. (N = 7, submitted), according to following criteria: (1) frozen material available, Distant Metastatic (DM) relapse as first event or no metastatic relapse (control group) with a follow up of more than 10 years (yr.); (3) ER or Progesterone‐receptor (PgR) positive and HER2 status negative. Patients who developed either contralateral breast cancer or a second primary breast tumor before the first metastatic event were excluded, as were patients who developed local or regional relapse before the distant metastatic relapse. Patients in the control group were event‐free during the follow up period (at least 10 yr).
A subgroup of 140 treatment naïve patients was subsequently selected from the 252 patient series. Patient tumors were classified in 3 groups: no relapse at 10 yr (M0, n = 57), DM relapses before 5yr (M ≤ 5, n = 42), or DM relapses after 5yr (M5‐15, n = 41). Patients with DM relapse after 15 years were excluded from the analysis. Clinical features of the patient group are summarized in Table 1. Due to the low number of patients with four or more positive lymph nodes, we grouped together all patients with at least one positive lymph node. Written informed consent was obtained from all patients included in the study. The ethics committee of the Netherlands Cancer Institute approved the study.
Table 1.
Clinical, pathological and molecular characteristics of the untreated ER+ (or PR+) and HER2− patients (N = 140).
| Variable | M0 | M ≤ 5 | M5‐15 | Total | |||
|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | ||
| Total | 57 | 40.7 | 42 | 30.0 | 41 | 29.3 | 140 |
| Age | |||||||
| <55 years | 29 (50.9%) | 42.6 | 24 (57.1%) | 35.3 | 15 (36.1%) | 22.1 | 68 (48.6%) |
| ≥55 years | 28 (49.1%) | 38.9 | 18 (42.9%) | 25.0 | 26 (63.4%) | 36.1 | 72 (51.4%) |
| Histology | |||||||
| IDC | 50 (87.7%) | 41.0 | 38 (90.5%) | 31.1 | 34 (82.9%) | 27.9 | 122 (87.1%) |
| ILC | 2 (3.5%) | 20.0 | 4 (9.5%) | 40.0 | 4 (9.8%) | 40.0 | 10 (7.1%) |
| Other | 5 (8.8%) | 62.5 | 0 (0.0%) | 0.0 | 3 (7.3%) | 37.5 | 8 (5.7%) |
| Hormonal status | |||||||
| ER positive, PR positive | 46 (80.7%) | 44.2 | 25 (61.0%) | 24.0 | 33 (80.5%) | 31.7 | 104 (74.8%) |
| ER positive, PR negative | 5 (8.8%) | 17.9 | 15 (36.6%) | 53.6 | 8 (19.5%) | 28.6 | 28 (20.1%) |
| ER positive, PR unknown | 6 (10.5%) | 85.7 | 1 (2.4%) | 14.3 | 0 (0.0%) | 0.0 | 7 (5.0%) |
| ER negative, PR positive | 0 (0.0%) | 0.0 | 1 (100.0%) | 100.0 | 0 (0.0%) | 0.0 | 1 (100.0%) |
| Diameter | |||||||
| ≤2 cm | 43 (75.4%) | 51.2 | 17 (40.5%) | 20.2 | 25 (58.5%) | 28.6 | 84 (60.0%) |
| >2 cm | 14 (24.6%) | 25.0 | 25 (59.5%) | 44.6 | 17 (41.5%) | 30.4 | 56 (40.0%) |
| Lymph node status | |||||||
| 0 | 54 (94.7%) | 49.1 | 26 (61.9%) | 23.6 | 30 (73.2%) | 27.3 | 110 (78.6%) |
| 1+ | 3 (5.3%) | 10.0 | 16 (38.1%) | 53.3 | 11 (26.8%) | 36.7 | 30 (21.4%) |
| Grade | |||||||
| 1 | 30 (52.6%) | 58.8 | 6 (14.3%) | 11.8 | 15 (29.4%) | 29.4 | 51 (36.4%) |
| 2 | 15 (26.3%) | 30.6 | 16 (38.1%) | 32.7 | 18 (36.7%) | 36.7 | 49 (35.0%) |
| 3 | 12 (21.1%) | 30.0 | 20 (47.6%) | 50.0 | 8 (20.0%) | 20.0 | 40 (28.6%) |
| MammaPrint | |||||||
| Good | 43 (75.4%) | 48.9 | 18 (42.9%) | 20.5 | 27 (65.9%) | 30.7 | 88 (62.9%) |
| Poor | 14 (24.6%) | 26.9 | 24 (57.1%) | 46.2 | 14 (34.1%) | 26.9 | 52 (37.1%) |
M0 no distant metastasis, M ≤ 5 distant metastasis between 0 and 5 yr, M5‐15 distant metastasis after 5 yr, and before 15 yr; ER = Estrogen Receptor, PR = Progesterone Receptor.
2.2. Gene expression profiling
Out of the 252 primary frozen tumors, 235 were analyzed at Agendia NV (Amsterdam, The Netherlands). RNA isolation was performed as described previously (van't Veer et al., 2002). RNA integrity was evaluated with the 2100 Bioanalyzer (Agilent) using the RNA 6000 Nano LabChip, following the manufacturer's protocol. RNA amplification, labeling and hybridization to a custom High Density (HD) 44K oligoarray (Agilent Technologies) were performed as described previously (Glas et al., 2006). Fluorescence intensities were Lowess normalized across the samples using Feature Extraction software version 7.5 and Log10 transformed.
The remaining 17 samples (17 out of 252) were analyzed at the Institute Gustave Roussy (IGR). RNA isolation, labeling and hybridization were performed with the same protocols as those used at Agendia NV. These samples were hybridized on commercial High Density 44K oligoarrays (Agilent Technologies) following the manufacturer's protocol, using the MammaPrint® Reference Pool (MRP), a breast tumor pool described in (van't Veer et al., 2002), as a reference signal. Fluorescence intensities were Lowess normalized across the samples using Feature Extraction software version 10.5.1.1 and then Log10 transformed. The Rosetta Resolver system version 7.2.2.0.SP1.31 was used for the data quality assessment. Data are MIAME compliant and have been submitted to ArrayExpress (E‐MTAB‐949).
In order to have a unique dataset of the 252 samples, we only considered the common probes (n = 39,859) between the two microarray platforms used, representing more than 90% of the all probes in both arrays. Probes that had more than 25% missing values were removed. If a probe was present more than once on the array, we retained the one that showed the highest variance across the samples and removed the others. These filtering steps resulted in a dataset with 32,840 probes.
After selection of the 140 treatment‐naïve patients used in the training set, probe intensities were median‐centered across the samples. Missing values were calculated using the k‐nearest neighbor algorithm, setting K to 10 (Troyanskaya et al., 2001).
2.3. ER, PgR and HER2 prognostic marker assessment
Immunohistochemistry for ER‐alpha, (Estrogen Receptor alpha), PgR (Progesterone Receptor alpha and HER2 (Human Epidermal growth factor Receptor 2) and additional chromogenic in situ hybridization (CISH) for HER2 was performed and scored as described previously (Hannemann et al., 2006; van de Vijver et al., 1988). Staining for ER and PgR was interpreted as positive when more than 10% of tumor cells were stained. HER2 status equal to 0 or 1 was considered as negative and HER2 status equal to 3 as positive. When HER2 status was not available, the microarray‐based gene expression test TargetPrint® (Roepman et al., 2009) was used for the assessment.
2.4. MammaPrint® profile
Patients were assessed for their MammaPrint status (Glas et al., 2006).
2.5. Statistics
Distant Metastasis Free Survival (DMFS) time was measured using the interval between the primary tumor diagnosis and the event of distant metastasis or death from any cause or last follow‐up. Survival curves were generated using the Kaplan–Meier method and p‐values of log‐rank tests were used. Univariate and multivariate analyses were conducted using, respectively, an uninomial or polynomial logistic regression model in which the variables included were tested to be independent predictor of early relapse (M0‐5 group) and late relapse (M5‐15 group) with respect to the control group (M0). Variables with a p‐value ≤0.3 at the univariate level were included in the subsequent multivariate analysis. The Wald statistic test was used to test the statistical significance. Analyses were performed using SPSS 18.0 and the survival package of the statistical language R (http://www.r‐project.org/).
2.6. Gene expression signature identification with PAM (prediction analysis of microarray)
In order to identify a gene expression signature predictive of the late metastatic relapse (M5‐15 group), we used the method implemented in the R package pamr from Tibshirani and colleagues, based on the nearest shrunken centroid classification (Tibshirani et al., 2002). First the 50% probes (out of the 32,840) with the highest variance across the 140 patients were selected (n = 16,420). Next, the nearest shrunken centroids method was applied to the NKI M0 and M5‐15 patients with a MammaPrint low‐risk profile (n = 70) and the classification performance was evaluated by 10‐fold‐cross validation repeated 10 times using the Bioconductor software package MRCestimate as described previously (Oberthuer et al., 2006). The final gene expression‐classifier was built using the ClassifierBuild function implemented in MRCestimate. The area under the receiver operating characteristic curve (AUC) was used as the prediction quality criterion. The ROC curves were calculated using the Bioconductor software package pROC (http://www.bioconductor.org/).
Next we performed a functional enrichment analysis using the DAVID Gene Functional Classification Tool 6.7 (Huang da et al., 2007) and gProfiler annotation tool (Reimand et al., 2007, 2011) in order to identify significantly enriched biological processes, cellular components, molecular functions, and pathways in the signature. Only GO terms that were statistically significant after multiple testing corrections (Benjamini Hochberg False Discovery Rate calculation, FDR) were selected (p‐value<0.05).
2.7. Identification of genes associated to late DMFS differences
In order to identify individual genes associated to late survival differences (using time as a continuous variable) in the 140 patient group, we used the survdiff function implemented in the R package survival and we set the parameter rho to −1 to give greater weight to the later part of the survival curves (Harrington and Fleming, 1982). Only the 50% probes (out of the 32,840) with highest variance across the 140 patients were considered (n = 16,420). The survdiff function was applied to each probe individually for DMFS time considering the probe as a covariate dichotomized into 2 groups (above and below the median expression across all samples). The log rank test p‐values were then corrected for multiple testing using the Benjamini Hochberg False Discovery Rate (FDR) calculation. The parameter “strata” was used to stratify the 140 patients based on additional clinico‐pathological parameters (Grade, Diameter, Lymph node status and MammaPrint), in order to find genes that add prognostic value to those parameters already known.
2.8. PARADIGM (Pathway Recognition Algorithm Using Data Integration On Genomic Models)
PARADIGM was used to estimate pathway activities to identify portions of the network models differentially active in the breast cancer samples (Vaske et al., 2010). PARADIGM is a probabilistic model that searches for altered pathways in the US National Cancer Institute Pathway Interaction Database. First we estimated pathway activities based on the gene expression levels of the 140 ER+ Her2− patients as described previously (Vaske et al., 2010). Next we applied a Wilcoxon statistical test to identify pathway components with activity levels that differed significantly between M0 and M5‐15 patients (uncorrected p‐values <0.05). The resulting subnetworks were then visualized using Cytoscape (Smoot et al., 2011) and analyzed for composite pathway enrichment using the EASE methodology (Hosack et al., 2003) including a multiple testing correction (Benjamini–Hochberg False Discovery Rate calculation, FDR) step. This methodology assumes a geometric distribution and uses a modified Fisher Exact Probability p‐value (so called EASE score) to test for significance.
3. Results
3.1. Patient characteristics
Patients, for whom frozen samples were available, were selected from the database of the Netherlands Cancer Institute (NKI) following the selection criteria including patient outcome, as described in the Methods. In order to capture the intrinsic aggressiveness of the tumor, excluding any effect of systemic treatment, we split the initial selection of ER+ HER2− patients (n = 252) into treated (n = 112) and untreated patients (n = 140). The untreated group was used as a training set (Figure 1), whereas the treated group was used as a validation set.
Figure 1.
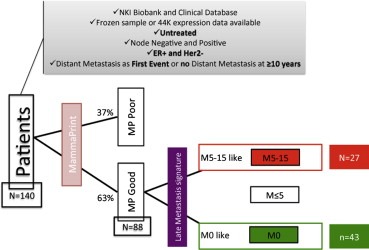
Patient selection strategy to identify the late metastasis gene expression signature. MP Poor = MammaPrint high‐risk profile, MP Good = MammaPrint low‐risk profile; M0 no distant metastasis, M ≤ 5 distant metastasis between 0 and 5 yr, M5‐15 distant metastasis after 5 yr.
Gene expression profiles of all the primary tumors were grouped in three classes: no DM (Distant Metastasis) relapse (M0) for at least 10 years of follow‐up, early DM relapse (M ≤ 5, DM event at ≤5 yr), late DM relapse (M5‐15, DM event >5 yr). All follow‐up events were censored at 15 years.
Clinico‐pathological and molecular data of this untreated patient group are summarized in Table 1: 83 out of 140 (59.3%) patients had a distant relapse, 42 (30.0%) patients before 5 years and 41 (29.3%) patients between 5 and 15 years.
In the treated group we observed more relapses, with a similar distribution over time: 77 relapses out of 112 patients (68.7%), 37 M ≤ 5 (33.0%) and 40 M5‐15 (35.7%). The characteristics of the treated group are summarized in Additional file 1, Table S1.
3.2. Lymph node status is the only clinico‐pathological variable associated with late metastatic relapses
Metastatic relapse has been associated with large tumor size, high‐grade, and positive lymph node status, characteristics believed to reflect a cancer's ability to proliferate rapidly, colonize other tissue types, and evade the immune system (Weigelt et al., 2005; Castano et al., 2011). To determine whether these variables or the other clinico‐pathological/established molecular features listed in Table 1 correlated with late metastasis occurrence versus no relapse, we performed univariate and multivariate analysis for their association to the M5‐15 group versus the M0 group (see Methods). As shown in Table 2A, in univariate analysis only lymph node status was significantly associated with late recurrence (M5‐15 class; p‐value = 0.01, OR = 0.15, CI 95% = 0.04–0.59). Table 2B shows the result of the multivariate analysis of age, tumor diameter, lymph node status, grade (1 vs. 2, 3). As in the univariate analysis, only lymph node status was significantly associated to the late DM class (M5‐15) compared to the control group (M0) (p‐value = 0.005, OR = 0.14, CI 95% = 0.03–0.56). In contrast, tumor grade retained significant association to early metastatic relapse M ≤ 5 in multivariate analysis (Additional file 1, Table S2B).
Table 2.
Univariate (A) and multivariate (B) analysis of different clinico‐pathological and genomic variables in relation to the M5‐15 class in the ER+ HER2− untreated group (N = 140).
| Class DMa | Variable | P value | OR | 95% CI |
|---|---|---|---|---|
| A. Univariate analysis of clinical pathological markers and MammaPrint in relation to Late Distant Metastasis (DM) class | ||||
| M5‐15 | Age (<55 yr vs ≥ 55 yr) | 0.16 | 0.56 | 0.24–1.27 |
| Histology (IDC vs others) | 0.50 | 0.68 | 0.22–2.11 | |
| Diameter (≤2 cm vs > 2 cm) | 0.08 | 0. 46 | 0.19–1.09 | |
| Lymph node status (0 vs 1+) | 0.01 | 0.15 | 0.04–0.59 | |
| Grade (1,2 vs 3) | 0.85 | 0.91 | 0.33–2.47 | |
| Grade (1 vs 2, 3) | 0.12 | 0.52 | 0.23–1.18 | |
| B. Multivariate analysis of clinical pathological markers and MammaPrint in relation to Late Distant Metastasis (DM) class | ||||
| M5‐15 | Age (<55 yr vs ≥ 55 yr) | 0.12 | 0.50 | 0.21–1.20 |
| Diameter (≤2 cm vs > 2 cm) | 0.24 | 0.57 | 0.23–1.45 | |
| Lymph node status (0 vs 1+) | 0.005 | 0.14 | 0.03–0.56 | |
| Grade (1 vs 2, 3) | 0.16 | 0.52 | 0.21–1.28 | |
Abbreviations: yr = years; OR = Odds Ratio; CI = Confidence Interval.
The reference class is M0.
3.3. Gene expression signature identification using PAM (Prediction Analysis of Microarray)
We set out to identify a predictor of late metastasis using our training set. Unsupervised Hierarchical Clustering (HCL) of the 140 patients using the highest variance selected probes (n = 16,420) did not reveal any obvious partitioning of the metastatic classes (Additional file 1, Figure S1). Therefore, we opted for a supervised approach based on nearest shrunken centroid algorithm (also known as PAM (Tibshirani et al., 2002)) to identify a gene expression signature predictive of late metastasis.
For this analysis we chose to exclude patients predicted to be at high risk of an early metastasis (MammaPrint poor profile), retaining only those patients predicted to be at low risk for subsequent events (MammaPrint good profile), reasoning that metastatic processes unique to late events are likely distinct from those captured by early relapse predictors.
Based on this hypothesis and the observation that the majority of late metastases patients were predicted to be MammaPrint low risk, we included M0 and M5‐15 untreated patients with MammaPrint low‐risk profile (43 and 27 cases respectively) to identify a predictive signature of distant metastasis (DM between 5 and 15 years). Figure 1 depicts our strategy for identifying a predictive signature of late metastasis.
A set of 241 predictive probes was identified using the PAM algorithm comparing M5‐15 versus M0 MammaPrint good patients (Additional file 1, Figure S2). The PAM vote matrix (Additional file 1, Figure S3A) estimated that an overall accuracy of 77% could be achieved with this classifier. Within the 10‐times‐repeated 10‐fold‐cross validation procedure, this classifier of 241 probes (threshold = 1.517) was able to correctly classify on average 17/27 M5‐15 patients and 37/43 M0 patients. Specifically, M5‐15 patients in the partitioned test sets were correctly identified with a sensitivity of 63% and a specificity of 86%. The median false discovery rate for the 241‐gene nearest shrunken centroid classifier, estimated after 1000 permutations, was below 1% (Additional file 1, Figure S3B). As expected, the area under the Receiver Operating Characteristic (ROC) curve (AUC) for the 241‐probe signature applied to the training set was close to 1 (AUC = 0.936, CI 95% = 0.866–1) confirming the good performance of the 241‐probe signature in the training set (Additional file 1, Figure S4).
3.4. Functional enrichment of the 241‐gene signature
After matching the 241 probe Agilent IDs with RefSeq ID and the Gene symbol ID we found 230 unique genes. Out of the 230 genes, 144 genes were well‐characterized protein coding genes; the remaining 86 genes were poorly annotated. The complete list of the 241 probes is reported in the Additional file 2.
In order to understand the biology behind the 241‐gene signature we performed a functional enrichment analysis of the 241 probes using DAVID and gProfiler (see Methods for details) (Huang da et al., 2007, 2007, 2011). Of the 241 probes, 150 matched to the corresponding DAVID/gProfiler ID's. Interestingly both tools gave similar results. Functional enrichment by DAVID revealed the 241‐probe signature to be dominated by genes active in the “extracellular region”, with functions relating to the “extracellular matrix” and “immune response” (p‐value <0.0001). Other highly ranked categories (p‐value ≤0.05) include “antigen binding” and “lectin and sugar binding sites”. A complete list of the Gene Ontology categories and the functional clusters found with DAVID are reported in the Additional file 3.
3.5. Performance assessment of the 241‐probe signature on an independent dataset
In order to validate the accuracy of the 241‐gene signature for detecting late metastases in the independent dataset, we tested the classifier on the M0 and M5‐15 MammaPrint low‐risk treated patients (n = 51) in the NKI test set. To predict the DM class of a patient we calculated the Pearson correlation between the 241‐probe centroid of the patient and the training 241‐probe centroids (M0 and M5‐15 centroids). The predicted DM class of the patient was defined as the one with the highest correlation coefficient to the training centroids. The 241‐gene signature classified the M5‐15 patients with a sensitivity of 77% (23/30) and a specificity of 33% (7/21). When we selected only hormonally treated patients, the 241‐gene classifier showed an increased sensitivity for the M5‐15 patients (85%, 17/20), but a decreased specificity (30%, 3/10). To summarize, the 241‐gene signature performed similarly on the hormonal treated only patients (AUC = 0.690, CI 95% = 0.486–0.892) and on the chemotherapy and/or hormonal treated patients (AUC = 0.654, CI 95% = 0.499–0.809) (Additional file 1, Figure S5).
3.6. Identification of individual genes associated with late distant metastasis using time as a continuous variable
In order to further investigate differences in gene expression related to outcome over time, we opted to identify individual genes able to predict late distant metastasis (DM) events. This approach differed from the previous analyses, because we used the distant‐metastasis free survival (DMFS) time as continuous variable without grouping the patients into different classes (i.e. M0, M ≤ 5 and M5‐15) and we aimed to predict the time to the DM event, using the expression of the genes as explanatory variable. We tested whether expression of each of the 16,420 most variable probes was significantly associated with late differences in DMFS time (see Methods for details) in the untreated patients (n = 140). In univariate analysis, MammaPrint risk class, tumor diameter, lymph node status and tumor grade were found to be significantly associated to late DMFS differences (Chi‐square test p‐values equal to 0.016, 0.004, <0.001 and 0.016 respectively) among all clinico‐pathological and molecular features listed in Table 1. Therefore we decided to identify genes associated to late DMFS differences after correcting for these variables. Two genes, cholesterol 25‐hydroxylase (CH25H) and follistatin‐like 4 (FSTL4) were selected as significantly associated to late DMFS after correction for multiple testing (p‐value = 0.01). Interestingly CH25H is part of the 241‐gene signature previously described. After Cox regression analysis, patients with high expression of CH25H (‘intensity above median’) showed a hazard ratio (HR) of 0.28 (95% CI = 0.17–0.44) compared to patients with a low expression of CH25H (‘intensity below median’). For the gene FSTL4 we observed an opposite trend: patients with high expression of FSTL4 showed an HR of 2.42 (95% CI = 1.54–3.80) compared to patients with low expression of FSTL4.
In order to independently validate the prognostic power of these two genes, we tested their performance in the validation set of treated patients (n = 112) and in three publicly available datasets (Desmedt et al., 2007; Harrell et al., 2011; Symmans et al., 2010). In the publicly available datasets we selected only ER+ patients in order to mimic our selection criteria. Unfortunately the HER2 status was only available for the Harrell series (Harrell et al., 2011), therefore for the two other series we only considered ER status for selection. As shown in Figure 2, the CH25H gene validated as being significantly associated to metastasis‐free survival time in all tested series (Harrell dataset p‐value = 0.024, Desmedt dataset p‐value = 0.048, Symmans dataset p‐value = 0.012). The gene FSTL4 was not significantly associated to metastasis‐free survival time in all four independent datasets (Additional file 1, Figure S6). Moreover in two out of four series (NKI treated patient group and Harrell series), high and low expression of FSTL4 was inversely associated to survival in respect to what observed in the NKI patient group, although the association was not significant. Since we were not able to reproduce the prognostic ability of FSTL4 in the independent datasets we excluded it from further investigation.
Figure 2.
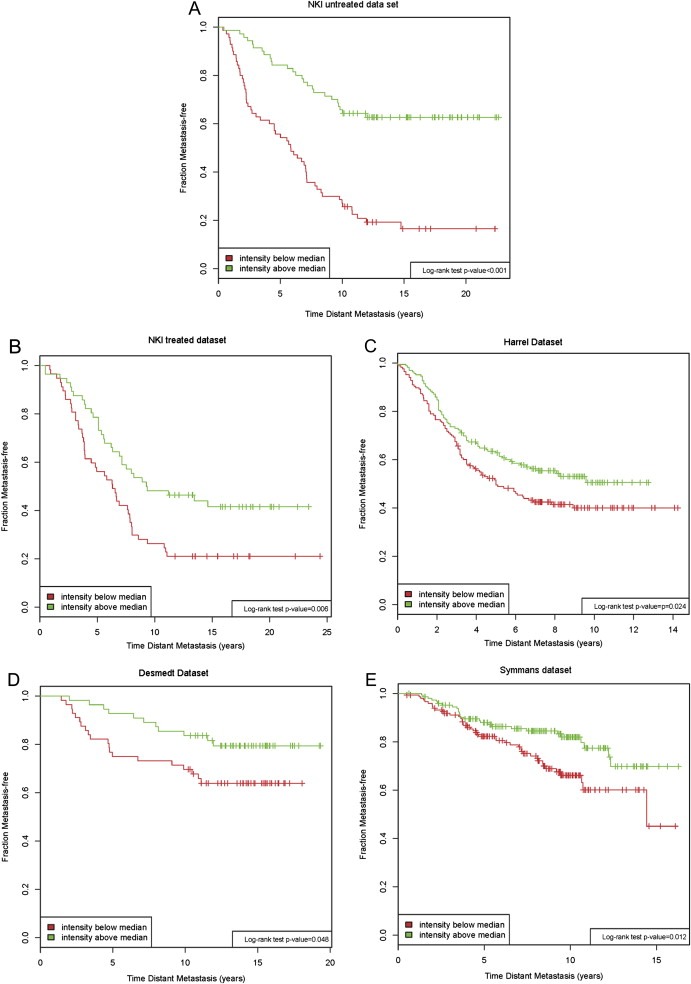
Kaplan Maier analysis of CH25H high/CH25H low in different datasets (NKI untreated dataset, n = 140; NKI treated dataset, n = 112; Harrell dataset (Harrell et al., 2011), n = 331; Desmedt dataset (Desmedt et al., 2007), n = 111; Symmans dataset (Symmans et al., 2010), n = 298). CH25H = cholesterol 25‐hydroxylase. Log rank test was performed to test for significance.
In order to specifically test the ability of CH25H to predict a late metastatic relapse (after 5 yr), we performed a multivariate analysis, using a multinomial logistic regression model, as was previously done for the clinico‐pathological and established molecular characteristics. We included age, diameter, lymph node status, grade (1 vs. 2,3), MammaPrint status and CH25H binary expression (above and below the median expression value). As expected, CH25H was confirmed as an independent predictor of the M5‐15 class in the untreated patient group (n = 140) (Table 3) as well as in the pooled series (n = 252, untreated and treated patients) (Additional file 1, Table S3). However, a significant association between CH25H expression and early metastasis (M ≤ 5) was also observed in the pooled series (Additional file 1, Table S3).
Table 3.
Multivariate analysis of Age, Diameter, Lymph node status, Grade, MammaPrint and CH25H expression in relation to the M5‐15 class in the ER+HER2− NKI untreated group (N = 140).
| Variable | P value | OR | 95% CI | |
|---|---|---|---|---|
| Multivariate analysis of clinical pathological markers and CH25H expression in relation to late DM (Distant Metastasis) | ||||
| M5‐15 | Age (<55 yr vs ≥ 55 yr) | 0.54 | 0.74 | 0.29–1.90 |
| Diameter (≤2 cm vs > 2 cm) | 0.75 | 0.85 | 0.31–2.35 | |
| Lymph node status (0 vs 1+) | 0.004 | 0.12 | 0.03–0.50 | |
| Grade (1 vs 2, 3) | 0.47 | 0.70 | 0.27–1.84 | |
| CH25H expression (Low vs High) | 0.001 | 5.43 | 2.00–14.72 | |
The reference class is M0.
Abbreviations: yr = years; OR = Odds Ratio; CI = Confidence Interval.
3.7. Pathway analysis using PARADIGM
Considering that analyses in different datasets show different genetic alterations involving common pathways, we used PARADIGM, a novel bioinformatics approach that identifies specific altered pathway activities and regulatory networks rather than single genes or profiles (Vaske et al., 2010).
To identify the pathway activities involved in early and late metastasis versus no metastasis, we performed comparisons between paired groups of outcome: early metastasis (M ≤ 5) versus late metastasis (M5‐15), early metastasis versus no relapse (M0) and late metastasis versus no relapse. We also analyzed for differences in pathway activities between MammaPrint good profile patients who did not recur (M0) and those with late recurrences (M5‐15), reasoning as we did for the PAM signature development that ‘low risk’ women with late recurrences might reflect distinct biology.
First we identified the PARADIGM pathway activities for the 140 untreated patients using the gene expression profiles as input to the algorithm described in (Vaske et al., 2010). After comparing the pathway activities of patients with different outcomes, we performed an EASE enrichment analysis in order to identify pathways with activity levels that were significantly differentially altered between the metastasis groups and visualized the resulting networks using Cytoscape. The number of significant pathways (EASE score <0.05) was similar between the three comparisons. Out of 1189 pathways tested 10 and 6 pathways remained significant in the M ≤ 5 vs. M0 and M ≤ 5 vs. M5‐15 comparisons respectively after Benjamini–Hochberg (BH) multiple testing correction was applied. However, after multiple testing corrections, no pathway was selected for the M5‐15 vs. M0 comparison (as shown in Additional file 1, Figure S7). The complete list of identified pathways is reported in the Additional File 4.
Two activated pathways recurred in early metastasis (M ≤ 5) compared to late metastasis (M5‐15) or no metastasis (M0): the FOXM1 transcription factor network, E2F signaling, Aurora B kinase signaling, and PLK1 signaling events.
Interestingly, when we restricted our analysis to patients with MammaPrint good (low risk) profiles (N = 70), and compared those with no metastasis (M0) to those with late recurrence (M5‐15), we found three pathways that significantly associated with late recurrence after BH multiple testing correction: IL12‐mediated immune signaling, FAS (CD95) apoptotic signaling, and retinoic acid signaling (vitamin D processing) (Figure 3). Significant genes in the IL12 immune pathway include granzyme genes GZMA and GZMB, NOS2, CCL3, and cytokines like TBX21 in addition to IL1B and IL1R1, all genes associated with activation of cytotoxic T lymphocytes and natural killer cells, and all expressed at lower levels in patients with late recurrence compared to those who did not recur. The abstract entities “natural killer cell activation” and “T cell proliferation” were also downregulated in late recurrence patients, reflecting the many immune genes in the PARADIGM super pathway with cohesive under‐expression in late recurring patients.
Figure 3.

PARADIGM analysis results considering only the untreated MammaPrint low‐risk patients (n = 70). The pathways significantly associated with late recurrences (M5‐15) after multiple testing corrections (p‐value<0.05) as compared with no recurrence (M0) are reported. For each pathway is reported the EASE score and the p‐value after Benjamini–Hochberg (BH) multiple testing correction.
The FAS (CD95) signaling pathway, part of the larger network of apoptosis related genes significantly downregulated in late recurrence MammaPrint good profile patients contains several key caspases (CASP3, CASP8) and other genes important to the apoptotic process including FAS and BID. Interestingly, these immune and apoptotic regulatory networks are connected, forming a larger network connected by links between the granzyme GZMB (immune) and apoptotic genes BID, CASP7 and CASP3 (Figure 4).
Figure 4.
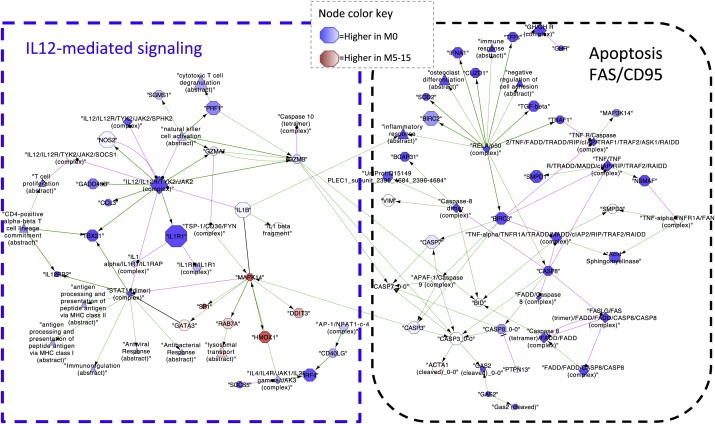
IL12‐mediated signaling and FAS (CD95) apoptotic signaling network visualized using Cytoscape (Smoot et al., 2011). Each node of the network is a gene. Red nodes represent genes overexpressed in M5‐15 patients and blue nodes genes overexpressed in M0 patients; the color intensity correlates with the size of the expression.
The third significant pathway, retinoic acid signaling, contains genes like RARA and RARS and various inferred vitamin D3 complexes, the latter of which appear to be downregulated in late recurrence patients compared to patients who did not recur. We note that genes expressed at lower levels in low risk women who recur late dominate all three pathways (Figure 5).
Figure 5.
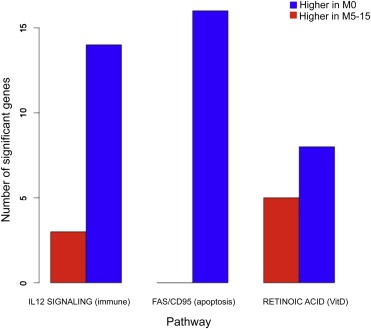
The bar plot reports for each of the three pathways significantly associated to late recurrence (M5‐15) as compared with no recurrence (M0), the number of significant genes that are higher expressed in the M0 and M5‐15 MammaPrint low‐risk patients.
4. Discussion
Late distant metastases represent a major cause of death for breast cancer patients. Numerous studies have focused on breast cancer metastases and how they might originate from primary breast tumors; however, few studies have addressed metastatic recurrence occurring many years after initial diagnosis.
Current breast cancer prognostic signatures effectively predict for outcome risk within five years of diagnosis. However, there still is a need for understanding which patients will relapse late to identify patients that may benefit from longer hormonal therapy regimen. Moreover, understanding the biological pathways behind this late metastasis process may help in the development of novel treatment strategies for this group of patients.
The Netherlands Cancer Institute has assembled a large database of breast cancer patients for which clinical data, treatment details, long‐term follow‐up, frozen samples or gene expression data are available. Therefore we were able to select breast cancer patients who underwent surgical treatment, with or without further adjuvant treatment. Consequently, we could analyze the expression profiles of primary tumors based on patient outcomes over long time periods that reflect the natural history of the initial breast cancer.
It has been shown previously that gene expression profiling of human primary breast tumors can predict metastasis risk, which indicates that the capacity to metastasize might be acquired early during tumor genesis, even if metastases develop after a long interval. Consequently we assumed that metastatic response profiles could be developed on primary tumors (Weigelt et al., 2005).
As shown recently (Esserman et al., 2011), risk of relapse for hormonal positive patients persists over 20 years, whereas the risk for both triple negatives and HER2+ patients spans up to 5 years approximately. Therefore, for this study we selected only ER+ HER2− breast cancer patients, in order to identify clinical and genomic characteristics predictive of late recurrences or long‐term remission. Patients with distant relapses after five years from first diagnosis (M5‐15 patients) were considered to be late metastatic patients and treated as the group of interest in our study.
Among all the clinico‐pathological features tested, only lymph node status was found to be a strong predictor of late metastasis, as emerged from the univariate and multivariate analysis of Table 2. This finding is in agreement with what was previously reported, that nodal status is the dominant characteristic predicting disease specific survival (Esserman et al., 2011). No other clinico‐pathological feature showed a correlation to late metastasis (M5‐15).
We used a supervised approach based on gene expression data to identify a gene expression signature specifically for late metastasis. The 241‐probe signature was shown to be a good predictor of late metastasis (M5‐15 patients) in the training group of untreated patients, with respect to the control group (M0 patients), with an overall accuracy of 77% as estimated by cross‐validation studies. When we applied the 241‐probe signature to the independent validation set, the treated patients, its overall accuracy decreased to 59%, with 23/30 M5‐15 patients correctly classified (77%), and with 7 out of 21 M0 patients correctly classified (33%).
The recently finalized work of the Cancer Genome Atlas Network provided key insights into previously defined gene expression subtypes and confirmed the existence of four main breast cancer classes when combining data from five platforms, each of which shows significant molecular heterogeneity (Comprehensive molecular, 2012). Occurrence of late metastases in each of these subgroups likely follows distinct pathways that might not be well represented by a single molecular profile. Our series allowed us to select only for ER+, HER2− and MammaPrint good profile patients. However this group likely remains heterogeneous in terms of the molecular and biological characteristics and pathways of late metastatic recurrence. The lack of predictive signal might point to dominating extrinsic factors, or for the need for other types of data, bigger sample size, or non‐genomic data, or different types of statistical approach to this time dependent analysis.
Interestingly, a functional enrichment analysis of the 241 probes using DAVID suggests that this genomic signature is significantly enriched in genes involved in the immune system and immune response (see Additional file 3). This finding reflects the possibility that late metastatic relapses might be explained by inflammatory events (with immune system activation) in patients, or the development of suppressed anti‐tumoral immunity, whereas early metastatic relapse might mostly result from high levels of proliferation in the primary tumor. The later hypothesis, that suppressed anti‐tumoral immunity might play a role, was supported by the results of a PARADIGM pathway analysis of MammaPrint good‐profile patients showing that late recurrence in this subgroup is associated with lower expression levels of the IL12 immune pathway including granzyme genes GZMA and GZMB, NOS2, CCL3, and cytokines like TBX21 in addition to IL1B and IL1R1, all genes associated with activation of cytotoxic T lymphocytes and natural killer cells. It is relevant to note here that interleukin dependent inflammatory signaling networks control responses to cellular senescence (Kuilman et al., 2008). Our finding that immune genes are associated with late metastases raises the interesting possibility that in the absence of anti‐tumoral immunity, dormant disseminated breast cancer cells could be reactivated to enter the cell cycle through interleukin signals or other extrinsic events. Concordantly with this idea, Esserman and colleagues recently proposed that late recurrences in hormone receptor negative and positive patients might be linked to altered immune function (specifically, to suppressed immunity) (Esserman et al., 2011).
Besides immune response, the other category that emerged from this analysis was the extracellular matrix. It is well established that to form a metastasis from the primary tumor, the cancer cells need to acquire additional properties that enable invasion of the extracellular matrix and, ultimately, invasion to a secondary site (Place et al., 2011; Spano and Zollo, 2012). Our data might thus reflect the role of the microenvironment in the late metastatic process, especially considering that late metastases often occur in the bone through the involvement of osteoblasts, an important component of the normal bone microenvironment as well as bone metastases (Place et al., 2011). Recently, using a global gene expression analysis, Rajski and colleagues showed that the interaction between breast cancer cells and osteoblasts plays a role in the bone metastasis formation (Rajski et al., 2012).
To summarize, our signature provides evidence that occurrence of late metastasis implies genes expressed in the primary tumor that are related to the immune system and the extra‐cellular matrix. This might suggest that beyond any intrinsic characteristic of the primary tumor, a triggering event from the external microenvironment (mediated by the immune system and/or the extra‐cellular matrix) might be needed to stimulate late metastatic proliferation of a dormant cell.
Furthermore, we performed an additional analysis to identify individual genes with expression levels that associated strongly with distant metastasis free survival time after stratification by clinico‐pathological variables and MammaPrint profile. Interestingly, we found that CH25H, a gene that codes for a cholesterol 25‐hydroxylase involved in cholesterol and lipid metabolism (Bauman et al., 2009), was significantly associated to late distant metastasis‐free survival (DMFS) time (p‐value = 0.01). As shown in the Kaplan–Meier curve of Figure 2, the expression of CH25H was able to stratify patients in two risk groups: high expression of CH25H was associated with longer DMFS (low‐risk group), conversely, low expression of CH25H was associated with worse DMFS (high‐risk group). The prognostic ability of CH25H was evaluated in three different independent series, and in all three datasets the CH25H expression was confirmed as being associated with the DMFS. Moreover, when we tested the ability of CH25H to specifically predict the M5‐15 class in a multivariate analysis including age, diameter, lymph‐node status, grade and MammaPrint, CH25H expression was shown to be an independent predictor of late metastasis (p‐value = 0.002). However, when we tested the ability of CH25H expression to predict early metastatic relapse (M ≤ 5 class), it appeared to be significant as well. This result suggests that CH25H may be an important general marker of distant metastasis, involved in all types of metastatic processes (early and late). Consequently, CH25H could be considered a useful clinical marker for predicting late metastasis for patients who are distant‐metastasis free until 5 years but who are still at risk of a later metastasis relapse. CH25H catalyzes the formation of 25‐hydroxycholesterol from cholesterol, leading to the repression of cholesterol biosynthetic enzymes (Bauman et al., 2009). In recent years, it has become increasingly clear that lipid metabolism plays an important role in breast cancer development and progression. In particular, it has been shown that increased cholesterol content is linked to more advanced and aggressive breast tumors (Llaverias et al., 2011). In our study, we showed that low expression of CH25H is associated with worse prognosis and this could be due to higher cholesterol concentration, a result of a lack of cholesterol biosynthetic enzyme repression (through CH25H). In addition to the role of CH25H in lipid metabolism, Bauman and colleagues showed that CH25H is an immunoregulatory lipid that negatively regulates the adaptive immune response (Llaverias et al., 2011); More recently, another study (Hannedouche et al., 2011) demonstrated that down‐regulated CH25H reduced plasma cell response after an immune challenge, confirming the important role of CH25H in the adaptive immune response. This role of CH25H in regulating immune responses is consistent with the proposed link between late recurrences in hormonal receptor negative patients and altered immune function. Taken together, these findings make the CH25H gene a potential target for distant metastasis control in breast cancer.
5. Conclusion
Our study allowed us to identify a molecular profile, a molecular target (CH25H gene) and immune and apoptotic biological pathways that may be further explored for a better understanding of the biological processes leading to late breast cancer relapses. Our data are consistent with a model in which suppressed anti‐tumoral immunity enables dormant tumor cells to re‐enter the cell cycle to form metastases in response to extrinsic events in the microenvironment.
Competing interests
Laura J van't Veer and René Bernards are shareholders in Agendia NV, the commercial entity that markets MammaPrint®.
Conflicts of interest
The other authors disclosed no potential conflicts of interest.
Authors' contributions
LM participated in the design of the study, experiments, data analysis, and drafted the manuscript. MS initiated the study, participated in its conception, design, data analysis, and helped to draft and revise the manuscript. DW participated in PARADIGM and DAVID data analysis and interpretation, and helped to draft and to revise the manuscript. SM participated in the statistical design of the study. PD performed Microarray data analysis. SD participated in the conception and design of the study. VL participated in the conception of the study and coordination of Microarray experiments. SB performed the pathway analysis. TT supervised the study, participated in its conception and design. RB participated in data analysis, interpretation and coordination of the research process, helped to draft the manuscript and revise it. LvV supervised the study, participated in its conception and design, data analysis, and coordination of the research process.
All authors read and approved the final manuscript.
Abbreviations
ER
estrogen receptor
HER2
Human Epidermal Growth Factor Receptor 2
DM
distant metastasis
NK
natural killer
AUC
area under the curve
NKI
Nederlands Cancer institute
IGR
Institut Gustave Roussy
HD
high‐density
PgR
progesterone receptor
DMFS
distant metastasis free survival
OR
odds ratio
CI
confidence interval
PAM
prediction analysis of MicroArray
MP
MammaPrint ®
FDR
false discovery rate
PARADIGM
pathway recognition algorithm using data integration on genomic models
HR
hazard ratio
BH
Benjamini–Hochberg
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
This work was supported by the European Union Research and Development 6th Framework Programme Transbig Project, the European Society of Medical Oncology Translational Research Grant, and the Top Institute Pharma (TI PHARMA) (project T3‐108‐1).
The authors thank Marjanka Schmidt and Sjoerd Elias for insights and valuable discussions.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.07.006.
Mittempergher Lorenza, Saghatchian Mahasti, Wolf Denise M., Michiels Stefan, Canisius Sander, Dessen Philippe, Delaloge Suzette, Lazar Vladimir, Benz Stephen C., Tursz Thomas, Bernards René, van't Veer Laura J., (2013), A gene signature for late distant metastasis in breast cancer identifies a potential mechanism of late recurrences, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.07.006.
References
- Bauman, D.R. , Bitmansour, A.D. , McDonald, J.G. , Thompson, B.M. , Liang, G. , Russell, D.W. , 2009. 25-Hydroxycholesterol secreted by macrophages in response to toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. U S A. 106, (39) 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein, H.J. , Griggs, J.J. , 2012. Deep time: the long and the short of adjuvant endocrine therapy for breast cancer. J. Clin. Oncol.. 30, (7) 684–686. [DOI] [PubMed] [Google Scholar]
- Buyse, M. , Loi, S. , van't Veer, L. , Viale, G. , Delorenzi, M. , Glas, A.M. , d'Assignies, M.S. , Bergh, J. , Lidereau, R. , Ellis, P. , 2006. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J. Natl. Cancer Inst.. 98, (17) 1183–1192. [DOI] [PubMed] [Google Scholar]
- Castano, Z. , Tracy, K. , McAllister, S.S. , 2011. The tumor macroenvironment and systemic regulation of breast cancer progression. Int. J. Dev. Biol.. 55, (7–9) 889–897. [DOI] [PubMed] [Google Scholar]
- Chia, S.K. , Wolff, A.C. , 2011. With maturity comes confidence: EBCTCG tamoxifen update. Lancet. 378, (9793) 747–749. [DOI] [PubMed] [Google Scholar]
- Comprehensive molecular portraits of human breast tumours. Nature. 490, (7418) 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, C. , Shah, S.P. , Chin, S.F. , Turashvili, G. , Rueda, O.M. , Dunning, M.J. , Speed, D. , Lynch, A.G. , Samarajiwa, S. , Yuan, Y. , 2012. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby, S. , McGale, P. , Correa, C. , Taylor, C. , Arriagada, R. , Clarke, M. , Cutter, D. , Davies, C. , Ewertz, M. , Godwin, J. , 2011. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet. 378, (9804) 1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, C. , Godwin, J. , Gray, R. , Clarke, M. , Cutter, D. , Darby, S. , McGale, P. , Pan, H.C. , Taylor, C. , Wang, Y.C. , 2011. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 378, (9793) 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt, C. , Piette, F. , Loi, S. , Wang, Y. , Lallemand, F. , Haibe-Kains, B. , Viale, G. , Delorenzi, M. , Zhang, Y. , d'Assignies, M.S. , 2007. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin. Cancer Res.. 13, (11) 3207–3214. [DOI] [PubMed] [Google Scholar]
- Esserman, L.J. , Moore, D.H. , Tsing, P.J. , Chu, P.W. , Yau, C. , Ozanne, E. , Chung, R.E. , Tandon, V.J. , Park, J.W. , Baehner, F.L. , 2011. Biologic markers determine both the risk and the timing of recurrence in breast cancer. Breast Cancer Res. Treat. 129, (2) 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas, A.M. , Floore, A. , Delahaye, L.J. , Witteveen, A.T. , Pover, R.C. , Bakx, N. , Lahti-Domenici, J.S. , Bruinsma, T.J. , Warmoes, M.O. , Bernards, R. , 2006. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 7, 278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannedouche, S. , Zhang, J. , Yi, T. , Shen, W. , Nguyen, D. , Pereira, J.P. , Guerini, D. , Baumgarten, B.U. , Roggo, S. , Wen, B. , 2011. Oxysterols direct immune cell migration via EBI2. Nature. 475, (7357) 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannemann, J. , Kristel, P. , van Tinteren, H. , Bontenbal, M. , van Hoesel, Q.G. , Smit, W.M. , Nooij, M.A. , Voest, E.E. , van der Wall, E. , Hupperets, P. , 2006. Molecular subtypes of breast cancer and amplification of topoisomerase II alpha: predictive role in dose intensive adjuvant chemotherapy. Br. J. Cancer. 95, (10) 1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbeck, N. , 2008. Never too late: reducing late breast cancer relapse risk. Curr. Med. Res. Opin.. 24, (12) 3295–3305. [DOI] [PubMed] [Google Scholar]
- Harrell, J.C. , Prat, A. , Parker, J.S. , Fan, C. , He, X. , Carey, L. , Anders, C. , Ewend, M. , Perou, C.M. , 2011. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res. Treat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington, D.P. , Fleming, T.R. , 1982. A class of rank test procedures for censored survival data. Biometrika. 69, 553–566. [Google Scholar]
- Hosack, D.A. , Dennis, G. , Sherman, B.T. , Lane, H.C. , Lempicki, R.A. , 2003. Identifying biological themes within lists of genes with EASE. Genome Biol.. 4, (10) R70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da, W. , Sherman, B.T. , Tan, Q. , Collins, J.R. , Alvord, W.G. , Roayaei, J. , Stephens, R. , Baseler, M.W. , Lane, H.C. , Lempicki, R.A. , 2007. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol.. 8, (9) R183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok, M. , Linn, S.C. , Van Laar, R.K. , Jansen, M.P. , van den Berg, T.M. , Delahaye, L.J. , Glas, A.M. , Peterse, J.L. , Hauptmann, M. , Foekens, J.A. , 2009. Comparison of gene expression profiles predicting progression in breast cancer patients treated with tamoxifen. Breast Cancer Res. Treat. 113, (2) 275–283. [DOI] [PubMed] [Google Scholar]
- Kuilman, T. , Michaloglou, C. , Vredeveld, L.C. , Douma, S. , van Doorn, R. , Desmet, C.J. , Aarden, L.A. , Mooi, W.J. , Peeper, D.S. , 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 133, (6) 1019–1031. [DOI] [PubMed] [Google Scholar]
- Llaverias, G. , Danilo, C. , Mercier, I. , Daumer, K. , Capozza, F. , Williams, T.M. , Sotgia, F. , Lisanti, M.P. , Frank, P.G. , 2011. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol.. 178, (1) 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mook, S. , Schmidt, M.K. , Viale, G. , Pruneri, G. , Eekhout, I. , Floore, A. , Glas, A.M. , Bogaerts, J. , Cardoso, F. , Piccart-Gebhart, M.J. , 2009. The 70-gene prognosis-signature predicts disease outcome in breast cancer patients with 1–3 positive lymph nodes in an independent validation study. Breast Cancer Res. Treat. 116, (2) 295–302. [DOI] [PubMed] [Google Scholar]
- Mook, S. , Schmidt, M.K. , Weigelt, B. , Kreike, B. , Eekhout, I. , van de Vijver, M.J. , Glas, A.M. , Floore, A. , Rutgers, E.J. , van 't Veer, L.J. , 2010. The 70-gene prognosis signature predicts early metastasis in breast cancer patients between 55 and 70 years of age. Ann. Oncol.. 21, (4) 717–722. [DOI] [PubMed] [Google Scholar]
- Oberthuer, A. , Berthold, F. , Warnat, P. , Hero, B. , Kahlert, Y. , Spitz, R. , Ernestus, K. , Konig, R. , Haas, S. , Eils, R. , 2006. Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J. Clin. Oncol.. 24, (31) 5070–5078. [DOI] [PubMed] [Google Scholar]
- Paik, S. , Shak, S. , Tang, G. , Kim, C. , Baker, J. , Cronin, M. , Baehner, F.L. , Walker, M.G. , Watson, D. , Park, T. , 2004. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med.. 351, (27) 2817–2826. [DOI] [PubMed] [Google Scholar]
- Peto, R. , Davies, C. , Godwin, J. , Gray, R. , Pan, H.C. , Clarke, M. , Cutter, D. , Darby, S. , McGale, P. , Taylor, C. , 2012. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet. 379, (9814) 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place, A.E. , Jin Huh, S. , Polyak, K. , 2011. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res.. 13, (6) 227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajski, M. , Vogel, B. , Baty, F. , Rochlitz, C. , Buess, M. , 2012. Global gene expression analysis of the interaction between cancer cells and osteoblasts to predict bone metastasis in breast cancer. PLoS One. 7, (1) e29743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand, J. , Kull, M. , Peterson, H. , Hansen, J. , Vilo, J. , 2007. g:Profiler – a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res.. 35, (Web Server issue) W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand, J. , Arak, T. , Vilo, J. , 2011. g:Profiler–a web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res.. 39, (Web Server issue) W307–W315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman, P. , Horlings, H.M. , Krijgsman, O. , Kok, M. , Bueno-de-Mesquita, J.M. , Bender, R. , Linn, S.C. , Glas, A.M. , van de Vijver, M.J. , 2009. Microarray-based determination of estrogen receptor, progesterone receptor, and HER2 receptor status in breast cancer. Clin. Cancer Res.. 15, (22) 7003–7011. [DOI] [PubMed] [Google Scholar]
- Smoot, M.E. , Ono, K. , Ruscheinski, J. , Wang, P.L. , Ideker, T. , 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 27, (3) 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano, D. , Zollo, M. , 2012. Tumor microenvironment: a main actor in the metastasis process. Clin. Exp. Metastasis. 29, (4) 381–395. [DOI] [PubMed] [Google Scholar]
- Symmans, W.F. , Hatzis, C. , Sotiriou, C. , Andre, F. , Peintinger, F. , Regitnig, P. , Daxenbichler, G. , Desmedt, C. , Domont, J. , Marth, C. , 2010. Genomic index of sensitivity to endocrine therapy for breast cancer. J. Clin. Oncol.. 28, (27) 4111–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani, R. , Hastie, T. , Narasimhan, B. , Chu, G. , 2002. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. U S A. 99, (10) 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanskaya, O. , Cantor, M. , Sherlock, G. , Brown, P. , Hastie, T. , Tibshirani, R. , Botstein, D. , Altman, R.B. , 2001. Missing value estimation methods for DNA microarrays. Bioinformatics. 17, (6) 520–525. [DOI] [PubMed] [Google Scholar]
- van't Veer, L.J. , Dai, H. , van de Vijver, M.J. , He, Y.D. , Hart, A.A. , Mao, M. , Peterse, H.L. , van der Kooy, K. , Marton, M.J. , Witteveen, A.T. , 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415, (6871) 530–536. [DOI] [PubMed] [Google Scholar]
- van de Vijver, M.J. , Peterse, J.L. , Mooi, W.J. , Wisman, P. , Lomans, J. , Dalesio, O. , Nusse, R. , 1988. Neu-protein overexpression in breast cancer. Association with comedo-type ductal carcinoma in situ and limited prognostic value in stage II breast cancer. N. Engl. J. Med.. 319, (19) 1239–1245. [DOI] [PubMed] [Google Scholar]
- van de Vijver, M.J. , He, Y.D. , van't Veer, L.J. , Dai, H. , Hart, A.A. , Voskuil, D.W. , Schreiber, G.J. , Peterse, J.L. , Roberts, C. , Marton, M.J. , 2002. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med.. 347, (25) 1999–2009. [DOI] [PubMed] [Google Scholar]
- Vaske, C.J. , Benz, S.C. , Sanborn, J.Z. , Earl, D. , Szeto, C. , Zhu, J. , Haussler, D. , Stuart, J.M. , 2010. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 26, (12) i237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt, B. , Peterse, J.L. , van 't Veer, L.J. , 2005. Breast cancer metastasis: markers and models. Nat. Rev. Cancer. 5, (8) 591–602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data