

Abstract
Background
Previous studies have shown successful antitumor effects of systemically delivered double‐deleted vaccinia virus (vvDD) against a number of adult tumor models, including glioma, colon and ovarian cancers. The purpose of this study was to investigate the oncolytic potential of vvDD against a panel of cell lines representative of pediatric solid tumors that are currently difficult to cure.
Methods
Cell lines derived from central nervous system atypical teratoid rhabdoid tumor (AT/RT) (BT12, BT16 and KCCF1), sarcoma (143B, HOS, RD and RH30), and neuroblastoma (SKNAS, SKNBE2, IMR‐5 and IMR‐32) were examined for vvDD mediated cytotoxicity defined by virus expansion followed by loss of tumor cell viability. The normal human fibroblast cell line HS68 was used as a control. Next, relevant orthotopic, subcutaneous and lung metastasis xenograft models were treated with intravenous doses of live vvDD or killed virus controls (DV). Tumor growth inhibition and viral replication were quantified and survival outcomes of these animals were assessed.
Results
vvDD was able to infect and kill nine of eleven of the pediatric tumor cells (81.8%) in vitro. In xenograft models, intravenous administration of a single dose of vvDD significantly inhibited the growth of tumors and prolonged the survival of intracranial and metastatic tumors.
Conclusions
Oncolytic vvDD administered i.v. shows activity in preclinical models of pediatric malignancies that are resistant to many currently available treatments. Our data support further evaluation of vvDD virotherapy for refractory pediatric solid tumors.
Keywords: Oncolytic virus, Double-deleted vaccinia virus, Pediatric tumors, Sarcoma, Neuroblastoma, Atypical teratoid rhabdoid tumors (AT/RT)
Highlights
This study aims to evaluate the feasibility of oncolytic virotherapy for chemotherapy resistant pediatric solid tumors.
In vitro studies show viral replication and cytotoxicity of double‐deleted vaccinia virus (vvDD) in pediatric tumor cells.
Experiments in xenograft models provide data to further evaluate vvDD in the treatment of refractory pediatric malignancies.
1. Introduction
Recent decades have seen significant advances in the overall survival rates of children with cancer. However, improving cure rates in several intractable pediatric solid tumors, particularly high risk neuroblastoma, infant brain tumors and metastatic sarcomas, still remains a challenge, with exceptionally high rates of failure under current treatment modalities. The escalation of conventional chemotherapeutic drugs and their combinations to induce clinical remission often leads to unacceptable toxicities. Consequently, such relapsed or refractory malignancies have become a leading cause of morbidity and mortality in children and young adults, requiring the search for differing therapeutic approaches (Macy et al., 2008).
A number of recent studies have provided evidence for the utility of oncolytic viruses (OVs) as means to deliver effective cytotoxic therapy to tumors that have become resistant to conventional treatment approaches (Russell et al., 2012; Bourke et al., 2011; Hammill et al., 2010). The strategy behind this approach is to develop viruses that can replicate specifically in cancer cells and cause cell lysis, while leaving non‐malignant cells unaffected. The ability of OVs to effectively spread and reach distant invasive metastatic tumor cells may further aid in their application in patients with highly aggressive tumor phenotypes. Recently completed clinical trials of several different OVs (adenovirus, reovirus, measles, herpes simplex, Newcastle disease virus, vaccinia virus and parvovirus) have demonstrated acceptable safety and tolerability of OVs in patients (Hammill et al., 2010; Donnelly et al., 2012). However, there are no substantive preclinical data on the expediency of OVs in pediatric tumors (Morton et al., 2010; Friedman et al., 2009; Studebaker et al., 2010). Although limited results have been observed in preclinical models of pediatric cancers, their translation into clinical trials has not occurred. Hence, research studies are needed to address the applicability of OVs to the unmet needs in refractory pediatric tumors, while drawing on the rapid advances in the field of viral oncolytics in adult trials to achieve practical benefits for these children in a timely manner.
Previously, we have described preclinical data using distinct xenograft models to study the utility of OVs such as Myxoma Virus (MYXV) and vesicular stomatitis virus (VSV, deltaM51) for activity against brain tumors (Lun et al., 2010, 2005, 2009, 2010, 2008). However, the double deleted vaccinia virus (vvDD) provides unique benefits and has been explored in the current study for therapeutic potential against refractory pediatric solid tumors. Vaccinia virus (VV) is a double‐stranded, enveloped, lytic DNA virus with several desirable attributes as a therapeutic agent over other OVs in development: 1. The clinical safety profile of the virus has been established from its accepted use as a vaccine in the smallpox eradication program. 2. Its contraindications and adverse reactions have been well identified and effective antivirals are available. 3. Previous studies have shown that VV is amenable for systemic (intravenous) delivery to distant tumors, making it an attractive agent for the treatment of pediatric tumors with metastases. Furthermore, there is significant evidence for its potential as an effective oncolytic agent from different preclinical studies in a number of adult tumor models (Lun et al., 2012, 2012, 2009, 2010, 2009). This has led to the initiation of early phase clinical trials using VV (JX‐594) against adult patients (Donnelly et al., 2012; Park et al., 2008). The generation and use of a ‘‘double‐deleted’’ version of the Western Reserve (WR) strain [double‐deleted VV (vvDD)] with deletions of the thymidine kinase and vaccinia growth factor genes further enhances the safety profile of this agent (Park et al., 2008). In addition, vvDD was demonstrated to be nontoxic following i.v. delivery in nonhuman primates (Naik et al., 2006).
Based on the feasibility data provided by adult tumor studies (He et al., 2012; Haddad et al., 2012; Merrick et al., 2009; Park et al., 2008), we wanted to investigate the activity of vvDD against cell lines from refractory pediatric solid tumors. The objectives of this study were to determine the susceptibility and efficacy of vvDD against pediatric solid tumors in vitro and in vivo. Here we show for the first time that diverse pediatric solid tumor cell lines are susceptible to infection and cytotoxicity by vvDD in vitro, and single i.v. administration of the vvDD is capable of significantly inhibiting tumor growth in tumor xenograft models. We also demonstrate that systemic administration of vvDD in tumor bearing immunocompromised mice is well tolerated. The results of this study suggest that further investigation of this virus in the treatment of pediatric solid cancers is warranted.
2. Materials and methods
2.1. Cell lines and cell culture
BT12 and BT16 cell lines were established from infants with atypical teratoid/rhabdoid tumors or the central nervous system (CNS AT/RT) and generously provided by Drs. Peter Houghton (Nationwide Children's Hospital, Columbus, OH) and Jaclyn Biegel (Children's Hospital of Philadelphia, PA). The cell line KCCF1 was established in our laboratory from the cerebral spinal fluid (CSF) cells of a two‐month‐old male infant with AT/RT. Characterization of this cell line has been described previously (Narendran et al., 2008). The HS68 primary skin fibroblast cells were provided by Dr. Peter Forsyth's laboratory at the University of Calgary. The neuroblastoma cell lines were provided by Dr. Herman Yeger (The Hospital for Sick Children, Toronto, ON). The remaining cell lines were obtained from ATCC. All cell lines were routinely tested before animal studies for mycoplasma contamination.
Cell lines were cultured in Opti‐MEM medium (Gibco, Invitrogen Corporation, Burlington, ON) containing 5% fetal bovine serum (FBS, Gibco), 100 units/ml penicillin and 100 units/ml streptomycin (Gibco). Cells were trypsinized with 0.25% Trypsin‐EDTA in Ca2+ and Mg2+ free balanced salt solution (Gibco) every three to five days and maintained in incubators at 37 °C in a humidified atmosphere with 5% CO2.
2.2. Double‐deleted vaccinia virus (vvDD)
A mutant attenuated “double‐deleted” version of the Western Reserve strain [double‐deleted vaccinia virus (vvDD)] with deletions of the thymidine kinase and vaccinia growth factor genes was created initially at the Ottawa Hospital Research Institute, Canada (JB). Propagation and titration in U2OS or BHK cells were carried out as described previously (Lun et al., 2009, 2010).
2.3. Cell viability assay
To determine whether pediatric tumor cells are susceptible to vvDD in vitro, AT/RT, sarcoma and neuroblastoma cells were plated in 96 well plates (Nunc, Rochester, NY) at a concentration of 5 × 103 cells per well and infected with different doses of vvDD (MOI = 0, 0.01, 0.1, 1 and 10). After four days in culture, the numbers of viable cells were determined by Alamar Blue assay (Invitrogen) according to the manufacturer's protocol.
2.4. In vitro viral replication assay
Tumor cells and control HS68 cells were infected with vvDD at a MOI of 0.01 and after 0, 24, 48, 72, 96 and 120 h of incubation, cell lysates were prepared by three cycles of freeze/thawing. Serial dilutions of supernatants and cell lysates were cultured on confluent layers of U2OS cells, viral plaques were counted, and plaque‐forming unit were calculated by the number of plaques multiplied by the dilution factor (Lun et al., 2009, 2010). All experiments were replicated at least three times.
2.5. Generation of BT16, SKNAS and 143B cells stably expressing enhanced firefly luciferase and mCherry or eGFP
BT16, SKNAS and 143B cell lines expressing enhanced firefly luciferase (effLuc) and mCherry were generated using a self‐inactivating lentiviral vector encoding the internal U3 region from mscv, effLuc, the IRES element from emcv, and eGFP (or mCherry) (Bai et al., 2011). Virus was packaged in 293‐FT cells using pMD2.G (VSV.G env) and pCMV‐deltaR8.91 and concentrated 50x using Amicon Ultra‐15 100,000 NMWL centrifugal concentration units (Millipore, Billerica, MA). Concentrated viral supernatants were used for transduction of BT16, SKNAS and 143B cell lines. After 72 h, eGFP or mCherry expression was observed via fluorescence microscopy. EffLuc based bioluminescent activity was calculated using an IVIS 200 (Caliper Life Sciences, Alameda, CA).
2.6. Animal experiments
All animal work procedures were carried out in accordance with the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals, issued by NIH. All protocols were reviewed and approved by the Animal Care Committee of the University of Calgary. Six to eight week‐old female CD‐1 mice and CB17 SCID mice (Charles River Laboratories, Wilmington, MA) were used in this study.
2.7. In vivo viral distribution studies in an AT/RT orthotopic xenograft model
AT/RT intracranial animal model established with BT16GFPFluc cells and the stereotactic techniques used to implant BT16GFPFluc cells in the right putamen have been described previously (Studebaker et al., 2010). Briefly, mice were anesthetized, a burr hole was drilled through a scalp incision, and BT16GFPFluc cells (1 × 105 cells/mouse) were inoculated under guidance of a stereotactic frame (Kopf Instruments). Three weeks later, animals were imaged to confirm the growth of tumor. The imaging process was carried out by the Xenogen IVIS 200 system to record bioluminescent signal emitted from tumors. Data were analyzed based on total photon flux emission (photons/s) in the region of interest over the intracranial space as reported previously (Alain et al., 2010). Following confirmation of tumor establishment, a single intravenous administration of 5 × 107 PFU/mouse of either vvDD or dead virus (DV) was given. Dead virus was prepared by UV exposure of live virus for 2 h.
Animals were sacrificed at 2 weeks after virus infection. At that time, the animals were perfused with sterile PBS and the brain tumor tissues were either saved frozen for viral culture or embedded for H&E staining and immunohistochemistry. Protein imaging of GFP tumor and mCherry virus were visualized using a Leica MZ‐FLIII fluorescence stereomicroscope equipped with 100‐W mercury‐vapor burner and mounted with a Kodak DC 2900 digital camera (Studebaker et al., 2010; Lun et al., 2005; Wu et al., 2008). Images were processed and analyzed by Photoshop 8.0 and Image‐Pro Plus software.
2.8. In vivo efficacy studies in BT16GFPFluc intracranial animal model in CD‐1 nude mice
Tumor progression in live virus and control dead virus treated animals was evaluated by the Xenogen IVIS 200 system (Xenogen Corporation, Alameda, CA). These mice were imaged to record bioluminescent signal emitted from tumors and data were analyzed based on total photon flux emission (photons/s) in the region of interest (ROI) over the intracranial space as per established methods (Lun et al., 2010; Szentirmai et al., 2006). Animals were sacrificed two weeks from the day of virus treatment, an arbitrary time point established based on our preliminary studies. Brain specimens from all experimental animals were prepared and gross and histological examinations were performed to confirm tumor dimension. For experiments assessing survival, animals were monitored for 100 days. However, animals were sacrificed before this time if body mass decreased by ≥20% or when difficulty with ambulation, feeding or grooming was reported.
2.9. In vivo antitumor efficacy of vvDD in neuroblastoma cell line‐SKNAS bearing subcutaneous tumor model
Female CB17 SCID mice (n = 10, 6–8weeks) were implanted with 5 × 106 cells/mouse of the SKNASmCherryFluc on the right flank to establish sub‐cutaneous (SC) tumors. Fourteen days after tumor implantation, tumor progression was measured by bioluminescence imaging (BLI) (Xenogen IVIS 200) or calipers (length × width) every week. Animals were then assigned randomly into two groups and treated with a single intravenous dose of 5 × 107 PFU/mouse of vvDD or an equivalent dose of killed virus. Five animals for each group were sacrificed after three weeks of treatment and tumor tissue was removed and subjected to growth measurements and histological examination.
2.10. In vivo activity of vvDD in the growth and lung metastasis of 143B sarcoma cells
Previous reports have shown that the osteosarcoma cell line 143B has significant cell motility, invasion and anchorage independent growth properties and provide an efficient xenograft model for invasive disease (Luu et al., 2005). In this study, CB17 SCID mice (n = 14) were given 3 × 105 143BmCherryFluc cells intravenously to establish a lung metastasis animal model. Intermittent imaging analysis indicated that lung metastasis is completed by ten to fourteen days. At this time, the animals (n = 10) were randomly assigned to two groups to receive either an intravenous dose of vvDD (5 × 107 PFU/mouse) or control killed virus. Progression of tumor growth was monitored weekly using BLI (Xenogen IVIS 200) for the first three weeks after virus treatment, then daily for the survival monitoring. As indicated above, animals were sacrificed sooner if significant physiological impairments were noted.
2.11. Data analysis
Statistical Analysis Software (SAS Institute, Cary, NC) and GraphPad Prism (Version 5; GraphPad Software, La Jolla, CA) were used for statistical analyses. Survival curves were generated by the Kaplan–Meier method. The Student's t test was used when appropriate. Data were expressed as means ± SD. The multiple group average data were analyzed with the two‐way analysis of variance. All reported P values were two‐sided and were considered to be statistically significant at P < 0.05.
3. Results
3.1. Infectivity of vvDD in pediatric solid tumor cell lines
As the first step in the evaluation of vvDD as an effective agent against refractory pediatric solid tumors, we investigated the potential of the virus to infect and replicate in tumor cell lines. The Hs68 cells, which do not permit the replication of vvDD, were used as a negative control. Cells were treated with 0.01 MOI of virus and the resulting virus titers were measured at various time intervals. Data presented in Figure 1 show an increase in viral titers in all of the cell lines tested with peak levels expressed at 72 h or 96 h for AT/RT and sarcoma cell lines. Decreases in titers beyond these time points are potentially due to the loss of available tumor cells for viral propagation. Maximum virus production occurred at the 120 h time point for NB cell lines under our experimental conditions (Figure 1C and Supplementary Fig S1). There was also a delay in the kinetics of vvDD replication in neuroblastoma cell lines (Figure 1C).
Figure 1.
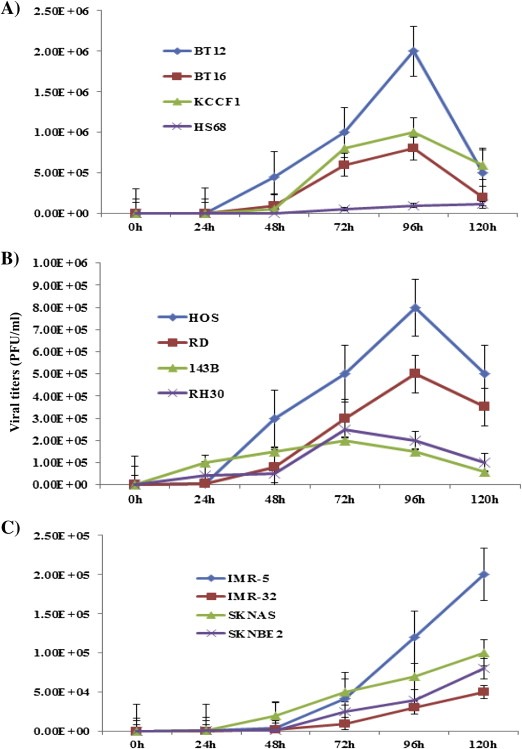
Analysis of the permissiveness and the replication kinetics of vvDD in pediatric solid tumor cells. AT/RT (A), HS68 human fibroblasts (A), sarcoma (B) and neuroblastoma (C) cells were infected with 0.01 MOI of vvDD and cell lysates were prepared by freeze‐thaw at the indicated time intervals. The viral titers were determined by plaque assay on confluent U2OS cells. Each assay was carried out in triplicate. Data are representative of two separate experiments.
3.2. Infection by vvDD leads to effective cytotoxicity in pediatric tumor cell lines
We then evaluated the ability of vvDD to induce cytotoxicity in the panel of tumor cells. Exponentially growing tumor cells were treated with increasing concentrations of vvDD and after four days in culture, viable cells were quantified. Significant growth inhibition was observed in a dose‐dependent manner in 10 of the 11 cell lines tested (Figure 2). The neuroblastoma cell line IMR32 appears to be resistant to cytotoxicity, showing 60% cell survival at high viral titers. This might be partially attributed to a delayed viral production in NB cells. When treated for a longer time, IMR32 cells were effectively killed by vvDD (Figure S1B). In contrast, killed virus (DV) did not induce cytotoxicity in selected tumor cells (Figure S2).
Figure 2.
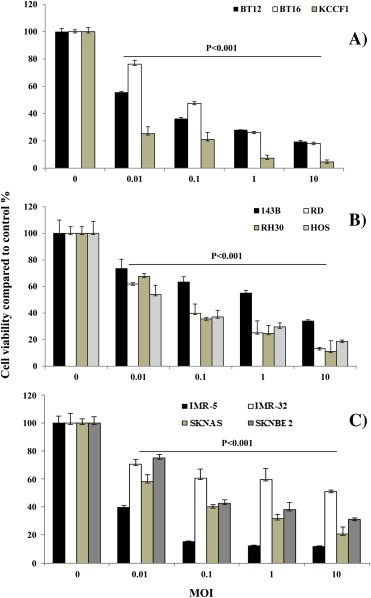
Dose dependent growth inhibition of pediatric solid tumor cell lines by vvDD. Three AT/RT cell lines (A), four osteosarcoma lines (B) and four neuroblastoma lines (C) were treated with increasing concentrations of vvDD. After four days in culture, cell viability under each condition was quantified by Alamar blue assay. The percentage of viable cells compared to untreated wells was plotted. Data in the figure are representative of two separate experiments and are shown as mean of triplicate wells ± standard deviation.
3.3. Infection and induction of cell death by vvDD in pediatric solid tumors in vivo
As a result of the data indicating that vvDD has infective and cytotoxic potential in vitro, we wanted to investigate how this may translate to tumor control in vivo. For these studies, we aimed to examine: 1. following the administration of the virus intravenously, its capacity to reach and subsequently inducing cytotoxicity in brain tumors; 2. the ability of the virus to induce significant regression of orthotropic tumors; and 3. the ability of the virus to suppress the spread of lesions in aggressive metastatic tumor phenotypes.
As a test of these concepts, we selected three experimental models from our panel of cell lines. The AT/RT cell line BT16 was used to establish an experimental model to evaluate the effects of virus homing and cytotoxicity in intracranial lesions. The SKNAS neuroblastoma cell line was used to examine the effects on orthotropic tumors. Lastly, the highly metastatic sarcoma cell line 143B was used to evaluate the ability of the virus to suppress the lung lesions formed by intravenous inoculation of tumor cells.
Data presented in Figure 3 show the successful establishment of brain lesions by BT16 cells and the effective suppression of these lesions by intravenous injection of vvDD (3A, 3B). Compared to the dead virus control, the tumors were smaller on day seven, with no additional growth observed on day fourteen after virus treatment. However, a continued increase in tumor volume was noted in control animals. In addition, we also found that viral replication occurs in areas occupied by the tumor and outside of the normal brain tissue. Total brain tissue from these animals was removed and subjected to simultaneous imaging with GFP (tumor) and RFP (viral replication). Representative photographs from two brain specimens presented in Figure 3C show the enhanced RFP activity in areas with GFP (white arrows).
Figure 3.
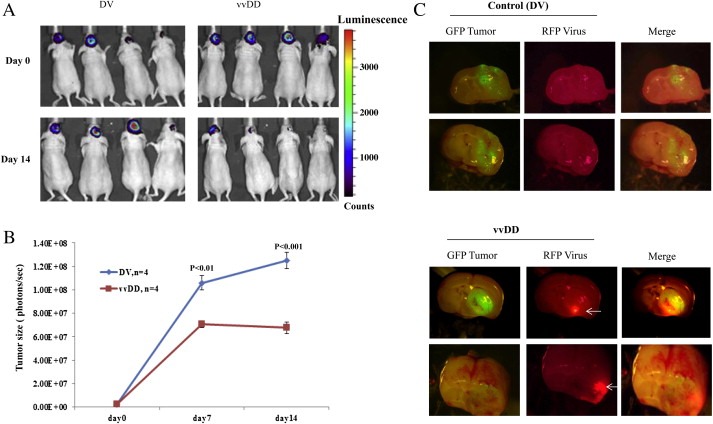
vvDD inhibits the growth of intracranial AT/RT tumors in CD‐1 nude mice. BT16mGFPfLuc cells were stereotactically implanted into the brain of CD‐1 nude mice. After allowing two weeks for tumors to establish, mice were randomized into two groups (four animals each) that received a single intravenous dose of either live (vvDD) or killed vvDD (DV). The sizes of the tumors were quantified by the bioluminescent signals emitted from the tumors using the Xenogen IVIS 200 system as described in methods (A). The growth of the tumors was monitored at weekly intervals and plotted (B). Two weeks after treatment, the animals were sacrificed and photomicrographs of the brain tissues were taken to detect GFP‐expression (green, tumor) and mCherry signals (red, virus). Representative pictures are given (C).
We also evaluated the effect of vvDD on SKNAS neuroblastoma tumors established in the flank of the animals (Figure 4). Loss of tumor growth was noted in all animals that received vvDD in comparison to animals that received dead virus. As seen with BT16 cells, an initial growth of the tumors on day seven did not advance further in treated animals compared to control.
Figure 4.
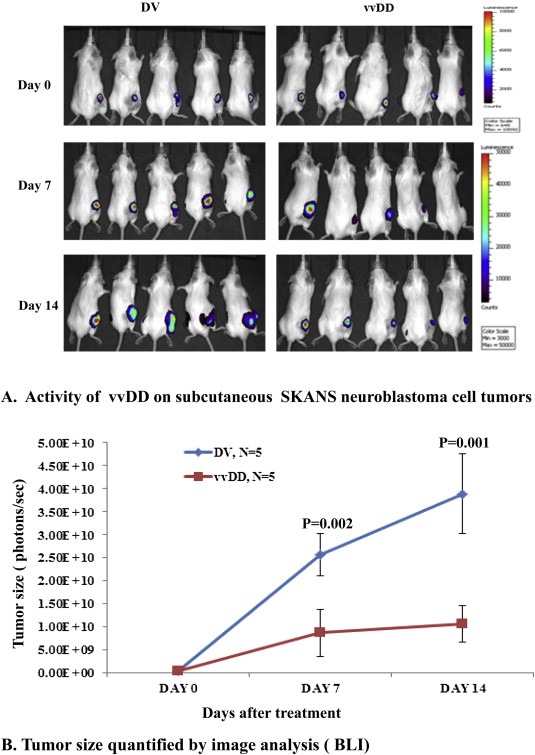
In vivo activity of vvDD against neuroblastoma xenografts. CB17 SCID mice received SKNASmCherryfLuc neuroblastoma cells in the right flank to establish subcutaneous tumors. Bioluminescence imaging was carried out weekly to confirm tumor growth. Two weeks after tumor implantation, animals were assigned randomly into two groups to receive a single intravenous dose of 5 × 107 PFU/mouse of vvDD or an equivalent dose of killed virus. Bioluminescent imaging was carried out at weekly intervals (A) and after approximately two weeks of receiving treatment, the animals were sacrificed and the size of tumor tissues was assessed (B).
To evaluate whether vvDD has effects against tumor metastasis, we established a lung metastasis model using 143B osteosarcoma cells (Figure 5). Tumor cells were given intravenously and formed, as described by other investigators, significant lung lesions (Figure 5A). The animals were then randomized to receive either vvDD or an equal concentration of dead virus control. These studies show that vvDD significantly reduced the growth of lung lesions formed by 143B osteosarcoma cells (Figure 5 A, B).
Figure 5.
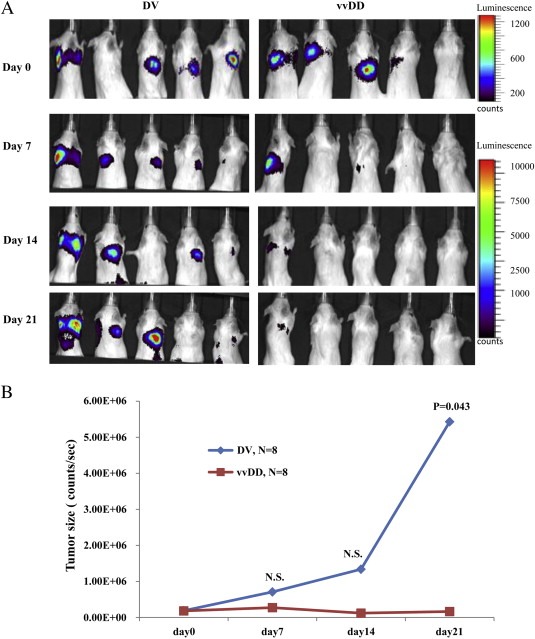
Evaluation of vvDD activity in the metastatic sarcoma experimental model. 143B cells labeled with mCherryFluc were injected intravenously through the tail vein of CB17 SCID mice to generate a commonly used model of lung metastatic lesions. Imaging analysis showed progressively increasing lung lesions in these animals. After allowing 14 days for the lesions to establish, the animals received a single dose of vvDD or killed virus (5 animals per group). Bioluminescent imaging was carried out at weekly intervals for up to 3 weeks (A) and corresponding tumor size measurements were made from the images (B) (N.S.: not statistically significant).
In addition, we also assessed the overall survival rates of tumor bearing animals treated with vvDD or dead virus control. Animals were followed for 80–150 days and their corresponding survival data are presented in Figure 6. In all three cases, survival rates were significantly increased in vvDD treated animals compared to controls.
Figure 6.
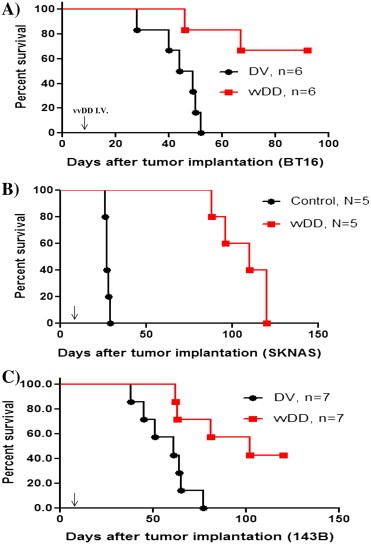
vvDD treatment prolongs the survival of tumor bearing mice. Immunocompromised mice were xenografted with AT/RT, neuroblastoma, or metastatic sarcoma cells. Animals were randomized and treated with a single dose of vvDD (5 × 107 PFUs/mouse) or an equivalent dose of killed virus. Animal survival was followed for 3–5 months. The data are presented as Kaplan–Meier curves and show that vvDD treatment increased median survival of animals bearing (A) AT/RT (nu mice, intracranial tumor model, P = 0.049, log‐rank test), (B) neuroblastoma (CB17 mice, subcutaneous model, P = 0.0018, log‐rank test) and (C) metastatic sarcoma (CB17 mice, lung metastatic model, P = 0.007, log‐rank test). Arrows indicate time of viral inoculation.
4. Discussion
In the last fifty years, clinical trials, combination therapy regimens and advances in surgical and radiation techniques have provided a significant increase in the overall survival of pediatric cancer patients. In spite of these improvements, cancer remains the most common cause of death between late infancy and early adulthood. In particular, patients with metastatic and/or relapsed disease continue to endure extremely poor outcomes, despite the use of intense treatment regimens that often carry significant risk, including acute toxicity and long term side effects. In many instances, further increases in dose intensity or drug combinations are no longer viable options due to uncertain efficacy and intolerable toxicities. Hence, novel approaches for the treatment of refractory pediatric malignancies are needed.
The purpose of this study was to determine if there is pre‐clinical evidence that i.v. vvDD be useful in selected pediatric solid tumors. Preferably, these preclinical models must determine: 1. is vvDD capable of replication and subsequent cytotoxicity in cell lines established from refractory pediatric solid tumors; and 2. is vvDD able to target and replicate in both the primary and metastatic tumors in vivo, following i.v. administration. We show here that vvDD infects, replicates and induces cell lysis in a majority of cell lines. Overall, 11 tumor cell lines were studied, representing a spectrum of molecular abnormalities and growth characteristics seen in their respective tumor types in patients. Interestingly, even within the same tumor type, significant variations in sensitivity were seen among cell lines. For example, among the NB cell lines, greater sensitivity to vvDD was seen in IMR5 cells, whereas IMR32 cells appear to show relative resistance to the virus. Similarly, the metastatic OS cell line 143B showed lower tumor lysis compared to other OS cell lines, although dose dependent cell killing was noted (Figure 2). In general, a correlation was noted between the ability of the virus to replicate (as shown by viral titers, Figure 1) and the relative tumor cytotoxicity in different cell lines, suggesting that the oncolytic effects are related to viral activity. Nevertheless, there were a few outliers which implicated an alternative mechanism of cell killing. For example, vvDD replicated in RH30 and 143B comparably, and yet, RH30 were sensitive to cytotoxicity. IMR32 had similar replication kinetics as SK‐N‐BE2, and yet, was more resistant to virus killing than the latter.
The molecular mechanisms responsible for the variations in susceptibility of tumor cells to oncolytic viruses have not been completely elucidated. Interestingly, a report by Tumilowicz and colleagues published more than forty years ago demonstrated the relative resistance of IMR32 cells to viruses, particularly in their experiments with the human enterovirus group (Tumilowicz et al., 1970). Some molecular characteristics of IMR32 cells as the possible critical determinant of viral infection comes from studies that showed that after treatment with all‐trans retinoic acid (RA), IMR32 cells became significantly more sensitive to Newcastle disease virus (NDV) (Reichard et al., 1992). Similarly, the potential mechanisms behind viral susceptibility of IMR5 cells have been investigated previously. For example, polio virus (PV) infection of IMR5 cells has been shown to result in c‐Jun N‐terminal kinase (JNK)‐mediated Bax dependent apoptosis (Autret et al., 2007). It has also been shown that an increase in mitochondrial Ca2+ concentration in IMR5 cells by PV contributes to mitochondrial dysfunction and apoptosis (Brisac et al., 2010). Although vaccinia virus infects most of the tumors studied, the molecular mechanisms that govern tumor selectivity are yet to be identified. vvDD depends on the availability of viral transformation genes present in the tumor cells for their replication (Kirn and Thorne, 2009). For example, thymidine kinase (TK) is almost undetectable in normal cells at G0 phase, whereas this kinase is constitutively over‐expressed regardless of cell cycle stages in many cancers (Hengstschlager et al., 1994). Such variation may be one of the factors that determine the efficacy of vvDD in different tumors. Identifying new markers of vvDD activity or resistance will help to stratify patients to maximize the effectiveness of these agents in clinical studies.
Brain tumors, such as AT/RT, constitute a group of most difficult to cure malignancies in pediatrics. OVs have been investigated in several preclinical and clinical trials (Russell et al., 2012; Donnelly et al., 2012). Initial clinical studies using oncolytic viruses to treat brain tumors clinical studies have not produced major improvements in outcome. Many of studies have used intratumoral administration of the virus which essentially confines the treatment to a single administration (Geletneky et al., 2012). Of consequence to clinical usefulness, poxviruses possess a number of attributes as a therapeutic agent, including their capability for rapid replication, systemic spread, and demonstrated clinical safety (Kirn et al., 2008). We have also demonstrated that i.v. vvDD localizes to intracranial tumors and metastatic lesions (Figures 3, 5). Previous studies have shown that intratumoral injection of poxviruses can lead to measurable tumor regression (Park et al., 2008; Mastrangelo et al., 1999). In addition, these viruses have the propensity to rapidly spread for effective target localization, thus providing an advantage for the treatment of multiple metastatic lesions.
Importantly, the ability of vaccinia virus with appropriate vectors to target tumors of distinct phenotypes provides a significant therapeutic advantage to their use in a range of distinct tumor types (Rojas and Thorne, 2012). Traditionally, the host antiviral immune response has been considered to be a major limitation to oncolytic therapies. However, it is becoming increasingly evident that by virtue of its ability to overcome immune suppression in the tumor locale, the antiviral immune response may be harnessed for considerable therapeutic advantage. For example, JX‐594 (Pexa‐Vec), that functions as an oncolytic as well as an immunotherapeutic vaccinia virus, has been evaluated in a number of clinical studies. Additionally, the immune regulatory components of virotherapy can also be extended to generate long term immune surveillance against tumor recurrence. However, the major experimental limitation of our analysis is the use of immune compromised animals. Nevertheless, a number of innovative experimental strategies are being developed to reduce the impact of this drawback (Chillón et al., 1998; Beer et al., 1998). Also, smallpox immunization was discontinued in 1978, thus essentially eliminating pre‐existing immunity to these viruses in the pediatric population.
In our study, a single administration of vvDD results in significantly decreased tumor progression and prolonged animal survival. It has been shown previously that the oncolytic poxvirus JX‐594 can selectively infect tumor tissue after intravenous infusion, in a dose‐related fashion. Normal tissues were not affected clinically (Breitbach et al., 2011). Whether multiple doses of vvDD will be more effective is unclear. It has been shown previously that in the glioma model, animals treated with multiple doses of vvDD did not demonstrate longer survival compared to those that received single dose (Lun et al., 2009). In a recent publication, Heo and colleagues have reported the findings of a randomized phase 2 dose‐finding trial (n = 30) of patients with advanced hepatocellular carcinoma (HCC). In this study JX‐594 demonstrated oncolytic and immunotherapeutic tumor responses and dose‐related survival in individuals with HCC (Heo et al., Feb 10 2013).
The in vivo studies presented in this report involved significantly immunocompromized hosts. In patients, the ultimate clinical utility of this approach can be compromised by the immune response generated against the virus. However, our previous studies have found evidence for activity of vvDD in immunocompetent animals (Lun et al., 2009). Furthermore, immunosuppressive agents such as rapamycin or cyclophosphamide are able to enhance vvDD replication and promote oncolysis (Lun et al., 2009). Additional studies are needed to evaluate the specific role of the host immune system and its interaction with vvDD in the net clinical efficacy of this treatment approach.
The rapid growth in knowledge of direct and immune system mediated antitumor activity of viruses in the past decade has helped to advance the field of viral oncolytics significantly and has brought about the realization of this approach as a feasible alternative in treating malignancies that are currently considered to be incurable. Consequently a number of viruses have already entered or are being actively evaluated for early phase clinical trials in children. Leading among these are modified forms of the vaccinia virus that carry a number of key attributes that make it a sound choice for active investigation for future therapeutics in pediatric patients. As the essential first step in this process, we show here that vvDD can infect and replicate in cell lines derived from pediatric tumors and demonstrate its tolerability and ability to localize in intracranial and metastatic tumors following intravenous administration. As viral infection is highly dependent on host tumor tissue properties, our data on multiple tumor lines from three distinct pediatric tumors provide supportive information for further studies to develop vvDD in future therapeutic opportunities in refractory childhood malignancies.
Supporting information
The following are the supplementary data related to this article:
Figure S1 Replication kinetics of vvDD in neuroblastoma cells. Three neuroblastoma cell lines (IMR5, IMR32, and SK‐N‐AS) were infected with 0.01 MOI of vvDD and cell lysates were prepared at the indicated time intervals. The viral titers were determined by plaque assay on U2OS cells. The replication of vvDD peaked at 120 h and started to decline afterward. In parallel, infected cells were monitored for viability using Alamar blue assay. Massive cell death were observed by day 5 and all cells were killed by virus by day 7.
Figure S2 Killed viruses do not present in vitro cytotoxicity on selected tumor cell lines. BT12 and IMR5 cells were treated with 1 MOI of killed virus (DV). After 96 h, the cell viability was measured using Alamar blue assay.
Acknowledgments
This research was supported by grants from the The Morgan Adams Foundation, Brain Tumour Foundation of Canada, Alberta Children's Hospital Foundation (ACHF) and the Kids Cancer Care Foundation of Alberta (KCCFA).
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.05.004.
Lun Xueqing, Ruan Yibing, Jayanthan Aarthi, Liu David J., Singh Anjali, Trippett Tanya, Bell John, Forsyth Peter, Johnston Randal N., Narendran Aru, (2013), Double‐deleted vaccinia virus in virotherapy for refractory and metastatic pediatric solid tumors, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.05.004.
References
- Alain, T. , Lun, X. , Martineau, Y. , Sean, P. , Pulendran, B. , Petroulakis, E. , Zemp, F.J. , Lemay, C.G. , Roy, D. , Bell, J.C. , Thomas, G. , Kozma, S.C. , Forsyth, P.A. , Costa-Mattioli, M. , Sonenberg, N. , 2010. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc. Natl. Acad. Sci. U S A. 107, (4) 1576–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autret, A. , Martin-Latil, S. , Mousson, L. , Wirotius, A. , Petit, F. , Arnoult, D. , Colbère-Garapin, F. , Estaquier, J. , Blondel, B. , 2007. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol.. 81, (14) 7504–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, X. , Yan, Y. , Coleman, M. , Wu, G. , Rabinovich, B. , Seidensticker, M. , Alt, E. , 2011. Tracking long-term survival of intramyocardially delivered human adipose tissue-derived stem cells using bioluminescence imaging. Mol. Imaging Biol.. 13, (4) 633–645. [DOI] [PubMed] [Google Scholar]
- Beer, S.J. , Matthews, C.B. , Stein, C.S. , Ross, B.D. , Hilfinger, J.M. , Davidson, B.L. , 1998. Poly (lactic-glycolic) acid copolymer encapsulation of recombinant adenovirus reduces immunogenicity in vivo. Gene Ther.. 5, (6) 740–746. [DOI] [PubMed] [Google Scholar]
- Bourke, M.G. , Salwa, S. , Harrington, K.J. , Kucharczyk, M.J. , Forde, P.F. , de Kruijf, M. , Soden, D. , Tangney, M. , Collins, J.K. , O'Sullivan, G.C. , 2011. The emerging role of viruses in the treatment of solid tumours. Cancer Treat. Rev.. 37, (8) 618–632. [DOI] [PubMed] [Google Scholar]
- Breitbach, C.J. , Burke, J. , Jonker, D. , Stephenson, J. , Haas, A.R. , Chow, L.Q. , Nieva, J. , Hwang, T.H. , Moon, A. , Patt, R. , Pelusio, A. , Le Boeuf, F. , Burns, J. , Evgin, L. , De Silva, N. , Cvancic, S. , Robertson, T. , Je, J.E. , Lee, Y.S. , Parato, K. , Diallo, J.S. , Fenster, A. , Daneshmand, M. , Bell, J.C. , Kirn, D.H. , 2011. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 477, (7362) 99–102. [DOI] [PubMed] [Google Scholar]
- Brisac, C. , Téoulé, F. , Autret, A. , Pelletier, I. , Colbère-Garapin, F. , Brenner, C. , Lemaire, C. , Blondel, B. , 2010. Calcium flux between the endoplasmic reticulum and mitochondrion contributes to poliovirus-induced apoptosis. J. Virol.. 84, (23) 12226–12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chillón, M. , Lee, J.H. , Fasbender, A. , Welsh, M.J. , 1998. Adenovirus complexed with polyethylene glycol and cationic lipid is shielded from neutralizing antibodies in vitro. Gene Ther.. 5, (7) 995–1002. [DOI] [PubMed] [Google Scholar]
- Donnelly, O.G. , Errington-Mais, F. , Prestwich, R. , Harrington, K. , Pandha, H. , Vile, R. , Melcher, A.A. , 2012. Recent clinical experience with oncolytic viruses. Curr. Pharm. Biotechnol.. 13, (9) 1834–1841. [DOI] [PubMed] [Google Scholar]
- Friedman, G.K. , Pressey, J.G. , Reddy, A.T. , Markert, J.M. , Gillespie, G.Y. , 2009. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol. Ther.. 17, (7) 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geletneky, K. , Huesing, J. , Rommelaere, J. , Schlehofer, J.R. , Leuchs, B. , Dahm, M. , Krebs, O. , von Knebel Doeberitz, M. , Huber, B. , Hajda, J. , 2012. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. 12, 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad, D. , Chen, N. , Zhang, Q. , Chen, C.H. , Yu, Y.A. , Gonzalez, L. , Aguilar, J. , Li, P. , Wong, J. , Szalay, A.A. , Fong, Y. , 2012. A novel genetically modified oncolytic vaccinia virus in experimental models is effective against a wide range of human cancers. Ann. Surg. Oncol.. 19, (Suppl 3) S665–S674. [DOI] [PubMed] [Google Scholar]
- Hammill, A.M. , Conner, J. , Cripe, T.P. , 2010. Oncolytic virotherapy reaches adolescence. Pediatr. Blood Cancer. 55, (7) 1253–1263. [DOI] [PubMed] [Google Scholar]
- He, S. , Li, P. , Chen, C.H. , Bakst, R.L. , Chernichenko, N. , Yu, Y.A. , Chen, N. , Szalay, A.A. , Yu, Z. , Fong, Y. , Wong, R.J. , 2012. Effective oncolytic vaccinia therapy for human sarcomas. J. Surg. Res.. 175, (2) e53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengstschlager, M. , Mullner, E. , Wawra, E. , 1994. Thymidine kinase is expressed differently in transformed versus normal-cells – a novel test for malignancy. Int. J. Oncol.. 4, (1) 207–210. [DOI] [PubMed] [Google Scholar]
- Heo, J. , Reid, T. , Ruo, L. , Breitbach, C.J. , Rose, S. , Bloomston, M. , Cho, M. , Lim, H.Y. , Chung, H.C. , Kim, C.W. , Burke, J. , Lencioni, R. , Hickman, T. , Moon, A. , Lee, Y.S. , Kim, M.K. , Daneshmand, M. , Dubois, K. , Longpre, L. , Ngo, M. , Rooney, C. , Bell, J.C. , Rhee, B.G. , Patt, R. , Hwang, T.H. , Kirn, D.H. , Feb 10 2013. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med.. 10.1038/nm.3089 ([Epub ahead of print]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn, D.H. , Thorne, S.H. , 2009. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer. 9, (1) 64–71. [DOI] [PubMed] [Google Scholar]
- Kirn, D.H. , Wang, Y. , Liang, W. , Contag, C.H. , 2008. Thorne SH. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res.. 68, (7) 2071–2075. [DOI] [PubMed] [Google Scholar]
- Lun, X. , Yang, W. , Alain, T. , Shi, Z.Q. , Muzik, H. , Barrett, J.W. , McFadden, G. , Bell, J. , Hamilton, M.G. , Senger, D.L. , Forsyth, P.A. , 2005. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res.. 65, (21) 9982–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun, X.Q. , Jang, J.H. , Tang, N. , Deng, H. , Head, R. , Bell, J.C. , Stojdl, D.F. , Nutt, C.L. , Senger, D.L. , Forsyth, P.A. , McCart, J.A. , 2009. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin. Cancer Res.. 15, (8) 2777–2788. [DOI] [PubMed] [Google Scholar]
- Lun, X. , Chan, J. , Zhou, H. , Sun, B. , Kelly, J.J. , Stechishin, O. , Bell, J.C. , Parato, K. , Hu, K. , Vaillant, D. , Wang, J. , Liu, T.C. , Breitbach, C. , Kirn, D. , Senger, D.L. , Forsyth, P.A. , 2010. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol. Ther.. 18, (11) 1927–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu, H.H. , Kang, Q. , Park, J.K. , Si, W. , Luo, Q. , Jiang, W. , Yin, H. , Montag, A.G. , Simon, M.A. , Peabody, T.D. , Haydon, R.C. , Rinker-Schaeffer, C.W. , He, T.C. , 2005. An orthotopic model of human osteosarcoma growth and spontaneous pulmonary metastasis. Clin. Exp. Metastasis. 22, (4) 319–329. [DOI] [PubMed] [Google Scholar]
- Macy, M.E. , Sawczyn, K.K. , Garrington, T.P. , Graham, D.K. , Gore, L. , 2008. Pediatric developmental therapies: interesting new drugs now in early-stage clinical trials. Curr. Oncol. Rep.. 10, (6) 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo, M.J. , Maguire, H.C. , Eisenlohr, L.C. , Laughlin, C.E. , Monken, C.E. , McCue, P.A. , Kovatich, A.J. , Lattime, E.C. , 1999. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther.. 6, (5) 409–422. [DOI] [PubMed] [Google Scholar]
- JX-594 Merrick, A.E. , Ilett, E.J. , Melcher, A.A. , 2009. A targeted oncolytic poxvirus for the treatment of cancer. Curr. Opin. Investig. Drugs. 10, (12) 1372–1382. [PubMed] [Google Scholar]
- Morton, C.L. , Houghton, P.J. , Kolb, E.A. , Gorlick, R. , Reynolds, C.P. , Kang, M.H. , Maris, J.M. , Keir, S.T. , Wu, J. , Smith, M.A. , 2010. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr. Blood Cancer. 55, (2) 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik, A.M. , Chalikonda, S. , McCart, J.A. , Xu, H. , Guo, Z.S. , Langham, G. , Gardner, D. , Mocellin, S. , Lokshin, A.E. , Moss, B. , Alexander, H.R. , Bartlett, D.L. , 2006. Intravenous and isolated limb perfusion delivery of wild type and a tumor-selective replicating mutant vaccinia virus in nonhuman primates. Hum. Gene Ther.. 17, (1) 31–45. [DOI] [PubMed] [Google Scholar]
- Narendran, A. , Coppes, L. , Jayanthan, A. , Coppes, M. , Teja, B. , Bernoux, D. , George, D. , Strother, D. , 2008. Establishment of atypical-teratoid/rhabdoid tumor (AT/RT) cell cultures from disseminated CSF cells: a model to elucidate biology and potential targeted therapeutics. J. Neurooncol.. 90, (2) 171–180. [DOI] [PubMed] [Google Scholar]
- Park, B. , Hwang, T. , Liu, T.C. , Sze, D.Y. , Kim, J.S. , Kwon, H.C. , Oh, S.Y. , Han, S.Y. , Yoon, J.H. , Hong, S.H. , Moon, A. , Speth, K. , Park, C. , Ahn, Y.J. , Daneshmand, M. , Rhee, B.G. , Pinedo, H.M. , Bell, J.C. , Kirn, D.H. , 2008. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol.. 9, (6) 533–542. [DOI] [PubMed] [Google Scholar]
- Reichard, R.W. , Lorence, R.M. , Cascino, C.J. , Peeples, M.E. , Walter, R.J. , Fernando, M.B. , Reves, H.M. , Greager, J.A. , 1992. Newcastle disase virus selectively kills human tumor cells. J. Surg. Res.. 52, (5) 448–453. [DOI] [PubMed] [Google Scholar]
- Rojas, J.J. , Thorne, S.H. , 2012. Theranostic potential of oncolytic vaccinia virus. Theranostics. 2, (4) 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, S.J. , Peng, K.W. , Bell, J.C. , 2012. Oncolytic virotherapy. Nat. Biotechnol.. 30, (7) 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studebaker, A.W. , Kreofsky, C.R. , Pierson, C.R. , Russell, S.J. , Galanis, E. , Raffel, C. , 2010. Treatment of medulloblastoma with a modified measles virus. Neuro Oncol.. 12, (10) 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentirmai, O. , Baker, C.H. , Lin, N. , Szucs, S. , Takahashi, M. , Kiryu, S. , Kung, A.L. , Mulligan, R.C. , Carter, B.S. , 2006. Noninvasive bioluminescence imaging of luciferase expressing intracranial U87 xenografts: correlation with magnetic resonance imaging determined tumor volume and longitudinal use in assessing tumor growth and antiangiogenic treatment effect. Neurosurgery. 58, 365–372. [DOI] [PubMed] [Google Scholar]
- Tumilowicz, J.J. , Nichols, W.W. , Cholon, J.J. , Greene, A.E. , 1970. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res.. 30, (8) 2110–2118. [PubMed] [Google Scholar]
- Wu, Y. , Lun, X. , Zhou, H. , Wang, L. , Sun, B. , Bell, J.C. , Barrett, J.W. , McFadden, G. , Biegel, J.A. , Senger, D.L. , Forsyth, P.A. , 2008. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin. Cancer Res.. 14, (4) 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1 Replication kinetics of vvDD in neuroblastoma cells. Three neuroblastoma cell lines (IMR5, IMR32, and SK‐N‐AS) were infected with 0.01 MOI of vvDD and cell lysates were prepared at the indicated time intervals. The viral titers were determined by plaque assay on U2OS cells. The replication of vvDD peaked at 120 h and started to decline afterward. In parallel, infected cells were monitored for viability using Alamar blue assay. Massive cell death were observed by day 5 and all cells were killed by virus by day 7.
Figure S2 Killed viruses do not present in vitro cytotoxicity on selected tumor cell lines. BT12 and IMR5 cells were treated with 1 MOI of killed virus (DV). After 96 h, the cell viability was measured using Alamar blue assay.