

Abstract
C4.4A is a metastasis‐associated molecule that functions appear to rely on associated alph6beta4 integrin. To corroborate the impact of the C4.4A‐alpha6beta4 integrin association on metastasis formation, C4.4A was knocked‐down in a highly metastatic rat pancreatic adenocarcinoma (ASML, ASML‐C4.4Akd).
Metastasis formation by ASML‐C4.4Akd cells after intrafootpad application was strongly retarded in draining nodes and lung colonization was rare. Furthermore, cisplatin treatment significantly prolonged the survival time only of ASML‐C4.4Akd‐bearing rats. ASML‐C4.4Akd cells display reduced migratory activity and impaired matrix protein degradation due to inefficient MMP14 activation; loss of drug‐resistance is due to mitigated PI3K/Akt pathway activation. These losses of function rely on the laminin receptor C4.4A recruiting activated alpha6beta4 integrin into rafts, where C4.4A cooperates with alpha6beta4 and via alpha6beta4 with MMP14. Within this raft‐located complex, MMP14 provokes focalized matrix degradation and mostly alpha6beta4 integrin promotes BAD phosphorylation and upregulated Bcl2 and BclXl expression.
Thus, metastasis‐promoting activities of C4.4A are not genuine characteristics of C4.4A. Instead, the raft‐located laminin receptor C4.4A recruits alpha6beta4 integrin and supports via the alpha6beta4 integrin MMP14 activation. Thereby C4.4A acts as a linker to facilitate several steps in the metastatic cascade. Taking the restricted C4.4A expression in non‐transformed tissue, this knowledge should pave the way toward the use of C4.4A as a therapeutic target.
Keywords: Metastasis, C4.4A, alpha6beta4, MMP14, Laminin 332, PI3K/Akt
Highlights
A knockdown of metastasis‐associated C4.4A impairs metastasis and drug resistance.
Invasiveness and drug resistance are not genuine characteristics of C4.4A.
The raft‐located laminin receptor C4.4A recruits alpha6beta4 and via alpha6beta4 MMP14.
C4.4A‐associated alpha6beta4 initiates MMP14 activation provoking laminin degradation.
Drug resistance by C4.4A‐associated alpha6beta4 relies on PI3K/AKT pathway activation.
Abbreviations
- α6β4
α6β4 integrin
- ASMLwt
BSp73ASML
- ASML-C4.4Akd
C4.4A shRNA transfected ASML cells
- GPI
glycosyl-phosphatidyl inositol
- IP
immunoprecipitation
- LN111/LN332
formerly laminin 1 and 5
- LN332
concentrated serum-free and vesicle-depleted culture supernatant of 804G cells
- MMP14
membrane type 1 matrix metalloproteinase
- MMP-Inh.II
MMP9/13-Inhibitor-II, WB: Western blot
1. Introduction
C4.4A, a molecule involved in metastasis and wound repair, is a glycosyl‐phosphatidyl‐inositol (GPI) (Rösel et al., 1998) anchored glycoprotein that, like uPAR, belongs to the Ly6 family (Jacobsen and Ploug, 2008; Rösel et al., 1998; Würfel et al., 2001). C4.4A expression is restricted to basal/suprabasal layers of squamous epithelium in non‐transformed tissue (Claas et al., 1996; Hansen et al., 2004; Jacobsen and Ploug, 2008; Kriegbaum et al., 2011; Smith et al., 2001), where it becomes up‐regulated during wound healing (Hansen et al., 2004; Smith et al., 2001). High C4.4A expression has been seen in several types of carcinoma like mammary, renal cell, colorectal (Fletcher et al., 2003; Konishi et al., 2010; Paret et al., 2007; Seiter et al., 2001) and most pronounced non‐small cell lung cancer (Hansen et al., 2007; Jacobsen et al., 2012). Its expression in other types of cancer, like esophageal cancer, becomes regulated during tumor progression (Hansen et al., 2008; Wang et al., 2006). C4.4A transcription requires C/EBP and is enhanced by JunD or c‐Jun (Fries et al., 2007), fitting up‐regulated expression during wound healing (Zahnow, 2009). Laminin (LN)111, LN332 and galectin3 are C4.4A ligands (Paret et al., 2005). LN111, formerly LN1, is composed of the LNalpha1beta1gamma1 chain; LN332, formerly LN5, is composed of the LNalpha3beta3gamma2 chain. In addition, under stress conditions, C4.4A associates with alpha6beta4 integrin and MMP14, which promoted motility and invasiveness (Ngora et al., 2012).
As MMP14, like C4.4A, is located in raft domains (Holmbeck et al., 2004) and was described to contribute to laminin degradation (Oku et al., 2006), a cooperation between C4.4A and MMP14 did not appear unlikely. Instead, activation of beta4 and recruitment into rafts has mostly been described to rely on cooperation with receptor tyrosine kinases (Giancotti, 2007; Lipscomb and Mercurio, 2005; Bon et al., 2007; Streuli and Akhtar, 2009) that phosphorylate tyrosine residues of the beta4 cytoplasmic tail (de Pereda et al., 2009; Wilhelmsen et al., 2006). This creates binding sites for several signaling proteins, such that alpha6beta4 promotes motility and invasion (Bertotti et al., 2005; Mercurio and Rabinovitz, 2001; Wilhelmsen et al., 2006), but also apoptosis resistance (Bon et al., 2006; Friedland et al., 2007).
In order to sustain the importance of a cooperation of C4.4A with alpha6beta4 on metastasis formation, C4.4A was stably knocked down in a highly metastatic rat adenocarcinoma line (Matzku et al., 1983). Metastasis formation of ASML‐C4.4Akd cells was significantly delayed and metastases grew as compact foci not invading the surrounding tissue. Exploring the underlying mechanism confirmed the essential contribution of C4.4A‐associated alpha6beta4 in promoting motility and invasiveness and revealed an additional contribution to apoptosis resistance.
2. Material and methods
Tumor lines: The highly metastatic pancreatic adenocarcinoma line BSp73ASML (ASMLwt) of the BDX strain (Matzku et al., 1983) was transfected with the pSuperGFP‐neo plasmid without (ASMLmock)/with containing the sequence for C4.4A shRNA (primers Suppl. Table 1) or 4 siRNA (Rn Itgb4‐5 Cat. No. SI03045203 and Rn Itgb4 7 Cat. No. SI03108112) (Qiagen, Hildesheim, Germany) following the supplier's suggestion. Efficiency of silencing was monitored after 48 h by WB. Stable ASML‐C4.4Akd clones were established by cloning in selection medium. Two clones were used throughout, clone 34c and clone 30c. Presented data mostly derived from clone 34c and ASMLwt cells. ASMLmock cells do not differ from ASMLwt cells (Klingbeil et al., 2009). Yet, ASMLmock cells were regularly used in repetitions including a repetition of the in vivo experiments with 3 rats/group. ASML and 804G (LN332 secreting rat bladder cancer line) (Homma et al., 1985) cells were maintained in RPMI 1640/10%FCS w/wo 0.5 mg/ml G418. Confluent cultures were trypsinized and split.
Antibodies, matrix proteins and inhibitors are listed in Suppl. Table 2.
LN332 enrichment: 804G cell culture supernatant was used as source of LN332. 804G cells were cultured (48 h) in serum‐free medium. Cleared supernatants (2 × 10 min, 500 g, 1 × 20 min, 2000 g, 1 × 30 min, 10000 g, 90 min, 100,000 g) were centrifuged for vesicle‐depletion and concentrated. These serum‐free, vesicle‐depleted supernatants, highly enriched for LN332, are for brevity referred to as LN332.
Flow‐cytometry: Cells (2–5 × 105) were incubated with 50 l primary antibody at pre‐tested concentration (30 min, 4 °C), washed 3‐times, incubated with dye‐labeled secondary antibody (30 min, 4 °C) and washed again. For intracellular staining, cells were fixed and permeabilized in advance. Apoptosis was evaluated by AnnexinV‐APC/PI staining following the supplier's suggestion (BD, Heidelberg. Germany). Cells were analyzed in a FACScan using the Cell Quest analysis program (BD, Heidelberg, Germany).
Immunoprecipitation, SDS‐PAGE and WB: Cell lysates (60 min, 4C, HEPES buffer, 1% Brij96 or Lubrol, protease inhibitor cocktail < Calbiochem, Darmstadt, Germany>) were centrifuged (13000 g, 10 min, 4C), incubated with antibody (overnight) and precipitated with ProteinG Sepharose (1 h, 4C). Washed immune complexes were dissolved in Laemmli buffer. Precipitates/lysates were resolved on 10%SDS‐PAGE. Gels were stained with Coomassie Blue or proteins were transferred to nitrocellulose membranes (30 V, 12 h, 4C). Membranes were blocked, blotted with primary and HRP‐conjugated secondary antibodies (1 h, RT) and developed with the ECL kit.
Sucrose density gradient centrifugation: Cell lysates in 2.5 M sucrose were overlaid by a continuous sucrose gradient from 0.25 M to 2M and centrifuged (15 h, 150,000 g). Twelve 1 ml fractions were collected.
In vitro kinase assay: Washed immune complexes were suspended in incomplete kinase buffer (100 μM NaCl, 20 mM HEPES, pH7.4). After centrifugation, beads were resuspended in 20 l complete kinase buffer (100 mM NaCl, 20 mM HEPES, pH7.4, 5 mM MnCl2, 5 mM MgCl2, 1M ATP) supplemented with 10 μCi [32P] ‐ATP, and incubated (15 min, 37C). The reaction was stopped by 10 μl non‐reducing 6 × Laemmli buffer. SDS‐PAGE was followed by autoradiography.
Immunofluorescence and immunohistochemistry: Cells seeded on LN332‐coated cover slides were fixed, permeabilized, blocked, incubated with primary antibody (60 min, 4 °C), fluorochrome‐conjugated secondary antibody (60 min, 4 °C), blocked, incubated with a second, dye‐labeled primary antibody (60 min, 4 °C) and washed. Where indicated, cells were removed by EDTA. Cover slides were mounted in Elvanol. Shock frozen tissue sections (7 m) were exposed to primary antibody, biotinylated secondary antibody and alkaline phosphatase‐conjugated avidin–biotin complex solutions. Sections were counter stained with H&E. Digitized images were generated using a Leica DMRBE microscope, a SPOT CCD camera and Software SPOT2.1.2.
Migration assays: Migration was evaluated in Boyden chambers seeding cells in the upper chamber (RPMI/1%BSA or RPMI/1%BSA/10−8 M PMA). Where indicated, cells were preincubated with antibody (10 μl/ml). The lower chamber, separated by an 8 μm pore size polycarbonate‐membrane, contained 804G culture supernatant. After 16 h (37 °C), cells at the lower membrane site were stained with crystal‐violet, measuring OD595 nm after lysis. Migration is presented as % input cells. For in vitro wound healing, a subconfluent monolayer was scratched. Wound closure, followed by light microscopy, is presented as % reduction of the freshly wounded area.
Apoptosis: Cells (1 × 105) were grown for 48 h in RPMI/10%FCS containing serial dilutions of cisplatin (Sigma, Munich, Germany). Survival was monitored by annexinV‐APC/PI staining, CFSE (Molecular Probes, Eugene, Oregon, USA) dilution and 3H‐thymidine uptake.
Soft agar assay: Tumor cells in 0.3% agar were seeded on a preformed 1% agar layer counting colonies after 3wk.
In vivo assays: Female, 8wk old BDX rats received tumor cells (1 × 106 or 5 × 106) intrafootpad (ifp) or intraperitoneally (ip). PBS or cisplatin (1 μg/g body weight) were given ip after 2d and 23d. Rats were controlled weekly for local, draining lymph node (LN) (sliding caliber) or ip tumor growth, ascites (palpation), short breathing or weight loss. Animals were sacrificed when the draining LN reached a mean diameter of 2 cm, ascites became obvious, rats became pale, fatigue, lost >10% weight or latest after 120d. Animal experiments were Government‐approved (Baden‐Wuerttemberg, Germany).
Statistics: P values <0.05 (two‐tailed Student's t‐test, Kruskal–Wallis test) were considered significant.
3. Results
C4.4A is a metastasis‐associated molecule, whose functional activity remains elusive (Jacobsen and Ploug, 2008; Rösel et al., 1998). We recently described that in hypoxia C4.4A associates with 6 4 and MMP14, which contributes to matrix degradation and increased motility (Ngora et al., 2012). To confirm the in vivo relevance of this association on metastasis formation, we generated a C4.4Akd of the highly metastatic ASML line.
3.1. C4.4A contributes to the metastatic spread
ASML and ASML‐C4.4Akd cells (Figure 1A), were injected intrafootpad and tumor growth was followed until animals became moribund. Distinct to ASML cells, ASML‐C4.4Akd cells transiently developed a small local tumor, but LN metastases developed with a significant delay. When ASML‐bearing rats became moribund 6wk after tumor cell application, ASML‐C4.4Akd‐bearing rats had not developed axillary or lung metastases, which, however, were recovered after 8–10wk. Due to the retarded metastatic spread, the mean survival time of ASML‐C4.4Akd‐bearing rats was significantly prolonged from 39d of ASML‐bearing rats to 60d and 65d, respectively (Figures 1B–1D).
Figure 1.
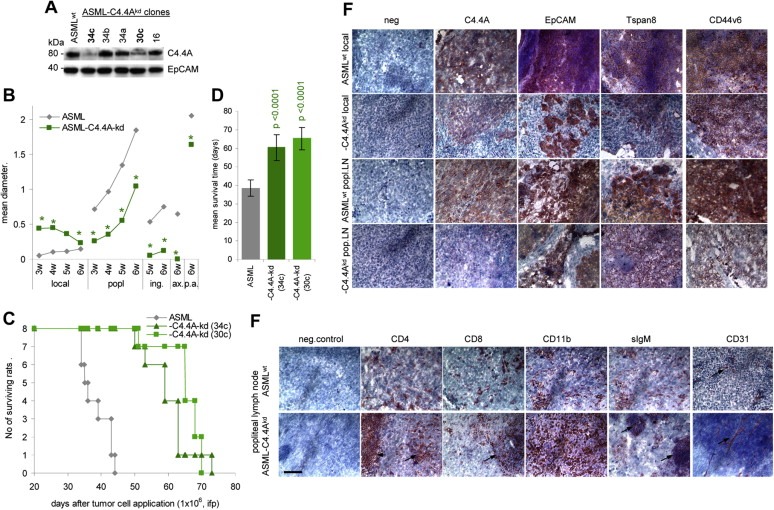
Retarded metastasis formation of ASML‐C4.4Akd cells: (A) WB of C4.4A in ASML and ASML‐C4.4Akd cells. EpCAM served as control. Clones 34c and 30c were used throughout, presented data mostly derived from clone 34c. (B‐D) BDX rats received 1 × 106 ASML or ASML‐C4.4Akd (clones 34c and 30c) cells, ifp. (B) Local tumor growth and growth in draining (popliteal) and distant (inguinal, paraaortic < p.a.>, axillary) LN during 6wk after tumor cell application. The mean diameter of 5 rats/group is shown. Significant differences between ASML and ASML‐C4.4Akd cells: *. (C) Survival time and survival rate of ASML and ASML‐C4.4Akd bearing rats (D) Mean survival timeSD of 8 rats/group; p‐values are shown. (E and F) Immunohistology of the local tumor and popliteal LN metastasis of ASML and ASML‐C4.4Akd‐bearing rats. Shock frozen footpad and popliteal LN sections were stained with the ASML markers C4.4A, EpCAM, Tspan8 and CD44v6 and (popliteal node) the leukocyte markers CD4, CD8, CD11b (M), sIgM (B cells) and the endothelial cell marker CD31. Scale bar: 100 m. In (F) staining of CD4+ and CD8+ cells in the perifollicular region and of B cell follicles in ASML‐C4.4Akd tumor bearers are indicated by an arrow. In ASML tumor bearers the lymph node structure is destroyed. Instead, ASML‐C4.4Akd tumor nodules are well vascularized, while only short stretches of endothelial cells are seen in ASML tumors (arrows). Metastasis formation of ASML‐C4.4Akd cells is delayed compared to ASML cells. ASML‐C4.4Akd cells do not invade the surrounding tissue and do not interfere with endothelial cell sprouting.
Immunohistology of local tumors, stained for the ASML markers C4.4A, EpCAM, Tspan8 and CD44v6 and of the popliteal node, stained in addition for leukocyte markers and an endothelial marker (CD31) and excised at late stages of tumor growth, confirmed a distinct growth profile of ASML and ASML‐C4.4Akd cells. While ASML cells grow dispersed between host cells, such that leukocytes are distributed between the tumor mass, the ASML‐C4.4Akd cells form tumor cell clusters that poorly penetrate the surrounding tissue, leaving e.g. B cells follicles (sIgM+) unattached. Notably, too, ASML‐C4.4Akd tumor nodules are better vascularized than ASML tumors (Figure 1E).
Thus, ASML‐C4.4Akd cells form a local tumor that regresses and metastasis formation is delayed, the capacity to invade surrounding tissue being strongly affected. Regression of the local tumor could be indicative for a loss in apoptosis resistance. Delayed metastasis formation and impaired invasiveness would be in line with the suggested co‐operativity of C4.4A with alpha6beta4 and MMP (Ngora et al., 2012).
3.2. Reduced motility and invasiveness of ASML‐C4.4Akd cells is a sequel of impaired focalization of alpha6beta4 and MMP14
We started to control for the impact of C4.4A on tumor cell motility. Transwell migration of ASML‐C4.4Akd cells was significantly reduced compared to ASML cell migration and was inhibited by B5.5 (anti‐ alpha6beta4). Notably, migration of PMA‐stimulated ASML cells was most strongly inhibited by B5.5, whereas PMA‐treated ASML‐C4.4Akd cell migration was poorly inhibited by B5.5 (Figure 2A). This indicated that particularly in stimulated ASML cells, C4.4A cooperates with alpha6beta4, which was confirmed in an in vitro wound healing assay. First to note, distinct to ASML cells, ASML‐C4.4Akd cell migration was hardly promoted by LN111 or LN332. Furthermore, whereas C4.4 and B5.5 inhibited ASML cell migration, poor migration of ASML‐C4.4Akd cells was not (C4.4/anti‐C4.4A) or minimally affected (B5.5) (Figure 2B, Suppl. Figure 1). Thus, the C4.4Akd does not only affect migratory activity on LN111 and LN332, which are joint ligands for C4.4A and alpha6beta4 (Paret et al., 2005), but hamper migration supporting alpha6beta4 activity, which is linked to alpha6beta4 phosphorylation upon stimulation (Frijns et al., 2010; Yang et al., 2010).
Figure 2.
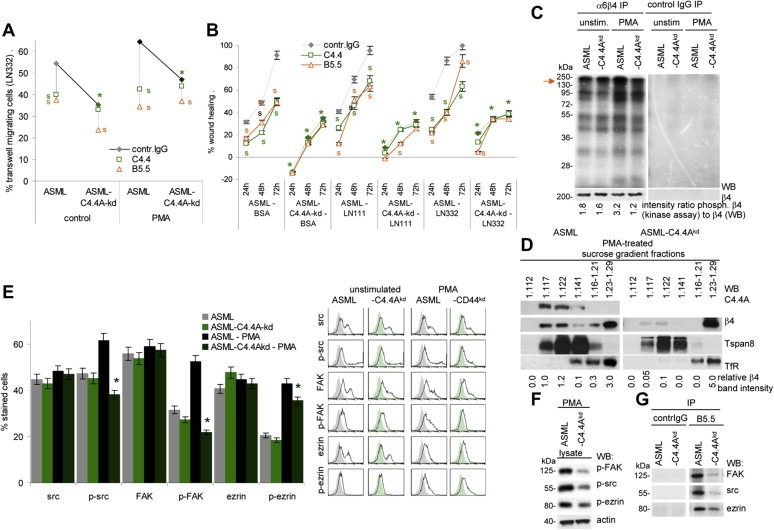
Reduced motility of ASML‐C4.4Akd cells is accompanied by impaired alpha6beta4 activation and raft recruitment: (A) Untreated orPMA treated ASML and ASML‐C4.4Akd cells were seeded in the upper part of a Boyden chamber. The lower chamber contained LN332 (804G supernatant). Where indicated cells were pre‐incubated with C4.4 (anti‐C4.4A) or B5.5 (anti‐alpha6beta4). Migration was evaluated after 16 h by staining the lowermembrane site with crystal violet. The % of migration cells (mean of triplicates) is shown. Significant differences between ASML and ASML‐C4.4Akd cells: *, significant antibody inhibition of migration: s. (B) ASML and ASML‐C4.4Akd cells were seeded in 24‐well plates coated with BSA, LN111 or LN332. When reaching subconfluence, the monolayer was scratched. “Wound healing” was evaluated for 72 h by light microscopy. Where indicated, the cultures contained C4.4 or B5.5. The mean percent ± SD(quadruplicates) of wound closure (as compared to the wound area at the time of scratching) is shown. Significant differences between ASML and ASML‐C4.4Akd cells: *; significant differences between control IgG, C4.4 and B5.5: s. (C) In vitro kinase assay of alpha6beta4 and control IgG immunoprecipitates of unstimulated and PMA‐stimulated ASML and ASMLC4.4Akd cell lysates. WB with anti‐beta4 served as loading control. The ratio of the signal strength of phosphorylated alpha6beta4 (kinase assay) to beta4 is indicated. (D) Lysates of PMA‐treated ASML and ASML‐C4.4Akd cells were separated by sucrose density gradient. Fractions were separated by SDS‐PAGE and after transfer blotted with C4.4 and anti‐beta4. Tspan8 (D6.1) serves as control raft marker and the transferin receptor (TfR (Ox26) as non‐raft marker. The relative 4 band intensity in sucrose fractions is shown. (E) Src, p‐src, FAK, p‐FAK, ezrin and p‐ezrin expression was evaluated by flow cytometry in untreated and PMA‐treated ASML and ASML‐C4.4Akd cells. Representative examples and mean values ± SD (triplicates) of stained cells are shown. Significant differences between ASML and ASML‐C4.4Akd cells: *. (F) Lubrol lysates of PMA‐treated ASML and ASML‐C4.4Akd cells were SDS‐PAGE separated and after transfer blotted with anti‐p‐FAK, anti‐p‐src and anti‐p‐ezrin and anti‐actin. (G) Lubrol lysates ofPMA‐treated ASMLand ASML‐C4.4Akd cells were precipitated with B5.5 (anti‐alpha6beta4) and control IgG. Immunoprecipitates were blotted with anti‐FAK, anti‐src and anti‐ezrin. (All experiments shown in Figure 2 were performed with ASMLwt and ASMLmock cells as well as with ASML‐C4.4Akd clone 30c and 34c cells revealing comparable results.) Stimulation‐promoted migration of ASMLwt cells is inhibited by anti‐C4.4A and anti‐alpha6beta4. Instead, poor migration of ASML‐C4.4Akd cells is hardly affected by alpha6beta4 blocking. Pronounced motility of ASML cells depends on recruitment of alpha6beta4 into rafts and beta4 phosphorylation, where alpha6beta4 supports src and FAK phosphorylation. Yet, beta4 may not directly associate with src and FAK as co‐immunoprecipitation was only observed under mild lysis conditions.
Indeed, an in vitro kinase assay revealed reduced PMA‐induced alpha6beta4 integrin phosphorylation in ASML‐C4.4Akd cells (Figure 2C). Furthermore, PMA treatment promoted alpha6beta4 recruitment into rafts (Merdek et al., 2007; Yang et al., 2004) only in ASML, but not in ASML‐C4.4Akd cells (Figure 2D). As Fyn‐induced phosphorylation of the beta4 cytoplasmic domain (Yang et al., 2010) causes recruitment of Shc and activation of downstream signaling molecules (Yang et al., 2010) and triggers src/FAK signaling (Danilkovitch‐Miagkova et al., 2000), we controlled for src, FAK and ezrin phosphorylation. Src, FAK and to a minor degree, ezrin phosphorylation was impaired in PMA‐treated ASML‐C4.4Akd cells (Figure 2E, F). Co‐immunoprecipitation under mild lysis conditions (Lubrol), which is not sufficient to disrupt raft‐located complexes, confirmed the association of alpha6beta4 with src and FAK in PMA‐stimulated ASML cells, which was strongly reduced in ASML‐C4.4Akd cells (Figure 2G).
Thus, the failure to recruit alpha6beta4 into rafts and the reduced beta4 phosphorylation may well account for impaired ASML‐C4.4Akd cell motility and could provide an explanation for the non‐invasive growth.
Raft‐recruited alpha6beta4 cooperates with constitutively raft‐located MMP14 (Yañez‐Mó et al., 2008) and upon stimulation C4.4A associates with alpha6beta4 and MMP14, which promotes LN111 and LN332 degradation (Ngora et al., 2012). Expression of MMP14 is not reduced in ASML‐C4.4Akd cells (Figure 3A). Expectedly, C4.4A showed strong and focalized co‐localization with alpha6beta4 and MMP14 and alpha6beta4 co‐localized with MMP14 in ASML cells seeded on LN332 in the presence of PMA. Co‐localization of alpha6beta4 and MMP14 was much weaker in PMA‐treated ASML‐C4.4Akd cells, which was confirmed by strongly reduced co‐immunoprecipitation of MMP14 with alpha6beta4 in PMA‐stimulated ASML‐C4.4Akd cells (Figure 3B,C). Furthermore, when ASML and ASML‐C4.4Akd cells were seeded on cover slides and stained after 24 h with anti‐LN gamma2, LN332 was degraded in the proximity of ASML, but not of ASML‐C4.4Akd cells. Degradation was inhibited in the presence of a broad range MMP inhibitor (Figure 3D). SDS‐PAGE of LN111 and LN332 and staining with anti‐LN beta1 or anti‐LN gamma2, confirmed reduced degradation by ASML‐C4.4Akd compared to ASML cells (Figure 3E).
Figure 3.
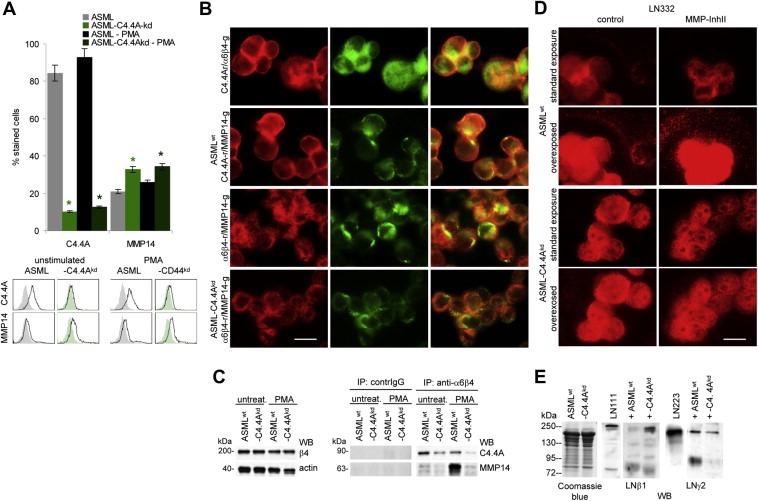
The cooperativity of alpha6beta4 with MMP14 is impaired in ASML‐C4.4Akd cells: (A) ASMLwt and ASML‐C4.4Akd cells were stained with C4.4 and anti‐MMP14. Expression was evaluated by flow cytometry. Mean values ± SD (triplicates) of stained cells and representative examples are shown. Significant differences between ASMLwt and ASML‐C4.4Akd cells: *. (B) ASMLwt and ASML‐C4.4Akd cells were seeded on LN332 (804G supernatant)‐coated slides. Cells were double stained for C4.4A and alpha6beta4 or MMP14 or alpha6beta4 and MMP14. Single fluorescence staining and digital overlays are shown (scale bar: 10 μm). (C) ASMLwt and ASML‐C4.4Akd cells were seeded on LN332‐coated plates and were stimulated, where indicated, o/n with PMA (10−8M). Cells were lysed, precipitated with B5.5 (anti‐alpha6beta4) or control IgG and after SDS‐PAGE and transfer blotted with C4.4 and anti‐MMP14. WB of beta4 and actin are included as controls. (D) ASMLwt and ASML‐C4.4Akd cells were cultured overnight in the presence of PMA and DMSO (control) or MMP‐InhII on glass cover slides. Cells were stained with anti‐LN gamma2. The standard exposure and overexposure are shown. (E) PMA stimulated ASMLwt and ASML‐C4.4Akd cells were co‐cultured overnight with LN111 or LN332 (804G supernatant). Cells were removed by centrifugation. Supernatants were separated by SDS‐PAGE and stained with anti‐LN beta1 and anti‐LN gamma2. Staining of supernatants with Coomassie Blue is included as control. Co‐localization and co‐immunoprecipitation of alpha6beta4 and MMP14 is reduced in PMA‐stimulated ASML‐C4.4Akd cells. This is accompanied by a reduction in laminin degradation.
Thus, C4.4A supports alpha6beta4 raft recruitment and phosphorylation. Raft‐located alpha6beta4 associates and cooperates with MMP14, such that by LN degradation ASML cells gain in focalized motility.
3.3. Drug resistance of ASML‐C4.4Akd cells
ASML‐C4.4Akd cells transiently develop a local tumor. Though this could have been due to reduced migratory capacity, it was surprising to see the local tumor vanishing with time (Figure 1B). As the locally growing mass of ASML‐C4.4Akd cells became necrotic (data not shown), we hypothesized that ASML‐C4.4Akd cells might be more apoptosis susceptible than ASML cells. To control this hypothesis, ASML and ASML‐C4.4Akd cells were ip injected and rats received 1 μg/g body weight cisplatin (ip) after 2d and 23d. Potential differences in migratory activity having no impact on intraperitoneal growth, growth retardation of ASML‐C4.4Akd cells will be indicative for increased drug susceptibility. Although cisplatin treatment led to a reduction of the ascitic load in ASML and ASML‐C4.4Akd‐bearing rats (data not shown), the survival time of ASML‐bearing rats was not, but that of ASML‐C4.4Akd‐bearing rats was >1.6‐fold increased (Figure 4A). In addition, while ip – as described for ifp – injected ASML cells metastasized to the lung, ASML‐C4.4Akd cells preferentially settled in the spleen and the liver. This was not observed in untreated ASML‐bearing rats and only in 1 of 5 cisplatin treated ASMLwt, but in 3 of 5 untreated and in 5 of 5 cisplatin‐treated ASML‐C4.4Akd‐bearing rats. Immunohistology revealed a pronounced reduction in alpha6beta4 expression in ASML‐C4.4Akd tumors (Figure 4B). The tendency of ASML‐C4.4Akd cells to grow non‐invasive was confirmed and became particularly obvious in spleen metastases, where tumors were surrounded by a LN111‐, coll I‐ and coll IV‐rich capsule. Lung metastases of ASML cells were not encapsuled (Figure 4C).
Figure 4.
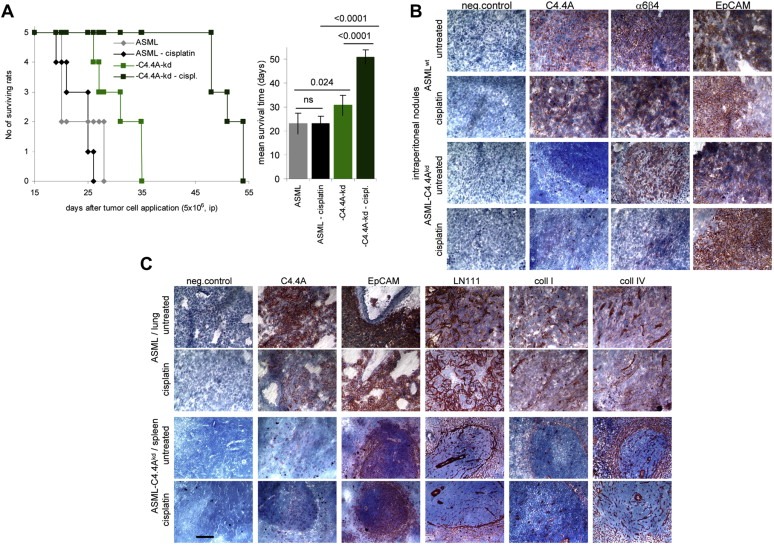
Impaired cisplatin resistance of ASML‐C4.4Akd cells: (A–C) BDX rats received ip 5 × 106 ASML or ASML‐C4.4Akd cells (clone 34c). After 2 and 23 days, they received cisplatin (1 μg/g body weight, ip). (A) Survival time and rate of 5 rats/group and the mean survival time; Significant differences between ASML‐ and ASML‐C4.4Akd‐bearing rats and between untreated and cisplatin‐treated rats are indicated. (B) Intraperitoneal tumor nodules were stained for the ASML markers C4.4A, 6 4 and EpCAM and (C) lung metastasis of untreated and cisplatin‐treated ASML and spleen metastases of untreated and cisplatin‐treated ASML‐C4.4Akd‐bearing rats were stained for C4.4A, EpCAM and the matrix proteins LN111, coll I and coll IV. Scale bar: 100 μm. ASML‐C4.4Akd cells respond better than ASML cells to cisplatin treatment. Notably, ip injected ASML‐C4.4Akd cells preferentially settle in spleen and liver, particularly in cisplatin‐treated rats, whereas ASML cells metastasize to the lung, irrespective of the injection site. Also distinct to ASML cells, surviving ASML‐C4.4Akd cells shield themselves from the surrounding tissue by a capsule mostly composed of LN111 and coll IV. Notably, too, particularly under cisplatin treatment, intraperitoneally grown ASML‐C4.4Akd cells reduce alpha6beta4 expression.
While the impaired matrix degradation in vivo corroborates the requirement of cooperative activity of C4.4A, alpha6beta4 and MMP14 in ASML cell invasiveness, the more striking effect of cisplatin on ASML‐C4.4Akd cell survival points toward C4.4A contributing to drug resistance.
3.4. Impaired drug resistance of ASML‐C4.4Akd cells and PI3K/Akt pathway activation
Impaired drug resistance of ASML‐C4.4Akd cells was confirmed in vitro, where 7.5 μg/ml cisplatin sufficed for a 50% reduction of the proliferative activity of ASML‐C4.4Akd cells as compared to ASML cells, which required 18 μg/ml cisplatin. Similar effects were seen evaluating CFSE dilution or the percentage of apoptotic cells by AnnV/PI staining (Suppl. Figure 2, Fig. 5A). The difference in cisplatin resistance between ASML‐C4.4Akd and ASML cells was even more striking considering anchorage‐independent growth. Only 0.6% of ASML‐C4.4Akd cells, but 30% of ASML cells formed colonies in soft agar in the presence of 1 μg/ml cisplatin (Figure 5B).
Figure 5.
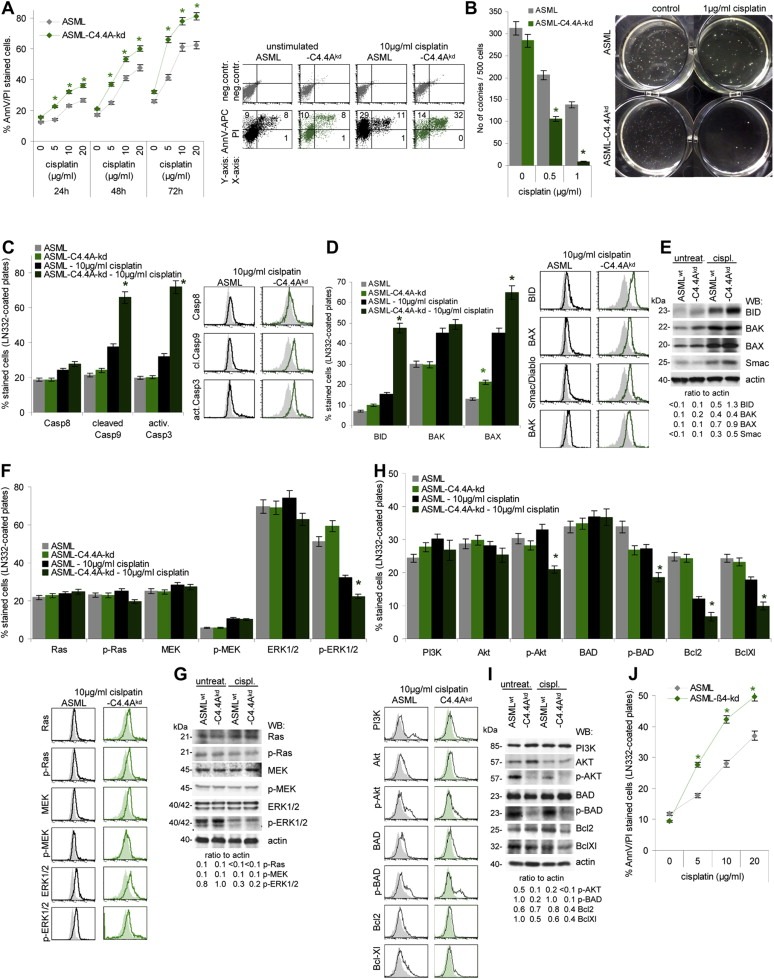
The contribution of the C4.4A‐alpha6beta4 cooperation to apoptosis resistance of ASML cells. (A,B) ASML and ASML‐C4.4Akd cells were cultured with titrated amounts of cisplatin for 48 h. (A) Apoptotic cells were determined by AnnV‐APC/PI staining after 48 h of culture (mean values ± SD of 3 experiments and representative example); (B) soft agar colony formation (mean valuesSD of 3 experiments and representative examples). In soft agar colony formation lower cisplatin concentrations were used, as growth in soft agar presents already a stress situation and cells are exposed to cisplatin for 3 weeks compared to 2 days in the apoptosis assays. Significant differences in cisplatin resistance between ASML and ASML‐C4.4Akd cells are indicated by *. (C‐I) ASML and ASML‐C4.4Akd cells, seeded on LN332, were cisplatin‐treated (10 μg/ml) for 48 h. Dead cells were discarded, performing the following analysis only with cells still alive. (C) Cells were stained with anti‐Casp8, anti‐cleaved Casp9 and anti‐activeCasp3; mean values ± SD of 3 experiments and representative examples are shown. (D) Cells were stained with anti‐BID, ‐BAK, ‐BAX, ‐Smac/Diablo; mean values ± SD of 3 experiments and representative examples are shown; (E) Lysates were SDS‐PAGE separated and blotted with the same antibodies as used in (D) and anti‐actin as control. The ratio of the signal intensity in comparison to actin is indicated. (F) Cells were stained with anti‐Ras, ‐p‐Ras, ‐MEK, ‐p‐MEK, ‐ERK1/2 and ‐p‐ERK1/2; mean values ± SD of 3 experiments and representative examples are shown. (G) Lysates were SDS‐PAGE separated and blotted with the same antibodies as in (F) and anti‐actin. The ratio of the signal intensity in comparison to actin is indicated. (H) Cells were stained with anti‐PI3K, ‐Akt, ‐p‐Akt, ‐BAD, ‐p‐BAD, ‐Bcl2, and ‐BclXl; mean values ± SD of 3 experiments and representative examples are shown. (I) Lysates were SDS‐PAGE separated and blotted with the same antibodies as in (H) and anti‐actin. The ratio of the signal intensity in comparison to actin is indicated. (A,B,C,D,F,H) Significant differences between untreated ASML and ASML‐C4.4Akd cells and between cisplatin‐treated ASML and ASML‐C4.4Akd cells: *. Repetition of the experiments with ASMLmock instead of ASMLwt and ASML‐C4.4Akd clone 30c instead of clone 34 cells revealed comparable results. (J) ASML and ASML‐ 4kd cells were seeded on LN332‐coated plates and cisplatin‐treated (10 μg/ml). Apoptosis was determined after 48 h by AnnexinV‐APC/PI staining. Significant differences between ASML and ASML‐ 4kd cells: *. ASML‐C4.4Akd cells show a significant decrease in drug resistance, most strongly in anchorage‐independent growth. Loss of drug resistance on LN332‐coated plates is not due to altered receptor‐mediated apoptosis, but proapoptotic molecules of the mitochondrial apoptosis pathway are upregulated. While activation of the MAPK pathway is hardly affected in cisplatin‐treated ASML‐C4.4Akd cells, phosphorylation of AKT and BAD is nearly abolished. As a transient beta4kd promotes a comparable loss in drug resistance, we hypothesize that C4.4A supports activation of the PI3K/Akt pathway via alpha6beta4 recruitment into rafts.
Cleaved casp9 and activated casp3 were stronger increased in cisplatin‐treated ASML‐C4.4Akd than ASML cells. Instead, casp8 expression did not differ (Figure 5C) excluding differences in receptor‐initiated apoptosis between ASML and ASML‐C4.4Akd cells. Focusing on the mitochondrial apoptosis pathway revealed reduced apoptosis‐resistance of ASML‐C4.4Akd cells to be accompanied by upregulation of BID, BAX and Smac/Diablo (Figure 5D). Activation of the MAPK pathway that promotes BAD phosphorylation and thereby the release of anti‐apoptotic Bcl2 and BclXl28, may not account for the reduced apoptosis resistance of ASML‐C4.4Akd cells; besides a stronger reduction in p‐ERK1/2, activation of the MAPK pathway was not impaired in cisplatin‐treated ASML‐C4.4Akd (Figure 5E). Instead, Akt and BAD phosphorylation, and concomitantly Bcl2 and BclXl expression was more strongly affected in cisplatin‐treated ASML‐C4.4Akd than ASML cells (Figure 5F). In tumor cells, activation of the PI3K/Akt pathway can be promoted by alpha6beta4 clustering via laminin binding (Merdek et al., 2007), suggesting that reduced drug resistance of ASML‐C4.4Akd cells may also rely on the impaired alpha6beta4 recruitment. To control the hypothesis, ASML‐ 4kd cells seeded on LN332 were cisplatin‐treated. Similar to ASML‐C4.4Akd cells, apoptosis susceptibility was strongly increased (Figure 5G).
Thus, reduced drug resistance of ASML‐C4.4Akd cells is due to a deficit in anti‐apoptotic protein activation that could again be a sequel of impaired alpha6beta4 recruitment.
4. Discussion
The metastatic cascade demands particular features from a tumor cell. Besides the capacity to detach from the primary tumor mass, tumor cells should have migratory and invasive potential and be apoptosis resistant to survive in a foreign environment (Mina and Sledge, 2011). C4.4A contributes to this process by supporting motility, matrix degradation and apoptosis resistance. None of these features are genuine characteristics of C4.4A. Instead, the laminin receptor C4.4A recruits and cooperates with alpha6beta4 and via alpha6beta4 with MMP14. Thus, by laminin binding C4.4A acts as a linker to facilitate several steps in the metastatic cascade.
4.1. The impact of C4.4A on metastasis formation
ASML cells metastasize exclusively via the lymphatic system and form miliary metastases (Matzku et al., 1983), which might be an escape mechanism due to very poor vascularization of ASML tumors. ASML‐C4.4Akd cells still metastasize via the lymphatic system, albeit with a significant delay. However, the growth profile differs from that of ASML tumors: (i) the ASML‐C4.4Akd tumor grows locally before metastases develop; (ii) the local tumor and metastases have a fibrous capsule such that hardly any dispersed tumor cells are detected in the surrounding tissue and (iii) tumors are well vascularized. The differences in the growth profile between ASML and ASML‐C4.4Akd cells could well rely on the association of C4.4A with MMP14 and, under stress conditions, the recruitment of alpha6beta4 toward raft‐located C4.4A.
4.2. C4.4A and tumor cell motility
The alpha6beta4 integrin, a component of hemidesmosomes in resting epithelial cells (Borradori and Sonnenberg, 1999), becomes recruited toward rafts in stimulatory conditions (Merdek et al., 2007; Mina and Sledge, 2011). This is accompanied by phosphorylation of the beta4 cytoplasmic domain (Yang et al., 2010), where tyrosine phosphorylation by Fyn causes recruitment of Shc and activation of downstream signaling molecules (Yang et al., 2010) like rac with a central role in directional cell migration (Pullar et al., 2006). Mostly serine phosphorylation by Ron accounts for formation of a 4‐14‐3‐3‐Ron complex, which relocalizes to lamellipodia and triggers src/FAK signaling (Danilkovitch‐Miagkova et al., 2000). We and others showed that C4.4A contributes to wound healing (Hansen et al., 2004; Jacobsen and Ploug, 2008; Paret et al., 2005), which can be inhibited by C4.4 and anti‐alpha6beta4 (Ngora et al., 2012). Accordingly, ASML‐C4.4Akd motility is strongly reduced and is poorly inhibited by anti‐alpha6beta4. In PMA‐treated ASML‐C4.4Akd cells, alpha6beta4 recruitment toward rafts is impaired and alpha6beta4, src and FAK phosphorylation are strongly mitigated. These findings are in line with reports on the contribution of alpha6beta4 to tumor cell motility (Frijns et al., 2010; Kariya et al., 2009; Marinkovich, 2007) that can be initiated via alpha6beta4 binding to LN332. alpha6beta4 signaling also accounts for LN332 track formation, which facilitates directed migration of metastasizing tumor cells (Sehgal et al., 2006). We do not know, whether alpha6beta4 becomes actively recruited by C4.4A and whether C4.4A contributes to beta4 phosphorylation. However, we rather speculate that alpha6beta4 becomes recruited toward raft‐located C4.4A due to their joint ligand LN332, where beta4 phosphorylation could proceed via raft‐associated signaling molecules (Staubach and Hanisch, 2011). Irrespective of this open question, the strong reduction in motility of ASML‐C4.4Akd cells and the concordant results obtained with a blockade of C4.4A and alpha6beta4 indicate that the association of C4.4A with alpha6beta4 is important for metastasizing tumor cell motility.
4.3. C4.4A and tumor cell invasion
The second peculiarity of ASML‐C4.4Akd cells, to form a fibrous capsule and not to penetrate into surrounding tissue also relies on C4.4A partners. MMP14 is located in lipid rafts (Yañez‐Mó et al., 2008), activates MMP2, degrades collagen (Holmbeck et al., 2004), LN gamma2 (Koshikawa et al., 2005) and LN beta3 (Udayakumar et al., 2003). Stress promotes the association of C4.4A and (activated) alpha6beta4 with MMP14, which is accompanied by focalized LN332 degradation (Ngora et al., 2012). As MMP14 hardly co‐immunoprecipitates with alpha6beta4 in ASML‐C4.4Akd cells, it is tempting to speculate that activated beta4 contributes to MMP14 activity and laminin degradation. Reduced collagen and laminin degradation, in turn, can well account for encapsulation of ASML‐C4.4Akd tumors and the failure to invade the surrounding tissue. Considering the better vascularization of ASML‐C4.4Akd tumors, we consider it possible that the poor vascularization of ASML tumors is a consequence of overshooting matrix degradation, thus destroying the path for sprouting capillaries. The presence of short capillary fragments in the ASML tumor supports our interpretation. Instead, reduced matrix degradation in ASML‐C4.4Akd tumors could allow for less disturbed capillary formation. Finally, we should mention that alpha6beta4 expression was downregulated in intraperitoneal nodules and spleen metastasis (data not shown) in cisplatin treated rats, though not in cultured tumor cells. We have no explanation for this finding. However, one could speculate that pronounced shedding/degradation in vivo might be less efficiently compensated in the absence of C4.4A.
Irrespective of this open question, by binding to LN332, LN111 and galectin3 (Paret et al., 2005), by raft localization, by activation‐induced association with alpha6beta4 accompanied by activation of raft‐located MMP14, C4.4A becomes central in coordinating tumor cell motility and invasiveness. The finding that in esophageal cancer C4.4A expression is reduced at early stages of tumor growth, but upregulated during metastasis formation (Hansen et al., 2008), supports the concept of tightly regulated expression to support migration and invasion of isolated or leading front tumor cells. Creation of space, LN332 track formation (Sehgal et al., 2006) and short fragments of LN gamma2 (Decline and Rousselle, 2001) can strengthen motility.
4.4. The contribution of C4.4A to drug resistance
ASML‐C4.4Akd cells are less cisplatin‐resistant than ASML cells, such that a low dose of cisplatin not exerting overt side effects allows for a significant prolongation of the survival time of ASML‐C4.4Akd bearing rats. In the absence of cisplatin, expression of pro‐ and anti‐apoptotic molecules did not differ from that of ASML cells, pointing again toward C4.4A to become engaged in apoptosis resistance under stress, which was confirmed in vitro, where upregulation of cleaved caspase9 and activated caspase3 was seen in cisplatin‐treated ASML‐C4.4Akd cells. Thus, it is the mitochondrial apoptosis pathway (Strasser et al., 2011) that is triggered in stressed ASML‐C4.4Akd cells. Having experienced that C4.4A becomes engaged in motility and invasiveness via its association with alpha6beta4, we speculated that the association with alpha6beta4 may also contribute to apoptosis resistance. The hypothesis is supported by our previous finding that high level PI3K, Akt and BAD phosphorylation and upregulation of Bcl2 and BclXl expression in ASML cells is triggered via CD44v6, but can be initiated via CD44, alpha6beta4 or Met ligand binding (Jung et al., 2011). We did not observe major differences in the activation of the MAPK pathway in cisplatin‐treated ASML versus ASML‐C4.4Akd cells seeded on LN332‐coated plates. Instead, Akt and BAD phosphorylation and Bcl and BclXl expression were strongly affected in cisplatin‐treated ASML‐C4.4Akd, but not ASML cells. Thus, C4.4A‐supported recruitment of alpha6beta4 into rafts might well account for beta4‐initiated activation of the PI3K/Akt pathway. PI3K/Akt activation by beta4 in cancer was repeatedly reported (Giancotti, 2007; Trusolino et al., 2001), where alpha6beta4 clustering by LN332 binding can be the initial trigger (Nguyen et al., 2000). Though the intracellular tail of beta4 does not suffice for Akt activation (Merdek et al., 2007) and PI3K unlikely becomes directly activated by phosphorylated beta4, besides activation via associated growth factors (Hintermann et al., 2001; Trusolino et al., 2001) or via insulin receptor substrate 1 and 2 (Shaw, 2001), raft recruitment and the interaction with raft‐located tyrosine kinases are considered to provide the initial trigger (Giancotti, 2007). As C4.4A recruits alpha6beta4 via the joint LN332 ligand into rafts, the latter mechanism most likely accounts for the contribution of C4.4A to apoptosis resistance.
4.5. Conclusion
C4.4A is a metastasis‐associated molecule with very restricted expression in non‐transformed tissue. We demonstrate using a C4.4A knockdown rat pancreatic cancer line that C4.4A‐promotes migration, invasion and apoptosis resistance. None of these features are genuine characteristics of C4.4A. Yet, the raft‐located laminin receptor C4.4A recruits and cooperates with alpha6beta4 and via alpha6beta4 with MMP14, provoking matrix degradation and PI3K/Akt activation. Taking the steps in the metastatic cascade benefiting from C4.4A, it appears of pathophysiological economy that C4.4A becomes selectively upregulated during wound repair and metastatic spread. C4.4A facilitating isolated tumor cell migration and supporting survival in foreign organs, we consider C4.4A a most promising therapeutic target that may hardly be burdened by side effects.
Authors' contribution
F. Thuma and H. Ngora performed and analyzed experiments, M. Zöller designed the experiments, performed experiments and wrote the manuscript.
Conflict of interest
Authors declare no conflict of interest.
Supporting information
Supplementary data
Acknowledgment
This work was supported by the Deutsche Krebshilfe (MZ, 10‐1821‐Z3). H.Ngora is a PhD grant recipient of the DAAD. We greatly acknowledge the help of Christine Niesik with animal experiments and immunohistology.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.05.002.
Thuma Florian, Ngora Honoré, Zöller Margot, (2013), The metastasis-associated molecule C4.4A promotes tissue invasion and anchorage independence by associating with the alpha6beta4 integrin, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.05.002.
References
- Bertotti, A. , Comoglio, P.M. , Trusolino, L. , 2005. Beta4 integrin is a transforming molecule that unleashes Met tyrosine kinase tumorigenesis. Cancer Res.. 65, 10674–10679. [DOI] [PubMed] [Google Scholar]
- Bon, G. , Folgiero, V. , Bossi, G. , Felicioni, L. , Marchetti, A. , Sacchi, A. , Falcioni, R. , 2006. Loss of beta4 integrin subunit reduces the tumorigenicity of MCF7 mammary cells and causes apoptosis upon hormone deprivation. Clin. Cancer Res.. 12, 3280–3287. [DOI] [PubMed] [Google Scholar]
- Bon, G. , Folgiero, V. , Di Carlo, S. , Sacchi, A. , Falcioni, R. , 2007. Involvement of alpha6beta4 integrin in the mechanisms that regulate breast cancer progression. Breast Cancer Res.. 9, 203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borradori, L. , Sonnenberg, A. , 1999. Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol.. 112, 411–418. [DOI] [PubMed] [Google Scholar]
- Claas, C. , Herrmann, K. , Matzku, S. , Möller, P. , Zöller, M. , 1996. Developmentally regulated expression of metastasis-associated antigens in the rat. Cell. Growth Differ.. 7, 663–678. [PubMed] [Google Scholar]
- Danilkovitch-Miagkova, A. , Angeloni, D. , Skeel, A. , Donley, S. , Lerman, M. , Leonard, E.J. , 2000. Integrin-mediated RON growth factor receptor phosphorylation requires tyrosine kinase activity of both the receptor and c-Src. J. Biol. Chem.. 275, 14783–14786. [DOI] [PubMed] [Google Scholar]
- Decline, F. , Rousselle, P. , 2001. Keratinocyte migration requires alpha2beta1 integrin-mediated interaction with the laminin 5 gamma2 chain. J. Cell. Sci.. 114, 811–823. [DOI] [PubMed] [Google Scholar]
- de Pereda, J.M. , Ortega, E. , Alonso-García, N. , Gómez-Hernández, M. , Sonnenberg, A. , 2009. Advances and perspectives of the architecture of hemidesmosomes: lessons from structural biology. Cell. Adh. Migr.. 3, 361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, G.C. , Patel, S. , Tyson, K. , Adam, P.J. , Schenker, M. , Loader, J.A. , Daviet, L. , Legrain, P. , Parekh, R. , Harris, A.L. , Terrett, J.A. , 2003. hAG-2 and hAG-3 human homologues of genes involved in differentiation are associated with oestrogen receptor-positive breast tumours and interact with metastasis gene C4.4a and dystroglycan. Br. J. Cancer. 88, 579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland, J.C. , Lakins, J.N. , Kazanietz, M.G. , Chernoff, J. , Boettiger, D. , Weaver, V.M. , 2007. alpha6beta4 integrin activates Rac-dependent p21-activated kinase 1 to drive NF-kappaB-dependent resistance to apoptosis in 3D mammary acini. J. Cell. Sci.. 120, 3700–3712. [DOI] [PubMed] [Google Scholar]
- Fries, F. , Nazarenko, I. , Hess, J. , Claas, A. , Angel, P. , Zöller, M. , 2007. CEBPbeta, JunD and c-Jun contribute to the transcriptional activation of the metastasis-associated C4.4A gene. Int. J. Cancer. 120, 2135–2147. [DOI] [PubMed] [Google Scholar]
- Frijns, E. , Sachs, N. , Kreft, M. , Wilhelmsen, K. , Sonnenberg, A. , 2010. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin {beta}4. J. Biol. Chem.. 285, 37650–37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G. , 2007. Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol. Sci.. 28, 506–511. [DOI] [PubMed] [Google Scholar]
- Hansen, L.V. , Gardsvoll, H. , Nielsen, B.S. , Lund, L.R. , Dano, K. , Jensen, O.N. , Ploug, M. , 2004. Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor. Biochem. J.. 380, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, L.V. , Laerum, O.D. , Illemann, M. , Nielsen, B.S. , Ploug, M. , 2008. Altered expression of the urokinase receptor homologue, C4.4A, in invasive areas of human esophageal squamous cell carcinoma. Int. J. Cancer. 122, 734–741. [DOI] [PubMed] [Google Scholar]
- Hansen, L.V. , Skov, B.G. , Ploug, M. , Rappot, H. , 2007. Tumour cell expression of C4.4A, a structural homologue of the urokinase receptor, correlates with poor prognosis in non-small cell lung cancer. Lung Cancer. 58, 260–266. [DOI] [PubMed] [Google Scholar]
- Hintermann, E. , Bilban, M. , Sharabi, A. , Quaranta, V. , 2001. Inhibitory role of alpha 6 beta 4-associated erbB-2 and phosphoinositide 3-kinase in keratinocyte haptotactic migration dependent on alpha 3 beta 1 integrin. J. Cell. Biol.. 153, 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmbeck, K. , Bianco, P. , Yamada, S. , Birkedal-Hansen, H. , 2004. MT1-MMP: a tethered collagenase. J. Cell. Physiol.. 200, 11–19. [DOI] [PubMed] [Google Scholar]
- Homma, Y. , Ozono, S. , Numata, I. , Seidenfeld, J. , Oyasu, R. , 1985. alpha-Difluoromethylornithine inhibits cell growth stimulated by a tumor-promoting rat urinary fraction. Carcinogenesis. 6, 159–161. [DOI] [PubMed] [Google Scholar]
- Jacobsen, B. , Ploug, M. , 2008. The urokinase receptor and its structural homologue C4.4A in human cancer: expression, prognosis and pharmacological inhibition. Curr. Med. Chem.. 15, 2559–2573. [DOI] [PubMed] [Google Scholar]
- Jacobsen, B. , Santoni-Rugiu, E. , Illemann, M. , Kriegbaum, M.C. , Laerum, O.D. , Ploug, M. , 2012. Expression of C4.4A in precursor lesions of pulmonary adenocarcinoma and squamous cell carcinoma. Int. J. Cancer. 130, 2734–2739. [DOI] [PubMed] [Google Scholar]
- Jung, T. , Gross, W. , Zöller, M. , 2011. CD44v6 coordinates tumor matrix-triggered motility and apoptosis resistance. J. Biol. Chem.. 286, 15862–15874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya, Y. , Kariya, Y. , Gu, J. , 2009. Roles of laminin-332 and alpha6beta4 integrin in tumor progression. Mini. Rev. Med. Chem.. 9, 1284–1291. [DOI] [PubMed] [Google Scholar]
- Klingbeil, P. , Marhaba, R. , Jung, T. , Kirmse, R. , Ludwig, T. , Zöller, M. , 2009. CD44 variant isoforms promote metastasis formation by a tumor cell-matrix cross-talk that supports adhesion and apoptosis resistance. Mol. Cancer Res.. 7, 168–179. [DOI] [PubMed] [Google Scholar]
- Konishi, K. , Yamamoto, H. , Mimori, K. , Takemasa, I. , Mizushima, T. , Ikeda, M. , Sekimoto, M. , Matsuura, N. , Takao, T. , Doki, Y. , Mori, M. , 2010. Expression of C4.4A at the invasive front is a novel prognostic marker for disease recurrence of colorectal cancer. Cancer Sci.. 101, 2269–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa, N. , Minegishi, T. , Sharabi, A. , Quaranta, V. , Seiki, M. , 2005. Membrane-type matrix metalloproteinase-1 (MT1-MMP) is a processing enzyme for human laminin gamma 2 chain. J. Biol. Chem.. 280, 88–93. [DOI] [PubMed] [Google Scholar]
- Kriegbaum, M.C. , Jacobsen, B. , Hald, A. , Ploug, M. , 2011. Expression of C4.4A, a structural uPAR homolog, reflects squamous epithelial differentiation in the adult mouse and during embryogenesis. J. Histochem. Cytochem.. 59, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb, E.A. , Mercurio, A.M. , 2005. Mobilization and activation of a signaling competent alpha6beta4integrin underlies its contribution to carcinoma progression. Cancer Metastasis Rev.. 24, 413–423. [DOI] [PubMed] [Google Scholar]
- Marinkovich, M.P. , 2007. Tumour microenvironment: laminin 332 in squamous-cell carcinoma. Nat. Rev. Cancer. 7, 370–380. [DOI] [PubMed] [Google Scholar]
- Matzku, S. , Komitowski, D. , Mildenberger, M. , Zöller, M. , 1983. Characterization of BSp73 a spontaneous rat tumor and its in vivo selected variants showing different metastasizing capacities. Invasion Metastasis. 3, 109–123. [PubMed] [Google Scholar]
- Mercurio, A.M. , Rabinovitz, I. , 2001. Towards a mechanistic understanding of tumor invasion–lessons from the alpha6beta 4 integrin. Semin. Cancer Biol.. 11, 129–141. [DOI] [PubMed] [Google Scholar]
- Merdek, K.D. , Yang, X. , Taglienti, C.A. , Shaw, L.M. , Mercurio, A.M. , 2007. Intrinsic signaling functions of the beta4 integrin intracellular domain. J. Biol. Chem.. 282, 30322–30330. [DOI] [PubMed] [Google Scholar]
- Mina, L.A. , Sledge, G.W. , 2011. Rethinking the metastatic cascade as a therapeutic target. Nat. Rev. Clin. Oncol.. 8, 325–332. [DOI] [PubMed] [Google Scholar]
- Ngora, H. , Galli, U.M. , Miyazaki, K. , Zöller, M. , 2012. Membrane-bound and exosomal metastasis-associated C4.4A promotes migration by associating with the ?(6)?(4) integrin and MT1-MMP. Neoplasia. 14, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, B.P. , Ryan, M.C. , Gil, S.G. , Carter, W.G. , 2000. Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr. Opin. Cell. Biol.. 12, 554–562. [DOI] [PubMed] [Google Scholar]
- Oku, N. , Sasabe, E. , Ueta, E. , Yamamoto, T. , Osaki, T. , 2006. Tight junction protein claudin-1 enhances the invasive activity of oral squamous cell carcinoma cells by promoting cleavage of laminin-5 gamma2 chain via matrix metalloproteinase (MMP)-2 and membrane-type MMP-1. Cancer Res.. 66, 5251–5257. [DOI] [PubMed] [Google Scholar]
- Paret, C. , Bourouba, M. , Beer, A. , Miyazaki, K. , Schnölzer, M. , Fiedler, S. , Zöller, M. , 2005. Ly6 family member C4.4A binds laminins 1 and 5 associates with galectin-3 and supports cell migration. Int. J. Cancer. 115, 724–733. [DOI] [PubMed] [Google Scholar]
- Paret, C. , Hildebrand, D. , Weitz, J. , Kopp-Schneider, A. , Kuhn, A. , Beer, A. , Hautmann, R. , Zöller, M. , 2007. C4.4A as a candidate marker in the diagnosis of colorectal cancer. Br. J. Cancer. 97, 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullar, C.E. , Baier, B.S. , Kariya, Y. , Russell, A.J. , Horst, B.A. , Marinkovich, M.P. , Isseroff, R.R. , 2006. beta4 integrin and epidermal growth factor coordinately regulate electric field-mediated directional migration via Rac1. Mol. Biol. Cell.. 17, 4925–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösel, M. , Claas, C. , Seiter, S. , Herlevsen, M. , Zöller, M. , 1998. Cloning and functional characterization of a new phosphatidyl-inositol anchored molecule of a metastasizing rat pancreatic tumor. Oncogene. 17, 1989–2002. [DOI] [PubMed] [Google Scholar]
- Sehgal, B.U. , DeBiase, P.J. , Matzno, S. , Chew, T.L. , Claiborne, J.N. , Hopkinson, S.B. , Russell, A. , Marinkovich, M.P. , Jones, J.C. , 2006. Integrin beta4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J. Biol. Chem.. 281, 35487–35498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiter, S. , Stassar, M. , Rappl, G. , Reinhold, U. , Tilgen, W. , Zöller, M. , 2001. Upregulation of C4.4A expression during progression of melanoma. J. Invest. Dermatol.. 116, 344–347. [DOI] [PubMed] [Google Scholar]
- Shaw, L.M. , 2001. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell. Biol.. 21, 5082–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B.A. , Kennedy, W.J. , Harnden, P. , Selby, P.J. , Trejdosiewicz, L.K. , Southgate, J. , 2001. Identification of genes involved in human urothelial cell-matrix interactions: implications for the progression pathways of malignant urothelium. Cancer Res.. 61, 1678–1685. [PubMed] [Google Scholar]
- Staubach, S. , Hanisch, F.G. , 2011. Lipid rafts: signaling and sorting platforms of cells and their roles in cancer. Expert Rev. Proteom.. 8, 263–277. [DOI] [PubMed] [Google Scholar]
- Strasser, A. , Cory, S. , Adams, J.M. , 2011. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J.. 30, 3667–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli, C.H. , Akhtar, N. , 2009. Signal co-operation between integrins and other receptor systems. Biochem. J.. 418, 491–506. [DOI] [PubMed] [Google Scholar]
- Trusolino, L. , Bertotti, A. , Comoglio, P.M. , 2001. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. 107, 643–654. [DOI] [PubMed] [Google Scholar]
- Udayakumar, T.S. , Chen, M.L. , Bair, E.L. , VonBredow, D.C. , Cress, A.E. , Nagle, R.B. , 2003. Bowden GT. Membrane type-1-matrix metalloproteinase expressed by prostate carcinoma cells cleaves human laminin-5 beta3 chain and induces cell migration. Cancer Res.. 63, 2292–2299. [PubMed] [Google Scholar]
- Wang, W. , Ding, Y.Q. , Li, Z.G. , Han, H.X. , Yang, L. , 2006. Expression and diagnostic application of C4.4A protein in squamous cell carcinoma and adenocarcinoma. Zhonghua Bing Li Xue Za Zhi. 35, 277–280. [PubMed] [Google Scholar]
- Wilhelmsen, K. , Litjens, S.H. , Sonnenberg, A. , 2006. Multiple functions of the integrin alpha6beta4 in epidermal homeostasis and tumorigenesis. Mol. Cell. Biol.. 26, 2877–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würfel, J. , Rösel, M. , Seiter, S. , Claas, C. , Herlevsen, M. , Weth, R. , Zöller, M. , 2001. Cloning of the human homologue of the metastasis-associated rat C4.4A. Gene. 262, 35–41. [DOI] [PubMed] [Google Scholar]
- Yañez-Mó, M. , Barreiro, O. , Gonzalo, P. , Batista, A. , Megías, D. , Genís, L. , Sachs, N. , Sala-Valdés, M. , Alonso, M.A. , Montoya, M.C. , Sonnenberg, A. , Arroyo, A.G. , Sánchez-Madrid, F. , 2008. MT1-MMP collagenolytic activity is regulated through association with tetraspanin CD151 in primary endothelial cells. Blood. 112, 3217–3226. [DOI] [PubMed] [Google Scholar]
- Yang, X. , Dutta, U. , Shaw, L.M. , 2010. SHP2 mediates the localized activation of Fyn downstream of the α6β4 integrin to promote carcinoma invasion. Mol. Cell. Biol.. 30, 5306–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Kovalenko, O.V. , Tang, W. , Claas, C. , Stipp, C.S. , Hemler, M.E. , 2004. Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J. Cell. Biol.. 167, 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahnow, C.A. , 2009. CCAAT/enhancer-binding protein beta: its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev. Mol. Med.. 11, e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data