

Abstract
Acquired tamoxifen (TAM) resistance limits the therapeutic benefit of TAM in patients with hormone‐dependent breast cancer. The switch from estrogen‐dependent to growth factor‐dependent growth is a critical step in this process. However, the molecular mechanisms underlying this switch remain poorly understood. In this study, we established a TAM resistant cell sub line (MCF‐7/TAM) from estrogen receptor‐α (ER‐α66) positive breast cancer MCF‐7 cells by culturing ER‐α66‐positive MCF‐7 cells in medium plus 1 μM TAM over 6 months. MCF‐7/TAM cells were then found to exhibit accelerated proliferation rate together with enhanced in vitro migratory and invasive ability. And the estrogen receptor‐α36 (ER‐α36), a novel 36‐kDa variant of ER‐α66, was dramatically overexpressed in this in vitro model, compared to the parental MCF‐7 cells. Meanwhile, the expression of epidermal growth factor receptor (EGFR) in MCF‐7/TAM cells was significantly up‐regulated both in mRNA level and protein level, and the expression of ER‐α66 was greatly down‐regulated oppositely. In the subsequent studies, we overexpressed ER‐α36 in MCF‐7 cells by stable transfection and found that ER‐α36 transfected MCF‐7 cells (MCF‐7/ER‐α36) similarly exhibited decreased sensitivity to TAM, accelerated proliferative rate and enhanced in vitro migratory and invasive ability, compared to empty vector transfected MCF‐7 cells (MCF‐7/V). Real‐time qPCR and Western blotting analysis revealed that MCF‐7/ER‐α36 cells possessed increased EGFR expression but decreased ER‐α66 expression both in mRNA level and protein level, compared to MCF‐7/V cells. This change in MCF‐7/ER‐α36 cells could be reversed by neutralizing anti‐ER‐α36 antibody treatment. Furthermore, knock‐down of ER‐α36 expression in MCF‐7/TAM cells resulted in reduced proliferation rate together with decreased in vitro migratory and invasive ability. Decreased EGFR mRNA and protein expression as well as increased ER‐α66 mRNA expression were also observed in MCF‐7/TAM cells with down‐regulated ER‐α36 expression. In addition, blocking EGFR/ERK signaling in MCF‐7/ER‐α36 cells could restore the expression of ER‐α66 partly, suggesting a regulatory function of EGFR/ERK signaling in down‐regulation of ER‐α66 expression. In conclusion, our results indicated for the first time a regulatory role of ER‐α36 in up‐regulation of EGFR expression and down‐regulation of ER‐α66 expression, which could be an underlying mechanism for the growth status switch in breast tumors that contribute to the generation of acquired TAM resistance. And ER‐α36 could be considered a potential new therapeutic target in breast tumors which have acquired resistance to TAM.
Keywords: Breast cancer, Acquired tamoxifen resistance, ER-α36, ER-α66 and EGFR
Highlights
-
►
MCF‐7/TAM cells possess overexpressed ER‐α36 and EGFR, but down‐regulated ER‐α66.
-
►
Overexpressing ER‐α36 in MCF‐7 cells up‐regulated EGFR and down‐regulated ER‐α66.
-
►
ER‐α36 is involved in maintaining malignant phenotype of MCF‐7/TAM cells.
1. Introduction
Breast cancer is the most common malignant tumor and is the leading cause of cancer‐related deaths in women in the United States (Siegel et al., 2011). Hormonal therapy to block the estrogen receptor‐α (ER‐α66, the classic estrogen receptor) pathway is highly effective for ER‐α66‐positive breast cancer and the selective estrogen receptor modulator (SERM) tamoxifen (TAM) has emerged as the most effective drug in this therapy (Jaiyesimi et al., 1995). However, the effectiveness of TAM therapy is limited as most advanced breast tumors eventually recur with acquired resistance despite initial responsiveness to TAM (Ali and Coombes, 2002; Clarke et al., 2003). A body of clinical and experimental studies suggests that molecular cross‐talks between ER‐α66 and other growth factors such as epidermal growth factor receptor (EGFR) might contribute to the development of acquired TAM resistance in breast cancer (Arpino et al., 2008; Fan et al., 2007; Knowlden et al., 2003; Massarweh and Schiff, 2006; Pancholi et al., 2008). Thus, ER‐α66 is not the only survival pathway driving breast tumors, and escape pathways when ER‐α66 is targeted are already functioning or begin to function during TAM treatment. As a matter of fact, the ER‐α66 positive breast tumors which possess low or normal levels of EGFR initially usually gain drastically overexpressed EGFR during development of acquired TAM resistance (Osborne and Schiff, 2011). However, the mechanisms underlying this switch are still not well established.
Recently, Wang et al. have identified and cloned a 36‐kDa variant of ER‐α66, ER‐α36. This truncated variant is the product of a transcript initiated from a previously unidentified promoter located in the first intron of ER‐α66 gene, suggesting that its expression is subjected to a transcription regulation different from ER‐α66. It lacks both transcriptional activation domains (AF‐1 and AF‐2) of ER‐α66, but retains a truncated ligand‐binding domain and an intact DNA‐binding domain (Wang et al., 2005). It is predominantly expressed on the plasma membrane and in the cytoplasm, modulates nongenomic estrogen signaling pathways that are resistant to antiestrogens (Kang et al., 2010; Lin et al., 2009, 2010; Tong et al., 2010; Wang et al., 2006; Zhang X et al., 2012). Clinical studies have reported that approximately 40% of ER‐α66‐positive breast cancer patients also expressed ER‐α36 in their tumors, and this subset of patients were less likely to benefit from TAM treatment compared with those with ER‐α66‐positive/ER‐α36‐negative tumors (Shi et al., 2009). All these findings raise the possibility that ER‐α36 expression may be involved in de novo TAM resistance in breast cancer. However, the molecular mechanisms for the association between ER‐α36 expression and acquired TAM resistance are still not resolved.
In this study, we established a TAM resistant cell sub line (MCF‐7/TAM) by culturing ER‐α66‐positive MCF‐7 cells in medium plus 1 μM TAM over 6 months, which was maintained in consistent medium continuously. We then found that MCF‐7/TAM cells possessed high levels of ER‐α36 and EGFR expression but nearly undetectable ER‐α66 expression. In addition, we revealed that ER‐α36 played an important role in this growth status switch via overexpressing ER‐α36 in MCF‐7 cells and knocking down ER‐α36 expression in MCF‐7/TAM cells, which contributed to the generation of acquired TAM resistance.
2. Materials and methods
2.1. Regents and antibodies
Geneticin (G418), Tamoxifen, EGFR tyrosine kinase inhibitor AG1478 and MAP kinase inhibitor PD098059 were ordered from Sigma–Aldrich (St. Louis, MO, USA). RPMI 1640 medium, fetal bovine serum (FBS), charcoal‐stripped fetal bovine serum (CFBS) were purchased from Thermo Scientific HyClone (South Logan, UT, USA). The monoclonal ER‐α36 antibody was developed by Abmart, Inc. (Shanghai, China) as a custom service, which were raised against a synthetic peptide antigen corresponding to the C‐terminal of ER‐α36. The polyclonal ERK1/2 antibody and phospho‐ERK1/2 (Thr202/Tyr204) antibody were purchased from Cell Signaling Technology, Inc. (Boston, MA, USA). The antibody against EGFR and ER‐α66 was purchased from Epitomics, Inc. (Burlingame, CA, USA). The antibody against β‐actin was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
2.2. Cell culture
Human breast cancer cell line MCF‐7 (ATCC, No.HTB‐22) was obtained from American Type Culture Collection (Manassas, VA, USA). These original cells were routinely cultured at 37 °C in the presence of 5% CO2 in RPMI 1640 supplemented with 10% FBS. MCF‐7/TAM cells were established by culturing ER‐α66‐positive, E2β‐responsive cell line MCF‐7 in medium plus 1 μM TAM over 6 months, which was maintained in consistent medium continuously. For neutralizing anti‐ER‐α36 antibody treatment, empty vector transfected MCF‐7 (MCF‐7/V) cells and ER‐α36 transfected MCF‐7 (MCF‐7/ER‐α36) cells were treated with 10 μg/ml of anti‐ER‐α36 antibody or equivalent normal mouse IgG for 4 days. MCF‐7/TAM cells were treated with 30 μg/ml anti‐ER‐α36 antibodies or equivalent normal mouse IgG for 10 days. For AG1478 and PD098059 treatment, MCF‐7/V and MCF‐7/ER‐α36 cells were starved in serum free medium for 12 h and then treated with 5 μM AG1478 (50 μM PD098059) or equivalent vehicle for 1 h, as previous studies described (Alessi et al., 1995; Dudley et al., 1995; Fox et al., 2008; Levitzki and Gazit, 1995; Zhu et al., 2001).
2.3. Plasmid preparation and transfection
The Coding sequence of ER‐α36 cDNA was successfully cloned, which was consistent with the NCBI database. On that basis, a eukaryotic expression vector of pcDNA3.1+ER‐α36 was constructed and verified by sequencing. Transfection was performed using Fugene HD Transfection Reagent (Roche Applied Science, Mannheim, Germany) as recommended by the manufacturer. After transfection, stable transfectants were selected via incubating cells with 600 ug/mL G418 for 2 weeks. Surviving single colonies were then picked and amplified. Two of established clonal cell lines that highly expressed ER‐α36 are described in detail in this study (MCF‐7/ER‐α36‐1 and ‐2). More than 30 individual clones transfected with the empty vector pcDNA3.1+ were pooled and used as a control (MCF‐7/V).
To knock down the expression of ER‐α36 in MCF‐7/TAM cells, an artificial microRNA‐expressing vector pcDNA3.1/6mi36 was designed before (Zhang J et al., 2012). Stable transfection and selection of ER‐α36 knock‐down cells was performed as described above. Two of established clonal cell lines that expressed decreased level of ER‐α36 are described in detail in this study (MCF‐7/TAM‐mi36‐1 and ‐2). Cells transfected with the vector pcDNA3.1+ were used as a control (MCF‐7/TAM‐V). For transient transfection with ER‐α66, a eukaryotic expression vector of pcDNA3.1+ER‐α66 was constructed and verified by sequencing. Transfection was performed as described above. About 48 h after transfection, cells were harvested to determine the expression of EGFR both in mRNA level and protein level.
2.4. RNA purification and quantitative reverse transcriptase‐PCR
Total RNA was extracted using TRIzol reagent according to the protocol provided by the manufacturer (Invitrogen, Carlsbad, Calif., USA). RNA concentrations were quantified by NanoDrop 1000 (Nanodrop, Wilmington, Del. USA). Reverse transcription reaction was performed using 2 μg of total RNA with Reverse Transcription System (Promega, Madison, WI, USA). The mRNA level of EGFR and ER‐α66 was analyzed using GoTaq® qPCR Master Mix Kit (Promega, Madison, WI, USA) in ABI PRISM 7500 Sequence Detection System (Applied Biosystems, CA, USA). The real time qPCR reaction was carried out in triplicate for each sample. The β‐actin gene was used as an endogenous control for normalization and the mRNA levels of EGFR and ER‐α66 were determined using the 2−ΔΔCt method (Livak and Schmittgen, 2001). Specific primer pairs are listed in Table 1.
Table 1.
Sequences of primers for real time qPCR.
| Name | Primer sequences |
|---|---|
| β‐actin | Forward primer 5′‐TGAGCGCGGCTACAGCTT‐3′ |
| Reverse primer 5′‐TCCTTAATGTCACGCACGATTT‐3′ | |
| EGFR | Forward primer:5′‐CGTCCGCAAGTGTAAGAA‐3′, |
| Reverse primer: 5′‐AGCAAAAACCCTGTGATT‐3′; | |
| ER‐α66 | Forward 5′‐AAGAAAGAACAACATCAGCAGTAAAGTC‐3′ |
| Reverse 5′‐GGGCTATGGCTTGGTTAAACAT‐3′; |
2.5. Western blotting analysis
Briefly, cell lysates for immunoblotting were prepared by adding lysis buffer (50 mM Tris–HCl (pH 7.4), 1% Nonidet P‐40, 0.5% sodium deoxycholate, 150 mM NaCl, 0.02% sodium azide, and 0.1% SDS) containing protease and phosphatase inhibitors (Sigma–Aldrich, St. Louis, MO, USA). Appropriate protein extracts of cell lysates were fractionated by SDS‐PAGE and electro‐transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocked at room temperature with 5% nonfat milk in TBS‐T (10 mmol/L Tris–HCl (pH 7.5), 0.5 mol/L NaCl, and 0.05% (w/v) Tween 20) buffer for 1 h, the membranes were incubated with primary antibodies overnight at 4 °C. The next day, the membranes were washed and then incubated with suitable peroxidase‐conjugated secondary antibodies for 1 h at room temperature. After washing thrice with TBS‐T, antibody binding was visualized using chemiluminescence detection system as described by the manufacturer (Millipore, Billerica, MA, USA). To show equal protein loading, the blots were stripped and reprobed for peroxidase‐conjugated β‐actin antibody. Molecular weights of the immunoreactive proteins were estimated based on PageRuler™ Prestained Protein ladder (MBI Fermentas, USA). Experiments were repeated for at least three times.
2.6. Methyl‐thiazolyl‐tetrazolium (MTT) assay
Cells grown to 70–80% confluence were harvested and seeded in 96‐well microtiter plates at 1000 cells per well and 5 wells were used for every experiment. The assay was begun after 12 h (day 0). When measuring cell growth, 0.5 mg/mL 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) (AMRESCO Inc, Solon, OH, USA) was added into the medium and cells were cultured for 4 h sequentially. Afterwards, the supernatant was removed and the formazan crystals were dissolved in 200 μL dimethyl sulfoxide (DMSO) at room temperature for 15 min. Absorbance of the solution was then measured at 570 nm wavelength using an ELx800 Absorbance Microplate Reader (Biotek, Winooski, VT, USA). Cell growth was measured daily and expressed as a multiple of the OD value at day 0. The experiments were performed in triplicate independently, mean ± standard error of mean (SEM) of which were presented in the Results section. For estrogen depletion, cells were maintained in phenol red‐free medium with 5% CFBS for 5 days to exhaust endogenous estrogen until the assay was begun.
2.7. Monolayer colony formation assay
500 cells were seeded into a well of 6‐well plate in triplicate, incubated in medium containing 1 μM TAM or equivalent DMSO (vehicle). Every 3–4 days the medium was replaced with fresh medium containing 1 μM TAM or equivalent DMSO. After 2 weeks, the colonies were fixed with 100% methanol, stained with 0.1% crystal violet and washed with phosphate buffer solution (PBS). Visible colonies (>=50 cells) were then counted for quantification. For anti‐ER‐α36 antibody treatment, anti‐ER‐α36 antibodies or equivalent normal mouse IgG were put into the culture on the basis of 1 μM TAM or equivalent DMSO.
2.8. In vitro migration and invasion assay
Cell migration and invasion was quantified using a previously described method (Zhang J et al., 2012). While performing the assays, cells were pre‐starved in serum‐free medium for 12 h. According to the protocol provided by the manufacturer (Millipore, Billerica, MA, USA), 900 μL of medium with 10% FBS was added into the wells of a 24‐well plate and 8‐μm pore transwell inserts were plated into those wells for 1 h rehydration at 37 °C. For invasion assay, the membranes of the inserts were coated with Matrigel (BD Bioscience, San Jose, CA, USA) at 37 °C for 30 min before the rehydration. Then, starved cells were harvested with serum‐free medium and 5 × 104 (1.5 × 105 for invasion assay) cells were seeded into the prepared inserts. After 24 h incubation at 37 °C with 5% CO2, the cells remaining inside of the inserts were removed using a cotton swab. Membranes were then fixed with 95% ethanol, stained with 0.1% crystal violet, washed with PBS. After that, they were cut from the inserts and fixed onto glass microscope slides using 50% glycerol with the cover glass. For quantification, the membranes were viewed at ×200 magnifications under light microscope. 5 separate fields per membrane were selected and the number of stained cells was counted in each field.
2.9. Statistical analysis
The data were presented as the means ± standard error of mean (SEM) of three independent experiments. One‐way ANOVA and Student's t‐test was used to determine the statistical differences between various experimental and control groups (GraphPad Prism v.5.0, La Jolla, CA, USA). Differences were considered statistically significant at a level of P < 0.05. * represents P < 0.05; ** represents P < 0.01.
3. Results
3.1. TAM‐resistant MCF‐7 (MCF‐7/TAM) cells exhibited significantly accelerated proliferation rate together with enhanced in vitro migratory and invasive ability
Acquired TAM resistance is a Gordian knot in current breast cancer treatment and the underlying molecular mechanisms are still unclear. To overcome this obstacle, a TAM resistant cell sub line (MCF‐7/TAM) was established by culturing ER‐α66‐positive MCF‐7 cells in medium plus 1 μM TAM over 6 months, which was maintained in consistent medium continuously. The TAM sensitivity of MCF‐7/TAM cells was then examined using monolayer colony formation assay. Compared to parental MCF‐7 cells, MCF‐7/TAM cells exhibited dramatically decreased sensitivity to TAM (13.5 ± 2.7% versus 62.3 ± 2%) (Figure 1A). Moreover, we found that MCF‐7/TAM cells proliferated much more rapidly than parental MCF‐7 cells using MTT assay (Figure 1B). Even after depleting estrogen from the medium, MCF‐7/TAM cells still possessed much faster proliferative rate than parental MCF‐7 cells (Figure 1C). Through an in vitro migration and invasion assay, we further discovered that MCF‐7/TAM cells possessed much stronger in vitro migratory and invasive ability than parental MCF‐7 cells. During seeded 5 × 104 cells, 151.4 ± 18.508 MCF‐7/TAM cells per field migrated through the membrane after 24 h incubation, versus 7.2 ± 1.281 MCF‐7 cells per field (P < 0.01). During seeded 1.5 × 105 cells, 106.8 ± 9.41 MCF‐7/TAM cells per field invaded through the membrane coated with matrigel after 24 h incubation, compared with 5.4 ± 0.872 MCF‐7 cells per field (P < 0.01) (Figure 1D and E). And MCF‐7/TAM cells were able to maintain this greatly enhanced in vitro migratory and invasive ability in depleted estrogen culture (Figure 1F and G).
Figure 1.
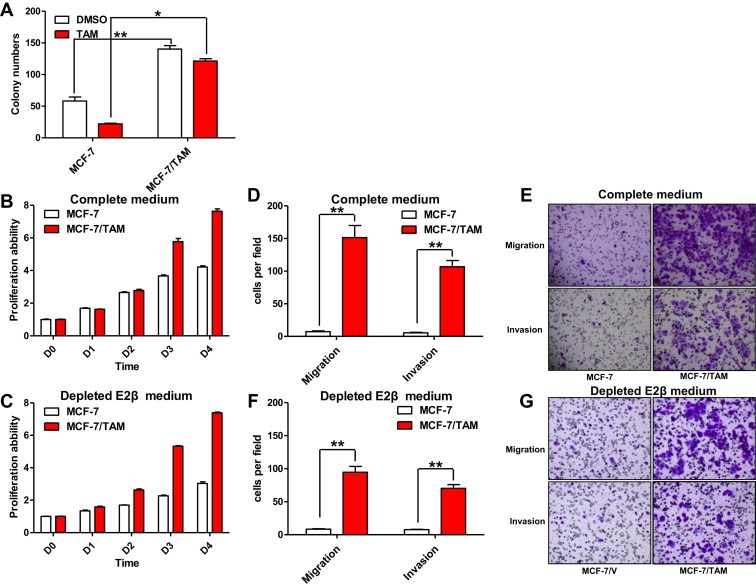
Tamoxifen‐resistant MCF‐7 cells (MCF‐7/TAM) exhibited accelerated proliferation rate together with enhanced in vitro migratory and invasive ability. (A). The TAM sensitivity in MCF‐7 cells and MCF‐7/TAM cells was examined using monolayer colony formation assay. Column: means of three independent experiments; bars, SEM. (B). Relative cell proliferation rate of parental MCF‐7 cells and MCF‐7/TAM cells were determined using MTT assay. Data presented are means ± SEM of three independent experiments. (C). Relative cell proliferation rate of parental MCF‐7 cells and MCF‐7/TAM cells was further determined via MTT assay after depleting E2β from the medium, as described in (B). (D & E). The in vitro migratory and invasive ability of MCF‐7 and MCF‐7/TAM cells was determined by in vitro migration and invasion assay. Cells migrated or invaded through the membrane were viewed at ×200 magnifications under light microscope, counted in 5 independent visual fields per transwell membrane. Photomicrographs were then taken. Cell numbers were presented as values of means ± SEM of triplicate experiments. (F & G). The in vitro migratory and invasive ability of MCF‐7 and MCF‐7/TAM cells was further determined by the same assay after depleting E2β from the medium, as described in (D & E).
3.2. MCF‐7/TAM cells possessed high levels of ER‐α36 and EGFR expression, but nearly undetectable ER‐α66 expression
Previous studies have showed that ER‐α36 was widely expressed in breast cancer cell lines, though there was only trace expression in the classical ER‐α66‐positive cell line MCF‐7 (Wang et al., 2006; Zhao et al., 2011; Zhang X et al., 2012). However, we observed that the expression level of ER‐α36 in MCF‐7/TAM cells was greatly increased compared to parental MCF‐7 cells. Meanwhile, the expression of EGFR in MCF‐7/TAM cells was significantly up‐regulated both in mRNA level and protein level, with increased basal levels of ERK1/2 phosphorylation which indicated activation of EGFR signaling (Figure 2A and B). In contrast, the ER‐α66 expression was lost at the protein level and greatly reduced at the mRNA level in MCF‐7/TAM cells (Figure 2A and C). However, we found no increased HER‐2 expression in MCF‐7/TAM cells here (Figure S1), though previous studies have reported that elevated HER‐2 was involved in the generation of acquired TAM resistance in breast cancer (Knowlden et al., 2003; Pancholi et al., 2008).
Figure 2.
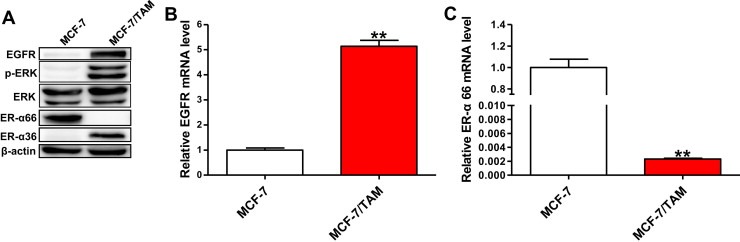
MCF‐7/TAM cells possessed increased levels of ER‐α36 and EGFR, together with reduced level of ER‐α66. (A). Western blotting analysis of the protein levels of ER‐α36, EGFR, ER‐α66, phosphorylated ERK1/2 and total ERK1/2 in MCF‐7 cells and MCF‐7/TAM cells. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B & C). Relative mRNA level of EGFR and ER‐α66 in MCF‐7/TAM cells was determined by real time qPCR. β‐actin gene was used as an endogenous control for normalization. Results showed are means ± SEM of three independent reactions.
3.3. Overexpressed ER‐α36 in MCF‐7 cells led to decreased sensitivity to TAM, accelerated proliferation rate together with enhanced in vitro migratory and invasive ability
The cDNA of ER‐α36 was first found and cloned by Wang et al. in 2005, which encoded a 310 amino acid open‐reading frame and a protein with a predicted molecular weight of 35.7 kDa (GeneBank: CAE45969.1) (Wang et al., 2005). To investigate the possible influence of ER‐α36 status on acquired TAM resistance in breast cancer, we up‐regulated the expression of ER‐α36 in MCF‐7 cells via stable transfection. In order to recognize the ER‐α36 protein, we developed a monoclonal anti‐ER‐α36 antibody raised against the C‐terminal of ER‐α36 (Materials and Methods). The anti‐ER‐α36 antibodies were used as a probe in Western blotting analysis of parental MCF‐7 cells, empty vector transfected MCF‐7/V cells and pcDNA3.1+ER‐α36 transfected MCF‐7/ER‐α36 cells. As shown in Figure 3A, MCF‐7 and MCF‐7/V cells expressed very low level of endogenous ER‐α36, but MCF‐7/ER‐α36 cells possessed significantly increased level of recombinant ER‐α36.
Figure 3.
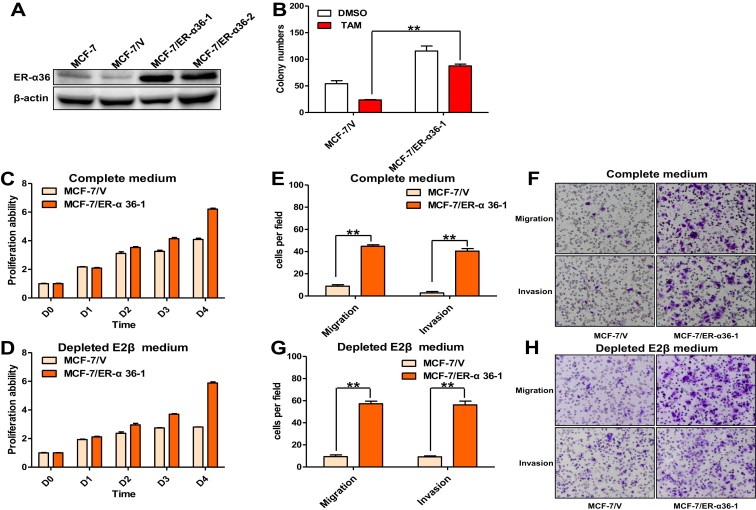
Overexpressed ER‐α36 in MCF‐7 cells caused resistance to TAM, accelerated proliferation rate together with enhanced in vitro migratory and invasive ability. (A) Whole cellular protein extracts of parent MCF‐7 cells and MCF‐7 cells transfected with empty vector or pcDNA3.1+ER‐α36 were subjected to Western blotting analysis using an anti‐ER‐α36 antibody. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B) The TAM sensitivity in MCF‐7/V cells and MCF‐7/ER‐α36‐1 cells was examined using monolayer colony formation assay. Column: means of three independent experiments; bars, SEM. (C) Relative cell proliferation rate of MCF‐7/V cells and MCF‐7/ER‐α36‐1 cells was determined using MTT assay. Data presented are means ± SEM of three independent experiments. (D). Relative cell proliferation rate of MCF‐7/V cells and MCF‐7/ER‐α36‐1 cells was further determined via MTT assay after depleting E2β from the medium, as described in (C). (E & F). The in vitro migratory and invasive ability of MCF‐7/V and MCF‐7/ER‐α36‐1 cells was determined by in vitro migration and invasion assay. Cells migrated or invaded through the membrane were viewed at ×200 magnifications under light microscope, counted in 5 independent visual fields per transwell membrane. Photomicrographs were then taken. Cell numbers were presented as values of means ± SEM of triplicate experiments (G & H). The in vitro migratory and invasive ability of MCF‐7/V and MCF‐7/ER‐α36 cells was further determined by the same assay after depleting E2β from the medium, as described in (E & F).
Lin et al. once demonstrated that MCF‐7 cells exhibited decreased sensitivity to TAM treatment after obtaining high expression of recombinant ER‐α36 (Lin et al., 2010). Here, the TAM sensitivity of MCF‐7/ER‐α36‐1 cells was determined using monolayer colony formation assay as mentioned above. Consistently, MCF‐7/ER‐α36‐1 cells were found to exhibit significantly decreased sensitivity to TAM compared to MCF‐7/V cells (24.2 ± 2.7% versus 56.2 ± 1.2%) (Figure 3B). Furthermore, MTT assay revealed that MCF‐7/ER‐α36‐1 cells proliferated much more rapidly than control MCF‐7/V cells both in complete medium and in depleted estrogen medium, which suggested that there may have other stimulations to the growth of MCF‐7/ER‐α36‐1 cells except estrogen only (Figure 3C and D). Similar to MCF‐7/TAM cells, MCF‐7/ER‐α36‐1 cells were also found to migrate and invade at a significantly higher rate than MCF‐7/V cells by in vitro migration and invasion assay (44.8 ± 1.319 versus 8.8 ± 1.356 cells per field in seeded 5 × 104 cells for migration; 40.4 ± 2.337 versus 2.8 ± 1.2 cells per field in seeded 1.5 × 105 for invasion) (Figure 3E and F). And MCF‐7/ER‐α36‐1 cells was also able to maintain this greatly enhanced in vitro migratory and invasive ability in culture without estrogen (Figure 3G and H).
3.4. MCF‐7/ER‐α36 cells possessed increased EGFR expression, but decreased ER‐α66 expression
In order to make the association between ER‐α36 and EGFR clear, we examined the expression of EGFR in ER‐α36 transfected MCF‐7 cells. Both mRNA level and protein level of EGFR, as well as basal levels of ERK1/2 phosphorylation, were found to be increased in MCF‐7/ER‐α36‐1 and MCF‐7/ER‐α36‐2 cells compared to MCF‐7/V cells (Figure 4A and B). In contrast, ER‐α66 expression in MCF‐7/ER‐α36‐1 and MCF‐7/ER‐α36‐2 cells was reduced both in mRNA level and protein level (Figure 4A and C). These results were consistent with what was showed in MCF‐7/TAM cells, suggesting that overexpressed ER‐α36 in MCF‐7 cells may play a crucial role in promoting EGFR expression and inhibiting ER‐α66 expression during development of acquired TAM resistance. Similar to MCF‐7/TAM cells, no increased HER‐2 expression was observed in MCF‐7/ER‐α36‐1 cells either (Figure S1).
Figure 4.

Overexpressing ER‐α36 in MCF‐7 cells up‐regulated EGFR expression and down‐regulated ER‐α66 expression. (A). Western blotting analysis of the protein levels of ER‐α36, EGFR, ER‐α66, phosphorylated ERK1/2 and total ERK1/2 in MCF‐7/V cells and MCF‐7/ER‐α36 cells. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B & C). Relative mRNA level of EGFR and ER‐α66 in MCF‐7/ER‐α36 cells was determined by real time qPCR. β‐actin gene was used as an endogenous control for normalization. Results showed are means ± SEM of three independent reactions.
3.5. Blocking of ER‐α36 by neutralizing antibodies inhibited growth of MCF‐7/ER‐α36‐1 cells but not MCF‐7/TAM cells
ER‐α36 is predominantly expressed on the plasma membrane (extracellular receptor) and in the cytoplasm, indicating that it could be blocked by specific antibodies. Wang et al. once developed an affinity‐purified rabbit polyclonal anti‐ER‐α36 antibody raised against C‐terminal of ER‐α36 (Wang et al., 2006), which has been proved to be able to block ER‐α36‐mediated biological function in breast cancer cells (Kang et al., 2010, 2011). Here, a monoclonal anti‐ER‐α36 antibody developed by ourselves were also raised against the C‐terminal of ER‐α36, which may be able to block ER‐α36‐mediated biological function similarly. In order to confirm this hypothesis, we treated MCF‐7/ER‐α36‐1 cells with 10 μg/ml anti‐ER‐α36 antibody for 4 days. And both the expression change of EGFR and ER‐α66 caused by overexpressed ER‐α36 in MCF‐7/ER‐α36‐1 cells were reversed to a certain extent by this treatment (Figure 5A–C). Furthermore, we discovered that anti‐ER‐α36 antibody treatment reduced the colony forming efficiency of MCF‐7/ER‐α36‐1 cells in monolayer colony formation assay, though it did not significantly increase their sensitivity to TAM (26.1 ± 4% in anti‐ER‐α36 antibody group versus 25.1 ± 2.9% in normal mouse IgG group) (Figure 5D). In addition, we tested the effects of monoclonal anti‐ER‐α36 antibodies on MCF‐7/TAM cells that possessed high levels of endogenous ER‐α36. Unfortunately, treating MCF‐7/TAM cells with even higher concentration of anti‐ER‐α36 antibodies could not obviously down‐regulate EGFR expression or up‐regulate ER‐α66 expression, as it did in MCF‐7/ER‐α36‐1 cells. Thus, the sensitivity of MCF‐7/TAM cells to TAM treatment could not be restored by treating them with neutralizing anti‐ER‐α36 antibodies (Figure 5E–H).
Figure 5.
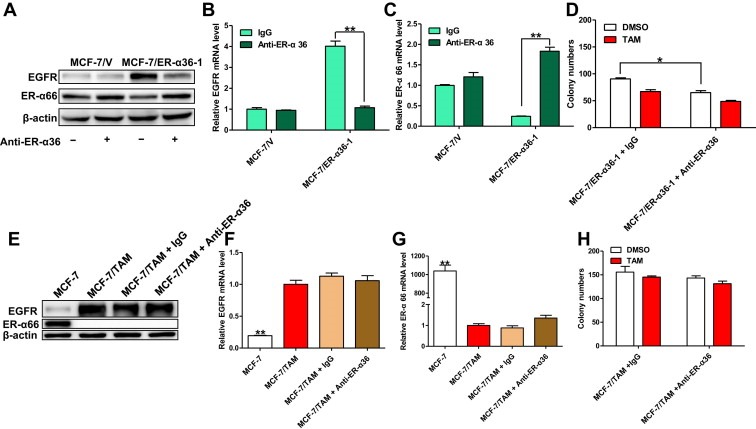
Inhibition of ER‐α36 with neutralizing anti‐ER‐α36 antibodies in MCF‐7/ER‐α36‐1 and MCF‐7/TAM cells. (A). MCF‐7/V cells and MCF‐7/ER‐α36‐1 cells were treated with 10 μg/ml anti‐ER‐α36 antibodies or equal control mouse IgG for 4 days. The survived cells were then harvested and the cell lysates were subjected to western blotting analysis. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown (B & C). The mRNA level of EGFR and ER‐α66 in treated MCF‐7/V and MCF‐7/ER‐α36‐1 cells were analyzed by real time qPCR using β‐actin gene as an endogenous control. Results showed are means ± SEM of three independent reactions. (D). The colony forming efficiency of MCF‐7/ER‐α36‐1 cells treated with 1 μM TAM plus 10 μg/ml anti‐ER‐α36 antibodies or equivalent normal mouse IgG was examined using monolayer colony formation assay. Column: Means of three independent experiments; bars, SEM. (E). MCF‐7/TAM cells were treated with 30 μg/ml anti‐ER‐α36 antibodies or equivalent normal mouse IgG for 10 days. The survived cells were then harvested and the cell lysates were subjected to western blotting analysis. β‐actin was used as the loading control. Untreated parental MCF‐7 cells were used as a control. All experiments were repeated at least three times, and the representative results are shown. (F & G). The mRNA level of EGFR and ER‐α66 in treated MCF‐7/TAM cells were analyzed by real time qPCR using β‐actin gene as an endogenous control. Untreated parental MCF‐7 cells were used as a control. Results showed are means ± SEM of three independent reactions. (H). The colony forming efficiency of MCF‐7/TAM cells treated with 1 μM TAM plus 30 μg/ml anti‐ER‐α36 antibodies or equivalent normal mouse IgG was examined using monolayer colony formation assay. Column: means of three independent experiments; bars, SEM.
3.6. Knock‐down of ER‐α36 expression in MCF‐7/TAM cells resulted in reduced proliferation rate as well as decreased in vitro migratory and invasive ability
As treating MCF‐7/TAM cells with neutralizing anti‐ER‐α36 antibodies could not block ER‐α36 mediated biological function, we further knocked down ER‐α36 expression in MCF‐7/TAM cells via stable transfection of an ER‐α36 multi‐hairpin vector (pcDNA3.1/mi36) (Zhang J et al., 2012). Western blotting analysis confirmed the significantly decreased expression of endogenous ER‐α36 in clones transfected with pcDNA3.1/6mi36 (Figure 6A). The TAM sensitivity of MCF‐7/TAM‐mi36‐2 cells was then determined using monolayer colony formation assay as mentioned above. Compared to MCF‐7/TAM cells and MCF‐7/TAM‐V cells, MCF‐7/TAM‐mi36‐2 cells exhibited significantly reduced colony forming efficiency, but no obvious increased sensitivity to TAM (12.8 ± 3% versus 8.3 ± 2.6%) (Figure 6B). MTT assay further revealed that MCF‐7/TAM‐mi36‐2 cells proliferated much more slowly than MCF‐7/TAM cells and MCF‐7/TAM‐V cells (Figure 6C). In addition, MCF‐7/TAM‐mi36‐2 cells were found to migrate and invade at a considerably lower rate than MCF‐7/TAM cells and MCF‐7/TAM‐V cells using in vitro migration and invasion assay (88.2 ± 12.091 versus 213.2 ± 12.153 cells per field during seeded 5 × 104 cells for migration; 9.4 ± 1.817 versus 111.2 ± 9.96 cells per field during seeded 1.5 × 105 cells for invasion) (Figure 6D and E).
Figure 6.

ER‐α36 depletion by microRNA in MCF‐7/TAM cells resulted in reduced proliferation rate together with decreased in vitro migratory and invasive ability. (A) Whole cellular protein extracts of parent MCF‐7/TAM cells and MCF‐7/TAM cells transfected with empty vector or pcDNA3.1/6mi36 were subjected to Western blotting analysis using an anti‐ER‐α36 antibody. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B). The TAM sensitivity in MCF‐7/TAM cells, MCF‐7/TAM‐V cells and MCF‐7/TAM‐mi36‐2 cells was examined using monolayer colony formation assay. Column: means of three independent experiments; bars, SEM. (C). Relative cell proliferation rate of MCF‐7/TAM cells, MCF‐7/TAM‐V cells and MCF‐7/TAM‐mi36‐2 cells was determined using MTT assay. Data presented are means ± SEM of three independent experiments. (D & E). The in vitro migratory and invasive ability of MCF‐7/TAM cells, MCF‐7/TAM‐V and MCF‐7/TAM‐mi36‐2 cells was determined by in vitro migration and invasion assay. Cells migrated and invaded through the membrane were viewed at ×200 magnifications under light microscope, counted in 5 independent fields per transwell membrane. Photomicrographs were then taken. Cell numbers were presented as values of means ± SEM of triplicate experiments.
3.7. MCF‐7/TAM‐mi36‐2 cells possessed decreased EGFR mRNA and protein expression, together with increased ER‐α66 mRNA expression
We further observed that both mRNA level and protein level of EGFR expression were decreased in MCF‐7/TAM‐mi36‐2 cells compared to MCF‐7/TAM‐V cells, which was still much higher than parental MCF‐7 cells (Figure 7A and B). Meanwhile, the mRNA expression level of ER‐α66 was more than doubled in MCF‐7/TAM‐mi36‐2 cells (Figure 7C). These findings proved that ER‐α36 indeed participated in the regulation of EGFR and ER‐α66 expression. However, the protein expression of ER‐α66 in MCF‐7/TAM‐mi36‐2 cells was still undetectable by Western blotting analysis (Figure 7A and C), which may explain why MCF‐7/TAM‐mi36‐2 cells that possessed significantly decreased proliferation ability could not regain sensitivity to TAM yet.
Figure 7.
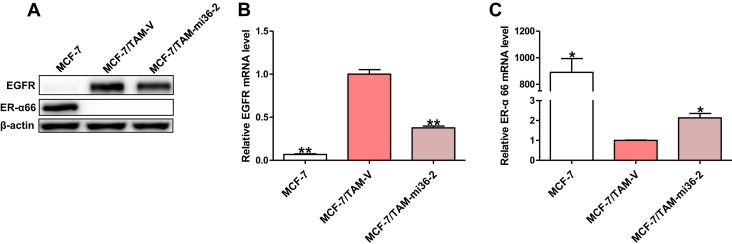
Knock‐down of ER‐α36 in MCF‐7/TAM cells resulted in decreased EGFR mRNA and protein expression, together with increased ER‐α66 mRNA expression. (A). Western blotting analysis of the protein levels of EGFR and ER‐α66 in MCF‐7/TAM‐V cells and MCF‐7/TAM‐mi36‐2 cells. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B & C). Relative mRNA level of EGFR and ER‐α66 in MCF‐7/TAM‐mi36‐2 cells was determined by real time qPCR. β‐actin gene was used as an endogenous control for normalization. Results showed are means ± SEM of three independent reactions.
3.8. Activated EGFR/ERK signaling was involved in the down‐regulation of ER‐α66
There existed an inverse relationship between the expression of ER‐α66 and EGFR in breast cancer (Arpino, 2004; Lee et al., 1990). Some studies demonstrated that ER‐α66 was involved in the repression of EGFR expression and decreased ER‐α66 expression could increase the expression of EGFR (deFazio et al., 1997; Yarden et al., 1996, 2001), but others declared that activation of EGFR mediated signaling contributed to the down‐regulation of ER‐α66 expression in breast cancer cells (Bayliss et al., 2007; Stoica et al., 2000). Here, to explore the effect of EGFR signaling on ER‐α66, MCF‐7/ER‐α36‐1 cells were treated with AG1478, a highly selective inhibitor of EGFR tyrosine kinase. Both mRNA level and protein level of ER‐α66 in treated MCF‐7/ER‐α36‐1 cells were then found to be increased, with reduced ERK1/2 phosphorylation (Figure 8A–C). It is well established that increased phosphorylated ERK1/2 indicated the activation of EGFR signaling pathway (Jorissen, 2003). And previous studies have shown that the phosphorylated ERK1/2 was involved in the down‐regulation of ER‐α66 (Creighton et al., 2006; Oh et al., 2001; Stossi et al., 2012). In the present study, to investigate the role of phosphorylated ERK1/2 in down‐regulation of ER‐α66, MCF‐7/ER‐α36‐1 cells were further treated with specific MAP kinase inhibitor PD098059. ERK1/2 phosphorylation was then found to be abolished and the expression of ER‐α66 was found to be increased both in the mRNA level and protein level in treated MCF‐7/ER‐α36‐1 cells (Figure 8D and E). These data supported a regulatory function of activated EGFR/ERK signaling in down‐regulation of ER‐α66 in MCF‐7/ER‐α36‐1 cells. On the other hand, in order to investigate if ER‐α66 was involved in repressing EGFR expression in MCF‐7 cells, we restored the expression of ER‐α66 in MCF‐7/ER‐α36‐1 cells via transient transfection. The mRNA level and protein level of EGFR expression in MCF‐7/ER‐α36‐1 cells were not found to be reduced after this transfection (Figure 8F and G).
Figure 8.
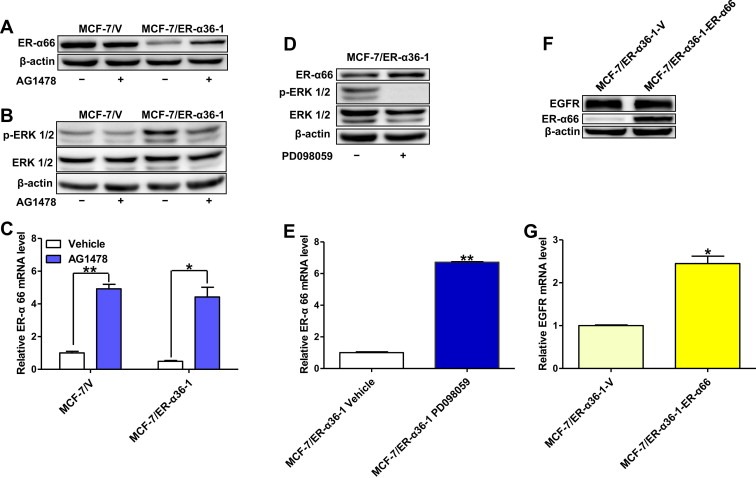
EGFR/ERK signaling participated in the down‐regulation of ER‐α66 in MCF‐7/ER‐α36‐1 cells. (A & B). MCF‐7/V and MCF‐7/ER‐α36‐1 cells were starved in serum free medium for 12 h and then treated with 5 μM AG1478 or equivalent vehicle for 1 h. The survived cells were then harvested and the cell lysates were subjected to western blotting analysis. β‐actin was used as the loading control. Data shown were representative of three separate experiments. (C). The mRNA level of ER‐α66 in AG1478 treated MCF‐7/V and MCF‐7/ER‐α36‐1 cells was analyzed by real time qPCR using β‐actin gene as an endogenous control. Results showed are means ± SEM of three independent reactions. (D). MCF‐7/ER‐α36‐1 cells were starved in serum free medium for 12 h and then treated with 50 μM PD098059 or equivalent vehicle for 1 h. The survived cells were then harvested and the cell lysates were subjected to western blotting analysis. β‐actin was used as the loading control. Data shown were representative of three separate experiments. (E). The mRNA level of ER‐α66 in PD098059 treated MCF‐7/ER‐α36‐1 cells was analyzed by real time qPCR using β‐actin gene as an endogenous control. Results showed are means ± SEM of three independent reactions. (F). MCF‐7/ER‐α36‐1 cells were transiently transfected with empty vector or pcDNA3.1+ER‐α66. The cell lysates were then subjected to Western blot analysis. β‐actin was used as the loading control. Data shown were representative of three separate experiments. (G). The mRNA level of EGFR in transiently transfected cells was analyzed by real time qPCR using β‐actin gene as an endogenous control. Results showed are means ± SEM of three independent reactions.
4. Discussion
TAM is able to bind to the ligand‐binding domain of the ER‐α66, effectively blocking the potential for estrogen stimulation and inhibiting the activity of ER‐α66, acting largely as an antagonist in ER‐α66‐positive breast cancer cells (Jaiyesimi et al., 1995; Osborne and Schiff, 2011). On the basis of published literatures, it is well established that overexpression of EGFR accompany with the loss of ER‐α66 after long term TAM treatment was responsible for development of acquired TAM resistance in ER‐α66‐positive primary tumors that initially respond to TAM therapy (Gutierrez et al., 2005; Johnston et al., 1995; Massarweh et al., 2008; Santen et al., 2009; Van den Berg et al., 1989; Zhang et al., 2009). However, the mechanisms underlying this growth status switch are largely unknown yet.
In the present study, we established a TAM‐resistant cell sub line MCF‐7/TAM from ER‐α66‐positive breast cancer MCF‐7 cells. Compared to parental MCF‐7 cells, MCF‐7/TAM cells were found to possess greatly increased ER‐α36 and EGFR expression, together with nearly undetectable ER‐α66 expression (Figure 2). ER‐α36, a variant of ER‐α66, was reported to be transcriptionally regulated differently from ER‐α66, and was found to be weakly expressed in ER‐α66‐positive breast cancer cells that expressed high levels of ER‐α66, but highly expressed in ER‐α66‐negative breast cancer cells that lack ER‐α66 expression (Wang et al., 2006; Zhao et al., 2011; Zhang, X. et al., 2012; Zou et al., 2009). Overexpression of ER‐α36 was demonstrated to be associated with poorer disease‐free survival and disease‐specific survival in patients with ER‐α66‐positive breast cancer who received TAM treatment (Shi et al., 2009). Some studies have demonstrated that the expression of ER‐α36 and EGFR exhibited a significant positive correlation (Tu et al., 2011; Zhang et al., 2011). Zhao et al. recently found that compared to parental MCF‐7 cells, TAM resistant MCF‐7/TAM cells possessed greatly overexpressed ER‐α36 but significantly down‐regulated ER‐α66, though the expression level of EGFR was not revealed (Zhao et al., 2011). Here, we revealed that overexpressing ER‐α36 in MCF‐7 cells decreased their sensitivity to TAM (Figure 3), which was consistent with previous study (Lin et al., 2010). We then found that MCF‐7/ER‐α36 cells possessed increased EGFR expression as well as decreased ER‐α66 expression compared to MCF‐7/V cells (Figure 4). And inhibiting ER‐α36 by neutralizing antibodies in MCF‐7/ER‐α36‐1 cells could reverse this expression change of EGFR and ER‐α66 partly (Figure 5A–C). Above all, we discovered that knocking down ER‐α36 expression in MCF‐7/TAM cells decreased EGFR mRNA and protein expression and increased ER‐α66 mRNA expression, though ER‐α66 protein expression was still undetectable (Figure 7). All these findings prompted that ER‐α36 play a regulatory role in the expression change of EGFR and ER‐α66 during the generation of acquired TAM resistance.
It has been revealed that most advanced breast tumors eventually recur with acquired resistance despite initial responsiveness to TAM (Ali and Coombes, 2002; Clarke et al., 2003). Previous studies have identified that upon development of resistance to TAM, MCF‐7 cells acquired an accelerated proliferation rate together with an enhanced capacity for in vitro migration and invasion (Hiscox et al., 2004, 2006; Kownlden et al., 2003). And our recent study also reported that ER‐α36 was correlated with the metastatic potential of ER‐α66‐negative breast cancer cells (Zhang, J. et al., 2012). In the present study, we found that MCF‐7/TAM cells exhibited accelerated proliferation rate together with enhanced in vitro migratory and invasive ability compared to parental MCF‐7 cells, properties associated with malignant transformation (Figure 1B, D and E). Furthermore, overexpressing ER‐α36 in MCF‐7 cells enhanced their multiplication capacity as well as in vitro migratory and invasive ability (Figure 3C, E and F). Chaudhri et al. once demonstrated that membrane estrogen signaling enhanced tumorigenesis and metastatic potential of breast cancer cells via ER‐α36 (Chaudhri et al., 2012). Here, we found that both MCF‐7/TAM and MCF‐7/ER‐α36‐1 cells were able to maintain increased proliferative activity together with in vitro migratory and invasive ability in depleted estrogen medium (Figure 1C, F and G; Figure 3D, G and H). This may be caused by overexpressed EGFR in MCF‐7/TAM and MCF‐7/ER‐α36‐1 cells, which was responsible for acquisition of an enhanced motile and invasive nature in TAM resistant breast cancer cells (Hiscox et al., 2004). Besides, we surprisingly found that both MCF‐7 and MCF‐7/V cells were also able to grow in estrogen‐depleted medium, though slower than in complete medium (Figure 1C; Figure 3D). This phenomenon may be induced by estrone‐sulfate, which was present in high amount in fetal calf serum and could not be totally removed by the charcoal stripping (Ruder et al., 1972). In addition, knock‐down of ER‐α36 expression in MCF‐7/TAM cells resulted in decreased proliferation capacity as well as in vitro migratory and invasive ability (Figure 6). These data indicated that ER‐α36 was involved in acquisition of an enhanced proliferative and invasive nature in TAM resistant breast cancer, which may explain why overexpression of ER‐α36 was associated with poorer disease‐free survival and disease‐specific survival in patients with ER‐α66‐positive breast cancer who received TAM treatment (Shi et al., 2009).
Current research has proved that ER‐α36‐mediated biological function could be blocked by anti‐ER‐α36 antibody raised against C‐terminal of ER‐α36 (Kang et al., 2010, 2011; Wang et al., 2006). In this study, we found that blocking ER‐α36 via a monoclonal anti‐ER‐α36 antibody developed by ourselves could decrease growth ability and reverse the expression change of EGFR and ER‐α66 in MCF‐7/ER‐α36‐1 cells (Figure 5A–C). Unfortunately, treating MCF‐7/TAM cells with even higher concentration of anti‐ER‐α36 antibodies for longer time could not obviously decrease EGFR expression or increase ER‐α66 expression, as it did in MCF‐7/ER‐α36‐1 cells. And the growth ability of MCF‐7/TAM cells was not decreased by this treatment either (Figure 5F–H). That may because the biological function mediated by ER‐α36 in MCF‐7/TAM could not be blocked completely by neutralizing anti‐ER‐α36 antibodies, as its expression level in MCF‐7/TAM was too high. Alternatively, the location of ER‐α36 on the plasma membrane or in the cytoplasm of MCF‐7/TAM cells might be different with MCF‐7/ER‐α36‐1 cells, which need further research. However, knocking down ER‐α36 expression via stable transfection in MCF‐7/TAM cells decreased EGFR expression both in mRNA level and protein level, though only increased ER‐α66 expression in mRNA level (Figure 7). This may be able to explain why MCF‐7/TAM‐mi36‐2 cells possessed significantly decreased proliferation rate than MCF‐7/TAM‐V cells, though did not regain sensitivity to TAM. Alternatively, the biological reprogramming during development of acquired TAM resistance was much more complicated than stable transfection of ER‐α36 in breast cancer cells, containing dynamic and widespread genomic changes (Aguilar et al., 2010; Osborne and Schiff, 2011). MCF‐7/TAM cells, unlike MCF‐7/ER‐α36‐1 cells, might have irreversibly turned into ER‐α66‐negative cells that were insensitivity to TAM therapy after long‐term exposure to TAM, as a published research once reported (Oesterreich et al., 2001).
Our results revealed that ER‐α36 could modulate the mRNA expression of EGFR. However, current studies demonstrated that ER‐α36 was predominantly expressed on the plasma membrane and in the cytoplasm, lacked both transcriptional activation domains of ER‐α66 (Wang et al., 2005, 2006). Previous studies once reported that GPR30, another membrane‐bound estrogen receptor, stimulated transactivation of EGFR in tamoxifen resistant MCF‐7 cells via nongenomic estrogen signaling (Filardo, 2002; Ignatov et al., 2010). But Kang et al. declared that ER‐α36, not GPR30, was involved in nongenomic estrogen signaling in breast cancer (Kang et al., 2010). Zhang et al. once demonstrated that EGFR protein was stabilized by ER‐α36 in ER‐α66‐negative breast cancer cells (Zhang et al., 2011). In fact, though lacking both transcriptional activation domains of ER‐α66, ER‐α36 retained a truncated ligand‐binding domain, suggesting that it may have a spectrum of ligand selectivity different from ER‐α66, and may mediate some other unknown signaling (Wang et al., 2005). Alternatively, ER‐α36 in cytoplasm may act indirectly by tethering to other transcription factors to modulate the mRNA expression of EGFR, as ER‐α36 retained the nuclear localization signals found in ER‐α66 (Wang et al., 2006). Anyway, further investigations were needed to elucidate the exact mechanisms underlying this regulation.
Both EGFR and ER‐α66 could regulate downstream genes via mediating themselves' signaling pathway (even each other). Our data here revealed that inhibiting EGFR tyrosine kinase with AG1478, as well as inhibiting MAP kinase with PD098059, could partly restore ER‐α66 expression in MCF‐7/ER‐α36‐1 cells (Figure 8A–E). These results supported a regulatory function of EGFR/ERK signaling in down‐regulation of ER‐α66 expression, which was consistent with previous reports (Bayliss et al., 2007; Creighton et al., 2006; Holloway et al., 2004; Oh et al., 2001). On the other hand, a role of ER‐α66 as a down‐modulator of EGFR could not be demonstrated here as re‐expression of ER‐α66 in MCF‐7/ER‐α36‐1 cells by transient transfection could not reduce EGFR expression (Figure 8F and G).
In summary, our findings strongly complement the current knowledge about the molecular mechanisms underlying acquired TAM resistance in breast cancer. Our data demonstrated for the first time that overexpressed ER‐α36 in breast cancer MCF‐7 cells could up‐regulate EGFR and down‐regulate ER‐α66, which contributed to the generation of acquired TAM resistance. Overexpressed ER‐α36 also enhanced the proliferation capacity as well as the in vitro migratory and invasive ability of breast cancer MCF‐7 cells, properties associated with cell malignancy. Furthermore, knock‐down of ER‐α36 expression in TAM resistant MCF‐7/TAM cells resulted in decreased EGFR expression, reduced proliferation rate together with decreased in vitro migratory and invasive ability, suggesting a critical role of ER‐α36 in maintaining malignant phenotype of MCF‐7/TAM cells. All these findings indicate that ER‐α36 can be used as a prognostic factor in breast cancer patients under TAM therapy, and can be considered a potential therapeutic target in tumors that have acquired resistance to TAM. Further study of the molecular mechanisms by which ER‐α36 is activated during development of acquired TAM resistance in breast cancer will provide more detailed insights for the biological function of ER‐α36 in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following is the supplementary data related to this article:
Supplemental Figure 1. The expression of HER‐2 in MCF‐7/TAM cells and MCF‐7/ER‐α36‐1 cells. (A) Western blotting analysis of the protein level of HER‐2 in MCF‐7 cells, MCF‐7/TAM cells and MCF‐7/ER‐α36‐1 cells. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B) Relative mRNA expression of HER‐2 in MCF‐7 cells, MCF‐7/V cells, MCF‐7/TAM cells and MCF‐7/ER‐α36‐1 cells was determined by real time qPCR (Forward primer 5′‐ACCGGCACAGACATGAAGCT‐3′; Reverse primer 5′‐AGGAAGGACAGGCTGGCATT‐3′). β‐actin gene was used as an endogenous control. Results showed are means ± SEM of three independent reactions.
Acknowledgments
This work was supported by the State Key Basic Research and Development Program of China (973 Program, Grant No. 2009CB521704), National High‐tech Research & Development Program of China (863 Program, Grant No. 2006AA02A245), National Natural Science Foundation of China (Grant No. 81000894), Natural Science Foundation of Zhejiang Province (Grant No. Y2090061) and Zhejiang Provincial Science and Technology Project (Grant No. 2009C13021).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.02.001.
Li Guangliang, Zhang Jing, Jin Ketao, He Kuifeng, Zheng Yi, Xu Xin, Wang Haohao, Wang Haiyong, Li Zhongqi, Yu Xiongfei, Teng Xiaodong, Cao Jiang, Teng Lisong, (2013), Estrogen receptor‐α36 is involved in development of acquired tamoxifen resistance via regulating the growth status switch in breast cancer cells, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.001.
Contributor Information
Jiang Cao, Email: caoj@zju.edu.cn.
Lisong Teng, Email: lsteng@zju.edu.cn, Email: fans_1988@163.com.
References
- Aguilar, H. , Sole, X. , Bonifaci, N. , Serra-Musach, J. , Islam, A. , Lopez-Bigas, N. , Mendez-Pertuz, M. , Beijersbergen, R.L. , Lazaro, C. , Urruticoechea, A. , Pujana, M.A. , 2010. Biological reprogramming in acquired resistance to endocrine therapy of breast cancer. Oncogene 29, 6071–6083. [DOI] [PubMed] [Google Scholar]
- Alessi, D.R. , Cuenda, A. , Cohen, P. , Dudley, D.T. , Saltiel, A.R. , 1995. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase in vitro and in vivo. Journal of Biological Chemistry 270, 27489–27494. [DOI] [PubMed] [Google Scholar]
- Ali, S. , Coombes, R.C. , 2002. Endocrine-responsive breast cancer and strategies for combating resistance. Nature Reviews Cancer 2, 101–112. [DOI] [PubMed] [Google Scholar]
- Arpino, G. , 2004. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a Southwest oncology group study. Clinical Cancer Research 10, 5670–5676. [DOI] [PubMed] [Google Scholar]
- Arpino, G. , Wiechmann, L. , Osborne, C.K. , Schiff, R. , 2008. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine Reviews 29, 217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss, J. , Hilger, A. , Vishnu, P. , Diehl, K. , El-Ashry, D. , 2007. Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clinical Cancer Research 13, 7029–7036. [DOI] [PubMed] [Google Scholar]
- Chaudhri, R.A. , Olivares-Navarrete, R. , Cuenca, N. , Hadadi, A. , Boyan, B.D. , Schwartz, Z. , 2012. Membrane estrogen signaling enhances tumorigenesis and metastatic potential of breast cancer cells via estrogen receptor-alpha36 (ERalpha36). The Journal of Biological Chemistry 287, 7169–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, R. , Liu, M.C. , Bouker, K.B. , Gu, Z. , Lee, R.Y. , Zhu, Y. , Skaar, T.C. , Gomez, B. , O'Brien, K. , Wang, Y. , Hilakivi-Clarke, L.A. , 2003. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 22, 7316–7339. [DOI] [PubMed] [Google Scholar]
- Creighton, C.J. , Hilger, A.M. , Murthy, S. , Rae, J.M. , Chinnaiyan, A.M. , El-Ashry, D. , 2006. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Research 66, 3903–3911. [DOI] [PubMed] [Google Scholar]
- deFazio, A. , Chiew, Y.E. , McEvoy, M. , Watts, C. , Sutherland, R.L. , 1997. Antisense estrogen receptor RNA expression increases epidermal growth factor receptor gene expression in breast cancer cells. Cell Growth & Differentiation: The Molecular Biology Journal of the American Association for Cancer Research 8, 903 [PubMed] [Google Scholar]
- Dudley, D.T. , Pang, L. , Decker, S.J. , Bridges, A.J. , Saltiel, A.R. , 1995. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proceedings of the National Academy of Sciences 92, 7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, P. , Wang, J. , Santen, R.J. , Yue, W. , 2007. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Research 67, 1352–1360. [DOI] [PubMed] [Google Scholar]
- Filardo, E.J. , 2002. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. The Journal of Steroid Biochemistry and Molecular Biology 80, 231–238. [DOI] [PubMed] [Google Scholar]
- Fox, E.M. , Bernaciak, T.M. , Wen, J. , Weaver, A.M. , Shupnik, M.A. , Silva, C.M. , 2008. Signal transducer and activator of transcription 5b, c-Src, and epidermal growth factor receptor signaling play integral roles in estrogen-stimulated proliferation of estrogen receptor-positive breast cancer cells. Molecular Endocrinology 22, 1781–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez, M.C. , Detre, S. , Johnston, S. , Mohsin, S.K. , Shou, J. , Allred, D.C. , Schiff, R. , Osborne, C.K. , Dowsett, M. , 2005. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. Journal of Clinical Oncology 23, 2469–2476. [DOI] [PubMed] [Google Scholar]
- Hiscox, S. , Morgan, L. , Barrow, D. , Dutkowski, C. , Wakeling, A. , Nicholson, R.I. , 2004. Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib (Iressa', ZD1839). Clinical and Experimental Metastasis 21, 201–212. [DOI] [PubMed] [Google Scholar]
- Hiscox, S. , Jiang, W.G. , Obermeier, K. , Taylor, K. , Morgan, L. , Burmi, R. , Barrow, D. , Nicholson, R.I. , 2006. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. International Journal of Cancer 118, 290–301. [DOI] [PubMed] [Google Scholar]
- Holloway, J.N. , Murthy, S. , El-Ashry, D. , 2004. A cytoplasmic substrate of mitogen-activated protein kinase is responsible for estrogen receptor-alpha down-regulation in breast cancer cells: the role of nuclear factor-kappaB. Molecular Endocrinology 18, 1396–1410. [DOI] [PubMed] [Google Scholar]
- Ignatov, A. , Ignatov, T. , Roessner, A. , Costa, S.D. , Kalinski, T. , 2010. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Research and Treatment 123, 87–96. [DOI] [PubMed] [Google Scholar]
- Jaiyesimi, I.A. , Buzdar, A.U. , Decker, D.A. , Hortobagyi, G.N. , 1995. Use of tamoxifen for breast cancer: twenty-eight years later. Journal of Clinical Oncology 13, 513–529. [DOI] [PubMed] [Google Scholar]
- Johnston, S. , Saccani-Jotti, G. , Smith, I. , Salter, J. , Newby, J. , Coppen, M. , Ebbs, S. , Dowsett, M. , 1995. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Research 55, 3331–3338. [PubMed] [Google Scholar]
- Jorissen, R. , 2003. Epidermal growth factor receptor: mechanisms of activation and signalling. Experimental Cell Research 284, 31–53. [DOI] [PubMed] [Google Scholar]
- Kang, L. , Zhang, X. , Xie, Y. , Tu, Y. , Wang, D. , Liu, Z. , Wang, Z.Y. , 2010. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Molecular Endocrinology 24, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, L. , Guo, Y. , Zhang, X. , Meng, J. , Wang, Z.Y. , 2011. A positive cross-regulation of HER2 and ER-alpha36 controls ALDH1 positive breast cancer cells. The Journal of Steroid Biochemistry and Molecular Biology 127, 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlden, J.M. , Hutcheson, I.R. , Jones, H.E. , Madden, T. , Gee, J.M.W. , Harper, M.E. , Barrow, D. , Wakeling, A.E. , Nicholson, R.I. , 2003. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 144, 1032–1044. [DOI] [PubMed] [Google Scholar]
- Lee, C.S.L. , Hall, R.E. , Alexander, I.E. , Koga, M. , Shine, J. , Sutherland, R.L. , 1990. Inverse relationship between estrogen receptor and epidermal growth factor receptor mRNA levels in human breast cancer cell lines. Growth Factors 3, 97–103. [DOI] [PubMed] [Google Scholar]
- Levitzki, A. , Gazit, A. , 1995. Tyrosine kinase inhibition: an approach to drug development. Science 267, 1782 [DOI] [PubMed] [Google Scholar]
- Lin, S.L. , Yan, L.Y. , Liang, X.W. , Wang, Z.B. , Wang, Z.Y. , Qiao, J. , Schatten, H. , Sun, Q.Y. , 2009. A novel variant of ER-alpha, ER-alpha36 mediates testosterone-stimulated ERK and Akt activation in endometrial cancer Hec1A cells. Reproductive Biology and Endocrinology: RB&E 7, 102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.L. , Yan, L.Y. , Zhang, X.T. , Yuan, J. , Li, M. , Qiao, J. , Wang, Z.Y. , Sun, Q.Y. , 2010. ER-α36, a variant of ER-α, promotes tamoxifen agonist action in endometrial cancer cells via the MAPK/ERK and PI3K/Akt pathways. PloS One 5, e9013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K.J. , Schmittgen, T.D. , 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Massarweh, S. , Schiff, R. , 2006. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocrine Related Cancer 13, (Suppl. 1) S15–S24. [DOI] [PubMed] [Google Scholar]
- Massarweh, S. , Osborne, C.K. , Creighton, C.J. , Qin, L. , Tsimelzon, A. , Huang, S. , Weiss, H. , Rimawi, M. , Schiff, R. , 2008. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Research 68, 826–833. [DOI] [PubMed] [Google Scholar]
- Oesterreich, S. , Zhang, P. , Guler, R.L. , Sun, X. , Curran, E.M. , Welshons, W.V. , Osborne, C.K. , Lee, A.V. , 2001. Re-expression of estrogen receptor alpha in estrogen receptor alpha-negative MCF-7 cells restores both estrogen and insulin-like growth factor-mediated signaling and growth. Cancer Research 61, 5771–5777. [PubMed] [Google Scholar]
- Oh, A.S. , Lorant, L.A. , Holloway, J.N. , Miller, D.L. , Kern, F.G. , El-Ashry, D. , 2001. Hyperactivation of MAPK induces loss of ERα expression in breast cancer cells. Molecular Endocrinology 15, 1344–1359. [DOI] [PubMed] [Google Scholar]
- Osborne, C.K. , Schiff, R. , 2011. Mechanisms of endocrine resistance in breast cancer. Annual Review of Medicine 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancholi, S. , Lykkesfeldt, A.E. , Hilmi, C. , Banerjee, S. , Leary, A. , Drury, S. , Johnston, S. , Dowsett, M. , Martin, L.A. , 2008. ERBB2 influences the subcellular localization of the estrogen receptor in tamoxifen-resistant MCF-7 cells leading to the activation of AKT and RPS6KA2. Endocrine Related Cancer 15, 985–1002. [DOI] [PubMed] [Google Scholar]
- Ruder, H.J. , Loriaux, L. , Lipsett, M. , 1972. Estrone sulfate: production rate and metabolism in man. Journal of Clinical Investigation 51, 1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen, R.J. , Fan, P. , Zhang, Z. , Bao, Y. , Song, R.X.D. , Yue, W. , 2009. Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids 74, 586–594. [DOI] [PubMed] [Google Scholar]
- Shi, L. , Dong, B. , Li, Z. , Lu, Y. , Ouyang, T. , Li, J. , Wang, T. , Fan, Z. , Fan, T. , Lin, B. , Wang, Z. , Xie, Y. , 2009. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. Journal of Clinical Oncology 27, 3423–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. , Ward, E. , Brawley, O. , Jemal, A. , 2011. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer Journal of Clinicians 61, 212–236. [DOI] [PubMed] [Google Scholar]
- Stoica, A. , Saceda, M. , Doraiswamy, V. , Coleman, C. , Martin, M. , 2000. Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. Journal of Endocrinology 165, 371–378. [DOI] [PubMed] [Google Scholar]
- Stossi, F. , Madak-Erdogan, Z. , Katzenellenbogen, B.S. , 2012. Macrophage-elicited loss of estrogen receptor-alpha in breast cancer cells via involvement of MAPK and c-Jun at the ESR1 genomic locus. Oncogene 31, 1825–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, J.S. , Zhang, Q.H. , Wang, Z.B. , Li, S. , Yang, C.R. , Fu, X.Q. , Hou, Y. , Wang, Z.Y. , Sheng, J. , Sun, Q.Y. , 2010. ER-α36, a novel variant of ER-α, mediates estrogen-stimulated proliferation of endometrial carcinoma cells via the PKCδ/ERK pathway. PloS One 5, e15408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, B.B. , Lin, S.L. , Yan, L.Y. , Wang, Z.Y. , Sun, Q.Y. , Qiao, J. , 2011. ER-alpha36, a novel variant of estrogen receptor alpha, is involved in EGFR-related carcinogenesis in endometrial cancer. American Journal of Obstetrics and Gynecology 205, (227) e221–226. [DOI] [PubMed] [Google Scholar]
- Van den Berg, H. , Lynch, M. , Martin, J. , Nelson, J. , Dickson, G. , Crockard, A. , 1989. Characterisation of a tamoxifen-resistant variant of the ZR-75-1 human breast cancer cell line (ZR-75-9a1) and ability of the resistant phenotype. British Journal of Cancer 59, 522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Zhang, X. , Shen, P. , Loggie, B.W. , Chang, Y. , Deuel, T.F. , 2005. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochemical and Biophysical Research Communications 336, 1023–1027. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Zhang, X. , Shen, P. , Loggie, B.W. , Chang, Y. , Deuel, T.F. , 2006. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proceedings of the National Academy of Sciences USA 103, 9063–9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden, R.I. , Lauber, A. , El-Ashry, D. , Chrysogelos, S. , 1996. Bimodal regulation of epidermal growth factor receptor by estrogen in breast cancer cells. Endocrinology 137, 2739–2747. [DOI] [PubMed] [Google Scholar]
- Yarden, R.I. , Wilson, M.A. , Chrysogelos, S.A. , 2001. Estrogen suppression of EGFR expression in breast cancer cells: a possible mechanism to modulate growth*. Journal of Cellular Biochemistry 81, 232–246. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Su, H. , Rahimi, M. , Tochihara, R. , Tang, C. , 2009. EGFRvIII-induced estrogen-independence, tamoxifen-resistance phenotype correlates with PgR expression and modulation of apoptotic molecules in breast cancer. International Journal of Cancer 125, 2021–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.T. , Kang, L.G. , Ding, L. , Vranic, S. , Gatalica, Z. , Wang, Z.Y. , 2011. A positive feedback loop of ER-alpha36/EGFR promotes malignant growth of ER-negative breast cancer cells. Oncogene 30, 770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Li, G. , Li, Z. , Yu, X. , Zheng, Y. , Jin, K. , Wang, H. , Gong, Y. , Sun, X. , Teng, X. , Cao, J. , Teng, L. , 2012. Estrogen-independent effects of ER-alpha36 in ER-negative breast cancer. Steroids 77, 666–673. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Ding, L. , Kang, L. , Wang, Z.Y. , 2012. Estrogen receptor-alpha 36 mediates mitogenic antiestrogen signaling in ER-negative breast cancer cells. PLoS One 7, e30174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Deng, C. , Lu, W. , Xiao, J. , Ma, D. , Guo, M. , Recker, R.R. , Gatalica, Z. , Wang, Z. , Guishan Xiao, G. , 2011. let-7MicroRNAs induce tamoxifen sensitivity by downregulation of estrogen receptor α signaling in breast cancer. Molecular Medicine 17, 1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X.F. , Liu, Z.C. , Xie, B.F. , Li, Z.M. , Feng, G.K. , Yang, D. , Zeng, Y.X. , 2001. EGFR tyrosine kinase inhibitor AG1478 inhibits cell proliferation and arrests cell cycle in nasopharyngeal carcinoma cells. Cancer Letters 169, 27–32. [DOI] [PubMed] [Google Scholar]
- Zou, Y. , Ding, L. , Coleman, M. , Wang, Z. , 2009. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Letters 583, 1368–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplemental Figure 1. The expression of HER‐2 in MCF‐7/TAM cells and MCF‐7/ER‐α36‐1 cells. (A) Western blotting analysis of the protein level of HER‐2 in MCF‐7 cells, MCF‐7/TAM cells and MCF‐7/ER‐α36‐1 cells. β‐actin was used as the loading control. All experiments were repeated at least three times, and the representative results are shown. (B) Relative mRNA expression of HER‐2 in MCF‐7 cells, MCF‐7/V cells, MCF‐7/TAM cells and MCF‐7/ER‐α36‐1 cells was determined by real time qPCR (Forward primer 5′‐ACCGGCACAGACATGAAGCT‐3′; Reverse primer 5′‐AGGAAGGACAGGCTGGCATT‐3′). β‐actin gene was used as an endogenous control. Results showed are means ± SEM of three independent reactions.