

Abstract
Cancer cells may use PARP enzymes and Homologous Recombination to repair single and double strand breaks caused by genotoxic insults. In this study, the PARP‐1 inhibitor Rucaparib was utilized to increase the sensitivity to chemoradiotherapy treatment in BRCA‐2‐deficient and ‐proficient pancreatic cancer cells. We used the pancreatic cancer cell lines, Capan‐1 with mutated BRCA‐2 and Panc‐1, AsPC‐1 and MiaPaCa‐2 with BRCA‐1/2 wild type. Cells were treated with Rucaparib and/or radiotherapy (4–10 Gy) plus Gemcitabine then the capability to proliferate was evaluated by colony formation, cell counting and MTT assays. Flow cytometry, immunocytochemistry and western blotting were utilized to assess cell response to Rucaparib plus irradiation. The antitumour effectiveness of combining the PARP‐1 inhibitor before, together and after radiotherapy evidenced the first as the optimal schedule in blocking cell growth. Pre‐exposure to Rucaparib increased the cytotoxicity of Gemcitabine plus radiotherapy by heavily inducing the accumulation of cells in G2/M phase, impairing mitosis and finally inducing apoptosis and authophagy. The upregulation of p‐Akt and downregulation of p53 were evidenced in MiaPaCa‐2 which displayed replication stress features. For the first time, the rationale of using a PARP inhibitor as chemoradiosensitizer in pancreatic cancer models has been hypothesized and demonstrated.
Keywords: PARP inhibitor, Pancreatic cancer, Chemoradiotherapy, Rucaparib, Gemcitabine
Highlights
-
►
Pre‐exposure to Rucaparib increased the cytotoxicity of radiotherapy plus Gemcitabine in pancreatic cancer models.
-
►
The formation of γH2AX foci after Rucaparib plus irradiation was evaluated to assess the DNA damage.
-
►
The formation of RAD51 foci after Rucaparib plus irradiation was evaluated to assess the HR system functionality.
-
►
The cell response to Rucaparib plus irradiation suggested the occurrence of different cell death mechanisms.
-
►
Akt‐1 activation plus p53 repression increase irradiation‐dependent cytotoxicity probably through mitotic catastrophe.
1. Introduction
Of all carcinomas, pancreatic cancer has the highest mortality rate, with a 1‐ and 5‐year survival of 25% and less than 5%, respectively (Gillen et al., 2010). Although surgery remains at the center of any potentially curable case, the need for other treatment modalities is paramount to improve survival. Both chemotherapy and radiotherapy have been widely used as an adjunct to surgery and/or as definitive treatment for unresectable locally advanced disease.
The standard of care as postoperative adjuvant therapy in this tumour setting is chemotherapy with Gemcitabine or 5‐FU. In the USA however, the combination of these drugs with radiotherapy is widely used, following large, single‐institute studies from the Johns Hopkins University and the Mayo Clinic, and a Gastro‐Intestinal Tumour Study Group (GITSG) trial performed in the early 1980s (van Tienhoven et al., 2011). Unfortunately neither the small GITSG trial (Kalser and Ellenberg, 1985), nor the randomized studies from the European Organisation for Research and Treatment of Cancer (EORTC) (Smeenk et al., 2007; Klinkenbijl et al., 1999), nor the ESPAC‐01 trial (Neoptolemos et al., 2004) validated this scheme. In summary, after R0 resection the current evidence supports the use of adjuvant chemotherapy rather than chemoradiotherapy followed by chemotherapy, even if the latter is regarded as the standard of care in North America. After R1 resection, adjuvant chemoradiotherapy should be considered (van Tienhoven et al., 2011).
Another subset of pancreatic cancer patients are those with resectable or borderline resectable pancreatic cancers. The definition of criteria for resectability of a pancreatic cancer varies among centers, and is often decided during exploratory laparotomy. However, preoperative modern imaging techniques, such as computed tomography scan or magnetic resonance imaging, have recently been developed (Phoa et al., 2005). Nevertheless, ‘making unresectable tumours resectable’ by chemotherapy or radiochemotherapy and consequently improving survival is one of the main goals in pancreatic cancer patients treatment. Recent studies of neoadjuvant radiochemotherapy suggests that this preoperative treatment provides better survival than postoperative one (Stessin et al., 2008) and randomized controlled trials are needed such as those started in 2007 by European partners (Brunner et al., 2007).
Unfortunately, the advantages of neoadjuvant treatment with Gemcitabine or 5‐FU in association with radiotherapy of pancreatic cancer patients are limited. Therefore clinicians and scientists are concentrating their efforts on finding both drugs with a low toxicological profile which could enhance the effectiveness of chemoradiotherapy and optimize the schedule to be used.
Assistance in this field comes from pharmacogenetics which allows to predict for drug response through the identification of genetic variants. Of particular interest are germ line BRCA‐2 and BRCA‐1 mutations responsible for an increased risk of developing pancreatic cancer of about 2.5‐ to 3.5‐fold higher than the general population (Lowery and O'Reilly, 2012). In details, Stadler et al. reported that the association between BRCA mutations and pancreatic cancer is not well defined and its prevalence varied between 6 and 17% (Stadler et al., 2012). Starting from this evidence and the well‐known relationship between BRCA mutation and PARP inhibitor effectiveness, we demonstrated for the first time in an in vitro model the role of Rucaparib as an anticancer agent in increasing the efficacy of Gemcitabine plus irradiation in “killing” pancreatic cancer cells.
Rucaparib is a PARP inhibitor which in turn results in inhibition of the repair of DNA single strand damage through the base excision‐repair pathway (BER). The exposure of cells to a PARP inhibitor leads to the accumulation of spontaneously occurring single strand breaks (SSBs) in DNA, since the latter cannot be repaired. When the cell divides and DNA replication takes place, these single strand breaks are converted to double strand breaks (DSBs) in one of the daughter strands. In cells that have functional Homologous Recombination (HR), these double strand breaks are repaired without errors, explaining the lack of toxicity of the PARP inhibitors towards the BRCA‐heterozygote and wild‐type cell lines. However if HR is deficient, as it is in the BRCA defective cell lines, these double strand breaks cannot be repaired, leading to collapse of the replication fork and cell death induction (Calvert and Azzariti, 2011).
The utilization of PARP inhibitors in HR‐deficient models has been demonstrated in mono and in combination with chemotherapeutics, and these drugs are currently undergoing evaluation in a variety of clinical settings. There has been successful translation from pre‐clinical to clinical use of PARP inhibitors as single agents in phase I and II trials for BRCA‐1 and ‐2‐deficient patients. Following encouraging pre‐clinical data, there is emerging clinical evidence for the use of PARP inhibitors in combination with chemotherapeutics, such as Temozolomide, Gemcitabine and Cisplatin and Topotecan (Calvert and Azzariti, 2011). The results are controversial as shown by the two following examples. Despite the promising results from a phase II trial of the PARP inhibitor Olaparib in patients with ovarian carcinoma or triple‐negative breast cancer (Gelmon et al., 2011) during 2011, AstraZeneca announced that the drug will not progress into phase III development for the treatment of ovarian cancer because an interim analysis of a phase II study did not confirm an overall survival benefit. In triple‐negative breast cancer patients, another PARP inhibitor, Iniparib, was administrated together with Gemcitabine and Carboplatin in an open‐label, phase II trial, providing to be more effective compared to chemotherapy alone (O'Shaughnessy et al., 2011a). However, in the same year during the ASCO Annual Meeting, O'Shaughnessy presented the preliminary results of the randomized, open‐label phase III study which, even if it confirmed the safety profile of the previous trial, seemed not to improve the endpoints of OS and PFS in patients with metastatic triple‐negative breast cancer (O'Shaughnessy et al., 2011b).
With respect to the therapeutic approach described above, literature data on the possibility of utilizing PARP inhibitors as radiosensitising agents are scarce. Today, radiation is combined with various PARP inhibitors, such as GPI‐15427, AZD2281 (Olaparib), ABT‐888, ET016 and AG14361, in different in vitro and in vivo cancer models. The radiosensitizing activity of these agents has been demonstrated by their ability to increase apoptosis and mitotic catastrophe, to reduce clonogenic survival and to inhibit endothelial tubule formation. Moreover, PARP inhibitors are evaluated as radiosensitizers in phase I and II clinical trials of treatment of head and neck cancers as well as CNS neoplasms. In particular, the first trial (NCT00649207) is ongoing and aims to determine the safety and pharmacokinetics of ABT‐888 (Veliparib) in combination with conventional whole brain irradiation in cancer patients with brain metastasis. The second (NCT00770471) conducted by Abbott with ABT‐888 recruits mainly patients with glioblastoma multiforme with the objective to assess pharmacokinetics, toxicity and efficacy when combined with standard radiation/TMZ treatment (Mangerich and Burkle, 2011; Verheij et al., 2010).
However, no data are available about the possibility to enhance the effectiveness of a chemoradiotherapeutic treatment with PARP inhibitors.
With particular respect to Rucaparib, data in the literature evidenced that it increases the effectiveness of chemotherapeutics, such as TMZ, Topotecan, Carboplatin, in different cancer in vitro and in vivo models (Ali et al., 2009; Drew et al., 2011; Thomas et al., 2007). Finally, only two original papers deal with the outcome of Rucaparib in combination with radiotherapy; the first highlighted that the combination significantly enhanced therapeutic response both in vitro and in vivo, and the second outlined that radiosensitization is due to the downstream inhibition that follows NF‐κB activation (Ali et al., 2011; Hunter et al., 2012).
The present study illustrates promising results on the role of Rucaparib as an enhancer of chemoradiotherapy effectiveness and suggests that our hypothesis could be particularly relevant in tumours which barely respond to the traditional anticancer therapy, such as pancreatic tumours, thus opening a new therapeutic window in this area.
2. Materials and methods
2.1. Drugs and chemicals
Rucaparib was provided by Pfizer/Clovis Oncology (USA). Stock solutions were prepared at 20 mM in DMSO and stored in aliquots at −20 °C. Gemcitabine (Gemzar®) was provided by Eli Lilly and further dilutions were made in medium supplemented with 10% foetal bovine serum, 2 mM glutamine, 50,000 UL−1 penicillin and 80 μM streptomycin.
2.2. Cell lines
Four pancreas cancer cell lines of human origin were used, MiaPaCa‐2 and Panc‐1 (from pancreas adenocarcinoma), and Capan‐1 and AsPC‐1 (from metastatic liver and ascites sites). Cells were routinely cultured in DMEM (Capan‐1, AsPC‐1 and Panc‐1) and 20 mM Hepes‐RPMI (MiaPaca‐2) supplemented with 10% foetal bovine serum, 2 mM glutamine, 50,000 UL−1 penicillin and 80 μM streptomycin in a humidified incubator at 37 °C with an atmosphere containing 5% CO2. Cells were trypsinized once a week with trypsin/EDTA (0.25%/0.02%) and medium was changed twice a week. Doubling time of MiaPaCA‐2 and Panc‐1 was 18 ± 1; of Capan‐1 was 48 ± 1 and of AsPC‐1 was 24 ± 1 h.
2.3. Irradiation exposure
An X‐irradiator (Model LINAC – Varian Medical Systems, Inc., USA) was used in this study. The dose rate we used was 3 Gy/min. Experiments were carried out at room temperature.
2.4. Cell proliferation assay
Determination of cell growth inhibition was performed using the 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazoliumbromide (MTT) assay and by cell counting. The MTT assay was used in the kinetic experiments carried out with the only Rucaparib both for the execution speed and the possibility of dosing a large number of samples simultaneously. The cell count was essential when we combined the drug with radiotherapy. In some cell lines, the IRR induced increase in ploidy detectable as an increase in size of the cells responsible for a distorted result if you used the MTT assay (Azzariti et al., 2011).
The MTT assay and the determination of the IC50 for Gemcitabine sensitivity were performed as described in Azzariti et al. (2004).
The evaluation of cell proliferation after the combined exposure to Rucaparib and IRR was carried out by exposing cells to Rucaparib (at concentration and time detailed in each experiment) and then irradiated with photon beam. The treated cells were incubated at 37 °C in the presence of CO2 for the reported time. When the combination therapy also included the administration of Gemcitabine, the drug was given concomitantly with radiotherapy.
For cell count determination, 1.5 × 105 cells were plated in 35 mm Petri dishes, exposed to therapeutic treatment, harvested in trypsin and counted. Rucaparib was given at concentrations of 0.1 μM, 0.5 μM, 1 μM for 3 days. In the combination studies, Rucaparib was given at 0.1 μM, 0.5 μM, 1 μM and the cells irradiated at 4 and 10 Gy. To define the best schedule for the combination, either simultaneous or sequential utilisation of the two drugs were tested. Each experiment was done in triplicate.
Results were analysed utilising Origin 8 software.
2.5. Cellular effectors analysis
Cells were exposed to Rucaparib and IRR as described in the text and protein level of the selected protein was analysed by fluorescence immunocytochemistry (ICC), western blotting and/or flow cytometry (CFM).
2.5.1. Fluorescence immunocytochemistry (ICC)
Cells were seeded onto coverslips. After overnight incubation, they were fixed in 3.7% paraformaldehyde, washed and permeabilized with 0.1% Triton X‐100. After saturation with 0.1% gelatin in PBS, cells were subsequently immunostained overnight with phospho‐Histone H2AX (Ser‐139) antibody (Millipore, USA) or anti‐RAD51 [14B4] (Abcam, UK) or anti‐LC3(APG8B) (ABGENT, USA). Cells were then incubated with FITC‐conjugated secondary antibody (Becton Dickinson‐USA) for 1 h. Nuclei were counterstained with 0.5 lg/ml 40,6‐diamidino‐2‐phenylindole (DAPI). The images were captured using a fluorescence microscope (Olympus BX40), equipped with X40 and X60 objectives with a SenSys 1401E‐Photometrics charge‐coupled device camera. FITC was excited using the 488 laserline and DAPI using the 568 laserline.
2.5.2. Western blot analysis
Protein extracts were obtained by homogenization in RIPA buffer (0.5 M NaCl, 1% Triton X‐100, 0.5% NP40, 1% deoxycolic acid, 3.5 mM SDS, 8.3 mM Tris HCl, pH 7.4, 1.6 mM Tris base and 1 mM phenylmethylsulfonyl fluoride). Total proteins were measured and analysed as described in Azzariti et al. (2004). In particular, 50 μg were electrophoretically separated on 10% acrylamide gel (SDS‐PAGE by Laemli). Signal was detected by chemoluminescence assay (ECL‐Plus, Amersham Life Science, UK). Expression levels were evaluated by densitometric analysis using Quantity One software (BioRad, Hercules, CA) and β‐actin expression levels were used to normalize the sample values. Antibodies: the monoclonal antibody anti‐Akt, anti‐p‐Akt (Ser473), anti Erk1/2, anti‐p‐Erk1/2, anti‐p53(DO‐1), anti‐β‐actin AC‐15 and anti‐LC3(APG8B) were provided by Cell signalling – USA, Santa Cruz Biotechnology, Sigma–Aldrich and ABGENT respectively. A mouse‐HRP and a rabbit‐HRP (Amersham Pharmacia Biotech, Uppsala, Sweden) were used as secondary antibody. All antibodies were utilized at the recommended dilutions.
2.5.3. Flow cytometry (FCM) analysis
Cells were harvested, washed twice in ice‐cold PBS pH 7.4, fixed in 4.5 ml of 70% ethanol and stored at −20 °C. Fixed cells were processed as described in our previous paper (Azzariti et al., 2006). The primary antibody was phospho‐Histone H2AX (Ser‐139) antibody (Millipore, USA) and the secondary the goat anti‐mouse IgG (H&L) fluorescein‐conjugated affinity purified secondary antibody (BD Pharmingen, USA).
2.6. 6‐well colony formation assay
Cells were trypsinized and plated in 6‐well dishes at density of 500 cells/well. Cells were allowed to attach overnight and then treated with Rucaparib at 0.1, 0.5 or 1 μM and incubated at 37 °C. Fourteen days later, the cells were fixed in ethanol 100% and stained with crystal violet 0.2%. The numbers of colonies were counted and the graphs were the composite results from three independent experiments.
2.7. Cell cycle perturbation
Treated cells were harvested, washed twice in ice‐cold PBS (pH 7.4), fixed in 4.5 ml of 70% ethanol at −20 °C, and washed once in ice‐cold PBS. The pellet was resuspended in PBS containing 1 mg/ml RNase, 0.01% NP40 and the cellular DNA was stained with 50 μg/ml propidium iodide (Sigma). Cells were stored in ice for 30 min prior to analysis. Cell cycle determinations were performed using a FACScan flow cytometer (Becton, Dickinson), and data were interpreted using the CellQuest and the ModFit software, provided by the manufacturer.
2.8. Cell apoptosis assay
Apoptosis detection was further investigated by the Cell Death ELISAPLUS kit (Roche Molecular Biochemicals, Milan, Italy). The test is based on the detection of mono‐ and oligonucleosomes in the cytoplasmic fraction of cell lysates by biotinylated antihistone‐coupled antibodies, and their enrichment in the cytoplasm is calculated as the absorbance of sample cells/absorbance of control cells. The enrichment factor was used as a parameter of apoptosis and shown on the Y‐axis as mean ± SE. Experiments were performed according to the manufacturer's instructions.
2.9. The cytokinesis‐block micronucleus (CBMN) assay
This assay was performed to evaluate micronuclei (MN) and nuclear buds (NBUDs) formation in cells that have completed nuclear division, after blocking cytokinesis with cytochalasin‐B according to the method developed by Fenech (2007). In vitro treated cells were incubated with cytochalasin‐B (4.5 μg/ml). At 24–28 h after addition of cytochalasin‐B, cells were harvested for slides preparation using cytocentrifugation. Cells were air‐dried then fixed in methanol for 15 min and stained in Giemsa 5%. After staining the slides were rinsed in water, air‐dried and then mounted with coverslips. Slides were examined with an optical microscope Leica DMLB at X40 magnification, equipped with acquisition software Leica TC300.
2.10. Statistical analysis
All in vitro experiments were performed in triplicate, and results have been expressed as the mean ± standard deviation (S.D) unless otherwise indicated. Statistical differences of in vitro data were assessed by the Student–Newman–Keuls test. p‐values lower than 0.05 were considered significant.
3. Results
The role of Rucaparib as enhancer of radiochemotherapy, in which Gemcitabine is added to the photonic therapy, has been explored in four pancreas cancer cell lines of human origin, MiaPaCa‐2 and Panc‐1 (from pancreas adenocarcinoma), and Capan‐1 and AsPC‐1 (from metastatic liver and ascites sites). Kinetic experiments were performed in the whole cell panel whereas, the investigation of the cell features responsible for drug(s) activity was carried out in two of them, more promising in terms of treatment response and taking into account their cellular characteristics: MiaPaCA‐2 cells are from a primary tumour and are BRCA‐1 wt; BRCA‐2 wt; p53 mt while Capan‐1 are from metastatic liver and are BRCA‐1 wt; BRCA‐2 mt; p53 mt.
3.1. Radiosensitivity of cells
The first phase of the study was the characterization of the radiosensitivity of our panel of cells; to irradiate cells, LINAC available in our hospital was utilized. Cells were irradiated and the absorbed doses were 2, 4, 6, 8 and 10 Gy; cell proliferation was determined after 1 and 4 days. The inhibition of cell growth was observed in function of photonic dose and of incubation time and the response to irradiation (IRR) was more marked in metastatic cells with mutated BRCA (Capan‐1) (Figure 1A).
Figure 1.
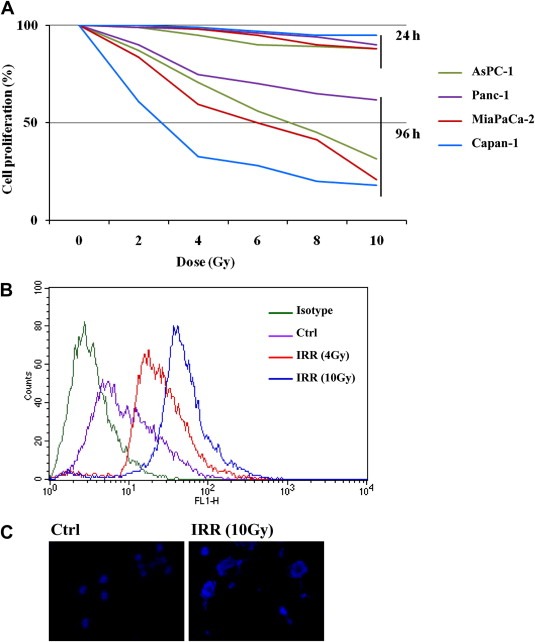
Radiosensitivity of pancreatic cancer cells. (A) Cells were exposed to photon beam utilising an LINAC. The absorbed doses were 2, 4, 6, 8 or 10 Gy and cell proliferation was determined after 1 and 4 days as described in the Material and methods section. (B) MiaPaCA‐2 cells were irradiated at 4 and 10 Gy and after 2 h the damage of genomic DNA was determined by flow cytometry analysis of γH2AX. (C) MiaPaCa‐2 cells were exposed to 10 Gy IRR and after 48 h polynucleate cells were evidenced by ICC (blue: DAPI).
The reduction of cell proliferation reflects the damage of the genomic DNA occurring within the cell and directly related to the increase of the phosphorylated histone H2AX (Ser‐139) (γH2AX). Results obtained in MiaPaCa‐2 are reported in Figure 1B; cells exposed to 4 and 10 Gy showed an increase of the γH2AX, compared to control (cells not irradiated), revealing by the shift of the fluorescence peak along the axis of X (Figure 1B). Similar results are obtained in other cells (data not shown). Other indications of increased DNA damage after IRR are nuclear abnormalities as the onset of nuclear fragmentation which is the hallmark of mitotic catastrophe (Ianzini et al., 1999; Meng et al., 2004). Figure 1C shows the polynucleate MiaPaCa‐2 cells collected after IRR with an absorbed dose of 10 Gy and the image is representative of the response of all cell lines.
3.2. Rucaparib activity
A prolonged exposure (7 days) of cells to Rucaparib, at concentration ranging between 0.1 and 1 μM, showed that the drug was not toxic with only a slight reduction of cell proliferation being observed (Figure 2A). Conversely, cell ability to produce colony was strongly reduced by the drug as shown in Figure 2B, where the modulation of Capan‐1 colony formation after continuous exposure to the PARP inhibitor for 14 days is illustrated and representative of the other cell lines response. The amount of colonies after each concentration of Rucaparib was statistically different from those without drug treatment (p < 0.05).
Figure 2.
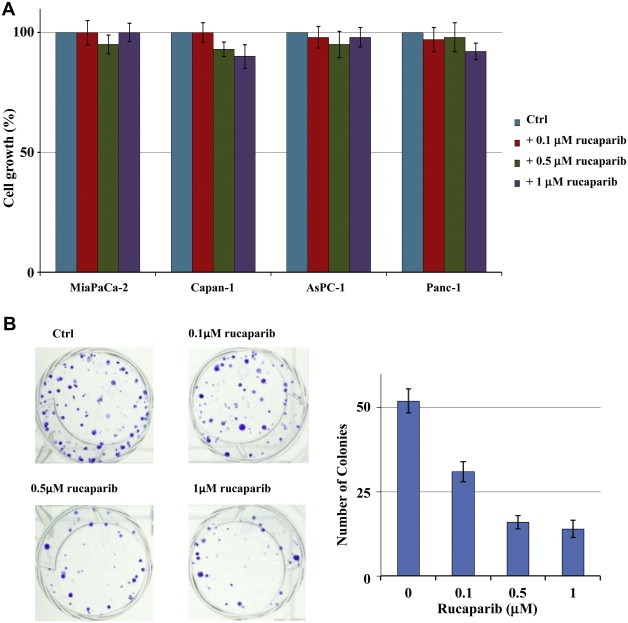
Modulation of proliferation by Rucaparib. (A) Cells were incubated with 0.1–1 μM Rucaparib for 4 days and cell growth was determined by cell counting. (B) Capan‐1 cells were incubated with 0.1–1 μM Rucaparib for 14 days and the effect on colony formation was evidenced as described in the Material and methods section. There were fewer colonies in drug treated cells than in non‐treated cells (P < 0.05).
3.3. Rucaparib plus chemoradiotherapy: effectiveness
Previous analysis provided doses of radiotherapy and Rucaparib to be utilized in the following experiments i.e., 4–10 Gy and 0.1, 0.5, 1 μM, respectively. Regarding Gemcitabine concentrations, both our own previous published data and a preliminary screening were considered (Azzariti et al., 2011). The IC50 of Gemcitabine in each cell lines after 3 days of continuous exposure was reduced 3‐fold as suggested for the dose given during radiochemotherapy regiment to patients with resectable or borderline resectable pancreatic cancers (Lombardi et al., 2012). In detail, 2, 2.5, 4.1 and 1.3 μM Gemcitabine were utilized in Panc‐1, AsPC‐1, MiaPaCa‐2 and Capan‐1, respectively.
Initially, the best way to combine Rucaparib with IRR was evaluated by analysing cell response to three different schedules, one simultaneous and two sequential. Results showed that the PARP inhibitor was more effective when administrated 1 day before radiotherapy (both 4 and 10 Gy) (in Figure 3 only the more promising schedule irradiated at 4 Gy for all cell lines are reported). The increase of absorbed dose is responsible for a marked effect but not for an increase of combination effectiveness (data not shown). Capan‐1 showed the highest sensitivity to IRR (with an inhibition of cell growth of about 50%) and to the sequential exposure to Rucaparib and IRR with a gain of effectiveness of about 10% (Figure 3).
Figure 3.
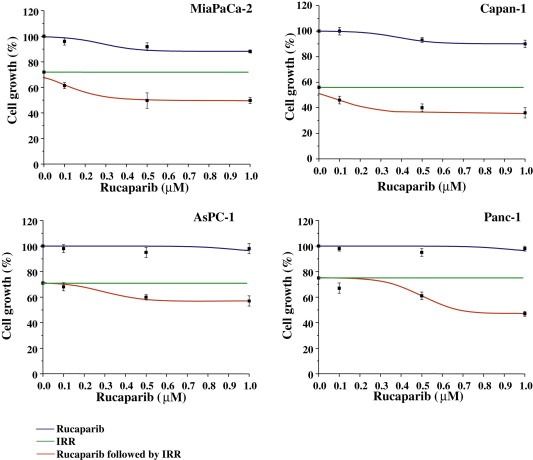
Cell growth inhibition by Rucaparib plus irradiation. Cells were incubated with 0.1–1 μM Rucaparib for 96 h and after the first 24 h, were irradiated with an absorbed dose of 4 Gy. The cell growth was measured by cell counting. Results from three independent experiments are shown (mean ± S.D).
Next, the capability of the PARP inhibitor to enhance chemoradiotherapy effectiveness was screened, and results are reported in Figure 4. Cells were exposed to 1 μM Rucaparib for 24 h followed by Gemcitabine plus IRR for 3 more days. The pre‐exposure to the PARP inhibitor results in a strong increase of cell death, suggesting a clear role for Rucaparib as a “chemoradio‐enhancer”. Proliferation of all cells was reduced; Rucaparib increased Capan‐1 cells death mainly at 10 Gy while MiaPaCa‐2 resulted more sensitive at 4 Gy, and the gap between chemoradio‐treatments with or without Rucaparib is statistically different.
Figure 4.
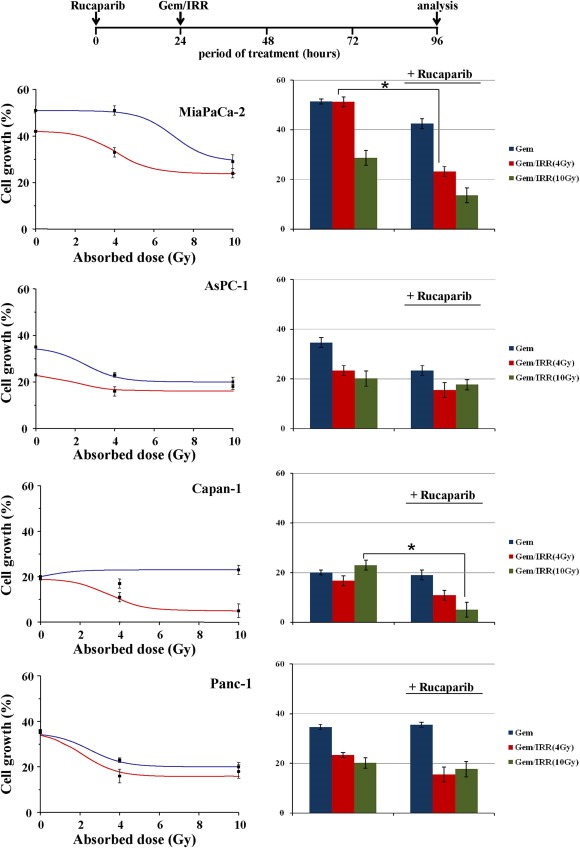
Cell growth inhibition by Rucaparib plus Gemcitabine/irradiation. Cell culture was incubated with 0.1–1 μM Rucaparib for 96 h, and after the first 24 h was added Gemcitabine and exposed to radiation (absorbed doses: 4–10 Gy). The cell growth was measured by cell counting. Results from three independent experiments are shown and data are expressed as mean ± S.D in both plots and histograms (::: Gemcitabine together with IRR; ::: Rucaparib followed by Gemcitabine together with IRR) (*p < 0.05).
3.4. Rucaparib plus chemoradiotherapy: mechanism of action
To identify determinants predicting the response to Rucaparib plus chemoradiotherapy, the comparison of results obtained in the two more responsive cell lines Capan‐1 (BRCA‐2 mutated) vs. MiaPaCa‐2 (BRCA wild type) was assessed. Attention was focused on the involvement of members of DNA damage and repair systems such as the formation of γH2AX and RAD51 foci, and on the analysis of cell cycle progression, autophagy and apoptosis induction, DNA misrepair landmarks and survival/proliferation targets modulation.
The presence of γH2AX foci correlates with DSBs (Pilch et al., 2003) and allows to determine the appearance of them in cells treated with Rucaparib as a consequence of the impairment of the BER pathway. Capan‐1 and MiaPaCa‐2 cells treated with Rucaparib together with IRR after 2 h were collected and stained. The ICC analysis evidenced the presence of γH2AX foci after both IRR and Rucaparib (Figure 5A). γH2AX increased in the presence of Rucaparib alone and this activation, also evident in cells exposed to photonradiation, increased when the PARP inhibitor was given together. As reported in Figure 5B, the PARP inhibitor induced a stronger phosphorylation of H2AX in Capan‐1 cells than in MiaPaCa‐2 cells whereas either the radiotherapy or the pre‐treatment with the PARP‐1 inhibitor Rucaparib increased the phosphorylation of H2AX at about the same extent in both cell lines, as demonstrated by the right shift of the picks in the pictures.
Figure 5.
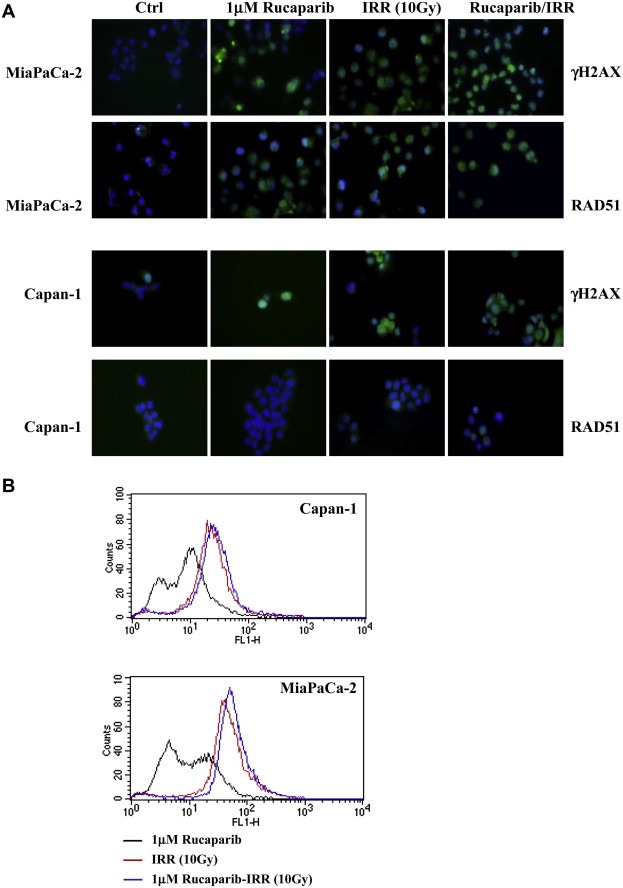
DNA damage and the involvement of HR pathway in cell response to Rucaparib plus irradiation. Cell culture was incubated with 1 μM Rucaparib and after 24 h, was exposed to a radiation dose of 4–10 Gy. After 2 h cells were characterised for the presence of γH2AX and RAD51 foci by ICC (blue: DAPI; green: γH2AX or RAD51 foci) (A). The quantification of the γH2AX expression in the same experimental conditions was obtained by CFM analysis (B).
The involvement of the HR pathway in determining cell response to Rucaparib plus irradiation was assessed by the analysis of RAD51 foci which relocalized within the nucleus in response to DNA damage and allowed to distinguish between HR‐proficient and HR‐deficient cell lines (Lee et al., 2009). As expected, Capan‐1 cells, characterised by BRCA‐2 mutation are not able to form RAD51 foci both after Rucaparib exposure and irradiation. Conversely, MiaPaCa‐2 cells showed RAD51 foci after irradiation and Rucaparib exposure. When they are simultaneously administrated RAD51 foci seem to be comparable to each treatment, suggesting the inability of cells to further increase DNA repair mechanism (Figure 5A). The higher sensitivity of Capan‐1 cells to IRR and combined treatment, evidenced in kinetic experiments, can depend on the impairment of the HR pathway in this cell line in which BRCA‐2 is mutated. Therefore Rucaparib, blocking PARP‐1 and the BER pathway, increases the accumulation of DNA DSB from SSB naturally occurring into cells causing a more pronounced cell death.
The analysis of cellular mechanisms related to drug(s) treatment response continued with the investigation of cell cycle progression. As expected, Rucaparib did not affect cell cycle progression whereas irradiation induced a marked accumulation of cell into G2/M phase (4N). In addition, in MiaPaCa‐2 cells, the exposure to a photon beam induced a supplemental round of DNA synthesis such that cells became also 8N. The pre‐exposure to Rucaparib markedly increased cell cycle perturbation (Figure 6 and Table 1) suggesting that the majority of cells can undergo death by apoptosis as a consequence of the prolonged arrest in G2 phase, while a small population are not arrested and enter abortive mitosis undergoing endoreduplication phenomena not followed by cell division.
Figure 6.
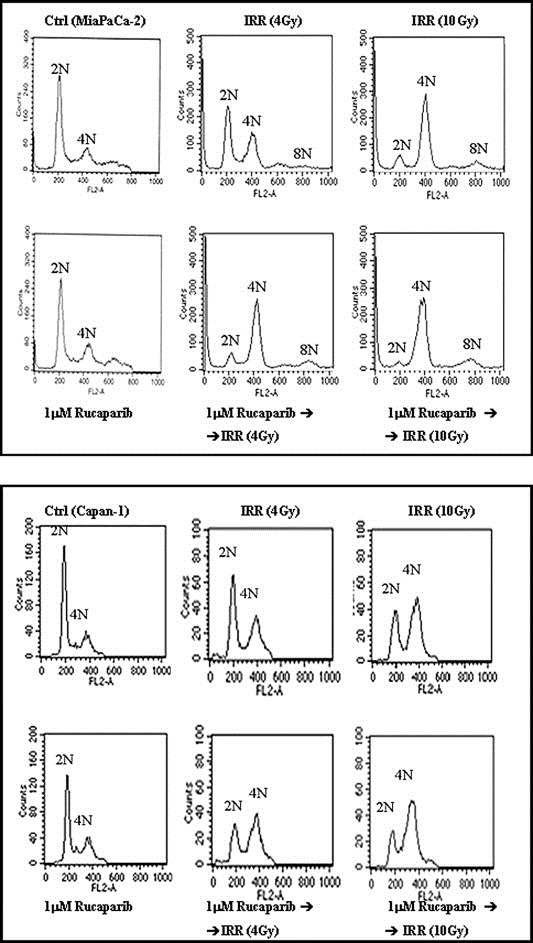
Rucaparib‐dependent cell cycle modification. Cells were incubated with 1 μM Rucaparib for 48 h, and after the first 24 h were exposed to a radiation dose of 4–10 Gy. Cell cycle was analysed by CFM as described in the Material and methods section.
Table 1.
Cell cycle distribution after exposure to Rucaparib plus IRR.
| Cell line | Drug(s) treatment | 2N(%) | 4N(%) | 8N(%) |
|---|---|---|---|---|
| MiaPaCa‐2 | 42.78 | 16.74 | 0 | |
| IRR (4 Gy) | 39.74 | 30.51 | 3.04 | |
| IRR (10 Gy) | 9.98 | 54.04 | 7.84 | |
| 1 μM Rucaparib | 39.42 | 18.83 | 0 | |
| 1 μM Rucaparib ➜ IRR (4 Gy) | 10.95 | 48.47 | 9.77 | |
| 1 μM Rucaparib ➜ IRR (10 Gy) | 4 | 56.73 | 10 | |
| Capan‐1 | 58.23 | 20.86 | 0 | |
| IRR (4 Gy) | 48.53 | 37.03 | 0 | |
| IRR (10 Gy) | 33.81 | 54.89 | 0 | |
| 1 μM Rucaparib | 47.65 | 26.48 | 0 | |
| 1 μM Rucaparib ➜ IRR (4 Gy) | 25.77 | 48.25 | 0 | |
| 1 μM Rucaparib ➜ IRR (10 Gy) | 19.18 | 65.40 | 0 |
To validate the hypothesis, apoptosis was analysed by DNA laddering assay. As showed in Figure 7A, results are in agreement with cell cycle determination with a marked apoptosis in MiaPaCa‐2 (BRCA wild‐type cells) rather than in Capan‐1 (the BRCA mutant HR‐deficient cell line) where the block in G2/M phase was less evident after irradiation alone at both 4 and 10 Gy. After combined treatment, the behaviour of the two cell lines was opposite with Rucaparib increasing and decreasing apoptosis in Capan‐1 and MiaPaCa‐2 cells, respectively.
Figure 7.
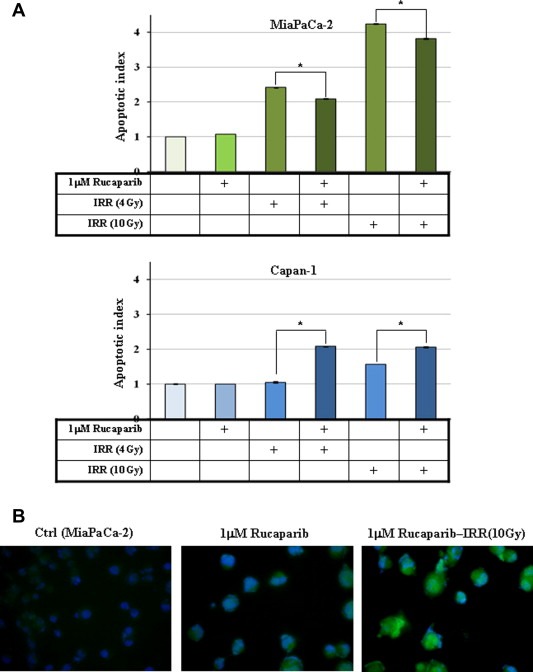
Effect of Rucaparib in combination with radiotherapy on apoptosis and autophagy. Cells were incubated with 1 μM Rucaparib for 72 h and after the first 24 h were exposed to a radiation dose of 4–10 Gy. (A) Apoptosis was measured as described in the Material and methods section and results of three independent experiments are reported as histograms. (B) ICC confirmation of autophagy induction in treated MiaPaCa‐2 cells through the determination of LC3‐stained autophagic compartments is reported as punctate dots (blue: DAPI; green LC3).
To solve the discrepancy, autophagy as other mechanism of cell death was investigated together with various cell markers of survival and cancer progression.
Autophagy is usually activated by radiation via the adaptive endoplasmic reticulum stress response as a result of the accumulation of misfolded proteins which induces LC3 conversion. Our question was if in MiaPaCa‐2 cells the lack of Rucaparib‐dependent apoptosis induction together with a trend of reduction of this programmed cell death when the drug was given together with IRR could be responsible for the shift of autophagic response from survival to cell death. A preliminary characterization of autophagy was carried out by western blotting and ICC. Both demonstrated that autophagy increased in samples in which Rucaparib was given before IRR, as reported in Figure 7B where the levels of punctate LC3‐positive autophagosomes in cells increased in the presence of the PARP inhibitor as while LC3 fluorescence was diffuse in the cytoplasm in the control (irradiated cells).
However, it is necessary to take into account that in MiaPaCa‐2 cells the exposure to photon beam and, more importantly, the preincubation with Rucaparib induced the formation of polynucleate cells (see Figure 1C) and other nuclear anomalies such as the formation of micronuclei (MN) and nuclear buds (NBUDs). In Figure 8A, MiaPaCa‐2 cells, after a combined treatment with Rucaparib and IRR, displayed both MNs and NBUDs, evident also in all other cell types (data not shown).
Figure 8.
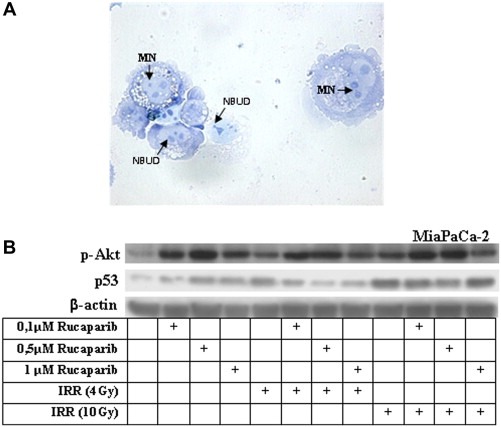
Rucaparib plus radiotherapy as inducer of mitotic catastrophe. MiaPaCa‐2 cells were incubated with 1 μM Rucaparib and after 24 h were exposed to a radiation dose of 10 Gy for 24 h. (A) MNs and NBUDs were highlighted by the cytokinesis‐block micronucleus assay (arrows). (B) Protein extracts were analysed by western blotting. The amount of p‐Akt and p53 was determined using β‐actin to normalize values.
The analysis of the involvement of specific cell effectors in determining the response to Rucaparib plus IRR included Akt, p‐Akt, Erk1/2, p‐Erk1/2, JNK, p38 and p53 characterization. In Figure 8B, only data of p‐Akt and p53 in MiaPaCa‐2 cells are reported because the other markers are not modulated by the drug. In Rucaparib/IRR treated cells, p‐Akt was always up‐expressed and this data together with the reduction of p53 expression could explains both the reduction of apoptosis and the ability of cells to endoreduplicate.
4. Discussion
In order to overcome the poor prognosis of patients affected by locally advanced pancreatic cancer, several studies have shown that the combination of chemotherapy plus radiotherapy confers a secondary resectability rate of 10–20% with a potential curative intent (van Tienhoven et al., 2011; Lombardi et al., 2012).
Gemcitabine is the drug most commonly used in combination with radiation therapy in borderline or locally advanced pancreatic cancer patients, appearing to decrease the ability of cancer cells to repair radiation‐induced DNA damage (Lawrence et al., 1996). Neoadjuvant Gemcitabine‐based chemoradiotherapy for this subset of patients can lead to a high rate of second resectability. A meta‐analysis by Zhu et al. considered the role of Gemcitabine plus radiotherapy in locally advanced pancreatic cancer patients and showed that this treatment seemed to be superior to 5‐FU‐based chemoradiotherapy (Zhu et al., 2011). The originality of the present study is that it uses a systematic and innovative approach to validate the opportunity to use the PARP inhibitor Rucaparib as a chemoradiosensitizer.
Recently, the interest of radiation oncologists focuses on the possibility to utilize this class of target‐oriented drugs as enhancers of chemoradiation effectiveness, since they have shown encouraging results in monotherapy and in combination with chemotherapy along with a low toxicity profile (Chalmers et al., 2010).
This report starts with the characterization of radiosensitivity of a suitable cancer cells panel which has been selected taking into account specific characteristics that have already been demonstrated to be responsible for the synthetic lethality of PARP inhibitors. Together with the Medical Physics Unit we defined the proper irradiation plan to be used on the LINAC. This characterization showed that cellular response to irradiation was in function of beam intensity and time after IRR. The most sensitive cells were Capan‐1. Mutated BRCA‐2, MiaPaCA‐2 and AsPC‐1 showed an intermediate behaviour while Panc‐1 cells were the most resistant ones. Cells were then exposed to Rucaparib and radiotherapy to define the best way to combine these two anticancer treatments, in function of the sequence they are administrated. The optimal schedule with the PARP‐1 inhibitor before IRR was utilized in the kinetic determination of cell response to Rucaparib plus chemoradiotherapy. PARP inhibitor increased the response to chemoradiotherapy mainly in Capan‐1 and MiaPaCa‐2 cells. In Capan‐1 cells it could be expected because they have the homologous recombination mechanism impaired for the presence of mutated BRCA‐2 and, consequently the contemporary presence of this mutation with the inhibition of PARP‐1 by Rucaparib is responsible for the well‐known establishment of synthetic lethality (Porcelli et al., 2012). For MiaPaCa‐2, we suggest a BRCAness phenotype which is a tumour with abnormal function of BRCA‐1 and BRCA‐2 genes and/or other genes implicated in DNA repair pathways even in the absence of reported mutations in BRCA‐1/2 (Rigakos and Razis, 2012). Our hypothesis is based on the capacity of Rucaparib to increase the cytotoxicity of chemoradiotherapy and on the absence of BRCA‐1/2 mutation.
The relevance of HR pathway functionality in response to photon beam exposure together or without the impairment of the BER pathway, through the addition of the PARP inhibitor, was evaluated by the analysis of γH2AX and RAD51 foci. As known, the phosphorylation of H2AX is performed by kinases belonging to the family of Pikk, including ATM (Ataxia Telangiectasia Mutated), and is crucial for the recruitment of molecules involved in the HR pathway, such as BRCA‐1/2. RAD51 is necessary to repair DSBs by NHEJ and HR. γH2AX increased in the presence of Rucaparib or IRR and more markedly when cells were exposed to Rucaparib/photon radiation. The increased phosphorylation of H2AX after treatment with Rucaparib in Capan‐1 cells can be explained by the fact that often the inhibition of PARP‐1, especially in cells defective in the HR system, stimulates the NHEJ (Non‐homologous end joining) to repair DNA. However the NHEJ is error prone thus induces the accumulation of DNA damage which could explain the increased sensitivity to radiation therapy in this cell line. RAD51 foci are evident only in MiaPACa‐2 samples and seem to be independent from the simultaneous administration of treatments. These data are in agreement with those reported by Drew et al. who described an increase of RAD51 and γH2AX foci after exposure to Rucaparib (Drew et al., 2011). We also focused our investigation on the cell response to Rucaparib plus irradiation with respect to each treatment alone to clarify if cell death occurs through apoptosis or others mechanisms. The analysis of the cell cycle progression, together with the appearance of endoreduplication, apoptosis and autophagy, suggests that the cells, characterized by mutated BRCA‐2 and consequently by HR impairment, die due to apoptosis. Conversely, cells with other phenotypes undergo autophagy when the BER pathway is also inhibited.
Finally, the onset of nuclear fragmentation and the analysis of cell proliferation and survival effectors suggest another possible mechanism of death. In fact, the former is a well‐known hallmark of mitotic catastrophe (Ianzini et al., 1999), while the increase in p‐Akt in parallel with the decrease in p53 could be explained considering the same model proposed by Lu et al. who demonstrated that the simultaneous activation of Akt‐1 and repression of p53 results in paradoxical enhancement of the effectiveness of cytotoxic chemotherapy through induction of mitotic catastrophe (Lu et al., 2009). The investigation of this topic is now ongoing.
Our report is the first systematic study relating the role of Rucaparib as an enhancer of chemoradiotherapy in a pancreatic cancer model, and is in agreement with the few published data reporting the combined administration of Rucaparib plus radiotherapy in other cancer types (Ali et al., 2011; Hunter et al., 2012).
In their paper, Ali et al. showed the capability of Rucaparib to be a radiosensitizer in a breast cancer cell line; in spite of small differences between their and our experimental approach, with reference to the absorbed dose (6 Gy vs. 4 and 10 Gy), the Rucaparib exposure time (3 vs. 24 h) and concentration (0.4 μM vs. 0.1–1 μM), the results of the two studies largely agree for the increased effectiveness of irradiation when combined with this PARP inhibitor, in terms of inhibition of both cell growth and colony formation.
On the other hand, Hunter et al. reported that radiosensitization by Rucaparib is due to inhibition of downstream activation of NF‐KB, which is ultimately responsible for the activation of apoptosis rather than for the impairment of DNA‐SSBs repair pathway (Hunter et al., 2012). Our results seem at odds with those reported by Hunter's, but one must keep in mind firstly that our in vitro model is different, and secondly, as pointed out by the author, that the variations between different forms of cancer are substantial. Furthermore, in Capan‐1 cells characterized by a defective DNA repair pathway, the exposure to Rucaparib before irradiation induces an increase of therapeutic response, in terms of enhanced induction of apoptosis, whereas in MiaPaCa cells a reduction of this phenomenon is observed. We propose in the near future to investigate more throughly the possible involvement of NF‐KB.
5. Conclusion
The combination of radiotherapy and chemotherapy is a promising approach that has led to improved treatment results in patients with borderline resectable pancreatic cancers. The concurrent application of both modalities resulted in the possibility to have surgery. We believe that the present evidences significantly reinforces the rationale of using PARP inhibitors as chemoradiosensitizers. Our approach has begun to identify part of the complex molecular networks involved in the increased chemoradiosensitivity after PARP inhibitor administration. Unfortunately, our data suggest that today a single biomarker predictive of response to combined therapy of PARP inhibitor with Gemcitabine/IRR has not yet been identified, and probably only a group of factors, a biomarker signatures, could provide useful information for stratification of patients who could benefit from the combination of a DNA repair system targeted agent plus chemoradiotherapy.
Conflicts of interest
The authors declare no conflict of interest.
Porcelli Letizia, Quatrale Anna E., Mantuano Paola, Leo Maria G., Silvestris Nicola, Rolland Jean F., Carioggia Enza, Lioce Marco, Paradiso Angelo, Azzariti Amalia, (2013), Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.10.002.
References
- Ali, M. , Kamjoo, M. , Thomas, H.D. , Kyle, S. , Pavlovska, I. , Babur, M. , Telfer, B.A. , Curtin, N.J. , Williams, K.J. , 2011. The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol. Cancer Ther. 10, 2320–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, M. , Telfer, B.A. , McCrudden, C. , O'Rourke, M. , Thomas, H.D. , Kamjoo, M. , Kyle, S. , Robson, T. , Shaw, C. , Hirst, D.G. , Curtin, N.J. , Williams, K.J. , 2009. Vasoactivity of AG014699, a clinically active small molecule inhibitor of poly(ADP-ribose) polymerase: a contributory factor to chemopotentiation in vivo?. Clin. Cancer Res. 15, 6106–6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzariti, A. , Bocci, G. , Porcelli, L. , Fioravanti, A. , Sini, P. , Simone, G.M. , Quatrale, A.E. , Chiarappa, P. , Mangia, A. , Sebastian, S. , Del Bufalo, D. , Del Tacca, M. , Paradiso, A. , 2011. Aurora B kinase inhibitor AZD1152: determinants of action and ability to enhance chemotherapeutics effectiveness in pancreatic and colon cancer. Br. J. Cancer 104, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzariti, A. , Colabufo, N.A. , Berardi, F. , Porcelli, L. , Niso, M. , Simone, G.M. , Perrone, R. , Paradiso, A. , 2006. Cyclohexylpiperazine derivative PB28, a sigma2 agonist and sigma1 antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol. Cancer Ther. 5, 1807–1816. [DOI] [PubMed] [Google Scholar]
- Azzariti, A. , Xu, J.M. , Porcelli, L. , Paradiso, A. , 2004. The schedule-dependent enhanced cytotoxic activity of 7-ethyl-10-hydroxy-camptothecin (SN-38) in combination with Gefitinib (Iressa, ZD1839). Biochem. Pharmacol. 68, 135–144. [DOI] [PubMed] [Google Scholar]
- Brunner, T.B. , Grabenbauer, G.G. , Meyer, T. , Golcher, H. , Sauer, R. , Hohenberger, W. , 2007. Primary resection versus neoadjuvant chemoradiation followed by resection for locally resectable or potentially resectable pancreatic carcinoma without distant metastasis. A multi-centre prospectively randomised phase II-study of the Interdisciplinary Working Group Gastrointestinal Tumours (AIO, ARO, and CAO). BMC Cancer 7, 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert, H. , Azzariti, A. , 2011. The clinical development of inhibitors of poly(ADP-ribose) polymerase. Ann. Oncol. 22, (Suppl. 1) i53–i59. [DOI] [PubMed] [Google Scholar]
- Chalmers, A.J. , Lakshman, M. , Chan, N. , Bristow, R.G. , 2010. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin. Radiat. Oncol. 20, 274–281. [DOI] [PubMed] [Google Scholar]
- Drew, Y. , Mulligan, E.A. , Vong, W.T. , Thomas, H.D. , Kahn, S. , Kyle, S. , Mukhopadhyay, A. , Los, G. , Hostomsky, Z. , Plummer, E.R. , Edmondson, R.J. , Curtin, N.J. , 2011. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J. Natl. Cancer Inst. 103, 334–346. [DOI] [PubMed] [Google Scholar]
- Fenech, M. , 2007. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2, 1084–1104. [DOI] [PubMed] [Google Scholar]
- Gelmon, K.A. , Tischkowitz, M. , Mackay, H. , Swenerton, K. , Robidoux, A. , Tonkin, K. , Hirte, H. , Huntsman, D. , Clemons, M. , Gilks, B. , Yerushalmi, R. , Macpherson, E. , Carmichael, J. , Oza, A. , 2011. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 12, 852–861. [DOI] [PubMed] [Google Scholar]
- Gillen, S. , Schuster, T. , Meyer Zum Buschenfelde, C. , Friess, H. , Kleeff, J. , 2010. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 7, e1000267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, J.E. , Willmore, E. , Irving, J.A. , Hostomsky, Z. , Veuger, S.J. , Durkacz, B.W. , 2012. NF-kappaB mediates radio-sensitization by the PARP-1 inhibitor, AG-014699. Oncogene 31, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianzini, F. , Cherubini, R. , Mackey, M.A. , 1999. Mitotic catastrophe induced by exposure of V79 Chinese hamster cells to low-energy protons. Int. J. Radiat. Biol. 75, 717–723. [DOI] [PubMed] [Google Scholar]
- Kalser, M.H. , Ellenberg, S.S. , 1985. Pancreatic cancer. Adjuvant combined radiation and chemotherapy following curative resection. Arch. Surg. 120, 899–903. [DOI] [PubMed] [Google Scholar]
- Klinkenbijl, J.H. , Jeekel, J. , Sahmoud, T. , van Pel, R. , Couvreur, M.L. , Veenhof, C.H. , Arnaud, J.P. , Gonzalez, D.G. , de Wit, L.T. , Hennipman, A. , Wils, J. , 1999. Adjuvant radiotherapy and 5-fluorouracil after curative resection of cancer of the pancreas and periampullary region: phase III trial of the EORTC gastrointestinal tract cancer cooperative group. Ann. Surg. 230, 776–782. discussion 782–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, T.S. , Chang, E.Y. , Hahn, T.M. , Hertel, L.W. , Shewach, D.S. , 1996. Radiosensitization of pancreatic cancer cells by 2′,2′-difluoro-2′-deoxycytidine. Int. J. Radiat. Oncol. Biol. Phys. 34, 867–872. [DOI] [PubMed] [Google Scholar]
- Lee, S.A. , Roques, C. , Magwood, A.C. , Masson, J.Y. , Baker, M.D. , 2009. Recovery of deficient homologous recombination in Brca2-depleted mouse cells by wild-type Rad51 expression. DNA Repair (Amst.) 8, 170–181. [DOI] [PubMed] [Google Scholar]
- Lombardi, L. , Troiano, M. , Silvestris, N. , Nanni, L. , Latiano, T.P. , Di Maggio, G. , Cinieri, S. , Di Sebastiano, P. , Colucci, G. , Maiello, E. , 2012. Combined modality treatments in pancreatic cancer. Expert Opin. Ther. Targets 16, (Suppl. 2) S71–S81. [DOI] [PubMed] [Google Scholar]
- Lowery, M.A. , O'Reilly, E.M. , 2012. Genomics and pharmacogenomics of pancreatic adenocarcinoma. Pharmacogenomics J. 12, 1–9. [DOI] [PubMed] [Google Scholar]
- Lu, J. , Kovach, J.S. , Johnson, F. , Chiang, J. , Hodes, R. , Lonser, R. , Zhuang, Z. , 2009. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl. Acad. Sci. U S A 106, 11697–11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangerich, A. , Burkle, A. , 2011. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int. J. Cancer 128, 251–265. [DOI] [PubMed] [Google Scholar]
- Meng, X. , Yuan, Y. , Maestas, A. , Shen, Z. , 2004. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. J. Biol. Chem. 279, 6098–6105. [DOI] [PubMed] [Google Scholar]
- Neoptolemos, J.P. , Stocken, D.D. , Friess, H. , Bassi, C. , Dunn, J.A. , Hickey, H. , Beger, H. , Fernandez-Cruz, L. , Dervenis, C. , Lacaine, F. , Falconi, M. , Pederzoli, P. , Pap, A. , Spooner, D. , Kerr, D.J. , Büchler, M.W. , 2004. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 350, 1200–1210. [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy, J. , Osborne, C. , Pippen, J.E. , Yoffe, M. , Patt, D. , Rocha, C. , Koo, I.C. , Sherman, B.M. , Bradley, C. , 2011. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 364, 205–214. [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy, J. , Schwartzberg, L.S. , Danso, M.A. , Rugo, H.S. , Miller, K. , Yardley, D.A. , Carlson, R.W. , Finn, R.S. , Charpentier, E. , Freese, M. , Gupta, S. , Blackwood-Chirchir, A. , Winer, E.P. , 2011. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC). J. Clin. Oncol. 29, (Suppl.) abstr 1007 [Google Scholar]
- Phoa, S.S. , Tilleman, E.H. , van Delden, O.M. , Bossuyt, P.M. , Gouma, D.J. , Lameris, J.S. , 2005. Value of CT criteria in predicting survival in patients with potentially resectable pancreatic head carcinoma. J. Surg. Oncol. 91, 33–40. [DOI] [PubMed] [Google Scholar]
- Pilch, D.R. , Sedelnikova, O.A. , Redon, C. , Celeste, A. , Nussenzweig, A. , Bonner, W.M. , 2003. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol. 81, 123–129. [DOI] [PubMed] [Google Scholar]
- Porcelli, L. , Quatrale, A.E. , Mantuano, P. , Silvestris, N. , Brunetti, A.E. , Calvert, H. , Paradiso, A. , Azzariti, A. , 2012. Synthetic lethality to overcome cancer drug resistance. Curr. Med. Chem. 19, 3858–3873. [DOI] [PubMed] [Google Scholar]
- Rigakos, G. , Razis, E. , 2012. BRCAness: finding the Achilles heel in ovarian cancer. Oncologist 17, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk, H.G. , van Eijck, C.H. , Hop, W.C. , Erdmann, J. , Tran, K.C. , Debois, M. , van Cutsem, E. , van Dekken, H. , Klinkenbijl, J.H. , Jeekel, J. , 2007. Long-term survival and metastatic pattern of pancreatic and periampullary cancer after adjuvant chemoradiation or observation: long-term results of EORTC trial 40891. Ann. Surg. 246, 734–740. [DOI] [PubMed] [Google Scholar]
- Stadler, Z.K. , Salo-Mullen, E. , Patil, S.M. , Pietanza, M.C. , Vijai, J. , Saloustros, E. , Hansen, N.A. , Kauff, N.D. , Kurtz, R.C. , Kelsen, D.P. , Offit, K. , Robson, M.E. , 2012. Prevalence of BRCA1and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer 118, 493–499. [DOI] [PubMed] [Google Scholar]
- Stessin, A.M. , Meyer, J.E. , Sherr, D.L. , 2008. Neoadjuvant radiation is associated with improved survival in patients with resectable pancreatic cancer: an analysis of data from the surveillance, epidemiology, and end results (SEER) registry. Int. J. Radiat. Oncol. Biol. Phys. 72, 1128–1133. [DOI] [PubMed] [Google Scholar]
- Thomas, H.D. , Calabrese, C.R. , Batey, M.A. , Canan, S. , Hostomsky, Z. , Kyle, S. , Maegley, K.A. , Newell, D.R. , Skalitzky, D. , Wang, L.Z. , Webber, S.E. , Curtin, N.J. , 2007. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 6, 945–956. [DOI] [PubMed] [Google Scholar]
- van Tienhoven, G. , Gouma, D.J. , Richel, D.J. , 2011. Neoadjuvant chemoradiotherapy has a potential role in pancreatic carcinoma. Ther. Adv. Med. Oncol. 3, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij, M. , Vens, C. , van Triest, B. , 2010. Novel therapeutics in combination with radiotherapy to improve cancer treatment: rationale, mechanisms of action and clinical perspective. Drug Resist. Updat. 13, 29–43. [DOI] [PubMed] [Google Scholar]
- Zhu, C.P. , Shi, J. , Chen, Y.X. , Xie, W.F. , Lin, Y. , 2011. Gemcitabine in the chemoradiotherapy for locally advanced pancreatic cancer: a meta-analysis. Radiother. Oncol. 99, 108–113. [DOI] [PubMed] [Google Scholar]