

Abstract
Obesity condition confers risks to breast cancer development and progression, and several reports indicate that the adipokine leptin, whose synthesis and plasma levels increase with obesity, might play an important role in modulating breast cancer cell phenotype. Functional crosstalk occurring between leptin and different signaling molecules contribute to breast carcinogenesis.
In this study, we show, in different human breast cancer cell lines, that leptin enhanced the expression of a chaperone protein Hsp90 resulting in increased HER2 protein levels. Silencing of Hsp90 gene expression by RNA interference abrogated leptin‐mediated HER2 up‐regulation. Leptin effects were dependent on JAK2/STAT3 activation, since inhibition of this signaling cascade by AG490 or ectopic expression of a STAT3 dominant negative abrogated leptin‐induced HER2 and Hsp90 expressions. Functional experiments showed that leptin treatment significantly up‐regulated human Hsp90 promoter activity. This occurred through an enhanced STAT3 transcription factor binding to its specific responsive element located in the Hsp90 promoter region as revealed by electrophoretic mobility shift assay and chromatin immunoprecipitation assay. Analysis of HER2, Akt and MAPK phosphorylation levels revealed that leptin treatment amplified the responsiveness of breast cancer cells to growth factor stimulation. Furthermore, we found that long‐term leptin exposure reduced sensitivity of breast cancer cells to the antiestrogen tamoxifen. In the same experimental conditions, the combined treatment of tamoxifen with the Hsp90 inhibitor 17‐AAG completely abrogated leptin‐induced anchorage‐independent breast cancer cell growth.
In conclusion, our results highlight, for the first time, the ability of the adipocyte‐secreted factor leptin to modulate Hsp90/HER2 expressions in breast cancer cells providing novel insights into the molecular mechanism linking obesity to breast cancer growth and progression.
Keywords: Leptin, Hsp90, HER2, Breast cancer, Tamoxifen resistance
Highlights
-
►
Leptin enhances HER2 protein levels by inducing Hsp90 expression in breast cancer.
-
►
The mechanism relies on enhanced STAT3 binding to a specific Hsp90 promoter region.
-
►
Leptin amplifies responsiveness to growth factors and reduces tamoxifen sensitivity.
-
►
Leptin/Hsp90/HER2 axis may represent a new target for breast cancer treatment.
1. Introduction
Breast cancer, a complex and heterogeneous disease, is one of the most common human malignancies in women worldwide. The development of human breast cancer is thought to depend on the accumulation of various genetic alterations, but it is now clear that modifiable factors such as overweight (Body Mass Index‐BMI 25–30 kg/m2) or obesity (BMI > 30 kg/m2) conditions also have an important role (Calle and Kaaks, 2004; Calle et al., 2003). Indeed, a number of epidemiological studies has suggested that obesity and high adipose‐tissue mass are associated with an increased risk of breast cancer development, as well as with an aggressive tumor phenotype and a poor survival (Harvie et al., 2003; Lahmann et al., 2004; Michels et al., 2006). Adipose tissue, initially considered only a fat‐storing tissue, is now known to have much more complex functions mainly mediated by a range of adipose‐tissue‐derived signaling molecules named adipokines. Recently, the adipokines are emerging as key mediators linking obesity with breast cancer occurrence and among them leptin, whose synthesis and plasma levels increase proportionally to total adipose‐tissue mass (Considine et al., 1996; Maffei et al., 1995), has been extensively examined at this regard.
Leptin, a 16 kDa polypeptide hormone encoded by the obese (Ob) gene, is a pleiotropic molecule that regulates food intake, inflammation, immunity, cell differentiation and proliferation of different cell types including cells of the breast (Andò and Catalano, 2011). Interestingly, leptin and both short and long isoforms of the leptin receptor (ObR) are overexpressed in breast cancer cells compared with healthy epithelium (Ishikawa et al., 2004). High expression of leptin receptor in breast cancer tissue predicts poor prognosis in patients with high serum leptin levels (Miyoshi et al., 2006), and over‐expression of both leptin and its specific receptors is correlated with distant metastasis (Ishikawa et al., 2004). Moreover, it has been reported that this adipokine enhances in vitro and in vivo breast cancer cell growth (Dieudonne et al., 2002; Hu et al., 2002; Mauro et al., 2007; Yin et al., 2004) through the activation of several signaling pathways, such as those involving Janus kinase 2‐signal transducer and activator of transcription 3 (JAK2‐STAT3), mitogen‐activated protein kinase (MAPK), and phosphatidylinositol 3‐kinase‐protein kinase B (PI3K‐AKT) (Ahima and Osei, 2004; Sweeney, 2002).
Leptin action is mainly mediated by the long and full‐functional isoform of ObR, but we and other authors have demonstrated that leptin can exert its activity also interacting with different signaling molecules. We have previously demonstrated that leptin promotes in situ estrogen production (Catalano et al., 2003a) and directly transactivates estrogen receptor (ER)α (Barone et al., 2012; Catalano et al., 2004) in human MCF‐7 breast cancer cells. Saxena et al. (2008) have reported the existence of a bidirectional crosstalk between leptin and insulin‐like growth factor I (IGF‐I) signaling, mediated by synergistic transactivation of epidermal growth factor receptor (EGFR), which influences breast cancer cell invasion and migration. In addition, two separate studies have reported an interplay between leptin signaling and the transmembrane tyrosine kinase receptor HER2, a member of epidermal growth factor receptor family. In vitro as well as preliminary in vivo studies have shown that ObR and HER2 are co‐expressed in breast cancer cell lines and tumors (Fiorio et al., 2008) and it has been demonstrated that leptin can transactivate HER2 in SKBR3 cells (Soma et al., 2008).
The HER2 gene is amplified and/or overexpressed in 20–25% of ERα‐positive breast cancers (Slamon et al., 1989), and multiple lines of evidences have suggested an important causal role of HER2 in the pathogenesis of breast carcinoma (Allred et al., 1992; Glockner et al., 2001). Indeed, HER2 over‐expression affects tumor growth, invasion and resistance to endocrine‐treatments and as recently reported increases the stem/progenitor cell population of both normal and malignant mammary cells (Korkaya et al., 2008). The regulation of HER2 expression is primarily a result of HER2 gene copy number amplification (Hurst, 2001), but it also occurs at protein level, through post‐transcriptional events mediated by the heat‐shock protein 90 (Hsp90) chaperone activity (Xu et al., 2001). Indeed, molecular chaperone Hsp90 protects HER2 from proteasome‐mediated degradation (Xu et al., 2001).
In the present study, we demonstrate that leptin by inducing Hsp90 expression enhances HER2 protein levels, providing a new molecular mechanism underlying the crosstalk between leptin and HER2 signaling pathways in breast cancer cells.
2. Materials and methods
2.1. Reagents, and antibodies
Dulbecco's Modified Eagle's Medium (DMEM), Fetal bovine serum (FBS), leptin and TRIzol by Invitrogen (Carlsbad, CA, USA). l‐glutamine, penicillin, streptomycin, phosphate‐buffered saline, aprotinin, leupeptin, phenylmethylsulfonyl fluoride (PMSF), bovine serum albumin (BSA), sodium orthovanadate, NP‐40, MTT, 4‐Hydroxytamoxifen, Epidermal growth factor and 17‐AAG were from Sigma (Milan, Italy). AG490 from Calbiochem. FuGENE 6, TaqDNA polymerase, RETROscript kit, Dual Luciferase kit and TK Renilla luciferase plasmid were provided by Promega (Madison, WI, USA). SYBR Green Universal PCR Master Mix by Bio‐Rad (Hercules, CA, USA). Antibodies against Hsp90, β‐Actin, by Santa Cruz Biotechnology (Santa Cruz, CA, USA), total MAPK, phosphorylated p42/44 MAPK (Thr202/Tyr204), total Akt, phosphorylated Akt (Ser473), total HER2 and phosphorylated HER2 (Tyr1248) from Cell Signaling Technology (Beverly, MA). ECL system and Sephadex G‐50 spin columns from Amersham Biosciences (Buckinghamshire, UK). [γ32P]ATP from PerkinElmer (Wellesley, MA, USA). Salmon sperm DNA/protein A agarose by UBI (Chicago, IL, USA).
2.2. Cell culture
Breast cancer epithelial cell line MCF‐7 were cultured in DMEM medium containing 10% fetal bovine serum, 1% l‐glutamine, 1% Eagle's nonessential amino acids, and 1 mg/ml penicillin–streptomycin at 37 °C with 5% CO2 air. SKBR3 cells were cultured in McCoy's 5A Medium modified containing 10% fetal bovine serum, 1% l‐glutamine, 1% Eagle's nonessential amino acids, and 1 mg/ml penicillin–streptomycin. MCF‐7/HER2‐18 were kindly provided by Dr. Schiff (Baylor College of Medicine, Houston, TX, USA) and maintained as described (Shou et al., 2004).
Before each experiment, cells were grown in phenol red‐free media, containing 5% charcoal‐stripped fetal bovine serum for 2 days and then treated as described.
2.3. Plasmids
The reporter construct containing the 5′‐flanking region of the human Hsp90α gene promoter (HSPLuc1430) was kindly provided by Professor KJ Wu (IBMB, National Yang‐Ming University, Taiwan). pSG5 vector containing the cDNA encoding dominant negative STAT3, which is a variant of the transcription factor STAT3 lacking an internal domain of 50 base pairs located near the C terminus (STAT−), was kindly provided by Dr. J. Turkson (University of South Florida College of Medicine, Tampa, FL).
2.4. Immunoprecipitation and immunoblot analysis
Cells were treated as indicated before lysis for total protein extraction (Catalano et al., 2009). For co‐immunoprecipitation experiments, we used 1 mg of total protein extract and 2 μg of Hsp90 or HER2 polyclonal antisera overnight, followed by protein A/G precipitation. Equal amounts of cell extracts and coimmunoprecipitated protein were subjected to SDS–polyacrylamide gel electrophoresis, as described (Catalano et al., 2010). The bands of interest were quantified by Scion Image laser densitometry scanning program.
2.5. Immunofluorescence
Cells were fixed with 4% paraformaldehyde, permeabilized with PBS 0.2% Triton X‐100 followed by blocking with 5% bovine serum albumin, and incubated with anti‐HER2 antibody and with fluorescein isothiocyanate‐conjugated secondary antibody. IgG primary antibody was used as negative control. 4′,6‐Diamidino‐2‐phenylindole (DAPI; Sigma) staining was used for nuclei detection. Fluorescence was photographed with OLYMPUS BX51 microscope, 100× objective.
2.6. Real‐time RT‐PCR assays
Analysis of HER2 and Hsp90 gene expression was performed by Real‐time reverse transcription‐PCR. Two micrograms of total RNA were reverse transcribed with the RETROscript kit; cDNA was diluted 1:3 in nuclease‐free water and 5 μl were analyzed in triplicates by real‐time PCR in an iCycler iQ Detection System (Bio‐Rad, USA) using SYBR Green Universal PCR Master Mix with 0.1 mmol/l of each primer in a total volume of 30 μl reaction mixture following the manufacturer's recommendations. Negative control contained water instead of first strand cDNA was used. Each sample was normalized on its GAPDH mRNA content. Primers used for the amplification were: forward 5′‐CACCTACAACACAGACACGTTTGA‐3′ and reverse 5′‐GCAGACGAGGGTGCAGGAT‐3′ (HER2); forward 5′‐ATTGCCCAGTTGATGTCATTGA‐3′ and reverse 5′‐ATGCATCTGATGAATTTGAAATGAG‐3′ (Hsp90); forward 5′‐CCCACTCCTCCACCTTTGAC‐3′ and reverse 5′‐TGTTGCTGTAGCCAAATTCGTT‐3′ (GAPDH). The relative gene expression levels were normalized as previously described (Catalano et al., 2010). ObR gene expression was evaluated by the RT‐PCR method using a RETROscript kit. The cDNAs obtained were amplified using the following primers: forward 5′‐AGAGAAGCACTTGGTGACTG‐3′ and reverse 5′‐GCCAACAACTGTGGTCTCTC‐3′ (ObR); forward 5′‐CTCAACATCTCCCCCTTCTC‐3′ and reverse 5′‐CAAATCCCATATCCTCGT‐3′(36B4). The PCR was performed for 35 cycles for ObR (94 °C for 1 min, 58 °C for 1 min, 72 °C for 1 min) and 18 cycles for 36B4 (94 °C for 1 min, 58 °C for 1 min and 72 °C for 1 min), as described (Catalano et al., 2010).
2.7. Soft‐agar growth assays
Soft‐agar anchorage‐independent growth assay was assessed as described (Giordano et al., 2010).
2.8. RNA silencing
MCF‐7 cells were transfected with RNA duplex of stealth siRNA targeted for the human ObR mRNA sequence (Ambion, ID:s224009), with siRNA targeted for the human Hsp90 mRNA sequence (Ambion, ID:119758) or with a control siRNA (Invitrogen ID:45‐2001) that does not match with any human mRNA, used as a control for non‐sequence‐specific effects to a final concentration of 50 nM using Lipofectamine 2000 as recommended by the manufacturer. After 5 h the transfection medium was changed with serum‐free medium and then the cells were exposed to treatments.
2.9. Transient transfection assay
MCF‐7 cells were transiently transfected using the FuGENE 6 reagent with HSPLuc1430 reporter gene. After transfection cells were treated with leptin in the presence or not of the JAK2/STAT3 inhibitor AG490 (1 μM) for additional 24 h and then luciferase activity was assayed as described (Catalano et al., 2007).
2.10. Electrophoretic mobility shift assay (EMSA)
Nuclear extracts from MCF‐7 cells, treated or not for 1 h with leptin, were prepared as previously described (Catalano et al., 2010). The probe was generated by annealing single‐stranded oligonucleotides, labeled with [γ32P]ATP using T4 polynucleotide kinase, and purified using Sephadex G‐50 spin columns. The DNA sequences used as probe or as cold competitors are the following (nucleotide motifs of interest are underlined and mutations are shown as lowercase letters): STAT3, 5′‐AGCACATGGCTTTCGGGGAACCAAAAGTAG‐3′; mutated STAT3, 5′‐GCACATGGCTTTaatttAACC AAAAGTAGGG‐3′. The protein‐binding reactions were carried out as described (Catalano et al., 2003b). For experiments involving anti‐STAT3 antibody, the reaction mixture was incubated with this antibody at 4 °C for 12 h before addition of labeled probe.
2.11. Chromatin immunoprecipitation assay
MCF‐7 cells were treated with leptin 500 ng/ml or left untreated for 1 h and then DNA/protein complexes were extracted as described (Lanzino et al., 2010). A 5 μl volume of each sample and input were used for real‐time PCR using the primers flanking STAT3 sequence in the human Hps90 promoter region: 5′‐TTCACCAAGTCCCCGATTCC‐3′ and 5′‐GTTCTCGGGGATTCTCCAGA‐3′. PCR reactions were performed as described above. Final results were calculated using the ΔΔCt method as explained above, using input Ct values instead of the GAPDH mRNA. The basal sample was used as calibrator.
2.12. Statistical analyses
Each datum point represents the mean ± S.D. of three different experiments. Data were analyzed by Student's t test using the GraphPad Prism 4 software program. P < 0.05 was considered as statistically significant.
3. Results
3.1. Leptin enhances HER2 protein levels in breast cancer cells
First, we investigated the effects of the obesity hormone leptin on HER2 expression in ERα‐positive MCF‐7 breast cancer cells. To this aim, we performed both immunoblotting and immunofluorescence (IF) analysis in cells treated with leptin at 500 ng/ml as indicated. Results obtained demonstrated that leptin exposure increased HER2 protein levels (Figure 1a) and induced a strong immunofluorescence at the plasma membrane (Figure 1b) compared to untreated cells.
Figure 1.
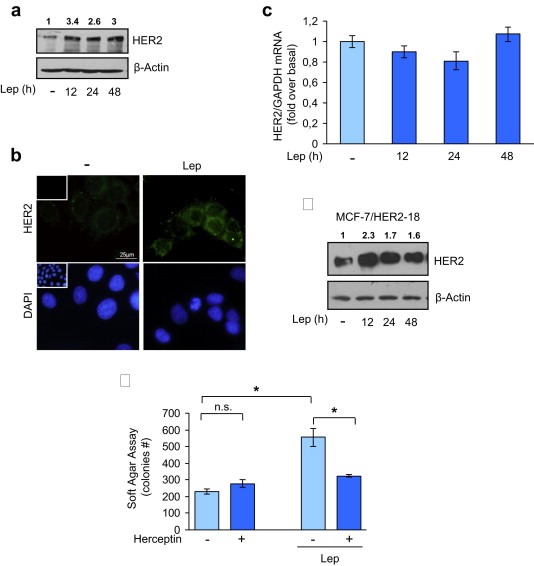
Leptin effects on HER2 expression. (a) MCF‐7 cells were untreated (−) or treated for 12, 24 and 48 h with leptin 500 ng/ml (Lep) before lysis. Equal amounts of total cellular extracts were analyzed for HER2 protein levels by immunoblotting analysis. β‐Actin was used as loading control. (b) Immunofluorescence of HER2 in MCF‐7 cells untreated (−) or treated with Lep for 24 h. Small squares, negative controls. 4′,6‐Diamidino‐2‐phenylindole (DAPI) staining was used to visualize the cell nucleus. Scale bar = 25 μm. (c) mRNA HER2 content, evaluated by real‐time RT‐PCR, after treatment with Lep as indicated. Each sample was normalized to its GAPDH mRNA content. The values represent the means ± s.d. of three different experiments each performed in triplicate. (d) Immunoblotting analysis of HER2 in total protein extracts from MCF‐7/HER2‐18 cells treated with Lep as indicated; β‐actin was used as loading control. (e) Soft‐agar growth assay in MCF‐7 cells treated with Lep in the presence or absence of herceptin (10 μg/ml). After 14 days of growth, colonies >50 μm diameter were counted. n.s., nonsignificant; *p < 0.05. Numbers on top of the blots represent the average fold change versus untreated cells normalized for β‐actin.
As shown in Figure 1c, HER2 mRNA levels were not affected by leptin treatment, at any time investigated, suggesting that a post‐translational mechanism could be involved in leptin‐mediated HER2 up‐regulation. To test this hypothesis, we evaluated leptin effect on HER2 protein expression in a cell line engineered to stably overexpress HER2 (MCF‐7/HER2‐18) in a manner independent by its own gene promoter activity. Leptin treatment up‐regulated HER2 protein content also in this cellular model (Figure 1d), confirming that the leptin effect was not dependent on a transcriptional regulation of HER2 gene expression. Since herceptin, a humanized monoclonal antibody directed against the extracellular domain of HER2, has been developed as an agent to specifically inhibit the growth of HER2‐overexpressing tumor cells, we performed anchorage‐independent growth assays in MCF‐7 cells treated with herceptin in the presence or not of leptin. As expected, herceptin did not affect MCF‐7 anchorage‐independent growth in basal condition, whereas significantly inhibited leptin‐induced effects (Figure 1e).
3.2. Leptin increases HER2 levels by inducing Hsp90 expression in breast cancer cells
HER2 protein expression can be regulated by the Hsp90 chaperone activity that is able to stabilize both nascent and mature form of this tyrosine kinase receptor (Citri et al., 2004). To investigate the potential involvement of Hsp90 in leptin‐induced HER2 expression, we evaluated leptin effects on Hsp90 expression in MCF‐7 and in MCF‐7/HER2‐18 breast cancer cells. We found that leptin treatment induced an increase in Hsp90 protein levels in both cell lines (Figure 2a). Concomitantly, co‐immunoprecipitation studies revealed a specific interaction between Hsp90 and HER2 that was further increased after leptin exposure in both cell lines (Figure 2b). To assess the role of Hsp90 in the leptin‐induced HER2 levels, we examined HER2 expression upon leptin treatment in the presence of increasing doses (0–50 nM) of a specific Hsp90 inhibitor, 17‐allylamino‐17‐demethoxygeldanamicin (tanespimycin; 17‐AAG). Immunoblotting analysis revealed that in leptin‐treated MCF‐7 cells low doses of 17‐AAG (10 nM) induced a marked down‐regulation of HER2 expression compared to leptin‐untreated cells (Figure 2c, upper panel). Similar results were obtained in MCF‐7/HER2‐18 cells (Figure 2c, lower panel). To further confirm the role of Hsp90 in leptin‐mediated HER2 up‐regulation, we knocked‐down Hsp90 in MCF‐7 cells with a specific siRNA (Ambion, ID:119758). As shown in Figure 2d, Hsp90 silencing abrogated the up‐regulatory effects induced by leptin on HER2 expression, whereas no changes were observed after transfection of cells with a control siRNA upon identical experimental conditions.
Figure 2.
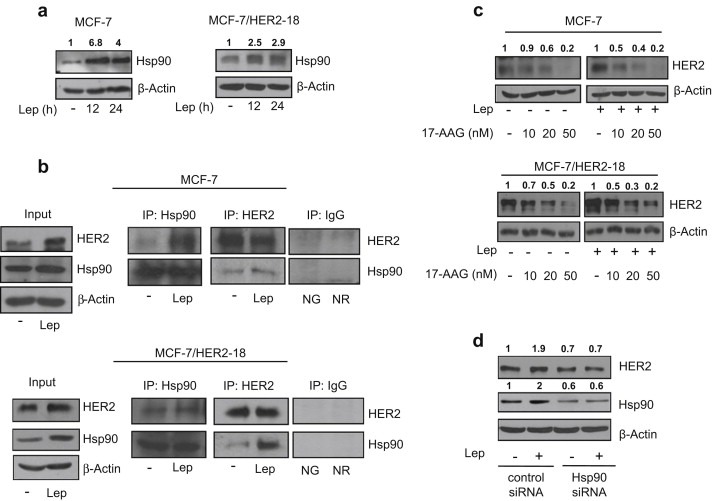
Leptin enhances Hsp90 expression. (a) Immunoblotting analysis of Hsp90 levels in total protein extracts from MCF‐7 (left panel) and MCF‐7/HER2‐18 (right panel) cells untreated (−) or treated with leptin 500 ng/ml (Lep) as indicated. (b) MCF‐7 (upper panel) and MCF‐7/HER2‐18 (lower panel) cells were untreated (−) or treated with Lep for 24 h before lysis. Hsp90 and HER2 proteins were immunoprecipitated using anti‐Hsp90 (IP:Hsp90) and anti‐HER2 (IP:HER2) antibodies respectively and resolved in SDS–polyacrylamide gel electrophoresis. Immunoblotting was performed using anti‐HER2 and anti‐Hsp90 antibodies respectively. Whole‐cell lysates (Input) were used as input controls. Negative control was performed by incubation of cell lysates with protein A/G agarose and normal goat (NG) or rabbit (NR) antisera. (c) Immunoblotting analysis of HER2 from total extracts of MCF‐7 (upper panel) and MCF‐7/HER2‐18 (lower panel) cells treated for 24 h with Lep (500 ng/ml) in the presence or not of growing doses (10–20 and 50 nM) of the selective Hsp90 inhibitor 17‐allylamino‐17‐demethoxygeldanamycin (17‐AAG). (d) Total cellular proteins were isolated from MCF‐7 cells transfected with Hsp90 siRNA or control siRNA and treated for 24 h with Lep. Equal amounts of total cellular extracts were analyzed for HER2 and Hsp90 protein levels by immunoblotting. β‐Actin was used as loading control. Numbers on top of the blots represent the average fold change versus untreated cells normalized for β‐actin.
3.3. Leptin up‐regulates at transcriptional level Hsp90 gene expression
To explore whether Hsp90 up‐regulation relies on transcriptional mechanisms, we evaluated Hsp90 mRNA levels after treatment with leptin for different times. Exposure to leptin resulted in a significative increase in Hsp90 mRNA levels in MCF‐7 breast cancer cells (Figure 3a). Then, to confirm the role of leptin/leptin receptor (ObR) signaling in the above mentioned effects, we knocked ObR by a specific siRNA (Ambion, ID:s224009). ObR mRNA expression was effectively silenced as revealed by reverse transcription‐PCR after 24, 48, and 72 h of siRNA transfection (Figure 3b). As shown in Figure 3c, silencing of the ObR gene completely reversed the leptin‐induced up‐regulation of Hsp90 mRNA and protein expression in breast cancer cells.
Figure 3.
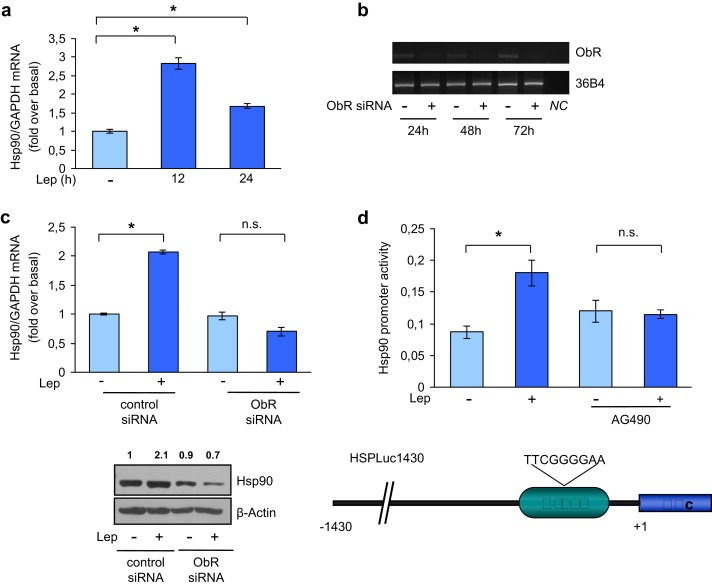
Leptin induces Hsp90 promoter activity. (a) mRNA Hsp90 content, evaluated by real‐time RT‐PCR, after 12 and 24 h with leptin 500 ng/ml (Lep). Each sample was normalized to its GAPDH mRNA content. The values represent the means ± s.d. of three different experiments each performed in triplicate. *p < 0.05. (b) ObR mRNA expression in MCF‐7 cells transfected with siRNA targeted human ObR mRNA sequence or with a control siRNA (−) for 24, 48, and 72 h 36B4 was used as loading control. NC, negative control, RNA sample without the addition of reverse transcriptase. (c) Hsp90 mRNA levels (upper panel) and protein levels (lower panel) in MCF‐7 cells transfected with ObR siRNA or control siRNA for 24 h followed by Lep treatment for 24 h. Each sample was normalized to its GAPDH mRNA content. The values represent the means ± SD of three different experiments each performed in triplicate. n.s., nonsignificant; *p < 0.05 compared to untreated cells (−). β‐Actin was used as loading control. Numbers on top of the blots represent the average fold change versus untreated cells normalized for β‐actin. (d) MCF‐7 cells transiently transfected with a HSPLuc1430 reporter gene were untreated (−) or treated with Lep for 12 h in the presence or not of AG490 (1 μM) and then luciferase activity was measured (upper panel). The values represent the means ± s.d. of three different experiments each performed in triplicate. n.s., nonsignificant; *p < 0.05. Schematic representation of Hsp90 promoter region used in this study (lower panel).
Next, we tested the transcriptional activity of a reporter plasmid containing the human Hsp90 promoter region (HSPLuc1430), and we found that exposure to leptin significantly induced Hsp90 promoter activity (Figure 3d, upper panel). The human Hsp90 promoter contains consensus sites for several transcription factors (Stephanou and Latchman) including STAT3, a well known mediator of leptin signaling (Ahima and Osei, 2004) (Figure 3d, lower panel). To verify the involvement of STAT3 signaling in the leptin‐mediated promoter activation, we tested luciferase activity in the presence of AG490, a specific JAK2/STAT3 inhibitor. Treatment with AG490 resulted in a complete abrogation of leptin effect (Figure 3e, upper panel). Furthermore, the specific involvement of the STAT3 motif in the transcriptional regulation of Hsp90 by leptin was investigated using EMSA. We observed the formation of a specific complex in nuclear extracts from MCF‐7 cells using synthetic oligodeoxyribonucleotides corresponding to the STAT3 motif located in the Hsp90 promoter region (Figure 4a, lane 1), which was abrogated by incubation with 100‐fold molar excess of unlabeled probe (Figure 4a, lane 2), demonstrating the specificity of the DNA‐binding complex. This inhibition was no longer observed when mutated oligodeoxyribonucleotide was used as competitor (Figure 4a, lane 3). Interestingly, treatment with leptin strongly increased the DNA‐binding protein complex compared with control samples (Figure 4a, lane 4). The inclusion of an anti‐STAT3 antibody in the reaction immunodepleted the specific band, confirming the presence of STAT3 in the complex (Figure 4a, lane 5). Moreover, the role of STAT3 in leptin‐mediated Hsp90 up‐regulation at the promoter level was analyzed by ChIP assays. Using specific antibodies against STAT3 and RNA‐polymerase II, protein–chromatin complexes were immunoprecipitated from cells cultured with or without leptin for 1 h. The resulting precipitated DNA was then quantified using (RT)‐PCR with primers spanning the STAT3‐binding element in the Hsp90 promoter region. As shown in Figure 4b recruitment of STAT3 (upper panel) as well as of RNA‐polymerase II (lower panel) to the Hsp90 regulatory region was significantly increased upon leptin treatment. Finally, the role of STAT3 as mediator of leptin signaling in the modulation of Hsp90/HER2 expression was tested in MCF‐7 cells transiently transfected with a dominant negative STAT3 (STAT−) plasmid. Over‐expression of the dominant negative STAT− plasmid, as revealed by western blot analysis (Figure 4c, left panel), completely abrogated the leptin‐mediated HER2 and Hsp90 up‐regulation (Figure 4c, right panel).
Figure 4.
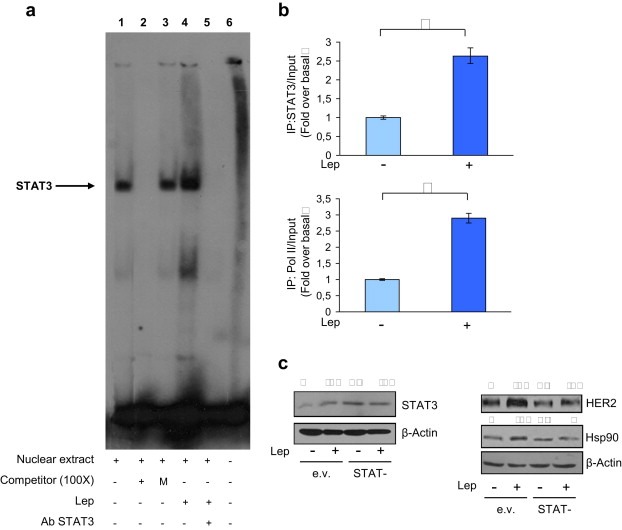
Leptin regulates Hsp90 promoter activity through STAT3‐binding site. (a) Nuclear extracts from MCF‐7 cells were incubated with a double‐stranded STAT3 specific sequence probe labeled with [γ32P]ATP and subjected to electrophoresis in a 6% polyacrylamide gel (lane 1). Competition experiments were performed adding as competitor a 100‐fold molar excess of unlabeled probe (lane 2) or a 100‐fold molar excess of unlabeled oligonucleotide containing a mutated STAT3 sequence (lane 3). Lane 4, nuclear extracts from MCF‐7 cells treated with leptin 500 ng/ml (Lep). Lanes 5, leptin‐treated nuclear extracts incubated with anti‐STAT3 antibody. Lane 6, probe alone. (b) MCF‐7 cells were treated or not (−) with Lep for 1 h, cross‐linked with formaldehyde and lysed. The precleared chromatin was immunoprecipitated with anti‐STAT3 (upper panel) and anti‐RNA‐polymerase II (lower panel) antibodies. A 5 μl volume of each sample and input was analyzed by real‐time PCR using specific primers to amplify Hsp90 promoter sequence, including the STAT3 site. Similar results were obtained in multiple independent experiments. (c) MCF‐7 cells were transiently transfected with either empty vector (e.v.) or STAT3 dominant negative plasmid (STAT−) and then treated or not (−) with Lep for 24 h. STAT3, HER2 land Hsp90 levels were evaluated by immunoblotting. β‐Actin was used as loading control. Numbers on top of the blots represent the average fold change versus untreated cells normalized for β‐actin.
3.4. Leptin modulates HER2/Hsp90 expression in SKBR3 breast cancer cells
As an additional model system to evaluate leptin effect on HER2 and Hsp90 expression, we used ERα‐negative and HER2‐overexpressing SKBR3 breast cancer cells. Treatment with leptin strongly increased HER2 immunofluorescence at the plasma membrane compared to untreated cells (Figure 5a). As expected, treatment with leptin‐induced both HER2 and Hsp90 protein levels, and the ectopic expression of the STAT− plasmid (Figure 5b, left panel) inhibited the above mentioned effects (Figure 5b, right panel). Moreover, leptin exposure induced a marked increase in Hsp90 mRNA levels (Figure 5c), and the specific interaction between Hsp90 and HER2 (Figure 5d). Finally, we demonstrated that, also in this cellular contest, low doses of the Hsp90 inhibitor 17‐AAG completely abrogated the leptin induction of HER2 protein expression (Figure 5e). These latter results clearly demonstrate that Hsp90 activity is involved in leptin‐mediated HER2 up‐regulation in different breast cancer cell lines, representing a general mechanism not related to cell specificity.
Figure 5.
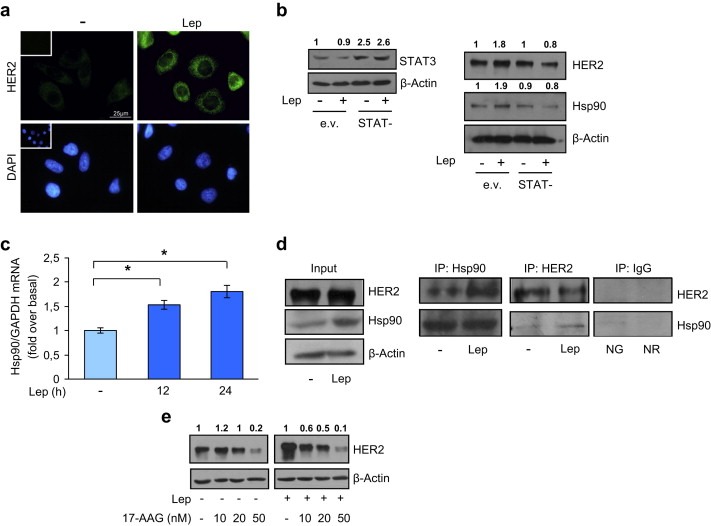
Leptin‐induced expression of HER2 and Hsp90 in ERα‐negative SKBR3 breast cancer cells. (a) SKBR3 cells were untreated (−) or treated with leptin 500 ng/ml (Lep) for 24 h and then HER2 expression was determined by immunofluorescence analysis. Small squares, negative controls. DAPI staining was used to visualize the cell nucleus. Scale bar = 25 μm. (b) Cells were transiently transfected with either empty vector (e.v.) or STAT3 dominant negative plasmid (STAT−) and then untreated (−) or treated with Lep for 24 h. STAT3, HER2 land Hsp90 levels were evaluated by immunoblotting. β‐Actin was used as loading control. (c) Hsp90 mRNA content, evaluated by real‐time RT‐PCR, after treatment with Lep as indicated. Each sample was normalized to its GAPDH mRNA content. The values represent the means ± s.d. of three different experiments each performed in triplicate. *p < 0.05. (d) SKBR3 cells were treated with vehicle (−) or Lep for 12 h before lysis. Hsp90 and HER2 proteins were immunoprecipitated using anti‐Hsp90 (IP:Hsp90) and anti‐HER2 (IP:HER2) antibodies respectively, the immunocomplexes were resolved in SDS–polyacrylamide gel electrophoresis. Immunoblotting was performed using anti‐HER2 and anti‐Hsp90 antibodies respectively. Whole‐cell lysates (Input) were used as input controls. Negative control was performed by incubation of cell lysates with protein A/G agarose and normal goat (NG) or rabbit (NR) antisera. (e) Immunoblotting analysis of HER2 from total extracts of SKBR3 cells treated for 12 h with Lep in the presence or not of growing doses (10–20 and 50 nM) of the selective Hsp90 inhibitor 17‐AAG. β‐Actin was used as loading control. Numbers on top of the blots represent the average fold change versus untreated cells normalized for β‐actin.
3.5. Leptin treatment reduces tamoxifen sensitivity in breast cancer cells
Since leptin‐induced HER2 expression, we next addressed whether this might result in an altered responsiveness of breast cancer cells to growth factor stimulation. To test this hypothesis, we performed immunoblot analysis to evaluate the phosphorylation levels of a number of growth factor signaling components (HER2, AKT, and MAPK) in cells pretreated or not with leptin for 24 h and then subjected to short‐term stimulation with epidermal growth factor (EGF). After leptin exposure, MCF‐7 cells exhibited an enhanced sensitivity to EGF treatment as evidenced by a rapid (within 1 min) and higher induction of phosphorylation levels of HER2 as well as of the downstream growth factor signaling molecules AKT and MAPK compared to untreated cells (Figure 6a). Moreover, to confirm that the leptin‐induced growth factor signal amplification depends on pHER2 we analyzed phosphorylation levels of HER2, AKT and MAPK in the presence of herceptin, a specific HER2 inhibitor. As shown in Figure 6b herceptin treatment reversed the leptin effects on the phosphorylation levels of the downstream growth factor signaling molecules.
Figure 6.
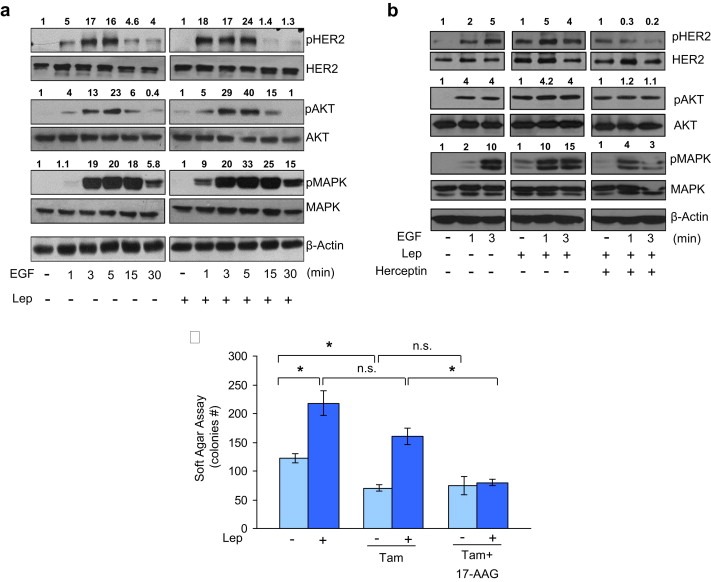
Effects of leptin on growth factor signaling and responsiveness to the antiestrogen tamoxifen. (a) MCF‐7 cells were treated with leptin 500 ng/ml (Lep) for 24 h and then treated with EGF 100 ng/ml as indicated. Levels of phosphorylated (p) HER2 (Tyr1248), Akt (Ser473), and MAPK (Thr202/Tyr204), at the indicated residues, and total non‐phosphorylated protein were measured in cellular extracts by immunoblot analysis. (b) MCF‐7 cells were treated with Lep for 24 h and then treated with EGF and herceptin (10 μg/ml) alone or in combination as indicated. Levels of phosphorylated (p) HER2 (Tyr1248), Akt (Ser473), and MAPK (Thr202/Tyr204), at the indicated residues, and total non‐phosphorylated protein were measured in cellular extracts by immunoblot analysis. β‐Actin was used as loading control. Numbers on top of the blots represent the average fold change versus untreated cells normalized for β‐actin. (c) Soft‐agar growth assay in MCF‐7 cells treated with Lep, tamoxifen (Tam, 1 μM) and 17‐AAG (20 nM) alone or in combination. After 14 days of growth, colonies >50 μm diameter were counted. n.s., nonsignificant; *p < 0.05.
Several preclinical and clinical studies indicate that over‐expression of EGFR or HER2, and/or high levels of phosphorylated Akt and MAPK in breast cancers contribute to Tamoxifen resistance (Arpino et al., 2004; Gee et al., 2001; Perez‐Tenorio and Stal, 2002; Shou et al., 2004). Thus, as a final step of this study, we investigated if leptin, through the described Hsp90/HER2 interplay, might influence the sensitivity of breast cancer cells to tamoxifen treatment. Therefore, we performed anchorage‐independent growth assay in MCF‐7 cells treated with or without leptin in the presence of tamoxifen alone or in combination of the specific Hsp90 inhibitor 17‐AAG. As expected, Tamoxifen treatment significantly reduced the number of colonies compared to untreated cells, but it was not able to significantly modify colony numbers in leptin‐treated cells. Moreover, the Hsp90 inhibitor 17‐AAG in combination with Tamoxifen did not exert, in basal condition, any additional inhibitory effects in colony formation. Importantly, the combined treatment with 17‐AAG and tamoxifen completely abrogated leptin‐induced MCF‐7 anchorage‐independent growth (Figure 6b), suggesting a crucial role of Hsp90/HER2 in mediating leptin effects in breast cancer cell growth proliferation.
4. Discussion
Here, we provide evidence, for the first time, that the adipokine leptin increased heat‐shock protein 90 (Hsp90) chaperone levels by inducing STAT3 binding to its response element located in the human Hsp90 promoter sequence in breast cancer cells. This results in an enhanced membrane tyrosine kinase receptor HER2 expression that reduces sensitivity of breast cancer cells to antiestrogen tamoxifen treatment. Because the recognized importance of Hsp90 and HER2 in promoting breast cancer progression, enhancing our understanding of the mechanisms able to regulate their expression in breast cancer cells, under different physiological and/or pathological conditions such as obesity, could provide new treatment options for breast cancer.
Heat‐shock proteins (Hsp‐s), first discovered as mediators of resistance to hyperthermia and other chemical and physical stresses in all cellular organisms, appear to play a role also in many pathophysiological conditions such as cancer. Indeed, Hsp‐s over‐expression results in increased incidence of cell transformation and is clinically correlated with poor prognosis and resistance to apoptosis in a wide range of human cancers including mammary carcinoma (Ciocca and Calderwood, 2005). Hsp‐s are the products of different gene families and are classified according to their molecular weight in: Hsp100, Hsp90, Hsp70, Hsp60 and the large family of small Hsp‐s (Craig et al., 1994; Johnson and Craig, 1997). Particularly, Hsp90, one of the most abundant proteins in the eukaryotic cells, may contribute to the resistant cancer cell phenotype by stabilizing the mutated and overexpressed oncogenes that can overcome the stresses of anticancer therapies (Calderwood, 2010). There are two major cytoplasmic forms of Hsp90: the inducible Hsp90α, and the Hsp90β constitutively expressed and encoded by a related gene (Csermely et al., 1998). Hsp90 belongs to the class of molecular chaperones that participate in the normal folding, intracellular localization and proteolytic turnover of more than 200 proteins, including cell cycle regulators (Cdk4, Cdk6), mutated signaling proteins (p53, v‐Src), metastable signaling proteins (Akt, Raf‐1 and IKK), and steroid hormone receptors (androgen, estrogen and progesterone receptors) (Li et al., 2009). In addition, the Hsp90 chaperone system regulates the mature form of the membrane tyrosine kinase HER2/ERBB2 which represents the most sensitive Hsp90 client (Xu et al., 2001).
First, we found that treatment of ERα‐positive MCF‐7 breast cancer cells with the obesity hormone leptin increased HER2 protein expression without affecting its mRNA levels, suggesting the possibility that an increase in protein stability could be involved. This hypothesis was confirmed by the results obtained in leptin‐treated stably transfected MCF‐7/HER2‐18 cells (Shou et al., 2004), in which HER2 over‐expression is not driven by its own gene promoter activity. It is well known that HER2 stability is critically dependent on Hsp90 activity. Indeed, pharmacologic disruption of Hsp90/HER2 association results in HER2 polyubiquitination and degradation (Mimnaugh et al., 1996). We have demonstrated that leptin treatment increased Hsp90 protein and mRNA levels in breast cancer cells, suggesting that leptin up‐regulates Hsp90 expression via a transcriptional mechanisms. Leptin exerts its actions by binding to a specific transmembrane receptor (ObR) (Tartaglia, 1997) by activation of RAS‐dependent MAPK pathway and signal transducers and activators of transcription (STAT) factors (Baumann et al., 1996; Yamashita et al., 1998). The involvement of leptin/leptin receptor signaling in the transcriptional regulation of Hsp90 expression was clearly demonstrated in MCF‐7 cells knocked‐down for ObR expression. In these experimental condition the leptin‐mediated Hsp90 up‐regulation was completely abrogated. Moreover, we found that in breast cancer cells treated with leptin the amount of HER2 bound to Hsp90 was significantly higher compared to untreated cells. These latter results suggest that leptin‐induced HER2 up‐regulation involves the Hsp90 chaperone activity.
Several small molecule inhibitors of Hsp90, able to reduce cellular levels of multiple oncogenic client proteins by enhancing their ubiquitination and proteasome‐mediated degradation, have been identified in the last years. One of these inhibitors the 17‐allylamino‐17‐demethoxygeldanamycin (17‐AAG), a geldanamycin analog that interferes with the ATP‐binding domain of Hsp90, has been subjected in clinical trials for different cancers (Lu et al., 2011). Although Hsp90 function is essential for normal cell viability and growth (Borkovich et al., 1989), targeting its activity in cancer cells is applicable, since it has been demonstrated that Hsp90 in tumor cells has approximately 100‐fold higher binding affinity for the 17‐AAG inhibitor than does Hsp90 in normal cells (Kamal et al., 2003). In addition, a phase II clinical trial of this inhibitor plus the monoclonal antibody targeting HER2 (trastuzumab) has shown promising results in HER2‐positive metastatic breast carcinoma (Modi et al., 2011). In this study, the role of Hsp90 on leptin‐mediated effects was highlighted by using either 17‐AAG or silencing Hsp90 expression in MCF‐7 breast cancer cells. In these experimental conditions, leptin effect on HER2 expression was completely lost.
The cellular levels of Hsp90 are mainly regulated by stress stimuli, through the activation of specific members of the transcription factor family heat‐shock factors (HSFs), which bind to the responsive element (heat‐shock element HSE) located in the Hsp90 promoter region (Akerfelt et al., 2010). In addition, Stephanou and Latchman (2010) have identified a separate group of transcription factors, such as STAT1, STAT3 and NF‐IL6, also able to modulate Hsp90 gene expression in non‐stress condition and in several disease states. In agreement with these latter observations, we found that leptin, in non‐stressful conditions, is able to transactivate a reporter plasmid containing the human Hsp90 promoter region (HSPLuc1430). Analysis of the human Hsp90 gene upstream sequence (NCBI GenBank U25822gi/793941) revealed the presence of a potential STAT‐binding DNA elements (nucleotides −1177 to 1185) that could bind STAT3 transcription factor (Decker et al., 1997). We have demonstrated in functional studies using a specific JAK2/STAT3 inhibitor AG490, that the activity of STAT3 transcription factor is essential for the leptin‐mediated effects on Hsp90 promoter. Moreover, EMSA revealed a marked increase in a specific DNA‐binding complex in nuclear extracts from MCF‐7 cells treated with leptin, when a synthetic oligodeoxyribonucleotides corresponding to the STAT3 motif located at −1177 to 1185 of the Hsp90 promoter region was used. The in vivo interaction between STAT3 and the Hsp90 promoter was further supported by ChIP assay, where upon leptin treatment we observed an enhanced recruitment of both STAT3 and RNA‐polymerase II, to the Hsp90 regulatory region bearing the STAT3‐binding element, supporting a positive transcriptional role for leptin‐activated STAT3 in modulating Hsp90 expression in breast cancer cells. Finally, the complete abrogation of HER2 and Hsp90 leptin‐mediated up‐regulation in the presence of a STAT3 dominant negative expression vector clearly demonstrated that STAT3 transcription factor was essential in this molecular mechanism. Leptin effect on both HER2 and Hsp90 expression were also reproduced in ERα‐negative and HER2‐overexpressing SKBR3 breast cancer cells, suggesting that it may represent a general mechanism not related to cell specificity.
Several mechanisms are responsible for the development of the endocrine resistance in breast cancer, and among these, increased expression and/or signaling of growth factor receptors have been extensively studied (Arpino et al., 2004; Barone et al., 2009; Schiff et al., 2003). Experimental and clinical studies have suggested that both de novo and acquired resistance to antiestrogen Tamoxifen in breast cancer can be associated with elevated levels of HER2 (Chung et al., 2002; Gutierrez et al., 2005; Meng et al., 2004; Shou et al., 2004). We demonstrated that leptin treatment amplified the responsiveness of breast cancer cells to growth factor stimulation, suggesting that increased leptin levels, as in obese women, could sustain growth factor signaling and thereby limit the efficacy of the endocrine therapy in breast cancer patients. Indeed, results from anchorage‐independent growth assays indicated that Tamoxifen was less effective in inhibiting colony formation in leptin‐treated breast cancer cells, and addition of the Hsp90 inhibitor 17‐AAG in combination to Tamoxifen completely reversed leptin‐induced growth.
In conclusion, the present study contributes to define the biological mechanisms linking obesity to breast cancer progression, showing, for the first time, a novel role for the adipocyte‐secreted factor leptin in sustaining HER2 protein levels through an up‐regulation of the Hsp90 chaperone expression. These findings could provide a strong rationale to support the development of new potential therapeutic agents to specifically target the leptin/Hsp90/HER2 axis that could be implemented in the adjuvant breast cancer clinical settings to improve patient outcome, especially in obese women.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) grant IG 11595. Reintegration AIRC/Marie Curie International Fellowship in Cancer Research to IB. We thank Professor KJ Wu (IBMB, National Yang‐Ming University, Taiwan) for providing the HSPLuc1430 plasmid and Dr. Rachel Schiff for providing the MCF‐7/HER2‐18 cells.
Giordano Cinzia, Vizza Donatella, Panza Salvatore, Barone Ines, Bonofiglio Daniela, Lanzino Marilena, Sisci Diego, De Amicis Francesca, Fuqua Suzanne A.W., Catalano Stefania, Andò Sebastiano, (2013), Leptin increases HER2 protein levels through a STAT3‐mediated up‐regulation of Hsp90 in breast cancer cells, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.11.002.
Contributor Information
Stefania Catalano, Email: stefcatalano@libero.it.
Sebastiano Andò, Email: sebastiano.ando@unical.it.
References
- Ahima, R.S. , Osei, S.Y. , 2004. Leptin signaling. Physiol. Behav. 81, (2) 223–241. [DOI] [PubMed] [Google Scholar]
- Akerfelt, M. , Morimoto, R.I. , Sistonen, L. , 2010. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, (8) 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred, D.C. , Clark, G.M. , Molina, R. , Tandon, A.K. , Schnitt, S.J. , Gilchrist, K.W. , Osborne, C.K. , Tormey, D.C. , McGuire, W.L. , 1992. Overexpression of HER-2/neu and its relationship with other prognostic factors change during the progression of in situ to invasive breast cancer. Hum. Pathol. 23, (9) 974–979. [DOI] [PubMed] [Google Scholar]
- Andò, S. , Catalano, S. , 2011. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 8, (5) 263–275. [DOI] [PubMed] [Google Scholar]
- Arpino, G. , Green, S.J. , Allred, D.C. , Lew, D. , Martino, S. , Osborne, C.K. , Elledge, R.M. , 2004. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin. Cancer Res. 10, (17) 5670–5676. [DOI] [PubMed] [Google Scholar]
- Barone, I. , Catalano, S. , Gelsomino, L. , Marsico, S. , Giordano, C. , Panza, S. , Bonofiglio, D. , Bossi, G. , Covington, K.R. , Fuqua, S.A. , Andò, S. , 2012. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 72, (6) 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone, I. , Iacopetta, D. , Covington, K.R. , Cui, Y. , Tsimelzon, A. , Beyer, A. , Andò, S. , Fuqua, S.A. , 2009. Phosphorylation of the mutant K303R estrogen receptor alpha at serine 305 affects aromatase inhibitor sensitivity. Oncogene 29, (16) 2404–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, H. , Morella, K.K. , White, D.W. , Dembski, M. , Bailon, P.S. , Kim, H. , Lai, C.F. , Tartaglia, L.A. , 1996. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. U.S.A. 93, (16) 8374–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich, K.A. , Farrelly, F.W. , Finkelstein, D.B. , Taulien, J. , Lindquist, S. , 1989. hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell. Biol. 9, (9) 3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood, S.K. , 2010. Heat shock proteins in breast cancer progression–a suitable case for treatment?. Int. J. Hyperthermia 26, (7) 681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle, E.E. , Kaaks, R. , 2004. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 4, (8) 579–591. [DOI] [PubMed] [Google Scholar]
- Calle, E.E. , Rodriguez, C. , Walker-Thurmond, K. , Thun, M.J. , 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 348, (17) 1625–1638. [DOI] [PubMed] [Google Scholar]
- Catalano, S. , Marsico, S. , Giordano, C. , Mauro, L. , Rizza, P. , Panno, M.L. , Andò, S. , 2003. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J. Biol. Chem. 278, (31) 28668–28676. [DOI] [PubMed] [Google Scholar]
- Catalano, S. , Pezzi, V. , Chimento, A. , Giordano, C. , Carpino, A. , Young, M. , McPhaul, M.J. , Andò, S. , 2003. Triiodothyronine decreases the activity of the proximal promoter (PII) of the aromatase gene in the mouse Sertoli cell line, TM4. Mol. Endocrinol. 17, (5) 923–934. [DOI] [PubMed] [Google Scholar]
- Catalano, S. , Mauro, L. , Marsico, S. , Giordano, C. , Rizza, P. , Rago, V. , Montanaro, D. , Maggiolini, M. , Panno, M.L. , Andò, S. , 2004. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J. Biol. Chem. 279, (19) 19908–19915. [DOI] [PubMed] [Google Scholar]
- Catalano, S. , Rizza, P. , Gu, G. , Barone, I. , Giordano, C. , Marsico, S. , Casaburi, I. , Middea, E. , Lanzino, M. , Pellegrino, M. , Andò, S. , 2007. Fas ligand expression in TM4 Sertoli cells is enhanced by estradiol “in situ” production. J. Cell. Physiol. 211, (2) 448–456. [DOI] [PubMed] [Google Scholar]
- Catalano, S. , Barone, I. , Giordano, C. , Rizza, P. , Qi, H. , Gu, G. , Malivindi, R. , Bonofiglio, D. , Ando, S. , 2009. Rapid estradiol/ERalpha signaling enhances aromatase enzymatic activity in breast cancer cells. Mol. Endocrinol. 23, (10) 1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano, S. , Malivindi, R. , Giordano, C. , Gu, G. , Panza, S. , Bonofiglio, D. , Lanzino, M. , Sisci, D. , Panno, M.L. , Andò, S. , 2010. Farnesoid X receptor, through the binding with steroidogenic factor 1-responsive element, inhibits aromatase expression in tumor Leydig cells. J. Biol. Chem. 285, (8) 5581–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, Y.L. , Sheu, M.L. , Yang, S.C. , Lin, C.H. , Yen, S.H. , 2002. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int. J. Cancer 97, (3) 306–312. [DOI] [PubMed] [Google Scholar]
- Ciocca, D.R. , Calderwood, S.K. , 2005. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10, (2) 86–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri, A. , Kochupurakkal, B.S. , Yarden, Y. , 2004. The achilles heel of ErbB-2/HER2: regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 3, (1) 51–60. [PubMed] [Google Scholar]
- Considine, R.V. , Sinha, M.K. , Heiman, M.L. , Kriauciunas, A. , Stephens, T.W. , Nyce, M.R. , Ohannesian, J.P. , Marco, C.C. , McKee, L.J. , Bauer, T.L. , Caro, J.F. , 1996. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 334, (5) 292–295. [DOI] [PubMed] [Google Scholar]
- Craig, E.A. , Weissman, J.S. , Horwich, A.L. , 1994. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell 78, (3) 365–372. [DOI] [PubMed] [Google Scholar]
- Csermely, P. , Schnaider, T. , Soti, C. , Prohaszka, Z. , Nardai, G. , 1998. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 79, (2) 129–168. [DOI] [PubMed] [Google Scholar]
- Decker, T. , Kovarik, P. , Meinke, A. , 1997. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res. 17, (3) 121–134. [DOI] [PubMed] [Google Scholar]
- Dieudonne, M.N. , Machinal-Quelin, F. , Serazin-Leroy, V. , Leneveu, M.C. , Pecquery, R. , Giudicelli, Y. , 2002. Leptin mediates a proliferative response in human MCF7 breast cancer cells. Biochem. Biophys. Res. Commun. 293, (1) 622–628. [DOI] [PubMed] [Google Scholar]
- Fiorio, E. , Mercanti, A. , Terrasi, M. , Micciolo, R. , Remo, A. , Auriemma, A. , Molino, A. , Parolin, V. , Di Stefano, B. , Bonetti, F. , Giordano, A. , Cetto, G.L. , Surmacz, E. , 2008. Leptin/HER2 crosstalk in breast cancer: in vitro study and preliminary in vivo analysis. BMC Cancer 8, 305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee, J.M. , Robertson, J.F. , Ellis, I.O. , Nicholson, R.I. , 2001. Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int. J. Cancer 95, (4) 247–254. [DOI] [PubMed] [Google Scholar]
- Giordano, C. , Cui, Y. , Barone, I. , Andò, S. , Mancini, M.A. , Berno, V. , Fuqua, S.A. , 2010. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast Cancer Res. Treat. 119, (1) 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glockner, S. , Lehmann, U. , Wilke, N. , Kleeberger, W. , Langer, F. , Kreipe, H. , 2001. Amplification of growth regulatory genes in intraductal breast cancer is associated with higher nuclear grade but not with the progression to invasiveness. Lab. Invest. 81, (4) 565–571. [DOI] [PubMed] [Google Scholar]
- Gutierrez, M.C. , Detre, S. , Johnston, S. , Mohsin, S.K. , Shou, J. , Allred, D.C. , Schiff, R. , Osborne, C.K. , Dowsett, M. , 2005. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 23, (11) 2469–2476. [DOI] [PubMed] [Google Scholar]
- Harvie, M. , Hooper, L. , Howell, A.H. , 2003. Central obesity and breast cancer risk: a systematic review. Obes. Rev. 4, (3) 157–173. [DOI] [PubMed] [Google Scholar]
- Hu, X. , Juneja, S.C. , Maihle, N.J. , Cleary, M.P. , 2002. Leptin–a growth factor in normal and malignant breast cells and for normal mammary gland development. J. Natl. Cancer Inst. 94, (22) 1704–1711. [DOI] [PubMed] [Google Scholar]
- Hurst, H.C. , 2001. Update on HER-2 as a target for cancer therapy: the ERBB2 promoter and its exploitation for cancer treatment. Breast Cancer Res. 3, (6) 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa, M. , Kitayama, J. , Nagawa, H. , 2004. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 10, (13) 4325–4331. [DOI] [PubMed] [Google Scholar]
- Johnson, J.L. , Craig, E.A. , 1997. Protein folding in vivo: unraveling complex pathways. Cell 90, (2) 201–204. [DOI] [PubMed] [Google Scholar]
- Kamal, A. , Thao, L. , Sensintaffar, J. , Zhang, L. , Boehm, M.F. , Fritz, L.C. , Burrows, F.J. , 2003. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, (6956) 407–410. [DOI] [PubMed] [Google Scholar]
- Korkaya, H. , Paulson, A. , Iovino, F. , Wicha, M.S. , 2008. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 27, (47) 6120–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmann, P.H. , Hoffmann, K. , Allen, N. , van Gils, C.H. , Khaw, K.T. , Tehard, B. , Berrino, F. , Tjonneland, A. , Bigaard, J. , Olsen, A. , Overvad, K. , Clavel-Chapelon, F. , Nagel, G. , Boeing, H. , Trichopoulos, D. , Economou, G. , Bellos, G. , Palli, D. , Tumino, R. , Panico, S. , Sacerdote, C. , Krogh, V. , Peeters, P.H. , Bueno-de-Mesquita, H.B. , Lund, E. , Ardanaz, E. , Amiano, P. , Pera, G. , Quiros, J.R. , Martinez, C. , Tormo, M.J. , Wirfalt, E. , Berglund, G. , Hallmans, G. , Key, T.J. , Reeves, G. , Bingham, S. , Norat, T. , Biessy, C. , Kaaks, R. , Riboli, E. , 2004. Body size and breast cancer risk: findings from the European Prospective Investigation into Cancer and Nutrition (EPIC). Int. J. Cancer 111, (5) 762–771. [DOI] [PubMed] [Google Scholar]
- Lanzino, M. , Sisci, D. , Morelli, C. , Garofalo, C. , Catalano, S. , Casaburi, I. , Capparelli, C. , Giordano, C. , Giordano, F. , Maggiolini, M. , Andò, S. , 2010. Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells–identification of a novel androgen response element. Nucleic Acids Res. 38, (16) 5351–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhang, T. , Schwartz, S.J. , Sun, D. , 2009. New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist. Updat. 12, (1–2) 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X. , Xiao, L. , Wang, L. , Ruden, D.M. , 2011. Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 83, (8) 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei, M. , Halaas, J. , Ravussin, E. , Pratley, R.E. , Lee, G.H. , Zhang, Y. , Fei, H. , Kim, S. , Lallone, R. , Ranganathan, S. , Kern, P.A. , Friedman, J.M. , 1995. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1, (11) 1155–1161. [DOI] [PubMed] [Google Scholar]
- Mauro, L. , Catalano, S. , Bossi, G. , Pellegrino, M. , Barone, I. , Morales, S. , Giordano, C. , Bartella, V. , Casaburi, I. , Andò, S. , 2007. Evidences that leptin up-regulates E-cadherin expression in breast cancer: effects on tumor growth and progression. Cancer Res. 67, (7) 3412–3421. [DOI] [PubMed] [Google Scholar]
- Meng, S. , Tripathy, D. , Shete, S. , Ashfaq, R. , Haley, B. , Perkins, S. , Beitsch, P. , Khan, A. , Euhus, D. , Osborne, C. , Frenkel, E. , Hoover, S. , Leitch, M. , Clifford, E. , Vitetta, E. , Morrison, L. , Herlyn, D. , Terstappen, L.W. , Fleming, T. , Fehm, T. , Tucker, T. , Lane, N. , Wang, J. , Uhr, J. , 2004. HER-2 gene amplification can be acquired as breast cancer progresses. Proc. Natl. Acad. Sci. U.S.A. 101, (25) 9393–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels, K.B. , Terry, K.L. , Willett, W.C. , 2006. Longitudinal study on the role of body size in premenopausal breast cancer. Arch. Intern. Med. 166, (21) 2395–2402. [DOI] [PubMed] [Google Scholar]
- Mimnaugh, E.G. , Chavany, C. , Neckers, L. , 1996. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J. Biol. Chem. 271, (37) 22796–22801. [DOI] [PubMed] [Google Scholar]
- Miyoshi, Y. , Funahashi, T. , Tanaka, S. , Taguchi, T. , Tamaki, Y. , Shimomura, I. , Noguchi, S. , 2006. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 118, (6) 1414–1419. [DOI] [PubMed] [Google Scholar]
- Modi, S. , Stopeck, A. , Linden, H. , Solit, D. , Chandarlapaty, S. , Rosen, N. , D'Andrea, G. , Dickler, M. , Moynahan, M.E. , Sugarman, S. , Ma, W. , Patil, S. , Norton, L. , Hannah, A.L. , Hudis, C. , 2011. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 17, (15) 5132–5139. [DOI] [PubMed] [Google Scholar]
- Perez-Tenorio, G. , Stal, O. , 2002. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br. J. Cancer 86, (4) 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, N.K. , Taliaferro-Smith, L. , Knight, B.B. , Merlin, D. , Anania, F.A. , O'Regan, R.M. , Sharma, D. , 2008. Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res. 68, (23) 9712–9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff, R. , Massarweh, S. , Shou, J. , Osborne, C.K. , 2003. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin. Cancer Res. 9, (1 Pt 2) 447S–454S. [PubMed] [Google Scholar]
- Shou, J. , Massarweh, S. , Osborne, C.K. , Wakeling, A.E. , Ali, S. , Weiss, H. , Schiff, R. , 2004. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 96, (12) 926–935. [DOI] [PubMed] [Google Scholar]
- Slamon, D.J. , Godolphin, W. , Jones, L.A. , Holt, J.A. , Wong, S.G. , Keith, D.E. , Levin, W.J. , Stuart, S.G. , Udove, J. , Ullrich, A. , Press, M.F. , 1989. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, (4905) 707–712. [DOI] [PubMed] [Google Scholar]
- Soma, D. , Kitayama, J. , Yamashita, H. , Miyato, H. , Ishikawa, M. , Nagawa, H. , 2008. Leptin augments proliferation of breast cancer cells via transactivation of HER2. J. Surg. Res. 149, (1) 9–14. [DOI] [PubMed] [Google Scholar]
- Stephanou, A. , Latchman, D.S. , 2010. Transcriptional modulation of heat-shock protein gene expression. Biochem. Res. Int. 2011, 238601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, G. , 2002. Leptin signalling. Cell. Signal. 14, (8) 655–663. [DOI] [PubMed] [Google Scholar]
- Tartaglia, L.A. , 1997. The leptin receptor. J. Biol. Chem. 272, (10) 6093–6096. [DOI] [PubMed] [Google Scholar]
- Xu, W. , Mimnaugh, E. , Rosser, M.F. , Nicchitta, C. , Marcu, M. , Yarden, Y. , Neckers, L. , 2001. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J. Biol. Chem. 276, (5) 3702–3708. [DOI] [PubMed] [Google Scholar]
- Yamashita, T. , Murakami, T. , Otani, S. , Kuwajima, M. , Shima, K. , 1998. Leptin receptor signal transduction: OBRa and OBRb of fa type. Biochem. Biophys. Res. Commun. 246, (3) 752–759. [DOI] [PubMed] [Google Scholar]
- Yin, N. , Wang, D. , Zhang, H. , Yi, X. , Sun, X. , Shi, B. , Wu, H. , Wu, G. , Wang, X. , Shang, Y. , 2004. Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res. 64, (16) 5870–5875. [DOI] [PubMed] [Google Scholar]