

Abstract
The SALL2 gene product and transcription factor p150 were first identified in a search for tumor suppressors targeted for inactivation by the oncogenic mouse polyoma virus. SALL2 has also been identified as a cellular quiescence factor, essential for cells to enter and remain in a state of growth arrest under conditions of serum deprivation. p150 is a transcriptional activator of p21Cip1/Waf1 and BAX, sharing important growth arrest and proapoptotic properties with p53. It also acts as a repressor of c‐myc. Restoration of SALL2 expression in cells derived from a human ovarian carcinoma (OVCA) suppresses growth of the cells in immunodeficient mice. Here we examine the pattern of p150 expression in the normal human ovary, in OVCA‐derived cell lines and in primary ovarian carcinomas. Immunohistochemical staining showed that p150 is highly expressed in surface epithelial cells of the normal human ovary. Expression is exclusively from the P2 promoter governing the E1A splice variant of p150. The P2 promoter is CpG‐rich and susceptible to methylation silencing. p150 expression was restored in OVCA cell lines following growth in the presence of 5‐azacytidine. In a survey of 210 cases of OVCA, roughly 90% across major and minor histological types failed to show expression of the protein. Immunological and biochemical approaches were used to show hypermethylation of the SALL2 P2 promoter in OVCA‐derived cell lines and in a majority of primary tumors. These results bring together molecular biological and clinical evidence in support of a role of SALL2 as a suppressor of ovarian cancers.
Keywords: SALL2, Ovarian cancer, Tumor suppressor, Promoter methylation, Polyoma T antigen
Highlights
-
►
p150, product of the SALL2 gene, is a target of the polyoma virus large T antigen.
-
►
p150 is a putative tumor suppressor with p53‐like functions.
-
►
p150 is highly expressed in surface epithelial cells of the normal human ovary.
-
►
p150 is absent or greatly diminished in a majority of human ovarian cancers.
-
►
Loss of p150 expression is due to methylation of the SALL2 P2 promoter.
Abbreviations
- OVCA
ovarian carcinoma
- FFPE
formalin-fixed paraffin-embedded
- HOSE cells
human ovarian surface epithelial cells
- Me-DIP
methylation sensitive DNA immunoprecipitation
1. Introduction
Epithelial ovarian cancers are a major cause of cancer‐related death among women. These cancers comprise a heterogeneous group with respect to tissue and cell type of origin and histological type (Karst and Drapkin, 2010; Kurman and Shih Ie, 2010; Vaughan et al., 2011). They may arise from the ovarian surface epithelium, from the epithelium of the distal fallopian tube, the secondary Mullerian system, or from the mesothelial lining of the peritoneal cavity (Bowen et al., 2009; Crum et al., 2012; Dubeau, 2008). A variety of genetic and epigenetic changes have been found in these cancers, some correlated with particular stage, grade or histological type (Asadollahi et al., 2010; Cannistra, 2004; Chen et al., 2003; Cho and Shih Ie, 2009; Etemadmoghadam et al., 2009; Jazaeri, 2009; Lengyel, 2010; Mok et al., 2009; Pearce et al., 2009; Tycko, 2000). Mutations in p53 are found in a majority of ovarian cancers as well as in precancerous lesions in the distal portion of the fallopian tube (Crum, 2009; Crum et al., 2007). In particular, 96% of high‐grade serous carcinomas, the most frequent and highly malignant type, harbor p53 mutations (2011; Engler et al., 2012).
First identified as a binding target of the SV40 large T (tumor) antigen (Lane and Crawford, 1979; Linzer and Levine, 1979), p53 is also targeted for inactivation or destruction by the high risk human papilloma viruses as agents of cervical cancer and by certain members of the adenovirus group that are oncogenic in experimental animals (Howley and Livingston, 2009). Surprisingly, the highly oncogenic mouse polyoma virus stands apart from other DNA tumor viruses, including the closely related SV40, in failing to target p53 (Dey et al., 2000). A ‘tumor host range’ selection procedure was devised with the aim of uncovering other possible tumor suppressor gene(s) with which polyoma virus must interact in order to replicate and induce tumors in the mouse. The p150 product of the SALL2 gene was uncovered as a binding partner of the polyoma virus large T antigen using this procedure (Li et al., 2001). Viral DNA replication is inhibited by p150 and binding by the large T protein overcomes this inhibition. A virus mutant unable to bind p150 is unable to replicate and fails to induce a broad spectrum of tumors in the mouse (Li et al., 2001).
Members of the SALL (Spalt‐like) gene family are evolutionarily conserved orthologues of the homeotic gene Spalt in Drosophila (Jurgens, 1988). They encode multi‐zinc finger transcription factors with roles in embryonic development in vertebrate as well as invertebrate species (de Celis and Barrio, 2009; Sweetman and Munsterberg, 2006). Mutations in SALL1 (Kohlhase et al., 1999) and SALL4 (Al‐Baradie et al., 2002; Kohlhase et al., 2005) give rise to developmental defects in man. SALL4 is important in maintaining a pluripotent state in mouse embryonic stem cells (Yang et al., 2010; Zhang et al., 2006; Zhou et al., 2007). The SALL2 transcription factor binds to the neurotrophin receptor and plays a role in neuronal development (Pincheira et al., 2009). SALL2 is essential for inducing and maintaining a quiescent state in human fibroblasts under conditions of serum deprivation (Liu et al., 2007). It is the only member of the SALL family implicated as a tumor suppressor (Li et al., 2001; Ma et al., 2001).
Though unrelated to p53 in amino acid sequence and differing in its DNA binding specificity (Gu et al., 2011), p150 overlaps functionally with p53 in inducing expression of p21Cip1/Waf1 and BAX (Li et al., 2004). Unlike p53, p150 is not a DNA damage response protein but is stably and highly expressed in certain terminally differentiated cells. In a survey of normal mouse tissues, the highest level of expression was found in the ovary (Li et al., 2001). Restoration of SALL2 expression in an OVCA‐derived cell line deficient in p150 expression resulted in partial suppression of tumor growth in SCID mice. Suppression of growth was accompanied by a decreased mitotic index and an increased apoptotic index along with induction of p21Cip1/Waf1 and BAX (Li et al., 2004). While p150 is highly expressed in the normal mouse ovary, the specific cell types within the ovary which express the protein and which of the two known isoforms (splice variants) (Ma et al., 2001) are expressed remain unknown. The present investigation was undertaken to determine the pattern of SALL2 expression in the normal human ovary and in epithelial ovarian cancers.
2. Material and methods
2.1. Cells
HOSE cells were telomerase‐immortalized, HPV E6‐transformed human ovarian surface epithelial cells (Clauss et al., 2010; Drapkin et al., 2005). OVCA‐derived cells SKOV‐3 and RMUGS were from the American Type Culture Collection. Cells were grown in DMEM with 10% fetal bovine serum. RMUGS and SKOV‐3 were subjected to DNA demethylation by incubation with 5‐azacytidine (2 μM) for 5 days.
2.2. Antisera
Polyclonal antisera were raised in rabbits against the N‐terminus (amino acids 1–550) and the C‐terminus (amino acids 717–1005) of human p150 purified as GST fusions. Antisera were purified by flow through over a GST column and binding to Staph A agarose beads. Polyclonal antisera were raised in chickens against peptides representing alternative exons E1A (AHESERSSRLGVPC) and E1 (QLISDCEGPSASEN).
2.3. Tissue samples and immunohistochemistry
After institutional review board approval, sections of formalin‐fixed, paraffin‐embedded (FFPE) human ovarian cancers were obtained from the Department of Pathology at the Brigham and Women's Hospital (Boston, MA). All major histological subtypes (serous, endometrioid, mucinous, clear cell, and transitional) were examined for p150 expression by immunohistochemistry (IHC) as previously described (Clauss et al., 2010; Drapkin et al., 2005). A high‐density tissue microarray (TMA) composed of high‐grade serous ovarian carcinomas (Clauss et al., 2010; Liu et al., 2009) was also utilized. Antibody to the C‐terminus of p150 was used at a dilution of 1:3000 with heat‐induced epitope retrieval. Negative controls included protein‐A purified preimmune serum, secondary antibody alone, and anti‐p150 antibody preincubated with recombinant p150 protein. Control sections were prepared from normal ovaries obtained from women with benign gynecologic diseases under protocols approved by the institutional review boards of the Brigham and Women's Hospital. None of the negative controls generated a positive signal in IHC.
2.4. Western blots
Immunoblotting was carried out on cell extracts using an Odyssey infrared imaging system (LI‐COR Biosciences, Lincoln, NE). Intensity values were determined with LI‐COR Odyssey software (Li‐COR Biosciences).
2.5. Assays for methylated DNA
Me‐DIP (methylated DNA immunoprecipitation) assay was carried out on purified DNAs (1 μg) with methylCpG‐specific monoclonal antibody conjugated magnetic beads from Active Motif (Carlsbad, CA) following manufacturer's suggestions. PCR amplification of P2 was carried out with primers F: 5′‐GCTCCACTGAGCCGGGCTACGTTTC and R: 5′‐CGAGACGAGAGCTCCTCTCGGATTC to give a 553 bp product. Amplified DNAs were run on agarose gels before and after digestion with MspI to give total and unmethylated amplicons, respectively.
2.6. Quantitative RT‐PCR
Total RNA was isolated using RNeasy® kit (Qiagen), reversed transcribed using QuantiTect Reverse Transcription Kit (Qiagen) and quantitated by RT‐PCR using primers: Aldolase A F: 5′‐CGCAGAAGGGGTCCTGGTGA‐3′, R: 5′‐CAGCTCCTTCTTCTGCTCCGGGGT‐3′. To distinguish between the alternatively spliced forms of p150, the following primers were used: E1‐SALL2 F – 5′‐CCAACAGTTAATCTCGGACTGCGAAG, E1A‐SALL2 F – 5′‐CACGAATCCGAGAGGAGCTCTC, and SALL2 R: 5′ – CACCATTACAGGAGGGTCAGTAG. RT‐PCR was carried out on a Roche LightCycler 480 using SYBR Green Master Mix. The data were analyzed by the comparative CT (ΔΔCT) method and quantitated relative to the aldolase A gene.
2.7. Pyrosequencing
The genomic DNA samples were extracted from various samples including normal ovarian surface epithelial cells, ovarian carcinoma cell lines, and formalin‐fixed, paraffin‐embedded (FFPE) human ovarian tumor tissue sections. QIAamp DNA FFPE Tissue Kit (Qiagen) was mainly used for the extractions following manufacture's protocols with a slight modification. The concentration of the extracted DNAs was measured with NanoDrop 1000 (Thermo), and the DNA samples were bisulfite converted and purified with Imprint DNA Modification Kit (Sigma–Aldrich). Primers for pyrosequencing assays (SALL2 P F, TAGATGGGGGGAGTTGGTTA; SALL2 P R Biotinylated, AATCTACCACAACCTCTACACCC; Sequencing primer, AAATTTTTTAGAGTTTTGAG) were designed using PyroMark Assay Design Software 2.0 (Qiagen), and PCR reactions (PyroMark PCR Kit; Qiagen) were performed using each converted DNA sample as a template. The size (348 bp) and purity of each amplicon was confirmed by agarose gel electrophoresis and then subjected to pyrosequencing assays (PyroMark Q24; Molecular and Integrative Physiological Sciences, Harvard School of Public Health). Briefly, PCR products were allowed to bind onto streptavidin‐coated Sepharose beads. After denaturation, the bioitinylated single‐stranded PCR fragments were isolated and annealed with the sequencing primer. The sequence variations of the target region (human SALL2 Promoter 2) were quantified by pyrosequencing assays using PyroMark Instrument and Software (Qiagen). Each tumor sample was run in duplicate.
3. Results
3.1. The E1A splice variant of p150 is expressed in established human ovarian surface epithelial cells
SALL2 is expressed from alternative promoters P1 and P2 upstream of short exons E1 and E1A (Ma et al., 2001), respectively, followed by a long common exon 2 (Figure 1A). Functional differences between the E1 and E1A splice variants have not been reported though it is likely that they differ in tissue distribution and developmental regulation. Antibodies specific to E1 and E1A were used to show that established human ovarian surface epithelial (HOSE) cells express only the E1A variant, while E1 is the predominant form expressed in P19 embryonal carcinoma cells (Figure 1B). RT‐PCR was used to confirm that transcription in HOSE cells is exclusively from the P2 promoter (Figure 1C).
Figure 1.
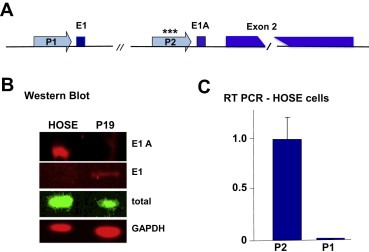
The E1A splice variant of p150 under control of the P2 SALL2 promoter is expressed in the established human ovarian surface epithelial (HOSE) cell line. (A) Schematic representation of the hSALL2 gene showing alternative first exons E1 (24 aa) and E1A (22aa) regulated by promoters P1 and P2, respectively, followed by the long common exon 2 (983 aa). Asterisks (***) indicate presence of a CpG island in P2. (B) Western blot of extracts from HOSE and P19 cells with antibodies to E1 and E1A. (C) RT‐PCR on RNA from HOSE cells with primers that bridge exon junctions.
3.2. The SALL2 P2 promoter contains a CpG island
Methylation of DNA on cytosines in CpG dinucleotides is well established as a basis of epigenetic regulation in development and in tumor suppressor gene silencing in a variety of cancers (Jones and Baylin, 2007). A comparison of sequences in the P1 and P2 promoters reveals the presence of a CpG island in P2 suggesting the possibility that loss of p150 expression in OVCA may be due to DNA methylation. Generally accepted criteria for a CpG island refer to regions of 200–500 bp with ≥55% GC content and an O/E (observed to expected) ratio of CpG dinucleotides ≥0.65 (Gardiner‐Garden and Frommer, 1987; Takai and Jones, 2002). In the ∼450 bp regions upstream of E1 and E1A, P1 has 52.1% GC and an O/E of 0.32 while P2 has 68% GC and an O/E of 0.86, establishing the latter as a CpG island (Figure 2).
Figure 2.

Sequences of hSALL2 P1 and P2 promoter regions. CpG dinucleotides are shaded in yellow. Translational start sites for E1 and E1A are shown in red. Primers used for amplification of a CpG‐rich region of P2 are underlined. The primer used for sequencing of the P2 amplicon and methylation analysis is in bold (see Figures 3d and 6c).
3.3. The SALL2 P2 promoter is hypermethylated in OVCA cell lines
Two OVCA‐derived cell lines were used to investigate whether P2 is hypermethylated relative to HOSE cells and whether methylation may underlie the loss or underexpression of SALL2. RMUGS expresses no detectible p150 while SKOV‐3 expresses a greatly reduced level compared to HOSE (Figure 3A). DNAs from these cells were first analyzed for methylation using 5‐methylCpG‐specific antibody (Me‐DIP assay). The amount of promoter methylation was estimated by PCR amplification of P2 present in the 5‐methylCpG antibody‐selected DNA relative to that using normal IgG. The highest level of P2 methylation was seen in RMUGS followed by SKOV‐3 with HOSE showing the least (Figure 3B). The inverse correlation between levels of p150 expression and 5‐methylCpG content in the P2 promoter suggests methylation silencing of SALL2 in the OVCA cell lines. To further test whether SALL2 expression is regulated by DNA methylation, RMUGS and SKOV‐3 were subjected to demethylation by growth for 5 days in the presence of the nucleoside analogue 5‐azacytidine (azaC). This resulted in greatly increased p150 expression in RMUGS and to a modest increase in SKOV‐3 (Figure 3C), consistent with methylation as a cause of loss of p150 expression in these cells.
Figure 3.
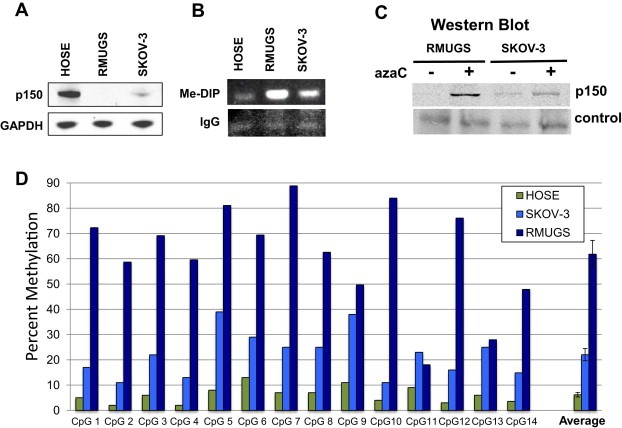
The SALL2 P2 promoter is hypermethylated in OVCA‐derived cell lines. (A) Western blot for p150 expression in HOSE cells and two OVCA‐derived cell lines (RMUGS and SKOV‐3). (B) Me‐DIP assay for P2 methylation using 5‐methylCpG antibody. (C) Restoration of p150 expression following growth of OVCA cell lines in 5‐azacytidine. (D) Percent methylation of CpG sites in the P2 promoter in HOSE, SKOV‐3, and RMUGS. DNAs were extracted, bisulfite treated, and a region within the CpG island in P2 amplified. Pyrosequencing was used to determine the percentage methylation across 14 CpG sites directly downstream of the primer binding site shown in Figure 2. Methylated and unmethylated controls were run concurrently to validate the bisulfite conversion efficiency and results. Error bars represent ±s.e.m for the average methylation across the 14 sites. P < 0.0001 for t tests of difference in average methylation for both SKOV‐3 and RMUGS compared to HOSE.
Recovery of p150 expression following azaC treatment suggests but does not necessarily imply direct silencing of SALL2 by P2 promoter methylation. To directly demonstrate the latter, bisulfite treatment followed by P2 amplification and sequencing was used. This procedure provides direct quantitative assessments of the degree of methylation at specific CpG sites. Bisufite is used to convert unmethylated CpGs to dUpGs leaving 5‐methylCpGs unmodified. PCR amplification of the converted DNA results in changes of CpG to TpG. The percent conversion at each site is revealed by pyrosequencng of the P2 amplicon (Dejeux et al., 2009; Frommer et al., 1992). Using this procedure on HOSE, RMUGS and SKOV‐3, the percent methylation at fourteen sites in the P2 promoter was determined (Figure 3D). These sites lie directly downstream of the sequencing primer shown in Figure 2. Increased methylation at each of the 14 sites is seen in both OVCA cell lines relative to HOSE. RMUGS shows the highest degree of methylation with an average of 62% across the 14 sites followed by SKOV‐3 with 22% and HOSE 7%. These results demonstrate P2 methylation in OVCA‐derived cell lines and support methylation silencing as a mechanism underlying loss of SALL2 expression in these tumor cells.
3.4. p150 is highly expressed in surface epithelial cells of the normal human ovary
Immunohistochemical staining for p150 in the normal human ovary was used to investigate the overall pattern of expression and to determine the specific cell type(s) within the ovary which express the protein. Using a polyclonal antibody to a C‐terminal fragment of human p150, staining was found to be confined almost exclusively to the surface epithelium (Figure 4A and B). Surface epithelial cells show strong nuclear staining ostensibly across the entire epithelium. Preadsorption of the antibody with recombinant p150 confirmed the specificity of staining (Figure 4C and D).
Figure 4.
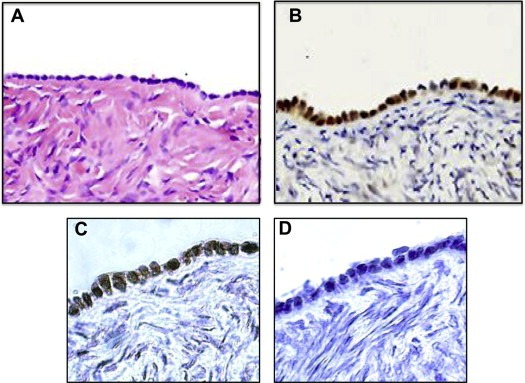
p150 is strongly expressed in surface epithelial cells of the normal human ovary. (A) Normal ovary – H&E‐stained. (B) Normal ovary – immunohistochemical staining (IHC) for p150. (C) Normal ovary – IHC for p150. (D) Normal ovary – IHC with anti‐p150 antibody pre‐adsorbed with p150.
3.5. p150 expression is absent or greatly diminished in ovarian carcinomas
To determine the status of p150 expression in primary ovarian carcinomas, an immunohistochemical survey was undertaken of 210 cases encompassing major and minor histological types. High‐grade serous tumors are the most frequent type and are associated with the highest morbidity and mortality (Karst and Drapkin, 2010). A papillary serous carcinoma devoid of staining is shown (Figure 5A and B). Three additional cases of serous carcinoma are shown, two uniformly negative (Figure 5C and D) and one with scattered p150‐positive cells (Figure 5E). A p150‐positive endometrioid carcinoma is also shown (Figure 5F). Results of the survey broken down according to histological type are shown in Table 1. Staining was absent or weak in most cases across major and minor histological types. The overwhelming majority, roughly 90% overall, showed no evidence of staining.
Figure 5.
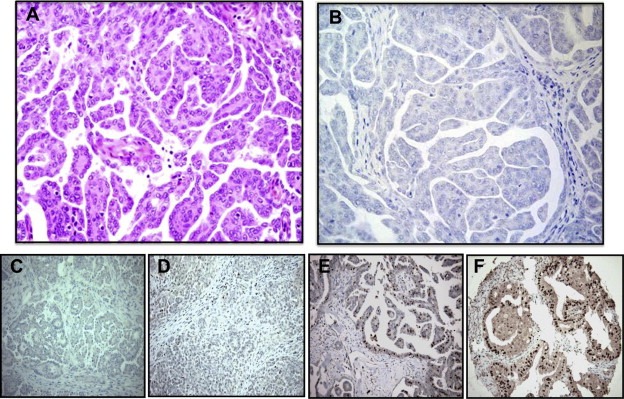
Ovarian carcinomas fail to express 150. Immunohistochemical staining (IHC) shows that p150 is absent in most ovarian carcinomas. (A) Serous carcinoma – H&E. (B) Serous carcinoma – p150 IHC. (C) Serous carcinoma – p150 IHC. (D) Serous carcinoma – p150 IHC. (E) Serous carcinoma– p150 IHC. (F) Endometrioid carcinoma– p150 IHC.
Table 1.
Survey of p150 expression in 210 cases of ovarian carcinomas by histological type.
| Histological types (n) | Intensity of immunostaining (%) | |||
|---|---|---|---|---|
| Strong | Moderate | Weak | Negative | |
| Papillary serous (155) | 4 (2.5%) | 3 (2%) | 3 (2%) | 145 (93.5%) |
| Mucinous (34) | 0 | 1 (3%) | 0 | 33 (97%) |
| Endometrioid (11) | 2 (18%) | 1 (9%) | 0 | 8 (73%) |
| Clear cell (6) | 0 | 0 | 1 (17%) | 5 (83%) |
| Transitional (4) | 0 | 0 | 0 | 4 (100%) |
3.6. The SALL2 P2 promoter is hypermethylated in a majority of serous ovarian carcinomas
P2 methylation was examined directly in human ovarian carcinomas. An attempt was first made to explore the feasibility of recovering and analyzing DNA from fixed tumor sections. An ovarian tumor metastasis growing on the surface of the intestine was examined using the Me‐DIP assay (Figure 6A). An H&E‐stained section was used to guide the microdissection of tissue from an adjacent unstained section. DNAs were isolated from the tumor (T1) and surrounding normal tissue (N1) and analyzed. Increased P2 methylation was evident in T1 relative to N1 (Figure 6B). As further efforts to recover sufficient quantities of DNA from single sections proved difficult, we turned to biochemical analysis of fixed paraffin‐embedded tumor tissues as a more direct procedure and one amenable to high throughput. Twenty‐six cases of serous carcinoma were selected from among those used in the immunohistochemical survey (Figure 5 and Table 1). Fifty micron cores were taken from these blocks guided by H&E‐stained sections as illustrated (see inset, Figure 6C). DNAs were extracted from the cores and analyzed by bisulfite treatment, P2 amplification, and pyrosequencing, as carried out for the OVCA‐derived cell lines (Figure 3D). Results from the twenty‐six tumors were pooled and compared with a pool of three independently established human ovarian surface epithelial (HOSE) cell lines as controls (Figure 6C). Hypermethylation was statistically significant at eleven of the fourteen CpG sites in the tumors relative to controls (p‐values <0.05). The average percent methylation across the 14 sites was roughly 3‐fold higher in the tumors compared to the HOSE cell lines (18.6% vs 6.6%, p‐value = 0.0054). The degree of methylation varied among tumors and among individual CpG sites. When averaged over the 14 sites, 18 of the 26 tumors (69%) showed 1.5‐fold or greater methylation compared to the controls.
Figure 6.
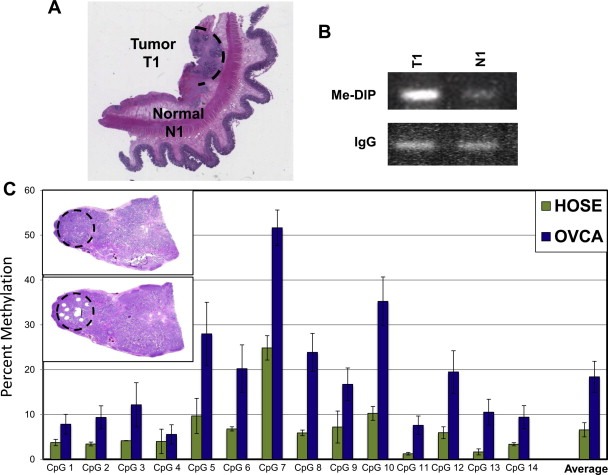
The SALL2 P2 promoter is hypermethylated in ovarian carcinomas. (A) H&E illustrating microdissection of a metastatic ovarian carcinoma on the intestinal wall. DNAs were isolated from the tumor (T1) and surrounding normal tissue (N1) and analyzed. (B) Me‐DIP assay shows hypermethylation of P2 in the tumor T1 relative to N1. (C) Percent methylation at 14 CpG sites in the P2 promoter of serous carcinomas (n = 26) and established HOSE cell lines (n = 3) as controls. Inset upper left: H&E sections of a serous carcinoma before and after cores were taken for DNA analysis. DNAs extracted from cores were bisulfite treated and amplified in the P2 promoter region.
4. Discussion
This investigation has origins in an experimental system based on the oncogenic mouse polyoma virus, specifically in the search for possible tumor suppressors with which this virus interacts. The rationale used to identify SALL2 as a target of polyoma virus embodies the idea that exploration of pathways altered by DNA tumor viruses stand to reveal genetic alterations that drive human cancers of non‐viral as well as viral etiology (Li et al., 2001; Rozenblatt‐Rosen et al., 2012). Here we have shown that the E1A splice variant of p150 is highly expressed in surface epithelial cells of the normal human ovary, that expression is lost or greatly diminished in most ovarian cancers, and that the SALL2 P2 promoter governing expression of the E1A variant of p150 is hypermethylated in a majority of these tumors.
Promoter methylation is a plausible mechanism for the absence of SALL2 expression in many of these cancers. Transcriptional silencing is likely not the only factor involved as roughly 30% of ovarian carcinomas show little or no evidence of promoter hypermethylation. Loss of heterozygosity in the region of 14q12 carrying the SALL2 gene has been reported in ovarian cancer (Bandera et al., 1997). SALL2 is sharply regulated at the posttranslational level. The pathway of degradation of p150 in normal cells as they re‐enter the cell cycle from a quiescent state has been partially elucidated (Sung et al., 2011). This ‘destruction pathway’ may operate in an uncontrolled manner in some cases of OVCA. Changes in micro RNA expression have been noted in OVCA (Yang et al., 2008) and these may also play a role.
Genome‐wide transcription profiling of normal human fibroblasts compared under conditions of serum stimulation and serum deprivation led to the identification of SALL2 as a major quiescence factor, essential for regulating the ability to enter and remain in a state of growth arrest. Downregulation of SALL2 with si‐RNA was found to interfere with the ability of these cells to arrest their growth when deprived of serum (Liu et al., 2007). Loss of SALL2 expression in ovarian surface epithelial cells likewise overcomes cell cycle arrest as SALL2 si‐RNA treatment promotes the G1‐>S transition in HOSE cells (Li et al., 2004). Specific pathways downstream of SALL2 that operate to impose a quiescent state are not fully understood.
Although a search for downstream targets of SALL2 has not been undertaken on a genome‐wide basis, three such targets have been identified. The cyclin cdk inhibitor p21Cip1/Waf1 and the proapoptotic factor BAX are transcriptionally activated by SALL2 (Gu et al., 2011; Li et al., 2004). p150 is a GC‐box binding protein (Gu et al., 2011) and sequences related to the consensus binding site in vitro are found in the promoters of these target genes. The c‐MYC protooncogene is frequently overexpressed in ovarian cancer due to transcriptional activation or DNA amplification. Consensus sequences for p150 binding are found in the nuclease hypersensitive control element of the c‐MYC promoter. p150 has recently been shown to bind to these sequences and to represses c‐MYC transcription (Sung et al., 2012). Following an apoptotic stimulus delivered to HOSE cells, p150 is recruited to the c‐MYC promoter. Analysis of data in The Cancer Genome Atlas shows inverse correlations in expression of SALL2 and c‐MYC in four solid tumor types deriving from tissues known to express p150 including ovarian. Based on our current understanding, silencing of SALL2 in OVCA is expected to result in the disruption of multiple pathways involved in regulating cell growth and survival. The molecular and physiological functions of SALL2 have only recently come under investigation. Much remains unknown about its normal functions and the full consequences of its loss as a suppressor of ovarian and possibly other forms of cancer.
5. Conclusions
p150, product of the SALL2 gene, was first identified as a putative tumor suppressor and binding target of the oncogenic mouse polyoma virus. p150 has growth arrest and proapoptotic properties akin to those of p53 and is essential for cells to enter a quiescent state. In this study for the first time we examine the expression of p150 in a human cancer and one of its normal tissues of origin. We show that p150 is highly expressed in surface epithelial cells of the normal human ovary under control of an alternative CpG‐rich promoter. Results of immunohistochemical staining showed that p150 is absent or undetectable in over 90% of ovarian carcinomas. Absence of p150 expression in these cancers is due in large part to methylation of the SALL2 promoter.
Conflict of interest
The authors declare no conflict of interests.
Acknowledgments
This work has been supported by a grant from the National Cancer Institute RO1 CA‐092520 to TB, grants P50 CA105009 and U01 CA152990, the Ovarian Cancer Research Fund, The Mary Kay Foundation, the Robert and Debra First Fund, and the Dana‐Farber Women's Executive Council Genetic Fingerprinting Award to RD.
Sung Chang K., Li Dawei, Andrews Erik, Drapkin Ronny, Benjamin Thomas, (2013), Promoter methylation of the SALL2 tumor suppressor gene in ovarian cancers, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.11.005.
References
- 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed]
- Al-Baradie, R. , Yamada, K. , St Hilaire, C. , Chan, W.M. , Andrews, C. , McIntosh, N. , Nakano, M. , Martonyi, E.J. , Raymond, W.R. , Okumura, S. , Okihiro, M.M. , Engle, E.C. , 2002. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 71, 1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadollahi, R. , Hyde, C.A. , Zhong, X.Y. , 2010. Epigenetics of ovarian cancer: from the lab to the clinic. Gynecol. Oncol. 118, 81–87. [DOI] [PubMed] [Google Scholar]
- Bandera, C.A. , Takahashi, H. , Behbakht, K. , Liu, P.C. , LiVolsi, V.A. , Benjamin, I. , Morgan, M.A. , King, S.A. , Rubin, S.C. , Boyd, J. , 1997. Deletion mapping of two potential chromosome 14 tumor suppressor gene loci in ovarian carcinoma. Cancer Res. 57, 513–515. [PubMed] [Google Scholar]
- Bowen, N.J. , Walker, L.D. , Matyunina, L.V. , Logani, S. , Totten, K.A. , Benigno, B.B. , McDonald, J.F. , 2009. Gene expression profiling supports the hypothesis that human ovarian surface epithelia are multipotent and capable of serving as ovarian cancer initiating cells. BMC Med. Genomics 2, 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannistra, S.A. , 2004. Cancer of the ovary. N. Engl. J. Med. 351, 2519–2529. [DOI] [PubMed] [Google Scholar]
- de Celis, J.F. , Barrio, R. , 2009. Regulation and function of Spalt proteins during animal development. Int. J. Dev. Biol. 53, 1385–1398. [DOI] [PubMed] [Google Scholar]
- Chen, L.M. , Yamada, S.D. , Fu, Y.S. , Baldwin, R.L. , Karlan, B.Y. , 2003. Molecular similarities between primary peritoneal and primary ovarian carcinomas. Int. J. Gynecol. Cancer 13, 749–755. [DOI] [PubMed] [Google Scholar]
- Cho, K.R. , Shih Ie, M. , 2009. Ovarian cancer. Annu. Rev. Pathol. 4, 287–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss, A. , Ng, V. , Liu, J. , Piao, H. , Russo, M. , Vena, N. , Sheng, Q. , Hirsch, M.S. , Bonome, T. , Matulonis, U. , Ligon, A.H. , Birrer, M.J. , Drapkin, R. , 2010. Overexpression of elafin in ovarian carcinoma is driven by genomic gains and activation of the nuclear factor kappaB pathway and is associated with poor overall survival. Neoplasia 12, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum, C.P. , Drapkin, R. , Miron, A. , Ince, T.A. , Muto, M. , Kindelberger, D.W. , Lee, Y. , 2007. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr. Opin. Obstet. Gynecol. 19, 3–9. [DOI] [PubMed] [Google Scholar]
- Crum, C.P. , McKeon, F.D. , Xian, W. , 2012. The oviduct and ovarian cancer: causality, clinical implications, and “targeted prevention”. Clin. Obstet. Gynecol. 55, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum, C.P. , 2009. Intercepting pelvic cancer in the distal fallopian tube: theories and realities. Mol. Oncol. 3, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejeux, E. , El abdalaoui, H. , Gut, I.G. , Tost, J. , 2009. Identification and quantification of differentially methylated loci by the pyrosequencing technology. Meth. Mol. Biol. 507, 189–205. [DOI] [PubMed] [Google Scholar]
- Dey, D.C. , Bronson, R.P. , Dahl, J. , Carroll, J.P. , Benjamin, T.L. , 2000. Accelerated development of polyoma tumors and embryonic lethality: different effects of p53 loss on related mouse backgrounds. Cell Growth Differ.: Mol. Biol. J. Am. Assoc. Cancer Res. 11, 231–237. [PubMed] [Google Scholar]
- Drapkin, R. , von Horsten, H.H. , Lin, Y. , Mok, S.C. , Crum, C.P. , Welch, W.R. , Hecht, J.L. , 2005. Human epididymis protein 4 (HE4) is a secreted glycoprotein that is overexpressed by serous and endometrioid ovarian carcinomas. Cancer Res. 65, 2162–2169. [DOI] [PubMed] [Google Scholar]
- Dubeau, L. , 2008. The cell of origin of ovarian epithelial tumours. Lancet Oncol. 9, 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler, D.A. , Gupta, S. , Growdon, W.B. , Drapkin, R.I. , Nitta, M. , Sergent, P.A. , Allred, S.F. , Gross, J. , Deavers, M.T. , Kuo, W.L. , Karlan, B.Y. , Rueda, B.R. , Orsulic, S. , Gershenson, D.M. , Birrer, M.J. , Gray, J.W. , Mohapatra, G. , 2012. Genome wide DNA copy number analysis of serous type ovarian carcinomas identifies genetic markers predictive of clinical outcome. PLoS One 7, e30996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemadmoghadam, D. , deFazio, A. , Beroukhim, R. , Mermel, C. , George, J. , Getz, G. , Tothill, R. , Okamoto, A. , Raeder, M.B. , Harnett, P. , Lade, S. , Akslen, L.A. , Tinker, A.V. , Locandro, B. , Alsop, K. , Chiew, Y.E. , Traficante, N. , Fereday, S. , Johnson, D. , Fox, S. , Sellers, W. , Urashima, M. , Salvesen, H.B. , Meyerson, M. , Bowtell, D. , 2009. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin. Cancer Res. 15, 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer, M. , McDonald, L.E. , Millar, D.S. , Collis, C.M. , Watt, F. , Grigg, G.W. , Molloy, P.L. , Paul, C.L. , 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U S A 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden, M. , Frommer, M. , 1987. CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261–282. [DOI] [PubMed] [Google Scholar]
- Gu, H. , Li, D. , Sung, C.K. , Yim, H. , Troke, P. , Benjamin, T. , 2011. DNA-binding and regulatory properties of the transcription factor and putative tumor suppressor p150(Sal2). Biochim. Biophys. Acta [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley, P.M. , Livingston, D.M. , 2009. Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazaeri, A.A. , 2009. Molecular profiles of hereditary epithelial ovarian cancers and their implications for the biology of this disease. Mol. Oncol. 3, 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.A. , Baylin, S.B. , 2007. The epigenomics of cancer. Cell 128, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens, G. , 1988. Head and tail development of the Drosophila embryo involves Spalt, a novel homeotic gene. EMBO J. 7, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst, A.M. , Drapkin, R. , 2010. Ovarian cancer pathogenesis: a model in evolution. J. Oncol. 2010, 932371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase, J. , Taschner, P.E. , Burfeind, P. , Pasche, B. , Newman, B. , Blanck, C. , Breuning, M.H. , ten Kate, L.P. , Maaswinkel-Mooy, P. , Mitulla, B. , Seidel, J. , Kirkpatrick, S.J. , Pauli, R.M. , Wargowski, D.S. , Devriendt, K. , Proesmans, W. , Gabrielli, O. , Coppa, G.V. , Wesby-van Swaay, E. , Trembath, R.C. , Schinzel, A.A. , Reardon, W. , Seemanova, E. , Engel, W. , 1999. Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am. J. Hum. Genet. 64, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase, J. , Chitayat, D. , Kotzot, D. , Ceylaner, S. , Froster, U.G. , Fuchs, S. , Montgomery, T. , Rosler, B. , 2005. SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum. Mutat. 26, 176–183. [DOI] [PubMed] [Google Scholar]
- Kurman, R.J. , Shih Ie, M. , 2010. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am. J. Surg. Pathol. 34, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, D.P. , Crawford, L.V. , 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263. [DOI] [PubMed] [Google Scholar]
- Lengyel, E. , 2010. Ovarian cancer development and metastasis. Am. J. Pathol. 177, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Dower, K. , Ma, Y. , Tian, Y. , Benjamin, T.L. , 2001. A tumor host range selection procedure identifies p150(sal2) as a target of polyoma virus large T antigen. Proc. Natl. Acad. Sci. U S A 98, 14619–14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Tian, Y. , Ma, Y. , Benjamin, T. , 2004. p150(Sal2) is a p53-independent regulator of p21(WAF1/CIP). Mol. Cell. Biol. 24, 3885–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linzer, D.I. , Levine, A.J. , 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Adler, A.S. , Segal, E. , Chang, H.Y. , 2007. A transcriptional program mediating entry into cellular quiescence. PLoS Genet. 3, e91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.F. , Hirsch, M.S. , Lee, H. , Matulonis, U.A. , 2009. Prognosis and hormone receptor status in older and younger patients with advanced-stage papillary serous ovarian carcinoma. Gynecol. Oncol. 115, 401–406. [DOI] [PubMed] [Google Scholar]
- Ma, Y. , Li, D. , Chai, L. , Luciani, A.M. , Ford, D. , Morgan, J. , Maizel, A.L. , 2001. Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J. Biol. Chem. 276, 48223–48230. [DOI] [PubMed] [Google Scholar]
- Mok, S.C. , Bonome, T. , Vathipadiekal, V. , Bell, A. , Johnson, M.E. , Wong, K.K. , Park, D.C. , Hao, K. , Yip, D.K. , Donninger, H. , Ozbun, L. , Samimi, G. , Brady, J. , Randonovich, M. , Pise-Masison, C.A. , Barrett, J.C. , Wong, W.H. , Welch, W.R. , Berkowitz, R.S. , Birrer, M.J. , 2009. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer Cell 16, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce, C.L. , Near, A.M. , Van Den Berg, D.J. , Ramus, S.J. , Gentry-Maharaj, A. , Menon, U. , Gayther, S.A. , Anderson, A.R. , Edlund, C.K. , Wu, A.H. , Chen, X. , Beesley, J. , Webb, P.M. , Holt, S.K. , Chen, C. , Doherty, J.A. , Rossing, M.A. , Whittemore, A.S. , McGuire, V. , DiCioccio, R.A. , Goodman, M.T. , Lurie, G. , Carney, M.E. , Wilkens, L.R. , Ness, R.B. , Moysich, K.B. , Edwards, R. , Jennison, E. , Kjaer, S.K. , Hogdall, E. , Hogdall, C.K. , Goode, E.L. , Sellers, T.A. , Vierkant, R.A. , Cunningham, J.M. , Schildkraut, J.M. , Berchuck, A. , Moorman, P.G. , Iversen, E.S. , Cramer, D.W. , Terry, K.L. , Vitonis, A.F. , Titus-Ernstoff, L. , Song, H. , Pharoah, P.D. , Spurdle, A.B. , Anton-Culver, H. , Ziogas, A. , Brewster, W. , Galitovskiy, V. , Chenevix-Trench, G. , 2009. Validating genetic risk associations for ovarian cancer through the international Ovarian Cancer Association Consortium. Br. J. Cancer 100, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincheira, R. , Baerwald, M. , Dunbar, J.D. , Donner, D.B. , 2009. Sall2 is a novel p75NTR-interacting protein that links NGF signalling to cell cycle progression and neurite outgrowth. EMBO J. 28, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen, O. , Deo, R.C. , Padi, M. , Adelmant, G. , Calderwood, M.A. , Rolland, T. , Grace, M. , Dricot, A. , Askenazi, M. , Tavares, M. , Pevzner, S.J. , Abderazzaq, F. , Byrdsong, D. , Carvunis, A.R. , Chen, A.A. , Cheng, J. , Correll, M. , Duarte, M. , Fan, C. , Feltkamp, M.C. , Ficarro, S.B. , Franchi, R. , Garg, B.K. , Gulbahce, N. , Hao, T. , Holthaus, A.M. , James, R. , Korkhin, A. , Litovchick, L. , Mar, J.C. , Pak, T.R. , Rabello, S. , Rubio, R. , Shen, Y. , Singh, S. , Spangle, J.M. , Tasan, M. , Wanamaker, S. , Webber, J.T. , Roecklein-Canfield, J. , Johannsen, E. , Barabasi, A.L. , Beroukhim, R. , Kieff, E. , Cusick, M.E. , Hill, D.E. , Munger, K. , Marto, J.A. , Quackenbush, J. , Roth, F.P. , DeCaprio, J.A. , Vidal, M. , 2012. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, C.K. , Dahl, J. , Yim, H. , Rodig, S. , Benjamin, T.L. , 2011. Transcriptional and post-translational regulation of the quiescence factor and putative tumor suppressor p150(Sal2). FASEB J. 25, 1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, C.K. , Yim, H. , Gu, H. , Li, D. , Andrews, E. , Duraisamy, S. , Li, C. , Drapkin, R. , Benjamin, T. , 2012. The polyoma virus large T binding protein p150 is a transcriptional repressor of c-MYC. PLoS One 7, e46486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetman, D. , Munsterberg, A. , 2006. The vertebrate Spalt genes in development and disease. Dev. Biol. 293, 285–293. [DOI] [PubMed] [Google Scholar]
- Takai, D. , Jones, P.A. , 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. U S A 99, 3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko, B. , 2000. Epigenetic gene silencing in cancer. J. Clin. Invest. 105, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, S. , Coward, J.I. , Bast, R.C. , Berchuck, A. , Berek, J.S. , Brenton, J.D. , Coukos, G. , Crum, C.C. , Drapkin, R. , Etemadmoghadam, D. , Friedlander, M. , Gabra, H. , Kaye, S.B. , Lord, C.J. , Lengyel, E. , Levine, D.A. , McNeish, I.A. , Menon, U. , Mills, G.B. , Nephew, K.P. , Oza, A.M. , Sood, A.K. , Stronach, E.A. , Walczak, H. , Bowtell, D.D. , Balkwill, F.R. , 2011. Rethinking ovarian cancer: recommendations for improving outcomes. Nature reviews. Cancer 11, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Kong, W. , He, L. , Zhao, J.J. , O'Donnell, J.D. , Wang, J. , Wenham, R.M. , Coppola, D. , Kruk, P.A. , Nicosia, S.V. , Cheng, J.Q. , 2008. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 68, 425–433. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Gao, C. , Chai, L. , Ma, Y. , 2010. A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PLoS One 5, e10766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Tam, W.L. , Tong, G.Q. , Wu, Q. , Chan, H.Y. , Soh, B.S. , Lou, Y. , Yang, J. , Ma, Y. , Chai, L. , Ng, H.H. , Lufkin, T. , Robson, P. , Lim, B. , 2006. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat. Cell Biol. 8, 1114–1123. [DOI] [PubMed] [Google Scholar]
- Zhou, Q. , Chipperfield, H. , Melton, D.A. , Wong, W.H. , 2007. A gene regulatory network in mouse embryonic stem cells. Proc. Natl. Acad. Sci. U S A 104, 16438–16443. [DOI] [PMC free article] [PubMed] [Google Scholar]