

Abstract
C13ORF18 is frequently hypermethylated in cervical cancer but not in normal cervix and might serve as a biomarker for the early detection of cervical cancer in scrapings. As hypermethylation is often observed for silenced tumor suppressor genes (TSGs), hypermethylated biomarker genes might exhibit tumor suppressive activities upon re‐expression. Epigenetic drugs are successfully exploited to reverse TSG silencing, but act genome‐wide. Artificial Transcription Factors (ATFs) provide a gene‐specific approach for re‐expression of silenced genes. Here, we investigated the potential tumor suppressive role of C13ORF18 in cervical cancer by ATF‐induced re‐expression.
Five zinc finger proteins were engineered to bind the C13ORF18 promoter and fused to a strong transcriptional activator. C13ORF18 expression could be induced in cervical cell lines: ranging from >40‐fold in positive (C13ORF18‐unmethylated) cells to >110‐fold in negative (C13ORF18‐methylated) cells. Re‐activation of C13ORF18 resulted in significant cell growth inhibition and/or induction of apoptosis. Co‐treatment of cell lines with ATFs and epigenetic drugs further enhanced the ATF‐induced effects. Interestingly, re‐activation of C13ORF18 led to partial demethylation of the C13ORF18 promoter and decreased repressive histone methylation. These data demonstrate the potency of ATFs to re‐express and potentially demethylate hypermethylated silenced genes. Concluding, we show that C13ORF18 has a TSG function in cervical cancer and may serve as a therapeutic anti‐cancer target. As the amount of epimutations in cancer exceeds the number of gene mutations, ATFs provide promising tools to validate hypermethylated marker genes as therapeutic targets.
Keywords: Methylation biomarkers, Artificial Transcription Factors, Tumor suppressor genes, DNA methylation/epigenetics, Cellular responses to anticancer drugs, Novel antitumor agents
Highlights
-
►
Gene‐specific re‐activation of putative TSG C13ORF18 by ATFs.
-
►
Re‐expression of C13ORF18 by ATFs inhibits tumor growth in cervical cancer.
-
►
Targeted reversal of epigenetic marks (DNA and histone methylation).
-
►
Synergistic effects of ATFs with epigenetic drugs.
-
►
Epigenetic regulation C13ORF18.
1. Introduction
Aberrant epigenetic regulation, including promoter hypermethylation of tumor suppressor genes (TSGs), is a significant event in the progression of cancer (Baylin and Jones, 2011). In the last decade, genome‐wide mapping of DNA methylation in normal and cancer genomes revealed that a large number of genes is associated with hypermethylated CpG islands in cancer; these epimutations exceed by far the number of genetic mutated genes (Baylin and Jones, 2011; Esteller, 2007). Genes specifically hypermethylated in cancer might provide diagnostic biomarkers, and the number of such methylated markers has increased dramatically in the last years (Cairns et al., 2001; Glockner et al., 2009; Roossink et al., 2012). A therapeutic application of such epigenetically silenced marker genes lies in the fact that they might exhibit tumor suppressive activities upon re‐expression. Indeed, re‐introduction of single genes into cancer cells where the gene is inactivated, such as CDKN2A (encoding p16Ink4A) or TP53, showed potent anti‐tumor effects (Hamada et al., 1996; Lu et al., 2012). However, the therapeutic efficacy of gene transfer in patients still faces significant challenges (Fukazawa et al., 2010).
Contrary to genes that are inactivated through genetic mutations, epigenetic deregulation is a reversible process and can be targeted by epigenetic drugs that affect DNA methylation and histone modification. Current clinical drugs affecting epimutations, such as the DNA methylation inhibitor 5‐aza‐deoxyCytosine (5‐aza‐dC) and the histone deacetylase inhibitor suberoylanilide hydroxamic acid, indeed induce re‐expression of a variety of hypermethylated silenced genes (Kelly et al., 2010). So far, four epigenetic drugs have been FDA approved for subtypes of leukemias and lymphomas, and ongoing clinical trials in solid tumors indicate some beneficial effects for these patients as well (Juergens et al., 2011). However, the disadvantages of these drugs include their lack of gene‐target specificity (Rius and Lyko, 2012). In that respect it is important to note that epigenetic drugs have been shown to drive tumor promotion and metastasis by enhanced expression of off‐target genes (Yu et al., 2010). Similarly, epigenetic drugs might also upregulate drug‐resistance genes, again causing unwanted side effects. Therefore, there is clearly a need for gene targeted re‐expression (Roossink et al., 2012).
To exploit reversibility of epigenetic mutations in a gene‐specific manner, Artificial Transcription Factors (ATFs) have been explored (Uil et al., 2003). ATFs consist of a transcriptional regulatory domain coupled to a DNA binding domain, generally an engineered Zinc Finger Protein, which can be designed to target virtually any gene in the human genome (Gommans et al., 2007; Sera, 2009). Advantages of ATFs include the potency to repress undruggable targets, and activate silenced genes which will occur from the endogenous gene locus (allowing all splice variants to be expressed, in natural ratios). The therapeutic potential of ATFs in cancer has been demonstrated eg by the re‐activation of TSG MASPIN in breast, lung and ovarian cancer (Beltran et al., 2007; Beltran and Blancafort, 2011) or down‐regulation of SOX2 (Stolzenburg et al., 2012), VEGF‐A (Snowden et al., 2003), ERBB2 (Beerli et al., 2000) and ERBB3 (Holbro et al., 2003), leading to apoptosis and/or cell growth inhibition.
For cervical cancer, the third most common malignancy among women worldwide, current screening programs rely on pap‐smear assays which have poor sensitivity. As alternative to the pap‐smear, our group and others reported several gene promoters that are specifically hypermethylated in cervical cancer and could thus serve as biomarkers for screening (Eijsink et al., 2012; Hoque et al., 2008; Wentzensen and von Knebel Doeberitz, 2007; Yang et al., 2009). The most potent hypermethylation marker we identified was C13ORF18, which was methylated in ∼70% of cervical cancer patients, compared to 3% in scrapings from healthy controls (Eijsink et al., 2012; Yang et al., 2009). As promoter hypermethylation is often seen for TSGs, we hypothesized that this gene might exhibit tumor suppressive activities upon re‐expression. In this study, we therefore aimed to specifically re‐express C13ORF18 in cervical cancer cell lines using ATFs and study the effect of re‐expression on in vitro cancer‐related cell biological features.
2. Materials and methods
2.1. Cell culture
Four cervical carcinoma cell lines (HeLa (cervical adenocarcinoma), SiHa (cervical squamous cell carcinoma), CaSki (epidermoid cervical carcinoma) and C33A (cervical epithelial carcinoma)), primary human dermal fibroblasts (adult) (ScienCell) and HEK293T cells (human embryonic kidney) were cultured in DMEM containing 10% FBS, 2 mM l‐glutamine and 50 μg/ml gentamycin. Cell lines were obtained from ATCC and authenticity of cervical cancer cell lines was verified by DNA short tandem repeat analysis (Baseclear). Treatment of cell lines with 5‐aza‐dC (Sigma) (500 nM or 5 μM) and trichostatin A (TSA) (Sigma) (500 nM) was performed as described previously (Ongenaert et al., 2008). ATF‐transduced cells were treated for 2 days with 5‐aza‐dC. Co‐treatment of ATF with TSA was performed for 24 h at day two after infection.
2.2. Engineering and delivery of ATFs
The promoter of C13ORF18 was analyzed using Ensemble and PubMed (NCBI‐id: NM_025113) databases for transcription start sites (TSS) (situated 66 bp upstream of the initiator (Inr) sequence ‘TCA (G/T) T (T/C)’) and transcription factor binding sites (Figure 1a and b). The region flanking the main TSS (from −410 bps till +610 bps) was screened for engineering the zinc finger proteins (ZFPs) consisting of 6 fingers fused together (6ZFP) to target 18 bps regions (www.zincfingertools.org) (Mandell and Barbas, 2006). ZFPs with predicted high affinity were selected and the uniqueness of the target sites was confirmed by a blast on NCBI. DS‐DNA Oligos (Mr Gene, Bio‐Basics) for each 6ZFP flanked with the restriction site SfiI were subcloned into the pMX‐IRES‐GFP retroviral vector (Beltran et al., 2007), which carries an HA‐tag, a nuclear localization signal and either the gene activator VP64 or no effector domain (NoEf) (Magnenat et al., 2004). A previously reported EpCAM specific ZFP (Gommans et al., 2007) was also cloned into the pMX‐IRES‐GFP retroviral vector and used as irrelevant ATF. To produce retroviruses pseudotyped with glycoprotein G from vesicular stomatitis virus (VSV‐G), HEK293T packaging cells were transfected with pMX‐IRES‐GFP with or without VP64 insert along with the viral packaging plasmids pMDg/p and pMDg in a 3:2:1 ratio using the calcium phosphate method. pMDg/p and pMDg were a kind gift from dr. R. Mulligan (Harvard Medical School, Boston, MA, USA). Virus‐containing supernatant was harvested at 24 and 48 h after transfection, cleared from cell debris by centrifugation, supplemented with 6 μg/ml polybrene (Sigma) and used to infect cervical cancer host cells. Efficient viral delivery was evaluated by the percentage of GFP‐positive cells at 2 days after transduction using flow cytometry using a FACSCalibur (BD Biosciences).
Figure 1.
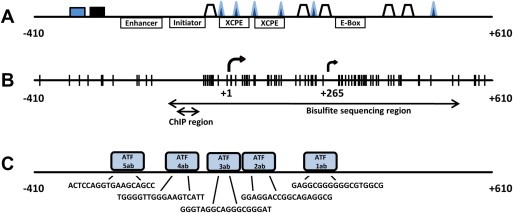
Schematic representations of the promoter region of the C13ORF18 gene. The analyzed part of the promoter encompasses a region that stretches from −410 to +610 bp relative to the main TSS (+1). An additional putative TSS is shown (+265). (A) Core promoter elements and putative transcription factors binding sites (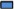 AP‐1R,
AP‐1R,  p53,
p53,  SP1 and
SP1 and  E2F (MatInspector)) linked to C13ORF18 transcription. (B) CpGs are indicated as vertical bars. DNA methylation status of the CpGs was analyzed using bisulfite sequencing and MSP for the indicated areas. Association of histone modifications with the C13ORF18 gene were characterized for the ChIP region. (C) Target sequences of 1ab‐VP64, 2ab‐VP64, 3ab‐VP64, 4ab‐VP64 and 5ab‐VP64 are shown, as well their schematic locations relative to the TSS.
E2F (MatInspector)) linked to C13ORF18 transcription. (B) CpGs are indicated as vertical bars. DNA methylation status of the CpGs was analyzed using bisulfite sequencing and MSP for the indicated areas. Association of histone modifications with the C13ORF18 gene were characterized for the ChIP region. (C) Target sequences of 1ab‐VP64, 2ab‐VP64, 3ab‐VP64, 4ab‐VP64 and 5ab‐VP64 are shown, as well their schematic locations relative to the TSS.
2.3. Quantitative real‐time PCR
Total RNA was extracted (RNeasy plus mini kit, Qiagen) from untreated, viral treated, or 5‐aza‐dC and TSA treated cells and 2.5 μg was used for reverse transcription. Subsequent real‐time PCR reactions (ABIPrism 7900HT, Applied Biosystems) for the quantification of C13ORF18, GAPDH and VP64 activation domain were performed with 20 ng cDNA, Rox enzyme mix (Thermo Scientific) and gene specific primer/probes (Table 1) for 40 cycles. PCR efficiency of every primer/probe pair was determined by performing a standard curve. RNA samples prepared without reverse transcriptase were included to rule out cross reaction with DNA, Data were analyzed with SDS 2.1 RQ software (Applied Biosystems) and expression levels relative to GAPDH were determined with the formula 2−ΔCt. Fold increase in gene‐expression compared to controls was calculated with the formula 2−ΔΔCt. Samples for which no amplification could be detected were assigned a Ct value of the total number of PCR cycles.
Table 1.
Sequence primers.
| Gene | Forward primer | Reverse primer | Probe | Tm (°/C) |
|---|---|---|---|---|
| C13ORF18 | qRT‐PCRGTGCTGCCTGTTGATGTAGA | TTCAGAGTCAGGCTGATCAC | CATTCTGAGTCAACAGCAGACAGAGAGC | 60 |
| VP64 | AAGCGACGCATTGGATGAC | GGAACGTCGTACGGGTAGTTAATT | TCGGCTCCGATGCT | 60 |
| GAPDH | CCACATCGCTCAGACACCAT | GCGCCCAATACGACCAAAT | GTTGACTCCGACCTTCACCTTCCC | 60 |
| C13ORF18 | ChIPTGCTAAATACTGCTGCTGGGGGC | CGCGAGACGCAACGACGCTT | GGTTGGGAAGTCATTTCTGCCAGGTCTTTG | 60 |
| C13ORF18 | Bisulfite sequencingATTTGGGGTTGGGAAGTTATTT | AAAAACCCCTTCCTACTATCTC | 60 | |
| C13ORF18 | Sequencing ATF target sitesACGCGGACAGTGAAGTGCTACA | CAGAAAGGGGAGGGAGGAGA | 60 |
2.4. (Bisulfite) sequencing
The methylation status of 65 CpGs in the C13ORF18 promoter was examined using bisulfite sequencing. For amplifying part of the C13ORF18 promoter, we used primers specific to the bisulfite modified promoter region of C13ORF18 (Figure 1, Table 1). Amplified PCR products were separated by electrophoresis and subsequently purified by gel extraction (Qiagen). The purified PCR products were cloned into the pCR 2.1‐TOPO Vector (Invitrogen) and transformed into TOP10 competent bacteria. Plasmids of positive recombinants were isolated (Qiagen) and subjected to sequencing (Baseclear) using M13 primers. Percentages of methylation for each clone were determined by dividing the amount of methylated CpGs by the total number of CpGs.
Sequencing of ATF target sites was performed using 1 primer pair spanning the 5 ZF binding sites (Table 1).
2.5. Chromatin ImmunoPrecipitation (ChIP)
Histone modifications associated with the C13ORF18 promoter (before and after ATF treatment) were determined by ChIP. In short, cells were fixed with 1% formaldehyde for 10 min and subsequently washed twice with PBS, lyzed and sonificated using a Bioruptor (High, 15 cycles of 30″ on, 30″ off, total time 15 min) (Diagnode). Sheared chromatin was cleared by centrifugation. Magnetic beads (Invitrogen) were pre‐coated by adding 3 μg of antibodies (HA‐tag (ab9110), H3‐core (ab1791), normal rabbit IgG (ab46540) (Abcam), acH3 (06‐599), acH4 (06‐598), H3K9me3 (07‐442) and H3K27me3 (07‐449) (Millipore)) to 50 μl of magnetic beads, followed by 15 min incubation and a washing step. Sheared chromatin of 1 million cells was added to the magnetic beads and incubated at a rotating platform at 4 °C O/N. Next day, magnetic bead complexes were washed with PBS three times and the DNA was subsequently eluted from the beads. Elutes were supplemented RNAse (Roche) followed by O/N incubation at 67 °C. Next day, DNA was purified by column purification (Qiagen) after a final incubation step at 45 °C for 1 h with Proteinase K (Roche). The recovered DNA was subsequently subjected to Quantitative real‐time PCR using primers and probes specific for the C13ORF18 promoter (Figure 1, Table 1). Relative enrichment in the recovered DNA was calculated with the formula: .
2.6. Growth assay
Following treatment, cells were plated in 96 wells plates (1000 cells per well) and incubated for 5 days at 37 °C. Every day, cell growth was measured using a thiazolyl blue tetrazolium blue (MTT) (Sigma) assay. MTT (5 mg/ml) was added to wells followed by 3.5 h of incubation at 37 °C. Then, plates were centrifuged, medium was aspirated and MTT crystals were dissolved in DMSO. The absorbance was determined at 520 nm with a Versamax microplate reader (Molecular Devices).
2.7. Clonogenic assay
Following treatment, cell were plated in 6‐wells plates (800 cells per well). After two weeks, medium was aspirated and colonies were stained with Coomassie brilliant blue (Bio‐Rad). The number of colonies was determined using phase‐contrast microscopy, only colonies with at least 50 cells were counted.
2.8. Apoptosis assay
Induction of apoptosis was quantified using a 1,1′,3,3,3′,3′‐Hexamethylindodicarbocyanine iodide (DilC) assay (Enzo Life Sciences). Following treatment, cells were trypsinized and incubated in culture medium supplemented with DilC (50 nM) for 15 min at 37 °C. After washing with PBS, DilC signal was analyzed using FACS Calibur (BD Biosciences). The percentage of apoptotic cells was determined as the number of viable cells with decreased DilC intensity, as reported before (Edel et al., 2011). As positive control, apoptosis was induced with Actinomycin D (ActD) at a final concentration of 20 μM and 40 μM. To inhibit apoptosis, cells were co‐treated with ActD (20 μM) and Z‐VAD‐fmk (Z‐VAD) (20 μM) or Caspase 8 inhibitor (50 μM).
2.9. Statistics
Statistical significant differences were determined by the Student's t‐test. A p‐value of less than 0.05 was considered statistically significant with *p < 0.05, **p < 0.01 and ***p < 0.001.
3. Results
3.1. C13ORF18 is epigenetically regulated in cervical cancer
We determined C13ORF18 expression in a panel of well‐established cervical cancer cell lines: HeLa, SiHa and CaSki cells show no or very low levels of C13ORF18 (Figure 2a), whereas C33A showed low expression levels of the gene. C13ORF18 gene silencing in the three cell lines was associated with near complete promoter hypermethylation (HeLa 89 ± 0.8%, SiHa 96 ± 2.1%, CaSki 94 ± 1.0%) (Figure 2b), while the C13ORF18 gene was almost completely unmethylated in C33A cells (0.5 ± 0.5%).
Figure 2.
The expression of C13ORF18 is associated with DNA methylation. (A) Quantification of C13ORF18 mRNA in the panel of cell lines. (B) Analysis of the methylation status of 65 CpGs in the C13ORF18 promoter of HeLa, SiHa, CaSki and C33A cells. Each circle represents a single CpG and 3 clones per cell line were analyzed (65 CpGs per clone). Shown are the TSS ( ) and the ATF target sites covering this area (
) and the ATF target sites covering this area ( ). (C) Relative C13ORF18 mRNA induction after treatment with 5‐aza‐dC (500 nM (+), 5 μM (++)) and TSA, normalized to untreated cells. Expression/induction levels were quantified with qRT‐PCR and the bars represent the mean of at least three independent experiments ± SEM.
). (C) Relative C13ORF18 mRNA induction after treatment with 5‐aza‐dC (500 nM (+), 5 μM (++)) and TSA, normalized to untreated cells. Expression/induction levels were quantified with qRT‐PCR and the bars represent the mean of at least three independent experiments ± SEM.
Treatment of HeLa, SiHa and CaSki with 5‐aza‐dC induced expression of C13ORF18 mRNA in a dose‐dependent way (Figure 2c). Highest re‐expression levels were obtained for high dose 5‐aza‐dC (5 μM) after co‐treatment with the histone de‐acetylase inhibitor TSA, (HeLa 34 ± 7.2 fold, SiHa 51 ± 11 fold, CaSki 21 ± 7.6). These findings confirm previous indications that DNA methylation and histone modifications play a role in the transcriptional regulation of C13ORF18 (Hoque et al., 2008).
3.2. C13ORF18 re‐expression by ATFs
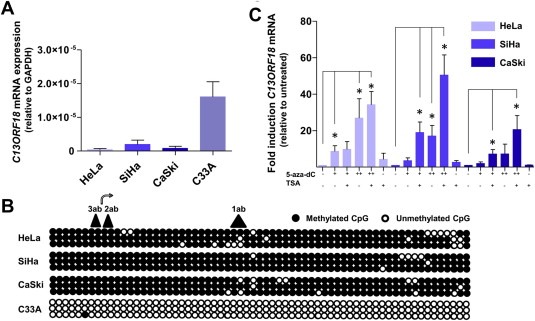
Retroviral constructs containing VP64 were engineered to express the different ATFs (pMX‐1ab‐VP64, pMX‐2ab‐VP64, pMX‐3ab‐VP64, pMX‐4ab‐VP64 and pMX‐5ab‐VP64). The zinc fingers contained 6 fingers and were constructed to target five 18 bp regions in the C13ORF18 promoter, surrounding the TSS (1ab, 2ab, 3ab, 4ab, 5ab) (Figure 1c). The five ZF target sites were intact, as no DNA mutations were detected in neither of the cervical cancer cells (Supplementary Figure 1a (HeLa), 1b (CaSki) and 1c (C33A)). Efficient transcription of ATF mRNA was confirmed for all cell lines (Supplementary Figure 2a). GFP positivity ranged from 60% for pMX‐1ab‐VP64 to 90% for pMX‐5ab‐VP64 (for C33A, Figure 3a).
Figure 3.
C13ORF18 specific ATFs upregulate/induce expression in cervical cancer cell lines. (A) Percentage of GFP positive cells after retroviral delivery of an empty vector (pMX empty), C13ORF18‐targeting ATFs (1ab‐VP64, 2ab‐VP64, 3ab‐VP64, 4ab‐VP64 and 5ab‐VP64), ZFPs without effector domain (3ab‐NoEf and 5ab‐NoEf) and an irrelevant ATF (EpCAM ATF) in C33A cells. (B) Relative upregulation of C13ORF18 mRNA after ATF treatment in C33A cells. (C) Relative induction of C13ORF18 mRNA after treatment with the different pMX constructs (1ab‐VP64, 2ab‐VP64, 3ab‐VP64, 4ab‐VP64 and 5ab‐VP64) in HeLa and CaSki cells. (D) Effect of co‐treatment of ATFs with 5‐aza‐dC (5 μM) or TSA on induction of C13ORF18 mRNA in HeLa cells. Quantification of mRNA was performed using qRT‐PCR and induction levels were normalized to untreated cells. Each bar represent the mean of at least three independent experiments (ATF‐NoEf and irrelevant ATF two experiments) measured in triplicate ± SEM.
First, we examined the ability of the 5 ATFs to upregulate endogenous C13ORF18 mRNA expression in the C13ORF18 low‐expressing cell line C33A (Figure 3b). C13ORF18 was significantly up‐regulated in this cell line by 3ab‐VP64 (43 ± 6.2 fold (p < 0.001)), 4ab‐VP64 (4.6 ± 1.3 fold (p < 0.05)) and 5ab‐VP64 (28 ± 5.4 fold (p < 0.001)), whereas the controls (3ab‐NoEf, 5ab‐NoEf and the irrelevant ATF) did not show any effect on C13ORF18 mRNA levels. 1 ab‐VP64 and 2ab‐VP64, although more efficiently expressed than 4ab‐VP64, (Supplementary Figure 2a), failed to up‐regulate C13ORF18 expression in C33A.
In the hypermethylated CaSki cells (Figure 3c, filled bars), both 3ab‐VP64 (44 ± 6.5 fold (p < 0.01)) and 5ab‐VP64 (29 ± 5.2 fold (p < 0.01)) were able to significantly re‐activate the C13ORF18 promoter. No re‐expression was induced by 1ab‐VP64, 2ab‐VP64 and 4ab‐VP64 in CaSki cells.
Despite the lack of effect of 1ab‐VP64 in CaSki and C33A, induction of C13ORF18 mRNA was most pronounced in HeLa cells by 1ab‐VP64, with an induction level of 112 ± 38.9 fold (p < 0.05) (Figure 3c). Interestingly, the induced C13ORF18 expression in the methylated cell line HeLa by 1ab‐VP64 was 3 fold higher than the endogenous expression of C13ORF18 in C33A cells (Figure 2a), which comprises an unmethylated promoter.
As it has been shown that epigenetic drugs synergize with ATF treatment (Beltran et al., 2008), we explored the possibility of ATF treatment in combination with epigenetic drugs to enhance or even induce the effects of the ATFs. In HeLa cells, co‐treatment of ATFs with epigenetic drugs further increased ATF induced expression of C13ORF18 mRNA (Figure 3d). Significant additive effects were reached with 3ab‐VP64 (up to 144 ± 22 fold (p < 0.001)) and 5ab‐VP64 (up to 124 ± 18 fold (p < 0.001)) after co‐treatment with both 5‐aza‐dC (5 μM) and TSA. Interestingly, ATF 2ab‐VP64, which had no effect by itself on gene activation in HeLa, consistently induced C13ORF18 re‐expression when co‐treated with TSA. The same consistent effect was seen for 1ab‐VP64 + TSA, which resulted in the highest observed induction of C13ORF18 re‐expression in this study (339 ± 106 fold), which is 9 fold higher than the endogenous expression of C13ORF18 in C33A cells.
To detect association of ATFs with the C13ORF18 promoter, a ChIP for HA‐tag was performed after ATF treatment alone (CaSki, Supplementary Figure 2b) or in combination with TSA (C33A, Supplementary Figure 2c). For CaSki, 3ab‐VP64 association with the C13ORF18 promoter was detectable in 1% of input DNA compared to no detection with pMX empty. Detection of 5ab‐VP64 could be observed in 2.5% of input DNA, which was increased by 2 fold when co‐treated with TSA.
3.3. ATF‐induced gene expression affects epigenetic marks
To determine whether the ATF‐induced C13ORF18 gene re‐expression was associated with DNA demethylation and/or histone modifications of its promoter, bisulfite sequencing and ChIP was performed. For HeLa, pMX‐1ab‐VP64‐transduced cells, which showed the strongest C13ORF18 re‐expression (Figure 3c), showed partial demethylation mainly around the target site of ATF‐1ab (+80 to +218), and around the TSS (−35 to +52) (Figure 4a left panel). Quantification of the methylated status of the total bisulfite sequencing region of the 5′ end of the C13ORF18 promoter (Figure 4a right panel) showed a significant decrease in methylation of 17 ± 0.6% compared to untreated cells (p < 0.001). Co‐treatment of 1ab‐VP64 with TSA, which even better enhanced C13ORF18 re‐expression (Figure 3d), did not further increase DNA demethylation compared to 1ab‐VP64 alone (1ab‐VP64: −17 ± 0.6%, 1ab‐VP64 + TSA: −14 ± 2.0%). Targeted DNA demethylation was also observed for CaSki after treatment with 3ab‐VP64 at its target site (which overlaps the TSS) (untreated cells: 94 ± 1.0%, 3ab‐VP64: 88 ± 1.5% p < 0.05) (Figure 4a right panel filled bars).
Figure 4.
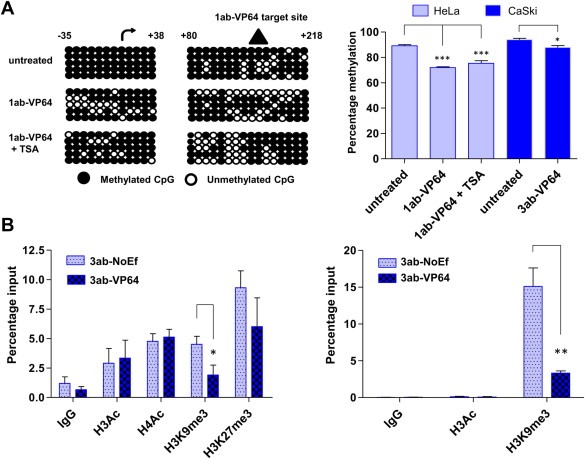
Changes in histone marks and DNA methylation after ATF induced re‐expression of C13ORF18. (A) DNA methylation status is shown for two subregions (from −35 to +52 and from +80 to +218) flanking the 5′ promoter of the C13ORF18 promoter in untreated, ATF treated and ATF + TSA treated HeLa cells (left panel). Shown are the TSS () and the 1ab‐VP64 target site (). Each circle represents a single CpG and 5 clones per condition were analyzed. The right panel shows the quantification of the DNA methylation of all 65 CpGs in the sequenced C13ORF18 promoter region before and after ATF treatment in HeLa (ATF alone or in combination with TSA) and CaSki cells: each bar represents the mean percentage of DNA methylation of five individual clones ±SEM (B) Quantitative ChIP for histone acetylation (H3Ac (and H4Ac)) and histone methylation (H3K9me3 (and H3K27me3)) after treatment with 3ab‐NoEf or 3ab‐VP64 in C33A cells (left panel) and CaSki cells (right panel). Values represent the mean of at least three independent experiments measured in triplicate ± SEM.
As DNA methylation is closely linked to histone modifications, ATF treatment might lead to a change in the pattern of histone modifications. We studied if forced C13ORF18 gene re‐expression by ATFs indeed changed the histone state to a more active state. As HeLa cells treated with 1ab‐VP64 did not yield sufficient surviving cells (see also Figure 5), C33A and CaSki cells were treated with 3ab‐VP64 and 3ab‐NoEf to express C13ORF18. Figure 4b (left panel) shows that re‐activation of C13ORF18 in C33A did not influence the active histone marks H3Ac and H4Ac, but significant less trimethylation of H3K9 was observed (3ab‐NoEf 4.5 ± 0.7, 3ab‐VP64 1.9 ± 0.8 (p < 0.05)). For H3K27me3, the decrease by ATF‐induced expression did not reach statistical significance (3ab‐NoEf 9.3 ± 1.4, 3ab‐VP64 6.0 ± 2.4 (p = 0.3)). For CaSki, significantly less trimethylation of H3K9me3 after 3ab‐VP64 treatment could be detected as well (3ab‐NoEf 15 ± 2.5, 3ab‐VP64 3.3 ± 0.3 (p < 0.01)) (Figure 4b right panel).
Figure 5.
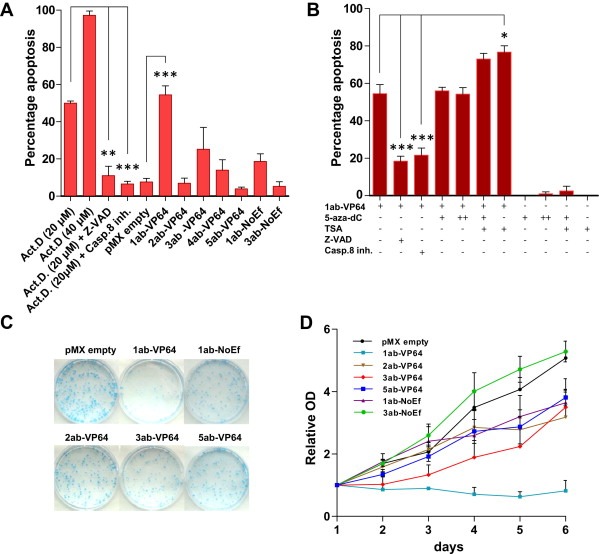
C13ORF18 targeting ATFs induce apoptosis and decrease cell growth. (A) Percentage of apoptotic cells after treatment with pMX empty and C13ORF18 directed ATFs in HeLa cells. In control cells, apoptosis was induced with Act‐D alone or in combination with Z‐VAD or Caspase 8 inhibitor (Casp.8 inh.). (B) Percentage of apoptotic cells after co‐treatment of 1ab‐VP64 with 5‐aza‐dC (500 nM (+), 5 μM (++)), TSA, Z‐VAD or Casp.8 inh. in HeLa cells. Quantification of apoptotic cells was assessed with a DilC assay reflecting a decrease in mitochondrial membrane potential. All bars represent the mean of at least 3 independent experiments ± SEM. (C) Visualization of colony forming potential of HeLa cells of a representative Coomassie blue staining after treatment with C13ORF18 specific ATFs or pMX empty. (D) Relative cell proliferation of HeLa cells after treatment with pMX empty, 1ab‐VP64, 2ab‐VP64, 3ab‐VP64, 5ab‐VP64, 1ab‐NoEf and 3ab‐NoEf normalized to day 1. Cell proliferation was assessed using a MTT assay and each data point represents the mean of in general three independent experiments measured in triplicate ± SEM. Significant growth differences between pMX empty and the ATFs were determined at day 6.
3.4. C13ORF18 functions as a tumor suppressor gene
Next, we studied whether ATF‐VP64‐induced re‐expression of C13ORF18 resulted in any functional effect on cell growth and apoptosis. In HeLa (Figure 5a), apoptosis was induced in 50 ± 1.1% of the cells by 20 μM of ActD and in 97 ± 2.3% of the cells by 40 μM ActD. The apoptotic induction was blocked with the pan‐caspase inhibitor Z‐VAD and Caspase 8 inhibitor to 11 ± 5.0% and 6.5 ± 1.5% of the cells, respectively. In 1ab‐VP64‐transduced HeLa cells with the highest upregulation of C13ORF18 mRNA (Figure 3c), apoptosis was significantly increased (7.1 ± 0.6 fold (p < 0.001)) compared to cells transduced with an empty vector (1ab‐VP64 54 ± 4.8% vs pMX empty 7.6 ± 2.0%). ATF‐ induced apoptosis by 1ab‐VP64 could be decreased by apoptosis inhibitors (Figure 5b); 3.2 ± 0.5 fold for Z‐VAD (p < 0.001) and 2.8 ± 0.4 fold for Caspase 8 inhibitor (p < 0.001). Contrary, a significant increase of apoptosis was detected by co‐treatment of 1ab‐VP64 with the histone de‐acetylase inhibitor TSA (1ab‐VP64 54 ± 4.8, 1ab‐VP64 + TSA 77 ± 3.4% (p < 0.05)), while TSA alone did not induce apoptosis in this dose (TSA: 2.5 ± 2.5%). Co‐treatment with 5‐aza‐dC did not further increase the apoptotic effect of 1ab‐VP64. The apoptotic effect of 1ab‐VP64 is also reflected in the colony formation potential of the HeLa cells, as treatment with 1ab‐VP64 strongly decreased the number of colonies compared to 1ab‐NoEf, 2ab‐VP64, 3ab‐VP64 and 5ab‐VP64 (Figure 5c). Cell growth in HeLa was completely prevented after treatment with the most potent C13ORF18 inducing ATF (1ab‐VP64) (Figure 5d) after 6 days of growth (p < 0.001). The constructs with moderate (3ab‐VP64, 5ab‐VP64) or no C13ORF18 re‐expression (2ab‐VP64; 1ab‐NoEf) induced far less effects on cell growth (2ab‐VP64 ‐37%, 3ab‐VP64 ‐31% and 5ab‐VP64 ‐25%) compared to pMX empty (100%) and 3ab‐NoEf (104%).
For CaSki, the best C13ORF18 mRNA‐inducing constructs (3ab‐VP64) significantly decreased cell proliferation (−59 ± 8.9% (p < 0.05)) (Supplementary Figure 3a) and colony formation (−73 ± 3.3% (p < 0.05)) (Supplementary Figure 3b) compared to pMX empty. The same effects on cell growth and colony formation in this cell line were observed for 5ab‐VP64 (−43 ± 19% and −52 ± 9.3% (p < 0.05) respectively) and not for 5ab‐NoEf.
The ATFs did not induce apoptosis in normal healthy cells (primary human adult fibroblasts) (Supplementary Figure 3c). However, co‐treatment of fibroblasts with 1ab‐VP64 and TSA, but not 5‐aza‐dC, did result in a small, although significant induction of apoptosis (1ab‐VP64 + TSA 5.8 ± 0.4%, pMX empty 3.8 ± 0.4% (p < 0.05)).
4. Discussion
Based on its differential methylation profile in cancer versus normal tissues and its predicted function as a phosphatase inhibitor, C13ORF18 has been suggested to function as a TSG in cervical cancer (Eijsink et al., 2012; Hoque et al., 2008; Yang et al., 2009). In the present study, we show that C13ORF18‐directed ATFs are able to restore expression of C13ORF18 and that its re‐expression is indeed associated with growth inhibition and apoptosis demonstrating its function as a tumor suppressor gene.
Few studies so far linked C13ORF18 to biological roles; C13ORF18 was linked to autophagy (Behrends et al., 2010) and identified as a marker to distinguish JAK2V617F‐negative thrombocythaemia patients (Puigdecanet et al., 2008), but provided only little mechanistic insights into how C13ORF18 might assert its tumor suppressive activities. In another study, C13ORF18 was associated with the cascade leading to cell senescence after down‐regulation of the human papillomavirus (HPV) E7 oncogene (Johung et al., 2007). It may well be that C13ORF18 silencing in cervical cancer is a consequence of introduction of HPV in cervical cells, which may facilitate in transforming normal cells into a malignant phenotype. This is also supported by the notion that the only C13ORF18‐expressing cell line used in this study (C33A) is HPV negative. Re‐activation of C13ORF18 might therefore indeed induce anti‐tumor effects as observed by us. Our results support the hypothesis that the methylation marker, C13ORF18, has TSG characteristics and may serve as a therapeutic target. Interestingly, the observed anti‐tumor effects seem to be cancer‐specific, as primary human fibroblasts cells are largely unaffected by ATF treatment in terms of apoptosis. Although co‐treatment of ATFs with TSA induced a slight apoptotic response in fibroblasts, the apoptotic effect was much more pronounced in cervical cancer cells co‐treated with TSA.
Epigenetically silenced genes are therapeutically exploited through re‐expression by epigenetic drugs (Kaiser, 2010; Rius and Lyko, 2012). Besides the use of epigenetic drugs in the treatment of patients with hematological malignancies, ongoing clinical trials for epigenetic drugs indicate beneficial effects for solid tumors as well (Juergens et al., 2011). Impressive results were achieved for lung cancer patients with a combination of epigenetic drugs (Juergens et al., 2011) and currently epigenetic drugs are in clinical trial for many cancers, including cervical cancer (Boumber and Issa, 2011). However, the disadvantages of these drugs include their lack of target specificity: non‐chromatin targets are also affected and although up‐regulation is expected, many genes get down‐regulated (Raynal et al., 2012). Furthermore, many cancers are relatively resistant to epigenetic therapy and adverse effects have been described leading to early study termination as reported for an ovarian cancer study (Matei and Nephew, 2010). The biggest problem with this study was that unwanted genes were also up‐regulated, which made the outcome of the treatment unpredictable and variable between tumors.
In order to achieve gene‐specific re‐expression of TSGs, ATF technology is promising and induction of gene expression has been reported for several endogenous genes, like ERBB2 (Beerli et al., 2000), VEGF‐A (Liu et al., 2001) and Utrophin (Onori et al., 2007). Despite the repressive chromatin environment, ATFs can gain access and re‐express epigenetically silenced genes (Sera, 2009), including the tumor suppressor gene MASPIN in breast (Beltran et al., 2007) and lung cancer cell lines (Beltran and Blancafort, 2011). Here, we extend these findings and show that ATFs can induce C13ORF18 gene expression even in the hypermethylated cell lines HeLa and CaSki. Variations were observed in the effectiveness of the ATFs between the cell lines, which could not be explained by genetic mutations in the ZF binding sites (Supplementary Figure 1). Also, differences in DNA methylation patterns do not underlie the differential effects observed, e.g. 1ab‐VP64 effectively induced expression of C13ORF18 in HeLa (methylated), but not in C33A (unmethylated) or CaSki (methylated). Characteristics of the chromatin, such as DNA methylation or histone modifications, however, are known to affect functionality of ATFs and epigenetic drugs have been reported to synergize with ATFs to enhance the ATF‐induced effects, as shown by us and others (Beltran et al., 2008). Apart from differential binding of ATFs in different cell lines, position effects (resulting in absence of transcriptional effects even though the ZF can access its target site) have been reported for effector domains, including VP64 (Lund et al., 2004). It may very well be that the enhanced effects of ATFs by epigenetic drugs are caused by increased binding of the ATF to its endogenous target sites as indicated by us here. Importantly, the ATF 2ab‐VP64, which had no detectable re‐expression activities in any cell line, was able to re‐activate the C13ORF18 promoter in combination with TSA. Another mechanism by which epigenetic drugs might enhance ATF‐induced effects is by protein acetylation. Acetylation of lysine residues within transcription factors is a mechanism by which cells can overcome gene repression (Bannister and Miska, 2000). However, acetylation of the C‐terminal zinc finger domain of the sequence‐specific DNA‐binding transcription factor YY1 has been shown to result in decreased DNA‐binding of YY1 (Yao et al., 2001). As our results show enhanced binding of ATFs after TSA treatment, increased acetylation of engineered ZFPs – if induced at all by TSA treatment – will most likely not affect binding negatively.
Affinity of ATFs to their target sites not only depends on the local structure of the chromatin, ATFs can have an impact on chromatin as well. As shown by us, specific re‐expression of C13ORF18 by ATFs was associated with site‐specific promoter DNA demethylation. Previously, the demethylating effect of ATFs was also reported for the MASPIN ATFs (Beltran and Blancafort, 2011). As a possible explanation, it was suggested that the demethylation is a result of a passive mechanism consequential to the recruitment of the RNA Pol II transcriptional complex to the MASPIN promoter. However, recent findings show that hypermethylated genes can be re‐expressed to high levels by treatment with HDAC inhibitors, without affecting the DNA methylation status of the promoter (Raynal et al., 2012). In line with this, we also show that loss of DNA methylation by the ATF is independent of the level of gene re‐expression, as co‐treatment with TSA greatly increased the level of re‐expression, but did not further increase the DNA demethylation status.
A direct relationship exists between the levels of DNA methylation and histone methylation and such crosstalk regulates chromatin structure (Cedar and Bergman, 2009). It is therefore likely that the ATF‐induced DNA demethylation is associated with decreased histone methylation as indeed found here and also recently observed in an artificial stable ATF‐system for p16 (Zhang et al., 2012). As some epigenetic changes might be inherited during cell divisions, ATF‐mediated demethylation may therefore even result in more permanent gene re‐expression. Some evidence for this notion is provided by the heritable regulation of AP3 by ATFs in plants over two subsequent generations (Guan et al., 2002). Also the MASPIN ATFs induced prolonged re‐expression of MASPIN for four cell generations in absence of the MASPIN ATF mRNA using Dox‐inducible constructs (Beltran et al., 2011). However, in the MASPIN study the ATF protein levels were not evaluated. For the long term, the induced heritability effects by ATFs are still unknown and might not even be expected as no enzymatic activity is targeted. A recent improvement to the field is the induction of sustained silencing of MASPIN and the oncogene SOX2 in breast cancer (Stolzenburg et al., 2012): ZFPs linked to a DNA methyltransferase induced methylation at the target site, enabling stable phenotypic reprogramming of the cancer cell. Such epigenetic editing approaches could also be designed by targeting activating histone modifiers or DNA demethylases to induce more sustained re‐expression of genes, as reviewed recently (de Groote et al., 2012).
In this study, we found that ATF‐induced levels of C13ORF18 repressed tumor growth. This is the first study to demonstrate tumor suppressive activity of a biomarker gene with unknown function, and shows that hypermethylated marker genes can be attractive targets for therapeutic strategies, although the use of ATFs in a clinical setting still faces some challenges (Eisenstein, 2012), novel delivery systems are being developed allowing for safer administration and higher efficacy (Gaj et al., 2012; Lara et al., 2012). Together, the specificity, biological effect, synergy with epigenetic drugs and “inheritability” effect of ATFs might offer great therapeutic potential for the treatment of cancer. As the amount of epimutations in cancer exceeds the number of gene mutations, ATFs provide promising tools to validate hypermethylated marker genes as therapeutic targets.
Conflict of interest
The authors of this manuscript have nothing to declare.
Support/grants
This work was supported by NWO/VIDI 91786373 (to M.G.R.).
Supporting information
Supplementary data
Acknowledgments
We thank Dr. Pilar Blancafort for the pMX‐IRES‐GFP vector and Dr. Saravanan Yuvaraj for his technical assistance with the retroviral transductions. Furthermore, we thank Jelleke Dokter for assistance with cell culturing.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.02.017.
Huisman Christian, Wisman G. Bea A., Kazemier Hinke G., van Vugt Marcel A.T.M., van der Zee Ate G.J., Schuuring Ed, Rots Marianne G., (2013), Functional validation of putative tumor suppressor gene C13ORF18 in cervical cancer by Artificial Transcription Factors, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.02.017.
Contributor Information
Christian Huisman, Email: c.huisman@umcg.nl.
G. Bea A. Wisman, Email: g.b.a.wisman@umcg.nl
Hinke G. Kazemier, Email: h.g.kazemier@umcg.nl
Ate G.J. van der Zee, Email: a.g.j.van.der.zee@umcg.nl
Ed Schuuring, Email: e.schuuring@umcg.nl.
Marianne G. Rots, Email: m.g.rots@umcg.nl
References
- Bannister, A.J. , Miska, E.A. , 2000. Regulation of gene expression by transcription factor acetylation. Cell. Mol. Life Sci. 57, 1184–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin, S.B. , Jones, P.A. , 2011. A decade of exploring the cancer epigenome – biological and translational implications. Nat. Rev. Cancer 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli, R.R. , Dreier, B. , Barbas, C.F. , 2000. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. U.S.A 97, 1495–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends, C. , Sowa, M.E. , Gygi, S.P. , Harper, J.W. , 2010. Network organization of the human autophagy system. Nature 466, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran, A. , Parikh, S. , Liu, Y. , Cuevas, B.D. , Johnson, G.L. , Futscher, B.W. , Blancafort, P. , 2007. Re-activation of a dormant tumor suppressor gene maspin by designed transcription factors. Oncogene 26, 2791–2798. [DOI] [PubMed] [Google Scholar]
- Beltran, A.S. , Blancafort, P. , 2011. Reactivation of MASPIN in non-small cell lung carcinoma (NSCLC) cells by artificial transcription factors (ATFs). Epigenetics 6, 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran, A.S. , Russo, A. , Lara, H. , Fan, C. , Lizardi, P.M. , Blancafort, P. , 2011. Suppression of breast tumor growth and metastasis by an engineered transcription factor. PLoS One 6, e24595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran, A.S. , Sun, X. , Lizardi, P.M. , Blancafort, P. , 2008. Reprogramming epigenetic silencing: artificial transcription factors synergize with chromatin remodeling drugs to reactivate the tumor suppressor mammary serine protease inhibitor. Mol. Cancer. Ther. 7, 1080–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumber, Y. , Issa, J.P. , 2011. Epigenetics in cancer: what's the future?. Oncol. (Williston Park) 25, 220–226. 228 [PubMed] [Google Scholar]
- Cairns, P. , Esteller, M. , Herman, J.G. , 2001. Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin. Cancer Res. 7, 2727–2730. [PubMed] [Google Scholar]
- Cedar, H. , Bergman, Y. , 2009. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. [DOI] [PubMed] [Google Scholar]
- de Groote, M.L. , Verschure, P.J. , Rots, M.G. , 2012. Epigenetic editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res. 40, 10596–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edel, M.J. , Menchon, C. , Vaquero, J.M. , Izpisua Belmonte, J.C. , 2011. A protocol to assess cell cycle and apoptosis in human and mouse pluripotent cells. Cell. Commun. Signal 9, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijsink, J.J. , Lendvai, A. , Deregowski, V. , 2012. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int. J. Cancer 130, 1861–1869. [DOI] [PubMed] [Google Scholar]
- Eisenstein, M. , 2012. Sangamo's lead zinc-finger therapy flops in diabetic neuropathy. Nat. Biotechnol. 30, 121–123. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , 2007. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 8, 286–298. [DOI] [PubMed] [Google Scholar]
- Fukazawa, T. , Matsuoka, J. , Yamatsuji, T. , Maeda, Y. , Durbin, M.L. , Naomoto, Y. , 2010. Adenovirus-mediated cancer gene therapy and virotherapy (Review). Int. J. Mol. Med. 25, 3–10. [PubMed] [Google Scholar]
- Gaj, T. , Guo, J. , Kato, Y. , Sirk, S.J. , Barbas, C.F. , 2012. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat. Meth. 9, 805–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glockner, S.C. , Dhir, M. , Yi, J.M. , 2009. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res. 69, 4691–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommans, W.M. , McLaughlin, P.M. , Lindhout, B.I. , Segal, D.J. , Wiegman, D.J. , Haisma, H.J. , van der Zaal, B.J. , Rots, M.G. , 2007. Engineering zinc finger protein transcription factors to downregulate the epithelial glycoprotein-2 promoter as a novel anti-cancer treatment. Mol. Carcinog. 46, 391–401. [DOI] [PubMed] [Google Scholar]
- Guan, X. , Stege, J. , Kim, M. , Dahmani, Z. , Fan, N. , Heifetz, P. , Barbas, C.F. , Briggs, S.P. , 2002. Heritable endogenous gene regulation in plants with designed polydactyl zinc finger transcription factors. Proc. Natl. Acad. Sci. U.S.A 99, 13296–13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada, K. , Alemany, R. , Zhang, W.W. , Hittelman, W.N. , Lotan, R. , Roth, J.A. , Mitchell, M.F. , 1996. Adenovirus-mediated transfer of a wild-type p53 gene and induction of apoptosis in cervical cancer. Cancer Res. 56, 3047–3054. [PubMed] [Google Scholar]
- Holbro, T. , Beerli, R.R. , Maurer, F. , Koziczak, M. , Barbas, C.F. , Hynes, N.E. , 2003. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. U.S.A 100, 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque, M.O. , Kim, M.S. , Ostrow, K.L. , 2008. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 68, 2661–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johung, K. , Goodwin, E.C. , DiMaio, D. , 2007. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J. Virol. 81, 2102–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juergens, R.A. , Wrangle, J. , Vendetti, F.P. , 2011. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 1, 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser, J. , 2010. Epigenetic drugs take on cancer. Science 330, 576–578. [DOI] [PubMed] [Google Scholar]
- Kelly, T.K. , De Carvalho, D.D. , Jones, P.A. , 2010. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 28, 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara, H. , Wang, Y. , Beltran, A.S. , 2012. Targeting serous epithelial ovarian cancer with designer zinc finger transcription factors. J. Biol. Chem. 287, 29873–29886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P.Q. , Rebar, E.J. , Zhang, L. , 2001. Regulation of an endogenous locus using a panel of designed zinc finger proteins targeted to accessible chromatin regions. Activation of vascular endothelial growth factor A. J. Biol. Chem. 276, 11323–11334. [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Zhang, X. , Zhang, J. , 2012. Inhibition of breast tumor cell growth by ectopic expression of p16/INK4A via combined effects of cell cycle arrest, senescence and apoptotic induction, and angiogenesis inhibition. J. Cancer 3, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, C.V. , Blancafort, P. , Popkov, M. , Barbas, C.F. , 2004. Promoter-targeted phage display selections with preassembled synthetic zinc finger libraries for endogenous gene regulation. J. Mol. Biol. 340, 599–613. [DOI] [PubMed] [Google Scholar]
- Magnenat, L. , Blancafort, P. , Barbas, C.F. , 2004. In vivo selection of combinatorial libraries and designed affinity maturation of polydactyl zinc finger transcription factors for ICAM-1 provides new insights into gene regulation. J. Mol. Biol. 341, 635–649. [DOI] [PubMed] [Google Scholar]
- Mandell, J.G. , Barbas, C.F. , 2006. Zinc finger tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 34, W516–W523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matei, D.E. , Nephew, K.P. , 2010. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 116, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongenaert, M. , Wisman, G.B. , Volders, H.H. , Koning, A.J. , Zee, A.G. , van Criekinge, W. , Schuuring, E. , 2008. Discovery of DNA methylation markers in cervical cancer using relaxation ranking. BMC Med. Genomics 1, 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onori, A. , Desantis, A. , Buontempo, S. , Di Certo, M.G. , Fanciulli, M. , Salvatori, L. , Passananti, C. , Corbi, N. , 2007. The artificial 4-zinc-finger protein Bagly binds human utrophin promoter A at the endogenous chromosomal site and activates transcription. Biochem. Cell. Biol. 85, 358–365. [DOI] [PubMed] [Google Scholar]
- Puigdecanet, E. , Espinet, B. , Lozano, J.J. , 2008. Gene expression profiling distinguishes JAK2V617F-negative from JAK2V617F-positive patients in essential thrombocythemia. Leukemia 22, 1368–1376. [DOI] [PubMed] [Google Scholar]
- Raynal, N.J. , Si, J. , Taby, R.F. , Gharibyan, V. , Ahmed, S. , Jelinek, J. , Estecio, M.R. , Issa, J.P. , 2012. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 72, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius, M. , Lyko, F. , 2012. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene 31, 4257–4265. [DOI] [PubMed] [Google Scholar]
- Roossink, F. , de Jong, S. , Wisman, G.B. , van der Zee, A.G. , Schuuring, E. , 2012. DNA hypermethylation biomarkers to predict response to cisplatin treatment, radiotherapy or chemoradiation: the present state of art. Cell. Oncol. (Dordr) 35, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sera, T. , 2009. Zinc-finger-based artificial transcription factors and their applications. Adv. Drug Deliv. Rev. 61, 513–526. [DOI] [PubMed] [Google Scholar]
- Snowden, A.W. , Zhang, L. , Urnov, F. , 2003. Repression of vascular endothelial growth factor A in glioblastoma cells using engineered zinc finger transcription factors. Cancer Res. 63, 8968–8976. [PubMed] [Google Scholar]
- Stolzenburg, S. , Rots, M.G. , Beltran, A.S. , Rivenbark, A.G. , Yuan, X. , Qian, H. , Strahl, B.D. , Blancafort, P. , 2012. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 40, 6725–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uil, T.G. , Haisma, H.J. , Rots, M.G. , 2003. Therapeutic modulation of endogenous gene function by agents with designed DNA-sequence specificities. Nucleic Acids Res. 31, 6064–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen, N. , von Knebel Doeberitz, M. , 2007. Biomarkers in cervical cancer screening. Dis. Markers 23, 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Eijsink, J.J. , Lendvai, A. , Volders, H.H. , Klip, H. , Buikema, H.J. , van Hemel, B.M. , Schuuring, E. , van der Zee, A.G. , Wisman, G.B. , 2009. Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer Epidemiol. Biomarkers Prev. 18, 3000–3007. [DOI] [PubMed] [Google Scholar]
- Yao, Y.L. , Yang, W.M. , Seto, E. , 2001. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol. 21, 5979–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Zeng, P. , Xiong, J. , Liu, Z. , Berger, S.L. , Merlino, G. , 2010. Epigenetic drugs can stimulate metastasis through enhanced expression of the pro-metastatic Ezrin gene. PLoS One 5, e12710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Xiang, S. , Zhong, Q. , Yin, Y. , Gu, L. , Deng, D. , 2012. The p16-specific reactivation and inhibition of cell migration through demethylation of CpG islands by engineered transcription factors. Hum. Gene Ther. 23, 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data