

Abstract
Despite intensifying multimodal treatments, children with central nervous system atypical teratoid/rhabdoid tumor (CNS ATRT) continue to endure unacceptably high mortality rates. At present, concerted efforts are focusing on understanding the characteristic INI1 mutation and its implications for the growth and survival of these tumors. Additionally, pharmaceutical pipeline libraries constitute a significant source of potential agents that can be taken to clinical trials in a timely manner. However, this process requires efficient target validation and relevant preclinical studies. As an initial screening approach, a panel of 129 small molecule inhibitors from multiple pharmaceutical pipeline libraries was tested against three ATRT cell lines by in vitro cytotoxicity assays. Based on these data, agents that have strong activity and corresponding susceptible cellular pathways were identified. Target modulation, antibody array analysis, drug combination and in vivo xenograft studies were performed on one of the pathway inhibitors found in this screening. Approximately 20% of agents in the library showed activity with IC50 values of 1 μM or less and many showed IC50 values less than 0.05 μM. Intra cell line variability was also noted among some of the drugs. However, it was determined that agents capable of affecting pathways constituting ErbB2, mTOR, proteasomes, Hsp90, Polo like kinases and Aurora kinases were universally effective against the three ATRT cell lines. The first target selected for further analysis, the inhibition of ErbB2‐EGFR pathway by the small molecule inhibitor lapatinib, indicated inhibition of cell migration properties and the initiation of apoptosis. Synergy between lapatinib and IGF‐IR inhibition was also demonstrated by combination index (CI) values. Xenograft studies showed effective antitumor activity of lapatinib in vivo. We present an experimental approach to identifying agents and drug combinations for future clinical trials and provide evidence for the potential of lapatinib as an effective agent in the context of the biology and heterogeneity of its targets in ATRT.
Keywords: Lapatinib, Teratoid Rhabdoid tumors, Targeted therapeutics
Highlights
-
►
A rationale for novel therapeutic approaches for the treatment of CNS ATRT is described.
-
►
Three representative cell lines have been used.
-
►
In depth analysis of lapatinib with target modulation and target validation assays.
1. Introduction
Primary CNS ATRT is a malignant embryonal tumor that commonly affects infants and very young children (Rorke et al., 1996). There are infrequent cases of long‐term survivors described in the literature following treatment with intensive multimodal therapy (Reddy, 2005). However, presently no standard or generally effective treatment protocols exist for the treatment of these children.
ATRT cells are distinguished by alterations of the INI1 tumor‐suppressor gene located on chromosome band 22q11.2 (Bikowska et al., 2011). Mechanistically, INI1/hSNF5 is a component of the ATP‐dependent chromatin remodeling SWI/SNF complex and shown to mediate cell cycle arrest due to the direct recruitment of HDAC activity to the cyclin D1 promoter, leading to its repression and subsequent G0‐G1 arrest (Fujisawa et al., 2005; Zhang et al., 2002). Currently, however, the pathways by which this molecular abnormality leads to the aggressive growth phenotype are not completely understood. Recent literature suggests that INI1 is capable of interacting with key signaling molecules and modifying processes such as cell cycle progression and growth factor response. For example, the interaction between the key signal transducer Akt and members of the hSWI/SNF chromatin remodeling complex leading to Akt activation has been demonstrated (Foster et al., 2006). A number of studies have also investigated specific cytokine driven growth regulatory pathways in ATRT cells. These include the growth dependency on IGF‐I and IGF‐II and the inhibition of these cytokines by small molecule inhibitors or antisense oligonucleotides (D'Cunja et al., 2007; Narendran et al., 2008; Ogino et al., 1999, 2001). Data from Foster and colleagues have shown the dependency of these cells on Akt activation, which may occur through aberrant stimulation of the IGF‐IR pathway (Foster et al., 2009). Similarly, autocrine signaling by insulin, via the PI3K/Akt pathway, leading to increased growth and survival of ATRT cell lines has also been demonstrated (Arcaro et al., 2007). These studies indicate that mechanistic associations exist between the distinctive genetic abnormalities of ATRT and altered sensitivity to specific growth factor mediated signaling processes. Hence, directed interference of these pathways provides unique opportunities to discover effective targets for future therapeutics.
In the recent past, efforts have intensified to identify molecular mechanisms that regulate ATRT cell growth and to detect targets for novel therapeutics. For example, supported by the previous finding that Cyclin D1 is a key target of INI1, Smith and colleagues have shown that the derivatives of fenretinide have the ability to down‐modulate Cyclin D1, inducing cytotoxicty in rhabdoid cell lines (Smith et al., 2011). Similarly, Knipstein and co‐workers demonstrated the utility of histone deacetylase inhibitors (HDI) to induce radiosensitization and apoptosis in ATRT cells (Knipstein et al., 2012). Our previous studies have provided evidence for an effective drug combination consisting of multi‐tyrosine kinase inhibitors with irinotecan (Jayanthan et al., 2011). Recently, the generation of genetically engineered INI1 +/− mice that spontaneously develop tumors, including CNS lesions, has provided valuable means to test new therapeutic agents and drug resistance mechanisms in ATRT (Guidi et al., 2001). The Pediatric Oncology Experimental Therapeutics Investigators' Consortium (POETIC) is currently evaluating libraries of pipeline agents to identify drugs that hold promise in future clinical trials for currently difficult to treat pediatric malignancies. This report describes the initial screening of such a drug panel against cell lines established from ATRT patients and the identification of agents that are able to induce cytotoxicity at sub‐micromolar concentrations. More detailed analysis of one of these agents, lapatinib, provides information on target modulation and evidence for effective drug synergy with IGF‐IR inhibition. Lapatinib also showed in vivo activity in a xenograft model of ATRT, validating an approach to develop future clinical studies in the treatment for ATRT.
2. Materials and methods
2.1. Cell lines and cell culture
BT12 and BT16 cell lines were established from infants with CNS ATRT and generously provided by Drs. Peter Houghton and Jaclyn Biegel (Nationwide Children's Hospital, Columbus, Ohio and The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania respectively). These cell lines have been used extensively in preclinical studies in ATRT. The cell line KCCF1 was established in our laboratory from the cerebral spinal fluid (CSF) cells of a two‐month‐old male infant with ATRT. Characterization of this cell line has been described previously (Jayanthan et al., 2011). The Hs68 primary skin fibroblast cells were provided by the Sung‐Woo Kim laboratory (University of Calgary) and the EGFR over‐expressing glioblastoma multiforme (GBM) cell line T98G was a gift from the laboratory of Dr. Greg Cairncross (University of Calgary). These cell lines were cultured in Opti‐MEM medium (Gibco, Invitrogen Corporation, Burlington, Ontario) containing 5% FBS (Fetal Bovine Serum), 100 units/ml penicillin and 100 units/ml streptomycin (Gibco). Cells were trypsinized with 0.25% Trypsin‐EDTA in Ca2+ and Mg2+ free balanced salt solution (Gibco) every three to five days. All cell cultures were maintained in incubators at 37 °C in a humidified atmosphere with 5% CO2.
2.2. Antineoplasic agents
All targeted therapeutic agents used in the screening analysis were synthesized, purity checked and provided by Chemi‐etek (Minneapolis, MN). Lapatinib was kindly provided by GlaxoSmithKline (Collegeville, PA). These agents were dissolved in DMSO to a final concentration of 10 mM and stored frozen at −20 °C and diluted appropriately in culture medium at the time of study.
2.3. Cell growth inhibition assays
ATRT cells were trypsinized and placed in 96 well plates (Grenier Bio One, Monroe, NC) at a concentration of 5 × 103 cells per well. Increasing concentrations of study agents were added to these wells to a final volume of 200 μl per well. Corresponding dilutions of the vehicle DMSO was used as control. After four days in culture, cell survival was quantified by automated cytometer (Celigo, Cyntellect Inc., San Diego, CA, USA), according to the manufacturer's protocol (Nabzdyk et al., 2011). The half maximal inhibitory concentration (IC50) values were calculated for each agent based on individual cytotoxicity plots. For drug combination studies involving lapatinib, IC25 concentration of lapatinib (i.e., the amount that induced 25% cell death by itself) was added to cultures containing increasing concentrations of the second agent. The new IC50 values corresponding to the combination were then calculated and used to derive combination index (CI) values as described previously (Chou, 2010). A CI of less than 1 indicates synergy between the two agents under the experimental conditions used.
2.4. Intracellular signaling studies
ATRT cells were grown to approximately two‐third confluence in six well culture plates (Nunc, Rochester, NY) and the culture medium was replaced with fresh medium containing lapatinib or drug combinations as indicated in individual experiments. After incubation for 12 h, the cells were washed with ice cold PBS and lysed in buffer containing 50 mM Tris, 5 mM EDTA, 0.1% SDS, 1% Triton X‐100, 0.5% sodium deoxycholate with phosphatase and protease inhibitors (Sigma, Oakville, ON). Protein concentrations of the lysates were quantified by BCA Protein Assay (Pierce, Rockford, IL, USA). Proteins were then separated on a 10% polyacrylamide gel electrophoresis and transferred onto nitrocellulose (NC) membranes (Bio‐Rad, Mississauga, ON). The membranes were blocked for 2 h at 4 °C with 5% skim milk powder in PBS containing 0.1% Tween‐20 (Sigma). The blots were incubated with primary antibodies (Cell Signaling Technology, Danvers, MA) overnight at 4 °C, washed and probed with appropriate secondary antibodies conjugated to horseradish peroxidase (HRPO) (Sigma), followed by a luminol based substrate (Mandel, Guelph, ON) and developed by exposure to x‐ray film (Christie InnoMed, Montreal, QC).
2.5. In vitro cell migration assay ("scratch" test)
The scratch test to quantify inhibition in cell migration was performed as described previously (Liang et al., 2007). ATRT cells were grown to confluence in six well culture plates (Nunc). On the day of the assay, the cell monolayer was scraped in a straight line with a 10 μl pipette tip and the culture medium was replaced with 3 ml of new medium or medium containing varying concentrations of lapatinib (0.001–1 μM). Pictures of the scratch at the same spot of all plates were taken at various time points (at 0 h, 8 h and 24 h) using an inverted microscope.
2.6. Tumor xenograft studies
2.6.1. Generation of BT16 cells stably expressing firefly luciferase and eGFP
BT16 cell line expressing enhanced firefly luciferase (effLuc)(Rabinovich et al., 2008) and eGFP were generated (BT16GFPFluc) using a self‐inactivating lentiviral vector encoding the internal U3 region from mscv, effLuc, the IRES element from emcv, and eGFP (Bai et al., 2011). Virus was packaged in 293‐FT cells using pMD2.G (VSV.G env) and pCMV‐deltaR8.91 and concentrated 50 times using Amicon Ultra‐15 100,000 NMWL centrifugal concentration units (Millipore, Billerica, MA). Concentrated viral supernatants were used for transduction in the BT16 cell line. After 72 h, eGFP expression was observed via fluorescent microscopy (Zeiss inverted microscope, Axiovert 200 M) and used to calculate transduction efficiency by flow cytometry on a FACS Calibur instrument (BD Biosciences, San Jose, CA). The effLuc based bioluminescent activity was calculated using an IVIS 200 (Caliper Life Sciences, Alameda, CA).
2.6.2. In vivo real‐time monitoring of tumor growth (bioluminescence imaging)
Six to eight week‐old female CD‐1 mice from Charles River Laboratories were used in this study. All protocols were reviewed and approved by the Animal Care Committee of the University of Calgary. All animal work procedures were in accordance with the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals issued by NIH.
Two groups of randomly assigned CD‐1 Nude mice (n = 5 per group, total 10 animals) were implanted in the flank with 5 × 106 BT16GFPLuc cells. After allowing approximately 2 weeks for the tumors to establish, tumor‐bearing animals were randomly divided into two groups for control vehicle treatment and for lapatinib treatment, which was given as a twice‐daily oral administration for 3 weeks (5‐days on, 2‐days off) at a dose of 160 mg/kg (320 mg/kg/day). Xenogen IVIS 200 system (Xenogen Corporation, Alameda, CA) was used to monitor the animal tumor growth in vivo. Every week after tumor implantation all mice were imaged to record bioluminescent signal emitted from tumors. Anesthesia was given in an induction chamber with 2.5% isoflurane in 100% oxygen at a flow rate of 1 L/min and maintained in the IVIS with a 1.5% mixture at 0.5 L/min. The mice were then injected with d‐luciferin (126 mg/kg, Xenogen Corp.) dissolved in PBS (15 mg/ml) by intraperitoneal administration. Subsequently, mice were placed in prone position in the IVIS instrument and bioluminescent acquisitions were collected until the maximum signal was reached. Data were analyzed based on total photon flux emission (photons/s) in the region of interest (ROI) over the tumor (Lun et al., 2010).
3. Results
3.1. Cytotoxicity profiling of a panel of novel therapeutic agents against ATRT cells
A panel of targeted small molecular weight inhibitors (n = 129) was evaluated against three ATRT cell lines using in vitro cytotoxicity assays. These agents were selected based on their known activities in other tumor cell systems and their potential to be used in human clinical trials. Table 1 provides the IC50 values obtained in cytotoxicity studies as described in Methods. Data provided in this table show a wide range of drug sensitivity values across the three cell lines. There were 16 agents that showed IC50 values of 0.1 μM or less in at least two out of the three cell lines. Of these, eight showed such activity in all three cell lines. Specifically, seven agents, INK‐128, carfilzomib, NVP‐AUY922, AV951, BI6727, AZD1152 and YM155, showed significant activity with IC50 values less than 0.05 μM in at least two out of the three cell lines, however carfilzomib, NVP‐AUY922 and BI6727 showed this activity in all three cell lines. Figure 1A is a diagrammatic representation of these data showing the distribution of IC50 values across all 129 agents for individual cell lines. To categorize the agents with relatively high cytotoxicity values, those with a median IC50 values less than the arbitrarily defined value of 1 μM, were selected and analyzed for cytotoxicity in an expanded range of drug dilutions (10–10−5 μM). Data obtained from these studies are summarized in Figure 1B. In this figure, for each drug treatment the vertical lines represent the lowest, median and highest values and the horizontal lines represent the range of the IC50 values. The identities of these agents are given along the Y axis of the graph presented. Agents inhibiting a multitude of targets are represented in this group with the proteasome inhibitor carfilzomib showing the lowest IC50 values. This figure also shows that certain agents such as bortezomib and NVP‐AUY922 are equally effective against all three cell lines, while others such as AZD‐4547 and BIBW 2992 carry differential sensitivity among the cell lines tested. These data have provided us with a short list of agents for further evaluation and more extensive pre‐clinical studies. Among the agents presented in Figure 1A, two of the molecules, lapatinib and CUDC‐101 are targeted inhibitors of HER2 and EGFR kinases. Based on currently available data demonstrating efficacy, tolerability and the potential for CNS penetration by lapatinib in adult clinical trials, we have selected this agent as the first in a series of additional studies for preclinical development.
Table 1.
Cytotoxicity profiling of a panel of targeted agents against three ATRT cell lines. Exponentially growing tumor cells were treated with four different concentrations of individual drugs (10 μM, 1 μM, 0.1 μM and 0.01 μM) in duplicate cultures. After four days in culture, cell growth inhibition was quantified by automated cytometer and percentage growth inhibition was calculated in comparison to vehicle‐only untreated control cells. The IC50 values are from a single complete study and trends observed are representative of three separate experiments.
| Mechanism of action | Number | Inhibitor | Cell line | ||
|---|---|---|---|---|---|
| BT12 | BT16 | KCCF1 | |||
| HER2/EGF‐R inhibition | 1 | Lapatinib | 5 | 0.1 | 0.1 |
| 2 | Canertinib (CI‐1033) | 10 | 0.1 | 0.1 | |
| 3 | BIBW 2992 (Tovok) | 5 | 0.01 | 0.01 | |
| 4 | BMS‐599626 | >10 | 1 | 1 | |
| 5 | Gefitinib (Iressa) | 10 | 0.5 | 1 | |
| 6 | Erlotinib, Hydrochloride | >10 | >10 | >10 | |
| 7 | CUDC‐101 | 0.08 | 0.7 | 0.9 | |
| 8 | WZ4002 | >10 | >10 | >10 | |
| mTOR inhibition | 9 | Pp242 | 3 | 10 | 1 |
| 10 | Rapamycin (Sirolimus) | >10 | 0.01 | 0.1 | |
| 11 | AZD8055 | 0.09 | 0.03 | 0.05 | |
| 12 | INK128 | 0.05 | 0.04 | 0.07 | |
| 13 | OSI‐027 | 1 | 0.5 | 7 | |
| 14 | PF‐04691502 | 0.7 | 1 | 0.8 | |
| IGF‐IR inhibition | 15 | OSI‐906 | 8 | 10 | 8 |
| 16 | BMS‐754807 | 10 | >10 | >10 | |
| HDAC inhibition | 17 | LBH‐589 (Panobinostat) | 0.1 | >10 | 0.08 |
| 18 | MS‐275 | >10 | >10 | 10 | |
| 19 | MGCD0103 | >10 | >10 | 10 | |
| 20 | Vorinostat (SAHA) | 8 | >10 | 10 | |
| 21 | Belinostat (PXD101) | 0.1 | 0.3 | 0.7 | |
| 22 | Tubacin | 1 | >10 | 8 | |
| 23 | Tubastatin A | 6 | >10 | >10 | |
| Akt inhibition | 24 | MK‐2206 | 8 | 10 | 8 |
| 25 | GSK690693 | >10 | 10 | >10 | |
| CRTH2‐R antagonist | 26 | Ramatroban (Bay u3405) | >10 | >10 | >10 |
| 27 | TM30089 | >10 | >10 | >10 | |
| c‐MET inhibition | 28 | PF‐2341066 | 7 | 8 | 8 |
| 29 | ARQ 197 (Tivantinib) | 6 | 10 | 10 | |
| 30 | BMS‐777607 | 7 | 6 | 10 | |
| 31 | EMD1214063 | 7 | 5.5 | >10 | |
| 32 | PF‐04217903 | >10 | >10 | >10 | |
| Topoisomerase inhibition | 33 | SN‐38 | 1 | 1 | 10 |
| 34 | Topotecan (Hycamtin) | 0.5 | 1 | >10 | |
| 35 | Etoposide | >10 | >10 | >10 | |
| NA Synthesis inhibition | 36 | Capecitabine (Xeloda) | >10 | >10 | >10 |
| 37 | Pemetrexed Disodium (Alimta) | >10 | >10 | >10 | |
| 38 | Gemcitabine, HCl (Gemzar) | 0.6 | 5 | 10 | |
| 39 | Doxorubicin (Adriamycin) | 0.05 | 0.1 | 0.5 | |
| Adenosine receptor antagonist | 40 | CVT‐6883 | >10 | >10 | >10 |
| 41 | CGS 21680 | >10 | >10 | >10 | |
| Proteasome inhibition | 42 | Bortezomib (Velcade) | 0.01 | 0.01 | 0.1 |
| 43 | Carfilzomib (PR‐171) | <0.01 | <0.01 | <0.01 | |
| HSP90 inhibition | 44 | 17‐DMAG | 0.01 | 5 | 5 |
| 45 | 17‐AAG | 0.01 | 0.01 | >10 | |
| 46 | NVP‐AUY922 (VER‐52296) | <0.01 | 0.02 | 0.03 | |
| JAK inhibition | 47 | CP‐690550 | >10 | >10 | >10 |
| 48 | AZD1480 | 0.6 | 0.1 | 2 | |
| 49 | CYT‐387 | 7 | 0.8 | >10 | |
| 50 | INCB018424 (Ruxolitinib) | >10 | >10 | >10 | |
| 51 | TG101348 | 1 | 1 | 0.8 | |
| Raf inhibition | 52 | PLX4032 (RG7204) | >10 | >10 | >10 |
| 53 | PLX4720 | 8 | 7 | 7 | |
| Tubulin stabilization | 54 | Docetaxel | >10 | >10 | >10 |
| 55 | Paclitaxel (Taxol) | >10 | >10 | >10 | |
| PI3K inhibition | 56 | TGX‐221 | >10 | >10 | >10 |
| 57 | PIK‐75, Hydrochloride | 0.07 | >10 | 0.1 | |
| 58 | GDC‐0941 | >10 | 1 | 5 | |
| 59 | CAL‐101 | >10 | >10 | >10 | |
| 60 | LY294002 | 6 | 9 | 7.5 | |
| 61 | TG100‐115 | >10 | >10 | >10 | |
| 62 | XL‐147 | >10 | >10 | 10 | |
| MEK inhibition | 63 | PD 0325901 | 1 | 0.1 | >10 |
| 64 | RDEA119 | 10 | 1 | 1 | |
| 65 | AZD 6244 (ARRY‐142886) | >10 | 10 | 10 | |
| 66 | ARRY‐162 | 0.01 | 5 | 5 | |
| 67 | GSK1120212 | 7 | >10 | <0.01 | |
| 68 | AS703026 (MSC 1936369B) | >10 | >10 | <0.01 | |
| Bcl‐2 inhibition | 69 | ABT‐263 | 8 | 10 | 8 |
| 70 | ABT‐737 | >10 | >10 | >10 | |
| VEGF‐R/PDGF‐R inhibition | 71 | Axitinib (AG‐013736) | 10 | >10 | >10 |
| 72 | Vandetanib (Zactima) | 7 | 7 | 5 | |
| 73 | Vatalanib Dihydrochloride | >10 | >10 | >10 | |
| 74 | Motesanib (AMG‐706) | >10 | >10 | >10 | |
| 75 | Sorafenib | 6 | 5 | 5 | |
| 76 | Sunitinib | 6 | 10 | 10 | |
| 77 | Tandutinib | >10 | >10 | >10 | |
| 78 | AV‐951 (Tivozanib) | 0.01 | 0.01 | >10 | |
| 79 | Pazopanib (Votrient) | >10 | >10 | >10 | |
| 80 | XL‐184 (Cabozantinib) | 6 | 8 | 5 | |
| Src inhibition | 81 | AZD05030 (Saracatinib) | 10 | 1 | 1 |
| 82 | Dasatinib | 4 | 1 | 1 | |
| Bcr‐Abl inhibition | 83 | Bosutinib (SKI‐606) | 6 | 1 | 1 |
| 84 | Nilotinib | >10 | >10 | >10 | |
| 85 | Imatinib | >10 | >10 | >10 | |
| 86 | Ponatinib (AP24534) | 0.5 | 1 | 5 | |
| Immunosuppression | 87 | FTY720, Hydrochloride | 5 | 5 | 0.6 |
| 88 | FK‐506 | >10 | >10 | >10 | |
| PARP inhibition | 89 | ABT‐888 (Veliparib) | >10 | >10 | >10 |
| 90 | AZD 2281 (Olaparib) | >10 | >10 | >10 | |
| 91 | AG014699 (PF‐01367338) | >10 | >10 | >10 | |
| 92 | BSI‐201 (Iniparib) | 9 | 9 | >10 | |
| 93 | MK‐4827 | 5 | 6 | 0.8 | |
| PLK inhibition | 94 | BI 2536 | 5 | 10 | 5 |
| 95 | BI6727 (Volasertib) | <0.01 | <0.01 | <0.01 | |
| 96 | GSK461364 | 0.6 | 0.7 | 0.6 | |
| Aurora kinase inhibition | 97 | ZM 447439 | >10 | >10 | >10 |
| 98 | VX‐680 | 9 | 8 | 10 | |
| 99 | AZD1152 | <0.01 | 0.1 | <0.01 | |
| FGFR inhibition | 100 | AZD4547 | 5 | 6 | 7.5 |
| Hedgehog pathway inhibition | 101 | GDC‐0449 | >10 | >10 | 10 |
| Androgen receptor inhibition | 102 | Bicalutamide (Casodex) | >10 | >10 | >10 |
| PGD2‐R antagonist | 103 | Laropiprant | >10 | >10 | >10 |
| HIV integrase inhibition | 104 | Raltegravir | >10 | >10 | >10 |
| Cathepsin inhibition | 105 | Odanacatib (MK‐0822) | >10 | >10 | >10 |
| NPM‐ALK inhibition | 106 | NVP‐TAE684 | 0.7 | 0.5 | 0.05 |
| p38 MAPK inhibition | 107 | VX702 | >10 | >10 | 1 |
| CCR5 antagonist | 108 | Maraviroc (UK‐427857) | >10 | >10 | >10 |
| PDE4 inhibition | 109 | AN2728 | >10 | >10 | >10 |
| RXR activation | 110 | Bexarotene (Targretin) | >10 | >10 | >10 |
| T cell activation inhibition | 111 | Hypothemycin | 6 | >10 | 10 |
| Antihistamine | 112 | Dimebolin Hydrochloride | >10 | >10 | >10 |
| LTR antagonist | 113 | Montelukast Sodium (Singulair) | >10 | >10 | >10 |
| COX‐2 inhibition | 114 | Rofecoxib (Vioxx) | >10 | >10 | >10 |
| p90 RSK inhibition | 115 | BI‐D1870 | >10 | >10 | >10 |
| AMPK activation | 116 | A‐769662 | >10 | 0.01 | 0.1 |
| FLT3 inhibition | 117 | AC220 (Quizartinib) | >10 | >10 | >10 |
| HMG‐CoA reductase inhibition | 118 | Atorvastatin Calcium (Lipitor) | >10 | >10 | >10 |
| 5α‐reductase inhibitor | 119 | Dutasteride (Avodart) | >10 | >10 | >10 |
| Angiotensin receptor inhibition | 120 | Eprosartan Mesylate (Teveten) | >10 | >10 | >10 |
| Immunomodulation | 121 | Lenalidomide (CC‐5013) | >10 | >10 | >10 |
| SMO inhibition | 122 | NVP‐LDE225 | >10 | >10 | >10 |
| CDK inhibition | 123 | PD‐0332991 | 0.1 | 10 | 0.6 |
| Multi‐kinase inhibition | 124 | Regorafenib (BAY 73–4506) | 6.5 | 10 | 5 |
| AHR antagonist | 125 | SR1 | 7.5 | >10 | 6 |
| Phospholipase A2 inhibition | 126 | Varespladib (LY315920) | >10 | >10 | >10 |
| ICE/Caspase‐1 inhibition | 127 | VX‐765 | >10 | >10 | >10 |
| Protease inhibition | 128 | VX‐950 (Telaprevir) | >10 | >10 | >10 |
| Survivin suppression | 129 | YM155 | 0.01 | 10 | <0.01 |
Figure 1.
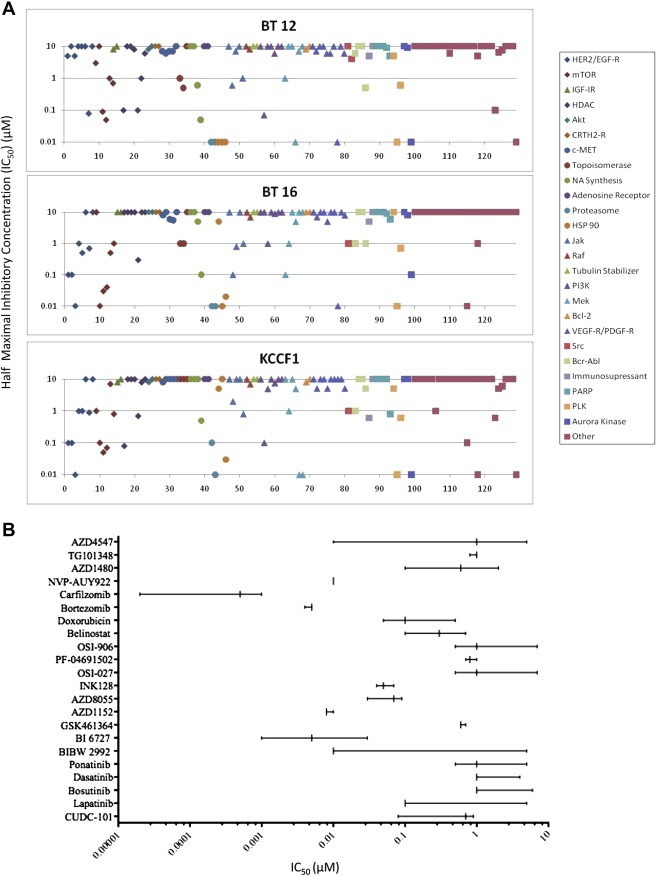
Differential sensitivity of ATRT cell lines against the drug panel. A: A schematic representation of the range of IC50 values presented in Table 1 to show that sensitivities differ greatly among the various agents tested. The top horizontal line indicates agents with IC50 values of 10 μM or higher (low sensitivity) and the lowest line indicates agents that showed IC50 of 0.01 μM or less (high sensitivity). B: Expanded studies of agents with relatively high cytotoxicity against ATRT cells in the screening assay. Drugs that showed IC50 values of 1 μM or lower in the screening assays were further evaluated in more expanded concentration range of 10–10−5 μM of each drug. Data obtained are summarized schematically. For each agent the vertical lines represent the lowest, median and highest values, and the horizontal line represents the range of IC50 values for the three cell lines tested. Names of the agents used in this analysis are given along the Y axis.
3.2. Activity of lapatinib against ATRT cell lines
ATRT cells and the (control) non‐malignant fibroblast cell line Hs68 were treated with increasing concentrations of lapatinib and cell viability was evaluated after four days in culture. Results presented in Figure 2 show that cell lines BT16 and KCCF1 are highly sensitive to lapatinib with IC50 of 0.1 μM and BT12 was sensitive for lapatinib activity at IC50 of 5 μM. The non‐malignant Hs68 cells showed no significant inhibitory effects by lapatinib at concentrations as high as 10 μM.
Figure 2.
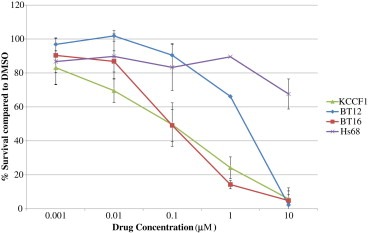
In vitro cytotoxicity of lapatinib against ATRT cell lines. A: Effect of Lapatinib alone in ATRT cell lines. Exponentially growing ATRT and control non‐malignant fibroblast cells were incubated with increasing concentrations of lapatinib or corresponding DMSO control in triplicate. After four days in culture, cell viability in each condition was measured by automated cytometer and percentages of cell survival were calculated with respect to DMSO control wells. Data presented above is representative of three separate experiments.
3.3. Target validation of lapatinib in ATRT cells
Next we aimed to look at the presence of the targets for lapatinib in ATRT cells that are highly and moderately sensitivity to its effects. We also wanted to identify the off‐target effects by lapatinib. The antibody array technique provides an effective tool to screen for inactivation of receptor tyrosine kinases by targeted therapeutic agents. BT16 (high sensitivity) and BT12 (moderate sensitivity) cells were treated with lapatinib and the resulting dephosphorylation of 42 separate receptor tyrosine kinases (RTKs) were analyzed. Results presented in Figure 3 show the loss of phosphorylation of both EGFR and other ErbB family of receptors in BT16 cells and the loss of activity of EGFR in BT12 cells, which did not show measurable ErbB activity at baseline. Both cell lines, however, expressed active IGF‐IR that was not affected by lapatinib. Figure 4 shows further analysis of the effect of lapatinib on EGFR with respect to phosphorylated tyrosine 1173 (Y1173), which has previously been shown to be a critical activation site. Interestingly, BT16 and KCCF1 cells expressed lower amounts of absolute EGFR than BT12 cells, but had highly phosphorylated Y1173, suggestive of an apparent correlation between receptor activity and lapatinib sensitivity.
Figure 3.
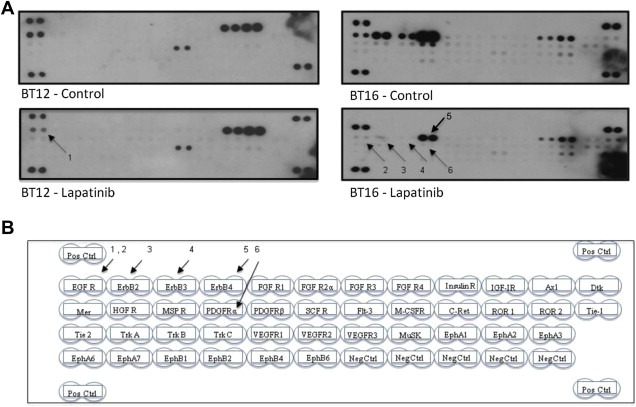
Identification of potential targets of lapatinib activity on ATRT cells. Antibody arrays to a panel of receptor tyrosine kinases were used to screen for dephosphorylation effects by lapatinib. Proteins were extracted from BT12 and BT16 cells treated with 20 mM of lapatinib and cell lysates were made with lysis buffer containing protease and phosphatase inhibitors. DMSO treated cells were used as control. Antibody arrays were incubated with 100 mg of proteins overnight at 4°C with gentle mixing and probed with HRPO conjugated anti‐phosphotyrosine antibodies as per manufacturer's protocol. A: Antibody array showing changes in phosphorylation as seen by a decrease or loss of signal as indicated by arrows. B: A diagrammatic representation of the array map showing the positions of capture antibodies to different RTKs. The arrows are numbered in sequence to indicate EGFR in BT12 cells and EGFR, ErbB2, ErbB3, ErbB4 and PDGFRα in BT16 cells.
Figure 4.
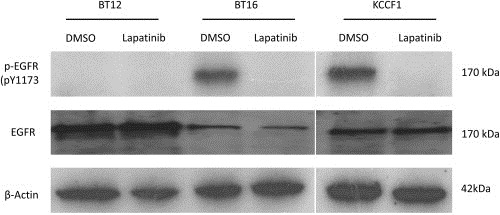
Expression and activity of EGFR in ATRT cells. Exponentially growing cells were treated with lapatinib (20 μM) or DMSO control for 4 h, washed and lysed in the presence of protease and phosphatase inhibitors and total cellular proteins were probed for total EGFR and pEGFR by Western blot analysis.
3.4. Target modulation effects of lapatinib on ATRT cells
Given that antibody array studies demonstrated that the primary targets of lapatinib are affected in treated ATRT cells, we then investigated the downstream consequences of this effect by looking at the modulation of critical signaling cascades linked to RTK mediated pathways. In this study, the activity modulation of three critical signaling nodes, Akt1/2/3, Erk1/2 and Stat‐3, were evaluated upon exposure to lapatinib. Proteins from untreated and lapatinib treated cells were extracted in the presence of protease and phosphatase inhibitors and the phosphorylation status of Akt1/2/3, Erk1/2 and Stat‐3 were probed with phospho‐specific antibodies by Western blot analysis. Data given in Figure 5 show that Erk 1/2 is phosphorylated in all three cell lines but demonstrate measurable dephosphorylation by lapatinib only in BT16 and KCCF1 cells. Secondly, measurable phosphorylation of Akt1/2/3 is seen in only BT16 and KCCF1 cells and in both cases becomes dephosphorylated in the presence of lapatinib. These data also categorize lapatinib activity in different ATRT cell lines with respect to modulations in distinct signaling pathways with corresponding drug sensitivity profile.
Figure 5.
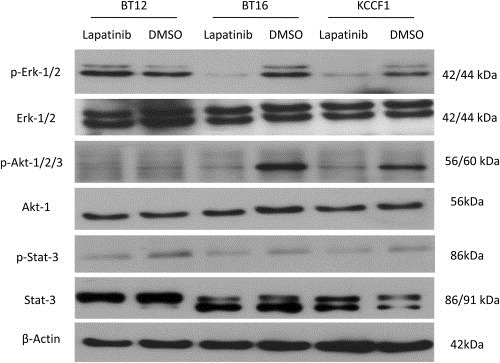
Intracellular signaling and lapatinib activity. ATRT cells were treated with lapatinib (20 μM) or DMSO control for 4 h, washed and lysed in the presence of protease and phosphatase inhibitors and total cellular proteins were probed for total and phosphorylated forms of Erk, Akt and Stat‐3. Findings presented above are representative of two experiments.
3.5. Synergistic activity of lapatinib with IGF‐IR inhibitors
Previous studies have demonstrated that IGF‐IR activity contributes to the growth and survival of ATRT cells. The antibody array analysis presented in Figure 3 also showed the activated status of IGF‐IR in both BT12 and BT16 cells. These data provided a mechanistic rationale to investigate the hypothesis that a combined inhibition of lapatinib with IGF‐IR inhibitors would show synergy against these cells. In the next set of experiments, we investigated lapatinib and the targeted IGF‐IR inhibitor AEW541 (Novartis Pharma AG, Basel, Switzerland) in drug combination studies.
Studies of lapatinib in combination with the targeted IGF‐IR inhibitor AEW541 were carried out as described in Materials and Methods. A graphic representation of cell survival when treated with drug combination is given in Figure 6. The combination indices (CI) (Chou, 2010) calculated from these experiments are given in Table 2. In this analysis, a CI value equals to 1, less than 1 and more than 1 indicates additive, synergistic and antagonistic effects, respectively, between the two agents. Values presented in Table 2 show synergy between lapatinib and AEW541. Lower CI values seen in BT16 and KCCF1 cells also indicate that these cell lines are most susceptible to the combined effect of lapatinib and IGF‐IR inhibition.
Figure 6.
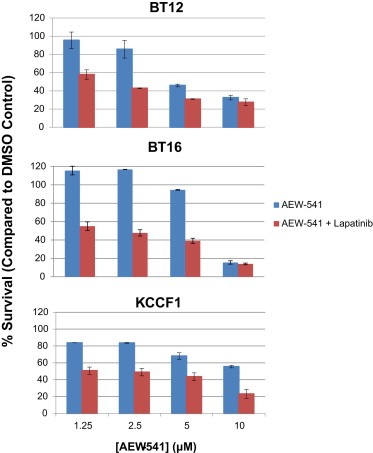
Drug combination study of Lapatinib with AEW‐541 in ATRT cell lines. A: In vitro synergy of lapatinib‐induced cytotoxicity with the IGF‐IR inhibitory agent AEW541. ATRT cells were incubated with increasing concentrations of AEW541 alone or increasing concentrations of AEW541 (X axis) plus a constant IC25 concentration of lapatinib. Cell growth inhibition was measured after four days in culture as describe above. The IC25 values of lapatinib used were 2 µM, 0.01 µM and 0.01 µM for BT12, BT16 and KCCF1 respectively that were calculated from Figure 2.
Table 2.
Activity of combined IGF‐1R inhibition and lapatinib against ATRT cells. IC50 values for single agent AEW541 and in combination with lapatinib were calculated from data presented in Figure 6 and used to calculate combination indices according to the method of Chou and Talalay (Chou, 2010). A CI value less than 1 indicates drug synergy under the specific experimental conditions used.
| Cell line | AEW‐541 + Lapatinib (CI) |
|---|---|
| BT12 | 0.7 |
| BT16 | 0.3 |
| KCCF1 | 0.22 |
3.6. Lapatinib inhibits BT16 cell migration in vitro
In addition to the cytotoxicity effects observed after four days of exposure to lapatinib, we evaluated the effects of lapatinib on the migration of ATRT cells during a shorter time period, presumably before the initiation of apoptosis. In the scratch assay, the movement of cells across a scratch line is evaluated as an indication of the capability of an agent to inhibit cell migration. Photographs presented in Figure 7A show a concentration‐dependent loss of cell migration over the scratch line when treated with lapatinib, demonstrating its potential ability to prevent BT16 cell migration. The similar trend was observed in KCCF1 cell line. Cell migration into the detection zone was quantified by counting cell number using ImageJ software (http://rsb.info.nih.gov/ij/) (version 1.4.3.67). To differentiate cell proliferation versus cell migration to account for the additional cells in the scratch zone, viable cell count was measured by Alamar blue assay and a graphic representation for cell migration with corresponding cell counts are given in Figure 7B.
Figure 7.
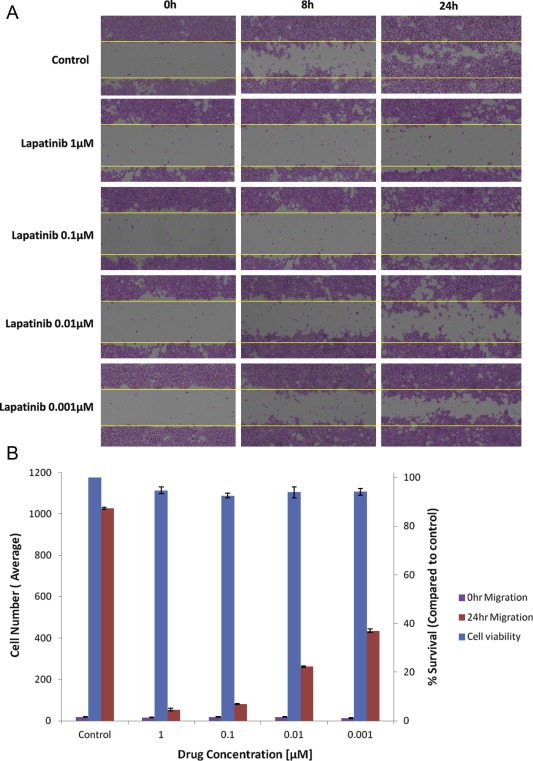
In vitro cell migration assay. A: BT16 cells were plated in 6‐well plate and a scratch was introduced when cells were 80% confluent. Images were acquired at 0 h, 8 h and 24 h following the in vitro scratch assay. The dotted lines define the areas lacking cells. B: The rate of migration was measured by quantifying the total distance that cells moved from the edge of the scratch toward the center of the scratch (marked by imaginary dotted lines). After 24 h in culture, the quantity of viable cells in each condition was measured by Alamar blue assay. Data presented above is representative of three separate experiments.
3.7. In vivo activity of lapatinib
It is likely that cells that express highly active ErbB2 will be more susceptible to treatment with lapatinib or similar agents. The BT16 cell line represents such a tumor phenotype. Hence, we selected this cell line for the next proof‐of‐concept in vivo studies. BT16 cells were labeled with luciferase and GFP (BT16GFPLuc) and used to generate xenografts in CD‐1 Nude mice. After allowing sufficient time for tumors to establish, randomly assigned groups received either lapatinib or vehicle control. Lapatinib was given as a twice‐daily oral administration for 3 weeks (5 days on, 2 days off) at a dose of 160 mg/kg (320 mg/kg/day). The tumor growth differences between the two groups were measured at weekly intervals by total flux emission photon/second as described in methods. Images presented in Figure 8A show luminescence imaging of the two groups of mice. The mice that received lapatinib show measurably less signal intensity indicating the inhibition of tumor growth. Quantitative analysis of this data presented in Figure 8B show growth inhibition of the tumors compared to the continuous increase seen in control animals. On day seventeen the animals were sacrificed and the tumors were removed. Figures 8C and D show the fluorescent and gross images of the tumors respectively, illustrating the antitumor effect of lapatinib on tumor xenografts.
Figure 8.
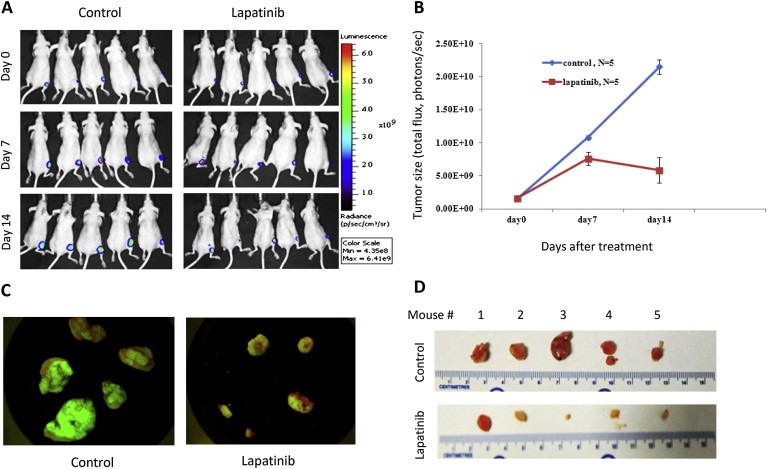
Lapatinib inhibits the tumor activity in vivo. Tumor cells labeled with luciferase and GFP (BT16GFPLuc) were implanted in the flank (5 × 106 cells per animal) of mice to generate xenografts. After allowing 10–14 days for tumors to establish, two randomly assigned groups received either lapatinib or vehicle control. The drug was given as a twice‐daily oral administration for 3 weeks (5 days on, 2 days off) at a dose of 160 mg/kg (320 mg/kg/day). Xenogen IVIS 200 system (Xenogen Corporation, Alameda, CA) was used to monitor and quantify tumor growth in vivo (A, B). Day 0 = first day of treatment, i.e. 2 weeks after implantation. After 17 days of treatment the animals were sacrificed and the tumors were removed and photographed under a dissection microscope with a fluorescent filter or (C) or white light, where a scale was included for size comparison (D).
4. Discussion
ATRT is currently considered to be one of the most malignant and difficult to cure tumors in the pediatric population. Although defects in the chromatin remodeling apparatus by the SWI/SNF complex is likely to be the key molecular feature in ATRT, the pathways and nodes that constitute deregulated growth regulatory mechanisms are critical for the identification of effective targets for future therapeutics. We report the initial screening of a comprehensive library of targeted therapeutic agents using in vitro cytotoxicity assays. Our data show that among these, there are individual agents, as well as the inhibition of specific growth regulatory pathways, that can significantly interfere with the growth and survival of these cells. Such an approach also provides essential initial data that can be further developed by different research groups with expertise in diverse experimental systems and new agents to complete additional preclinical studies without delay.
Among the agents that have been found to be effective, we selected lapatinib for further studies for a number of reasons. Recent clinical trials in adults have shown tolerability of this agent (de Souza et al., 2012) and it has been suggested to possess the ability to cross the blood–brain barrier, which supports its use in patients with CNS tumors (Mukohara, 2011). However, drug distribution during treatment for brain metastases of breast cancer appears to be partially restricted by blood‐tumor barrier permeability (Taskar et al., 2012). Importantly, in one of the earlier studies, the targets of lapatinib, EGFR and ErbB2, have been found in 3/7 and 6/7 of ATRT primary tumor specimens, respectively (Patereli et al., 2010). The contribution of off‐target effects notwithstanding, three independent agents that target ErbB2 family, lapatinib, CUDC101 and canertinib, showed significant cytotoxicity in our screening, suggesting the utility of targeting these receptors in ATRT (Table 1, Figure 1). The IC50 values of lapatinib in the ATRT cells were similar for two of the three cell lines, BT16 and KCCF1, but higher for the BT12 cell line (Table 1, Figure 2). These values are within the range reported for susceptible Her2‐positive breast cancer cell lines (O'Donovan et al., 2011). The higher IC50 of BT12 cells is closer to the values described for lapatinib hypo‐responsive breast cancer cell lines MDA‐MB‐468 and T47D, which express low basal levels of ErbB2 and the IC50 seen for BT16 and KCCF1 cells are similar to the lapatinib‐responsive lines, BT474 and SKBr3, that constitutively over‐express ErbB2 (Hegde et al., 2007). These findings are in line with the molecular and phenotypic heterogeneity of ATRT and underscore the importance of target validation studies in the stratification of patients for Her2 EGFR‐based therapies in the future.
Data from the antibody array studies showed loss of activation signals of the previously described lapatinib targets EGFR, ErbB2 and ErbB4 (Figure 3). We have also noted loss of signals with respect to ErbB3 and PDGFRα. However, the exact mechanisms for the additional activity against ErbB3 and PDGFα are currently unclear. It is possible that these effects may be due to the ability of lapatinib to interfere with dimerization of Her family of receptors and to disrupt previously formed receptor dimers (Sanchez‐Martin and Pandiella, 2011), or due to previously not described off‐target effects of this agent.
Autophosphorylation at the tyrosine residue 1173 (Tyr 1173) has been shown to be a significant event and important for signal transduction following ligand binding and receptor dimerization of EGFR (Chattopadhyay et al., 1999). Data presented in Figure 4 show effective dephosphorylation of Y1173 by lapatinib in the two highly sensitive lines BT12 and KCCF1, although there appears to be EGFR phosphorylation in all three cell lines, as determined by pan‐phospho antibodies in Figure 3. This prompted us to further evaluate the effect of lapatinib on intracellular signaling pathways in relation to EGFR and ErbB2 activities (Figure 5). In these studies, a loss of Erk and Akt activities was seen in the highly sensitive and ErbB active cells (BT16 and KCCF1). However, although all three cell lines expressed Stat‐3 with detectable phosphorylated bands, no significant changes in response to lapatinib was seen in Stat‐3. The major signaling cascades that are initiated as a consequence of EGFR and ErbB activation are thought to be mediated by PI3, Ras‐Raf (MAPK), JNK and PLCγ kinases (Eccles, 2011). Consequently, these activities lead to a multitude of cellular functions necessary for the growth and survival of tumor cells. It is known that Akt and Erk functions are critical for the flow of many of these pathways. Our findings are also in agreement with the microarray and phosphorylated protein findings of Hedge and colleagues, in which phosphorylation and gene expression changes in breast cancer cell lines in response to lapatinib were explored (Hegde et al., 2007). This study showed that the cells highly responsive to lapatinib significantly down‐regulated a number of transcripts, including Akt1, whereas the non‐responsive lines only weakly down‐regulated the Akt pathway. Phosphorylated Akt also decreased in response to lapatinib. Furthermore, gene expression profiling showed that lapatinib modulated many of the genes involved in cell cycle control and in the regulation of metabolic pathways such as glycolysis and fatty acid metabolism (Hegde et al., 2007). Metabolomic studies are currently in progress in our laboratory to evaluate such an effect in ATRT cells, especially the ways in which such changes can be monitored in the CSF of patients who may receive ErbB2‐directed treatments in the future.
Findings from the cell motility inhibition studies (scratch tests) demonstrated that lapatinib inhibits the movement of BT16 cells in a concentration‐dependent manner within hours (Figure 7). Previous reports have provided evidence that, in addition to their positive contribution to cell proliferation, activation of EGFR‐ErbB2 receptors also promote cell adhesion and motility (Freudenberg et al., 2009). In the breast cancer model, a multitude of studies have shown that HER2 in metastatic cells promote cell motility (Eccles, 2011). A recent investigation by Siedel and colleagues has shown that breast cancer cells that express HER2 neu+ and not the HER2‐neu (+/−) phenotype show decreased invasion and loss of anchorage when treated with lapatinib. This appears to be independent of the surface receptor CUB domain‐containing protein 1 (CDCP1) activity (Seidel et al., 2011). In the glioma tumor model, it has been found that lapatinib interferes with cellular migration through the interruption of EGFR‐integrin β(1) complex formation (Dimitropoulos et al., 2010). Frequently ATRT presents with significant infiltration into brainstem, making tumor resection a difficult task. Recently, a case report by Beschorner and colleagues described a child who received incomplete resection, followed by multi‐modal therapy, but presented with a second tumor, possibly a late metastasis of the original ATRT. This lesion appearing along the right trigeminal nerve was not in continuity with the primary tumor (Beschorner et al., 2006). Although the rare possibility of a radiation‐induced second ATRT cannot be ruled out, this case reveals the potential of aggressive ATRT cells for invasion and migration.
Lapatinib has shown promising result in trastuzumab‐refractory metastatic breast cancer in Phase I, II and III studies and a number of trials are currently in progress to further understand its utility in CNS metastasis. Further studies are needed to define the contribution of inhibition of tumor migration in the context of surgical and overall management of ATRT in the future.
Generation of resistance to RTK‐targeted therapeutics has been a major obstacle in the utility of this family of agents. In addition, tolerability concerns have also limited the effectiveness of single agent RTK‐targeted therapies in the past. Our current results (Figure 6, Table 2) as well as previously published studies have alluded to the critical role of IGF‐IR activity in ATRT cells (D'Cunja et al., 2007). As lapatinib has shown strong growth inhibition in only two of the three cell lines studied, we wanted to investigate the effect of combining IGF‐IR inhibition with lapatinib, especially for tumors that may have lower Erb expression. Our in vitro studies show enhanced activity in all three cell lines with combination indices less than 1, suggesting drug synergy under the experimental conditions used (Table 2). In the recent past, effective drug combinations with lapatinib have been explored in a number of tumor models. For example, combining lapatinib with the notch inhibitor (γ‐secretase inhibitor) MRK‐003 GSI showed significant reduction of tumor growth in ErbB‐2‐positive breast cancer xenografts (Pandya et al., 2011). Treatment of animals carrying orthotopic CNS tumor isolates with lapatinib and Bcl‐2 homology domain‐3 (BH3) mimetic obatoclax (GX15‐070) prolonged survival of these animals (Cruickshanks et al., 2012). The ability of lapatinib to synergize with HDAC inhibitors has been shown in previous studies (LaBonte et al., 2011). Interestingly, in breast cancer cells, the failure of the EGFR inhibitor transtuzumab appears to be mediated by the upregulation of IGF‐IR and lapatinib may actively block Erb2 and IGF‐IR cross talk in transtuzumab‐resistant cells (Nahta et al., 2007). It is conceivable that lapatinib also blocks IGF‐IR and ErbB2 cross talk in ATRT cells, thus providing an additional mechanism to enhance the effect of a combined inhibition of these two pathways. Additional studies are needed to experimentally demonstrate this interesting activity. Such information including in vivo studies using combined lapatinib and AEW‐541, is crucial for the development of future drug combination therapies to optimize cell killing and reducing toxicity and the potential for drug resistance.
In the following set of experiments, in vitro cytotoxic activity of lapatinib was further evaluated using in vivo xenograft experiments (Figure 8). Our initial studies used the cell line that expressed all targets as we envision that such tumors will be most suitable for future clinical studies with lapatinib. We used a regimen of twice‐daily oral administration for 3 weeks (5 days on, 2 days off) at a dose of 160 mg/kg (320 mg/kg/day). This dose was based on previous studies including a report by Gorlick and co‐workers who evaluated lapatinib for activity in pediatric tumor xenografts (Gorlick et al., 2009). Our findings are in agreement with previous xenograft studies of other ErbB2 and EGFR over‐expressing tumor models (Konecny et al., 2006; Rusnak et al., 2001). Future xenograft studies are needed to evaluate drug combinations that would benefit the complete spectrum of EGFR ErbB‐expressing ATRT tumors. In our studies, lapatinib alone gave significant tumor kill at low and non‐toxic concentrations, making data from in vivo drug combination studies difficult to interpret. It has been suggested that the utility of a IGF‐IR inhibitors will be of significance for patients who have generated treatment resistance (Abraham et al., 2011; Jin and Esteva, 2008). We are currently in the process of generating variants of ATRT cell lines to test this hypothesis in future studies.
Abnormal expression and activity of ErbB family of proteins have been described in a number of tumors and are central in the development, metastasis and treatment of breast cancer. Our report provides evidence for the first time that these molecules present an effective target for therapeutics in at least a sub group of CNS ATRT. Although there are reports showing the existence of ErbB family of proteins in rhabdoid tumors, additional studies are needed in an expanded cohort of specimens to precisely define the incidence of ErbB expression and activation in ATRT. Studies are currently in progress in our laboratory using immunohistochemical analysis of tissue microarrays (TMA) of CNS ATRT specimens. In addition to the effects on ErbB family of proteins, potential off‐target effects of lapatinib need to be evaluated. For example, a recent report by Dolloff and colleagues has shown the effect of lapatinib on TRAIL death receptor expression and signaling that is independent of EGFR and HER2 inhibition (Dolloff et al., 2011). Importantly, information is also needed on the pathways that may link the loss of INI1 to the activity of these molecules leading to an aggressive tumor physiology. For instance, recent reports indicate that EGFR may act as a transcriptional regulator of cyclin D1 (Burness et al., 2010; Eccles, 2011), and the loss of INI1 has been known to lead to derepression of cyclin D1 in rhabdoid tumors (Smith et al., 2011). Targeting ErbB should be further evaluated in the context of patients who are receiving other treatment modalities such as surgical debulking. This is important as it has been postulated that ErbB2 positive breast cancers show proliferative responses to growth factors found in postsurgical wound fluids and this can be blocked by anti‐HER2 antibodies (Tagliabue et al., 2003). Recent studies using high‐resolution genome‐wide analysis have failed to show recurrent genomic alterations other than SMARCB1 in ATRT (Hasselblatt et al., 2012). In addition Kieran and colleagues have examined tumor specimens with INI1 alterations for changes in oncogenes and tumor suppressor genes and found no evidence of mutations in canonical pathways critical for adult cancers (Kieran et al., 2012). Although these findings may limit the possibility of identifying single effective agents based on highly prevalent molecular abnormalities, an extension of the approach of screening diverse drug libraries can provide an avenue to identify effective agents based on operational growth regulatory pathways and individually tailored, tumor‐defined therapeutic interventions. Approaches to target epigenetic modifiers with small molecule inhibitors have led to the availability of novel candidate epidrugs which can also be used in similar studies (Andreoli et al., 2012). Interestingly, a recent study demonstrating the suppression of HER2 receptor in subgroups of breast cancer after treatment with 5‐aza‐2′‐deoxycytidine (DAC) (Radpour et al., 2011) indicates the feasibility of effective evaluation such agents in novel therapeutic opportunities.
In summary, as a proof‐of‐concept, we present a preclinical study pathway to identify novel therapeutic regimens for rare pediatric malignancies such as CNS ATRT. The selection of agents to screen comes from a library of drugs that have been validated in a range of more common adult tumors. The safety and dosing data that are becoming available on these drugs provides the feasibility of the successful candidates to be considered for early phase clinical trials in a timely manner. Most often these agents already have key information such as CNS penetration data, potential toxicities and pharmacokinetic profiles. Secondly, initial in vitro studies in representative cell lines help to gain preliminary data on potential drug combination and treatment schedules that can be further validated in additional xenograft studies. Intracellular target modulation experiments provide information that can be incorporated into selecting the most targetable patient population based on the analysis of pre‐treatment biopsy specimens. Importantly, the identification of multiple active agents and survival pathways in early preclinical studies is practical and expedient for “pick the winner” trial designs that are needed for the treatment of rare cancers such as ATRT.
Acknowledgment
This research was supported by grants from the Kids Cancer Care Foundation of Alberta (KCCFA) Alberta Children's Hospital Foundation (ACHF) and the Brain Tumour Foundation of Canada. AJ holds a Canadian Institutes for Health Research (CIHR) Training Program in Genetics, Child Development and Health Graduate Studentship awarded by Alberta Children's Hospital for Child and Maternal Health (ACHRI) and a McCarthy Tetrault Graduate Studentship awarded by ACHF.
Singh Anjali, Lun Xueqing, Jayanthan Aarthi, Obaid Halah, Ruan Yibing, Strother Douglas, Chi Susan N., Smith Amy, Forsyth Peter, Narendran Aru, (2013), Profiling pathway‐specific novel therapeutics in preclinical assessment for central nervous system atypical teratoid rhabdoid tumors (CNS ATRT): Favorable activity of targeting EGFR‐ ErbB2 signaling with lapatinib, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.01.001.
References
- Abraham, J. , Prajapati, S.I. , Nishijo, K. , Schaffer, B.S. , Taniguchi, E. , Kilcoyne, A. , McCleish, A.T. , Nelon, L.D. , Giles, F.G. , Efstratiadis, A. , LeGallo, R.D. , Nowak, B.M. , Rubin, B.P. , Malempati, S. , Keller, C. , 2011 Apr. Evasion mechanisms to Igf1r inhibition in rhabdomyosarcoma. Molecular Cancer Therapeutics 10, (4) 697–707. Epub 2011 Mar 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoli, F. , Barbosa, A.J. , Parenti, M.D. , Del Rio, A. , 2012 Sep 26. Modulation of epigenetic targets for anticancer therapy: clinicopathological relevance, structural data and drug discovery perspectives. Current Pharmaceutical Design [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaro, A. , Doepfner, K.T. , Boller, D. , Guerreiro, A.S. , Shalaby, T. , Jackson, S.P. , Schoenwaelder, S.M. , Delattre, O. , Grotzer, M.A. , Fischer, B. , 2007. Novel role for insulin as an autocrine growth factor for malignant brain tumour cells. The Biochemical Journal 406, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, X. , Yan, Y. , Coleman, M. , Wu, G. , Rabinovich, B. , Seidensticker, M. , Alt, E. , 2011. Tracking long-term survival of intramyocardially delivered human adipose tissue-derived stem cells using bioluminescence imaging. Molecular Imaging and Biology: MIB: The Official Publication of the Academy of Molecular Imaging 13, 633–645. [DOI] [PubMed] [Google Scholar]
- Beschorner, R. , Mittelbronn, M. , Koerbel, A. , Ernemann, U. , Thal, D.R. , Scheel-Walter, H.G. , Meyermann, R. , Tatagiba, M. , 2006. Atypical teratoid-rhabdoid tumor spreading along the trigeminal nerve. Pediatric Neurosurgery 42, 258–263. [DOI] [PubMed] [Google Scholar]
- Bikowska, B. , Grajkowska, W. , Jozwiak, J. , 2011. Atypical teratoid/rhabdoid tumor: short clinical description and insight into possible mechanism of the disease. European Journal of Neurology: The Official Journal of the European Federation of Neurological Societies 18, 813–818. [DOI] [PubMed] [Google Scholar]
- Burness, M.L. , Grushko, T.A. , Olopade, O.I. , 2010. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker?. Cancer Journal 16, 23–32. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A. , Vecchi, M. , Ji, Qs , Mernaugh, R. , Carpenter, G. , 1999 Sep 10. The role of individual SH2 domains in mediating association of phospholipase C-gamma1 with the activated EGF receptor. Journal of Biological Chemistry 274, (37) 26091–26097. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Research 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Cruickshanks, N. , Hamed, H. , Bareford, M.D. , Poklepovic, A. , Fisher, P.B. , Grant, S. , Dent, P. , 2012. Lapatinib and Obatoclax kill tumor cells through blockade of ERBB1/3/4 and through inhibition of BCL-XL and MCL-1. Molecular Pharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Cunja, J. , Shalaby, T. , Rivera, P. , von Buren, A. , Patti, R. , Heppner, F.L. , Arcaro, A. , Rorke-Adams, L.B. , Phillips, P.C. , Grotzer, M.A. , 2007. Antisense treatment of IGF-IR induces apoptosis and enhances chemosensitivity in central nervous system atypical teratoid/rhabdoid tumours cells. European Journal of Cancer 43, 1581–1589. [DOI] [PubMed] [Google Scholar]
- de Souza, J.A. , Davis, D.W. , Zhang, Y. , Khattri, A. , Seiwert, T.Y. , Aktolga, S. , Wong, S. , Kozloff, M.F. , Nattam, S. , Lingen, M.W. , Kunnavakkam, R. , Stenson, K.M. , Blair, E.A. , Bozeman, J. , Dancey, J.E. , Vokes, E.E. , Cohen, E.E. , 2012. A phase II study of lapatinib in recurrent/metastatic squamous cell carcinoma of the head and neck. Clinical Cancer Research [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitropoulos, K. , Giannopoulou, E. , Argyriou, A.A. , Zolota, V. , Petsas, T. , Tsiata, E. , Kalofonos, H.P. , 2010. The effects of anti-VEGFR and anti-EGFR agents on glioma cell migration through implication of growth factors with integrins. Anticancer Research 30, 4987–4992. [PubMed] [Google Scholar]
- Dolloff, N.G. , Mayes, P.A. , Hart, L.S. , Dicker, D.T. , Humphreys, R. , El-Deiry, W.S. , 2011. Off-target lapatinib activity sensitizes colon cancer cells through TRAIL death receptor up-regulation. Science Translational Medicine 3, 86ra56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles, S.A. , 2011. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. The International Journal of Developmental Biology 55, 685–696. [DOI] [PubMed] [Google Scholar]
- Foster, K. , Wang, Y. , Zhou, D. , Wright, C. , 2009. Dependence on PI3K/Akt signaling for malignant rhabdoid tumor cell survival. Cancer Chemotherapy and Pharmacology 63, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, K.S. , McCrary, W.J. , Ross, J.S. , Wright, C.F. , 2006. Members of the hSWI/SNF chromatin remodeling complex associate with and are phosphorylated by protein kinase B/Akt. Oncogene 25, 4605–4612. [DOI] [PubMed] [Google Scholar]
- Freudenberg, J.A. , Wang, Q. , Katsumata, M. , Drebin, J. , Nagatomo, I. , Greene, M.I. , 2009. The role of HER2 in early breast cancer metastasis and the origins of resistance to HER2-targeted therapies. Experimental and Molecular Pathology 87, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa, H. , Misaki, K. , Takabatake, Y. , Hasegawa, M. , Yamashita, J. , 2005. Cyclin D1 is overexpressed in atypical teratoid/rhabdoid tumor with hSNF5/INI1 gene inactivation. Journal of Neuro-Oncology 73, 117–124. [DOI] [PubMed] [Google Scholar]
- Gorlick, R. , Kolb, E.A. , Houghton, P.J. , Morton, C.L. , Phelps, D. , Schaiquevich, P. , Stewart, C. , Keir, S.T. , Lock, R. , Carol, H. , Reynolds, C.P. , Maris, J.M. , Wu, J. , Smith, M.A. , 2009. Initial testing (stage 1) of lapatinib by the pediatric preclinical testing program. Pediatric Blood and Cancer 53, 594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi, C.J. , Sands, A.T. , Zambrowicz, B.P. , Turner, T.K. , Demers, D.A. , Webster, W. , Smith, T.W. , Imbalzano, A.N. , Jones, S.N. , 2001 May. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Molecular and Cellular Biology 21, (10) 3598–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselblatt, M. , Isken, S. , Linge, A. , Eikmeier, K. , Jeibmann, A. , Oyen, F. , Nagel, I. , Richter, J. , Bartelheim, K. , Kordes, U. , Schneppenheim, R. , Frühwald, M. , Siebert, R. , Paulus, W. , 2012 Oct 17. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 10.1002/gcc.22018 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hegde, P.S. , Rusnak, D. , Bertiaux, M. , Alligood, K. , Strum, J. , Gagnon, R. , Gilmer, T.M. , 2007. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Molecular Cancer Therapeutics 6, 1629–1640. [DOI] [PubMed] [Google Scholar]
- Jayanthan, A. , Bernoux, D. , Bose, P. , Riabowol, K. , Narendran, A. , 2011. Multi-tyrosine kinase inhibitors in preclinical studies for pediatric CNS AT/RT: evidence for synergy with Topoisomerase-I inhibition. Cancer Cell International 11, 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Q. , Esteva, F.J. , 2008 Dec. Cross-talk between the ErbB/HER family and the type I insulin-like growth factor receptor signaling pathway in breast cancer. Journal of Mammary Gland Biology and Neoplasia 13, (4) 485–498. [DOI] [PubMed] [Google Scholar]
- Kieran, M.W. , Roberts, C.W. , Chi, S.N. , Ligon, K.L. , Rich, B.E. , Macconaill, L.E. , Garraway, L.A. , Biegel, J.A. , 2012 Dec 15. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatric Blood and Cancer 59, (7) 1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipstein, J.A. , Birks, D.K. , Donson, A.M. , Alimova, I. , Foreman, N.K. , Vibhakar, R. , 2012. Histone deacetylase inhibition decreases proliferation and potentiates the effect of ionizing radiation in atypical teratoid/rhabdoid tumor cells. Neuro-Oncology 14, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konecny, G.E. , Pegram, M.D. , Venkatesan, N. , Finn, R. , Yang, G. , Rahmeh, M. , Untch, M. , Rusnak, D.W. , Spehar, G. , Mullin, R.J. , Keith, B.R. , Gilmer, T.M. , Berger, M. , Podratz, K.C. , Slamon, D.J. , 2006. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Research 66, 1630–1639. [DOI] [PubMed] [Google Scholar]
- LaBonte, M.J. , Wilson, P.M. , Fazzone, W. , Russell, J. , Louie, S.G. , El-Khoueiry, A. , Lenz, H.J. , Ladner, R.D. , 2011 May 15. The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Research 71, (10) 3635–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, C.C. , Park, A.Y. , Guan, J.L. , 2007. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature Protocols 2, 329–333. [DOI] [PubMed] [Google Scholar]
- Lun, X. , Alain, T. , Zemp, F.J. , Zhou, H. , Rahman, M.M. , Hamilton, M.G. , McFadden, G. , Bell, J. , Senger, D.L. , Forsyth, P.A. , 2010. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Research 70, 598–608. [DOI] [PubMed] [Google Scholar]
- Mukohara, T. , 2011. Role of HER2-targeted agents in adjuvant treatment for breast cancer. Chemotherapy Research and Practice 2011, 730360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabzdyk, C.S. , Chun, M. , Pradhan, L. , Logerfo, F.W. , 2011. High throughput RNAi assay optimization using adherent cell cytometry. Journal of Translational Medicine 9, 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahta, R. , Yuan, L.X. , Du, Y. , Esteva, F.J. , 2007. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Molecular Cancer Therapeutics 6, 667–674. [DOI] [PubMed] [Google Scholar]
- Narendran, A. , Coppes, L. , Jayanthan, A. , Coppes, M. , Teja, B. , Bernoux, D. , George, D. , Strother, D. , 2008. Establishment of atypical-teratoid/rhabdoid tumor (AT/RT) cell cultures from disseminated CSF cells: a model to elucidate biology and potential targeted therapeutics. Journal of Neuro-Oncology 90, 171–180. [DOI] [PubMed] [Google Scholar]
- O'Donovan, N. , Byrne, A.T. , O'Connor, A.E. , McGee, S. , Gallagher, W.M. , Crown, J. , 2011. Synergistic interaction between trastuzumab and EGFR/HER-2 tyrosine kinase inhibitors in HER-2 positive breast cancer cells. Investigational New Drugs 29, 752–759. [DOI] [PubMed] [Google Scholar]
- Ogino, S. , Cohen, M.L. , Abdul-Karim, F.W. , 1999. Atypical teratoid/rhabdoid tumor of the CNS: cytopathology and immunohistochemistry of insulin-like growth factor-II, insulin-like growth factor receptor type 1, cathepsin D, and Ki-67. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc 12, 379–385. [PubMed] [Google Scholar]
- Ogino, S. , Kubo, S. , Abdul-Karim, F.W. , Cohen, M.L. , 2001. Comparative immunohistochemical study of insulin-like growth factor II and insulin-like growth factor receptor type 1 in pediatric brain tumors. Pediatric and Developmental Pathology: The Official Journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 4, 23–31. [DOI] [PubMed] [Google Scholar]
- Pandya, K. , Meeke, K. , Clementz, A.G. , Rogowski, A. , Roberts, J. , Miele, L. , Albain, K.S. , Osipo, C. , 2011. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. British Journal of Cancer 105, 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patereli, A. , Alexiou, G.A. , Stefanaki, K. , Moschovi, M. , Doussis-Anagnostopoulou, I. , Prodromou, N. , Karentzou, O. , 2010. Expression of epidermal growth factor receptor and HER-2 in pediatric embryonal brain tumors. Pediatric Neurosurgery 46, 188–192. [DOI] [PubMed] [Google Scholar]
- Rabinovich, B.A. , Ye, Y. , Etto, T. , Chen, J.Q. , Levitsky, H.I. , Overwijk, W.W. , Cooper, L.J. , Gelovani, J. , Hwu, P. , 2008. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proceedings of the National Academy of Sciences of the United States of America 105, 14342–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radpour, R. , Barekati, Z. , Kohler, C. , Schumacher, M.M. , Grussenmeyer, T. , Jenoe, P. , Hartmann, N. , Moes, S. , Letzkus, M. , Bitzer, J. , Lefkovits, I. , Staedtler, F. , Zhong, X.Y. , 2011. Integrated epigenetics of human breast cancer: synoptic investigation of targeted genes, microRNAs and proteins upon demethylation treatment. PLoS One 6, (11) e27355 Epub 2011 Nov 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, A.T. , 2005. Atypical teratoid/rhabdoid tumors of the central nervous system. Journal of Neuro-Oncology 75, 309–313. [DOI] [PubMed] [Google Scholar]
- Rorke, L.B. , Packer, R.J. , Biegel, J.A. , 1996. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. Journal of Neurosurgery 85, 56–65. [DOI] [PubMed] [Google Scholar]
- Rusnak, D.W. , Lackey, K. , Affleck, K. , Wood, E.R. , Alligood, K.J. , Rhodes, N. , Keith, B.R. , Murray, D.M. , Knight, W.B. , Mullin, R.J. , Gilmer, T.M. , 2001. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Molecular Cancer Therapeutics 1, 85–94. [PubMed] [Google Scholar]
- Sanchez-Martin, M. , Pandiella, A. , 2011. Differential action of small molecule HER kinase inhibitors on receptor heterodimerization: therapeutic implications. International journal of cancer. Journal international du cancer [DOI] [PubMed] [Google Scholar]
- Seidel, J. , Kunc, K. , Possinger, K. , Jehn, C. , Luftner, D. , 2011. Effect of the tyrosine kinase inhibitor lapatinib on CUB-domain containing protein (CDCP1)-mediated breast cancer cell survival and migration. Biochemical and Biophysical Research Communications 414, 226–232. [DOI] [PubMed] [Google Scholar]
- Smith, M.E. , Das, B.C. , Kalpana, G.V. , 2011. In vitro activities of novel 4-HPR derivatives on a panel of rhabdoid and other tumor cell lines. Cancer Cellular International 11, 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabue, E. , Agresti, R. , Carcangiu, M.L. , Ghirelli, C. , Morelli, D. , Campiglio, M. , Martel, M. , Giovanazzi, R. , Greco, M. , Balsari, A. , Menard, S. , 2003. Role of HER2 in wound-induced breast carcinoma proliferation. Lancet 362, 527–533. [DOI] [PubMed] [Google Scholar]
- Taskar, K.S. , Rudraraju, V. , Mittapalli, R.K. , Samala, R. , Thorsheim, H.R. , Lockman, J. , Gril, B. , Hua, E. , Palmieri, D. , Polli, J.W. , Castellino, S. , Rubin, S.D. , Lockman, P.R. , Steeg, P.S. , Smith, Q.R. , 2012. Lapatinib distribution in HER2 overexpressing experimental brain metastases of breast cancer. Pharmaceutical Research 29, 770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.K. , Davies, K.P. , Allen, J. , Zhu, L. , Pestell, R.G. , Zagzag, D. , Kalpana, G.V. , 2002. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Molecular and Cellular Biology 22, 5975–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]