

Abstract
Genetically engineered stem cells (GESTECs) exhibit a potent therapeutic efficacy via their strong tumor tropism toward cancer cells. In this study, we introduced the human parental neural stem cells, HB1.F3, with the human interferon beta (IFN‐β) gene which is a typical cytokine gene that has an antitumor effect and the cytosine deaminase (CD) gene from Escherichia coli (E. coli) that could convert the non‐toxic prodrug, 5‐fluorocytosine (5‐FC), to a toxic metabolite, 5‐fluorouracil (5‐FU). Two types of stem cells expressing the CD gene (HB1.F3.CD cells) and both the CD and human IFN‐β genes (HB1.F3.CD.IFN‐β) were generated. The present study was performed to examine the migratory and therapeutic effects of these GESTECs against the colorectal cancer cell line, HT‐29. When co‐cultured with colorectal cancer cells in the presence of 5‐FC, HB1.F3.CD and HB1.F3.CD.IFN‐β cells exhibited the cytotoxicity on HT‐29 cells via the bystander effect. In particular, HB1.F3.CD.IFN‐β cells showed the synergistic cytotoxic activity of 5‐FU and IFN‐β. We also confirmed the migration ability of HB1.F3.CD and HB1.F3.CD.IFN‐β cells toward HT‐29 cells by a modified migration assay in vitro, where chemoattractant factors secreted by HT‐29 cells attracted the GESTECs. In a xenograft mouse model, the volume of tumor mass was decreased up to 56% in HB1.F3.CD injected mice while the tumor mass was greatly inhibited about 76% in HB1.F3.CD.IFN‐β injected mice. The therapeutic treatment by these GESTECs is a novel strategy where the combination of the migration capacity of stem cells as a vector for therapeutic genes towards colorectal cancer and a synergistic antitumor effect of CD and IFN‐β genes can selectively target this type of cancer.
Keywords: Therapeutic stem cells, Cytosine deaminase, Interferon-beta, Colorectal cancer, Xenograft model
Highlights
-
►
Migratory effect of the stem cells was confirmed towards HT‐29 colorectal cancer.
-
►
Therapeutic effect of the stem cells was observed against HT‐29 colorectal cancer.
-
►
The volume of colorectal tumor mass was inhibited in xenografted mice.
-
►
A remarkable synergistic antitumor effect of CD and IFN‐b genes can selectively target this type of cancer.
1. Introduction
Colorectal malignancy is one of the most common types of tumors in humans. Major causes of death associated with this cancer could be attributed to metastasis in liver, abdominal lymph nodes, and lungs (Bird et al., 2006). Conventional therapies such as surgical removal (colectomy abdomino‐perineal resection), radiotherapy, and chemotherapy are typical treatment for colorectal cancer (Milsom et al., 1998). For instance, 5‐fluorouracil (5‐FU) has been used as a chemotherapeutic agent for treating the majority of solid tumors as well as colorectal cancer. Although most patients are currently treated with chemotherapy of 5‐FU, prognosis is still poor (Pessino and Sobrero, 2006). It is also known that the side effects such as gastrointestinal or hematological toxicities of this drug are serious (Douillard et al., 2000). Since conventional 5‐FU treatments or therapies generally have low selectivity for tumors and result in unwanted effects to normal and highly proliferative healthy tissue (parentheses should only be used for citations and acronyms), novel therapeutic strategies are needed to enhance selective therapeutic efficiency and effectiveness to treat colorectal cancer (Pessino and Sobrero, 2006; Saukkonen and Hemminki, 2004) such as the recently proposed molecular chemotherapy (Kucerova et al., 2007; Reiser et al., 2005). A molecular chemotherapy is based on the theory of targeting therapeutic suicide gene delivery using a cell‐based system (Conrad et al., 2007; Kaliberov et al., 2007).
As an approach of molecular chemotherapy known as gene‐directed enzyme/prodrug therapy (GEPT) has received much attention because it can be designed to selectively target tumor cells more than normal cells which can minimize side‐effects (Saukkonen and Hemminki, 2004). The cytosine deaminase (CD)/5‐fluorocytosine (5‐FC) system is one of the GEPTs where CD is tagged as a suicide enzyme to eradicate cancer cells (Crystal et al., 1997; Hirschowitz et al., 1995; Kanai et al., 1997). The CD gene could convert 5‐FC to 5‐FU, which inhibits DNA synthesis in cancer cells (Yi et al., 2011a). This CD/5‐FC GEPT system has been used as experimental treatments for several types of cancers, including prostate and ovarian cancers (Freytag et al., 2002; Kim et al., 2010). Whereas the application of GEPT systems seem to reduce the toxicity in normal tissues than other therapies, problems still exist that have yet to be solved in an effective delivery of exogenous enzymes to tumor cells (Freytag et al., 2002).
The stem cells have recently received a great deal of attention for their clinical and therapeutic potential for treating human cancers with their strong tumor tropic properties (Altaner, 2008; Yazawa et al., 2002). For instance, human neural stem cells (NSCs) are able to target tumor cells (Yi et al., 2011b). GEPT using NSCs can serve as a potent delivery system to target and eradicate tumor cells following systemic prodrug administration (Aboody et al., 2008) specifically medulloblastomas and gliomas, and could have a great tropic and therapeutic potential for these human malignant tumors (Aboody et al., 2006; Kim et al., 2006). Although tumor tropism of NSCs such as moving toward the tumor sites, is yet to be understood completely, it is used to deliver therapeutic molecules in the patients who have had disseminated metastatic cancer (Fodde, 2006; Muller et al., 2006). In this study, we introduced HB1.F3 immortalized NSCs for treating colorectal cancer. These HB1.F3 cells are human NSCs generated from fetal telencephalon and immortalized using a retroviral vector carrying v‐myc (Kim et al., 2002). This clonally isolated and multi‐potent human NSC line has the ability to self‐renew and differentiate into cells of neuronal and glial lineages both in vivo and in vitro (Kim, 2004). When these cells were cultured in in vitro, they are homogeneous and can be expanded to large numbers. Since these NSCs have their inherent migratory and tumor‐tropic properties, they can represent a novel and potentially powerful approach for the treatment of invasive tumors (Kim et al., 2006; Kim, 2007; Nakamura et al., 2004). It was of interest that the tropism of NSCs can be used to target intra‐ and extra‐cranial tumors of both neural and non‐neural origins (Brown et al., 2003; Fodde, 2006). Other studies have shown that HB1.F3 cells can migrate to subcutaneous xenografts of diverse types of solid tumors including prostate and breast tumors, melanomas, gliomas, and neuroblastomas, which indicates that these cells do not possess a tissue‐specific homing tendency but could be useful therapeutically nonetheless due to their tendency to migrate to tumor tissues in general (Aboody et al., 2006; Dietrich et al., 2008; Freytag et al., 2002; Kim).
As a delivery vehicle that targets tumor cells, coupled with the ability to selectively infiltrate tumor masses and disseminate therapeutic gene products throughout tumor sites, these therapeutic stem cells may solve major obstacles that current gene therapy strategies are facing (Kim et al., 2011). For the delivery of therapeutic genes, HB1.F3 cells were engineered to HB1.F3.CD and HB1.F3.CD.IFN‐β cells to express a therapeutic suicide gene such as the CD or IFN‐β gene. In the previous studies, the combination of interferon (IFN) and chemotherapeutic drugs resulted in excellent outcomes (Kim et al., 2012; Nagano et al., 2007; Ren et al., 2008; Studeny et al., 2004). SH Juang et al., also demonstrated the effect of IFN‐β in the HT‐29 human colorectal cancer cell line (Juang et al., 2004). Type I IFNs, such as IFN‐alpha (IFN‐α) or IFN‐beta (IFN‐β), have been shown to have highly suppressive effects on carcinogenesis by inhibiting cell cycle progression at the G1, S, or G2/M phase with or without induction of apoptosis (Kashiwagi et al., 2003; Ren et al., 2008).
In the present study, we investigated whether these genetically engineered stem cells (GESTECs) have a significant migrating capacity for selective targeting as well as the therapeutic value of a suicide gene/prodrug system in colorectal cancer therapy. We also confirmed the synergetic and therapeutic effects in the CD/5‐FC and IFN‐β using a double‐punch method which involves chemotherapy consisting of the suicide gene/prodrug coupled with immunotherapy using IFN‐β for the treatment of human colorectal cancer.
2. Materials and methods
2.1. Cell culture
A human colon adenocarcinoma cell line, HT‐29, was purchased from Korean Cell Line Bank (KCLB, Seoul, Korea). Three types of GESTECs, HB1.F3, HB1.F3.CD, and HB1.F3.CD.IFN‐β generated by us and primary human fibroblast (human FB) were employed in this study. All of these cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories Inc., Logan, UT, USA), 1% antibiotics (penicillin G and streptomycin; Cellgro Mediatech Inc., Manassas, VA, USA), 1% HEPES (Invitrogen Life Technologies, Calsbad, CA, USA), and 0.1% of the anti‐mycoplasmal agent Plasmocin, (Invivogen, San Diego, CA, USA) at 37 °C in a humidified atmosphere of 5% CO2‐95% air. The cells were trypsinized with 0.05% trypsin/0.02% EDTA (PAA Laboratories Int., Dartmouth, MA, USA).
2.2. RNA extraction and RT‐PCR
Total RNA extracts were prepared using TriZol Reagent (Invitrogen Life Technologies) and reverse transcribed from 1 μg of total RNA into cDNA using M‐MLV RT (iNtRON Biotechnology, Sungnam, Kyeonggido, Korea) following the manufacturer's protocols. PCR was conducted to amplify the bacterial CD and IFN‐β genes from GESTECs using the respective primers. To confirm the expression of the chemoattractant ligands and receptors including stem cell factor (SCF), c‐Kit, chemokine receptor 4 (CXCR4), vascular endothelial growth factor (VEGF) and VEGF receptor2 (VEGFR2), PCR was also conducted using the cDNA of cancer cells with sense and antisense primers based on the published sequence of the genes respectively. Human glyceraldehydes‐3‐phosphate dehydrogenase (GAPDH) gene was used as a positive control and PCR mixture without a cDNA template for a negative control. The sense and antisense oligo‐sequences and the expected size of RT‐PCR products are described in Table 1. PCR amplification was performed for 30 cycles. The reaction products were analyzed on a 1.5% agarose gel pre‐stained with ethidium bromide (EtBr; Sigma–Aldrich Corp., St. Louis, MO, USA). These results were scanned by Gel Doc 2000 (Bio‐Rad Laboratories Inc., Hercules, CA, USA) and compared to 100 bp ladder of DNA (iNtRON Biotechnology).
Table 1.
The oligonucleotide sequences of the primers used in this study and the predicted sizes of the PCR products.
| mRNA | Oligo‐sequences (5′–3′) | Expected size (bp) | |
|---|---|---|---|
| CD | Forward | GCGCGAGTCACCGCCAGCCACACCACGGC | 559 |
| Reverse | GTTTGTATTCGATGGCTTCTGGCTGC | ||
| SCF | Forward | ACTTGGATTCTCACTTGCATTT | 505 |
| Reverse | CTTTCTCAGGACTTAATGTTGAAG | ||
| c‐Kit | Forward | GCCCACAATAGATTGGTATTT | 570 |
| Reverse | AGCATCTTTACAGCGACAGTC | ||
| CXCR4 | Forward | CTCTCCAAAGGAAAGCGCAGGTGGACAT | 558 |
| Reverse | AGACTGTACACTGTAGGTGCTGAAATCA | ||
| IFN‐β | Forward | AAAGAAGCAGCAATTTTCAG | 296 |
| Reverse | TTTCTCCAGTTTTTCTTCCA | ||
| VEGF | Forward | AAGCCATCCTGTGTGCCCCTGATG | 377 |
| Reverse | GCTCCTTCCTCCTGCCCGGCTCAC | ||
| VEGFR2 | Forward | ACGCTGACATGTACGGTCTAT | 438 |
| Reverse | GCCAAGCTTGTACCATGTGAG | ||
| GAPDH | Forward | ATGTTCGTCATGGGTGTGAACCA | 351 |
| Reverse | TGGCAGGTTTTTCTAGACGGCAG |
2.3. Cell viability assay
To investigate the effect of 5‐FC and 5‐FU on HT‐29 colorectal cancer cells, the cancer cells were seeded in 96‐well plates (4000 cells/well) and cultured in 0.1 ml medium. After 24 h of pre‐incubation, HB1.F3.CD, or HB1.F3.CD.IFN‐β cells (8000 cells/well or do increase cell number 8000, 16000 and 24000 cells/well) in the medium containing 5% FBS were added to the cancer cell cultures and incubated for 24 h before the treatment with 5‐FC or 5‐FU. After 24 h, 5‐FC and 5‐FU (Sigma–Aldrich Corp.) were serially diluted with PBS (final concentration, 100, 200, 300, 400, and 500 μg/ml or constant concentration 500 ug/ml), and were added to each well in the plates and treated for 4 days. After the treatment of 5‐FC or 5‐FU, MTT (3‐(4,5‐dimethyl‐thiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assays were performed to measure the cancer cell viability. MTT solution (10 μl; 5 mg/ml) was added to each well and incubated for 4 h at 37 °C. Supernatants were removed and 100 μl of dimethyl sulfoxide (DMSO, 99.0%; Junsei Chemical Co., Ltd, Tokyo, Japan) was added to each well to dissolve the resultant formazan crystals. Optical densities were measured at 540 nm using an ELISA reader (VERSA man, Molecular Devices, CA, USA). MTT assay was carried out in duplicate.
2.4. In vitro migration assay
To investigate whether GESTECs are capable of migrating to HT‐29 (1 × 105 cells/well), HT‐29 and human FB were plated in 24‐well plates and incubated in culture media for 24 h at 37 °C. The cells were then incubated with new serum‐free media and incubated for 24 h. Transwell plates (8 μm; BD Biosciences, Franklin lakes, NJ, USA) coated with fibronectin (250 μg/ml; Sigma–Aldrich Corp.) were placed in the 24‐well plates and incubated overnight. According to the manufacturer's instruction, chloromethyl‐benzamido‐1,1′‐dioctadecyl‐3,3,3′‐tetramethylindocarbocyanin perchlorate (CM‐DiI; Invitrogen Lift Technologies) was used to labeled HB1.F3.CD or HB1.F3.CD.IFN‐β cells (1 × 105 cells/well) that were plated in the upper chambers of the transwell plates and cultured in serum‐free medium for 24 h at 37 °C. HT‐29 and human FB were stained by adding 4′,6‐diamidino‐2‐phenylindole (DAPI; 200 ng/ml, Invitrogen Life Technologies) and the plate was incubated for 20 min at 37 °C. Each well was washed with PBS and the upper side of the transwell was then scraped to remove cells that had not migrated into the transwell. The cells stained with CM‐DiI and DAPI were examined by fluorescence microscopy (IX71 Inverted Microscope, Olympus, Japan).
2.5. Colorectal cancer xenograft model
Twenty‐one 6 week old male BALB/c nude mice were purchased from Central Laboratory Animal (SLC, Shizuoka, Japan) and the experiment was conducted according to the protocols approved by the Animal Care Committee of Chungbuk National University. The mice were acclimatized in a controlled room where the temperature was set at 23 ± 1 °C and humidity was set at 40–60% under a 12 h light/dark cycle for at least 1 week prior to the experiments. The colorectal cancer cells, HT‐29 (2 × 106 cells/mouse) were injected into the right flank of the mice. Tumor size of these heterotopic xenograft models was measured by using calipers once a week and tumor volume was calculated by 0.5236 × length × width × height.
2.6. In vivo therapeutic effect of HB1.F3.CD and HB1.F3.CD.IFN‐β
To identify the therapeutic effect of HB1.F3.CD and HB1.F3.CD.IFN‐β, xenograft mouse model was employed as indicated in the aforementioned paragraph. The mice were divided into 3 groups; group 1 was treated with 0.85% saline as a negative control, group 2 was treated with HB1.F3.CD cells with 5‐FC (500 mg/kg/day), and group 3 was treated with HB1.F3.CD.IFN‐β in the presence of 5‐FC at the same dose. When the tumor volume reached 200 mm3, HB1.F3.CD or HB1.F3.CD.IFN‐β (4 × 106) cells were injected into xenografted mice next to the colorectal tumor mass subcutaneously at 0.5 cm distance. These stem cells were pre‐labeled with a CM‐DiI cell tracker prior to injection for post‐sacrifice cell detection. These pre‐labeled stem cells were injected to the adjacent colorectal cancer mass two times during the experimental period. Two days after injecting NSCs, the mice of group 1 were injected with normal saline (100 μl, i.p. explain acronym), while the mice of group 2 and 3 were injected with 5‐FC (500 mg/kg/day in 100 μl, i.p.) every 24 h. The mice were sacrificed and tumor or skin tissues were isolated 48 h after the last 5‐FC treatment for histopathological analysis. Before the mice were dead or euthanized where tumor mass reached at 20 mm size in any dimension, the survival rate and volume of tumors were measured. The survival rate was drawn by Graphpad Prism (v5.0; Graphpad software, San Diego, CA, USA) and a Chi‐square test was used to test statistical significance.
2.7. Histopathology
For histopathological analysis, tumor mass and surrounding tissue excised from colorectal cancer xenografts following necropsy were fixed at 10% normal formalin solution (Sigma–Aldrich Corp.). These fixed samples were cut into 4–6 mm thick sections, embedded in paraffin and sectioned using microtome at a 5 μm thickness. Prepared slides were stained using the hematoxylin and eosin (H&E, Sigma–Aldrich Corp.) staining method. In stained slides, skin and surrounding tissue basis structure and morphology of cancer cells in tumor mass or structure of surrounding tissues was observed by light microscope using a BX51 microscope model.
2.8. DAPI staining
To observe the distribution of NSCs (CM‐DiI fluorescent red) in the colorectal cancer mass, DAPI (nuclei fluorescent blue) staining was performed in prepared tumor section slides. After rehydration, the slides were fixed with 10% formalin (Sigma–Aldrich Corp.) solution for 10 min, washed using PBS two times, and treated with DAPI for 10 min at 37 °C. The slides were mounted with coverslips and observed by fluorescent microscopy using an IX71 inverted microscope (Olympus).
2.9. Statistical analysis
The results from most experiments are presented as the mean ± SD except survival rate. A statistical analysis was performed with a one‐way ANOVA test and/or paired t test using Graphpad Prism. P < 0.05 was considered statistically significant.
3. Results
3.1. Confirmation of the introduced genes and chemoattractant factors
Confirmation of the expression of CD and IFN‐β genes in HB1.F3.CD or HB1.F3.CD.IFN‐β cells was performed by RT‐PCR. CD gene (559 bp) was expressed in HB1.F3.CD and HB1.F3.CD.IFN‐β cells, while human IFN‐β gene (296 bp) was only detected in HB1.F3.CD.IFN‐β cells as shown in Figure 1. GAPDH was used as an internal control detected in HB1.F3.CD and HB1.F3.CD.IFN‐β cells at 351 bp. To examine whether colorectal cells express chemoattractant factors, the expressions of several chemoattractant ligands and their associated receptors were examined in HT‐29 cells by RT‐PCR. These chemoattractant ligands and receptors, i.e., SCF, CXCR4, and VEGF, were shown to be expressed in the HT‐29 cells (Figure 2).
Figure 1.
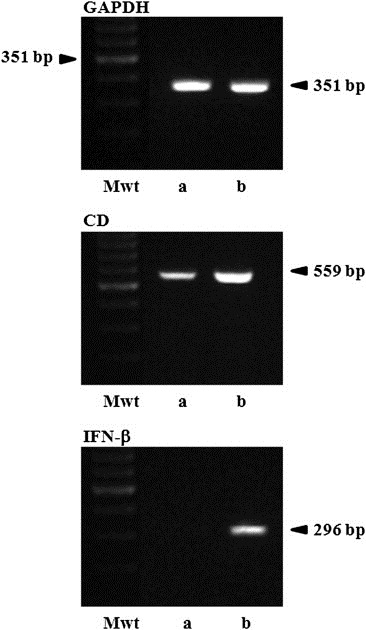
Expressions of E. coil CD and human IFN‐β genes in GESTECs. Expected products of E. coli CD or human IFN‐β genes in HB1.F3.CD and HB1.F3.CD.IFN‐β were shown at 559 bp and 296 bp, respectively. The cDNAs were synthesized from extracted RNAs of HB1.F3.CD and HB1.F3.CD.IFN‐β by RT and amplified by PCR. Next, the sizes of the PCR products were confirmed by 1.5% agarose gel electrophoresis. GAPDH was used as a control. a; HB1.F3.CD cells, b; HB1.F3.CD.IFN‐β cells, Mwt; molecular weight marker.
Figure 2.
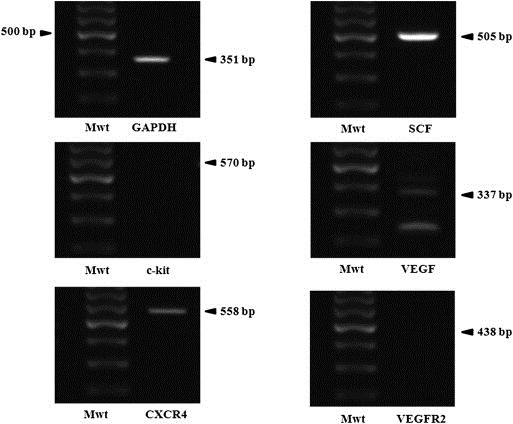
Expression of chemoattractant factors in colorectal cancer cells. After extracting total RNA from HT‐29 cells, RT‐PCR was performed. The expressions of chemoattractant ligands and receptors including SCF, VEGF, VEGFR2, CXCR4, and c‐kit, were identified by 1.5% agarose gel electrophoresis. Chemoattractant ligands and receptors, i.e., CXCR4, SCF, and VEGF, were highly expressed in HT‐29 cells. GAPDH was used an internal control Mwt: Molecular weight marker, NC: Negative control without cDNA template.
3.2. In vitro cell migration assay
A modified transwell migration assay was performed to measure the ability of GESTECs to migrate toward colorectal cancer cells. We confirmed that CM‐DiI stained stem cells, HB1.F3.CD cells, and HB1.F3.CD.IFN‐β cells significantly migrated to DAPI‐stained HT‐29 cells compared to DAPI‐stained human FB as seen in Figure 3a. The ratio of migrated stem cells was shown in Figure 3b. Migration ratio of stem cells cultured with HT‐29 cells was increased seven times compared to stem cells cultured with human FB. These results was indicated that GESTECs may have a tumor‐tropic effect towards colorectal cancer cells.
Figure 3.
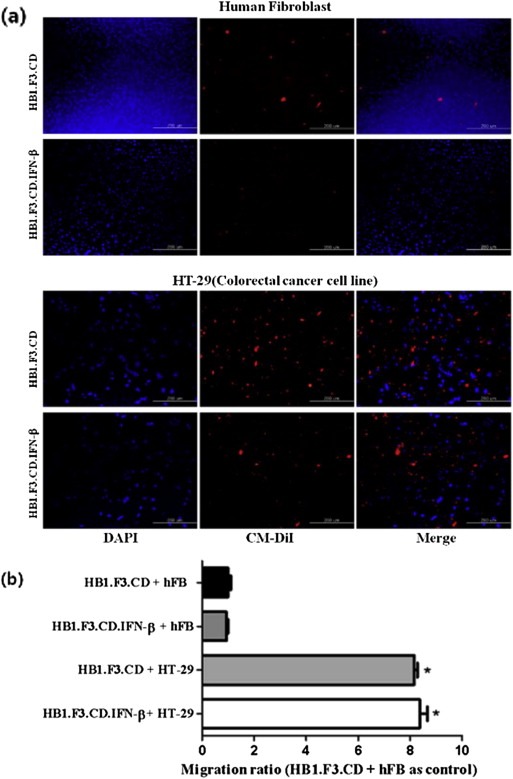
Migratory ability of GESTECs toward colorectal cancer cells. (a) Human fibroblast or HT‐29 (colorectal cancer cell; 1 × 105 cells/well) were seeded in the lower wells of 24‐well plates. HB1.F3.CD cells (1 × 105 cells/well) were stained with CD‐DiI and seeded in the fibronectin pre‐coated upper wells of 24‐well plates. DAPI staining solution was added to lower wells to observe colorectal cancer and human fibroblast. Blue stained cells indicated HT‐29 or human fibroblast as a control in the lower wells. Red stained cells indicated HB1.F3.CD or HB1.F3.CD.IFN‐β cells migrated from the upper wells toward HT‐29 colorectal or human fibroblast cells. (b) The migration ratio of stem cells was quantitated.
3.3. Effect of 5‐FC and 5‐FU on colorectal cancer cells and GESTECs
The cytotoxic effects of HB1.F3.CD and HB1.F3.CD.IFN‐β cells were confirmed by a co‐culture system using a MTT assay. To determine the effects of 5‐FC and 5‐FU on colorectal cancer cells, these cells were seeded and treated with 5‐FC and 5‐FU at increasing concentrations (100, 200, 300, 400, and 500 μg/ml). In this case, 5‐FC treatment did not decrease the growth of HT‐29 cells, whereas 5‐FU significantly inhibited the cancer cell proliferation (Figure 4a). We also examined the cytotoxic effects of HB1.F3.CD and HB1.F3.CD.IFN‐β cells on HT‐29 cells in the presence of 5‐FC. After 5‐FC (500 μg/ml) treatment, the viability of HT‐29 cells was decreased by 35.7% in the co‐culture system with HB1.F3.CD cells while HB1.F3.CD.IFN‐β cells significantly reduced the cell viability by 56.4% as shown in Figure 4b.
Figure 4.
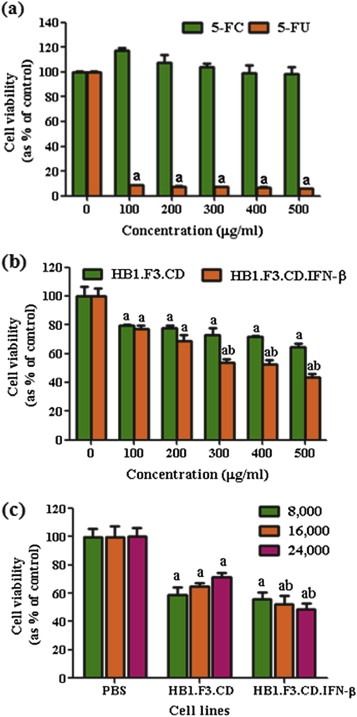
Effects of 5‐FC and 5‐FU on colorectal cancer cell growth. Proliferative rates at each concentration of 5‐FC or 5‐FU are expressed as relative fold‐change compared to the controls. (a) HT‐29 cells (4 × 103 cells/well) were seeded in 96‐well plates and treated with 5‐FC and 5‐FU at various concentrations (100, 200, 300, 400 or 500 μg/ml). (b) HB1.F3.CD and HB1.F3.CD.IFN‐β cells (8 × 103 cells/well) cultured with HT‐29 cells (4 × 103 cells/well) were treated with different concentrations of 5‐FC (100, 200, 300, 400, and 500 μg/ml). (c) HT‐29 cells (4 × 103 cells/well) were seeded in 96‐well plates and increasing numbers of HB1.F3.CD and HB1.F3.CD.IFN‐β cells (8 × 103, 1.6 × 104, or 2.4 × 104 cells/well) were added to the plates. The cells were then treated with 5‐FC or PBS (control) at a concentration of 500 μg/ml. Values are the mean ± SD for three independent experiments. a; P < 0.05 compared to the control. b; P < 0.05 compared the value of HB1.F3.CD.IFN‐β to HB1.F3.CD with identical number of cells.
In both cases, the treatment with 5‐FC resulted in a dose‐dependent inhibition of cancer cell growth. In addition, when co‐cultured with different numbers of HB1.F3.CD or HB1.F3.CD.IFN‐β cells (8 × 103, 1.6 × 104, or 2.4 × 104 cells/well), HT‐29 cell growth appeared to decrease in the presence of a high dense population of HB1.F3.CD or HB1.F3.CD.IFN‐β cells (2.4 × 104 cells/well) (Figure 4c). These results demonstrate that HB1.F3.CD or HB1.F3.CD.IFN‐β cells induced growth inhibition of colorectal cancer cells when 5‐FC is converted to 5‐FU.
3.4. Effectiveness in reducing tumor burden and enhancing survival in the colorectal cancer xenograft model
All animals injected with the colorectal cancer cell line, HT‐29 cell suspension, developed tumor mass after 4 weeks from the injection. A schematic experimental schedule was presented in Figure 5a. During the experimental period, we measured xenografted colorectal tumor mass by calipers. The growth of each tumor mass was inhibited by HB1.F3.CD and HB1.F3.CD.IFN‐β cells with 5‐FC as a prodrug. The calculated tumor mass was inhibited by HB1.F3.CD and HB1.F3.CD.IFN‐β approximately 56% and 76% respectively when compared to a control as shown in Figure 5b. These results showed that these GESTECs have the therapeutic efficacy and synergistic effect of IFN‐β in vivo. In addition, it was of interest that the mice treated with HB1.F3.CD and HB1.F3.CD.IFN‐β in the presence of 5‐FC showed a prolonged survival rate compared with the untreated control group during the animal study (Figure 5c).
Figure 5.
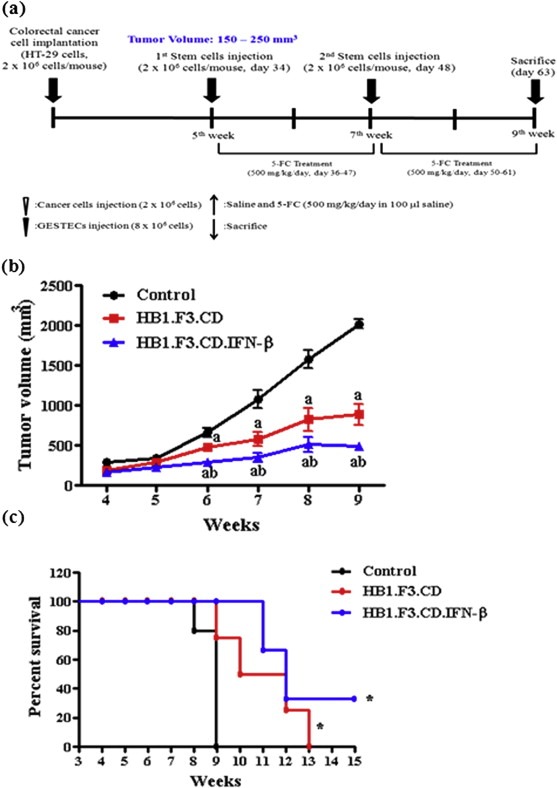
Scheme of xenograft mouse model and the changes in tumor volume following GESTEC treatment. A xenograft model was employed implanting HT‐29 (2 × 106 cells) to male BALB/c nu/nu mice. (a) During 5 weeks, tumor mass volume mostly reached at 200 mm3. At day 34 and 48, pre‐stained with CM‐DiI human NSCs (4 × 106 cells/mouse) were injected surrounding xenografted tumor mass. After 2 days injected with GESTECs, 5‐FC (500 mg/kg/day) was treated every 24 h. At day 63, the mice were sacrificed. (b) The tumor volume of xenografted mice was measured caliper twice a week. Tumor volume was calculated by length × width × high × .5236. The volume of tumor mass was decreased up to 56% in HB1.F3.CD injected mice compared to a control. In addition, the tumor mass was inhibited about 74% in HB1.F3.CD.IFN‐β treated mice. (c) The Kaplan–Meier analysis and log rank test of mouse survival showed improved survival curve in the mice injected with GESTECs in the presence of 5‐FC compared to a control (P = 0.0437, by Log rank test for trend). a, P < 0.05 compared to the control; b, P < 0.05 compared the value of HB1.F3.CD only.
3.5. Histopathological analysis of xenografted model
Histopathological examination of xenografted tumor mass and its surrounding skin at the 6th week after first treatment was used to assess alteration of tumors. There was no significant change in circumferential skin tissues (Data not shown). Although the tumor burden of a control group appeared to be more compact with tumor cells, there was a significant suppression of colorectal cancer cells in the presence of HB1.F3.CD and HB1.F3.CD.IFN‐β plus 5‐FC as shown in Figure 6a–c. Moreover, extensive tumor necrosis was observed in the tumor burden of both GESTECs‐treated mice. In HB1.F3.CD.IFN‐β treated group, the vacuoles indicating tumor necrosis were higher in number compared to a control and HB1.F3.CD treated group as shown in Figure 6 (b and c). It should also be noted that the mice treated with HB1.F3.CD.IFN‐β cells had relatively small and less pyknotic nuclei, implying the process of necrosis (Figure 6c).
Figure 6.
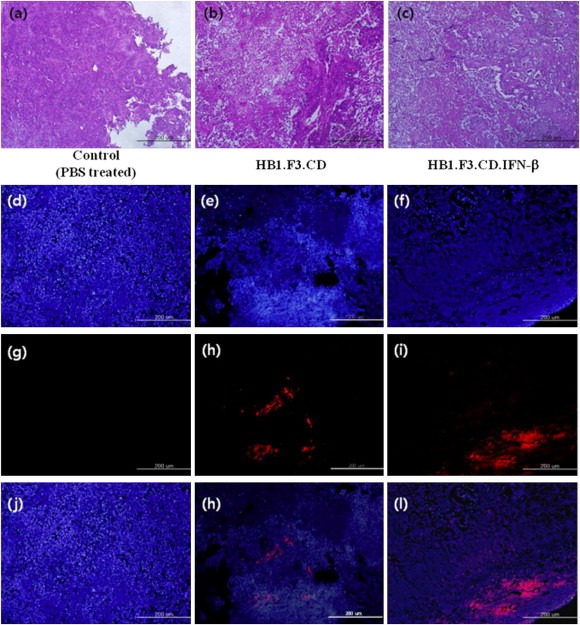
Histological analysis of xenografted tumor mass and fluorescent analysis of xenografted tumor mass. The xenografted tumor mass isolated at day 63 were fixed 10% neutral formalin and cut into 4–6 mm thick. Then, they were embedded in paraffin, sectioned 5 μm thick by a microtome. Tissue slides were stained with hematoxylin and eosin (H&E); (a)–(c). Injected GESTECs were pre‐labeled with CM‐DiI as cell tracker and prepared tissue slides were counterstained with DAPI solution. The stained tissue slides were observed under a microscope (Magnification: ×100, Blue: DAPI stained nuclei of colon cancer cells and GESTECs; (d), (e), (f). Red: CM‐DiI labeled GESTECs; (g), (h), (i). Merged; (j), (k), (l).). We observed red pre‐labeled stem cells surrounding or inside of tumor mass, indicating these GESTECs have a migratory ability in vivo. In addition less number of DAPI stained cells were observed at the surrounding areas located near red fluorescent cells.
A fluorescent analysis was further conducted to explain these changes using CM‐DiI pre‐labeled GESTECs (Figure 6d–l). We confirmed that the red fluorescent GESTECs (Figure 6h and i) were present inside of the tumor mass as seen in Figure 6h and i. These results demonstrate that these GESTECs have the migratory capability through colorectal tumor mass and effectively exert an antitumor effect by delivering therapeutic genes.
4. Discussion
Although systemic administration of 5‐FU has been used in the treatment of cancer patients for a long time, it also has severe side effects, including myelosuppression and stomatitis (Hartmann and Heidelberger, 1961). As an alternative in cancer therapy, GESTECs have advantages in the gene therapy against various types of cancers (Yi et al., 2011b; You et al., 2009; Zhao et al., 2008). The GESTECs that are employed in GEPT system can convert non‐toxic prodrug into the toxic active form. In particular, the CD/5‐FC GEPT system has been tested as an anticancer therapy for several types of cancers including breast, prostate, and colon cancers (Boucher et al., 2006; Chung‐Faye et al., 2001). In addition, as GESTECs take advantage of the tumor tropic capacity that originated from stem cells, they can be efficient delivery vehicles of exogeneous therapeutic genes to tumor sites, leading to the reduction of side effects (Kim et al., 2006; Yi et al., 2011a).
In this study, we have presented GESTECs which have the capability of therapeutic gene delivery for colorectal cancer chemotherapy. HB1.F3.CD and HB1.F3.CD.IFN‐β cells are the GESTECs manufactured to express CD and human IFN‐β genes. First, we confirmed the expression of CD and human IFN‐β genes in these GESTECs by RT‐PCR. In the in vitro modified migration assay, HB1.F3.CD and HB1.F3.CD.IFN‐β cells appeared to effectively migrate toward HT‐29 cells compared to non‐tumorigenic human fibroblasts cells. This selective migratory ability of GESTECs to cancer cells was considered by the responsiveness of GESTECs to chemoattractant factors secreted by colorectal cancer cells. In previous studies, SCF and VEGF secreted from tumor cells caused the tumor tropic effect of several stem cells (Sun et al., 2006, 2004). Also, recent studies suggested that the tumor‐targeting behavior of NSCs was mediated by chemoattractant molecules and their respective receptors, which includes SCF/c‐Kit (Sun et al., 2004), CXC chemokine receptor 4 (CXCR4) (Ehtesham et al., 2004), and VEGF and VEGF receptor (VEGFR)‐2 (Schmidt et al., 2005). By RT‐PCR, we also confirmed that these chemoattractant factors were highly expressed in HT‐29 cells. These chemoattractant molecules and their individual receptors may play a role in the intrinsic tumor specific migration of these GESTECs, which is a crucial factor in selectively delivering a therapeutic enzyme to the tumor site (Kim et al., 2006; Nakamizo et al., 2005). However, the molecular mechanisms underlying the tumor‐tropism of GESTECs through the chemoattractant factors is not clearly understood (Kucerova et al., 2007; You et al., 2009) and further study is required to confirm the role of these factors in the mechanisms of tumor cell recognition and/or tumor tropism of GESTECs.
In this study, we also examined the cytotoxic activity of these GESTECs. When co‐cultured with HT‐29 cells, HB1.F3.CD and HB1.F3.CD.IFN‐β cells decreased cancer cell growth in the presence of 5‐FC. Although colorectal cancer cells by themselves are not sensitive to a prodrug of 5‐FC (500 μg/ml), the viability of cancer cells on co‐culture system was decrease by 50% at the concentration of 5‐FC (500 μg/ml). In our previous study, the viability of HB1.F3.CD cells were decrease by nearly 75% at 100 μg/ml of 5‐FC (Kim et al., 2010). Therefore, these therapeutic stem cells appear to be mostly transduced with CD gene in this study. By increasing the number of treated HB1.F3.CD.IFN‐β cells, the proliferation of HT‐29 cells decreased more rapidly at the constant concentration of 5‐FC. When the number of GESTECs was constant, 5‐FC at various concentrations (100–500 μg/ml) inhibited the cancer cell growth in a dose‐dependent manner. It should be noted that HB1.F3.CD.IFN‐β gene cells expressing the CD gene and IFN‐β decreased cell growth of HT‐29 cells more than HB1.F3.CD cells alone. This result demonstrates the synergistic effect of HB1.F3.CD.IFN‐β cells by the combined effect of two fused gene expression, CD and IFN‐β, even though the individual therapeutic actions appear to be different. CD acts as a prodrug activating enzyme and IFN‐β can enhance anti‐angiogenic effects and immune responses. The anticancer activity of these GESTECs on colorectal cancer cells can be attributed to the cytotoxic effect of their gene products and the bystander effect (Huber et al., 1994; Mullen et al., 1992).
An in vivo xenograft mouse model was further employed to prove the efficiency of these GESTECs in vitro. In in vivo assays, GESTECs that express CD and IFN‐β genes significantly inhibited tumor growth. The volume of the tumor mass was decreased up to 56% in HB1.F3.CD injected mice when compared to a control. In contrast, the tumor mass was further inhibited about 76% in HB1.F3.CD.IFN‐β injected mice which supports the hypothesis that HB1.F3.CD.IFN–β expressing both the CD and IFN‐β genes may have a synergistic anticancer effect compared to HB1.F3.CD only against HT‐29 cells. Although the tissue structure from a control group appeared to be more compact with tumor cells, there was a significant suppression of colorectal cancer cells in the presence of HB1.F3.CD and HB1.F3.CD.IFN‐β plus 5‐FC. Moreover, extensive tumor necrosis was observed in the tumor burden of both GESTECs‐treated mice, and the mice treated with HB1.F3.CD.IFN‐β cells have relatively small and less pyknotic nucleus. These results indicate that GESTECs expressing both CD and IFN‐β has a synergistic anti‐tumor effect for selectively target human colorectal cancer cells (Kohase et al., 1986). In addition, these therapeutic stem cells should be examined following intravenous application as well as xenografted application for clinical use.
In summary, these results suggest that the GESTECs expressing CD and/or IFN‐β genes may selectively migrate toward colorectal cancer cells and exert a synergistic anti‐tumor effect by the fusion gene. Therefore, the GESTECs derived from HB1.F3 cells have a therapeutic potential for selectively targeting colorectal cancer as an efficient delivery vehicle for therapeutic genes and can provide a progressive approach in GEPT.
Disclosure statement
The authors do not have any conflict of interest to publish this paper.
Acknowledgments
This work was supported by two National Research Foundation of Korea (NRF) grants funded by the Ministry of Education, Science and Technology (MEST) of Korea Government (no. 2010‐0003093 and 2011‐0015385). In addition, the authors appreciated Mr. Chang‐Hwang Ahn for his excellent technical support to perform the study.
Yi Bo-Rim, Park Min-Ah, Lee Hye-Rim, Kang Nam-Hee, Choi Kelvin J., Kim Seung U., Choi Kyung-Chul, (2013), Suppression of the growth of human colorectal cancer cells by therapeutic stem cells expressing cytosine deaminase and interferon‐β via their tumor‐tropic effect in cellular and xenograft mouse models, Molecular Oncology, 7, doi: 10.1016/j.molonc.2013.01.004.
References
- Aboody, K.S. , Bush, R.A. , Garcia, E. , Metz, M.Z. , Najbauer, J. , Justus, K.A. , Phelps, D.A. , Remack, J.S. , Yoon, K.J. , Gillespie, S. , Kim, S.U. , Glackin, C.A. , Potter, P.M. , Danks, M.K. , 2006. Development of a tumor-selective approach to treat metastatic cancer. PLoS One 1, e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboody, K.S. , Najbauer, J. , Danks, M.K. , 2008. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 15, 739–752. [DOI] [PubMed] [Google Scholar]
- Altaner, C. , 2008. Prodrug cancer gene therapy. Cancer Lett. 270, 191–201. [DOI] [PubMed] [Google Scholar]
- Bird, N.C. , Mangnall, D. , Majeed, A.W. , 2006. Biology of colorectal liver metastases: a review. J. Surg. Oncol. 94, 68–80. [DOI] [PubMed] [Google Scholar]
- Boucher, P.D. , Im, M.M. , Freytag, S.O. , Shewach, D.S. , 2006. A novel mechanism of synergistic cytotoxicity with 5-fluorocytosine and ganciclovir in double suicide gene therapy. Cancer Res. 66, 3230–3237. [DOI] [PubMed] [Google Scholar]
- Brown, A.B. , Yang, W. , Schmidt, N.O. , Carroll, R. , Leishear, K.K. , Rainov, N.G. , Black, P.M. , Breakefield, X.O. , Aboody, K.S. , 2003. Intravascular delivery of neural stem cell lines to target intracranial and extracranial tumors of neural and non-neural origin. Hum. Gene Ther. 14, 1777–1785. [DOI] [PubMed] [Google Scholar]
- Chung-Faye, G.A. , Chen, M.J. , Green, N.K. , Burton, A. , Anderson, D. , Mautner, V. , Searle, P.F. , Kerr, D.J. , 2001. In vivo gene therapy for colon cancer using adenovirus-mediated, transfer of the fusion gene cytosine deaminase and uracil phosphoribosyltransferase. Gene Ther. 8, 1547–1554. [DOI] [PubMed] [Google Scholar]
- Conrad, C. , Gupta, R. , Mohan, H. , Niess, H. , Bruns, C.J. , Kopp, R. , von Luettichau, I. , Guba, M. , Heeschen, C. , Jauch, K.W. , Huss, R. , Nelson, P.J. , 2007. Genetically engineered stem cells for therapeutic gene delivery. Curr. Gene Ther. 7, 249–260. [DOI] [PubMed] [Google Scholar]
- Crystal, R.G. , Hirschowitz, E. , Lieberman, M. , Daly, J. , Kazam, E. , Henschke, C. , Yankelevitz, D. , Kemeny, N. , Silverstein, R. , Ohwada, A. , Russi, T. , Mastrangeli, A. , Sanders, A. , Cooke, J. , Harvey, B.G. , 1997. Phase I study of direct administration of a replication deficient adenovirus vector containing the E. coli cytosine deaminase gene to metastatic colon carcinoma of the liver in association with the oral administration of the pro-drug 5-fluorocytosine. Hum. Gene Ther. 8, 985–1001. [DOI] [PubMed] [Google Scholar]
- Dietrich, J. , Imitola, J. , Kesari, S. , 2008. Mechanisms of disease: the role of stem cells in the biology and treatment of gliomas. Nat. Clin. Pract. Oncol. 5, 393–404. [DOI] [PubMed] [Google Scholar]
- Douillard, J.Y. , Cunningham, D. , Roth, A.D. , Navarro, M. , James, R.D. , Karasek, P. , Jandik, P. , Iveson, T. , Carmichael, J. , Alakl, M. , Gruia, G. , Awad, L. , Rougier, P. , 2000. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 355, 1041–1047. [DOI] [PubMed] [Google Scholar]
- Ehtesham, M. , Yuan, X. , Kabos, P. , Chung, N.H. , Liu, G. , Akasaki, Y. , Black, K.L. , Yu, J.S. , 2004. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia 6, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde, R. , 2006. Stem cells and metastatic cancer: fatal attraction?. PLoS Med. 3, e482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag, S.O. , Khil, M. , Stricker, H. , Peabody, J. , Menon, M. , DePeralta-Venturina, M. , Nafziger, D. , Pegg, J. , Paielli, D. , Brown, S. , Barton, K. , Lu, M. , Aguilar-Cordova, E. , Kim, J.H. , 2002. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 62, 4968–4976. [PubMed] [Google Scholar]
- Hartmann, K.U. , Heidelberger, C. , 1961. Studies on fluorinated pyrimidines. XIII. Inhibition of thymidylate synthetase. J. Biol. Chem. 236, 3006–3013. [PubMed] [Google Scholar]
- Hirschowitz, E.A. , Ohwada, A. , Pascal, W.R. , Russi, T.J. , Crystal, R.G. , 1995. In vivo adenovirus-mediated gene transfer of the Escherichia coli cytosine deaminase gene to human colon carcinoma-derived tumors induces chemosensitivity to 5-fluorocytosine. Hum. Gene Ther. 6, 1055–1063. [DOI] [PubMed] [Google Scholar]
- Huber, B.E. , Austin, E.A. , Richards, C.A. , Davis, S.T. , Good, S.S. , 1994. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc. Natl. Acad. Sci. U. S. A. 91, 8302–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang, S.H. , Wei, S.J. , Hung, Y.M. , Hsu, C.Y. , Yang, D.M. , Liu, K.J. , Chen, W.S. , Yang, W.K. , 2004. IFN-beta induces caspase-mediated apoptosis by disrupting mitochondria in human advanced stage colon cancer cell lines. J. Interferon. Cytokine Res. 24, 231–243. [DOI] [PubMed] [Google Scholar]
- Kaliberov, S.A. , Market, J.M. , Gillespie, G.Y. , Krendelchtchikova, V. , Della Manna, D. , Sellers, J.C. , Kaliberova, L.N. , Black, M.E. , Buchsbaum, D.J. , 2007. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 14, 1111–1119. [DOI] [PubMed] [Google Scholar]
- Kanai, F. , Lan, K.H. , Shiratori, Y. , Tanaka, T. , Ohashi, M. , Okudaira, T. , Yoshida, Y. , Wakimoto, H. , Hamada, H. , Nakabayashi, H. , Tamaoki, T. , Omata, M. , 1997. In vivo gene therapy for alpha-fetoprotein-producing hepatocellular carcinoma by adenovirus-mediated transfer of cytosine deaminase gene. Cancer Res. 57, 461–465. [PubMed] [Google Scholar]
- Kashiwagi, K. , Furusyo, N. , Kubo, N. , Nakashima, H. , Nomura, H. , Kashiwagi, S. , Hayashi, J. , 2003. A prospective comparison of the effect of interferon-alpha and interferon-beta treatment in patients with chronic hepatitis C on the incidence of hepatocellular carcinoma development. J. Infect. Chemother. 9, 333–340. [DOI] [PubMed] [Google Scholar]
- Kim, K.Y. , Kim, S.U. , Leung, P.C. , Jeung, E.B. , Choi, K.C. , 2010. Influence of the prodrugs 5-fluorocytosine and CPT-11 on ovarian cancer cells using genetically engineered stem cells: tumor-tropic potential and inhibition of ovarian cancer cell growth. Cancer Sci. 101, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.Y. , Yi, B.R. , Lee, H.R. , Kang, N.H. , Jeung, E.B. , Kim, S.U. , Choi, K.C. , 2012. Stem cells with fused gene expression of cytosine deaminase and interferon-beta migrate to human gastric cancer cells and result in synergistic growth inhibition for potential therapeutic use. Int. J. Oncol. 40, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.K. , Kim, S.U. , Park, I.H. , Bang, J.H. , Aboody, K.S. , Wang, K.C. , Cho, B.K. , Kim, M. , Menon, L.G. , Black, P.M. , Carroll, R.S. , 2006. Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression. Clin. Cancer Res. 12, 5550–5556. [DOI] [PubMed] [Google Scholar]
- Kim, S.U. , 2011. Neural stem cell-based gene therapy for brain tumors. Stem Cell Rev. 7, 130–140. [DOI] [PubMed] [Google Scholar]
- Kim, S.U. , 2004. Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology 24, 159–171. [DOI] [PubMed] [Google Scholar]
- Kim, S.U. , 2007. Genetically engineered human neural stem cells for brain repair in neurological diseases. Brain Dev. 29, 193–201. [DOI] [PubMed] [Google Scholar]
- Kim, S.U. , Jeung, E.B. , Kim, Y.B. , Cho, M.H. , Choi, K.C. , 2011. Potential tumor-tropic effect of genetically engineered stem cells expressing suicide enzymes to selectively target invasive cancer in animal models. Anticancer Res. 31, 1249–1258. [PubMed] [Google Scholar]
- Kim, S.U. , Nakagawa, E. , Hatori, K. , Nagai, A. , Lee, M.A. , Bang, J.H. , 2002. Production of immortalized human neural crest stem cells. Methods Mol. Biol. 198, 55–65. [DOI] [PubMed] [Google Scholar]
- Kohase, M. , Henriksen-DeStefano, D. , May, L.T. , Vilcek, J. , Sehgal, P.B. , 1986. Induction of beta 2-interferon by tumor necrosis factor: a homeostatic mechanism in the control of cell proliferation. Cell 45, 659–666. [DOI] [PubMed] [Google Scholar]
- Kucerova, L. , Altanerova, V. , Matuskova, M. , Tyciakova, S. , Altaner, C. , 2007. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 67, 6304–6313. [DOI] [PubMed] [Google Scholar]
- Milsom, J.W. , Bohm, B. , Hammerhofer, K.A. , Fazio, V. , Steiger, E. , Elson, P. , 1998. A prospective, randomized trial comparing laparoscopic versus conventional techniques in colorectal cancer surgery: a preliminary report. J. Am. Coll. Surg. 187, 46–54. discussion 54–45 [DOI] [PubMed] [Google Scholar]
- Mullen, C.A. , Kilstrup, M. , Blaese, R.M. , 1992. Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5-fluorocytosine: a negative selection system. Proc. Natl. Acad. Sci. U. S. A. 89, 33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, F.J. , Snyder, E.Y. , Loring, J.F. , 2006. Gene therapy: can neural stem cells deliver?. Nat. Rev. 7, 75–84. [DOI] [PubMed] [Google Scholar]
- Nagano, H. , Miyamoto, A. , Wada, H. , Ota, H. , Marubashi, S. , Takeda, Y. , Dono, K. , Umeshita, K. , Sakon, M. , Monden, M. , 2007. Interferon-alpha and 5-fluorouracil combination therapy after palliative hepatic resection in patients with advanced hepatocellular carcinoma, portal venous tumor thrombus in the major trunk, and multiple nodules. Cancer 110, 2493–2501. [DOI] [PubMed] [Google Scholar]
- Nakamizo, A. , Marini, F. , Amano, T. , Khan, A. , Studeny, M. , Gumin, J. , Chen, J. , Hentschel, S. , Vecil, G. , Dembinski, J. , Andreeff, M. , Lang, F.F. , 2005. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 65, 3307–3318. [DOI] [PubMed] [Google Scholar]
- Nakamura, K. , Ito, Y. , Kawano, Y. , Kurozumi, K. , Kobune, M. , Tsuda, H. , Bizen, A. , Honmou, O. , Niitsu, Y. , Hamada, H. , 2004. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 11, 1155–1164. [DOI] [PubMed] [Google Scholar]
- Pessino, A. , Sobrero, A. , 2006. Optimal treatment of metastatic colorectal cancer. Expert Rev. Anticancer Ther. 6, 801–812. [DOI] [PubMed] [Google Scholar]
- Reiser, J. , Zhang, X.Y. , Hemenway, C.S. , Mondal, D. , Pradhan, L. , La Russa, V.F. , 2005. Potential of mesenchymal stem cells in gene therapy approaches for inherited and acquired diseases. Expert Opin. Biol. Ther. 5, 1571–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, C. , Kumar, S. , Chanda, D. , Kallman, L. , Chen, J. , Mountz, J.D. , Ponnazhagan, S. , 2008. Cancer gene therapy using mesenchymal stem cells expressing interferon-beta in a mouse prostate cancer lung metastasis model. Gene Ther. 15, 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saukkonen, K. , Hemminki, A. , 2004. Tissue-specific promoters for cancer gene therapy. Expert Opin. Biol. Ther. 4, 683–696. [DOI] [PubMed] [Google Scholar]
- Schmidt, N.O. , Przylecki, W. , Yang, W. , Ziu, M. , Teng, Y. , Kim, S.U. , Black, P.M. , Aboody, K.S. , Carroll, R.S. , 2005. Brain tumor tropism of transplanted human neural stem cells is induced by vascular endothelial growth factor. Neoplasia 7, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studeny, M. , Marini, F.C. , Dembinski, J.L. , Zompetta, C. , Cabreira-Hansen, M. , Bekele, B.N. , Champlin, R.E. , Andreeff, M. , 2004. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 96, 1593–1603. [DOI] [PubMed] [Google Scholar]
- Sun, L. , Hui, A.M. , Su, Q. , Vortmeyer, A. , Kotliarov, Y. , Pastorino, S. , Passaniti, A. , Menon, J. , Walling, J. , Bailey, R. , Rosenblum, M. , Mikkelsen, T. , Fine, H.A. , 2006. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9, 287–300. [DOI] [PubMed] [Google Scholar]
- Sun, L. , Lee, J. , Fine, H.A. , 2004. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J. Clin. Invest. 113, 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa, K. , Fisher, W.E. , Brunicardi, F.C. , 2002. Current progress in suicide gene therapy for cancer. World J. Surg. 26, 783–789. [DOI] [PubMed] [Google Scholar]
- Yi, B.R. , Kang, N.H. , Hwang, K.A. , Kim, S.U. , Jeung, E.B. , Choi, K.C. , 2011. Antitumor therapeutic effects of cytosine deaminase and interferon-beta against endometrial cancer cells using genetically engineered stem cells in vitro. Anticancer Res. 31, 2853–2861. [PubMed] [Google Scholar]
- Yi, B.R. , O, S.N. , Kang, N.H. , Hwang, K.A. , Kim, S.U. , Jeung, E.B. , Kim, Y.B. , Heo, G.J. , Choi, K.C. , 2011. Genetically engineered stem cells expressing cytosine deaminase and interferon-beta migrate to human lung cancer cells and have potentially therapeutic anti-tumor effects. Int. J. Oncol. 39, 833–839. [DOI] [PubMed] [Google Scholar]
- You, M.H. , Kim, W.J. , Shim, W. , Lee, S.R. , Lee, G. , Choi, S. , Kim, D.Y. , Kim, Y.M. , Kim, H. , Han, S.U. , 2009. Cytosine deaminase-producing human mesenchymal stem cells mediate an antitumor effect in a mouse xenograft model. J. Gastroenterol. Hepatol. 24, 1393–1400. [DOI] [PubMed] [Google Scholar]
- Zhao, D. , Najbauer, J. , Garcia, E. , Metz, M.Z. , Gutova, M. , Glackin, C.A. , Kim, S.U. , Aboody, K.S. , 2008. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol. Cancer Res. 6, 1819–1829. [DOI] [PubMed] [Google Scholar]