

Abstract
In this proof‐of‐principle study, we sought to define whether targeted capture massively parallel sequencing can be employed to determine the origin of metastatic deposits in cases of synchronous primary malignancies and metastases in distinct anatomical sites. DNA samples extracted from synchronous tumor masses in the breast, adnexal, and pelvic‐peritoneal regions from a 62‐year‐old BRCA1 germline mutation carrier were subjected to targeted massively parallel sequencing using a platform comprising 300 cancer genes known to harbor actionable mutations. In addition to BRCA1 germline mutations, all lesions harbored somatic loss of the BRCA1 wild‐type allele and TP53 somatic mutations. The primary breast cancer displayed a TP53 frameshift (p.Q317fs) mutation, whereas and the adnexal lesion harbored a TP53 nonsense (p.R213*) mutation, consistent with a diagnosis of two independent primary tumors (i.e. breast and ovarian cancer). The adnexal tumor and all pelvic‐peritoneal implants harbored identical TP53 (p.R213*) and NCOA2 (p.G952R) somatic mutations. Evidence of genetic heterogeneity within and between lesions was observed, both in terms of somatic mutations and copy number aberrations. The repertoires of somatic genetic aberrations found in the breast, ovarian, and pelvic‐peritoneal lesions provided direct evidence in support of the distinct origin of the breast and ovarian cancers, and established that the pelvic‐peritoneal implants were clonally related to the ovarian lesion. These observations were consistent with those obtained with immunohistochemical analyses employing markers to differentiate between carcinomas of the breast and ovary, including WT1 and PAX8. Our results on this case of a patient with BRCA1‐mutant breast and ovarian cancer demonstrate that massively parallel sequencing may constitute a useful tool to define the relationship, clonality and intra‐tumor genetic heterogeneity between primary tumor masses and their metastatic deposits in patients with multiple primary malignancies and synchronous metastases.
Keywords: Massively parallel sequencing, Clonality, Breast cancer, Ovarian cancer, Metastasis, BRCA1
Highlights
Massively parallel sequencing can help define the origin of metastatic deposits in patients with multiple primary tumors.
Genetic heterogeneity was observed within and between lesions from the same patient.
Synchronous metastatic deposits may differ in their repertoire of somatic genetic aberrations.
1. Introduction
Massively parallel sequencing analysis is reshaping the way the genomic, transcriptomic and epigenomic analyses of human tumors are performed (McDermott et al., 2011; Natrajan and Reis‐Filho, 2011; Sandoval and Esteller, 2012; Yates and Campbell, 2012). This technology, when applied to the study of germline DNA, has resulted in the identification of genes whose mutations result in increased cancer predisposition (Natrajan and Reis‐Filho, 2011; Yates and Campbell, 2012) or are causative of hereditary diseases (Poduri et al., 2013). In the context of somatic genetics and cancer transcriptomics, massively parallel sequencing analyses have not only led to the characterization of the repertoire of somatic genetic aberrations in human cancers, but also to the unraveling of intra‐tumor genetic heterogeneity, mutational signatures in human cancer, identification of mutations that can explain response or resistance to specific therapeutic agents, and characterization of novel fusion genes and ‘read‐throughs’ in epithelial malignancies (Alexandrov et al., 2013; Aparicio and Caldas, 2013; Castellarin et al., 2013; Crockford et al., 2013; Li and Durbin, 2009; McDermott et al., 2011; Natrajan and Reis‐Filho, 2011; Turner and Reis‐Filho, 2012; Yap et al., 2012).
Massively parallel sequencing approaches have recently been successfully employed in the characterization of the repertoire of mutations in biological materials other than primary or metastatic tumor tissue, including circulating cell‐free plasma DNA and cervical cytological preparations, where the genetic information provided by these analyses may constitute a way of monitoring disease progression and response to specific therapies (De Mattos‐Arruda et al., 2013; Kinde et al., 2013). Furthermore, this technology is rapidly being incorporated into the diagnostic armamentarium of oncologists and pathologists, in particular in the form of targeted sequencing approaches (i.e. massively parallel sequencing approaches based on the sequencing of a limited number of clinically relevant or biologically important genes or mutations) (Domchek et al., 2013; Wagle et al., 2012).
One of the potential applications of massively parallel sequencing is establishing the clonality between neoplastic lesions. The establishment of clonality between different tumor masses is particularly important in cases where patients present with synchronous primary disease and metastatic deposits, and there is uncertainty as to whether some of the metastatic deposits may constitute a second primary cancer. For example, in BRCA1 and BRCA2 germline mutation carriers, patients may present with both breast, abdominal and pelvic disease, and establishing whether the patient has two primaries (breast and ovarian) or metastatic disease is of clinical importance, given the therapeutic implications these diagnoses would have. Although histopathological and immunohistochemical analysis of primary tumors and metastatic deposits remain the lynchpins of diagnostic pathology and the usual means to define the primary site of a carcinoma of unknown primary site or whether a metastatic deposit stemmed from a given primary tumor, these approaches are by no means infallible, given potential changes in immunohistochemical markers as disease progresses from primary to metastatic (Amir et al., 2012; Niikura et al., 2012), overlap in the distribution of markers between metastatic tumors from different sites (Amir et al., 2012), and technical issues (Pusztai et al., 2010).
In this proof‐of‐principle study, we describe the use of targeted massively parallel sequencing as a means to define the site of origin of multiple metastases in a patient with two primary malignancies. Using a combination of massively parallel sequencing and bioinformatic methods, we analyzed a set of paraffin‐embedded tumor tissue samples from a patient with synchronous breast and adnexal masses and pelvic‐peritoneal metastases, whose histopathological and immunohistochemical features were insufficient to determine the origin of the metastatic deposits. Based on the repertoire of mutations found in each sample analyzed, not only the site of origin of the metastatic deposits could be defined, but also the clonal relatedness of the lesions biopsied, demonstrating the potential of this approach to provide answers for challenging clinical scenarios.
2. Material and methods
This study was approved by the IRB of Vall d'Hebron Institute of Oncology (Barcelona, Spain).
2.1. Clinical history
A 62‐year‐old Caucasian woman was referred in early 2012 to the Vall d'Hebron Institute of Oncology with history of a palpable 6 cm mass (largest diameter) in the right breast and a pelvic mass, which have grown in size over the last 12 months. The patient had no personal history of cancer. Family history was investigated and an unconfirmed history of pelvic cancer of the patient's mother was inferred.
Mammography revealed abnormal radiological features (Figure 1A and B), and a core biopsy of the breast mass was performed. Histological examination revealed a high‐grade invasive ductal carcinoma of no special type of the breast. Immunohistochemical analysis was performed using antibodies against estrogen receptor (ER) (Novocastra, Leica Biosystems Newcastle Ltd, UK, 6F11, 7 ml Bond ready‐to‐use, Heat Induced Epitope Retrieval (HIER) pH 9), progesterone receptor (PR) (Novocastra, Leica Biosystems Newcastle Ltd, UK, SAN27, 7 ml Bond ready‐to‐use, HIER pH 6) and HER2 (PATHWAY® HER2, clone 4B5; Ventana Medical Systems Inc., Tucson, AZ, USA), which demonstrated that approximately 1% of the neoplastic cells expressed ER, but lacked PR and HER2 expression (Supplementary Figure S1).
Figure 1.

Radiological assessment of the breast and ovarian tumors, and metastatic deposits. Cranial‐caudal (left) and mediolateral‐oblique (right) mammograms depicting a spiculated 60 × 45 mm mass (*) at the upper inner quadrant of the right breast at diagnosis (A1 and B1) and after 11 months of systemic therapy (A2 and B2). Note the substantial tumor shrinkage. Abdominal/pelvic magnetic resonance imaging depicting the presence of a heterogeneous left‐adnexal mass (10 × 9 cm, T2‐weighted sequence in coronal plane (C) and sagittal plane (D). Pelvic‐peritoneal implants are shown (arrowhead, ►) contacting the superior wall of the urinary bladder (E, coronal plane), descending colon (F, axial plane), peri‐sigmoid colon (G, coronal plane), and pelvis (H, axial plane). Arrowheads (►) depict the tumor masses and open arrowheads (➤) depict the descending (F) and sigmoid (G) colons.
Clinical and radiological work‐up was performed. Chest, abdominal and pelvic computerized tomography (CT) scans, and abdominal and pelvic magnetic resonance imaging (MRI) revealed the presence of a heterogeneous left‐adnexal mass (10 × 9 cm), pelvic‐peritoneal implants and ascites (Figure 1C–H). A positron emission tomography (PET)/CT scan was subsequently performed and depicted hypermetabolic breast and pelvic‐peritoneal masses and enlarged paraaortic lymph nodes. Complete blood count, chemistry profile and tumor markers were obtained, including CA‐125 (2321 U/mL; upper limit of 35 U/mL) and CA15.3 (442.7 U/mL; upper limit of 35 U/mL). Biopsies of the left‐adnexal mass and pelvic‐peritoneal implants were performed. Histopathological analysis of these lesions revealed poorly differentiated adenocarcinomas (Figure 2). Immunohistochemical analysis of the left‐adnexal mass and pelvic‐peritoneal implants demonstrated that these lesions, in a way akin to the high‐grade invasive ductal carcinoma of no special type of the breast, were ER positive (60%), but lacked PR and HER2 expression. Based on these results, the histological and immunohistochemical findings were inconclusive as to whether the adnexal mass and the pelvic‐peritoneal implants would constitute metastatic deposits of the breast cancer, or two independent synchronous primary cancers, namely a high‐grade breast cancer and a primary high‐grade serous carcinoma with peritoneal metastases.
Figure 2.

Representative micrographs of the breast and ovarian tumors and the pelvic‐peritoneal implants. Representative micrographs of the breast (A), ovarian (B), urinary bladder (C), descending colon (D), sigmoid colon (E), and pelvic (F) lesions. Note that all lesions are composed of solid masses of atypical cells, with high nuclear‐cytoplasmic ratio and pleomorphic nuclei. Necrosis and mitotic figures were present. Hematoxylin & eosin staining; original magnification ×20.
Optimal surgical debulking was performed and included total abdominal hysterectomy, bilateral salpingo‐oophorectomy, and resection of implants on the urinary bladder, descending colon, sigmoid colon and pelvic peritoneum. The results of the histological and immunohistochemical analyses of the surgical specimens were identical to those obtained from the analyses of the biopsies from the breast, left‐adnexal and pelvic‐peritoneal lesions. Initial assessment based on the pathological features, the left‐adnexal and pelvic‐peritoneal tumor deposits were considered consistent with breast cancer metastases. A first‐line paclitaxel‐based chemotherapy, intended to treat the metastatic breast cancer, was initiated, and the patient was referred to genetic counseling for BRCA1 and BRCA2 germline testing, which was performed as previously described (Schouten et al., 2002). In brief, exons 2 to 24 (except 4) of BRCA1 and exons 2 to 27 of BRCA2 and flanking intronic sequences were analyzed using the Multiplex Ligation‐dependent Probe Amplification (MLPA) assay (MRC Holland, Amsterdam, The Netherlands). Written informed consent was obtained from the patient for the BRCA1 and BRCA2 testing and further genetic analyses of the samples. A large, germline deletion of exons 1 and 2 of the BRCA1 gene was confirmed using clinically validated methods for BRCA1 and BRCA2 testing.
Following the results of the BRCA1 testing, the therapy was subsequently changed to a regimen based on carboplatin and paclitaxel, intended as neoadjuvant and adjuvant treatment for the breast and ovarian lesions, respectively. Radiological and clinical evaluation demonstrated a reduction in size of the breast tumor by 12% (stable disease as per RECIST criteria) and a dramatic decrease in CA15.3 (56 U/mL; baseline 442.7 U/mL). After 20 weeks of therapy, the patient achieved a partial response of the breast cancer and no evidence of recurrence of the ovarian lesion. Subsequently, the patient underwent a breast wide local excision, which confirmed the results of the initial histological and immunohistochemical analyses. Local radiotherapy to the breast was performed and the patient is undergoing clinical and radiological follow‐up.
Following the results of the BRCA1 and BRCA2 testing and the targeted massively parallel sequencing analysis of the lesions (see below), representative 4 μm‐tick sections of the breast lesion, adnexal mass, and pelvic‐peritoneal implants were subjected to immunohistochemical analysis using antibodies against paired box gene 8 (PAX8), Wilms' Tumor 1 (WT1), cancer antigen 125 (CA125), gross cystic disease fluid protein 15 (GCDFP‐15), Cytokeratin (CK) 7 and CK20, essentially as previously described (Nonaka et al., 2008; Tornos et al., 2005) in the Immunohistochemistry Laboratory at the Department of Pathology of Memorial Sloan‐Kettering Cancer Center.
2.2. DNA extraction
Representative histological sections from all tissue samples were reviewed by a histopathologist (P.N.) and tumor cellularity was assessed. Five 10 μm‐thick sections of the primary breast lesion, left adnexal lesion, and four pelvic‐peritoneal implants were cut, stained with nuclear fast red and subjected to microdissection with a needle under a stereomicroscope when possible, to maximize the tumor cell content (Hernandez et al., 2012). DNA from microdissected samples and from peripheral blood leukocytes was extracted with RecoverAll™ Total Nucleic Acid Isolation Kit (Ambion, Austin, TX, USA) for formalin‐fixed paraffin‐embedded tissue and QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA), respectively, according to the manufacturer's instructions.
2.3. Targeted massively parallel sequencing
DNA samples from each lesion and germline DNA obtained from peripheral blood leukocytes were subjected to targeted capture massively parallel sequencing using the IMPACT platform (Wagle et al., 2012). Custom oligonucleotides (NimblegenSeqCap) were designed for hybridization capture of all protein‐coding exons of 300 key cancer‐associated genes (Supplementary Table S1). Barcoded sequence libraries were prepared (New England Biolabs, KapaBiosystems) using 36–250 ng DNA and pooled at equimolar concentrations into a single exon capture reaction as previously described (Wagle et al., 2012). Sequencing was performed in a single lane of an Illumina HiSeq2000 (San Diego, CA), and reads were aligned to the reference human genome hg19 using the Burrows‐Wheeler Aligner (BWA) (DePristo et al., 2011; Li and Durbin, 2009). Somatic mutations were called using muTect (Cibulskis et al., 2013) for single base substitutions, and Somatic Indel Detector (McKenna et al., 2010) for small insertions and deletions (indels), and all candidate mutations were reviewed manually using the Integrative Genomics Viewer (Robinson et al., 2011). Mutations with allelic frequency of <1% and/or supported by <5 reads were disregarded.
The mean sequence coverage of each target exon was subjected to a loess normalization to adjust for bias in nucleotide composition (G + C) and compared to the diploid normal sample. Gene copy number profiles were generated using circular binary segmentation; the CGH call package was employed to define somatic gene copy number gains and losses (van de Wiel et al., 2007).
A phylogenetic tree was generated from the mutant allele frequencies, from the presence/absence of mutations or smoothed gene copy number data using the neighbor‐joining method (Saitou and Nei, 1987) implemented in the APE package (Paradis et al., 2004). Mutations with mutant allele frequencies <1% were not included in the analysis. The tree was rooted by a ‘normal tissue’ which did not have any of the somatic mutations or copy number aberrations.
3. Results
Given the inconclusive nature of the results of the histological and immunohistochemical ER, PR and HER2 analyses, we sought to define whether targeted massively parallel sequencing of the breast, ovarian and pelvic‐peritoneal lesions would provide direct evidence i) to determine whether the breast and ovarian lesions would constitute synchronous primary malignancies or would be clonally related (i.e. one would constitute a metastatic deposit from the other), and ii) to ascertain whether the pelvic‐peritoneal implants would originate from the breast or ovarian cancer, or would constitute independent peritoneal carcinomas in the context of a patient with a BRCA1 germline mutation.
Targeted capture massively parallel sequencing yielded average read depths ranging from 117× to 463× in the archival primary and spatially distinct metastatic tumor samples, and 465× in the normal sample. This analysis confirmed the presence of a germline deletion of exons 1 and 2 of BRCA1, and revealed that all lesions harbored somatic loss of the BRCA1 wild‐type allele and TP53 somatic mutations in the modal population of cancer cells (Figure 3A, Supplementary Table S2).
Figure 3.
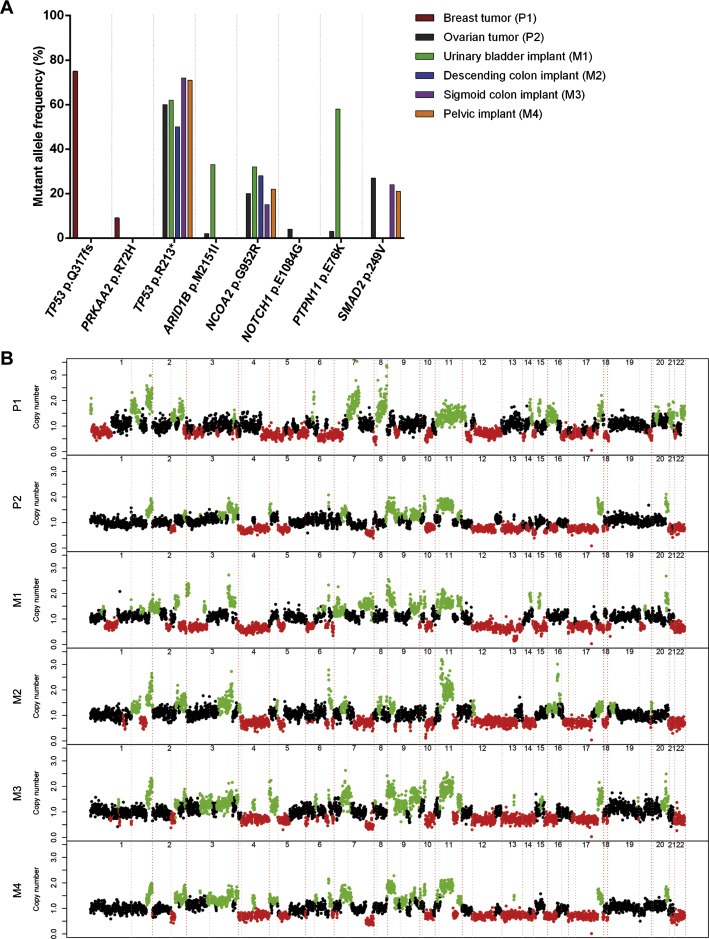
Genomic profiles of the breast and ovarian tumors, and metastatic deposits. (A) Mutant allele frequency of the somatic mutations. The mutant allele frequency of each somatic mutation is shown according to the tumor location (breast, ovary or peritoneum‐pelvic implants). (B) Copy‐number profiles generated with circular binary segmentation (cbs)‐processed targeted massively parallel sequencing data of the breast and ovarian tumors, and metastatic deposits. Gene copy number gains are highlighted in green and gene copy number losses are highlighted in red. P1, breast tumor; P2, ovarian tumor; M1, urinary bladder implant; M2, descending colon implant; M3, sigmoid colon implant; M4, pelvic implant.
Most importantly, the repertoire of somatic genetic aberrations provided by massively parallel sequencing demonstrated that the breast and ovarian lesions were not clonally related (Figure 3A), given that the modal population of cancer cells from the breast cancer harbored a TP53 frameshift mutation (p.Q317fs), whereas cells from the ovarian lesion displayed a distinct TP53 nonsense mutation (p.R213*). Detailed analyses of the sequencing data revealed no reads harboring the TP53 p.R213* mutation in the breast lesion, and no reads displaying the TP53 p.Q317fs mutation in the ovarian lesion (Supplementary Table S2). Taken together, these results provided direct evidence to support the contention that the patient harbored two distinct primary malignancies: a high‐grade breast carcinoma and a poorly differentiated high‐grade serous ovarian carcinoma.
The pelvic‐peritoneal implants (i.e. urinary bladder, descending colon, sigmoid colon and pelvic peritoneum) harbored TP53 mutations identical to that found in the ovarian lesion (i.e. p.R213*), In addition, the ovarian tumor and all pelvic‐peritoneal implants displayed NCOA2 (p.G952R) somatic mutations (Figure 3A, Supplementary Table S2). These observations demonstrate that the pelvic‐peritoneal implants are clonally related to the ovarian lesion, and provide strong circumstantial evidence to demonstrate that they constitute metastatic deposits of the ovarian rather than the breast cancer.
The somatic copy number changes determined by massively parallel sequencing analysis found in the breast and ovarian lesions were distinct, whereas the pelvic‐peritoneal implants displayed copy number aberrations similar to those found in the ovarian lesion (Figure 3B), consistent with the notion that the breast and ovarian lesions were distinct primary malignancies, and that the pelvic‐peritoneal implants stemmed from the ovarian carcinoma.
In line with previous reports demonstrating intra‐tumor genetic heterogeneity in human solid malignancies, evidence of intra‐tumor genetic heterogeneity was observed in the samples analyzed in this study. Not only mutations of SMAD2 (p.249V), ARID1B (p.M2151I), NOTCH1 (p.E1084G) and PTPN11 (p.E76K) were found in the ovarian lesion and in only two, two, one and one pelvic‐peritoneal implants, respectively, but also the mutant allele frequencies varied according to the lesion (Figure 3A, Supplementary Table S2). TP53 and NCOA2 allele frequencies were similar in the ovarian and pelvic‐peritoneal implants, whereas the mutant allele frequencies of PTPN11 and ARID1B were substantially higher in the urinary bladder implant as compared to the ovarian cancer (Supplementary Table S2). It should be noted that some of the somatic mutations identified were likely restricted to a subclone within each lesion (e.g. PRKAA2 p.R72H present in 9% of the alleles of the breast lesion, and NOTCH1 p.E1084G and PTPN11 p.E76K present in 4% and 3% of the alleles of the ovarian cancer, respectively), providing circumstantial evidence to suggest that the breast and ovarian lesions also displayed intra‐tumor genetic heterogeneity. Gene copy number analysis also confirmed the heterogeneity amongst the lesions analyzed here (e.g. high‐level, focal amplifications on 14q32.33–14q32.33 and 15q23 found in the urinary bladder implant M1, but not in the ovarian primary or remaining metastatic deposits; Figure 3B).
Phylogenetic trees were constructed to infer the genetic relatedness of the lesions subjected to targeted capture massively parallel sequencing on the basis of mutant allele frequencies (Figure 4A), the presence/absence of specific somatic mutations (Figure 4B) and gene copy number aberrations (Figure 4C). These analyses confirmed independently that the breast and ovarian cancers were not clonally related, whereas pelvic‐peritoneal tumors were clonally related to the ovarian cancer.
Figure 4.

Phylogenetic trees depicting the clonal relationship among the breast tumor, ovarian tumor and the pelvic‐peritoneal implants. The ‘normal tissue’, which does not harbor any somatic mutation, represents the root of the tree. Based on the (A) mutant allele frequencies, (B) presence/absence of somatic mutations, and (C) copy number alterations, it can be inferred that the breast and ovarian cancers were not clonally related, whereas pelvic‐peritoneal tumors were clonally related to the ovarian cancer. Desc, descending; sigm, sigmoid.
Comprehensive immunohistochemical analysis revealed that the breast lesion displayed an immunohistochemical profile distinct from that of the left adnexal mass and pelvic peritoneal implants (Supplementary Table S3 and Supplementary Figure S2). While the breast lesion expressed CK7 and CA125, and displayed focal and weak expression of PAX8, the left adnexal mass and pelvic peritoneal implants displayed strong and diffuse expression of CK7, CA125, WT1 and PAX8. None of the lesions expressed CK20 or GCDFP‐15. Taken together, the results of the immunohistochemical analyses are consistent with the notion that the adnexal lesion is a high‐grade serous adenocarcinoma and that the pelvic‐peritoneal lesions were metastatic deposits of the ovarian rather than the breast lesion (Nonaka et al., 2008; Tornos et al., 2005).
4. Discussion
In this proof‐of‐principle study, we demonstrate that massively parallel sequencing can be of clinical value to determine the relationship between synchronous primary tumor masses and their metastatic deposits, and to provide additional, complementary information to that obtained from histological and immunohistochemical analyses (Nonaka et al., 2008; Tornos et al., 2005). The repertoires of somatic mutations and gene copy number aberrations found in the breast, ovarian, and pelvic‐peritoneal lesions provided direct evidence in support of the distinct origin of the breast and ovarian cancers, and established that the pelvic‐peritoneal implants are clonally related to the ovarian lesion. These results were consistent with those of the immunohistochemical analyses using antibodies that have proven to be useful to distinguish breast from ovarian lesions (Nonaka et al., 2008; Tornos et al., 2005).
It should be noted that the case presented here, albeit rare, exemplifies a diagnostic challenge in patients with BRCA1 germline mutations, given that the histological and immunohistochemical profiles of BRCA1 breast and ovarian cancers may overlap, in particular in metastatic sites. Although immunohistochemical markers, such as WT1 may be used as an ancillary marker to help distinguish between breast and ovarian cancers, up to 7% of breast cancers (Hwang et al., 2004) can be WT1 positive and approximately 7–26% of serous ovarian cancers (Hwang et al., 2004; Tornos et al., 2005) lack expression of this marker; in this case, WT1 was expressed by neoplastic cells of the ovarian and pelvic‐peritoneal lesions and absent in the cells of the breast lesion, confirming the results of the targeted capture massively parallel sequencing analysis. It should be noted that WT1 together with the additional markers employed in this study, namely PAX8, CA125, GCDFP‐15, CK7 and CK20, can reliably differentiate between primary breast and ovarian lesions. In the context of BRCA1 and BRCA2 germline mutation carriers, however, these markers would not suffice to determine whether the adnexal and pelvic‐peritoneal lesions would constitute a primary high‐grade ovarian serous carcinoma and pelvic‐peritoneal metastases or a primary high‐grade ovarian serous carcinoma and a primary peritoneal carcinoma. Importantly, however, sequencing data can provide the clonal relatedness of the lesions on the basis of the mutational repertoire of the lesions.
Massively parallel sequencing analyses have the potential to establish the clonality between lesions. This proof‐of‐principle study, however, has several limitations. The analyses were performed utilizing materials from a single patient and this patient is a BRCA1 germline mutation carrier. The case analyzed in this study could be considered as an ideal setting to test the use of targeted sequencing to define clonality of multiple tumors from the same patient, given that tumors arising in BRCA1 germline mutation carriers often harbor somatic mutations of TP53, PTEN and RB1 (Weigelt and Reis Filho, 2013). Therefore, although targeted massively parallel sequencing was conclusive in the case analyzed here, it is plausible that in other cancer types, this approach may not be sufficient, as cancer cells may not harbor mutations in any of the genes included in a given targeted capture panel. Importantly, however, clonality between lesions may also be established through the analyses of gene copy number aberrations inferred by this methodology, provided that the targeted capture panel has an adequate distribution throughout the genome.
The results of the massively parallel sequencing analysis of this case also corroborate previous studies demonstrating that breast (Shah et al., 2012) and ovarian (Bashashati et al., 2013; Castellarin et al., 2013) cancers display intra‐tumor genetic heterogeneity, and that spatially distinct areas of a primary tumor and/or metastatic deposits may harbor private genetic aberrations in addition to the founder genetic events (i.e. in the present case, the loss of BRCA1 wild‐type allele and TP53 mutations) (Gerlinger et al., 2012; Navin et al., 2011; Shah et al., 2012; Weigelt and Reis Filho, 2013).
In conclusion, our findings demonstrate that massively parallel sequencing analyses, even when restricted to a limited number of genes (n = 300), can yield information to help define the clonality between lesions and the origin of metastatic deposits in patients with multiple primary malignancies, providing a proof‐of‐principle of another application of massively parallel sequencing in current oncology and pathology practice.
Disclosure
The authors have no conflicts of interest to declare.
Supporting information
The following is the supplementary data related to this article.
Supplementary data
Acknowledgments
We thank the patient and her family for her participation in this study. We also thank Alexandra Arias (Gene Expression and Cancer Laboratory, Vall d'Hebron Institute of Oncology, VHIO, Barcelona, Spain) and Jose Jimenez (Molecular Pathology Laboratory, VHIO, Barcelona, Spain) for technical assistance; Judith Balmaña (VHIO, Barcelona, Spain) for genetic counseling and Susana Gispert Herrero (Institut Diagnòstic Imatge‐Ressonància Magnètica, Vall d'Hebron University Hospital, Barcelona, Spain) for assisting with the interpretation of radiologic findings. The authors acknowledge Fundación Rafael del Pino (to LDMA) and Asociación Española Contra el Cancer AECC grants (to Joan Seoane) for financial support. Francois‐Clement Bidard is supported by a fellowship from the Nuovo‐Soldati Foundation for Cancer Research, Britta Weigelt is supported in part by the Kaleidoscope of Hope Foundation.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.10.006.
De Mattos-Arruda Leticia, Bidard Francois-Clement, Won Helen H., Cortes Javier, Ng Charlotte K.Y., Peg Vicente, Nuciforo Paolo, Jungbluth Achim A. Weigelt Britta, Berger Michael F., Seoane Joan and Reis-Filho Jorge S., (2014), Establishing the origin of metastatic deposits in the setting of multiple primary malignancies: The role of massively parallel sequencing, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.10.006.
Contributor Information
Joan Seoane, Email: jseoane@vhio.net.
Jorge S. Reis-Filho, Email: reisfilj@mskcc.org
References
- Alexandrov, L.B. , Nik-Zainal, S. , Wedge, D.C. , Aparicio, S.A. , Behjati, S. , Biankin, A.V. , Bignell, G.R. , Bolli, N. , Borg, A. , Borresen-Dale, A.L. , Boyault, S. , Burkhardt, B. , Butler, A.P. , Caldas, C. , Davies, H.R. , Desmedt, C. , Eils, R. , Eyfjord, J.E. , Foekens, J.A. , Greaves, M. , Hosoda, F. , Hutter, B. , Ilicic, T. , Imbeaud, S. , Imielinsk, M. , Jager, N. , Jones, D.T. , Jones, D. , Knappskog, S. , Kool, M. , Lakhani, S.R. , Lopez-Otin, C. , Martin, S. , Munshi, N.C. , Nakamura, H. , Northcott, P.A. , Pajic, M. , Papaemmanuil, E. , Paradiso, A. , Pearson, J.V. , Puente, X.S. , Raine, K. , Ramakrishna, M. , Richardson, A.L. , Richter, J. , Rosenstiel, P. , Schlesner, M. , Schumacher, T.N. , Span, P.N. , Teague, J.W. , Totoki, Y. , Tutt, A.N. , Valdes-Mas, R. , van Buuren, M.M. , van 't Veer, L. , Vincent-Salomon, A. , Waddell, N. , Yates, L.R. , Australian Pancreatic Cancer Genome, I. , Consortium, I.B.C. , Consortium, I.M.-S. , PedBrain, I. , Zucman-Rossi, J. , Futreal, P.A. , McDermott, U. , Lichter, P. , Meyerson, M. , Grimmond, S.M. , Siebert, R. , Campo, E. , Shibata, T. , Pfister, S.M. , Campbell, P.J. , Stratton, M.R. , 2013. Signatures of mutational processes in human cancer. Nature. 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir, E. , Miller, N. , Geddie, W. , Freedman, O. , Kassam, F. , Simmons, C. , Oldfield, M. , Dranitsaris, G. , Tomlinson, G. , Laupacis, A. , Tannock, I.F. , Clemons, M. , 2012. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol.. 30, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio, S. , Caldas, C. , 2013. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med.. 368, 842–851. [DOI] [PubMed] [Google Scholar]
- Bashashati, A. , Ha, G. , Tone, A. , Ding, J. , Prentice, L.M. , Roth, A. , Rosner, J. , Shumansky, K. , Kalloger, S. , Senz, J. , Yang, W. , McConechy, M. , Melnyk, N. , Anglesio, M. , Luk, M.T. , Tse, K. , Zeng, T. , Moore, R. , Zhao, Y. , Marra, M.A. , Gilks, B. , Yip, S. , Huntsman, D.G. , McAlpine, J.N. , Shah, S.P. , 2013. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol.. 231, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellarin, M. , Milne, K. , Zeng, T. , Tse, K. , Mayo, M. , Zhao, Y. , Webb, J.R. , Watson, P.H. , Nelson, B.H. , Holt, R.A. , 2013. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol.. 229, 515–524. [DOI] [PubMed] [Google Scholar]
- Cibulskis, K. , Lawrence, M.S. , Carter, S.L. , Sivachenko, A. , Jaffe, D. , Sougnez, C. , Gabriel, S. , Meyerson, M. , Lander, E.S. , Getz, G. , 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol.. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockford, A. , Jamal-Hanjani, M. , Hicks, J. , Swanton, C. , 2013. Implications of intratumour heterogeneity for treatment stratification. J. Pathol.. 10.1002/path.4270 Epub ahead of print [DOI] [PubMed] [Google Scholar]
- De Mattos-Arruda, L. , Cortes, J. , Santarpia, L. , Vivancos, A. , Tabernero, J. , Reis-Filho, J.S. , Seoane, J. , 2013. Circulating tumour cells and cell-free DNA as tools for managing breast cancer. Nat. Rev. Clin. Oncol.. 10, 377–389. [DOI] [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. , del Angel, G. , Rivas, M.A. , Hanna, M. , McKenna, A. , Fennell, T.J. , Kernytsky, A.M. , Sivachenko, A.Y. , Cibulskis, K. , Gabriel, S.B. , Altshuler, D. , Daly, M.J. , 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet.. 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domchek, S.M. , Bradbury, A. , Garber, J.E. , Offit, K. , Robson, M.E. , 2013. Multiplex genetic testing for cancer susceptibility: out on the high wire without a net?. J. Clin. Oncol.. 31, 1267–1270. [DOI] [PubMed] [Google Scholar]
- Gerlinger, M. , Rowan, A.J. , Horswell, S. , Larkin, J. , Endesfelder, D. , Gronroos, E. , Martinez, P. , Matthews, N. , Stewart, A. , Tarpey, P. , Varela, I. , Phillimore, B. , Begum, S. , McDonald, N.Q. , Butler, A. , Jones, D. , Raine, K. , Latimer, C. , Santos, C.R. , Nohadani, M. , Eklund, A.C. , Spencer-Dene, B. , Clark, G. , Pickering, L. , Stamp, G. , Gore, M. , Szallasi, Z. , Downward, J. , Futreal, P.A. , Swanton, C. , 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med.. 366, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, L. , Wilkerson, P.M. , Lambros, M.B. , Campion-Flora, A. , Rodrigues, D.N. , Gauthier, A. , Cabral, C. , Pawar, V. , Mackay, A. , A'Hern, R. , Marchio, C. , Palacios, J. , Natrajan, R. , Weigelt, B. , Reis-Filho, J.S. , 2012. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J. Pathol.. 227, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, H. , Quenneville, L. , Yaziji, H. , Gown, A.M. , 2004. Wilms tumor gene product: sensitive and contextually specific marker of serous carcinomas of ovarian surface epithelial origin. Appl. Immunohistochem. Mol. Morphol.. 12, 122–126. [DOI] [PubMed] [Google Scholar]
- Kinde, I. , Bettegowda, C. , Wang, Y. , Wu, J. , Agrawal, N. , Shih Ie, M. , Kurman, R. , Dao, F. , Levine, D.A. , Giuntoli, R. , Roden, R. , Eshleman, J.R. , Carvalho, J.P. , Marie, S.K. , Papadopoulos, N. , Kinzler, K.W. , Vogelstein, B. , Diaz, L.A. , 2013. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci. Transl Med.. 5, 167ra164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Durbin, R. , 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott, U. , Downing, J.R. , Stratton, M.R. , 2011. Genomics and the continuum of cancer care. N. Engl. J. Med.. 364, 340–350. [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , DePristo, M.A. , 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res.. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natrajan, R. , Reis-Filho, J.S. , 2011. Next-generation sequencing applied to molecular diagnostics. Expert Rev. Mol. Diagn.. 11, 425–444. [DOI] [PubMed] [Google Scholar]
- Navin, N. , Kendall, J. , Troge, J. , Andrews, P. , Rodgers, L. , McIndoo, J. , Cook, K. , Stepansky, A. , Levy, D. , Esposito, D. , Muthuswamy, L. , Krasnitz, A. , McCombie, W.R. , Hicks, J. , Wigler, M. , 2011. Tumour evolution inferred by single-cell sequencing. Nature. 472, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura, N. , Liu, J. , Hayashi, N. , Mittendorf, E.A. , Gong, Y. , Palla, S.L. , Tokuda, Y. , Gonzalez-Angulo, A.M. , Hortobagyi, G.N. , Ueno, N.T. , 2012. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J. Clin. Oncol.. 30, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka, D. , Chiriboga, L. , Soslow, R.A. , 2008. Expression of pax8 as a useful marker in distinguishing ovarian carcinomas from mammary carcinomas. Am. J. Surg. Pathol.. 32, 1566–1571. [DOI] [PubMed] [Google Scholar]
- Paradis, E. , Claude, J. , Strimmer, K. , 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Poduri, A. , Evrony, G.D. , Cai, X. , Walsh, C.A. , 2013. Somatic mutation, genomic variation, and neurological disease. Science. 341, 1237758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusztai, L. , Viale, G. , Kelly, C.M. , Hudis, C.A. , 2010. Estrogen and HER-2 receptor discordance between primary breast cancer and metastasis. Oncologist. 15, 1164–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdottir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. , Mesirov, J.P. , 2011. Integrative genomics viewer. Nat. Biotechnol.. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou, N. , Nei, M. , 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol.. 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sandoval, J. , Esteller, M. , 2012. Cancer epigenomics: beyond genomics. Curr. Opin. Genet. Dev.. 22, 50–55. [DOI] [PubMed] [Google Scholar]
- Schouten, J.P. , McElgunn, C.J. , Waaijer, R. , Zwijnenburg, D. , Diepvens, F. , Pals, G. , 2002. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res.. 30, e57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, S.P. , Roth, A. , Goya, R. , Oloumi, A. , Ha, G. , Zhao, Y. , Turashvili, G. , Ding, J. , Tse, K. , Haffari, G. , Bashashati, A. , Prentice, L.M. , Khattra, J. , Burleigh, A. , Yap, D. , Bernard, V. , McPherson, A. , Shumansky, K. , Crisan, A. , Giuliany, R. , Heravi-Moussavi, A. , Rosner, J. , Lai, D. , Birol, I. , Varhol, R. , Tam, A. , Dhalla, N. , Zeng, T. , Ma, K. , Chan, S.K. , Griffith, M. , Moradian, A. , Cheng, S.W. , Morin, G.B. , Watson, P. , Gelmon, K. , Chia, S. , Chin, S.F. , Curtis, C. , Rueda, O.M. , Pharoah, P.D. , Damaraju, S. , Mackey, J. , Hoon, K. , Harkins, T. , Tadigotla, V. , Sigaroudinia, M. , Gascard, P. , Tlsty, T. , Costello, J.F. , Meyer, I.M. , Eaves, C.J. , Wasserman, W.W. , Jones, S. , Huntsman, D. , Hirst, M. , Caldas, C. , Marra, M.A. , Aparicio, S. , 2012. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 486, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornos, C. , Soslow, R. , Chen, S. , Akram, M. , Hummer, A.J. , Abu-Rustum, N. , Norton, L. , Tan, L.K. , 2005. Expression of WT1, CA 125, and GCDFP-15 as useful markers in the differential diagnosis of primary ovarian carcinomas versus metastatic breast cancer to the ovary. Am. J. Surg. Pathol.. 29, 1482–1489. [DOI] [PubMed] [Google Scholar]
- Turner, N.C. , Reis-Filho, J.S. , 2012. Genetic heterogeneity and cancer drug resistance. Lancet Oncol.. 13, e178–185. [DOI] [PubMed] [Google Scholar]
- van de Wiel, M.A. , Kim, K.I. , Vosse, S.J. , van Wieringen, W.N. , Wilting, S.M. , Ylstra, B. , 2007. CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics. 23, 892–894. [DOI] [PubMed] [Google Scholar]
- Wagle, N. , Berger, M.F. , Davis, M.J. , Blumenstiel, B. , Defelice, M. , Pochanard, P. , Ducar, M. , Van Hummelen, P. , Macconaill, L.E. , Hahn, W.C. , Meyerson, M. , Gabriel, S.B. , Garraway, L.A. , 2012. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov.. 2, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt, B. , Reis Filho, J.S. , 2013. Epistatic interactions and drug response. J. Pathol.. 10.1002/path.4265 Epub ahead of print [DOI] [PubMed] [Google Scholar]
- Yap, T.A. , Gerlinger, M. , Futreal, P.A. , Pusztai, L. , Swanton, C. , 2012. Intratumor heterogeneity: seeing the wood for the trees. Sci. Transl. Med.. 4, 127ps110 [DOI] [PubMed] [Google Scholar]
- Yates, L.R. , Campbell, P.J. , 2012. Evolution of the cancer genome. Nat. Rev. Genet.. 13, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article.
Supplementary data