

Abstract
Aberrant activation of the canonical Wnt signal transduction pathway is involved in a large number of human diseases. β‐catenin, the key effector protein of the canonical Wnt pathway, functions in the nucleus with T‐cell factor/lymphoid enhancer factor (TCF/LEF) to activate expression of Wnt target genes. Here we show that members of the 14‐3‐3 protein family bind disheveled‐2 (Dvl‐2) and glycogen synthase‐3β (GSK‐3β) to attenuate the interaction between GSK‐3β and β‐catenin. Importantly, 14‐3‐3 and β‐catenin form “bleb‐like” structures and are secreted via extracellular vesicles to induce Wnt signaling activity in target cells. Our data suggest a novel way of transducing the oncogenic Wnt signal in which β‐catenin is regulated by 14‐3‐3ζ through the formation of “oncosomes” that contain both the 14‐3‐3 and β‐catenin proteins.
Keywords: Wnt signaling, Extracellular vesicles, 14‐3‐3, Exosomes, β‐catenin
Highlights
A controversy exists regarding the role of the 14‐3‐3 proteins and the oncogenic Wnt pathway.
14‐3‐3 activates the pathway through interaction with a number of Wnt signaling components.
14‐3‐3 and β‐catenin form “bleb‐like structures”.
14‐3‐3 and β‐catenin transmit the oncogenic signal via extracellular vesicles.
1. Introduction
The canonical Wnt/β‐catenin signaling pathway is an evolutionarily conserved key pathway controlling both embryonic development and tissue homeostasis (Nusse and Varmus, 2012). As such, mutations or deregulated expression of different components of the Wnt pathway result in aberrant constitutive activation of the latter and may lead to the development of different diseases including cancer (Polakis, 2012) (Klaus and Birchmeier, 2008) (MacDonald et al., 2009). The key effector of the canonical Wnt cascade is the β‐catenin protein, whose levels are normally kept low through continuous proteasome‐mediated degradation. β‐catenin degradation is regulated by the “β‐catenin destruction complex” that contains, among others, the tumor suppressor protein adenomatous polyposis coli (APC), glycogen synthase kinase‐3β (GSK‐3β) and Axin. This complex promotes phosphorylation of β‐catenin by casein kinase 1α (CK1α) and GSK‐3β, marking it for ubiquitylation and subsequent proteasomal degradation. When the Wnt pathway is activated, the Wnt ligand binds to the Frizzled (Fz) transmembrane receptor and the co‐receptor low‐density lipoprotein receptor‐related 5 (LRP5) or LRP6 (Tamai et al., 2000). The cytoplasmic tail of LRP6 is then phosphorylated by CK1α and GSK‐3β, which are sequestered together with Axin away from the “β‐catenin destruction complex” rendering its disassembly. These events also trigger the polymerization of Dishevelled (Dvl), in membrane associated punctate protein clusters named “signalosomes” (Schwarz‐Romond et al., 2007) (Bilic et al., 2007) (Zeng et al., 2008) (Metcalfe et al., 2010) (Angers and Moon, 2009). Interestingly, it has been proposed that the signalosomes can give rise to Wnt induced multi vesicular bodies (MVBs) (Taelman et al., 2010). Recent data suggest that activation of the Wnt cascade does not cause the disruption of the “degradation complex” but rather leads to saturation of the complex with phosphorylated‐β‐catenin (Li et al., 2012). Nevertheless, according to both models, as a result of Wnt stimulation, β‐catenin accumulates in the cytoplasm and translocates into the nucleus where it associates with members of the T‐cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors and up‐regulates the transcription of Wnt target genes. Over‐expression of Wnt‐induced target genes such as c‐myc, cyclin‐D1 and Sox‐9 (He et al., 1998) (Tetsu and McCormick, 1999) (Blache et al., 2004) is one of many aberrations that likely contribute to abnormal cellular transformation (Morrish et al., 2008) (Wolfer and Ramaswamy, 2011) (Meyer and Penn, 2008) (Levens, 2010) (Alao, 2007) (Matheu et al., 2012).
The 14‐3‐3 proteins are conserved regulatory molecules expressed in all eukaryotic cells and participate in a wide range of cellular processes through interactions with numerous proteins (Obsil and Obsilova, 2011) (Fu et al., 2000) (Yaffe et al., 1997) (Morrison, 2009). In mammals, seven isoforms of 14‐3‐3 (β, ɛ, η, γ, τ, ζ and σ) have been identified and their ability to dimerize has been established. Usually the 14‐3‐3 dimer binds to a target protein via a conserved binding motif containing serine or threonine residues that become phosphorylated in response to cellular signals (Fu et al., 2000). Several studies indicate that members of the 14‐3‐3 family, especially 14‐3‐3ζ, play a pro‐oncogenic role in multiple tumor types and that over‐expression of 14‐3‐3 proteins is generally associated with poor survival of cancer patients (Zhao et al., 2011). The 14‐3‐3 protein family has been previously linked to the Wnt pathway in different ways. Although a number of studies suggest that 14‐3‐3 proteins negatively regulate the Wnt pathway through association with chibby (Li et al., 2008) (Li et al., 2010) (Fischer et al., 2012), other studies have found that 14‐3‐3 activates the Wnt signal (Tian et al., 2004) (Fang et al., 2007) (Chen et al., 2011) (Chang et al., 2012). In the present study we show that the 14‐3‐3ζ protein specifically interacts with Dvl‐2 and other components of the Wnt cascade. We propose that 14‐3‐3ζ enhances Wnt/β‐catenin signaling by forming a cellular complex with Dvl‐2 and GSK‐3β thus weakening the interaction between β‐catenin and the latter. Importantly, co‐expression of β‐catenin and 14‐3‐3ζ leads to extracellular vesicles formation that shuttle between cells to induce Wnt signaling and enhanced oncogenic phenotypes.
2. Results
2.1. 14‐3‐3 protein family members form cellular complexes with members of the Wnt signaling pathway
In experiments aimed at identifying proteins that interact with Dvl‐2, HEK293T cells expressing FLAG‐tagged human Dvl‐2 were used. Lysates from cells transfected with FLAG‐Dvl‐2 or from cells transfected with an empty FLAG vector were incubated with anti‐FLAG M2 antibodies conjugated to agarose beads. Following SDS–PAGE of the captured proteins, colloidal coomassie blue stains revealed a protein band that co‐precipitated with FLAG–Dvl‐2 and not with the control vector. The identity of these proteins was determined by tandem mass spectrometry. Through this analysis, we identified six of the known seven 14‐3‐3 isoforms (β; γ; ζ; θ; η and σ) as potential binding partners of Dvl‐2 (Figure 1A, Supplementary Table 1). This experiment was repeated three times with similar results. To verify the cellular interaction between 14‐3‐3ζ and Dvl‐2, lysates from HEK293T cells were subjected to IP assay using specific antibodies. As shown in Figure 1B endogenous Dvl‐2 and 14‐3‐3ζ indeed co‐immunoprecipitate. The interaction between the two proteins was not affected by Wnt stimuli as in cells incubated with Wnt3a CM (secreted Wnt3a is shown in Figure 1B lower right panel) the Dvl‐2‐14‐3‐3ζ interaction was not affected (Figure 1B upper panel). The Wnt signaling induction was examined by measuring active β‐catenin protein levels (Figure 1B lower left panel). 14‐3‐3 proteins typically bind to their target proteins through one of the two consensus sequences, RSXpSXP or RXXXpSXP, (Muslin et al., 1996) (Yaffe et al., 1997) (Li et al., 2008). Examining the human Dvl‐2 amino acid sequence revealed a potential 14‐3‐3 binding motif, 705RDLGSVP711 which is unique to the Dvl‐2 protein and is not found in the other Dvl isoforms. To study whether this putative 14‐3‐3 binding motif is important for the 14‐3‐3‐Dvl‐2 interaction, site‐directed mutagenesis was performed to replace the serine, which is predicted to be a crucial residue, with an alanine. A similar mutation was introduced into the Chibby protein to abolish its interaction with 14‐3‐3 (Li et al., 2008). Results show that the S709A mutation disrupted the 14‐3‐3ζ‐Dvl‐2 interaction (Figure 1C) indicating that 14‐3‐3ζ binds Dvl‐2 through the 705RDLGSVP711 motif. GSK‐3β, another component of the canonical Wnt pathway, was previously shown to interact with 14‐3‐3ζ in brain extracts (Agarwal‐Mawal et al., 2003) (Yuan et al., 2004). We have confirmed this interaction in HEK293T cells using both exogenous and endogenous proteins (Figure 1D, E). The previously established interaction between 14‐3‐3‐ and β‐catenin, the key component of the canonical Wnt pathway, (Tian et al., 2004) (Fang et al., 2007) was also confirmed (Figure 1F). Interestingly, the core region of APC, an important member of the Wnt cascade that contains the β‐catenin binding sites, did not bind 14‐3‐3ζ (APCcore, Figure 1G lower panel). However, the armadillo repeat region of APC co‐immunopercipitated with over‐expressed 14‐3‐3ζ protein (APCarm, Figure 1G upper panel).
Figure 1.

Wnt signaling members associate with 14‐3‐3 proteins. (A) Identification of 14‐3‐3 proteins as Dvl‐binding partners by mass spectrometry. HEK293T cells expressing FLAG‐tagged‐Dvl‐2 or FLAG empty vector were immunoprecipitated with anti‐FLAG M2‐agarose affinity gel. Associated proteins were separated by SDS‐PAGE, and stained by coomassie blue. Specific bands were excised from the gel and analyzed by mass spectrometry. (B) Co‐IP between endogenous Dvl‐2 and 14‐3‐3ζ in Wnt stimulated and unstimulated cells. Cell lysates from non‐transfected HEK293T cells incubated with media derived from L or L‐Wnt3a cells (as indicated) were immunoprecipitated using control sera or anti‐Dvl‐2 antibodies and immunoblotted for 14‐3‐3ζ and Dvl‐2 (upper panel). Western blotting analysis of lysates or cells growing medium was conducted to indicate the presence of the Wnt3a ligand in the medium of L‐Wnt3a cells. Increased expression of active β‐catenin is seen in the L‐Wnt3a cells (lower panel). Tubulin was used as a loading control (C) Co‐IP between FLAG‐Dvl‐2; FLAG‐Dvl‐2(S709A) and HA‐14‐3‐3ζ. HEK293T cells were co‐transfected with the indicated vectors. Forty eight hours later, cells were harvested and lysates were co‐immunoprecipitated using anti‐FLAG M2 Affinity beads, subjected to SDS gel and immunoblotted. (D) Co‐IP between FLAG‐GSK‐3β and HA‐14‐3‐3ζ. HEK293T cells were transfected with the indicated plasmids and co‐IP experiment was conducted as in C. (E) Co‐IP between endogenous 14‐3‐3ζ and GSK‐3β. Cell lysates from non‐transfected HEK293T cells were immunoprecipitated with control sera or anti‐GSK‐3β antibodies and immunoblotted for GSK‐3β and 14‐3‐3ζ specific antibodies. (F) Co‐IP between endogenous β‐catenin and HA‐14‐3‐3ζ. HEK293T cells were transfected with HA‐14‐3‐3ζ and IP experiment was conducted 48 h later using an anti‐HA antibody. Endogenous β‐catenin was detected using a specific antibody. (G) Co‐IP between different domains of APC and HA‐14‐3‐3ζ. Lysates from HEK293T cells transfected with the indicated plasmids were immunoprecipitated using FLAG‐M2 affinity beads or an anti‐GFP antibody as indicated. Detection was performed using the indicated antibodies. IP: immunoprecipitation; IB: immunoblot. Molecular mass markers are indicated on the right. All blots are representative results from at least three independent experiments.
2.2. The 14‐3‐3 protein family activates Wnt/β‐catenin mediated transcription
As our results show that 14‐3‐3ζ interacts with different members of the Wnt pathway, we next tested the effect of different 14‐3‐3 isoforms on Wnt signaling levels by using the pTOPFLASH/pFOPFLASH assay that measures TCF/β‐catenin mediated transcription (Korinek et al., 1997). Our data show that although ectopic expression of different 14‐3‐3 isoforms (ζ, η and ε) alone had no effect on Wnt/β‐catenin mediated transcription, these 14‐3‐3 proteins strongly enhance the positive effect of Dvl‐2 on Wnt signaling (Figure 2A, B). Interestingly, 14‐3‐3ζ had a reduced effect when co‐expressed with the S709A Dvl‐2 mutant (Figure 2A) suggesting that although the S709A Dvl‐2 mutant cannot bind 14‐3‐3ζ (Figure 1D) it, partly, activates the Wnt signal (Figure 2A). One possible explanation for these results may be that when un‐bound by Dvl‐2 the 14‐3‐3ζ protein is recruited through other Wnt components to enhance signaling activity. We then examined the effect of 14‐3‐3ζ depletion on the Wnt cascade by using specific siRNA oligos. Results from these experiments show that decreasing the levels of 14‐3‐3ζ leads to a reduced effect of Dvl‐2 on the Wnt signal (Figure 2C). These results are in agreement with recent findings showing that 14‐3‐3β inhibits the degradation of Dvl (Chen et al., 2011).
Figure 2.

14‐3‐3 augments the activity of Dvl. Luciferase assay was performed in cells transfected with the indicated 14‐3‐3 constructs, WT and mutated FLAG‐Dvl‐2 along with the pTOPFLASH reporter and either β‐gal or renilla plasmids. Lysates were obtained 48 h post transfection and subjected to luciferase assay. Relative luciferase values are shown (upper panels). Values (mean ± SD) in this and subsequent figures are from triplicate wells calculated from at least three independent experiments. Extracts were also subjected to SDS‐PAGE analysis using the appropriate antibodies. Representative blots are shown (lower panels). (A) HEK293T cells were co‐transfected with WT or mutated Flag‐Dvl‐2, HA‐14‐3‐3ζ and the reporter constructs as indicated. Asterisk denotes statistical significance in an un‐paired Student t test *P < 0.05. (B) The 2 isoforms of 14‐3‐3 were transfected as before along with the FLAG‐Dvl‐2 and reporter plasmids as in A. (C) 14‐3‐3ζ was silenced using specific siRNA oligos in HEK293T cells that were later subjected to both luciferase test and WB analysis.
2.3. 14‐3‐3ζ interferes with the GSK‐3β‐β‐catenin interaction
As mentioned above, 14‐3‐3ζ forms a complex with both GSK‐3β and β‐catenin. β‐catenin is tightly regulated and its levels and activity are attenuated mainly by GSK‐3β that binds and phosphorylates β‐catenin, marking it for degradation by the proteasome (Valenta et al., 2012). Our results show that over‐expression of 14‐3‐3ζ interferes with the GSK‐3β‐β‐catenin complex (Figure 3A), whereas silencing 14‐3‐3ζ leads to enhanced binding of the two (Figure 3B). Stabilization of β‐catenin by either LiCl or MG‐132 abolished this effect (Figure 3C). As binding of GSK‐3β to β‐catenin leads to phosphorylation and degradation of the latter, disrupting the GSK‐3β‐β‐catenin complex may result in elevated levels of free β‐catenin that activate the Wnt pathway. Indeed, expression of 14‐3‐3ζ leads to an increase in β‐catenin protein levels (Figure 3D).
Figure 3.

14‐3‐3ζ interferes with the GSK‐3β‐β‐catenin interaction. (A) HEK293T cells were co‐transfected with FLAG‐GSK‐3β and HA‐14‐3‐3ζ or HA empty vector. Cell lysates were immunoprecipitated with FLAG‐M2 affinity beads. Western blot was performed using anti‐HA, anti‐FLAG and anti‐total β‐catenin antibodies. (B) 14‐3‐3ζ was silenced by specific siRNA oligos in HEK293T cells. Cell lysates were immunoprecipitated with anti‐GSK‐3β and WB was performed using specific antibodies as indicated. (C) HEK293T cells were transfected with FLAG‐GSK‐3β and HA‐14‐3‐3ζ or HA empty vector and either left untreated or treated with LiCl (30 mM, 24 h) or MG‐132 (25 μM, 5 h). Immunoprecipitation and WB analysis were performed as described using the appropriate antibodies. (D) HEK293T cells were co‐transfected with constant amount of GFP‐β‐catenin and increasing concentrations of HA‐14‐3‐3ζ. Lysates were subjected to SDS gel and immunoblotted as indicated. Representative blots of three experiments are shown.
2.4. The kinase activity of GSK‐3β is important for 14‐3‐3ζ‐GSK‐3β and 14‐3‐3ζ‐Dvl‐2 complexes stability
The interaction between 14‐3‐3ζ and GSK‐3β has been proposed to be important for the catalyzed phosphorylation induced by GSK‐3β (Agarwal‐Mawal et al., 2003). To explore which of the GSK‐3β properties affect its interaction with 14‐3‐3ζ, three GSK‐3β mutants previously described were used; A kinase‐dead GSK‐3β protein (K85R), a GSK‐3β protein that does not bind Axin (3GR) and a double mutant (K85M) (Fraser et al., 2002). Results show that disrupting the ability of GSK‐3β to bind Axin did not affect the GSK‐3β‐14‐3‐3ζ interaction (Figure 4A, lane 1 compared to lane 5). However, when the kinase activity of GSK‐3β was abolished (in both K85R and K85M, Figure 4A, lanes 2, 3) the complex stability was compromised (Figure 4A). The intensity of the interaction between the different GSK‐3β constructs bound by 14‐3‐3ζ is shown (Figure 4A right panel). GSK‐3β in turn affected the 14‐3‐3ζ‐Dvl‐2 interaction; silencing GSK‐3β by specific siRNA oligos or inhibiting its kinase activity with LiCl or SB (Davies et al., 2000) led to a decrease in 14‐3‐3ζ‐Dvl‐2 complex stability (Fig. 4B, C).
Figure 4.

The kinase activity of GSK‐3β is important for 14‐3‐3ζ‐GSK‐3β and 14‐3‐3ζ‐Dvl‐2 complexes stability. (A) HEK293T cells were co‐transfected with FLAG‐14‐3‐3ζ and one of four HA‐GSK‐3β vectors expressing; a WT protein, a kinase‐dead form of HA‐GSK‐3β protein (K85R), an HA‐GSK‐3β protein that does not bind axin (3 GR) and double mutant (K85M). The HA empty vector was used as control. Forty eight hours later, cells were harvested and lysates were co‐immunoprecipitated using anti‐FLAG‐M2 Affinity beads and subjected to western blotting as indicated. TINA analysis of the intensity of the different GSK‐3β constructs bound by 14‐3‐3ζ is shown. (B) HEK293T cells co‐transfected with HA‐14‐3‐3ζ and FLAG‐Dvl‐2 were left untreated or treated with LiCl (30 mM, 24 h) or SB (10 μM, 4 h). Forty eight hours later the cells were subjected to co‐IP experiment as described above (upper panel). TINA analysis of the intensity of the HA‐14‐3‐3ζ bands is shown (lower panel). (C) HEK293T cells were subjected to GSK‐3β depletion using siRNA oligonucleotide as indicated and then transfected with FLAG‐Dvl‐2 and HA‐14‐3‐3ζ. Co‐IP was performed as described. TINA analysis of the intensity of the HA‐14‐3‐3ζ bands is shown. Molecular mass markers are indicated on the right. All blots are representative results from at least three independent experiments.
2.5. 14‐3‐3ζ and β‐catenin enhance cell motility
Over activation of β‐catenin is widely implicated in cancer development and one of the hallmarks of cancer cells is their enhanced mobility and migratory properties. Furthermore, 14‐3‐3ζ and β‐catenin were recently shown to be involved in migration of lung cancer metastasis (Chen et al., 2012). Thus, we used Transwell filters to study the effect of β‐catenin and 14‐3‐3ζ on cell motility. As shown in Figure 5A, transfection of either GFP‐β‐catenin or Cherry‐14‐3‐3ζ alone, led to only a few cells migrating to the bottom chamber. However, when cells were co‐transfected with both β‐catenin and 14‐3‐3ζ there was a threefold increase in the number of transfected cells that migrated to the bottom chamber (Figure 5A). The effect of β‐catenin and 14‐3‐3ζ expression on cellular migration ability was also tested by a wound healing assay. Cells transfected with both GFP‐β‐catenin and Cherry‐14‐3‐3ζ migrated across a wound and created a monolayer faster than the cells transfected with each of these proteins separately or empty vectors (Figure 5B, C). In both types of experiments, the combined expression of 14‐3‐3ζ and β‐catenin led to enhanced cell migration, although not all migrating cells showed visible expression of both proteins indicating that the effect may be non‐cell autonomies or that one plasmid is expressed at higher levels as compared to the other. Though migration was clearly accelerated when 14‐3‐3ζ and β‐catenin were co‐expressed, proliferation rates were seemingly un‐affected (Figure 5D).
Figure 5.
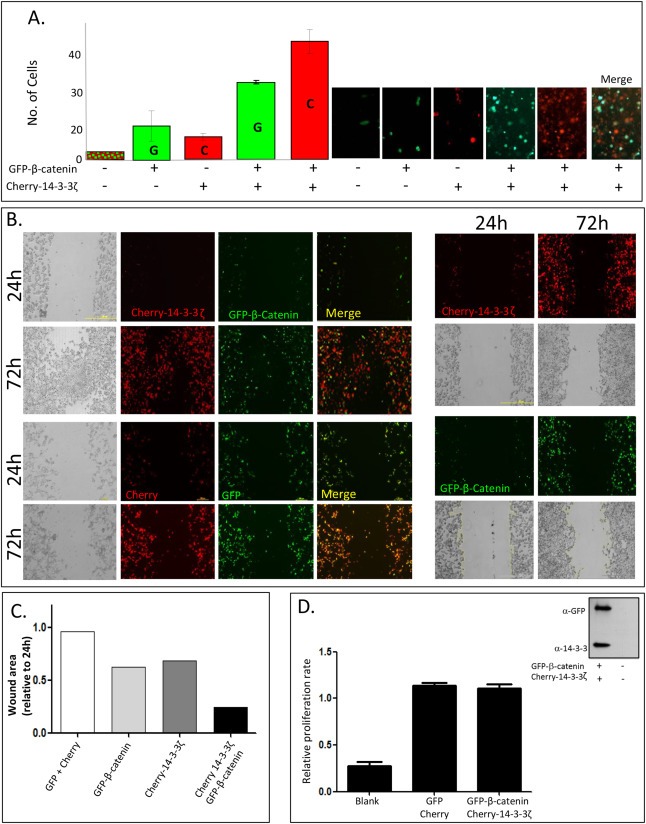
14‐3‐3ζ and β‐catenin enhance cell motility (A) A Transwell motility assays using empty Cherry and GFP vectors, Cherry‐14‐3‐3ζ, GFP‐β‐catenin and double Cherry‐14‐3‐3ζ and GFP‐β‐catenin transfected cells were performed. Shown are representative images of migrating cells over‐expressing vectors, Cherry‐14‐3‐3ζ, GFP‐β‐catenin or both proteins (right panel). Number of GFP (G) or Cherry (C) expressing cells counted in single or double transfection in three independent experiments (left panel) is shown. (B) HEK293T cells were plated in 6‐well culture plates, grown to 70–80% confluence and transfected with Cherry‐14‐3‐3ζ, GFP‐β‐catenin or both, control cells were transfected with the two empty vectors. After aspirating the medium, a thin "wound", constant in width was introduced by scratching with a pipette tip. The right panel depicts migrating cells after a single transfection as indicated and the left panel is of cells co‐transfected with both proteins (upper panel) and empty vectors (lower panel). The images were taken at 24 and 72 h after wound application. The experiment was repeated 5 times with similar results (C) Wound areas for each transfection were measured using the ImageJ software http://rsb.info.nih.gov/ij/. (D) Proliferation rate ‐ HEK293T cells grown in a 24‐well plate were co‐transfected with either GFP‐β‐catenin and Cherry‐14‐3‐3 or GFP and Cherry vectors. Twenty‐four hours post transfection cells were re‐plated in a 96‐well plate for additional 24 h. Proliferation substrate (CellTiter 96 proliferation kit; Promega) was added and samples were measured according to the manufacturer's instructions. Western blot shows expression levels of the transfected proteins.
2.6. 14‐3‐3 and β‐catenin form “bleb‐like” structures and are secreted via extracellular vesicles
To gain further insight regarding the role of the 14‐3‐3 proteins, the human colorectal cancer (CRC) cell line SW480 which express high levels of nuclear β‐catenin (Korinek et al., 1997) was transfected with different 14‐3‐3 isoforms (ζ, η and ε). Immunofluorescence analysis revealed extensive formation of “bleb‐like” cell protrusions that contained both the exogenous 14‐3‐3ζ and endogenous β‐catenin proteins (Figure 6A). Similar results were obtained when HEK293T cells were co‐transfected with Cherry‐14‐3‐3ζ and GFP‐β‐catenin (Figure 6B upper panel, supplementary movie 1). However, formation of these “bleb‐like” structures was not detected when 14‐3‐3ζ was co‐transfected with a constitutively active form of β‐catenin (Figure 6B lower panel). “Bleb‐like” protrusions often indicate apoptotic blabbing (Coleman et al., 2001). Nonetheless, ectopic expression of Cherry‐14‐3‐3ζ and GFP‐β‐catenin did not lead to cleavage of PARP (Figure 6C). Furthermore, caspase‐3 cleavage or activity was not observed when Cherry‐14‐3‐3ζ and GFP‐β‐catenin were co‐transfected in HEK293T cells (Figure 6D, E) ruling out the possibility that the expression of 14‐3‐3ζ and β‐catenin results in apoptotic “blebing”. Previous studies have shown that similar “bleb‐like” structures can shed off the parental cells and affect neighboring cells (Di Vizio et al., 2009). Interestingly, among the proteins found in these vesicles were both 14‐3‐3ζ and β‐catenin (Di Vizio et al., 2009). Moreover, β‐catenin was already shown to be expressed in secreted exosomes (Chairoungdua et al., 2010). To follow this line, conditioned medium (CM) from cells expressing 14‐3‐3ζ and β‐catenin was collected and examined for the presence of secreted vesicles. Our results show that 14‐3‐3ζ and β‐catenin were recovered from the cells growing media. Importantly 14‐3‐3ζ and β‐catenin were purified with the exosomal markers: Heat shock protein 70, CD9 and CD63 (Figure 6F, G). Treating the transfected cells with GW4869 that significantly decreases exosome release (Yuyama et al., 2012) leads to reduced levels of 14‐3‐3ζ and β‐catenin in the extracellular vesicles pellet (Figure 6G upper panel).
Figure 6.

14‐3‐3 proteins and β‐catenin form "blebs" and are partly secreted via extracellular vesicles. (A) SW480 cells were transfected with HA‐14‐3‐3ζ, HA‐14‐3‐3ε or HA‐14‐3‐3η. Forty eight hours post transfection cells were fixed and stained using an anti‐HA and anti‐β‐catenin antibodies. DAPI marks the nucleus. (B) Live imaging of GFP‐β‐catenin or GFP‐β‐catenin S33A and Cherry‐14‐3‐3ζ. HEK293T cells were co‐transfected with GFP‐β‐catenin constructs and Cherry14‐3‐3ζ. Twenty four hours post transfection cells were visualized using a confocal microscope. (C) HEK293T cells were co‐transfected with either GFP‐β‐catenin and Cherry‐14‐3‐3ζ or empty vectors. Lysates were subjected to WB analysis using anti‐PARP. (D) HEK293T cells were co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ or empty vectors, control cells were un‐transfected. Lysates were subjected to WB analysis using anti‐caspase‐3. SSP (an apoptosis inducing reagent, 18 h) was used as a positive control. (E) Caspase 3 activity was determined in the HEK293T cells transfected with the indicated plasmids. Cycloheximide (25 μM, 4 h) served as a positive control. (F) HEK293T cells were co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ or empty vectors. CM was collected and microvesicles were then pelleted by ultracentrifugation. The recovered material was resuspended and blotted with an anti‐heat shock protein 70 (HSP70) antibody. (G) HEK293T cells were co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ. The exosome inhibitor GW4869 was added for 16 h (5 μM). CM was collected and purified as in F. Cell lysates (lower panel) and the recovered material (upper panel) were blotted with anti‐GFP, anti‐14‐3‐3ζ, anti‐CD63 and anti‐CD9 antibodies.
2.7. Secreted 14‐3‐3ζ and β‐catenin are detected in neighboring cells
Once secreted, the exosomes can be up‐taken by the surrounding cells. Thus COS‐7 (Figure 7B) and SW480 (Figure 7C) cells were incubated for 24 h with extracellular vesicles secreted from HEK293T cells expressing 14‐3‐3ζ and β‐catenin (Figure 7A). Results obtained from Immunofluorescence and Western blot analyses show the presence of both 14‐3‐3ζ and β‐catenin in receiving cells (Figure 7D; supplementary movie 2). In these receiving cells 14‐3‐3ζ and β‐catenin partly co‐localize and are frequently found in close vicinity of the nucleus (Figure 7E). Nuclear extracts derived from these receiving cells reveal that a small portion of the exogenous β‐catenin resides within the nucleus itself (Figure 7F). Both the “bleb‐like” structures in the secreting cells and the appearance of 14‐3‐3ζ and β‐catenin in the accepting cells are much more visible in live cells as compared to fixated cells. Thus, to gain further information on the nature of these structures LysoTracker that traces acidic vesicles in living cells was used. Interestingly, the co‐localization of 14‐3‐3ζ, β‐catenin and the LysoTracker was detected only in the receiving cells and not in the secreting cells (Figure 7A, compared to 7B–C). Taken together these results imply that 14‐3‐3ζ and β‐catenin are secreted on extracellular vesicles that can be up‐taken by neighboring cells where they are associated with acidic vesicles.
Figure 7.

Secreted 14‐3‐3ζ and β‐catenin are detected in neighboring cells and co‐localize with a lysosomal marker in acceptor cells. HEK293T cells were co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ (A). Extracellular vesicles were isolated as described and resuspended in PBS which then was added to COS‐7 (B) or SW480 (C) acceptor cells. Twenty four hours post transfection or addition of the extracellular vesicles, the cells were incubated with LysoTracker blue (Invitrogene 1:1000; 1 h 37 °C) and visualized using live cell confocal microscopy. Lower panel depicts computerized co‐localization analysis in SW480 cells. Bars = 10 μm (D) Western blot analysis of extracts derived from HEK293T cells co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ or empty vectors (left) or HEK293T acceptor cells that were incubated with purified extracellular vesicles (right). (E) SW480 acceptor cells cultivated with extracellular vesicles derived from HEK293T cells co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ were fixed and visualized using a confocal microscope. Bottom panel is a z‐section of the above. (F) Extracellular vesicles (as indicated) derived from transfected HEK293T cells were added to SW480 acceptors for 24 h. The cells were then harvested and subjected to nuclear extraction and WB analysis. All experiments were repeated 3 times.
2.8. Purified 14‐3‐3ζ β‐catenin extracellular vesicles enhance cell migration and lead to cytoskeleton rearrangement
We then tested the effect of the extracellular vesicles on cellular migration and on the cells cytoskeleton. Our results show that incubating HEK293T cells with 14‐3‐3ζ‐β‐catenin containing extracellular vesicles lead to enhanced cell motility in both Transwell chambers and wound healing assays (Figure 8A). Once again proliferation rates remained un‐affected (Figure 8B). The cytoskeleton in both HEK293T and COS‐7 cells exposed to the extracellular vesicles was examined by staining for actin and tubulin. Results show large differences in the actin cytoskeleton arrangement between cells treated with control or 14‐3‐3ζ‐β‐catenin extracellular vesicles. As can be seen from Figure 8C, exposing COS‐7 cells (that have low migration ability) to the 14‐3‐3ζ‐β‐catenin extracellular vesicles, lead to assembly of actin stress fibers in thick bundles at the periphery of the cell. Similarly, treating HEK293T cells with the 14‐3‐3ζ‐β‐catenin extracellular vesicles lead to formation of long, condensed stress fibers and changes in the cells shape (Figure 8D).
Figure 8.

The activity of the β‐catenin‐14‐3‐3ζ extracellular vesicles (A). Purified extracellular vesicles were prepared as described and added to HEK293T acceptor cells to perform Transwell motility assay where migrating cells were stained using H&E staining (upper panel) or a "wound healing" experiment, in which the migration rate was evaluated by measuring the distance between the edges 24 h later (lower panel). (B) Proliferation rates of HEK293T treated with the indicated extracellular vesicles were determined as described above. (C–D) COS‐7 (C) or HEK293T (D) cells were incubated with extracellular vesicles as indicated for 24 h. The cells were then fixed and stained with anti‐tubulin antibody and FITC‐conjugated phalloidine and visualized using a confocal microscope. The control panel depicts stained cells prior to extracellular vesicle addition. Bars = 25 μm.
2.9. Purified extracellular vesicles can induce Wnt signaling activity
We next investigated whether 14‐3‐3ζ and β‐catenin that enter the cell via extracellular vesicles up‐take can activate the Wnt signal. Our results show that in comparison to basal reporter level, addition of extracellular vesicles from cells transfected with both 14‐3‐3ζ and β‐catenin led to an increase in Wnt/β‐catenin mediated transcription in a dose dependent manner (Figure 9A). A similar experiment was performed in other cancerous (Huh7, HeLa) and non‐cancerous (COS‐7) cells (Figure 9B). To further validate the idea that extracellular vesicles obtained from cell expressing 14‐3‐3ζ and β‐catenin can transduce the Wnt signal the HEK293‐EBNA‐PurR cells (Skalka et al., 2013) were used. Specific activation of the Wnt cascade in this cell line results in expression of the puromycin gene, rendering resistance and cell survival (Skalka et al., 2013). Addition of extracellular vesicles obtained from cells expressing 14‐3‐3ζ and β‐catenin led to a dramatic increase in cell survival as compared to the control (Figure 9C). Wnt3a CM served as a positive control (Figure 9C, right panel). Taken together these experiments indicate that purified extracellular vesicles from 14‐3‐3ζ and β‐catenin expressing cells have the capacity to induce Wnt signaling and affect cell migration. As Wnt3a is also known to be secreted via extracellular vesicles, we tested our extracellular vesicles for the presence of Wnt3a along with the 14‐3‐3ζ and β‐catenin proteins. Wnt3a was not detected in extracellular vesicles derived from HEK293T cells co‐transfected with Cherry‐14‐3‐3ζ and GFP‐β‐catenin (Figure 9D left panel) or SW480 expressing exogenous 14‐3‐3 (Figure 9D right panel).
Figure 9.

β‐catenin‐14‐3‐3ζ extracellular vesicles activate the Wnt pathway. (A) HEK293T transfected with the pTOPFLASH reporter vectors and β‐gal were incubated with increasing amounts of purified extracellular vesicles (derived from double transfected cells as described above). Relative luciferase values are shown. (B) Luciferase assay was performed in HeLa, Huh7 and COS‐7 acceptor cells (incubated with purified extracellular vesicles – as described above) transfected with the pTOPFLASH reporter system and renilla CMV. Relative luciferase values are shown. (C) Purified extracellular vesicles were resuspended in PBS and then added to 2 × 106/per 60 mm plate HEK293‐EBNA‐PurR acceptor cells (Skalka et al., 2013). Twenty four hours later, the growth media was replaced with puromycin CM (1 μg/ml) or with medium collected from L‐wnt3a cells. Thirty h later the cells were fixed and stained using methylene blue. (D) Extracellular vesicles from HEK293T cells (left panel) or SW480 (right panel) do not contain the Wnt3a ligand. Growing medium from L‐Wnt3a cells served as a positive control. Cell lysates or extracellular vesicles were incubated with the indicated antibodies.
3. Materials and methods
3.1. Cell culture, transfection
HEK293T, COS‐7, SW480, HeLa, Huh7, HEK293‐EBNA‐PurR and L‐Wnt3a cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FCS and 100 U/ml penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere. Huh7 were supplemented with 1% Non‐essential amino acids solution. Transfection of HEK293T cells was performed by either CaPO4 precipitation or Polyethyleneimine “MAX” (Polysciences, Warrington, Pennsylvania) following manufacturer's protocols. All other cells were transfected using the DNA transfection reagents jet PEI (Polyplus Transfection) or Lipofectamine 2000 (Invitrogen), following the manufacturers protocols.
3.2. Plasmids and reagents
14‐3‐3ζ constructs: HA‐14‐3‐3ζ, HA‐14‐3‐3ε and HA‐14‐3‐3η (pCS2+) were kindly provided by Ken‐Ichi Takemaru (Stony Brook, NY 11794). Cherry‐14‐3‐3ζ was constructed by inserting the 14‐3‐3ζ cDNA into pmCherry–C1 (Clontech) using BglII and EcoRI restriction sites. FLAG‐14‐3‐3ζ construct was prepared by inserting the 14‐3‐3ζ cDNA into a FLAGX3‐pcDNA3.1(−) vector using XbaI and EcoRI restriction sites. GFP‐β‐catenin was constructed by inserting the β‐catenin cDNA into pEGFP‐C1 (Clontech) using BamHI and XbaI restriction sites. This expression vector was used as a template for mutagenesis of β‐catenin by the QuikChange site‐directed mutagenesis kit (Stratagene) to replace serine 33 with alanine to generate GFP‐S33A‐β‐catenin. The HA‐GSK‐3β, HA‐GSK‐3β GR, HA‐GSK‐3β K85R and HA‐GSK‐3β M85 expression vectors, were kindly provided by Dr. T. C. Dale (Developmental Biology, Chester Beatty Laboratories, Institute of Cancer Research, London, UK). FLAG‐GSK‐3β was kindly provided by Dr. Hagit Eldar‐Finkelman (Tel‐Aviv University, Israel). FLAG‐Dvl‐2 was kindly provided by Dr. Arnona Gazit and Dr. Abraham Yaniv (Tel‐Aviv University, Israel). This expression vector was used as a template for mutagenesis of Dvl‐2 by the QuikChange site‐directed mutagenesis kit (Stratagene) to replace serine 709 with alanine to generate FLAG‐S709A‐Dvl‐22. The Wnt‐responsive TCF‐dependent luciferase constructs pTOPFLASH (containing multi TCF‐binding sites linked to a luciferase reporter) and its mutated version pFOPFLASH were kindly provided by Dr. H. Clevers (Center for Biomedical Genetics, Hubrecht, The Netherlands) and were described previously (Morin et al., 1997). pCMV‐β‐galactosidase expression plasmid, used to evaluate transfection efficiency, was purchased from Clontech. APCarm encodes the human APC armadillo repeat region spanning amino acids 188–774 whereas the APC core spans amino acids 1379‐2080. Specific RNA oligonucleotides for silencing 14‐3‐3ζ or GSK‐3β were purchased from Thermo Scientific Dharmacon (siGENOME SMART pool M‐003332‐01‐0005, Human YWHAZ and M‐003010‐03‐0005, hGSK3β NM‐002093 respectively) as well as the transfection reagent Dharmafect‐1. Transfections were performed according to manufacturer's protocol using 100 nM siRNA oligonucleotides. Non targeting RNA oligonucleotides (Thermo Scientific Dharmacon) were used as controls. Twenty‐four h later cells were transfected with the indicated plasmids and subjected to further experiments (either IP or luciferase assay).
MG‐132 (Calbiochem, San Diego, CA, USA; 25 μM, 5 h), LiCl (Sigma, Israel; 30 mM, 24 h), GW4869 (Sigma, Israel; 5 μM, 16 h), SB (Sigma, Israel; 10 μM; 4 h), LysoTracker blue (Invitrogene, 1:1000; 1 h). Cycloheximide (Sigma, Israel 25 μM; 4 h), staurosporine (SSP, Sigma, Israel 1 μM; 18 h)
3.3. Antibodies
The following antibodies were used: mouse anti‐β‐catenin (IB: 1:5000; IF: 1:300; BD Transduction Laboratories), rabbit anti‐GFP (1:1000; Santa Cruz Biotechnology), rat anti‐HA (IB: 1:2500; IF: 1:300; Roche), mouse anti‐FLAG (1:5000; Sigma), rabbit anti‐FLAG (1:500; Sigma), rabbit anti‐14‐3‐3ζ (IB: 1:500; Santa cruz), rabbit anti‐Dvl‐2 (IB: 1:250; Santa‐cruz), mouse anti–GSK‐3β (IB: 1:1000; IF: 1:300; BD Transduction Laboratories), mouse anti PARP (1:5000; Biomol), mouse anti‐CD63 (1:100; Abcam), rabbit anti‐CD9 (1:50; Abcam), mouse anti‐active β‐catenin(1:1000; Milipore), rabbit anti‐HSP70 (1:2000; Santa Cruz Biotechnology), rabbit anti‐wnt3a (1:1000; Cell Signaling), rabbit anti‐lamin B1 (1:2500; Abcam), rabbit anti‐caspase 3 (1:1000; Cell Signaling), mouse‐anti‐Cyclin D1 (1:1000; Santa Cruz Biotechnology), rabbit anti‐alpha tubulin (IF: 1:1000, Abcam). FITC‐conjugated phalloidine (IF: 1:500; Sigma, Israel). Mouse anti‐tubulin (1:10,000; Sigma) was used as loading controls. Anti‐rat horseradish peroxidase‐conjugated secondary antibodies were obtained from Santa Cruz Biotechnology (1:5000). Anti‐mouse and anti‐rabbit HRP‐conjugated secondary antibodies were obtained from Jackson Immuno Research (1:10,000). For IF Alexa red and green (1:500; Molecular Probes) were used.
3.4. Western blot analysis and immunoprecipitation (IP)
Forty‐eight hours following transfection, cells were washed with PBS and solubilized in reporter lysis buffer (luciferase buffer, Promega), or M2 lysis buffer (100 mM NaCl, 50 mM Tris, pH7.5, 1% Triton X‐100, 2 mM EDTA) containing protease inhibitor cocktail (Sigma). Extracts were clarified by centrifugation at 12,000× g for 15 min at 4 °C. Following SDS polyacrylamide gel electrophoresis (SDS‐PAGE) separation, proteins were transferred to nitrocellulose membranes and blocked with 5% low fat milk. Membranes were incubated with specific primary antibodies, washed with PBS containing 0.001% Tween‐20 (PBST) and incubated with the appropriate horseradish peroxidase‐conjugated secondary antibody. After washing in PBST, membranes were subjected to enhanced chemiluminescence detection analysis. For IP analysis, cells were solubilized in lysis buffer (see above). Cell lysates were incubated with anti‐FLAG M2‐agarose affinity gel (Sigma), with rotation for 2–18 h at 4 °C. Alternatively, cell lysates were incubated with the specific antibody for 1–2 h at 4 °C prior to 2–18 h rotated incubation with protein A/G agarose (Santa Cruz Biotechnology) at 4 °C. Beads were collected by slow centrifugation, washed 4 times with lysis buffer and analyzed by SDS‐PAGE followed by detection with specific antibody. Band intensity was measured by TINA ‐ computer‐assisted densitometer program (TINA 2.0c; Fuji BAS, Tokyo, Japan) for measuring the intensity of protein bands.
3.5. Extracellular vesicle purification
HEK293T cells were co‐transfected with GFP‐β‐catenin and Cherry‐14‐3‐3ζ or with the empty vectors. Alternatively, SW480 cells that express high levels of β‐catenin were transfected with Cherry‐14‐3‐3ζ. Twenty four hours later, extracellular vesicles were collected from conditioned medium (CM) and purified. Briefly, medium was centrifugations at 580× g 10 min at 4 °C to eliminate cells debris. Microvesicles were then pelleted by ultracentrifugation for 70 min at 4 °C, 100,000× g and recovered material was suspended in 100 μl M2 lysis buffer containing protease inhibitor. Lysates were subjected to SDS PAGE gel as describe or alternatively, recovered microvesicles were re‐suspended in ice‐cold PBS containing protease inhibitor and incubated with pre‐treated serum‐free HEK293T, SW480, COS‐7 or HEK293‐EBNA‐PurR recipient cells, for 24 h. The recipient cells were then harvested and analyzed by SDS PAGE gel as describe or used as indicated in different activity assays and cell imaging.
3.6. Luciferase reporter assays
To assay TCF‐mediated transcription, cells were seeded at 1 × 105 cells per well in a 24‐well plate 24 h before transfection. Cells were transfected with the specific vectors, along with pTOPFLASH/pFOPFLASH and either β‐gal (HEK293T) or Renilla CMV (SW480, HeLa, Huh7 and COS‐7) plasmids. Forty‐eight hours post‐transfection the cells were harvested and subjected to luciferase assay according to the manufacturer's instructions. In all assays, FOPFLASH activity was measured by replacing the pTOPFLASH with pFOPFLASH under equivalent conditions. To assess extracellular activity activity, the transfected cells were incubated with purified extracellular vesicles for 24 h prior to preforming the luciferase assay.
3.7. Immunofluorescence (IF) and live cell imaging
SW480 cells were grown on coverslips and fixed 48 h post transfection for 20 min in PBS containing 4% paraformaldehyde. After 3 washes with PBS, the fixed cells were permeabilized with 0.1% Triton X‐100 for 10 min and blocked with bovine serum albumin for 1 h. Subsequently, cells were incubated at room temperature with primary and secondary antibodies for 60 and 30 min, respectively. 4–6′ diamidino‐2 phenylindole (DAPI, Sigma) was used to stain cell nuclei.) HEK293T or COS‐7 cells were incubated with extracellular vesicles for 24 h. The cells were then fixed and stained with anti‐tubulin antibody and FITC‐conjugated phalloidine. Cherry and GFP were detected without staining. Cells were visualized by Confocal Microscopy. For live imaging, SW480 or COS‐7 recipient cells were serum‐starved for 6 h prior to incubation with purified extracellular vesicles. Twenty four hours post incubation, live imaging analysis was performed using confocal microscope designed for that application.
3.8. Puromycin selection assay
HEK293‐EBNA‐PurR acceptor cells were seeded on 60 mm plate at a concentration of 2 × 106 and incubated with purified extracellular vesicles for twenty‐four hours, after which, the growth media was replaced with puromycin CM (1 μg/ml) or with medium collected from L‐wnt3a cells. Thirty hours later the cells were fixed with methanol and stained using Methylene blue staining.
3.9. Wound healing assay
HEK293T cells were plated in 60 mm culture plates, grown to 70–80% confluence and transfected with GFP‐β‐catenin, Cherry‐14‐3‐3ζ or both. Alternatively, the cells were incubated with isolated extracellular vesicles (described earlier) for 24 h. After aspirating the medium, a thin “wound”, constant in width was introduced by scratching with a pipette tip. Cells at the wound edge polarize and migrate into the wound space. The cells were photographed by Nikon eclipse Ti microscope and migration rates were evaluated by either measuring the distance between the edges at the indicated time points or calculating the wound areas using the ImageJ software (http://rsb.info.nih.gov/ij/).
3.10. Transwell migration assay
Cell migration was assayed in 24‐well, 8‐mm pore membrane Transwell cell culture chambers (Costar, Cambridge, MA). HEK293T Cells were transfected with Cherry‐14‐3‐3ζ, GFP‐β‐catenin or both. Twenty four hours later, 2 × 105 cells from each transfection type were seeded in the upper chamber in DMEM medium with no FCS. Growing medium was added to the lower chamber after 2 h. Twenty four hours post seeding, cells were washed twice with PBS and fixed in 3.7% formaldehyde for 20 min at R.T. The non‐migrating cells were scraped with cotton swab and cells that had migrated to the lower surface of the membrane were visualized by Nikon eclipse TE‐2000E microscope.
Alternatively, 2 × 105 HEK293T cells were seeded in the upper chamber in DMEM medium with no FCS and incubated with purified extracellular vesicles (described above) for 24 h. Growing medium was added to the lower chamber after 2 h. Twenty‐four hours post seeding, the cells were fixed in ice‐cold methanol for 5 min, stained with Eosin and Haematoxylin Solutions for 5 min each and finally washed twice with DDW. The non‐migrating cells were scraped with cotton swab and cells that had migrated to the lower surface of the membrane were visualized by Nikon eclipse TE‐2000E microscope.
3.11. Nuclear extracts
Sw480 cells were plated in a 6‐well plate at 50% confluence and grown over night. The media was then replaced by 500 μl serum free medium for 2 h after which extracellular vesicles were added (500 μl) as indicated. Twenty‐four hours later the cells were lyzed in 200 μl of “low salt” buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA) containing protease inhibitor cocktail (Sigma). Cells were incubated for 30 min on ice with vortexing and centrifugation at 10,000× rpm for 10 min. The supernatant was discarded and nuclei were resuspended in 40 μl “high salt” buffer (20 mM HEPES pH 7.9, 0.42 M NaCl, 1 mM EDTA, 1 mM EGTA) containing protease inhibitor cocktail (Sigma). Cells were incubated for 30 min on ice, followed by centrifugation at 14,000× rpm for 10 min. Sample buffer was added and the nuclear lysates were boiled for 30 min before WB analysis.
3.12. Proliferation assay
HEK293T cells grown in a 24‐well plate were co‐transfected with either GFP‐β‐catenin and Cherry‐14‐3‐3 or GFP and Cherry vectors. Twenty‐four hours post transfection cells were trypsinized and re‐plated in a 96‐well plate (100 μl, 1:4 ratio). Alternatively, HEK293T cells were grown in a 96‐well plate and incubated with extracellular vesicles as indicated. After additional 24 h proliferation substrate (CellTiter 96 proliferation kit; Promega) was added and samples were measured according to manufacturer protocol.
3.13. Caspase‐3 activity
HEK293T cells grown in a 6‐well plate were co‐transfected with either GFP‐β‐catenin and Cherry‐14‐3‐3 or GFP and Cherry vectors. Forty‐eight h post transfection cells were harvested and caspase activity was measured using the Caspase‐3 Colorimetric Assay kit (ab39401 Abcam) according to manufacturer's protocol. SSP or cycloheximide were used as positive controls. Lysates from this assay were also analyzed via WB to detect caspase‐3 cleavage using a specific antibody.
3.14. Statistical analysis
Data are expressed as means ± standard deviation (SD). Significant differences in mean values were assessed by un‐paired Student t test. A P value of ≤0.05 was considered significant. All experiments were performed at least three times.
4. Discussion
Up‐regulation of the canonical Wnt signaling pathway is linked to numerous different diseases, including cancer. Attenuating the levels of the Wnt cascade key effector, β‐catenin, is complex and occurs through several distinct mechanisms with the most robust being the phosphorylation of the latter by CK1α and GSK‐3β marking it for degradation by the proteasome system (Valenta et al., 2012). In an attempt to identify new Wnt signaling component we performed immunopercipitation assays using Dvl‐2, that is a component of both the noncanonical and canonical Wnt pathways, as bait. Our experiments revealed that Dvl‐2 forms a complex with different members of the 14‐3‐3 protein family. The 14‐3‐3 family comprises at least seven isoforms and is a class of highly conserved proteins involved in regulating signal transduction pathways, cellular proliferation, differentiation and survival (Mhawech, 2005) (Fu et al., 2000) (Morrison, 2009). Different members of the 14‐3‐3 protein family have been shown to regulate the Wnt cascade. In a number of studies the 14‐3‐3 proteins attenuate the pathway, mostly through Chibby (Li et al., 2008) (Li et al., 2010) (Fischer et al., 2012) while in other the 14‐3‐3 proteins activate the Wnt cascade (Tian et al., 2004) (Fang et al., 2007) (Chen et al., 2011) (Chang et al., 2012) demonstrating the complex relationship between Wnt signaling and the 14‐3‐3 protein family. In this study we show that 14‐3‐3ζ, previously shown to bind β‐catenin (Tian et al., 2004) (Li et al., 2008) forms a complex with Dvl‐2. Activation of the Wnt pathway did not affect this interaction indicating that the 14‐3‐3ζ and Dvl‐2 proteins interact constitutively. Interestingly, this interaction depends on a conserved 14‐3‐3 binding site that is found in Dvl‐2 but not in Dvl‐1 or Dvl‐3. All three Dvl isoforms have been suggested to have roles in the canonical Wnt pathway. However, the most abundant Dvl isoform‐Dvl‐2 (Lee et al., 2008), has been suggested to function as the main “building‐block” of the Wnt induced signalosomes (Wang and Malbon, 2012). Although somewhat controversial (Matthews and Johnson, 2005), former studies (Agarwal‐Mawal et al., 2003), as well as our results indicate that 14‐3‐3ζ binds GSK‐3β. Our data also suggest that the 14‐3‐3ζ‐GSK‐3β interaction inhibits the association between GSK‐3β and β‐catenin. Similarly, it has recently been shown that in mouse embryonic stem cells 14‐3‐3σ disrupts β‐catenin binding to the “β‐catenin degradation complex” (Chang et al., 2012). Taken together, these results suggest that 14‐3‐3ζ forms a complex with GSK‐3β, Dvl‐2 and β‐catenin to sequester β‐catenin away from the “degradation complex” resulting in increased levels of Wnt signaling. Up‐regulation of the canonical Wnt signaling levels is associated with oncogenesis mostly through cell proliferation and renewal. However activation of the pathway is also involved in enhanced cell migration (Dwyer et al., 2010) (Cho et al., 2013) which is usually characterized by increased cell mobility. The Wnt pathway is also involved in cell migration from another aspect as the key component of this signaling pathway‐β‐catenin is an integral component of the cadherin complex which controls cell–cell adhesion and is involved in cell migration (Nelson and Nusse, 2004). Indeed, the data provided in this work show that 14‐3‐3ζ and β‐catenin enhance cell motility. Similarly, it has previously been shown that in lung cancer the interaction between 14‐3‐3 and β‐catenin leads to stabilization of β‐catenin and enhanced cell migration (Chen et al., 2012). As opposed to GSK‐3β and β‐catenin, the Dvl proteins are core components of both the canonical and non‐canonical Wnt signaling cascades (Grumolato et al., 2010) and it has recently been shown that exosomes secreted from fibroblasts induce breast cancer cells motility and invasion in a non‐canonical Wnt signaling dependent manner. Moreover, as a result of exposure to exosomes the cancer cells show changes in the cytoskeleton rearrangement as well as in Dvl's subcellular organization which eventually result in enhanced cell migration (Luga et al., 2012). The 14‐3‐3 proteins have also been shown to have important roles in enhanced cell migration, invasion and cytoskeleton changes (Boudreau et al., 2013) (Powell et al., 2003). Our results show that expression of 14‐3‐3ζ leads to formation of “bleb‐like” structures in SW480 cells that express high levels of endogenous β‐catenin. Similarly, co‐expression of 14‐3‐3ζ and β‐catenin in HEK293T cells results in the formation of large “bleb‐like” structures that contain both 14‐3‐3ζ and β‐catenin. Following formation of these protrusions the two proteins were secreted from the parental cells and could be co‐isolated with exosomal markers. Once released, exosomes can be internalized into target cells by fusion, adhesion or direct binding (Raposo et al., 1996) (Denzer et al., 2000) (Morelli et al., 2004). According to our results the extracellular vesicles that contained the 14‐3‐3ζ and β‐catenin proteins are up‐taken by neighboring cells and associate with an acidic vesicle marker. Since cells exposed to these extracellular vesicles exhibit enhanced motility, it is plausible to speculate that expression of 14‐3‐3ζ and Dvl‐2 in the receiving cells enhances cell migration. Cell migration is largely associated with changes in the actin cytoskeleton and enhanced cell proliferation. However, our results show that although the 14‐3‐3ζ‐β‐catenin extracellular vesicles induce large changes in the actin cytoskeleton, namely the re‐assembly of actin stress fibers in thick bundles at the periphery of the cell and the formation of long, condensed stress fibers and changes in cell shape the rates of cell proliferation do not change as has been show in other systems (Gong et al., 2014) (Ponugoti et al., 2013) (Chitanuwat et al., 2013). The main conclusion drown from our data is that extracellular vesicles containing 14‐3‐3ζ and β‐catenin (but not the Wnt3a ligand) activate Wnt signaling in the receiving cells. The results suggest a novel mechanism by which the 14‐3‐3ζ protein, via extracellular vesicles, shuttles β‐catenin to acceptor cells in which β‐catenin enters the nucleus to transduce the oncogenic Wnt signal. Interestingly, it has recently been shown that oncosomes that transfer oncogenic signals between cells contain among others both the 14‐3‐3 and the β‐catenin protein (Di Vizio et al., 2009). Exosomes originated from multi vesicular bodies (MVBs) (Thery et al., 2002) and β‐catenin has been shown to be secreted via theses exosomes. Secretion of β‐catenin on exosomes was suggested to reduce the intracellular pool of β‐catenin and Wnt signaling levels (Chairoungdua et al., 2010). Here we show that extracellular vesicles containing β‐catenin can be up‐taken by neighboring cells suggesting that β‐catenin can travel between cells using extracellular routes. Taken together, it is reasonable to speculate that secretion of β‐catenin on exosomes may lead to reduced levels of Wnt signaling in the secreting cells, while in cells receiving theses extracellular vesicles the pathway is activated. 14‐3‐3 has been shown to chaperone different proteins. For example, it modulates the secretion of Endopeptidase 24.15 (Carreno et al., 2005) and interacts with Chibby leading to nuclear export of β‐catenin (Li et al., 2008). It was also demonstrated that the secretion of LRKK2 on exosomes is regulated by 14‐3‐3 (Fraser et al., 2013) much like the data presented in this study regarding its effect on β‐catenin. A number of studies have shown that both the canonical and non‐canonical Wnt ligands can transport via exosomes to activate the Wnt cascade in receiving cells (Gross et al., 2012) (Beckett et al., 2013) (Menck et al., 2013) (Luga et al., 2012) and that Rab11 is important for exosomes secretion (Beckett et al., 2013). Interestingly, it has also been shown that the 14‐3‐3 proteins are important for correct positioning of Rab11 in recycling endosomes (Winter et al., 2012). As different types of exosomes are expressed in different systems and function in diverse biological processes (Zhang and Grizzle, 2014) we speculate that the extracellular vesicles described in this study differ from the Wnt‐including exosomes and transmit the Wnt signal via the β‐catenin protein. Activation of the Wnt pathway leads to formation of MVBs that contain a large number of Wnt signaling components, including β‐catenin (Taelman et al., 2010) and it has been suggested that the proteins in these vesicles may be re‐cycled (Dobrowolski and De Robertis, 2012). Based on our data we propose that the 14‐3‐3 proteins participate in a mechanism responsible for shuttling β‐catenin between cells using extracellular vesicles. Following the internalization of β‐catenin in accepting cells and the formation of acidic vesicles such as MVBs, the vesicles disintegrate; β‐catenin is released and can initiate signaling in the target cells without dependents on the activation of the pathway by the Wnt ligands.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Supplementary movie 1
Supplementary movie 2
Acknowledgments
This work was supported by the Israel Science Foundation (ISF), by grant no. 20120016 from the Public Committee for Allocation of Estate Funds, Ministry of Justice, Israel, the Recanati Foundation, Israel Cancer Association through the Estate of the late Alexander Smidoda, U.S. – Israel Binational Science Foundation (BSF).
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.03.011
Dovrat Shiri, Caspi Michal, Zilberberg Alona, Lahav Lital, Firsow Anastasia, Gur Hila and Rosin-Arbesfeld Rina, (2014), 14‐3‐3 and β‐catenin are secreted on extracellular vesicles to activate the oncogenic Wnt pathway, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.03.011.
References
- Agarwal-Mawal, A. , Qureshi, H.Y. , Cafferty, P.W. , Yuan, Z. , Han, D. , Lin, R. , 2003. 14-3-3 connects glycogen synthase kinase-3 beta to tau within a brain microtubule-associated tau phosphorylation complex. J. Biol. Chem.. 278, 12722–12728. [DOI] [PubMed] [Google Scholar]
- Alao, J.P. , 2007. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol. Cancer. 6, 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers, S. , Moon, R.T. , 2009. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol.. 10, 468–477. [DOI] [PubMed] [Google Scholar]
- Beckett, K. , Monier, S. , Palmer, L. , Alexandre, C. , Green, H. , Bonneil, E. , 2013. Drosophila S2 cells secrete wingless on exosome-like vesicles but the wingless gradient forms independently of exosomes. Traffic. 14, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilic, J. , Huang, Y.L. , Davidson, G. , Zimmermann, T. , Cruciat, C.M. , Bienz, M. , 2007. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 316, 1619–1622. [DOI] [PubMed] [Google Scholar]
- Blache, P. , van de Wetering, M. , Duluc, I. , Domon, C. , Berta, P. , Freund, J.N. , 2004. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J. Cell Biol.. 166, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau, A. , Tanner, K. , Wang, D. , Geyer, F.C. , Reis-Filho, J.S. , Bissell, M.J. , 2013. 14-3-3sigma stabilizes a complex of soluble actin and intermediate filament to enable breast tumor invasion. Proc. Natl. Acad. Sci. U S A. 110, E3937–E3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno, F.R. , Goni, C.N. , Castro, L.M. , Ferro, E.S. , 2005. 14-3-3 epsilon modulates the stimulated secretion of endopeptidase 24.15. J. Neurochem.. 93, 10–25. [DOI] [PubMed] [Google Scholar]
- Chairoungdua, A. , Smith, D.L. , Pochard, P. , Hull, M. , Caplan, M.J. , 2010. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol.. 190, 1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, T.C. , Liu, C.C. , Hsing, E.W. , Liang, S.M. , Chi, Y.H. , Sung, L.Y. , 2012. 14-3-3sigma regulates beta-catenin-mediated mouse embryonic stem cell proliferation by sequestering GSK-3beta. PloS One. 7, e40193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.H. , Chuang, S.M. , Yang, M.F. , Liao, J.W. , Yu, S.L. , Chen, J.J. , 2012. A novel function of YWHAZ/beta-catenin axis in promoting epithelial-mesenchymal transition and lung cancer metastasis. Mol. Cancer Res. MCR. 10, 1319–1331. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Liu, L. , Ma, B. , Ma, T.M. , Hou, J.J. , Xie, G.M. , 2011. Protein kinase A-mediated 14-3-3 association impedes human Dapper1 to promote dishevelled degradation. J. Biol. Chem.. 286, 14870–14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitanuwat, A. , Laosrisin, N. , Dhanesuan, N. , 2013. Role of HMGB1 in proliferation and migration of human gingival and periodontal ligament fibroblasts. J. Oral Sci.. 55, 45–50. [DOI] [PubMed] [Google Scholar]
- Cho, S.W. , Lee, E.J. , Kim, H. , Kim, S.H. , Ahn, H.Y. , Kim, Y.A. , 2013. Dickkopf-1 inhibits thyroid cancer cell survival and migration through regulation of beta-catenin/E-cadherin signaling. Mol. Cell Endocrinol.. 366, 90–98. [DOI] [PubMed] [Google Scholar]
- Coleman, M.L. , Sahai, E.A. , Yeo, M. , Bosch, M. , Dewar, A. , Olson, M.F. , 2001. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol.. 3, 339–345. [DOI] [PubMed] [Google Scholar]
- Davies, S.P. , Reddy, H. , Caivano, M. , Cohen, P. , 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J.. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzer, K. , van Eijk, M. , Kleijmeer, M.J. , Jakobson, E. , de Groot, C. , Geuze, H.J. , 2000. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol.. 165, 1259–1265. [DOI] [PubMed] [Google Scholar]
- Di Vizio, D. , Kim, J. , Hager, M.H. , Morello, M. , Yang, W. , Lafargue, C.J. , 2009. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res.. 69, 5601–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski, R. , De Robertis, E.M. , 2012. Endocytic control of growth factor signalling: multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Biol.. 13, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer, M.A. , Joseph, J.D. , Wade, H.E. , Eaton, M.L. , Kunder, R.S. , Kazmin, D. , 2010. WNT11 expression is induced by estrogen-related receptor alpha and beta-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res.. 70, 9298–9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, D. , Hawke, D. , Zheng, Y. , Xia, Y. , Meisenhelder, J. , Nika, H. , 2007. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem.. 282, 11221–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, V. , Brown-Grant, D.A. , Li, F.Q. , 2012. Chibby suppresses growth of human SW480 colon adenocarcinoma cells through inhibition of beta-catenin signaling. J. Mol. Signal.. 7, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, E. , Young, N. , Dajani, R. , Franca-Koh, J. , Ryves, J. , Williams, R.S. , 2002. Identification of the Axin and Frat binding region of glycogen synthase kinase-3. J. Biol. Chem.. 277, 2176–2185. [DOI] [PubMed] [Google Scholar]
- Fraser, K.B. , Moehle, M.S. , Daher, J.P. , Webber, P.J. , Williams, J.Y. , Stewart, C.A. , 2013. LRRK2 secretion in exosomes is regulated by 14-3-3. Hum Mol Genet. 22, 4988–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H. , Subramanian, R.R. , Masters, S.C. , 2000. 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol.. 40, 617–647. [DOI] [PubMed] [Google Scholar]
- Gong, X.H. , Chen, C. , Hou, P. , Zhu, S.C. , Wu, C.Q. , Song, C.L. , 2014. Overexpression of miR-126 Inhibits the Activation and Migration of HSCs through Targeting CRK. Cell. Physiol. Biochem. Int. J. Exp. Cellular Physiol. Biochem. Pharmacol.. 33, 97–106. [DOI] [PubMed] [Google Scholar]
- Gross, J.C. , Chaudhary, V. , Bartscherer, K. , Boutros, M. , 2012. Active Wnt proteins are secreted on exosomes. Nat. Cell Biology. 14, 1036–1045. [DOI] [PubMed] [Google Scholar]
- Grumolato, L. , Liu, G. , Mong, P. , Mudbhary, R. , Biswas, R. , Arroyave, R. , 2010. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 24, 2517–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, T.C. , Sparks, A.B. , Rago, C. , Hermeking, H. , Zawel, L. , da Costa, L.T. , 1998. Identification of c-MYC as a target of the APC pathway. Science. 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Klaus, A. , Birchmeier, W. , 2008. Wnt signalling and its impact on development and cancer. Nature Rev. Cancer. 8, 387–398. [DOI] [PubMed] [Google Scholar]
- Korinek, V. , Barker, N. , Morin, P.J. , van Wichen, D. , de Weger, R. , Kinzler, K.W. , 1997. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- Lee, Y.N. , Gao, Y. , Wang, H.Y. , 2008. Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell Signal.. 20, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levens, D. , 2010. You don't muck with MYC. Genes Cancer. 1, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F.Q. , Mofunanya, A. , Harris, K. , Takemaru, K. , 2008. Chibby cooperates with 14-3-3 to regulate beta-catenin subcellular distribution and signaling activity. J. Cell Biol.. 181, 1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F.Q. , Mofunanya, A. , Fischer, V. , Hall, J. , Takemaru, K. , 2010. Nuclear-cytoplasmic shuttling of Chibby controls beta-catenin signaling. Mol. Biol. Cell. 21, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, V.S. , Ng, S.S. , Boersema, P.J. , Low, T.Y. , Karthaus, W.R. , Gerlach, J.P. , 2012. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 149, 1245–1256. [DOI] [PubMed] [Google Scholar]
- Luga, V. , Zhang, L. , Viloria-Petit, A.M. , Ogunjimi, A.A. , Inanlou, M.R. , Chiu, E. , 2012. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 151, 1542–1556. [DOI] [PubMed] [Google Scholar]
- MacDonald, B.T. , Tamai, K. , He, X. , 2009. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheu, A. , Collado, M. , Wise, C. , Manterola, L. , Cekaite, L. , Tye, A.J. , 2012. Oncogenicity of the developmental transcription factor Sox9. Cancer Res.. 72, 1301–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, T.A. , Johnson, G.V. , 2005. 14-3-3Zeta does not increase GSK3beta-mediated tau phosphorylation in cell culture models. Neurosci. Lett.. 384, 211–216. [DOI] [PubMed] [Google Scholar]
- Menck, K. , Klemm, F. , Gross, J.C. , Pukrop, T. , Wenzel, D. , Binder, C. , 2013. Induction and transport of Wnt 5a during macrophage-induced malignant invasion is mediated by two types of extracellular vesicles. Oncotarget. 4, 2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe, C. , Mendoza-Topaz, C. , Mieszczanek, J. , Bienz, M. , 2010. Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J Cell Sci. 123, 1588–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, N. , Penn, L.Z. , 2008. Reflecting on 25 years with MYC. Nature Rev. Cancer. 8, 976–990. [DOI] [PubMed] [Google Scholar]
- Mhawech, P. , 2005. 14-3-3 proteins–an update. Cell Res.. 15, 228–236. [DOI] [PubMed] [Google Scholar]
- Morelli, A.E. , Larregina, A.T. , Shufesky, W.J. , Sullivan, M.L. , Stolz, D.B. , Papworth, G.D. , 2004. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 104, 3257–3266. [DOI] [PubMed] [Google Scholar]
- Morin, P.J. , Sparks, A.B. , Korinek, V. , Barker, N. , Clevers, H. , Vogelstein, B. , 1997. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- Morrish, F. , Neretti, N. , Sedivy, J.M. , Hockenbery, D.M. , 2008. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 7, 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, D.K. , 2009. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol.. 19, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslin, A.J. , Tanner, J.W. , Allen, P.M. , Shaw, A.S. , 1996. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 84, 889–897. [DOI] [PubMed] [Google Scholar]
- Nelson, W.J. , Nusse, R. , 2004. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 303, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse, R. , Varmus, H. , 2012. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J.. 31, 2670–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil, T. , Obsilova, V. , 2011. Structural basis of 14-3-3 protein functions. Semin. Cell Dev. Biol.. 22, 663–672. [DOI] [PubMed] [Google Scholar]
- Polakis, P. , 2012. Wnt signaling in cancer. Cold Spring Harbor Persp. Biol.. 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponugoti, B. , Xu, F. , Zhang, C. , Tian, C. , Pacios, S. , Graves, D.T. , 2013. FOXO1 promotes wound healing through the up-regulation of TGF-beta1 and prevention of oxidative stress. J. Cell Biol.. 203, 327–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, D.W. , Rane, M.J. , Joughin, B.A. , Kalmukova, R. , Hong, J.H. , Tidor, B. , 2003. Proteomic identification of 14-3-3zeta as a mitogen-activated protein kinase-activated protein kinase 2 substrate: role in dimer formation and ligand binding. Mol. Cell Biol.. 23, 5376–5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo, G. , Nijman, H.W. , Stoorvogel, W. , Liejendekker, R. , Harding, C.V. , Melief, C.J. , 1996. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med.. 183, 1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz-Romond, T. , Metcalfe, C. , Bienz, M. , 2007. Dynamic recruitment of axin by Dishevelled protein assemblies. J. Cell Sci.. 120, 2402–2412. [DOI] [PubMed] [Google Scholar]
- Skalka, N. , Caspi, M. , Caspi, E. , Loh, Y.P. , Rosin-Arbesfeld, R. , 2013. Carboxypeptidase E: a negative regulator of the canonical Wnt signaling pathway. Oncogene. 32, 2836–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman, V.F. , Dobrowolski, R. , Plouhinec, J.L. , Fuentealba, L.C. , Vorwald, P.P. , Gumper, I. , 2010. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 143, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai, K. , Semenov, M. , Kato, Y. , Spokony, R. , Liu, C. , Katsuyama, Y. , 2000. LDL-receptor-related proteins in Wnt signal transduction. Nature. 407, 530–535. [DOI] [PubMed] [Google Scholar]
- Tetsu, O. , McCormick, F. , 1999. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 398, 422–426. [DOI] [PubMed] [Google Scholar]
- Thery, C. , Zitvogel, L. , Amigorena, S. , 2002. Exosomes: composition, biogenesis and function. Nature Rev. Immunol.. 2, 569–579. [DOI] [PubMed] [Google Scholar]
- Tian, Q. , Feetham, M.C. , Tao, W.A. , He, X.C. , Li, L. , Aebersold, R. , 2004. Proteomic analysis identifies that 14-3-3zeta interacts with beta-catenin and facilitates its activation by Akt. Proc. Natl. Acad. Sci. U S A. 101, 15370–15375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta, T. , Hausmann, G. , Basler, K. , 2012. The many faces and functions of beta-catenin. EMBO J.. 31, 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H.Y. , Malbon, C.C. , 2012. Dishevelled C-terminus: prolyl and histidinyl motifs. Acta Physiol (Oxf). 204, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, J.F. , Hopfner, S. , Korn, K. , Farnung, B.O. , Bradshaw, C.R. , Marsico, G. , 2012. Caenorhabditis elegans screen reveals role of PAR-5 in RAB-11-recycling endosome positioning and apicobasal cell polarity. Nat. Cell Biol.. 14, 666–676. [DOI] [PubMed] [Google Scholar]
- Wolfer, A. , Ramaswamy, S. , 2011. MYC and metastasis. Cancer Res.. 71, 2034–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, M.B. , Rittinger, K. , Volinia, S. , Caron, P.R. , Aitken, A. , Leffers, H. , 1997. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 91, 961–971. [DOI] [PubMed] [Google Scholar]
- Yuan, Z. , Agarwal-Mawal, A. , Paudel, H.K. , 2004. 14-3-3 binds to and mediates phosphorylation of microtubule-associated tau protein by Ser9-phosphorylated glycogen synthase kinase 3beta in the brain. J. Biol. Chem.. 279, 26105–26114. [DOI] [PubMed] [Google Scholar]
- Yuyama, K. , Sun, H. , Mitsutake, S. , Igarashi, Y. , 2012. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem.. 287, 10977–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, X. , Huang, H. , Tamai, K. , Zhang, X. , Harada, Y. , Yokota, C. , 2008. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 135, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H.G. , Grizzle, W.E. , 2014. Exosomes: a novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am. J. Pathol.. 184, 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Meyerkord, C.L. , Du, Y. , Khuri, F.R. , Fu, H. , 2011. 14-3-3 proteins as potential therapeutic targets. Semin. Cell Dev. Biol.. 22, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data
Supplementary movie 1
Supplementary movie 2