

Abstract
The tumor suppressor p53 regulates the expression of genes involved in cell cycle progression, senescence and apoptosis. Here, we investigated the effect of single point mutations in the oligomerization domain (OD) on tetramerization, transcription, ubiquitylation and stability of p53. As predicted by docking and molecular dynamics simulations, p53 OD mutants show functional defects on transcription, Mdm2‐dependent ubiquitylation and 26S proteasome‐mediated degradation. However, mutants unable to form tetramers are well degraded by the 20S proteasome. Unexpectedly, despite the lower structural stability compared to WT p53, p53 OD mutants form heterotetramers with WT p53 when expressed transiently or stably in cells wild type or null for p53. In consequence, p53 OD mutants interfere with the capacity of WT p53 tetramers to be properly ubiquitylated and result in changes of p53‐dependent protein expression patterns, including the pro‐apoptotic proteins Bax and PUMA under basal and adriamycin‐induced conditions. Importantly, the patient derived p53 OD mutant L330R (OD1) showed the more severe changes in p53‐dependent gene expression. Thus, in addition to the well‐known effects on p53 stability, ubiquitylation defects promote changes in p53‐dependent gene expression with implications on some of its functions.
Keywords: p53, Ubiquitylation, Oligomerization, Transcription, Proteasome
Highlights
p53 OD mutants show defects on Mdm2‐dependent ubiquitylation and degradation.
Oligomerization efficient p53 molecules are degraded by 26S and 20S proteasomes.
p53 OD mutants unable to form tetramers are mainly degraded by the 20S proteasome.
p53 OD mutants interfere with ubiquitylation and expression of p53‐dependent genes.
p53 OD mutant L330R found in patients showed severe changes gene expression.

Abbreviations
- OD
Oligomerization Domain
- Bax
Bcl2-associated X protein
- Bcl-XL
B-cell lymphoma-extralarge
- PUMA
p5pregulated Modulator of Apoptosis
- Noxa
Noxious stress induced protein
- Bid
BH3 interacting domain death agonist
- FAS/CD95
Fas (TNF receptor superfamily, member 6)
- p21
Chromosome 6p21.2 located Cyclin-dependent kinase inhibitor 1A
- E2F1
E2F transcription factor 1
- Mdm2
Mouse double minute 2 homolog ubiquitin ligase
- PCNA
Proliferating Cell Nuclear Antigen
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- 14.3.3σ
14-3-3 sigma protein eluted in the 14th fraction of bovine brain homogenate and migrating on position 3.3 after electrophoresis
- MD
Molecular dynamics
- FTDOCK
FFT-based docking program
- ADR
Adriamycin
- RMSD
Root mean square deviation
1. Introduction
The tumor suppressor p53 is a major gatekeeper of the genome that tightly controls critical processes in the cell, acting as a central player within a large network of proteins involved in DNA‐repair, apoptosis or cell cycle (Levine, 1997; Vogelstein et al., 2000; Zilfou and Lowe, 2009). P53 is a transcription factor that binds its DNA consensus sequence as a tetramer to activate transcription. The C‐terminus of p53 contains the oligomerization domain (OD) that is required for the formation of the transcriptionally active tetramer. The C‐terminus also contains sub‐cellular localization signals and amino acids targeted by a wide variety of posttranslational modifications, including phosphorylation, acetylation, methylation, SUMOylation (Alarcon‐Vargas and Ronai, 2002; Chuikov et al., 2004; Rodriguez et al., 1999) and ubiquitylation that control its degradation by the 26S proteasome (Maki, 1999; Rodriguez et al., 2000) and its function (Funk et al., 1992; Maki and Howley, 1997; Maltzman and Czyzyk, 1984). Data from in vivo experiments in mouse models have suggested that ubiquitylation of lysines located in C‐terminal may play a role in the regulation of transcriptional activity in a manner of “fine‐tuning” the expression of specific p53‐dependent genes (Krummel et al., 2005) (Feng et al., 2005). Among the most important p53‐dependent genes, the ubiquitin protein‐ligase Mdm2, has been shown to be crucial for regulation of p53 function (Fang et al., 2000) and its proteasomal degradation (Chowdary et al., 1994; Maki, 1999; Maki et al., 1996). Other well‐known p53‐target‐genes are Bax, PUMA, Noxa, Bid, Fas/CD95, which are involved in apoptosis, while p21 and E2F1 control cell cycle arrest and proliferation (Riley et al., 2008).
P53 is found mutated in about half of human cancers (Hollstein et al., 1991; Soussi and Beroud, 2001) with most of the mutations being missense and mainly located in the core DNA‐Binding domain. To date, the database of TP53 maintained at IARC, has registered more than 30,000 mutations in humans (www‐p53.free.fr/www‐p53.iarc.fr) (Olivier et al., 2002). Few mutations have been registered in its OD (Milner and Medcalf, 1991; Sturzbecher et al., 1992). After epidemiologic studies, 17% of germline mutations in patients with Li‐Fraumeni syndrome occurred in the p53 OD (Petitjean et al., 2007). Transcriptionally silent p53 mutants generate transdominant negative effects over a WT allele through mechanisms that are not well understood. The aberrant accumulation of mutant p53 in tumor cells suggests an alteration in its degradation pathway. Intrigued by these aspects, we have introduced single point mutations in the β‐strand of the p53 OD, mimicking the alterations in L330 observed in cancer, which is one of the crucial amino acids for correct tetramerization (Chene and Bechter, 1999). NMR and X‐Ray crystallography analysis revealed that OD (residues 323–356) contains one β‐strand (326–333) and one α‐helix (335–354) with a V‐shaped structure (Chene, 1997; Clore et al., 1995; Jeffrey et al., 1995; Lee et al., 1994; Miller et al., 1996; Mittl et al., 1998). The hydrophobic amino‐acid L330 together with seven other residues (F328, I332, R337, F338, F341, N345 and D352) form a critical hydrophobic pocket, allowing association of two dimers, necessary for the formation of a transcriptionally active tetramer and for preservation of p53 stability (Clore et al., 1995; Friedman et al., 1993; Jeffrey et al., 1995; Kato et al., 2003; Kawaguchi et al., 2005; Mateu and Fersht, 1998).
2. Materials and methods
2.1. Modeling of the p53 dimer structure by docking simulation
We used the monomeric structure of the p53tet PDB 1AIE; 1.5 Å in its active tetrameric form as starting structure for the WT dimer model and as scaffold to build all the structures of the mutants described herein, using SCWRL3.0 program (Canutescu et al., 2003). The rigid‐body docking simulations were performed using the FFT‐based docking program FTDOCK with grid resolution of 0.7 Å and electrostatics (Gabb et al., 1997), which generated a set of 10,000 docking poses that were evaluated by the energy‐based pyDock scoring scheme (Cheng et al., 2007). For each docking run, we selected the lowest‐energy solution. RMSD was calculated for a monomer respect to its equivalent in the p53 dimer structure (as found in the p53 biological assembly) after superposition of the interacting partner monomer.
2.2. Molecular dynamics simulation of WT and mutant p53 dimer structures
The WT p53 dimer structure for the Molecular dynamics (MD) simulation was extracted from PDB code 1AIE. The mutants were built in silico from the WT structure, using the AMBER module LEAP. Each system was then minimized solvated and equilibrated in a five‐step at the same conditions as previously described for the MoDEL database (Meyer et al., 2010). The AMBER parm99SB force field parameters in AMBER 10 package were used (Case et al., 2005). Root mean square deviation (RMSD) and residue–residue minimal interatomic distances were calculated with ICM‐browser program (www.molsoft.com) on a total of 4000 snapshots generated every 100ps along the MD trajectories.
2.3. Pairwise residue–residue decomposition of binding energy
The MMPBSA.py script in AMBER12 (Steinbrecher et al., 2012) was used to estimate the binding free energy of each dimer by calculating the interaction energy and solvation free energy for the complex, receptor and ligand (MM‐GBSA method). All energy components (van der Waals contribution from MM, electrostatic energy as calculated by the MM force field, the electrostatic contribution to the solvation free energy calculated by GB, nonpolar contribution to the solvation free energy calculated by an empirical model) were calculated using 200 snapshots extracted from the last 20ns of each MD trajectory and then averaged. Finally, in order to identify the key L330 contacts responsible for binding, we computed the energetic contribution to the total binding free‐energy of all the contacts between the L330 residue (or equivalent mutated residue) of one monomer and the other residues from the other monomer, using the MM‐GBSA free energy decomposition process in AMBER12 (the average for the two values obtained for the L330 residue in each monomer is given in Table 2, in which we have highlighted those pairwise contacts with more than 1 kcal/mol change in the mutant with respect to WT).
Table 2.
Free energy of dimer residue–residue contacts (Kcal/mol).
| ResB‐ResA | WT | OD1 | OD2 | OD3 | OD4 | OD5 |
|---|---|---|---|---|---|---|
| L330‐T329 | −2.39 | −2.25 | −2.37 | −2.89 | −2.47 | −2.53 |
| L330‐L330 | −4.22 | −3.55 | −3.64 | −3.03 | −4.58 | −0.65 |
| L330‐I332 | −1.16 | −0.77 | −0.86 | −0.94 | −1.22 | −1.39 |
| L330‐F338 | −1.67 | −3.22 | −3.75 | −1.06 | −1.67 | −0.86 |
| L330‐F341 | −1.26 | −3.40 | −0.75 | −1.31 | −1.49 | −2.21 |
| L330‐R342 | −1.43 | −1.56 | −0.89 | −11.52 | −1.12 | −0.08 |
| L330‐N345 | −0.46 | 0.22 | 0.00 | −5.90 | −0.47 | −0.02 |
| L330‐E346 | −0.06 | −4. 04 | −0.26 | 0.29 | −0.05 | −0.02 |
| L330‐E349 | −0.04 | −0.10 | −4.92 | 0.01 | −0.03 | −0.01 |
Pairwise contacts with more than 1 kcal/mol change indicated in bold.
2.4. Reagents, plasmids and DNA manipulations
The following reagents were used at the indicated concentrations: MG132 (Sigma, 5 μM, overnight), Z‐VAD (Calbiochem, 50 μM, 24 h), Chloroquine (Sigma, 200 μM, 24 h), Cycloheximide (Sigma, 50 μM), Adriamycin (ADR) (Sigma, 1 μM). Plasmid encoding Mdm2 has been previously described (Blattner et al., 1994). WT p53 and p53 OD mutant fragments were cloned into the vector pcDNA3‐SV5 using the restriction sites KpnI and EcoRI. For the amplification of p53 molecules, the following sense primer: 5′‐cc ggt acc atg gag gag ccg cag tca g‐3′ and anti sense primer were used: 5′‐cg gaa ttc gtc tga gtc agg ccc ttc tg‐3′.
2.5. Site directed mutagenesis
For mutagenesis, the QuickChange® Site‐Directed Mutagenesis Kit from Stratagene was used with the following sense and anti‐sense primers: OD1 (L330R), sense primer: 5′‐ctg gat gga gaa tat ttc acc CGT cag atc cgt ggg‐3′ and anti sense primer 5′‐ccc acg gat ctg acg ggt gaa ata ttc tcc atc cag‐3′. OD2 (F328V + L330R) sense primer: 5′‐ca ctg gat gga gaa tat GTC acc CGT cag atc cgt ggg cg‐3′. OD3 (L330E) sense primer: 5′‐ctg gat gga gaa tat ttc acc GAA cag atc cgt ggg‐3′ and anti sense primer 5′‐ccc acg gat ctg acg ggt gaa ata ttc tcc atc cag‐3′. OD4 (L330M), sense primer, 5′‐ctg gat gga gaa tat ttc acc ATG cag atc cgt ggg‐3′ and anti sense primer 5′‐ccc acg gat ctg acg ggt gaa ata ttc tcc atc cag‐3′. OD5 (L330P), sense primer 5′‐ctg gat gga gaa tat ttc acc CCT cag atc cgt ggg‐3′ and anti sense primer 5′‐ccc acg gat ctg acg ggt gaa ata ttc tcc atc cag‐3′.
2.6. Cell culture and transfection
P53 null H1299 human lung cancer cells and WT p53 U2OS human osteosarcoma cell line were grown in DMEM with 10% FBS and antibiotics. Population U2OS stable cells were established by transfecting pcDNA3/WT p53 or pcDNA3/OD mutants to obtain equal levels of expression and maintained under genetic selection during all experiments. Cells were transiently transfected with indicated plasmids using lipofectamine (Invitrogen). For most degradation experiments, a plasmid ratio 1:5 p53: Mdm2 was used, with the exception of the Figure 3A where it was used a ratio 1:10. After transfection, cells were grown in six‐well plates for 24 h, and harvested for Western blotting analysis or immunofluorescence. For measurement of transcriptional activity, H1299 cells were transiently co‐transfected with plasmids expressing WT p53 or OD mutants with reporter plasmid luciferase gene under the control of promoter p21 or Bax and control β‐galactosidase. Luciferase and β‐galactosidase activities were measured as previously described (Rodriguez et al., 1996). P53 WT and OD mutant half‐life quantifications in H1299 were performed as described (Blattner et al., 1994).
Figure 3.

Proteolytical pathways driving WT p53 and OD mutants to degradation. A) Role of ubiquitylation on the degradation of OD mutants. Mdm2 was expressed to high levels together with WT p53 or OD mutants in H1299 cells. Mdm2 mutant (C464) was used as indicated. GAPDH was used as charge control. Western blot detection with the indicated antibodies. B) Alternative proteolytical pathways driving WTp53 and OD mutants to degradation. H1299 cells were co‐transfected with plasmids expressing WT p53 or OD mutants together with plasmid expressing Mdm2. The indicated treatments were used during 24 h: Chloroquine (CQ), Z‐VAD (Z) and MG‐132 (MG). GAPDH was used as charge control. Analysis by Western blots with the indicated antibodies.
2.7. Tetramerization assay
In order to analyze the oligomerization status of p53, tetramerization assays using glutaraldehyde cross‐linking were performed as reported (Hjerpe et al., 2010). Assays were performed using cells transiently (H1299) or stably (U2OS) expressing WT p53 and/or OD mutants. For transient experiments constant levels of WTp53‐SV5 (100 ng) and increasing amounts (100 or 250 ng) of untagged OD mutants were used. When required, constant levels (100 ng) of untagged WT p53 and increasing amounts (100 or 250 ng) of SV5 tagged OD mutants were also expressed. Briefly, cells were grown 24 h before being treated or not 1 h with 1 μM ADR (stable cells), and lysed in a buffer containing 100 mM NaCl, 100 mM Tris pH 8, 0.5% NP40 (Igepal). Samples were split in two and treated or not with 0.1% of the crosslinking agent glutaraldehyde. The reaction was quenched after 10 min with 100 mM PBS‐Glycine. Laemmli sample buffer was added to each sample and boiled for 5 min before SDS‐PAGE. Western‐blot analysis with the indicated antibodies is shown.
2.8. Chemotherapy simulation assay
This assay was adapted from a previous procedure aiming to mimic a sequential treatment of chemotherapy using ADR (Kopp et al., 2012). MCF‐7 cells were plated and exposed or not (CTR) to 1 μM of ADR during 1 h and maintained in fresh DMEM medium for 24 h. Cells were collected at day 2 or maintained in culture. When indicated, cells were treated or not at day 6 and collected at day 7. Finally cells were also treated or not at day 10 and collected at day 11. Thus D2 was exposed or not to a single ADR treatment, whereas D7 and D11 cells were treated or not for two or three times with ADR, respectively (Figure 7A). Duplicate samples were prepared for Western‐blot analysis and tetramerization assays.
Figure 7.

OD defective mutants modify the expression of p53‐dependent genes. U2OS stably expressing the different p53 mutants were stimulated with ADR during 1 h and kept in culture for the indicated periods of time (day 2, day 7 and day 11). A) Schema of the chemotherapy‐like treatment used in this experiment. Monitoring of protein levels of both endogenous and exogenous p53 molecules was performed by Western blot. B) Formation of tetramers in stable cell lines expressing OD mutants. Basal and ADR induced formation of p53 oligomers at days 2, 7 and 11. C) The expression of p53‐dependent genes involved in apoptosis and cell cycle regulation. Western blot analysis using the indicated antibodies.
2.9. RNA extraction and real time quantitative PCR
RNA was isolated by using NucleoSpin RNA kit (Macherey–Nagel, following manufacturer's instructions). cDNA was subsequently obtained using the qScript cDNA Synthesis Kit (Quanta) with random hexanucleotides. Real time quantitative PCR was performed in a VIIA7 (life technologies) using the 384 standard PCR program, and data was normalized by the housekeeping gene and relativized to untreated WT p53 expressing cells. VIC fluorescent PCR probes for the housekeeping gene (GAPDH) were purchased from Life Technologies (Hs02758991_g1) and PCR probes for the target genes were generated using the Universal Probe Library (Roche) of forward and reverse primers as follows: 14‐3‐3σ gacacagagtccggcattg & atggctctggggacacac (probe 27); p21 tcactgtcttgtacccttgtgc & ggcgtttggagtggtagaaa (probe 32); MDM2 gactccaagcgcgaaaac & cagacatgttggtattgcacatt (probe 68); E2F1 tccaagaaccacatccagtg & ctgggtcaacccctcaag (probe 5); BAX agcaaactggtgctcaagg & tcttggatccagcccaac (probe 69).
2.10. Preparation of cell extracts and western blot analysis
Harvested cells were lysed in Laemmli buffer and proteins were then analyzed by Western blot. Immunodetection was performed using the following primary antibodies: anti‐p53 (clone DO1, kindly given by RTH); anti‐SV5 (MCA1360, Serotec, Germany); anti‐Mdm2 (Calbiochem, EMD Chemicals, USA). PCNA and E2F‐1 antibodies (Santa Cruz Technology, USA). P21, PUMA and Bax antibodies from Cell Signalling Technology (MA, USA), GAPDH antibody used as a control of charge from Sigma, 14.3.3σ (Abcam, UK), Bax and Bcl‐XL from Life Technologies (USA), anti‐ubiquitin lys‐K48 specific from Millipore, (MA, USA) and anti‐ubiquitin lys‐K63 specific from Enzo Life Sciences (NY, USA).
2.11. Pull‐downs of polyubiquitylated proteins using TUBES followed by immunoprecipitation
Cells were plated the day before and stimulated according to the experiment. Then cells were lysed using TUBEs lysis Buffer as previously described (Hjerpe et al., 2009; Aillet et al., 2012). Captured polyubiquitylated proteins were submitted to an immunoprecipitation assay using Protein A dynabeads cross‐linked with 4 μg specific antibody. After 2 h of binding at 4 °C, beads were washed three times with lysis buffer and resuspended in Laemmli buffer X3. Proteins were then submitted to Western blot analysis.
2.12. In vitro degradation of OD mutants by 20S and 26S proteasomes
In vitro transcribed/translated p53 WT and OD were incubated in a degradation mixture containing ATP regenerating system [25 mM Tris pH 7.6, 5 mM MgCl2, 2 mM ATP, 10 mM creatine phosphate (Sigma), 5 mM NaCl2, 3.5 U/ml of creatine kinase (Sigma) and 0.6 U/ml of inorganic pyrophosphatase (Sigma)], 10 μg Ubiquitin (Sigma), 10 ng human E1 Ubiquitin activating enzyme (Biomol), 500 ng E2 Ubiquitin conjugating enzyme UbcH5b (Biomol), as previously described (Hjerpe et al., 2010) with 1 μg of purified 26S or 20S proteasome (Boston Biochem). For 26S proteasome reactions, in vitro transcribed/translated MDM2 was supplemented in the degradation mixture. The degradation mixture was incubated at 30 °C for 2 h and the reaction was stopped by addition of SDS sample buffer containing β−mercaptoethanol. Reaction products were fractionated by SDS‐PAGE (10%) and analyzed by Western‐blotting.
3. Results
3.1. Effect of point mutations in the dimerization‐folding nucleus
To better understand the structural determinants of how mutations in the p53 OD affect tetramerization, and consequently transcriptional activity, ubiquitylation and stability, we performed docking and molecular dynamics (MD) studies. The following mutants were analyzed: L330R (OD1), L330R + F328V (OD2), L330E (OD3), L330M (OD4) and L330P (OD5) (Figure 1A). We performed one docking simulation between two WT p53 monomers (PDB 1AIE; 1.5 Å), and five other simulations between mutant monomers (in which the same mutations were introduced in both monomers). The docking simulation well predicted the WT p53 dimer orientation (Table 1, Figure 1B). The mutants OD1 and OD2 showed significantly worse binding energies (Table 1) and structurally worse predictions (Figure 1B), while the OD3, OD4 and OD5 mutants were predicted to be structurally correct, with only slightly worse binding energies. In order to evaluate the effect of the mutations on the conformational stability of the p53 dimeric structure, we performed MD on the WT and mutant variants. The mutants OD1 and OD5 showed the highest general deviation from initial structure along the MD (Figure 1C, Table 1). Overall, MD shows low stability in OD1 and OD5 mutants, while docking shows poor binding energy in OD1, and OD2 mutants. Only OD3 and especially OD4 show binding energy and stability values similar to WT.
Figure 1.

Effect of oligomerization domain point mutations on the stability of p53 dimers. A) Single point mutations were introduced in the OD of p53 as indicated. B) Lowest‐energy docking solutions obtained for WT (dark gray) and the different mutants (OD1 in blue; OD2 in red; OD3 in magenta; OD4 in yellow; OD5 in green), as compared to the X‐ray structure of the p53 dimer (in grey). C) RMSD for Cα atoms of WT and different mutants (same color code as above). D) Minimal pairwise residue distance between L330 (or equivalent residue if mutated) and E346 from different monomers in WT and mutants OD1 (L330R), and OD2 (L330R + F328V) (same color code as above). E) Minimal pairwise residue distance between L330 (or equivalent residue if mutated) and E349 from different monomers in WT and mutants L330R, and L330R + F328V (same color code as above).
Table 1.
Docking simulation of homodimers and heterodimers of p53 OD mutants and WT p53. Predicted dimer binding energy and ligand RMSD values of the lowest‐energy solution from the p53 dimer complex structure.
| p53 OD form | Lowest‐energy docking solution | MD Avg Tot‐RMSD | |
|---|---|---|---|
| Energy | RMSD (Å) | (10–40 ns) | |
| WT | −61.2 | 3.5 | 2.0 |
| OD1 | −38.4 | 6.1 | 4.9 |
| OD2 | −26.4 | 14.0 | 3.5 |
| OD3 | −50.5 | 2.8 | 2.6 |
| OD4 | −53.2 | 3.1 | 2.8 |
| OD5 | −55.7 | 1.1 | 5.1 |
To understand the structural rearrangements that are responsible for the loss of stability or changes in the relative dimer orientation in the mutants, we used the MD data to compute the free energy of all possible contacts between each 330 residue and the other monomer (Table 2). Interestingly, in L330M mutant, all the studied contacts are similar to WT, which indicates that no major energetic effect is seen along the dynamics. In the other mutants, there are always contacts whose energy changes in the mutant. In the L330P mutant, there is a loss of binding energy in several of the contacts. In the case of L330E mutant, in spite of not showing major differences in docking energy or dynamics stability, it is clear that the mutation introduces new highly favorable interactions like E330‐R342 or E330‐N345, which can affect dimerization. In mutants L330R and L330R + F328V there are also several new favorable interactions as well. Interestingly, the new residue R330 forms a favorable interaction with E346 in L330R mutant, but not in L330R + F328V and, on the contrary, the same new residue R330 forms a favorable interaction with E349 in L330R + F328V but not in the single L330R mutant (Table 2; Figure 1D, E). All mutants except L330M significantly affect the network of contacts formed by the core residues involved in dimerization.
3.2. The hydrophobicity of the OD conditions p53 tetramerization and transcription
To evaluate the characteristics of the L330 mutants, transient transfections of p53 null H1299 cells were performed. Tetramer formation was analyzed using glutaraldehyde (GA) crosslinking (Hjerpe et al., 2010) and assessed by two criteria, detection of high molecular weight forms of around 175 kDa and disappearance of p53 monomeric forms. The patient derived mutation L330R (OD1) exhibited severe defects in tetramer formation (Figure 2A and Duddy et al., 2000; Hjerpe et al., 2010; Kawaguchi et al., 2005) and transcriptional activity (Figure 2B and Hjerpe et al., 2010; Kato et al., 2003; Kawaguchi et al., 2005). Introduction of a second patient‐derived mutation, F328V, did not have any additional effect on tetramerization of the resulting L330R/F328V double mutant (Kato et al., 2003). Changing L330 to the negatively charged amino acid E (OD3) or to the less hydrophobic amino acid P (OD5) equally impaired the formation of tetramers and transcription of p21 and Bax genes measured using luciferase reporter assays (Figure 2A and B). However, mutation of L330 to the bulky hydrophobic amino acid M (OD4) allows tetramer formation and transcription of p53‐dependent genes (Figure 2A and B). The observed defects were not due to a mislocalization of the p53 mutants compared to WT p53 (Figure 2C). Thus, the hydrophobic nature of the p53 oligomerization domain conditions the formation of transcriptionally active p53 homotetramers without affecting the nuclear localization of p53 monomers (OD1, OD2, OD3 and OD5 mutants).
Figure 2.

Oligomerization domain controls p53 tetramerization and transcription. A) P53 OD mutants present severe tetramerization defects after glutaraldehyde (GA) crosslinking. H1299 cells were transfected with WT p53 or OD mutants. B) Tetramerization defective p53 OD mutants do not activate transcription of target genes. H1299 cells were co‐transfected with the indicated luciferase reporters and WTp53 or OD mutants. C) Localization of WT p53 and OD mutants by immunofluorescence in H1299 cells. Anti‐p53 (clone DO1) antibody was used for detection.
3.3. Alternative proteolytical pathways contribute to degrade WT p53 and OD mutants
As ubiquitylation is known to play an important role in regulating the stability of WT p53, we investigated if the degradation of the OD mutants was affected. In order to evaluate potential differences in degradability, low levels of Mdm2 expression were used in p53 null H1299 cells. Tetramerizable p53 molecules (WT and mutant OD4) were more efficiently degraded than monomeric mutants OD1, OD2, OD3 and OD5, showing various levels of degradation (Figure 3A). The observed degradation was ubiquitin‐proteasome‐dependent and could be fully or partially inhibited when using a catalytically inactive Mdm2 mutant (C464) or the proteasome inhibitor MG132 (Figure 3A).
The possible contribution of alternative proteolytical pathways in the degradation of non‐tetramerizable mutants was explored in H1299 cells (Figure 3B) and U2OS (Supplementary Figure 1). To better evaluate the effects of distinct protease inhibitors, degradation driven by Mdm2 was improved by increasing its expression in cells co‐transfected with WT p53 or OD mutants. After verification that Mdm2 was equally expressed (data not shown), cells were treated during 24 h with different combinations of protease inhibitors: 1) chloroquine (CQ) that inhibits autophagy and lysosomal protein degradation (Mehrpour et al., 2010) 2) pan‐caspase Z‐VAD‐fmk (Z) an inhibitor that irreversibly binds to the catalytic site of caspase proteases (Slee et al., 1996) and 3) MG132 (MG). MG efficiently protects WTp53 and p53 OD mutants from proteasomal degradation without reaching 100% inhibition (Figure 3B). In contrast Z modestly (but consistently) protected WTp53 and p53 OD mutants. However, the combination of Z with MG increased its protective effect on all analyzed p53 molecules. Chloroquine showed no protective effect when used alone but enhanced the protective effect of MG/Z, allowing the recovery of almost 100% of all p53 molecules. With small variations, similar results were obtained with U2OS cells transiently transfected with different mutants or WT p53 (Supplementary Figure 1). From these experiments we conclude that proteasomal degradation is a major pathway for the Mdm2‐mediated degradation of WTp53 and p53 OD mutants. Additionally, caspases and lysosomal pathways might also be implicated in the Mdm2‐mediated turnover of these p53 molecules.
3.4. Oligomerization determines ubiquitin‐dependent or ubiquitin‐independent proteasomal degradation of p53
Given that p53 OD mutants are degraded through the proteasome, we further investigated their turnover by the ubiquitin‐proteasome system (UPS). First we analyzed the ubiquitylation pattern of WT p53 or p53 OD mutants using ubiquitin traps known as TUBEs (Hjerpe et al., 2009) in H1299 cells (Figure 4A). We found that p53 molecules that cannot tetramerize (OD1, OD2, OD3 and OD5) have defects in ubiquitylation compared to wild type p53 and OD4 mutant (2, 4A). Using in vitro degradation assays, we confirmed that tetramerizable p53 molecules were more efficiently degraded by the 26S proteasome in a reaction containing ubiquitin and Mdm2 while non‐tetramerizable p53 OD mutants were preferentially degraded by the 20S proteasome in the absence of Mdm2 (Figure 4B) (Hjerpe et al., 2010). To investigate if the defects in ubiquitylation and the degradation by 26S and 20S proteasome had any impact on the half‐life of p53 OD mutants, we performed cycloheximide experiments in H1299 cells (Figure 4C). We observed that OD mutants were more unstable than WT p53 at early times after cycloheximide treatment. OD1, OD2, OD3, and OD5 mutants show a similar degradation profile in the in vitro assay executed in presence of the 20S proteasome (Figure 4B). Unexpectedly, OD4 mutant was more unstable than WT p53 despite the fact that those molecules share most biochemical properties. Differences in stability between WT p53 and OD mutants remain at late stages of cycloheximide treatment even if these were significantly reduced.
Figure 4.

Oligomerization determines ubiquitin‐dependent or ‐independent proteasomal degradation of p53. A) Ubiquitylation pattern of WT p53 and OD mutants. H1299 cells were transfected with the indicated p53 molecules and Mdm2. TUBEs‐captured material was analyzed by Western blot with the indicated antibodies. B) In vitro degradation assays of p53 OD mutants using either 26S or 20S proteasomes in the presence or absence of Mdm2. Western blot detections with the indicated antibodies. Quantifications from 4 different experiments were performed and plotted to the right. Standard deviations are indicated. C) Half‐life of p53 OD mutants in H1299 cells. Cells were treated with cycloheximide during the indicated times to analyze OD mutants half‐life. Western blot analysis with anti‐p53 DO1. GAPDH was used as charge control. Quantification were plotted in the graph (right panel).
3.5. Oligomerization defective p53 mutants form heterotetramers with WT p53
Next, we wanted to investigate if p53 OD mutants could exert an effect on the wild type molecule, to analyze what could occur in a model of autosomal dominant inheritance (Petitjean et al., 2007). For this purpose, we first evaluated the capacity of the untagged OD mutants to form tetramers in H1299 cells co‐expressing constant low level of SV5 tagged WT p53 (Figure 5A). Using GA crosslinking, we found that OD1, OD2, OD3 and OD5 mutants promote a dose dependent formation of WT p53 tetramers suggesting that these mutants were integrated into heterotetramers. To confirm our interpretation a similar experiment was performed using SV5 tagged OD mutants and constant levels of untagged WT p53 (Figure 5B). We observed that all mutants were integrated but in different proportions following the order OD4 > WT > OD5 > OD1 > OD2 > OD3. Difficulties to integrate heterotetramers of OD1, OD2, OD3, and OD5 mutants were also observed after ADR stimulation of U2OS cells (Figure 7B and Supplementary Figure 2). Altogether our tetramerization assays indicate that all OD mutants integrate heterotetramers in different proportions and have an impact on the stability of the WT p53 tetramer. To further support these observations, p53‐dependent reporter assays were performed by co‐expressing similar levels of WT p53 together with p53 OD mutants (Figure 5B). Interestingly, WT p53 and p53 OD4 mutant activate transcription of Bax‐Luc or p21‐Luc reporters. In contrast, p53 OD1, OD2, OD3 and OD5 heterotetramers repress the gene expression driven by WT p53, thus acting as dominant negative mutants (Figure 5B).
Figure 5.

Oligomerization defective p53 mutants out compete WT p53 transcription. A) Capacity of p53 OD mutants to integrate tetramers with WT p53. H1299 cells were transiently transfected with constant WT p53‐SV5 together with increasing concentrations of untagged OD mutants. Tetramerization assays were performed and detection was done by Western blot with anti‐SV5 antibodies. B) Untagged WT p53 was co‐transfected with increasing concentrations of OD‐SV5 mutants into H1299 cells. Tetramerization assays were performed and detection was done by Western blot with anti‐SV5 antibodies. C) Effect of p53 OD mutants on transcription mediated by WTp53. Luciferase reporter assays were performed in H1299 cells using the Bax and p21 promoters. n = 4.
3.6. WT p53 and OD mutants are modified by distinct polyubiquitin chains
The effect of p53 OD mutants on the ubiquitylation pattern of endogenous WT p53 was investigated using U2OS cells. Cells were transiently transfected with plasmids expressing SV5 tagged versions of WT p53 or OD mutants in the presence or absence of Mdm2 (Figure 6A). In the absence of Mdm2, few polyubiquitin chains were observed in the TUBEs‐captured fraction with WT p53 and OD4 mutant. In presence of Mdm2, an accumulation of ubiquitylated forms of WT p53 and OD4 mutant were captured while a mild accumulation was observed for OD1, OD2, OD3 and OD5 (Figure 6A). When WT p53 or OD4 mutant were transfected, endogenous WT p53 was efficiently captured by TUBEs suggesting that these molecules can be well integrated into the ubiquitylated p53 complexes. Furthermore, TUBEs can also capture endogenous p53 into complexes with OD1, OD2, OD3 and OD5 mutants but to a lesser extend (Figure 6A lower panels), thus supporting our previous conclusion. To characterize these chains, a similar experiment was done in presence of Mdm2, now with the aim to perform immunoprecipitations from TUBE‐captured material using p53, ubiquitin K48 or ubiquitin K63 specific antibodies (Figure 6B). In the presence of WT p53 and OD4, we can observe an accumulation of K48 polyubiquitin chains. The presence of OD2 and OD3 mutants show an intermediate level of K48 ubiquitin chains compared to the low level found with OD1 and OD5 mutants. Surprisingly, only WTp53 allows an accumulation of the K63 polyubiquitin chains. The amount of K63 ubiquitin chains is severely affected in all mutants including OD4 that shared most molecular characteristics with WT p53 (Figure 6B). Thus, distinct patterns of ubiquitin chains can be formed on p53 in the presence of OD mutants, and only WT p53 shows efficient K48 and K63 polyubiquitylation.
Figure 6.

Ubiquitylation pattern of p53 OD mutants in presence of WT p53. A) U2OS cells were transfected with WT p53 or OD mutants in the presence or absence of Mdm2. Twenty‐four hours after transfection, ubiquitylated forms were captured using TUBEs and revealed by Western blot using anti‐p53 antibody. B) Ubiquitin chain types formed in the presence of p53 OD mutants. TUBEs capture was followed by an immunoprecipitation using an anti‐p53, anti‐K48 or anti‐K63 antibodies revealed by Western blot using anti‐p53 antibody DO1.
3.7. OD defective mutants differentially affect expression of p53‐dependent genes
To better evaluate the repressive effects of OD mutants on endogenous WT p53, the response to ADR was analyzed using U2OS cells stably expressing the different p53 mutants. ADR stimulations during short periods of time and up to 24 h did not provide important differences in the expression of p53‐dependent genes (Supplementary Figure 3). For this reason, we have implemented a protocol to mimic chemotherapy treatments used for patients were established by adapting a previously reported procedure (Kopp et al., 2012). Cells were exposed for 1 h of ADR (1 μM) then incubated for 24 h and harvested at three different times: day 2, day 7 and day 11 (see schematic of the procedure in Figure 7A). Cell number counting, tetramerization assays and extracts for Western blot analysis were prepared for each time point. The level of expression of both endogenous and exogenous p53 molecules was monitored by Western blot (Figure 7A) as well as their capacity to form heterodimers and heterotetramers (Figure 7B). The expression of several p53‐dependent genes including those involved in apoptosis and regulation of cell cycle was also analyzed using the same samples (Figure 7C). Under comparable GAPDH loading conditions, the expression of p21, 14.3.3σ, Bax, PUMA and Bcl‐XL was increased after each pulse of ADR until day 11, with only minor differences in some mutants (Figure 7C). While the expression of p21 was similar for most p53 OD mutants compared to WT p53, the expression of Bax was affected and showed different levels of expression. Interestingly, OD3 did not favor the expression of Bax on the third pulse of ADR (D11) and OD5 delayed the expression of Bax until D11. It is important to underline that U2OS cells do not express the anti‐apoptotic Bcl‐2 protein that usually heterodimerizes with Bax during apoptosis (data not shown). While the expression of 14.3.3σ was severely reduced at D11 in the presence of OD1, OD2 and OD3 mutants (Figure 7C), Bcl‐XL showed low expression levels at D2 in presence of OD2, OD3, OD4 and OD5. PUMA showed high basal levels of expression with OD1 and OD2 mutants while PCNA and Mdm2 proteins levels showed minor changes in presence of all mutants. E2F was expressed to basal levels in WT and most OD mutants but at D7 and D11, after ADR stimulation, expression was decreased except for OD5. To confirm that these effects on transcription are also reflected at mRNA expression level, we performed qPCR analysis using the same U2OS cells stably expressing OD mutants or WT p53 (Supplementary Figure 4). We could observe that although the pattern of mRNA expression of p21, MDM2, E2F1, BAX and 14.3.3σ is not exactly the same than the pattern of protein expression before and after stimulation with ADR at days 2 and 7 according to our protocol (Figure 7C), all OD mutants promote a different pattern of gene expression compared to WT p53. This was especially evident for E2F1 and 14.3.3σ. Altogether, our results indicate that p53 OD mutants alter WT p53‐dependent gene expression.
Thus, the negative effects on basal transcription of all OD mutants were different from their capacity to inhibit the expression of p53‐dependent genes after genotoxic insult with ADR. Unlike the overexpression, the equivalent level of WT p53 and OD mutants does not exert a dominant negative effect but rather affects the pattern of expression of p53‐dependent genes that appears to be unique for each OD mutant. Remarkably, the p53 OD1 mutant, found in patients appears to be the one with the more severe changes in gene expression. Altogether these results indicate that mutations in the OD domain of p53 affect the capacity to oligomerize, altering ubiquitylation, stability and transcription mediated by this tumor suppressor (Figure 8).
Figure 8.
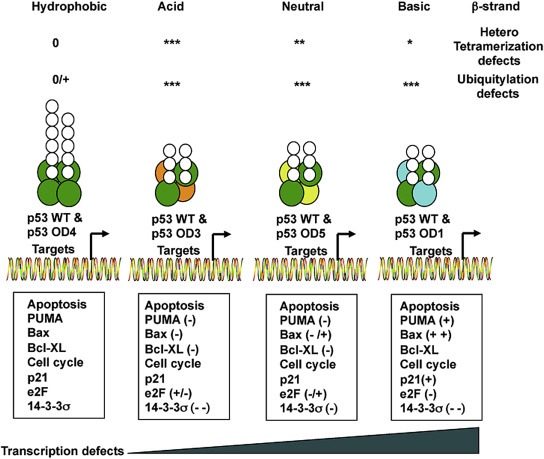
Integrated view of the effect of p53 OD mutants on tetramerization, ubiquitylation and p53 mediated transcription. According to the nature of the amino acid change, mutants can be grouped in hydrophobic, acid, neutral or basic and resulting in a different expression of p53 dependent genes. Due to the double mutation of OD2, the pattern of expression of this mutant shows a mixed phenotype OD1/OD3/OD4. Defects of ubiquitylation and tetramerization are indicated as follows: 0, no defect; + poor defects; ++ medium defects; +++ severe defects. Changes in genes expression are indicated as follows: (−) reduction; (−−) significant reduction; −/+ alterations; increase (+); significant increase (++).
4. Discussion
Early studies underlined the importance of the oligomerization domain of p53 in the regulation of transcription and tumor suppressor potential of this crucial cellular factor (Chene and Bechter, 1999; Chene et al., 1997). The connection between ubiquitylation and several p53 properties, including tetramerization and susceptibility to the 26S proteasome degradation has been studied by several groups (Hjerpe et al., 2010; Maki, 1999; Maki and Howley, 1997). More recently, ubiquitylation on the C‐terminal region of p53 has been proposed to regulate gene expression rather than stability (Feng et al., 2005). To shed some light on different conclusions, we focused our efforts on studying p53 mutants that affect the dimerization‐“folding nucleus” (Figure 1). Our first approach used docking and molecular dynamic simulations to predict binding energy and stability of these mutants. We found that only OD3 (L330E) and especially OD4 (L330M) show binding energy and stability values similar to WT. We also observed poor binding energy for L330R (OD1), and L330R + F328V (OD2) mutants and low stability for OD1 and OD5 (L330P) mutants. The loss of stability or changes in the relative dimer orientation studies revealed that all mutants except L330M introduced significant energy changes in the network of contacts formed by the core residues that can affect dimerization. Our results show that the good integration of OD1 and OD2 into heterotetramers (Figure 5A) could be related to their capacity to form homodimers when expressed alone (Figure 2A). On the other hand, OD3 and OD5 are not in favor of forming either homodimers or homotetramers (Figure 2A). Therefore, one can speculate that there is a predisposition to form heterotetramers containing only one molecule of OD3 or OD5, while two mutant molecules might exist within the heterotetramer formed by OD1 or OD2 (2, 5A). Coherently with our interpretation, we found that, with the exception of the hydrophobic OD4 mutant, the inability for p53 OD mutants to form homotetramers was dramatically affecting their ubiquitylation and transcriptional activity capacity (2, 4). Also, OD1, OD2, OD3 and OD5 p53 mutants were mainly degraded by the ubiquitin‐independent degradation mediated by the 20S proteasome (Figure 4C) while WT and OD4 mutant were good substrates for the 26S proteasome. These results suggest the intervention of the so‐called “default pathway” proposed by Shaul and collaborators where the 20S proteasome contribute to the degradation of unstructured monomers (Asher et al., 2006). Indeed, the lack of integration of p53 OD mutants into a more stable tetrameric structure could facilitate the action of the 20S proteasome in vitro and bypass the requirement of ubiquitylation (Figure 4C, D). Furthermore, we found that other proteolytical pathways including caspases or the lysosome might contribute to p53 degradation when Mdm2 is overexpressed (Figure 3B and Supplementary Figure 1).
Interestingly, we observed that p53 OD mutants unable to form homotetramers could form heterotetramers with WT p53 thus affecting p53‐dependent transcription in overexpression conditions (Figure 5). Those defects were also reflected in the ubiquitylation pattern of endogenous p53 detected in U2OS cells (Figure 6). While we cannot exclude that some of the efficient ubiquitylation observed might come from exogenous WT p53 or OD4, one can clearly observe that endogenous WT p53 was efficiently captured by TUBEs (Figure 6A low panels). In the same experiment we confirmed that other OD mutants can also integrate endogenous p53 but to a lesser extend. These results indicate that endogenous WT p53 molecules can be well integrated into the ubiquitylated exogenous p53 complexes. Furthermore the pattern of ubiquitin chains is not the same when immunoprecipitated with antibodies against K48 or K63 chains, arguing in favor of an OD mutant‐dependent formation of heterologous ubiquitin chains.
Despite the similar pattern of K48 ubiquitin chains found when overexpressing WT p53 or OD4 mutant, the pattern of K63 ubiquitin chains was significantly reduced for OD4 mutant. This absence of ubiquitin K63 chains, here appears to change the expression of some apoptotic and cell cycle arrest genes induced by WT p53 at least the ones that we analyzed. More dramatically, we found that defects in the formation of K48 or K63 ubiquitin chains in all other OD mutants affect gene expression in different proportions. All together, these results indicate that changing a single amino acid in OD can affect the pattern of ubiquitylation and gene expression.
Most features and properties of WT p53 and OD4 mutant are shared due to the hydrophobic nature of both forms including its capacity to form homotetramers and activate transcription of luciferase reporters. However some differences between WT p53 and OD4 mutant can be observed at the level of protein stability (Figure 4C) and ubiquitin chain pattern (4, 6). The preference of OD4 to form K48 chains in the presence of WT p53 is compatible with the fact that OD4 is more instable than WT p53 (4, 5A). Interestingly, in the presence of OD4, the protein levels of E2F1, PUMA and 14.3.3σ were often higher that those found when WT p53 was expressed. These observations support the possibility that K48 chains on OD4 mutant are implicated in the formation of transcriptionally active complexes coherent with the transcriptional activation by destruction mechanism of regulation recently discussed by McShane and Selbach (2014) and deeply studied by Caltic and collaborators (Catic et al., 2013). Such mechanism might be masking a more important role for this post‐translational modification in the formation of active p53 transcriptional complexes. In that sense, our evidence are not the first ones that associate K48 chain formation to activation of transcription (Le Cam et al., 2006).
We have shown that overexpression of p53 OD mutants exert a dominant negative effect on WT counterpart. This is coherent with the notion that in cancer cells where p53 is mutated, this form is overexpressed as a consequence of the elimination of the wt allele. Therefore overexpression conditions most likely reflect what happens in those tumoral cells. However, to better evaluate the impact of OD mutants on heterotetramerization and transcriptional activity, we generate U2OS cells stably expressing equivalent or lower levels of OD mutants than endogenous WT p53. We found that formation of heterotetramers interferes with the capacity of endogenous WT p53 to activate transcription of p53‐dependent genes after ADR stimulation. While the induction of p21 is comparable in all cases, Bax and PUMA expression drops in some cases, with the patient‐derived OD1 (L330R) mutant having the most severe reduction of protein levels (Figure 8). In apparent contradiction with observations obtained using luciferase reporters and overexpression systems (Figure 5B), the stable expression of OD mutants at levels similar to endogenous WT p53 still allows the expression of p53‐dependent genes (Figure 7C). As it occurs in many cancer types, the overexpression of p53 mutants show more drastic repressive effects on p53‐dependent genes due to several molecular events including saturation of signaling pathways, promoter occupancy or basal transcription factors quenching. We have also observed important differences on the formation of heterotetramers between transient transfection experiments and U2OS cells stable expressing OD mutants. Under overexpression conditions OD mutants might favor the formation of complexes up to saturating levels and therefore no differences could be appreciated. The establishment of a system where OD mutants are expressed to fairly equal levels than endogenous WT p53 was required to assess differences between mutants. However, the molecular characterization of tightly regulated systems such as p53 is impossible with high protein concentrations. Thus, our work shows that the inhibitory potential of p53 OD mutants could be better assessed using a stable expression system and differences could be better evaluated in response to a genotoxic insult such as ADR.
Figure 7.

(continued)
Thus, collected data allow us to correlate the effects of oligomerization deficient OD mutants on p53‐mediated transcription with their ubiquitylation pattern. One can speculate that ubiquitin proteases or ubiquitin‐ligases such as USP7/HAUSP or E4F1 might contribute to modulate the ubiquitylation status of p53 controlling its functions (Le Cam et al., 2006). Further exploration of the ubiquitin‐mediated transcription processes will be crucial to understand how transcriptionally silent p53 OD mutants end up suppressing WT p53 functions and result in tumor development. This could be particularly important in pathologies such as the Li‐Fraumeni syndrome also known as the Sarcoma, breast, leukemia and adrenal gland (SBLA) syndrome, where there is a predisposition to develop tumors due to the presence of a mutated p53 allele. Understanding the role of ubiquitylation in the control of p53‐mediated transcription and tumor development process will open new possibilities for treatment development.
5. Conclusion
In this work, we found that p53 OD mutants unable to form tetramers show ubiquitylation defects and are not transcriptionally active. However, when co‐transfected with WT p53, p53 OD mutants are able to integrate into non‐functional heterotetramers, acting as dominant‐negative proteins over WT p53. Further exploration of the mechanism of OD mutants trans‐dominance allowed us to establish a link between ubiquitylation defects and changes in the pattern of p53‐dependent gene expression. The study presented herein is of physiological relevance and might help to explain in part, the role of cancer‐related p53 missense mutations within the oligomerization domain (OD) that occur in patients with Li‐Fraumeni syndrome.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
This work was funded by the MINECO‐Spain grants BFU2008‐01108/BMC and BFU2011‐28536 (MSR), and BIO2010‐22324, and the Department of Industry of the Government of the Basque Country, Etortek Research Programmes 2011/2012. Our group at Inbiomed is supported by the Obra Social KUTXA and the Diputación Foral de Gipuzkoa. MT‐R was supported with a fellowship from the Autonomous Community of the Basque Country. A.C. is supported by the Ramón y Cajal award Spanish Ministry of Education, the Basque Department of Industry, Tourism and Trade (Etortek), Marie Curie Reintegration grant (277043), Movember Global Action Plan, ISCIII (PI13/00031) and the Basque Government of Health (2012111086) and Basque Government of Education (PI2012‐03) and the European Research Council (336343).
We sincerely thank Adriana Rojas for her advice on structural biology aspects.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.04.002.
Lang Valérie, Pallara Chiara, Zabala Amaia, Lobato-Gil Sofia, Lopitz-Otsoa Fernando, Farrás Rosa, Hjerpe Roland, Torres-Ramos Monica, Zabaleta Lorea, Blattner Christine, Hay Ronald T., Barrio Rosa, Carracedo Arkaitz, Fernandez-Recio Juan, Rodríguez Manuel S. and Aillet Fabienne, (2014), Tetramerization‐defects of p53 result in aberrant ubiquitylation and transcriptional activity, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.04.002.
Contributor Information
Valérie Lang, Email: vlang@inbiomed.org.
Chiara Pallara, Email: chiara.pallara@bsc.es.
Amaia Zabala, Email: azabala@cicbiogune.es.
Sofia Lobato-Gil, Email: slobato@inbiomed.org.
Fernando Lopitz-Otsoa, Email: flopitz@cicbiogune.es.
Rosa Farrás, Email: rfarras@cipf.es.
Roland Hjerpe, Email: n.r.e.hjerpe@dundee.ac.uk.
Monica Torres-Ramos, Email: monica.atorres@gmail.com.
Lorea Zabaleta, Email: lzabaleta@inbiomed.org.
Christine Blattner, Email: christine.blattner@kit.edu.
Ronald T. Hay, Email: r.t.hay@dundee.ac.uk
Rosa Barrio, Email: rbarrio@cicbiogune.es.
Arkaitz Carracedo, Email: acarracedo@cicbiogune.es.
Juan Fernandez-Recio, Email: juanf@bsc.es.
Manuel S. Rodríguez, Email: msrodriguez@inbiomed.org
Fabienne Aillet, Email: faillet@inbiomed.org.
References
- Aillet, F. , Lopitz-Otsoa, F. , Hjerpe, R. , Torres-Ramos, M. , Lang, V. , Rodriguez, M.S. , 2012. Isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. Methods Mol Biol. 832, 173–183. [DOI] [PubMed] [Google Scholar]
- Alarcon-Vargas, D. , Ronai, Z. , 2002. SUMO in cancer–wrestlers wanted. Cancer Biol. Ther.. 1, 237–242. [DOI] [PubMed] [Google Scholar]
- Asher, G. , Reuven, N. , Shaul, Y. , 2006. 20S proteasomes and protein degradation “by default”. BioEssays: News Rev. Mol. Cell. Dev. Biol.. 28, 844–849. [DOI] [PubMed] [Google Scholar]
- Blattner, C. , Knebel, A. , Radler-Pohl, A. , Sachsenmaier, C. , Herrlich, P. , Rahmsdorf, H.J. , 1994. DNA damaging agents and growth factors induce changes in the program of expressed gene products through common routes. Environ. Mol. Mutagen.. 24, 3–10. [DOI] [PubMed] [Google Scholar]
- Canutescu, A.A. , Shelenkov, A.A. , Dunbrack, R.L. , 2003. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci.: Publ. Protein Soc.. 12, 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D.A. , Cheatham, T.E. , Darden, T. , Gohlke, H. , Luo, R. , Merz, K.M. , Onufriev, A. , Simmerling, C. , Wang, B. , Woods, R.J. , 2005. The Amber biomolecular simulation programs. J. Comput. Chem.. 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catic, A. , Suh, C.Y. , Hill, C.T. , Daheron, L. , Henkel, T. , Orford, K.W. , Dombkowski, D.M. , Liu, T. , Liu, X.S. , Scadden, D.T. , 2013. Genome-wide map of nuclear protein degradation shows NCoR1 turnover as a key to mitochondrial gene regulation. Cell. 155, 1380–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chene, P. , 1997. Semiquantitative comparison of the DNA-binding activity of in vitro-synthesized proteins. BioTechniques. 23, 792–794. [DOI] [PubMed] [Google Scholar]
- Chene, P. , Bechter, E. , 1999. Cellular characterisation of p53 mutants with a single missense mutation in the beta-strand 326-333 and correlation of their cellular activities with in vitro properties. J. Mol. Biol.. 288, 891–897. [DOI] [PubMed] [Google Scholar]
- Chene, P. , Mittl, P. , Grutter, M. , 1997. In vitro structure-function analysis of the beta-strand 326-333 of human p53. J. Mol. Biol.. 273, 873–881. [DOI] [PubMed] [Google Scholar]
- Cheng, T.M. , Blundell, T.L. , Fernandez-Recio, J. , 2007. pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins. 68, 503–515. [DOI] [PubMed] [Google Scholar]
- Chowdary, D.R. , Dermody, J.J. , Jha, K.K. , Ozer, H.L. , 1994. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol. Cell. Biol.. 14, 1997–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov, S. , Kurash, J.K. , Wilson, J.R. , Xiao, B. , Justin, N. , Ivanov, G.S. , McKinney, K. , Tempst, P. , Prives, C. , Gamblin, S.J. , Barlev, N.A. , Reinberg, D. , 2004. Regulation of p53 activity through lysine methylation. Nature. 432, 353–360. [DOI] [PubMed] [Google Scholar]
- Clore, G.M. , Ernst, J. , Clubb, R. , Omichinski, J.G. , Kennedy, W.M. , Sakaguchi, K. , Appella, E. , Gronenborn, A.M. , 1995. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat. Struct. Biol.. 2, 321–333. [DOI] [PubMed] [Google Scholar]
- Duddy, P.M. , Hanby, A.M. , Barnes, D.M. , Camplejohn, R.S. , 2000. Improving the detection of p53 mutations in breast cancer by use of the FASAY, a functional assay. J. Mol. Diagn.: JMD. 2, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, S. , Jensen, J.P. , Ludwig, R.L. , Vousden, K.H. , Weissman, A.M. , 2000. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem.. 275, 8945–8951. [DOI] [PubMed] [Google Scholar]
- Feng, L. , Lin, T. , Uranishi, H. , Gu, W. , Xu, Y. , 2005. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol.. 25, 5389–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, P.N. , Chen, X. , Bargonetti, J. , Prives, C. , 1993. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. U S A. 90, 3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk, W.D. , Pak, D.T. , Karas, R.H. , Wright, W.E. , Shay, J.W. , 1992. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol.. 12, 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabb, H.A. , Jackson, R.M. , Sternberg, M.J. , 1997. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol.. 272, 106–120. [DOI] [PubMed] [Google Scholar]
- Hjerpe, R. , Aillet, F. , Lopitz-Otsoa, F. , Lang, V. , England, P. , Rodriguez, M.S. , 2009. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep.. 10, 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe, R. , Aillet, F. , Lopitz-Otsoa, F. , Lang, V. , Torres-Ramos, M. , Farras, R. , Hay, R.T. , Rodriguez, M.S. , 2010. Oligomerization conditions Mdm2-mediated efficient p53 polyubiquitylation but not its proteasomal degradation. Int. J. Biochem. Cell Biol.. 42, 725–735. [DOI] [PubMed] [Google Scholar]
- Hollstein, M. , Sidransky, D. , Vogelstein, B. , Harris, C.C. , 1991. p53 mutations in human cancers. Science. 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Jeffrey, P.D. , Gorina, S. , Pavletich, N.P. , 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science. 267, 1498–1502. [DOI] [PubMed] [Google Scholar]
- Kato, S. , Han, S.Y. , Liu, W. , Otsuka, K. , Shibata, H. , Kanamaru, R. , Ishioka, C. , 2003. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. U S A. 100, 8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi, T. , Kato, S. , Otsuka, K. , Watanabe, G. , Kumabe, T. , Tominaga, T. , Yoshimoto, T. , Ishioka, C. , 2005. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene. 24, 6976–6981. [DOI] [PubMed] [Google Scholar]
- Kopp, F. , Oak, P.S. , Wagner, E. , Roidl, A. , 2012. miR-200c sensitizes breast cancer cells to doxorubicin treatment by decreasing TrkB and Bmi1 expression. PloS One. 7, e50469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel, K.A. , Lee, C.J. , Toledo, F. , Wahl, G.M. , 2005. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc. Natl. Acad. Sci. U S A. 102, 10188–10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cam, L. , Linares, L.K. , Paul, C. , Julien, E. , Lacroix, M. , Hatchi, E. , Triboulet, R. , Bossis, G. , Shmueli, A. , Rodriguez, M.S. , Coux, O. , Sardet, C. , 2006. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell. 127, 775–788. [DOI] [PubMed] [Google Scholar]
- Lee, W. , Harvey, T.S. , Yin, Y. , Yau, P. , Litchfield, D. , Arrowsmith, C.H. , 1994. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Biol.. 1, 877–890. [DOI] [PubMed] [Google Scholar]
- Levine, A.J. , 1997. p53, the cellular gatekeeper for growth and division. Cell. 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Maki, C.G. , 1999. Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. J. Biol. Chem.. 274, 16531–16535. [DOI] [PubMed] [Google Scholar]
- Maki, C.G. , Howley, P.M. , 1997. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol. Cell. Biol.. 17, 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki, C.G. , Huibregtse, J.M. , Howley, P.M. , 1996. In vivo ubiquitination and proteasome-mediated degradation of p53(1). Cancer Res.. 56, 2649–2654. [PubMed] [Google Scholar]
- Maltzman, W. , Czyzyk, L. , 1984. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell. Biol.. 4, 1689–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu, M.G. , Fersht, A.R. , 1998. Nine hydrophobic side chains are key determinants of the thermodynamic stability and oligomerization status of tumour suppressor p53 tetramerization domain. EMBO J.. 17, 2748–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane, E. , Selbach, M. , 2014. Gene expression: degrade to derepress. EMBO J.. 33, 407–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrpour, M. , Esclatine, A. , Beau, I. , Codogno, P. , 2010. Overview of macroautophagy regulation in mammalian cells. Cell Res.. 20, 748–762. [DOI] [PubMed] [Google Scholar]
- Meyer, T. , D'Abramo, M. , Hospital, A. , Rueda, M. , Ferrer-Costa, C. , Perez, A. , Carrillo, O. , Camps, J. , Fenollosa, C. , Repchevsky, D. , Gelpi, J.L. , Orozco, M. , 2010. MoDEL (Molecular Dynamics Extended Library): a database of atomistic molecular dynamics trajectories. Structure. 18, 1399–1409. [DOI] [PubMed] [Google Scholar]
- Miller, M. , Lubkowski, J. , Rao, J.K. , Danishefsky, A.T. , Omichinski, J.G. , Sakaguchi, K. , Sakamoto, H. , Appella, E. , Gronenborn, A.M. , Clore, G.M. , 1996. The oligomerization domain of p53: crystal structure of the trigonal form. FEBS Lett.. 399, 166–170. [DOI] [PubMed] [Google Scholar]
- Milner, J. , Medcalf, E.A. , 1991. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. 65, 765–774. [DOI] [PubMed] [Google Scholar]
- Mittl, P.R. , Chene, P. , Grutter, M.G. , 1998. Crystallization and structure solution of p53 (residues 326-356) by molecular replacement using an NMR model as template. Acta Crystallograph. Sec D Biolog. Crystallogr.. 54, 86–89. [DOI] [PubMed] [Google Scholar]
- Olivier, M. , Eeles, R. , Hollstein, M. , Khan, M.A. , Harris, C.C. , Hainaut, P. , 2002. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat.. 19, 607–614. [DOI] [PubMed] [Google Scholar]
- Petitjean, A. , Mathe, E. , Kato, S. , Ishioka, C. , Tavtigian, S.V. , Hainaut, P. , Olivier, M. , 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat.. 28, 622–629. [DOI] [PubMed] [Google Scholar]
- Riley, T. , Sontag, E. , Chen, P. , Levine, A. , 2008. Transcriptional control of human p53-regulated genes. Nature reviews. Mole. Cell Biol.. 9, 402–412. [DOI] [PubMed] [Google Scholar]
- Rodriguez, M.S. , Desterro, J.M. , Lain, S. , Lane, D.P. , Hay, R.T. , 2000. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol.. 20, 8458–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, M.S. , Desterro, J.M. , Lain, S. , Midgley, C.A. , Lane, D.P. , Hay, R.T. , 1999. SUMO-1 modification activates the transcriptional response of p53. EMBO J.. 18, 6455–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, M.S. , Wright, J. , Thompson, J. , Thomas, D. , Baleux, F. , Virelizier, J.L. , Hay, R.T. , Arenzana-Seisdedos, F. , 1996. Identification of lysine residues required for signal-induced ubiquitination and degradation of I kappa B-alpha in vivo. Oncogene. 12, 2425–2435. [PubMed] [Google Scholar]
- Slee, E.A. , Zhu, H. , Chow, S.C. , MacFarlane, M. , Nicholson, D.W. , Cohen, G.M. , 1996. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J.. 315, (Pt. 1) 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi, T. , Beroud, C. , 2001. Assessing TP53 status in human tumours to evaluate clinical outcome. Nature reviews. Cancer. 1, 233–240. [DOI] [PubMed] [Google Scholar]
- Steinbrecher, T. , Latzer, J. , Case, D.A. , 2012. Revised AMBER parameters for bioorganic phosphates. J. Chem. Theory Comput.. 8, 4405–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturzbecher, H.W. , Brain, R. , Addison, C. , Rudge, K. , Remm, M. , Grimaldi, M. , Keenan, E. , Jenkins, J.R. , 1992. A C-terminal alpha-helix plus basic region motif is the major structural determinant of p53 tetramerization. Oncogene. 7, 1513–1523. [PubMed] [Google Scholar]
- Vogelstein, B. , Lane, D. , Levine, A.J. , 2000. Surfing the p53 network. Nature. 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Zilfou, J.T. , Lowe, S.W. , 2009. Tumor suppressive functions of p53. Cold Spring Harbor Perspectives in Biology. 1, a001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data