

Abstract
Tumor evaluation in pathology is more and more based on a combination of traditional histopathology and molecular analysis. Due to the rapid development of new cancer treatments that specifically target aberrant proteins present in tumor cells, treatment decisions are increasingly based on the molecular features of the tumor. Not only the number of patients eligible for targeted precision medicine, but also the number of molecular targets per patient and tumor type is rising. Diagnostic molecular pathology, the discipline that determines the molecular aberrations present in tumors for diagnostic, prognostic or predictive purposes, is faced with true challenges. The laboratories have to meet the need of comprehensive molecular testing using only limited amount of tumor tissue, mostly fixed in formalin and embedded in paraffin (FFPE), in short turnaround time. Choices must be made for analytical methods that provide accurate, reliable and cost‐effective results. Validation of the test procedures and results is essential. In addition, participation and good performance in internal (IQA) and external quality assurance (EQA) schemes is mandatory. In this review, we critically evaluate the validation procedure for comprehensive molecular tests as well as the organization of quality assurance and assessment of competence of diagnostic molecular pathology laboratories within Europe.
Keywords: Molecular pathology, Next generation sequencing, Proficiency testing, Quality assessment, Quality assurance, Tumor testing
Highlights
Molecular pathology testing should be performed in ISO 15189‐accredited laboratories.
Appropriately educated and dedicated personnel are essential.
One dedicated organization that coordinates all EQA schemes will improve efficiency.
The actions for laboratories after poor performance need standardization.
1. Molecular diagnostics in pathology
Routine molecular diagnostic determinations of tumor specimens in the pathology laboratory have been performed since the late 1990's and concerned mainly classification of tumors, clonality determinations and tests such as microsatellite instability analysis (MSI) to select patients for referral to clinical geneticists. New biological agents that target specific molecular alterations or act to block activated pathways present in individual tumors have become available and enable treatment decisions based on the molecular features of the malignancy. This precision medicine has rapidly gained access to daily practice and it has become a challenge for molecular biologists and pathologists to provide relevant information on the predictive markers in the shortest timeframe possible.
In this review we will highlight aspects on choice and validation of comprehensive molecular assays including assays using next generation sequencing (NGS) technology, and on internal and external quality assurance of molecular tests in Europe. Before the challenges for the molecular pathology and validation and quality assurance issues will be discussed, we will first give a brief overview of the different molecular applications.
1.1. Overview of different molecular applications in pathology
The identification of mutations or chromosomal rearrangements that are characteristic for disease entities can assist the pathologist in the differential diagnosis of these entities. For example, fusion transcripts are seen in the majority of sarcomas and can be, in the right pathological and clinical context, helpful as highly specific molecular diagnostic markers with significant impact on the classification of the tumor (Bovee and Hogendoorn, 2010; Demicco, 2013). Likewise, clonality assessment of the highly polymorphic immunoglobulin and T‐cell receptor gene rearrangements is an important tool in the diagnostics of lymphomas. The clonality results should be interpreted with knowledge of the guidelines and the pathological and clinical context (van Krieken et al., 2007; Groenen et al., 2012; Langerak et al., 2012). Analysis of microsatellite instability (MSI) as a hallmark of Lynch syndrome‐associated tumors is used to select patients suspected of having Lynch syndrome before referral to a clinical geneticist. Subsequent analysis of MLH1 and MSH2 promoter methylation is part of this diagnostics (van Lier et al., 2010), but also somatic mutation analyses of mismatch repair genes if no germ line mutation has been found in these patients after referral to a clinical geneticist (Mensenkamp et al., 2014; Geurts‐Giele et al., 2014).
The identification of specific molecular characteristics may guide therapy. Genetic aberrations can discriminate if morphological similar or asynchronous tumors in one patient represent one or two entities or not e.g. whether a secondary tumor is indeed an independent tumor or a metastasis of a primary (van der Sijp et al., 2002; Blokx et al., 2007). Furthermore, analysis of the MGMT promoter methylation status in glioblastoma has become important for predicting outcome to treatment with temozolomide (Weller et al., 2013).
The observation that some genetic aberrations make tumor cells dependent on or “addicted to” a gene product or cellular pathway has powered the development of drugs that specifically target these aberrations allowing treatment based on the genetic makeup of a tumor, also called precision medicine (Weinstein, 2002). At present there are several genetic changes leading to targetable proteins. Examples are overexpression of ErbB2 (HER2) due to ERBB2 amplification in e.g. breast cancer, and treatment with trastuzumab (Herceptin) (Piccart‐Gebhart et al., 2005) and activating KIT and PDGFRA mutations in gastrointestinal stromal tumors (GIST) as targets for the tyrosine kinase inhibitors (TKI) (Joensuu et al., 2001; Lasota and Miettinen, 2008). More recently, high volume screening for EGFR and KRAS mutations and ALK, ROS1 and RET rearrangements in non‐small cell lung cancer, KRAS and NRAS mutations in colon cancer and BRAF, NRAS and KIT mutations in metastasized melanomas has become an essential part of daily molecular pathology diagnostics to select patients for targeted treatment options (Chapman et al., 2011; Douillard et al., 2013; Lindeman et al., 2013).
2. Challenges for molecular diagnostic tests in a pathology laboratory
The efforts of The Cancer Genome Atlas (TCGA) initiative have led to a still growing body of information on acquired somatic genomic changes in different cancer genomes (Cancer Genome Atlas Research, 2008). Due to the fast increase of available targeted therapies as well, the TCGA efforts rapidly result in a growing demand for routine molecular tumor diagnostics and screening for actionable mutations in a wide variety of tumor types. To offer patients the best treatment options for a certain tumor, diagnostic tests should be reliable, reproducible, of sufficient high sensitivity, and able to investigate all potential targets with the constrains of limited amount of tissue, time and budget. These criteria for comprehensive molecular testing require permanent development of new assays, awareness of their potentials and drawbacks, continuous quality assessment to improve testing of diagnostic tissues, consciousness of budget and costs and clinical demands such as turnaround time. Apart from these issues there are tissue‐ and technological challenges as well.
2.1. Tissue challenges
There are many challenges typically for molecular pathology diagnostics of solid tumors. The vast majority of the DNA to be analyzed is retrieved from routine formalin‐fixed, paraffin‐embedded (FFPE) tissue, which leads to suboptimal DNA quality for the required assays due to fixation and histoprocessing procedures (see also Groenen et al., 2011). For the design of the molecular test it should be taken into account that suboptimal DNA samples (isolated from routine FFPE tissues) allow only amplification of small‐sized PCR‐amplicons (100–200 bp). In addition, the DNA is isolated from (dissected) tissue fragments composed of mixed populations of normal and neoplastic cells reducing the mutant allele frequency, and frequently only a limited amount of (biopsied) tumor tissue is available containing a low percentage of neoplastic cells, therefore the test developed must be able to accurately detect low levels of mutations.
2.2. Technological challenges: a multitude of different molecular tests
To detect the various genomic DNA alterations in the tumor cells including point mutations, large insertions and deletions, complex indels, genomic rearrangements, MSI, and promoter methylation, a diversity of methods has been developed for daily routine pathology molecular diagnostics. It can be anticipated that in the era of precision medicine the number of molecular markers, which need to be assessed, will steadily increase per sample. The challenge for the diagnostic laboratory is to select high‐performing technological methodologies that enable reliable detection of all mutations requested, at a high sensitivity, with a limited amount of tissue (biopsies, cytological preparations), within short turnaround times and at low costs.
2.2.1. Mutation detection: low throughput assays
The majority of diagnostic tests for precision medicine involve a wide range of PCR‐based assays that amplify short DNA fragments for analysis of a single gene (Bellon et al., 2011; Deans et al., 2011; Deans et al., 2013; van den Bent et al., 2013; Deans et al., 2014). Each of these methods has its own unique strengths and challenges. For example, laboratory‐developed gold standard Sanger sequencing, still the method of choice by most laboratories for detection of EGFR, KRAS, BRAF and KIT mutations, allows detection of essentially all diagnostically‐relevant base substitutions, insertions and deletions, but has a relatively modest limit of detection (Tsiatis et al., 2011, 2013, 2014, 2010, 2012). Other commonly used methods like mutation‐ or allele‐specific PCR (AS‐PCR), pyrosequencing, and high resolution melting (HRM) are all considerably more sensitive, have a faster turnaround time and less hands‐on time than Sanger sequencing. However, AS‐PCR and pyrosequencing only identify mutations at predefined positions and are not suitable for detection of novel mutations, and HRM requires often, and pyrosequencing occasionally, confirmation by another method (Tsiatis et al., 2010). Similarly, commercial tests like the EGFR, KRAS and BRAF mutation tests from Roche (COBAS test) and Qiagen (Therascreen test), have high sensitivity, while analyzing only predefined DNA positions and are incapable to detect novel mutations. In addition, commercial tests are not flexible, since addition of tests for new actionable targets is entirely dependent on the manufacturer.
Applied technologies in molecular diagnostics should allow fast implementation of new tests for actionable targets to be able to offer optimal patient care. The need for this is illustrated by the recent trial on treatment of metastatic colorectal cancer, showing that besides KRAS exon 2 mutations, another 17% of colon cancers of patients that do not respond to anti‐EGFR treatment harbor mutations in KRAS exon 3 and 4 and NRAS exon 2, 3 and 4 (Douillard et al., 2013). Importantly, when using the described low throughput assays, these extra analyses require more material and overall costs and an organized testing pipeline.
FDA‐approved or CE‐marked tests suggest a high reliability in clinical testing. However, although the tests themselves may be sensitive, work properly and are standardized, these tests might not cover all clinically relevant mutations. For example, the FDA‐approved COBAS BRAF mutation test is particularly developed for detection of BRAF p.V600E and may also detect p.V600K and p.V600D mutations albeit less reliable. However, in approximately 25% of the melanomas, clinically relevant BRAF codon 600 mutations other than p.V600E occur (da Rocha Dias et al., 2013). Using the COBAS BRAF mutation test results in lack of detection of other codon 600 mutations, which withholds patients from appropriate treatment. Although it can be assumed that the laboratories are aware of these shortcomings they still can pass external quality assessment, as long as these laboratories indicate the shortcomings of the test in their Standard Operating Procedure (SOP). It is questionable whether this procedure should be considered as sufficiently high quality and competent patient care.
2.2.2. Mutation detection: comprehensive analysis
None of the tests described above is suitable for high throughput mutational analysis and may prove insufficient to detect all clinically relevant mutations in a cost‐effective fashion in the future. For diagnostic molecular pathology, whole genome sequencing is not yet affordable, requires too much DNA and still has a long turnaround time. At present, there are few options to perform comprehensive analysis for precision medicine, including the OncoCarta panels of Bioscience and targeted NGS approaches. The OncoCarta panels are multiplexed PCR systems using a mass spectrometry‐based read‐out for fast screening of more than 200 hotspot mutation sites across 20 cancer genes. Although the method is slightly more sensitive than Sanger sequencing and tests for more mutations, the DNA input to obtain all this information is relatively high (∼500 ng), only predefined positions are screened and detected mutations require follow‐up conventional sequencing to confirm the presence of the mutation (Beadling et al., 2011).
NGS‐based methods using the Ion Torrent Personal Genome Machine (IT‐PGM) from Life Technologies and the MiSeq Benchtop Sequencer from Illumina are now applied for analysis of gene‐panels for diagnostic purposes (Endris et al., 2013; Geurts‐Giele et al., 2013; McCourt et al., 2013; Tops et al., 2014). Both platforms use a sequencing‐by‐synthesis approach, but the underlying sequencing technology differs. The IT‐PGM uses semiconductor sequencing detecting hydrogen ion release during base incorporation by DNA polymerase, the Miseq detects emission of fluorescent signal released from labeled nucleotides after incorporation (Ulahannan et al., 2013). The sensitivity of NGS is higher than Sanger sequencing (detection of 2–10% versus 15–25% allele frequency). Moreover, the amount of DNA that is needed for the analysis of gene panels is very low, only 10–50 ng for all amplicons (IT‐PGM and Illumina, respectively) versus 10 ng per amplicon needed for Sanger sequencing. The turnaround time and costs can be competitive with respect to low throughput technologies in centers that have sufficient number of samples. The use of small, dedicated gene panels and efficient loading of the chips for IT‐PGM also significantly reduce costs per case. Both IT‐PGM and MiSeq systems allow the use of commercially established gene panels as well as custom‐designed gene panels for amplicon sequencing. The major benefit of (targeted) NGS is that it uncovers all kinds of mutations in selected genomic regions instead of only mutations at predefined positions. An additional advantage of NGS is that in the same assay mutations and allelic imbalances can be detected e.g. EGFR amplification and loss of heterozygosity by single nucleotide polymorphism (SNP) analyses. Unfortunately, it is not yet possible to detect both mutations and gene rearrangements in one assay by the IT‐PGM and MiSeq. Currently, a major disadvantage of NGS in implementation in molecular diagnostics is that the data‐generating and data‐processing technologies are not yet fully developed which regularly leads to equipment and software improvements. Below, we will further discuss these aspects and the consequences for validation of NGS. We are most experienced with IT‐PGM analysis therefore we will further focus on validation using this platform.
2.2.3. Detection of chromosomal rearrangements: in situ hybridization
At present, genomic rearrangements in e.g. NSCLC are mainly detected by in situ hybridization (ISH) on FFPE material or cytology preparations with commercially available, CE‐marked and FDA‐approved probes. The advantage of FISH is that it is relatively fast and can be performed semi or fully automated. However, subsequent microscopic analysis of the results is relatively labor‐intensive: it can be hard to discriminate normal from tumor cells in particular in cytology preparations and interpretation of the signals might be complex, especially for intrachromosomal rearrangements in NSCLC leading to EML4‐ALK fusions (Thunnissen et al., 2012). The number of FISH determinations per NSCLC case increases, as apart from ALK, also ROS1 and RET rearrangements yield actionable products. Consequently, also the time spent and costs per case expand. Detection of chromosomal rearrangements in e.g. NSCLC might benefit from an RNA‐based sequencing approach that allows simultaneous detection of different chromosomal rearrangements in a limited amount of tissue.
3. Validation of comprehensive molecular assays
Since the number of actionable mutations to be screened for per tumor rapidly increases and the accuracy, speed and cost enables clinical use of comprehensive molecular assays, there is an urgent need for development of consensus validation procedures. Implementation of new technology in the laboratory, for example NGS, needs determination of test‐conditions including DNA‐input, setup of SOPs, determination of coverage needed and testing software applications. The desired sensitivity of the test in diagnostic samples determines the read‐depth, which reflects how often a genomic region has been sequenced. In our centers, we have established NGS assay designs resulting in a read‐depth per amplicon of 500× at minimum, which enables accurate detection of low frequency allelic variants. In general, the observed mutation frequency will correlate with the estimated percentage of neoplastic cells, but tumor heterogeneity may account for the presence of low‐abundance mutations. A pathologists that is familiar with basic molecular testing should score the percentage of neoplastic cells. The relevance for treatment of mutations present in a low percentage of the malignant cells due to tumor heterogeneity is not clear as yet (Ulahannan et al., 2013).
Currently, validation occurs according to the criteria of the local laboratory Quality Control management system and there is no consensus for validation of NGS‐tests in Europe. In the Netherlands, modern pathology laboratories work with specially trained clinical scientists in molecular pathology (CSMP) who are educated in design, analysis and evaluation of molecular pathology tests and have knowledge on basic surgical pathology (see also 4.2). Also technology‐ and process validation benefit from the knowledge and expertise from these CSMPs.
Validation of broad‐spectrum mutation detection requires essentially the same process as for other conventional mutational analysis i.e. addressing technology specificity by analysis of different known mutations in parallel with a “golden standard”, for example Sanger sequencing, and assessing sensitivity and reproducibility by replicating the analyses of samples with different mutant allele frequencies. Commercially available pre‐designed reference standards might be used especially for detection of low abundance mutations. For example, Horizon Diagnostics (www.horizondx.com) offers multiplex reference standards either purified or to be purified from FFPE cell line material to determine the limit of detection of the used NGS system and identification of various mutations. At least part of the NGS‐method validation should include analysis of a series of diagnostic samples, isolated according to the standards of the laboratory, with a variety of known mutations, comprising missense mutations, simple and complex deletions and insertions. Representative DNA samples from FFPE tissue are essential to include, because DNA quality strongly affects NGS performance i.e. poor quality DNA yields higher error rate (Hofreiter et al., 2001). During the validation process the laboratory must not only demonstrate that the test works well but also that reliable results are provided within the desired turnaround time.
An essential difference with standard Sanger sequencing is that during targeted NGS tens to hundreds amplicons are amplified in a multiplex PCR that are subsequently simultaneously analyzed by massive parallel sequencing. As a consequence, NGS of 50 amplicons at a read depth of 500× yields 25,000 independent sequence reads of one sample. The complexity of one NGS analysis multiplies when simultaneously running multiple samples on the same chip. In this strategy in which samples are pooled, data identification is achieved by incorporating a sample‐specific DNA barcode in the amplified DNA fragments. Due to the enormous complexity of these data one has to rely much more than with Sanger sequencing on bioinformatic analysis using appropriate software. Validation of software in the NGS‐pipeline is required, as is described by the working group of Next‐Generation Sequencing: standardization of Clinical Testing (Gargis et al., 2012) and United Kingdom, Association of Clinical Genetic Science Practice guidelines for Targeted Next Generation Sequencing Analysis and Interpretation (www.acgs.uk.com/media/774802/bpg_for_targeted_next_generation_sequencing_final.pdf). In our opinion, exchange of raw datasets between laboratories that preferentially use different software packages should be part of software validation in order to establish that the participating laboratories detect identical gene mutations. In addition, new software updates need to be validated, by analysis of prior NGS‐datasets covering various simple and complex mutations. Finally, we recommend the use of raw NGS datasets to become part of in silico external assessment schemes.
The validation process should assess the entire workflow including the molecular report. The general information that is needed in the report is described by van Krieken et al. (2013a). In addition, an NGS‐test report should include which genes or regions of the genes are investigated, information about the gene coverage, the sensitivity of the detection and the frequency of the detected mutation. It is highly recommended to evaluate the mutant allele frequency in the context of the percentage of neoplastic cells estimated by the pathologist.
4. Quality assurance for diagnostic laboratories
Given the implications of molecular analyses on treatment of patients a high quality of test‐ and laboratory performance is required. Procedures for continuous measurement and improvement of laboratory performance should be fully integrated in the laboratory internal quality assurance system that will ensure a consistently high standard of performance. External quality assurance (EQA) programs, also known as proficiency testing (PT) are inevitable for monitoring of performance.
4.1. Internal quality assurance (IQA)
IQA is necessary to ensure high assay reproducibility and performance and enable detection and correction of errors in daily practice. Assays should be performed according to standard operating procedures (SOP) using appropriate positive and negative controls. Implementation of new tests requires a validation procedure using predefined parameters. Dedicated trained personnel (laboratory technicians and members of the medical staff) as well as a manageable, easy to use quality management system are necessary to maintain high level of performance and improve test results or logistics whenever needed. A laboratory quality manager is essential to take charge of participation and performance in quality assurance schemes and to organize internal and external audits.
4.2. Competence of personnel
IQA also involves personnel competency. The wide variety of tests in a molecular pathology laboratory, the rapid technological advances and high complexity of the tests requires experienced personnel. Both technicians and supervisors should have an adequate theoretical and technical training in molecular biology techniques and should remain up‐to‐date by knowledge about the peer reviewed literature and by regularly attending meetings and symposia. According to the guidelines of the Dutch Society for Pathology (www.pathology.nl), each pathology laboratory performing molecular diagnostics is recommend to have a certified CSMP, who is responsible for development and supervision of the molecular pathology diagnostics. This CSMP is a PhD or MD/PhD in molecular biology and/or molecular pathology and/or genetics, accomplished a post‐doctoral training in this field, and subsequently has completed a 2‐year training in molecular pathology, covering design, analysis and evaluation of molecular tests, tissue/cell based diagnostic possibilities and quality management. Since 2012, the Dutch Society for Pathology has officially recognized the training program for the CSMP. We are not aware of special requirements and training programs in other European countries, although in France trained molecular biologists are associated with pathologists for optimal molecular testing (Nowak et al., 2013). In the United Kingdom there is a Royal College of Pathologists five year Clinical Scientist specialist training programme in Molecular Pathology of acquired disease which focuses on service delivery and development specifically for diagnosis of solid tumors within health care science (http://www.rcpath.org/).
4.3. External quality assurance (EQA)/proficiency testing (PT)
One of the requirements to become accredited according to e.g. ISO 15189 is to participate in EQA programs also known as PT, but in this paper the term EQA will be used. EQA is an inevitable tool to periodically assess the analytical performance of molecular tests by inter‐laboratory comparison, which will assist laboratories in monitoring their assays and improve assay performance as well as evaluation of results whenever needed. Frequent assessment of European laboratories improved the quality of EGFR mutation detection in non‐small‐cell lung cancer (Deans et al., 2013), BRAF testing of melanoma (Emile et al., 2013), and KIT mutation testing in GIST (Wong et al., 2012). The need for EQA schemes is illustrated by the fact that 10–15% of laboratories do not carry out according to the standard set by the EQA provider (van Krieken et al., 2013b).
4.4. EQA providers in Europe
At present, there are several providers for European EQA schemes for mutation detection in solid tumors. These include the European Society for Pathology (ESP, www.esp‐pathology.org), and the European Molecular Genetics Quality Network (EMQN, www.emqn.org) (Table 1). Since 2009, the ESP offers KRAS EQA of colon cancer on a yearly basis and the 2014 round will also include NRAS and BRAF testing (Table 2). As of 2012, lung EQA schemes for EGFR and KRAS mutation analysis and for detection of ALK rearrangements by RT‐PCR, ISH and via digital fluorescent ISH cases are provided. Previously, the EMQN provided EQA schemes particularly for inherited disorders for laboratories worldwide. In 2014, there is also a scheme available for KRAS, NRAS and BRAF testing in colon cancer samples.
Table 1.
External quality assessment providers for molecular pathology schemes in Europe.
| Name | Abbreviation | Website | |
|---|---|---|---|
| European | European Society for Pathology | ESP | www.esp‐pathology.org |
| European Molecular Genetics Quality Network | EMQN | www.emqn.org | |
| National | Dutch Foundation for Quality Assessment in Medical Laboratories | SKML | www.skml.nl |
| UK National External Quality Assurance Services | UK NEQAS | http://www.ukneqas.org.uk |
Table 2.
External quality assessment schemes for molecular pathology in Europe.
| Provider | Tissue | Scheme | Type | Starting year |
|---|---|---|---|---|
| European Society for Pathology | Colon cancer | KRAS | Mutation detection | 2009 |
| Colon cancer | KRAS, NRAS and BRAF | Mutation detection | 2014 | |
| Lung cancer | EGFR, KRAS | Mutation detection | 2012 | |
| Lung cancer | ALK | Rearrangement (ISH, FISH) | 2012 | |
| Lung cancer | Digital ALK | Rearrangement, digital cases | 2012 | |
| European Molecular Genetics Quality Network | Colon cancer | KRAS, NRAS and BRAF | Mutation detection | 2014 |
| UK National External Quality Assessment Services | Adult Molecular Neuropathology | 1p/19q FISH, MGMT promoter methylation, IDH | Translocations, methylation and mutation detection | 2012 |
| Colon cancer | KRAS | Mutation detection | 2009 | |
| Colon cancer | KRAS, BRAF, PIK3CA and NRAS | Mutation detection | 2013 | |
| Gastrointestinal stromal tumor | KIT, PDGFRA | Mutation detection | 2008 | |
| Lung cancer | EGFR | Mutation detection | 2010 | |
| Lung cancer | EGFR, KRAS, BRAF and PIK3CA | Mutation detection | 2013 | |
| Lung cancer | ALK | Rearrangement (ISH, FISH, RT‐PCR) | 2013 | |
| Melanoma | BRAF | Mutation detection | 2012 | |
| Melanoma | BRAF, NRAS, KIT | Mutation detection | 2013 | |
| Sarcoma | Common translocations | Translocations (FISH, RT‐PCR) | 2014 | |
| Dutch Foundation for Quality Assessment in Medical Laboratoriesa | B‐cell clonality | IG heavy and light chain gene rearrangements | Rearrangement detection (Fragment analysis) | 2003, 2004, 2008, 2010 |
| Breast cancer | ERBB2 (HER2) | Amplification | 2005, yearly | |
| Colon cancer | KRAS | Mutation detection | 2012 | |
| Lymphoma | BCL2, BCL6 and MYC translocations | Translocation detection | 2005, 2011 | |
| Lynch prescreening | MSI | Fragment analysis | 2006 | |
| Lung cancer | EGFR, KRAS | Mutation detection | 2009 | |
| Lung cancer | EGFR | Mutation detection | 2010 | |
| Melanoma | BRAF | Mutation detection | 2012 | |
| Sarcoma | Common translocations | Translocations (FISH, RT‐PCR) | 2002 and 2009 | |
| Solid tumor clonality | TP53 | Mutation, LOH detection | 2001 | |
| Tissue identification | Polymorphic markers | Fragment analysis | 2002–2007 | |
| French nationwide initiative | Melanoma | BRAF | Mutation detection | 2012 |
EQA schemes provided in the years indicated.
The United Kingdom National External Quality Assessment Services (UK NEQAS) for Molecular Pathology is open for laboratories outside the UK (www.ukneqas.org.uk and www.ukneqas‐molgen.org.uk) and offers a variety of EQA schemes, including gene panel molecular pathology EQAs for prediction of therapy response in GISTs, lung cancer, colon cancer and melanoma, and MSI testing (Table 2). In these EQA schemes, the participating laboratories perform the test that is used in the routine diagnostic setting, which can vary from either high resolution melting, mutation‐specific TaqMan tests, Sanger sequencing to NGS technologies for mutation detection. The interpretation of the test results as well as the evaluation of the observed genotype is described in a diagnostic report and presented to the EQA provider for assessment. The EQA assessors evaluate the performance of the participating laboratories, by scoring of: identification of a mutation, the correct description of the genotype (coding sequence and protein) according to the nomenclature guidelines of human genome variation society (www.hgvs.org), interpretation of the observed genotype for the diagnostic question and clerical accuracy of the report. The EQA assessors produce a scheme report in which is presented: the correct genotype as scored by professional consensus, interpretation and clinical data of the EQA‐cases, an overview of the results reported by the participating laboratories and a critical evaluation of the observed difficulties or pitfalls, supported by references whenever needed. In our opinion, participation in these EQA schemes is not only obligatory for maintaining a good quality management, but also absolutely useful, since many EQAs include at least one “difficult” case that give laboratories insight in their performance. In addition, after introduction and validation of a newly introduced test or technology such as NGS, participation in EQAs can be instrumental to evaluate the performance of the new technology on validated EQA samples.
In addition to the European EQA schemes there are national EQA schemes, among which are the Dutch Foundation for Quality Assessment in Medical Laboratories (SKML, www.skml.nl) and Nationwide EQA in France according to recommendations of the French National Institute for Cancer (INCa) as described in Table 2. A BRAF testing of melanoma samples EQA was a first successful nationwide effort to improve molecular testing on FFPE tumor tissue in France (Emile et al., 2013). The 46 French laboratories involved in molecular testing, may be stimulated by the need to become ISO 15189‐accredited by 2016.
Apart for these official organizations for EQA there are “private” initiatives to perform inter‐laboratory comparisons of molecular tests not yet being part of EQA schemes. Examples are an international evaluation on IDH1 and IDH2 mutation detection (van den Bent et al., 2013) and a German‐Austrian‐Dutch ring test on MGMT promoter methylation (Felsberg et al., 2013).
In conclusion, laboratories in Europe have several opportunities to participate in EQA schemes. Some EQA providers, like the UK NEQAS and the ESP, already run the same program for several years and improve their service by increasing the number of targets to be analyzed per tissue along with the changes in the field. In addition, their number of molecular programs slightly increases with time. Continuous assessment with the same program is essential for laboratories to be able to maintain and improve their competence.
5. Laboratory performance
5.1. Accreditation
In the Netherlands, accreditation of the quality system and competence of medical laboratories, including (molecular) pathology laboratories, is organized by an external organization called CCKL (National Coordination Committee for Quality Assurance for Health Care Laboratories in The Netherlands, www.cckl.com), which since 2010 is part of the Dutch National Accreditation Body (www.rva.nl). The CCKL guidelines are based upon but not yet fully conform the International Organization for Standardization (ISO) 15189 norm. The national accreditation body is now in transition towards NEN‐EN‐ISO 15189:2012 (www.iso.org) in order to commit to internationally recognized standards. All currently CCKL‐accredited laboratories have to fulfill the new standard by July 2019 at the latest. Although accreditation is strongly recommended it is not yet mandatory, neither is the participation in EQAs.
The policy towards accreditation differs in the European countries. For example, in Belgium only laboratories that are accredited according to the ISO 15189 standard can get reimbursement, i.e. each test requires separate accreditation to qualify for reimbursement. The Belgian laboratories are allowed to perform unaccredited molecular test, but these financial incentives accelerate laboratories to apply for accreditation and thus improving their quality and competence (Raymaekers et al., 2011). In contrast, in France all medical laboratories performing clinically‐relevant tests are mandatory to obtain an accreditation to the ISO 15189 standard before 2016 in order to be able to continue their clinical activity (Nowak et al., 2012, 2013).
5.2. Actions following poor laboratory performance
EQA providers are not by themselves able to penalize laboratories after poor performance. In fact, solving the underlying cause of the inferior performance is the responsibility of the concerning laboratory. In this respect, it is of interest that the ESP requires successful participation to be listed on their website, thus stimulating laboratories to keep on improving their standards. As part of this process, providers will advise and support laboratories how to improve procedures and protocols and provide extra reference material (van Krieken et al., 2013b).
Also the policy of the UK NEQAS is to encourage participants with performance difficulties to improve by education rather than penalty. If any participant has fallen below the acceptable performance standard, the EQA scheme director will inform the laboratory about the poor performance status and help and advice will be made available on request. This is the policy for UK and non‐UK participants. If a UK‐laboratory persists in poor performing (designated code red), the UK Royal College of Pathologists National Quality Assurance Advisory Panel (NQAAP) for Genetics will be informed with the details of the laboratory's performance issues. The NQAAP will consider the best approach to improve the situation and may arrange an urgent visit to the laboratory, review of the service provided and offer help and advice on corrective and preventative actions. If persistent poor performance remains (classed as code black) the NQAAP is obliged to notify the Joint Working Group for Quality Assessment in Pathology (JWG), which includes the United Kingdom Accreditation Service and Care Quality Commission (regulator of health services) and the JWG who in turn will take further appropriate actions tailored to the laboratory specific problems. In the end, after several opportunities for improvement and other measures, but remaining incapability to resolve poor performance, according to the Genetics NQAAP‐Annual Report 2011–12, the ultimate outcome might be closure of the laboratory. Comprehensive information on the NQAAP and the JWG is available via: www.rcpath.org/committees/intercollegiate‐and‐joint‐committees/joint‐working‐group‐for‐quality‐assessment‐in‐pathology and the UK NEQAS Molecular Pathology management procedure (www.ukneqas‐molgen.org.uk/ukneqas/index/participantsManual/poorPerformanceCriteria.html).
The actions following identification of a persistent poor performing non‐UK laboratory are different. The scheme director will contact the laboratory informing them of the laboratory's persistent poor performance and offers help and advice in order to improve the service. Should no satisfactory response be given within a defined period, the scheme director will discuss the situation and the suitable actions with the Molecular Pathology Specialist Advisory Group. Experiences with EQA schemes have shown that the majority of the laboratories will correct any deficiencies before reaching the status of a persistent poor performance laboratory.
6. Towards regional specialized centers
The use of molecular tests on tumor tissue for therapeutic decision making will increase and requires that the introduction of mutation detection for new (panels of) genes in the routine pathology practice is efficient and fast, while also providing robust and accurate test results within a short turnaround time (days). In order to do so, sufficient cases, personnel and expertise is necessary. In our opinion, it is highly unlikely that small laboratories that have limited numbers of samples and provide a limited number of tests can maintain their service. Clearly, collaboration and forming regional centers for molecular pathology is needed. In France, molecular pathology is organized in 28 regional genetics centers. This network, dedicated to molecular oncology tests, is initiated and funded by the INCa and the French Ministry of Health (Nowak et al., 2012, 2013). Nationwide organization of molecular diagnostics including funding yields a powerful instrument to enforce ISO 15189 accreditation by all laboratories, to make it mandatory to participate in nationwide EQA programs (Emile et al., 2013), and to actively monitor and maintain high quality molecular diagnostics if necessary by adequate penalties in case of poor performance. In the Netherlands, the foundation for oncological collaboration (Stichting Oncologische Samenwerking, SONCOS) consisting of surgical, medical and radiotherapeutical oncologists generated a standardization report with the minimal requirements for the treatment of different tumor types (www.soncos.org) in 2012. There is a need for specialized centers for treatment (and diagnostics) of oncology patients. The ministry for health in the Netherlands recently has decided that targeted therapies for patients with metastasized melanoma can only be provided in a limited number of specialized centers in the Netherlands. An important (and wise) prerequisite is the registry of all metastasized melanoma patients, of which clinical data, therapies, pathology and molecular genotyping data are being registered. The DICA (Dutch Institute for Clinical Auditing) will play an important role in the registration and data‐assessment (www.clinicalaudit.nl). Pathology laboratories of the specialized “Melanoma” centers need to have expertise on melanoma and on the technologies for mutation detection of the genes of interest (not only BRAF).
7. Critical appraisal of laboratory performance and quality assurance
The successful introduction of modern molecular technologies in diagnostic pathology is in our opinion regulated by the profession of the CSMP itself, namely the requirements for training and continuous education, as well as the accreditation of the pathology laboratory (see also van Krieken et al., 2010). The first guarantees the quality of the CSMP, using their knowledge, expertise and skills to come to optimal results and interpretation of the data. The second guarantees that the laboratory techniques and processes are performed standardized and yield high quality results. Successful implementation of NGS in pathology molecular diagnostics clearly benefits from a close collaboration between CSMPs and pathologists, as the latter are trained in cell/tissue‐based pathology and can provide essential information with respect to neoplastic cells and tissue heterogeneity.
Clearly, ensuring high quality molecular testing starts within the laboratory, with appropriately educated and dedicated personnel, analyses performed by SOPs, quality assurance schemes and accreditation. IQA and EQA are essential methods to monitor the quality, performance and competence of a laboratory. To improve the current standard we propose that all European diagnostic molecular pathology laboratories are obliged to become accredited according to the international ISO 15189 standard for medical laboratories. Similar to the situation in the United Kingdom, we propose that each European country has a National Body with a mandate to enforce laboratories to become ISO 15189‐accredited and to participate in EQA schemes (Figure 1). In case of poor performance or lack of participation the National Body should have the authority to undertake adequate steps and when patient care cannot be guaranteed anymore should be able to stop funding or reimbursement and ultimately can force the laboratory to stop clinical activity. The multiple European EQA providers and the overlap in EQA schemes between providers would benefit from a structural solution to improve efficiency: e.g. one dedicated organization that coordinates all EQA schemes via one provider or via multiple already existing providers and the EQA schemes should be open for all European laboratories (Figure 2).
Figure 1.
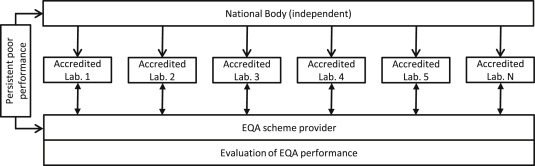
Proposed model for the interaction between the EQA providers, the National Body and ISO 15189‐accredited laboratories. Each European country has an independent National body with a mandate to enforce laboratories to become ISO 15189 accredited and to participate in EQA schemes. In case of poor performance, there will be an encouraging interaction between the EQA provider and the laboratory. In case of persistent poor performance, the EQA provider will communicate this to the National Body who will undertake adequate steps.
Figure 2.
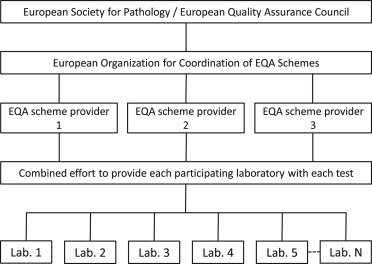
Proposed framework for the organization of EQA schemes in Europe. EQA programmes are organized under supervision of the European Society for Pathology and/or a European Quality Assurance Council (van Krieken et al., 2008). A European organization for the coordination of EQA schemes determines in collaboration with representatives from the different participating countries and the EQA providers which schemes should be provided. One or several EQA provider(s) coordinate(s) collecting representative tissues or in silico data and distribution of a scheme as well as evaluation and feedback of the performance to all participating laboratories and in case of persistent poor performance to a National Body as depicted in Figure 1.
In conclusion, in our opinion molecular testing in pathology should be performed in an ISO 15189‐accredited laboratory that participates in EAQ schemes with a good performance and a laboratory with appropriately educated and dedicated personnel and with close collaboration between CSMPs and pathologists.
Dubbink Hendrikus J., Deans Zandra C., Tops Bastiaan B.J., van Kemenade Folkert J., Koljenović S., van Krieken Han J.M., Blokx Willeke A.M., Dinjens Winand N.M., Groenen Patricia J.T.A., (2014), Next generation diagnostic molecular pathology: Critical appraisal of quality assurance in Europe, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.03.004.
References
- Beadling, C. , Heinrich, M.C. , Warrick, A. , 2011. Multiplex mutation screening by mass spectrometry evaluation of 820 cases from a personalized cancer medicine registry. J. Mol. Diagn.. 13, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellon, E. , Ligtenberg, M.J. , Tejpar, S. , 2011. External quality assessment for KRAS testing is needed: setup of a European program and report of the first joined regional quality assessment rounds. Oncologist. 16, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokx, W.A. , Lesterhuis, W.J. , Andriessen, M.P. , 2007. CDKN2A (INK4A-ARF) mutation analysis to distinguish cutaneous melanoma metastasis from a second primary melanoma. Am. J. Surg. Pathol.. 31, 637–641. [DOI] [PubMed] [Google Scholar]
- Bovee, J.V. , Hogendoorn, P.C. , 2010. Molecular pathology of sarcomas: concepts and clinical implications. Virchows Arch.. 456, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, P.B. , Hauschild, A. , Robert, C. , 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med.. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha Dias, S. , Salmonson, T. , van Zwieten-Boot, B. , 2013. The European Medicines Agency review of vemurafenib (Zelboraf(R)) for the treatment of adult patients with BRAF V600 mutation-positive unresectable or metastatic melanoma: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur. J. Cancer. 49, 1654–1661. [DOI] [PubMed] [Google Scholar]
- Deans, Z.C. , Tull, J. , Beighton, G. , 2011. Molecular genetics external quality assessment pilot scheme for KRAS analysis in metastatic colorectal cancer. Genet. Test. Mol. Biomarkers. 15, 777–783. [DOI] [PubMed] [Google Scholar]
- Deans, Z.C. , Bilbe, N. , O'Sullivan, B. , 2013. Improvement in the quality of molecular analysis of EGFR in non-small-cell lung cancer detected by three rounds of external quality assessment. J. Clin. Path. 66, 319–325. [DOI] [PubMed] [Google Scholar]
- Deans, Z.C. , Wallace, A. , O'Sullivan, B. , 2014. External quality assessment of BRAF molecular analysis in melanoma. J. Clin. Path. 67, 120–124. [DOI] [PubMed] [Google Scholar]
- Demicco, E.G. , 2013. Sarcoma diagnosis in the age of molecular pathology. Adv. Anat. Pathol.. 20, 264–274. [DOI] [PubMed] [Google Scholar]
- Douillard, J.Y. , Oliner, K.S. , Siena, S. , 2013. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med.. 369, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Emile, J.F. , Tisserand, J. , Bergougnoux, L. , 2013. Improvement of the quality of BRAF testing in melanomas with nationwide external quality assessment, for the BRAF EQA group. BMC Cancer. 13, 472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endris, V. , Penzel, R. , Warth, A. , 2013. Molecular diagnostic profiling of lung cancer specimens with a semiconductor-based massive parallel sequencing approach: feasibility, costs, and performance compared with conventional sequencing. J. Mol. Diagn.. 15, 765–775. [DOI] [PubMed] [Google Scholar]
- Felsberg, J. , Malzkorn, B. , Bujan, B. , 2013. Molecular diagnostics of glioma – results of the first interlaboratory comparison of MGMT promoter methylation testing at twenty-three academic centers in Germany, Austria and the Netherlands. Clin. Neuropathol.. 32, 414–415. (Abstract) [Google Scholar]
- Gargis, A.S. , Kalman, L. , Berry, M.W. , 2012. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol.. 30, 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts-Giele, W.R. , Dirkx-van der Velden, A.W. , Bartalits, 2013. Molecular diagnostics of a single multifocal non-small cell lung cancer case using targeted next generation sequencing. Virchows Arch.. 462, 249–254. [DOI] [PubMed] [Google Scholar]
- Geurts-Giele, W.R. , Leenen, C.H. , Dubbink, H.J. , 2014. Somatic Aberrations of Mismatch Repair Genes as a Cause of Microsatellite-Instable Cancers. (submitted for publication) [DOI] [PubMed] [Google Scholar]
- Groenen, P.J. , Blokx, W.A. , Diepenbroek, C. , 2011. Preparing pathology for personalized medicine: possibilities for improvement of the pre-analytical phase. Histopathology. 59, 1–7. [DOI] [PubMed] [Google Scholar]
- Groenen, P.J. , van Raaij, A. , van Altena, 2012. A practical approach to diagnostic Ig/TCR clonality evaluation in clinical pathology. J. Hematopathol.. 5, 17–25. [Google Scholar]
- Hofreiter, M. , Jaenicke, V. , Serre, D. , 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res.. 29, 4793–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensuu, H. , Roberts, P.J. , Sarlomo-Rikala, M. , 2001. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N. Engl. J. Med.. 344, 1052–1056. [DOI] [PubMed] [Google Scholar]
- Langerak, A.W. , Groenen, P.J. , Bruggemann, M. , 2012. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 26, 2159–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasota, J. , Miettinen, M. , 2008. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 53, 245–266. [DOI] [PubMed] [Google Scholar]
- Lindeman, N.I. , Cagle, P.T. , Beasley, M.B. , 2013. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn.. 15, 415–453. [DOI] [PubMed] [Google Scholar]
- McCourt, C.M. , McArt, D.G. , Mills, K. , 2013. Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PloS One. 8, e69604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensenkamp, A.R. , Vogelaar, I.P. , van Zelst-Stams, 2014. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in lynch syndrome-like tumors. Gastroenterology. 146, 643–646. [DOI] [PubMed] [Google Scholar]
- Nowak, F. , Soria, J.C. , Calvo, F. , 2012. Tumour molecular profiling for deciding therapy-the French initiative. Nat. Rev. Clin. Oncol.. 9, 479–486. [DOI] [PubMed] [Google Scholar]
- Nowak, F. , Calvo, F. , Soria, J.C. , 2013. Europe does it better. Am. Soc. Clin. Oncol. Educ. Book. 332–337. [DOI] [PubMed] [Google Scholar]
- Piccart-Gebhart, M.J. , Procter, M. , Leyland-Jones, B. , 2005. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med.. 353, 1659–1672. [DOI] [PubMed] [Google Scholar]
- Raymaekers, M. , Bakkus, M. , Boone, E. , 2011. Reflections and proposals to assure quality in molecular diagnostics. Acta Clin. Belg.. 66, 33–41. [DOI] [PubMed] [Google Scholar]
- Thunnissen, E. , Bubendorf, L. , Dietel, M. , 2012. EML4-ALK testing in non-small cell carcinomas of the lung: a review with recommendations. Virchows Arch.. 461, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tops, B.B.J. , Normanno, N. , Kurth, H. , 2014. Rapid and Cost-effective Genotyping of Colon and Lung Cancer Using Semi-conductor Sequencing. (Submitted for publication) [Google Scholar]
- Tsiatis, A.C. , Norris-Kirby, A. , Rich, R.G. , 2010. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J. Mol. Diagn.. 12, 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulahannan, D. , Kovac, M.B. , Mulholland, P.J. , 2013. Technical and implementation issues in using next-generation sequencing of cancers in clinical practice. Br. J. Cancer. 109, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bent, M.J. , Hartmann, C. , Preusser, M. , 2013. Interlaboratory comparison of IDH mutation detection. J. Neurooncol.. 112, 173–178. [DOI] [PubMed] [Google Scholar]
- van der Sijp, J.R. , van Meerbeeck, J.P. , Maat, A.P. , 2002. Determination of the molecular relationship between multiple tumors within one patient is of clinical importance. J. Clin. Oncol.. 20, 1105–1114. [DOI] [PubMed] [Google Scholar]
- van Krieken, J.H. , Langerak, A.W. , Macintyre, E.A. , 2007. Improved reliability of lymphoma diagnostics via PCR-based clonality testing: report of the BIOMED-2 concerted action BHM4-CT98-3936. Leukemia. 21, 201–206. [DOI] [PubMed] [Google Scholar]
- van Krieken, J.H. , Jung, A. , Kirchner, T. , 2008. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch.. 453, 417–431. [DOI] [PubMed] [Google Scholar]
- van Krieken, J.H. , Jansen, C. , Hebeda, K.M. , Groenen, P.J. , 2010. Biomarkers as disease definition: mantle cell lymphoma as an example. Proteomics Clin. Appl.. 4, 922–925. [DOI] [PubMed] [Google Scholar]
- van Krieken, J.H. , Normanno, N. , Blackhall, F. , 2013. Guideline on the requirements of external quality assessment programs in molecular pathology. Virchows Arch.. 462, 27–37. [DOI] [PubMed] [Google Scholar]
- van Krieken, J.H. , Siebers, A.G. , Normanno, N. , 2013. European consensus conference for external quality assessment in molecular pathology. Ann. Oncol.. 24, 1958–1963. [DOI] [PubMed] [Google Scholar]
- van Lier, M.G. , Wagner, A. , van Leerdam, M.E. , 2010. A review on the molecular diagnostics of Lynch syndrome: a central role for the pathology laboratory. J. Cell Mol. Med.. 14, 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein, I.B. , 2002. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Weller, M. , Pfister, S.M. , Wick, W. , 2013. Molecular neuro-oncology in clinical practice: a new horizon. Lancet Oncol.. 14, e370–379. [DOI] [PubMed] [Google Scholar]
- Wong, N.A. , Deans, Z.C. , Ramsden, S.C. , 2012. The UK NEQAS for molecular genetics scheme for gastrointestinal stromal tumour: findings and recommendations following four rounds of circulation. J. Clin. Pathol.. 65, 786–790. [DOI] [PubMed] [Google Scholar]