

Abstract
Reprogramming of NK cells with a chimeric antigen receptor (CAR) proved an effective strategy to increase NK cell reactivity and recognition specificity toward tumor cells. To enhance the cytotoxicity of NK cells against CD138‐positive multiple myeloma (MM) cells, we generated genetically modified NK‐92MI cells carrying a CAR that consists of an anti‐CD138 single‐chain variable fragment (scFv) fused to the CD3ζ chain as a signaling moiety. The genetic modification through a lentiviral vector did not affect the intrinsic cytolytic activity of NK‐92MI toward human erythroleukemic cell line K562 cells or CD138‐negative targets. However, these retargeted NK‐92MI (NK‐92MI‐scFv) displayed markedly enhanced cytotoxicity against CD138‐positive human MM cell lines (RPMI8226, U266 and NCI‐H929) and primary MM cells at various effector‐to‐target ratios (E:T) as compared to the empty vector‐transfected NK‐92MI (NK‐92MI‐mock). In line with the enhanced cytotoxicity of NK‐92MI‐scFv, significant elevations in the secretion of granzyme B, interferon‐γ and proportion of CD107a expression were also found in NK‐92MI‐scFv in response to CD138‐positive targets compared with NK‐92MI‐mock. Most importantly, the enhancement in the cytotoxicity of NK‐92MI‐scFv did not attenuate with 10Gy‐irradiation that sufficiently blocked cell proliferation. Moreover, the irradiated NK‐92MI‐scFv exerted definitely intensified anti‐tumor activity toward CD138‐positive MM cells than NK‐92MI‐mock in the xenograft NOD‐SCID mouse model. This study provides the rationale and feasibility for adoptive immunotherapy with CD138‐specific CAR‐modified NK cells in CD138‐positive plasmacytic malignancies, which potentially further improves remission quality and prolongs the remission duration of patients with MM after upfront chemotherapy.
Keywords: Multiple myeloma, Adoptive immunotherapy, NK cells, Chimeric antigen receptor, CD138 (syndecan‐1)
Highlights
We generated genetically modified NK cells targeting CD138 positive myeloma cells.
The retargeted NK cells exerted markedly enhanced ex vivo anti‐myeloma activities.
The enhancement in cytotoxicity may be due to elevated NK cell degranulation.
Irradiation of retargeted NK cells did not attenuate their cytotoxicity.
The retargeted NK cells after irradiation had potent anti‐MM effect in xenografts.
Abbreviations
- α-MEM
alpha modification of Eagle's minimum essential medium
- ATCC
American type culture collection
- CAR
chimeric antigen receptor
- GvM
graft-vs-myeloma effect
- IMiDs
immunomodulatory drugs
- KIRs
immunoglobulin-like receptors
- MFI
mean fluorescence intensity
- MM
multiple myeloma
- PBMNC
peripheral blood mononuclear cell
- PCL
plasma cell leukemia
- RACE
rapid amplification of cDNA ends
- scFv
single-chain variable fragment
- SP
signal peptide
- TCR
T-cell receptor
1. Introduction
Multiple myeloma (MM) remains an incurable malignant plasma cell disorder. Despite the markedly improved outcome in recent years, the vast majority of patients with MM could not escape disease relapse or progression (Hart et al., 2012). Thus, novel strategies are urgently needed to further increase the response frequency and quality, and prolong the overall survival and disease free survival of patients with MM.
Human natural killer (NK) cells play an essential role in innate immune defense against malignant cells, potentiating it to be a satisfying effector cells for adoptive immunotherapy (Vivier et al., 2011). It has been demonstrated that NK cells mediate graft‐vs‐myeloma effect (GvM) in MM patients undergoing allogenic hematopoietic cell transplantation (Reynolds et al., 2001). A recent study indicated that NK cells were capable to kill clonogenic MM cells in vitro and in vivo (Swift et al., 2012). The striking efficacy of the upfront immunomodulatory drugs (IMiDs) in maintenance therapy of MM, have been identified to be closely related to its positive influence on NK cell function (McDaniel et al., 2012). All these results suggested that adoptive immunotherapy with NK cells provides a promising treatment modality for eradication or control of the residual MM cells, potentially complementing the first‐line therapies.
However, adoption of primary allogeneic or autologous NK cells is largely limited by difficulties in ex vivo cell expansion as well as the variation in NK cell activity from different patients (Tonn et al., 2001), which made the established NK cell lines an attractive option as effector cells for immunotherapy. NK‐92 is the only NK cell line to be tested in clinical trials for immunotherapy of malignancies, and its safety and expansion feasibility have been validated in phase I trial in renal cell cancer or melanoma (Arai et al., 2008). NK‐92 cells lack almost all inhibitory killer cell immunoglobulin‐like receptors (KIRs) except KIR2DL4, which inhibit NK cell activation by binding to HLA molecule on target cells (Tonn et al., 2001). The lack of KIRs on NK‐92 cells may, at least in part, account for its marked anti‐tumor activity against a broad spectrum of tumor targets (Morett et al., 2001). NK‐92MI is an interleukin‐2 (IL‐2) independent derivative cell line of NK‐92 by transfection of human IL‐2 cDNA, with the same characteristics of activated NK cells as its parental NK‐92 cells (Favors et al., 2012). Reprogramming of NK cells with a chimeric antigen receptor (CAR) proved an effective strategy to enhance their reactivity against the antigen‐expressing tumor cells or overcome resistance (Boissel et al., 2012, 2009, 2012).
CD138 (syndecan‐1) is an integral membrane protein widely expressed on differentiated plasma cells, and has been taken as a primary diagnostic marker of MM (Lutz and Whiteman, 2009). It acts as a receptor for the extracellular matrix through its extracellular domain, mediating MM development and proliferation (Dhodapkar et al., 1998; Bataille et al., 2006). The high expression of CD138 on MM cells potentiates it to be a specific immunotherapeutic target for MM. To enhance the cytotoxicity of NK‐92MI to CD138 expressing MM cells, we transfected NK‐92MI cells with a lentiviral vector encoding a recombinant CAR termed scFv (4B3)‐CD3ζ that is CD138‐specific single‐chain antibody fragments (scFv) genetically fused to the CD3ζ chain of the T‐cell receptor (TCR) complex (another signaling molecule known to trigger cytotoxicity of NK cells) (André et al., 2004; Imai et al., 2005), via a flexible hinge region of CD8. Then we detected the expression of CAR on the transfected NK cells, and examined their anti‐MM potential in vitro and in vivo.
2. Materials and methods
2.1. Cell culture
Human myeloma‐derived cell line RPMI8226 secreting λ light chain, U266 secreting IgE λ light chain, NCI‐H929 secreting IgA κ light chain, plasma cell leukemia (PCL) cell line ARH‐77 secreting IgG κ light chain, human erythroleukemic cell line K562 and NK‐92MI were introduced from the American Type Culture Collection (Manassas, VA). NK‐92MI and transduced NK‐92MI were incubated in alpha modification of Eagle's minimum essential medium (α‐MEM; Invitrogen, Carlsbad, CA) supplemented with 2 mM l‐glutamine, 0.2 mM inositol, 0.02 mM folic acid, 0.01 mM 2‐mercaptoethanol, 12.5% FBS and 12.5% horse serum (Sigma–Aldrich Corporation, St Louis, MO). All the other cell lines were cultured in RPMI‐1640 medium containing 10% FBS, 2 mM l‐glutamine, penicillin (10 IU/ml) and streptomycin (100 μg/ml) (Invitrogen, Carlsbad, CA) at 37 °C in a humidified atmosphere with 5% CO2. Cells in the logarithmic growth phase were used for all experiments.
Peripheral blood samples collected from three healthy volunteers were processed by Ficoll–Paque density gradient centrifugation to obtain PBMNC. Primary myeloma cells were isolated from bone marrow aspirates of five newly diagnosed MM patients approved by the Institutional Ethics Committee (Shanghai Changzheng Hospital Institutional Ethics Committee). All participants provided their written informed consent. The patients with MM were hospitalized in Shanghai Changzheng Hospital from October 2009 to December 2012, including three males and two females with a median age of 53 years (ranging from 40 to 69 years) (Table 1). The mononuclear cells from the bone marrow aspirates were isolated using CD138 MACS MicroBeads and an automatic MACS magnetic cell sorter machine (Miltenyi, Bergisch Gladbach, Germany) according to the manufacturer's instructions and as described by Baumann et al. (2012). The primary myeloma cells were grown in the same medium as that for MM cell lines.
Table 1.
Clinical features of patients.
| Case number | Sex | Age | M‐protein | D‐S stagea | ISSb stage | Percentage of plasma cells in BMc at diagnosis |
|---|---|---|---|---|---|---|
| 1 | Female | 50 | IgA λ | IIA | I | 31% |
| 2 | Male | 40 | λ | IIIA | I | 55% |
| 3 | Female | 69 | IgG λ | IIIB | II | 59.5% |
| 4 | Male | 63 | κ | IIIA | I | 40.5% |
| 5 | Male | 53 | IgG λ | IIIB | III | 28.5% |
D‐S stage, Durie–Salmon stage.
ISS, International Staging System.
BM, bone marrow.
2.2. Flow cytometry
For CD138 staining, tumor cells were harvested, stained with anti‐CD138‐PE (Miltenyi, Bergisch Gladbach, Germany), and analyzed using a Navios flow cytometer (Beckman Coulter).
2.3. Generation of CD138 specific‐CAR modified NK cells
2.3.1. Construction of the CAR‐scFv (4B3)‐CD3ζ
cDNA of variable domains of heavy‐chain (VH) and light‐chain (VL) of CD138‐specific monoclonal antibody 4B3 were derived by RT‐PCR of mRNA from hybridoma cells generating 4B3 (Sun et al., 2007). The universal primers for amplification of VH and VL domain were as follows: VH: forward 5′‐AGGTG/CA/CAG/ACTGCAGG/CAGTCT/AGG‐3′; reverse 5′‐TCTTGTCCACCTTGGTGCTGCTG‐3′. VL: forward 5′‐CAA/CAT TG/CT/AGATGACCCAGTCTCCA‐3′; reverse 5′‐GTTAGATCTCCAGCTTGGTCC C‐3′. The PCR condition was 94° C for 10 min followed by 35 cycles at 94° C for 30 s, 67° C for 30 s and 72° C for 3 min plus a final extension period at 72° C for 7 min. The PCR products were confirmed and subcloned into pMD18‐T vector (Takara, Dalian, China). Positive clones were sequenced by ABI 3730xl sequencer. Then, the full length cDNA sequences of VH and VL were obtained by the 5′‐ rapid amplification of cDNA ends (RACE) PCR as previously described (Maruyama et al., 1995). For the RACE PCR of VH and VL, the first and second round of PCR amplifications were carried out using primers (VH 1st forward 5′‐ CTACAGACAGTGTGGCCTTGCCCTTG ‐3′, reverse 5′‐CAGCCTACGTGGATCTCAGCAGCCTTA‐3′; VH 2nd forward 5′‐GGTGTAG CCAGAAGTCTTACAGGACAG‐3′, reverse 5′‐CCAAGGCACCACTCTCACAGTCTCC‐3′; VL 1st forward 5′‐ AAGCGGTTGGAAACTCTGTAGATCAGGAGT ‐3′, reverse 5′‐ AGGTTCAGTGGC AGTGGATCAGGGAC ‐3′; VL 2nd forward 5′‐ TGATCTCCAAGACTG ACAGGTAGGGAG ‐3′, reverse 5′‐ GGAGGCTGAGGATCTGGGAGTTTATT ‐3′). The 1st and 2nd round PCR were carried out for 30 cycles with the following conditions: 94 °C for 30 s denaturation, 55 °C for 30s annealing and 72 °C for 1 min extension reaction. The PCR products were purified and sequenced by ABI 3730xl sequencer.
The putative signal peptide sequence of heavy chain was identified by SignalP 3.0 Server. Subsequently, the integrated cDNA of the fusion protein was artificially synthesized, comprising the sequence encoding the SP of heavy chain, VH and VL linked by a (G4S)3‐linker (scFv (4B3)), in conjunction with human CD3ζ chain (QSFGLLDPKLCYLLDGILFIYGVILTALFLRVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPQRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR) by human CD8α hinge region (LSNSIMYFSHFVPVFLPAKPTTTPAPRPPTPAPTIASQPLSLRPEACRPAAGGAVHTRGLDFA). The construct was then cloned into pCDNA3.1 (+)/myc plasmid (invitrogen) and sequenced for identification.
2.3.2. Transduction of NK‐92MI cells and establishment of a stable transfected NK‐92MI cell line
The complete CAR sequence was cloned into the SalI, BamHI restriction sites of pLenO‐GTP lentiviral Vector (Invabio, Shanghai, China), yielding pL‐scFv (4B3)‐ CD3ζ. Lentiviruses were generated by transfection of 80% confluent HEK293T cells with pL‐scFv (4B3)‐CD3ζ or control plasmid, together with pRsv‐REV, pMDlg‐pRRE and pMD2G (Tronolab) using 2.5 mol/L CaCl2. Lentivirus was harvested at 72 h post transfection, centrifuged to remove cell debris, and then filtered through a 0.45 μm cellulose acetate filter followed by ultracentrifugation at 50,000 × g 16 °C for 120 min. Then, the pellet was resuspended with 1 × PBS. The Lentiviral titer was then determined by detecting the EGFP using flow cytometry‐based method (Savan et al., 2010). In brief, 293T cells were plated at 1 × 105/well with DMEM‐10 in a 6‐well plate and incubated overnight. The next day, the medium was removed and the cells were transduced with a 10‐fold dilution series of virus in the DMEM‐10 with 8 μg/ml polybrene, generating 1 ml, 10−1, 10−2, 10−3, 10−4, 10−5 ml of virus/well. After incubation overnight at 37 °C, replace the media with fresh DMEM‐10. Forty‐eight hours after infection, cells were detached by adding trypsin/EDTA and resuspended in 1 × PBS, and analyzed for EGFP expression using FACS. Use a well that has between 0.1% and 10% of cells expressing EGFP to determine titer. The titer of virus was calculated as follows.
For lentivirus infection, NK‐92MI cells in logarithmic growth phase were adjusted to 1 × 105 cells/ml using NK cell medium, followed by addition of lentivirus supernatant at the MOI of 50–100 in the presence of 8 μg/ml polybrene overnight at 37 °C. Then the cells were grown for 40 h in fresh NK‐92MI medium before puromycin (Calbiochem, Bad Soden, Germany) was added to a final concentration of 5 μg/ml for selection of transgene‐positive cells.
Transfected NK‐92MI cells were repeatedly selected by addition of 5 μg/ml puromycin to the culture medium. After 2–3 week selection, the dead NK‐92MI cells were removed by density gradient centrifugation using Lymphoprep TM (1.077 g/ml, Axis‐Shield PoC AS, Oslo, Norway). Mock (empty vector with EGFP) transfected NK‐92MI were transfected with the plasmid pLenO‐GTP and selected with puromycin. The transfection efficiency of NK‐92MI by lentiviral vector was determined by flow cytometry. Single‐cell suspensions (5 × 105) of scFv (4B3)‐CD3ζ‐transfected NK‐92MI (NK‐92MI‐scFv), empty vector‐transfected NK‐92MI (NK‐92MI‐mock) or parental NK‐92MI were washed twice in PBS, and the expression proportion and mean fluorescence intensity (MFI) of EGFP were detected by a Beckman Coulter Navios flow cytometer.
2.3.3. Identification of CAR expression in NK‐92MI‐scFv
2.3.3.1. RNA Extraction and RT‐PCR
Total RNA was extracted from NK‐92MI‐scFv, NK‐92MI‐mock or parental NK‐92MI cells using the TRI Reagent (Sigma) according to the manufacturer's protocol. RNA was then treated with DNaseI (Roche Diagnostics), purified through a RNeasy column (Qiagen) and electrophoresed to determine the integrity of the extracted RNA. Complementary DNA (cDNA) was synthesized from 2 μg of total RNA using random hexamers (Proligo) and SuperScript III Reverse Transcriptase (Invitrogen). RT‐PCR was performed using primers specific for scFv (4B3) sequence: forward 5′‐ AGCCTGACAAGCGAGGATAGC‐3′, reverse 5′‐ GTCTGGGTCATCACCACATCG‐3′. The primers for GAPDH: forward 5′‐ GGAGTCCACTGGCGTCTTC‐3′, reverse 5′‐ GCTGATGATCTTGAGGCTGTTG‐3′. The RT‐PCR conditions were as follows: 10 min at 94 °C, denaturation at 94 °C for 20 s, annealing at 59 °C for 30 s, and extension at 72 °C for 60 s.
2.3.3.2. Immunofluorescence assay
To identify the membrane surface expression of the constructed CAR on NK‐92MI‐scFv cells, the cells were labeled with 200 nM CFSE (Sigma) in a volume of 1 ml 1 × PBS for 1 × 106 cells for 8 min at 37 °C as described previously (Olson et al., 2009). After washed twice in PBS, the cells were resuspended in 1 × PBS (4 × 105/ml) and cytospinned, followed by direct immunofluorescence staining as described previously (Heinz et al., 2010). In summary, the transfected NK‐92MI cells on slides were incubated in 0.05% Tween 20 in 1 × PBS (PBST) for 25 min at room temperature. After removal of Tween 20, the cells were incubated with blocking solution (2% bovine serum albumin in 1 × PBS) for 10 min at room temperature. Then, blocking solution was replaced with 1:64 dilution of tetramethylrhodamine isothiocyanate (Tritc)‐labeled goat anti‐mouse IgG (Fab specific) (t7782, Sigma). After incubation for 1 h in the dark at room temperature, cells were washed three times with 1 × PBST and stained with DAPI (Invitrogen). The slices were analyzed by fluorescence microscopy (Olympus BX51).
2.4. Cytotoxicity assays
The LDH release assay was performed by using CytoTox 96® Non‐Radioactive Cytotoxicity Assay (Promega Corporation, USA) according to the manufacturer's instructions. Briefly, transfected or parental NK‐92MI (effector cells) were co‐cultured for 4 h with a constant number of target cells (2 × 104) at effector‐to‐target (E:T) ratio of 10:1, 5:1, 1:1 in a total volume of 150 μl. In the competition binding assay, the target cell (U266 and RPMI8226) were pretreated with 30 μg/ml CD138 mAb (clone BB4, Immunotech, Beckman Coulter, Inc.) or an isotype‐matched control antibody (mouse anti‐human IgG1) for 30 min, followed by co‐culture with various effector cells for 4 h. The supernatants were collected, and the LDH release in the supernatants was evaluated by a colorimetric reaction (absorbance at 490 nm). The spontaneous release of effector and target cells and maximum release of target cells were also measured. The percentage specific lysis was calculated as follows:
In the apoptotic assay, effector cell NK‐92MI‐scFv, NK‐92MI‐mock or paretal NK‐92MI were labeled with CFSE and immediately co‐cultured with a constant number of target cells (5 × 104), at E:T ratios of 10:1, 5:1 and 1:1. Cells were incubated in 48‐well microplates in a total volume of 150 μl RPMI1640 complete medium for 4 h in a 5% CO2 atmosphere at 37 °C. Cell mixtures were then washed twice in PBS, and apoptotic cells were detected with Annexin V–APC (BD Biosciences) and propidium iodide (PI) (Beckman Coulter), and analyzed on a Beckman Coulter Navios flow cytometer. The CFSE fluorescence distinguished the effector cells from the target cells, and the apoptotic target cells, including the annexin V+/PI+ and annexin V+/PI− cells in CFSE‐negative gated target cells were detected. The cytotoxicity was calculated as the proportion of annexin V+ target cells to the total target cells. Analysis was performed using Navios software. Data presented are the mean ± SD of three separate experiments.
2.5. ELISA for secreted IFN‐γ and granzyme B by NK cells
The ability of NK‐92MI‐scFv and NK‐92MI‐mock to produce IFN‐γ and granzyme B in response to CD138‐positive target cells were analyzed by ELISA as previously described (Hoover et al., 2009). The effector cells were co‐cultured with a constant number of target cells (1 × 105) at various E:T ratios in 48‐well microplates within a final volume of 150 μl RPMI1640 complete medium. After incubation for 4 h, the supernatant were collected and used for ELISA assays. Human IFN‐γ ELISA kit (catalog: VAL104) and granzyme B ELISA kit (catalog: BMS2027) were respectively obtained from R&D Systems, Inc. (MN, USA) and eBioscience (San Diego, CA, USA). The results represented the mean ± SD of three separate experiments.
2.6. CD107a and Fas‐L assays
The scFv (4B3)‐CD3ζ‐ or mock‐transfected NK‐92MI cells were co‐cultured with different target cells (1 × 106) at the E:T ratio of 1:1 and anti‐CD107a‐PECy5 (BD Bioscience, San Jose, CA) or an isotype‐matched control was added to the tubes at 20 μl/ml at the beginning of the cultures. Cells were incubated for 1 h at 37 °C in 5% CO2 after which monensin (Golgi‐Stop, BD Biosciences) was added to a final concentration of 6 μg/ml and incubated for an additional 3 h at 37 °C in 5% CO2. Cells were then stained with Alex fluor 647‐conjugated anti‐CD178 (Fas‐ligand, Fas‐L) mAb or Alex fluor 647‐conjugated mouse IgG1 isotype control (AbD Serotec). The CD107a and Fas‐L expression were measured in EGFP‐gated NK cells using Beckman Coulter Navios flow cytometer, and data analysis was performed using Navios software.
2.7. Murine models
Five to 7‐week old male nonobese diabetic (NOD)/severe combined immunodeficient (SCID) mice (Shanghai SLAC Laboratory Animal CO. LTD) were subcutaneously injected into the right flank with 5 × 106 RPMI8226 or ARH‐77 cells in a mixture of 100 μl PBS and 100 μl matrigel (BD Biosciences). One to 2‐week later when palpable tumors (≥5 mm in diameter) developed, mice inoculated with RPMI8226 or ARH‐77 were assigned into three treatment groups (6 per group) receiving intravenously injection of 4 × 107 10Gy‐irradiated NK‐92MI‐scFv or NK‐92MI‐mock or the vehicle alone (PBS) via tail vein every other day for a total of three injections. Then, the tumors were measured with a caliper at their greatest length and width every alternate day to estimate the tumor volume, using the following formula: 4π/3 × (tumor width/2)2 × (tumor length/2) (Frost et al., 2004). Mice were humanely sacrificed when subcutaneous tumors reached 15 mm in diameter or when moribund. Survival was evaluated from the first day of treatment until death. Mice studies were approved by the Institutional Animal Care and Use Committees of the second military medical University.
2.8. Immunohistochemistry
Immunohistochemical studies were performed using routinely fixed and processed, paraffin‐embedded sections of the xenograft subcutaneous tumors, and heat‐induced epitope retrieval was performed as described previously (Khoury et al., 2003). In brief, the tissue sections were placed in plastic Coplin jars containing preheated target retrieval solution (DAKO) heated in a household vegetable steamer (Sunbeam‐Oster, Model Sunbeam 4713/5710, 900 W) for 35 min, and allowed to cool at room temperature for at least 15 min. We used the primary antibody specific for CD20 (clone L26, Abcam), CD138 (clone B‐A38, Abcam), κ (clone EPR5367‐8, Epitomics), λ (clone EPR5367, Epitomics), CD2 (clone L26, Abcam), at a dilution of 1:250. The primary antibody was incubated at 4 °C for 6–8 h. Detection of primary antibody was achieved using the LSAB + kit (DAKO, Carpinteria, CA, USA), which contains secondary biotinylated antibody and streptavidin/horseradish peroxidase complex, according to the manufacturer's recommendations. Nuclei were counterstained with hematoxylin and sections were evaluated by light microscopy using an Olympus BH‐2 microscope (Olympus, Melville, NY, USA).
2.9. Statistical analysis
Statistical analysis was performed using the GraphPad Prism and Statistical Package for Social Sciences 15.0 (SPSS Inc., USA). In vitro data represent means of three replicates and results are representative of at least three independent experiments. Significance levels were determined by two‐tailed Student's t test analysis. A p value of 0.05 or less was considered statistically significant. For in vivo experiments, tumor volumes were compared using One‐Way Anova test for multiple comparison. Student's t test was used for comparison between two groups. Survival was assessed using Kaplan–Meier curves and log‐rank analysis.
3. Results
3.1. Expression of CD138 in myeloma and plasma cell leukemia cell lines
We first evaluated the CD138 expression in MM cell line U266, RPMI8226, NCI‐H929, and plasma cell leukemia (PCL) cell line ARH‐77. Flow cytometric analyses demonstrated that CD138 is highly expressed on all MM cell lines tested except ARH‐77 (Supplementary Figure S1).
3.2. Generation of NK‐92MI cells carrying a CD138‐specific CAR
A CD138‐specific CAR was generated by synthesizing the cDNA sequence encoding the N‐terminal heavy‐chain signal peptide (SP) (MGWSSIILFLVATATGVHS), scFv (4B3) consisting of VH‐linker‐VL, human CD8α hinge region, and CD3ζ chain. A schematic representation of the resulting CAR construct was shown in Figure 1A.
Figure 1.
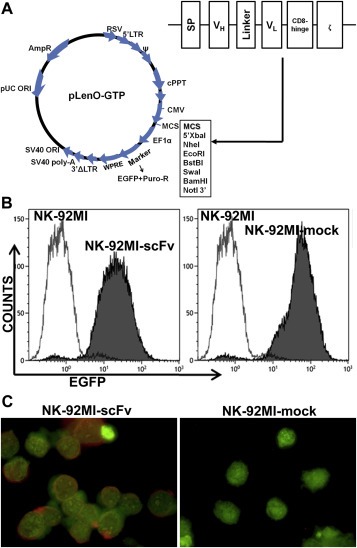
Transduction of NK‐92MI cells with a lentiviral vector carrying a CD138‐specific CAR‐encoding gene (A) Schematic representation of the CD138‐specific CAR construct and the lentiviral vector. A CD138‐specific CAR was generated by synthesizing the cDNA sequence encoding the N‐terminal heavy‐chain signal peptide (SP), scFv (4B3) consisting of VH‐linker‐VL, CD8α hinge region, and CD3ζ chain. (B) The CAR‐ or empty vector‐transfected NK‐92MI cells were repeatedly selected by puromycin, resulting in a stable transfected cell population with an EGFP expression proportion exceeding 95%, as determined by flow cytometry. (C) The membrane expression of CAR on NK‐92MI‐scFv was identified by immunofluoresence assay using a Tritc‐labeled goat anti‐mouse IgG (Fab specific) antibody. The whole cell was counterstained with CFSE. Magnification: × 1000.
Following repeated selection of transfected NK‐92MI cells with puromycin, the expression proportion of EGFP in both NK‐92MI‐scFv and NK‐92MI‐mock exceeded 95%, as shown in Figure 1B. The high‐level transgene expression remained unchanged during the present study, and was routinely confirmed by FACS analysis.
3.3. Expression of CAR by NK‐92MI‐scFv
The mRNA expression of CD138‐specific CAR‐encoding gene in NK‐92MI‐scFv was confirmed by reverse transcription‐polymerase chain reaction (RT‐PCR) (Supplementary Figure S2). Importantly, the CAR protein expression on the cell surface of NK‐92MI‐scFv was demonstrated by immunofluorescence staining using Tritc‐labeled goat anti‐mouse IgG (Fab specific) antibody (Figure 1C). The specific recognition of scFv sequence derived from mouse monoclonal IgG (4B3) on the cell surface by this goat anti‐mouse IgG antibody confirmed the CAR membrane expression by NK‐92MI‐scFv.
3.4. Enhanced cytotoxicity of NK‐92MI‐scFv cells against CD138‐positive MM cells
To investigate whether genetic modification resulted in an alteration of the intrinsic cytotoxic properties, the cytotoxicity of transduced NK‐92MI and parental NK‐92MI toward CD138 positive myeloma cells were compared by LDH release assays and flow cytometric analysis of cell apoptosis. Considering the human erythroleukemic cell K562 has been taken as a standard target control for its high sensitivity to NK‐92 mediated killing (Uherek et al., 2002), the cytotoxicity of both NK‐92MI‐scFv and NK‐92MI‐mock against K562 equaled that of parental NK‐92MI even at low E/T ratios in LDH release assay (Figure 2A), indicating that the genetic modification did not alter the intrinsic cytotoxicity of transfected NK‐92MI cells against NK‐92MI‐sensitive targets. Concurrently, no obvious cytotoxicity of the transfected NK‐92MI cells was observed against PBMNC derived from three healthy donors (Figure 2A).
Figure 2.
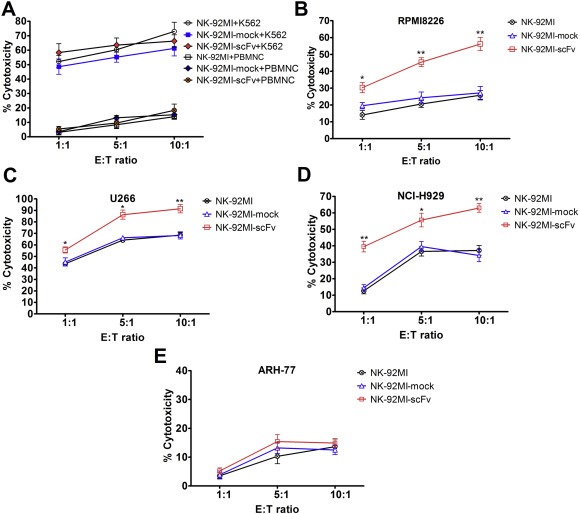
Cytolytic activity of NK‐92MI‐scFv against CD138‐positive and negative targets determined by LDH release assay. The transfected or parental NK‐92MI cells were co‐cultured for 4 h with a constant number of target cells (2 × 104) at E:T ratio of 10:1, 5:1, 1:1 in a total volume of 150 μl, respectively. The supernatants were collected, and the LDH release in the supernatants was evaluated by a colorimetric reaction (absorbance at 490 nm). The cytolytic activity of transfected or parental NK‐92MI toward K562 and PBMNC derived from three healthy donors (A), RPMI8226 (B), U266 (C), NCI‐H929 (D) and ARH‐77 (E). Data presented are the mean ± SD of three separate experiments. *p < 0.05, **p < 0.01 compared with NK‐92MI‐mock at the same E:T ratio.
Toward RPMI8226, there was still no significant difference in the cytotoxicity between NK‐92MI‐mock and parental NK‐92MI. In contrast, NK‐92MI‐scFv exerted markedly enhanced anti‐MM activity compared with NK‐92MI‐mock (19.6 ± 3.3% vs 30.3 ± 5.3% for E:T = 1:1 p = 0.041, 24.3 ± 5.8% vs 45.4 ± 4.6% for E:T = 5:1 p = 0.008, 27.2 ± 6.6% vs 56.2 ± 6.8% for E:T = 10:1 p = 0.006) (Figure 2B). The significantly increased cytotoxicity of NK‐92MI‐scFv toward U266 and NCI‐H929 were also found at all E:T ratios, compared with NK‐92MI‐mock or NK‐92MI (Figure 2C and D). For example, the cytotoxicity of NK‐92MI‐mock and NK‐92MI‐scFv were 68.3 ± 5.4% vs 91.5 ± 6.4% (p = 0.009) toward U266, 34.2 ± 6.6% vs 63.0 ± 4.6% (p = 0.003) toward NCI‐H929 at the E:T = 10:1. Whereas, no difference in the cytotoxicity between NK‐92MI‐mock and NK‐92MI‐scFv against ARH‐77 was detected at all E:T ratios tested (e.g., 13.2 ± 3.3% vs 15.4 ± 4.2% for E:T = 5:1 p = 0.515, 12.5 ± 2.8% vs 14.9 ± 2.6% for E:T = 10:1 p = 0.338) (Figure 2E). In the five primary MM cells, the cytotoxicities of NK‐92MI‐scFv were also significantly higher than those of NK‐92MI‐mock nearly at all E:T ratios, e.g., 32.4 ± 5.2% vs 70.7 ± 6.3% (p = 0.001), 26.2 ± 3.3% vs 48.8 ± 6.5% (p = 0.006), 22.1 ± 5.5% vs 46.8 ± 6.9% ( p = 0.008), 16.9 ± 2.1% vs 55.4 ± 6.1% (p < 0.001), 29.9 ± 4.7% vs 63.8 ± 6.1% (p = 0.002) in Pt 1–5 at E:T of 10:1, respectively, except at the E:T of 1:1 in Pt 2 and Pt 5 (Supplementary Figure S3).
To confirm the results from LDH release assays, we performed apoptotic assays with Annexin V‐PI dual staining by flow cytometry. Apoptotic analyses verified the markedly enhanced anti‐MM activity of NK‐92MI‐scFv in contrast to NK‐92MI‐mock toward MM cell line U266, RPMI8226, NCI‐H929 (Figure 3A and B), together with five primary MM cells (Pt 1‐Pt 5) (Figure 3C). Meanwhile, the cytotoxicity of NK‐92MI‐scFv toward CD138 negative PCL cell ARH‐77 was comparable to that of NK‐92MI‐mock (Figure 3B), in line with the results from LDH release assay.
Figure 3.
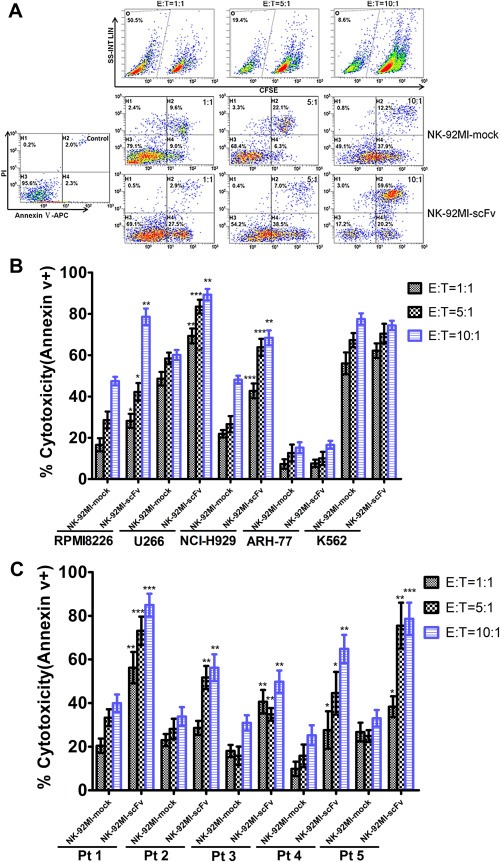
Cytotoxicity of NK‐92MI‐scFv against CD138 positive and negative MM cells determined by apoptotic analysis. CFSE‐labeled effector cells were cocultured with a constant number of target cells (5 × 104) at E:T ratios of 10:1, 5:1 and 1:1, and the apoptotic cells in CFSE‐negative gated target cells were detected by FACS analysis using Annexin V‐APC and PI. (A) A representative density plots of discrimination of effector and target cells by CFSE and the cytotoxicities of NK‐92MI‐scFv and NK‐92MI‐mock against RPMI8226 at various E:T ratios. (B, C) Cytotoxicity of NK‐92MI‐scFv and NK‐92MI‐mock toward RPMI8226, U266, NCI‐H929, ARH‐77, K562 and five primary MM cells sorted by CD138 magnetic microbeads (Pt1–Pt5). Data presented are the mean ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the cytotoxicity of NK‐92MI‐mock at the same E:T ratios.
These results strongly suggested that the improvement in cytotoxicity of NK‐92MI‐scFv toward MM cells was strictly dependent on the CD138 expression on the target cells.
3.5. The mAb BB4 markedly attenuated the enhanced anti‐MM activities of NK‐92MI‐scFv
To ascertain whether the increase in cytotoxicity of NK‐92MI‐scFv was directly related to the CD138 specific CAR stimulation, we preincubated U266 and RPMI8226 with CD138 mAb BB4 for 30 min, and then detected the cytotoxicity of NK‐92MI‐scFv toward these pretreated targets at an E/T ratio of 10:1 by LDH release assay. As shown in Figure 4, NK‐92MI‐scFv–mediated killing of MM cells was markedly attenuated by the pretreatment of CD138 mAb BB4 as compared to the group pretreated by irrelevant control mAb (79.6 ± 4.4% vs 93.5 ± 2.6% in U266 p = 0.009, 32.3 ± 7.4% vs 65.8 ± 4.9% in RPMI8226 p = 0.003). These results confirmed the dependence of enhanced cytotoxicity induced by NK‐92MI‐scFv toward CD138 positive target cells on the expression of CD138 antigen.
Figure 4.
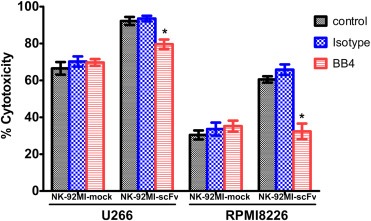
Recognition specificity of NK‐92MI‐scFv toward target cells expressing CD138. U266 and RPMI8226 cells were preincubated with 30 μg/ml CD138 mAb (clone BB4) or an isotype‐matched control antibody for 30 min, followed by co‐culture with NK‐92MI‐scFv or NK‐92MI‐mock for 4 h. Then, the cytotoxicities of NK‐92MI‐scFv and NK‐92MI‐mock toward these pretreated targets at an E/T ratio of 10:1 were determined by LDH release assay. Data presented are the mean ± SD of three separate experiments. *p < 0.05, **p < 0.01 compared with the isotype antibody‐pretreated group in NK‐92MI‐scFv.
3.6. Elevated secretion of IFN‐γ and granzyme B by NK‐92MI‐scFv in response to CD138‐positive target cells
To determine whether the scFv (4B3)‐CD3ζ–mediated killing resulted in an increased cytokine/chemokine secretion, we incubated the effector cell NK‐92MI‐scFv or NK‐92MI‐mock with MM cell line RPMI8226, U266, NCI‐H929 and 3 primary MM cells (Pt1, Pt4 and Pt5) at different E/T ratios for 4 h, and the supernatants were collected for ELISA assays. The results showed that NK‐92MI‐scFv contributed to a markedly increased production of both IFN‐γ (e.g., 308.33 ± 35.32 pg/ml vs 49.57 ± 26.17 pg/ml, p < 0.001 at the E/T ratio of 1:1; 1160.15 ± 136.45 pg/ml vs 316.02 ± 28.66 pg/ml, p < 0.001 at the E/T ratio of 10:1 toward U266) and granzyme B (e.g., 1620.86 ± 144.41 pg/ml vs 443.85 ± 69.98 pg/ml, p < 0.001 at the E/T ratio of 1:1; 5311.67 ± 352.54 pg/ml vs 2305.87 ± 358.26 pg/ml, p < 0.001 at the E/T ratio of 10:1 toward U266) in response to CD138‐positive target cells at all ratios tested, compared to NK‐92MI‐mock. The representative data at the E/T ratio of 10:1 were shown in Figure 5A, B.
Figure 5.
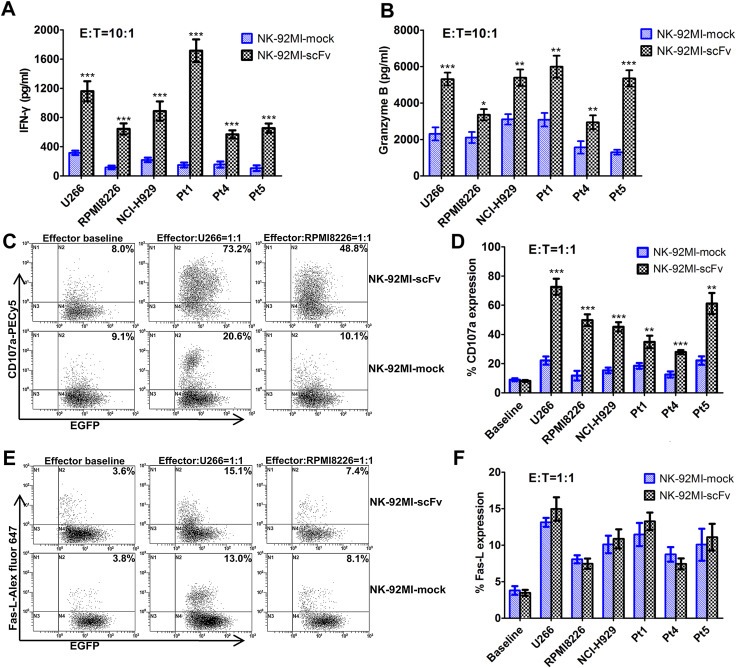
Enhanced activation of NK‐92MI‐scFv in response to CD138‐positive MM cells. The effector cell NK‐92MI‐scFv or NK‐92MI‐mock were co‐cultured with a constant number of target cells (1 × 105) at various E:T ratios in 48‐well microplates within a final volume of 150 μl RPMI1640 complete medium. After incubation for 4 h, the supernatant were collected and used for detection of IFN‐γ and granzyme B by ELISA assays. The representative data of the secretion of IFN‐γ and granzyme B by NK‐92MI‐scFv or NK‐92MI‐mock in response to U266, RPMI8226, NCI‐H929 and three primary MM cells (Pt1, Pt4, Pt5) at the E:T ratio of 10:1 were shown in (A) and (B). (C, D) NK‐92MI‐scFv or NK‐92MI‐mock were cocultured with the foresaid myeloma cells at the E:T ratio of 1:1 in the presence of anti‐CD107a‐PECy5 or an isotype‐matched control for 1 h, followed by addition of monensin and incubation for additional 3 h. The CD107a were measured in EGFP‐gated NK cells by flow cytometry. The representative dot plots (U266 and RPMI8226) and proportions of CD107a expression on NK‐92MI‐scFv and NK‐92MI‐mock in the presence or absence of MM cells were shown. (E, F) NK‐92MI‐scFv or NK‐92MI‐mock were cocultured with MM cells at E:T ratio of 1:1 for 4 h, and then stained with Alex fluor 647‐conjugated anti‐CD178 (Fas‐L) mAb or Alex fluor 647‐conjugated mouse IgG1 isotype control (AbD Serotec). The Fas‐L expression was measured in EGFP‐gated NK cells by flow cytometry. The representative dot plots (U266 and RPMI8226) and proportions of Fas‐L expression on NK‐92MI‐scFv and NK‐92MI‐mock in the presence or absence of MM cells were shown. Data presented are the mean ± SD of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with NK‐92MI‐mock at the same E/T ratio.
3.7. Increased CD107a expression and comparable Fas‐Ligand expression of NK‐92MI‐scFv compared with NK‐92MI‐mock in response to CD138‐positive target cells
CD107a has been shown to be a sensitive marker for NK cell activation and correlates strongly with both cytokine secretion and NK cell‐mediated lysis of target cells (Alter et al., 2004; De Santis et al., 2012). Fas‐L/Fas interaction also plays a significant role in NK cell‐induced cytotoxicity (Lugini et al., 2012; Glässner et al., 2012). To investigate whether the CD107a and Fas‐L expression of NK‐92MI‐scFv also exceed those of NK‐92MI‐mock upon stimulation, we incubated NK‐92MI‐scFv or NK‐92MI‐mock with RPMI8226, U266, NCI‐H929 and three primary MM cells at the E/T ratio of 1:1 for 1 h or 4 h, and determined the CD107a and Fas‐L expression on NK cells by FACS analysis. A significant difference was observed in the proportion of CD107a‐expressing effector cells between NK‐92MI‐scFv and NK‐92MI‐mock in response to all the above‐mentioned MM cells (e.g., 72.6 ± 5.5% vs 22.1 ± 2.8%, p < 0.001 in U266, 49.8 ± 4.0% vs 11.7 ± 3.3%, p < 0.001 in RPMI8226, 45.2 ± 3.2% vs 15.3 ± 1.9%, p < 0.001 in NCI‐H929 (Figure 5C, D). Whereas, the Fas‐L expression on NK‐92MI‐scFv was similar to that of NK‐92MI‐mock upon activation by these MM cells (Figure 5E and F, p > 0.05), which suggested that the Fas‐L/Fas signaling may not be involved in the CAR‐mediated enhancement of anti‐MM activity.
3.8. Transfected NK‐92MI cells retained their cytotoxicities upon irradiation with 10Gy
To avoid the risk of NK cell line in vivo proliferation in adoptive immunotherapy which may result in engraftment and secondary lymphoma in immunocompromised patients, we irradiated the transfected NK‐92MI cells with 10Gy and compared their cytotoxicity with their non‐irradiated counterparts. We found that 10Gy irradiation completely blocked the further proliferation of NK‐92MI‐mock and NK‐92MI‐scFv by [3H] thymidine incorporation and cell viability assays (data not shown). However, the cytotoxicity of irradiated NK‐92MI‐mock or NK‐92MI‐scFv was comparable with their non‐irradiated counterparts toward U266, RPMI8226, ARH‐77 and primary MM cells (Pt 5) at the E/T ratios of 5:1 and 10:1 by FACS apoptotic analysis (Supplementary Figure S4, p > 0.05). Moreover, the viabilities of transfected NK‐92MI‐mock and NK‐92MI‐scFv remained as high as 62–65% 72 h after irradiation with 10Gy assessed by trypanblue dye exclusion test (data not shown).
3.9. Anti‐MM efficacy of irradiated NK‐92MI‐scFv in xenografts
To compare the anti‐tumor activity of irradiated NK92MI cells expressing scFv (4B3)‐CD3ζ with that of the mock‐transduced cells in vivo, we established a CD138‐positive (RPMI8226) or CD138 negative (ARH‐77) human plasma cell malignancy xenograft model. The subcutaneous injection of RPMI8226 or ARH‐77 led to rapid tumor formation in one to two weeks after inoculation, as determined by immunohistochemistry staining (Figure 6A). As shown in Figure 6B, the injection with NK‐92MI‐scFv or NK‐92MI‐mock after irradiation with 10Gy markedly inhibited the RPMI8226 tumor growth since 17 days after treatment compared with PBS control (p < 0.01, One‐Way Anova test). A significantly enhanced antitumor effects of irradiated NK‐92MI‐scFv on RPMI8226 tumor growth was evident since 21days after treatment, compared with irradiated NK‐92MI‐mock (i.e., tumor volume, 526.30 ± 164.12 mm3 vs 886.24 ± 234.23 mm3, p = 0.015, on day 21; 765.41 ± 67.15 mm3 vs 1126.03 ± 179.39 mm3, p = 0.001, on day 28, Student's t‐test). Correspondingly, the survival of mice treated with NK‐92MI‐scFv was significantly prolonged compared with NK‐92MI‐mock‐treated group (p < 0.001, log‐rank (Mantel Cox) Test, Figure 6D). Whereas, NK‐92MI‐scFv only exerted similar in vivo anti‐tumor efficacy as NK‐92MI‐mock in inhibition of ARH‐77 tumor (p = 0.093, log‐rank (Mantel Cox) Test, Figure 6C and E). We found that the NK‐92MI‐scFv cells were detectable in appreciable numbers in the MM tumor bed more than 20 days after adoptive transfer, as shown in Figure 6F. Furthermore, there were many apoptotic cells surrounding the labeled effector cells.
Figure 6.
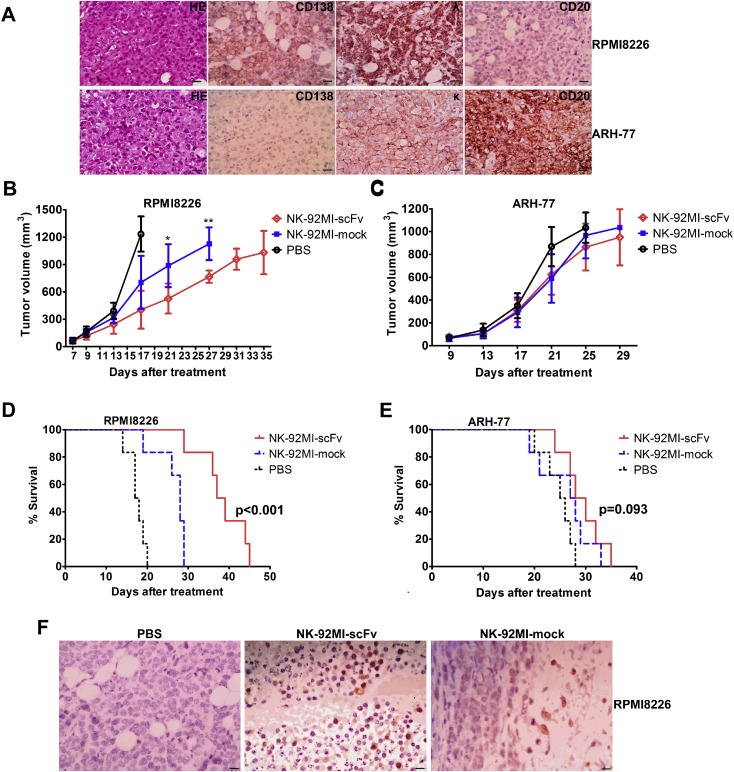
Improved anti‐MM efficacy induced by irradiated NK‐92MI‐scFv against CD138‐positive MM cell in xenograft model. Mice challenged subcutaneously with RPMI8226 or ARH‐77 cells (5 × 106). When palpable tumors (≥5 mm in diameter) developed, mice inoculated with RPMI8226 or ARH‐77 were randomly assigned into three treatment groups (6 per group) receiving intravenously injection of 4 × 107 10Gy –irradiated NK‐92MI‐scFv or NK‐92MI‐mock or the vehicle alone (PBS) via tail vein every other day for a total of three injections. (A) Representative immunohistochemistry slides of subcutaneous xenograft sections for RPMI8226‐ and ARH‐77‐challenged mice. Magnification: × 400. (Scale bar: 20 μm); Caliper measurements of the greatest length and width were performed on alternate days to estimate the tumor volume using the following formula: 4π/3 × (tumor width/2)2 × (tumor length/2). The tumor volume of mice challenged with RPMI8226 (B) or ARH‐77 (C) was expressed as mean ± SD. Mice were humanely sacrificed when subcutaneous tumors reached 15 mm in diameter. Survival was evaluated from the first day of treatment until death. Survival were shown for mice challenged with RPMI8226 (D) or ARH‐77 (E). (F) Human NK cells were detectable in appreciable numbers in the MM tumor bed of sacrificed mice 29 days after adoptive transfer. Immunohistochemical analysis of subcutaneous xenograft sections of RPMI8226‐ challenged mice 29 days after injection of NK‐92MI‐scFv or NK‐92MI‐mock. The NK cells were stained brown with rabbit anti‐human CD2 antibody using the LSAB + kit (DAKO, Carpinteria, CA, USA). Nuclei are stained with hematoxylin. Magnification: × 400. (Scale bar: 20 μm) *p < 0.05, **p < 0.01 compared with mice treated with NK‐92MI‐mock.
These results were in line with those from in vitro experiments, confirming that the intensified in vivo anti‐MM activity of NK‐92MI‐scFv is dependent on the binding of scFv (4B3)‐CD3ζ to CD138 antigen expressed by MM cells.
4. Discussion
NK cells are important effectors of innate immunity, and function as a first‐line defense in the control of malignancies (Long et al., 2013). However, many tumors escape this immune surveillance, possibly due to either the dysfunction of NK cells, or to inherent features of tumor cells that interfere their recognition by NK cells and the subsequent NK cell activation (Maki et al., 2008; Hasenkamp et al., 2006). NK cell activation is regulated by the combined effect of suppressive and stimulatory signals delivered through surface receptors (Hudspeth et al., 2013). Previous studies have showed that NK cell activity is reduced in MM, and MM cells may develop mechanisms to escape NK cell immunosurveillance by up‐regulating classical or nonclassical HLA class I molecule to inhibit NK cell function upon ligation with certain KIRs on NK cell surface (Maki et al., 2008). In addition, Costello et al. (2013) recently demonstrated that three major activating NK receptors (NCR3/NKp30, NKG2D and CD244/2B4/p38) were drastically down‐regulated at the site of tumor, i.e., BM in MM. Hence, enhancing the activating signaling of NK cells would be an optimal strategy to overcome the inhibitory signaling, which is probably adaptively up‐regulated in the setting of MM.
Transduction of NK cells with a CAR has been shown to facilitate the generation of antigen‐specific NK cells and directly trigger the intrinsic cytolytic effector functions through the signaling molecule CD3ζ linked to antigen recognition region (Boissel et al., 2012; Chang et al., 2013; Shimasaki and Campana, 2013). CD138 (syndecan‐1) is highly expressed on MM cells, making it an attractive immunotherapeutic target for MM (Lutz and Whiteman, 2009). Herein, the genetically modified NK‐92MI‐scFv cells harbors the scFv sequence specific for CD138 mAb 4B3 on membrane, which recognizes epitopes similar to the commercialized CD138 mAb BB4 as determined by competition assay (Sun et al., 2007). Western blot assay also showed that 4B3 could recognize the same molecular weight protein band of U266 cell lysis as BB4 mAb (Sun et al., 2007). It is clear that BB4 binds to a linear epitope of the core protein of CD138, and preferentially binds to membrane‐bound vs soluble CD138; In addition, BB4 strongly reacts with CD138 highly expressing MM cell line, but not with endothelial cells (Wijdenes et al., 2002). Considering the similarity in epitopes recognized by BB4 and 4B3, all the above‐mentioned characteristics of BB4 probably endow the constructed scFv (4B3) with binding sensitivity and specificity to membrane‐bound CD138 expressed by MM cells, while their off‐target effects, e.g., on CD138‐expressing endothelial cells, is likely minimal.
After antibiotic selection, we established a stably transfected NK‐92MI cell population that displays homogenous and high‐level surface expression of the CAR, which has not changed substantially during continuous propagation for more than one year. Most importantly, transduction of NK‐92MI with the lentiviral vector does not alter its intrinsic features, as indicated by the unchanged cytotoxocity of the resulting NK‐92MI‐scFv against K562 as compared to parental NK‐92MI.
The in vitro cytotoxicity of NK‐92 against a panel of MM cell lines has been demonstrated by chromium release assay or apoptotic analysis by FACS in separated studies (Swift et al., 2012; Maki et al., 2008). Despite the discrepancies in cytotoxicities of NK‐92 toward the same MM cell line between different groups or methods, the MM cell lines were mostly insensitive to the cytotoxic activity of NK‐92 compared with K562. Moreover, the patient‐derived primary MM cells were shown to be consistently resistant to NK‐92‐mediated killing with less than 15% cytotoxicity even at a high E:T ratio of 10:1 in a calcein assay (Maki et al., 2008). NK‐92MI has the same biologic characteristics as NK‐92. In our study, all the MM cell lines and primary MM cells demonstrated much lower sensitivity to NK‐92MI‐mediated cytotoxic activity, with a mean cytotoxicity of not higher than 50% even at E:T ratio of 10:1 except for U266, in contrast to the high sensitivity of K562 which exceeds 60% at a low E:T ratio of 1:1. In contrast, the transduced NK‐92MI‐scFv exerted significantly increased cytotoxicity toward all the CD138‐positive MM cell lines and primary MM cells tested so far, as compared to NK‐92MI‐mock. Even at a low E/T ratio of 1:1, the anti‐MM activity of NK‐92MI‐scFv was also markedly elevated in the majority (4/5) of primary MM cells. Whereas, the CD138‐negative target ARH‐77 remained insensitive to NK‐92MI‐scFv. Furthermore, the enhanced cytotoxicity of NK‐92MI‐scFv was significantly inhibited if interaction of the CAR with CD138 antigen was blocked by the competitive mAb BB4, further confirming the specific recognition of NK‐92MI‐scFv toward CD138 antigen expressed by MM cells, and the increased cytotoxicity of NK‐92MI‐scFv was dependent on the binding of CAR with CD138 antigen.
NK cell killing occurs predominantly via the perforin‐granzyme pathway and the Fas‐L pathway, as well as secretion of cytokines and chemokine, such as IFN‐γ (Finton and Strong, 2012). In this study, in response to the same CD138‐positive target cell, NK‐92MI‐scFv produced much more IFN‐γ and granzyme B when compared with NK‐92MI‐mock. Whereas, the Fas‐L expression of NK‐92MI‐scFv was comparable to that of NK‐92MI‐mock, which both elevated to a similar extent in response to the same target cell. These results suggested that the enhanced cytolytic activity of NK‐92MI‐scFv was mainly mediated by perforin‐granzyme pathway and IFN‐γ, but not the Fas‐L pathway.
CD107a is emerging as a sensitive marker of NK cell degranulation, which correlates strongly with both cytokine secretion and target cell lysis. Furthermore, CD107a proves to represent a more comprehensive marker of NK activity as it is expressed on a larger proportion of functionally active cells that are not detected using simple assessment of cytokine secretion (Schönberg et al., 2012). Herein, in accord with the increased secretion of IFN‐γ and granzyme B by NK‐92MI‐scFv, these CAR‐expressing NK‐92MI cells had a significantly higher proportion of CD107a expression than NK‐92MI‐mock upon activation by the same target cell even at a low E:T ratio, strongly indicating that the CD138 specific CAR upgraded NK cell activation in response to a CD138‐expressing target.
Finally, NK‐92MI cells were originally established from large granular lymphoma cells, and adoptive transfer of irradiated NK‐92MI cells would ensure the safety of such an immunotherapeutic strategy in immunocompromised hosts. Irradiation of NK‐92 cells with 5–10 Gy has been shown to block further cell division, but exerted negligible effects on the NK cell cytotoxicity (Uherek et al., 2002). Our results confirmed that transfected NK‐92MI cells after irradiation with 10 Gy could retain their substantial ex vivo cytotoxicity. More importantly, the in vivo anti‐MM activities of irradiated NK‐92MI‐scFv were demonstrated in xenograft MM mouse model in the present study. The specific anti‐MM effects of irradiated NK‐92MI‐scFv were observed when these cells were intravenously administered following the formation of subcutaneous xenograft, which markedly delayed tumor growth and prolonged the survival in mice inoculated with CD138 positive target cells compared with irradiated NK‐92MI‐mock.
Nowadays, CD138 mAb‐based therapies represent a promising strategy for the treatment of MM. For example, anti‐CD138 antibody‐maytansinoid conjugates have been shown to exert potent and selective cytotoxicity against CD138‐positive MM cells (Ikeda et al., 2009; Rousseau et al., 2012). NK‐92 is the first NK cell line to be tested in several clinical trials, and its safety for clinical application has been validated. There were at least two earlier studies demonstrating that repeated adoptive transfer of NK‐92 cells did neither result in permanent engraftment in immunocompromised SCID or NK cell–deficient pfp‐Rag‐2 mice, nor induce any organ toxicity in spleen, kidney, lung, bone marrow or brain (Tam et al., 1999a; Yan et al., 1998). Irradiation might also be adopted to prevent in vivo proliferation of genetically modified NK‐92‐MI cells. Irradiation of NK‐92 cells with 5–10 Gy was shown to efficiently block further cell division, but only exert trivial effect on cytotoxicity (Tam et al., 1999b). The tolerability of irradiated NK‐92 cell transfusion was also confirmed by the earlier phase I clinical trial. Herein, we redirected the NK‐92 derivative NK‐92MI to CD138‐positive malignant cells by a CD138‐specific CAR fused to intrinsic signaling molecule, thus upgrading the intrinsic cytotoxic potential of the effector cells and their capacity to extravasate and home to tumor sites. Our work provides the rationale and feasibility for adoptive immunotherapy with CD138‐specific CAR‐modified NK cells in CD138‐expressing plasma cell malignancies. It is certain that this strategy could also be applied to modify the expanded allogeneic or autologous NK cells by electroporation to guarantee the safety of clinical application. Altogether, we offer a new approach to enhance the anti‐MM efficacy of NK cell therapy and to widen its application.
Disclosure
There is no potential conflict of interest to disclose.
Supporting information
The following is the supplementary data related to this article.
Supplementary data
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.12.001
Jiang Hua, Zhang Wenhao, Shang Peipei, Zhang Hui, Fu Weijun, Ye Fei, Zeng Tianmei, Huang Hejing, Zhang Xueguang, Sun Wanping, Man-Yuen Sze Daniel, Yi Qing and Hou Jian, (2014), Transfection of chimeric anti‐CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.12.001.
This study was supported by funds from National Natural Science Foundation of China (NSFC, grant number: 30800489).
References
- Alter, G. , Malenfant, J.M. , Altfeld, M. , 2004. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods. 294, 15–22. [DOI] [PubMed] [Google Scholar]
- André, P. , Castriconi, R. , Espéli, M. , Anfossi, N. , Juarez, T. , Hue, S. , 2004. Comparative analysis of human NK cell activation induced by NKG2D and natural cytotoxicity receptors. Eur. J. Immunol.. 34, 961–971. [DOI] [PubMed] [Google Scholar]
- Arai, S. , Meagher, R. , Swearingen, M. , Myint, H. , Rich, E. , Martinson, J. , 2008. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 10, 625–632. [DOI] [PubMed] [Google Scholar]
- Bataille, R. , Jégo, G. , Robillard, N. , Barillé-Nion, S. , Harousseau, J.L. , Moreau, P. , 2006. The phenotype of normal, reactive and malignant plasma cells. Identification of “many and multiple myelomas” and of new targets for myeloma therapy. Haematologica. 91, 1234–1240. [PubMed] [Google Scholar]
- Baumann, P. , Schneider, L. , Mandl-Weber, S. , Oduncu, F. , Schmidmaier, R. , 2012. Simultaneous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in multiple myeloma. Anticancer Drugs. 23, 131–138. [DOI] [PubMed] [Google Scholar]
- Boissel, L. , Betancur, M. , Lu, W. , Wels, W.S. , Marino, T. , Van Etten, R.A. , 2012. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk. Lymphoma. 53, 958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissel, L. , Betancur, M. , Wels, W.S. , Tuncer, H. , Klingemann, H. , 2009. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res.. 33, 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y.H. , Connolly, J. , Shimasaki, N. , Mimura, K. , Kono, K. , Campana, D. , 2013. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res.. 73, 1777–1786. [DOI] [PubMed] [Google Scholar]
- Costello, R.T. , Boehrer, A. , Sanchez, C. , Mercier, D. , Baier, C. , Le Treut, T. , 2013. Differential expression of natural killer cell activating receptors in blood versus bone marrow in patients with monoclonal gammopathy. Immunology. 139, 338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis, D. , Foley, B. , Witt, C.S. , Christiansen, F.T. , 2012. The detection of NK cell alloreactivity by flow cytometric CD107a assay. Methods Mol. Biol.. 882, 477–489. [DOI] [PubMed] [Google Scholar]
- Dhodapkar, M.V. , Abe, E. , Theus, A. , Lacy, M. , Langford, J.K. , Barlogie, B. , 1998. Syndecan-1 is a multifunctional regulator of myeloma pathobiology: control of tumor cell survival, growth, and bone cell differentiation. Blood. 91, 2679–2688. [PubMed] [Google Scholar]
- Favors, S.E. , Curd, L.M. , Gregg, R.K. , 2012. Use of the anti-inflammatory cytokine interleukin-11 to reverse HIV-1gp120 repression of a natural killer cell line. Cell. Immunol.. 276, 1–5. [DOI] [PubMed] [Google Scholar]
- Finton, K.A. , Strong, R.K. , 2012. Structural insights into activation of antiviral NK cell responses. Immunol. Rev.. 250, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost, P. , Moatamed, F. , Hoang, B. , Shi, Y. , Gera, J. , Yan, H. , 2004. In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood. 104, 4181–4187. [DOI] [PubMed] [Google Scholar]
- Glässner, A. , Eisenhardt, M. , Krämer, B. , Körner, C. , Coenen, M. , Sauerbruch, T. , 2012. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Invest.. 92, 967–977. [DOI] [PubMed] [Google Scholar]
- Hart, A.J. , Jagasia, M.H. , Kim, A.S. , Mosse, C.A. , Savani, B.N. , Kassim, A. , 2012. Minimal residual disease in myeloma: are we there yet?. Biol. Blood Marrow. Transpl.. 18, 1790–1799. [DOI] [PubMed] [Google Scholar]
- Hasenkamp, J. , Borgerding, A. , Wulf, G. , Uhrberg, M. , Jung, W. , Dingeldein, S. , 2006. Resistance against natural killer cell cytotoxicity: analysis of mechanisms. Scand. J. Immunol.. 64, 444–449. [DOI] [PubMed] [Google Scholar]
- Heinz, E. , Rockey, D.D. , Montanaro, J. , Aistleitner, K. , Wagner, M. , Horn, M. , 2010. Inclusion membrane proteins of protochlamydia amoebophila UWE25 reveal a conserved mechanism for host cell interaction among the chlamydiae. J. Bacteriol.. 192, 5093–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover, R.G. , Gullickson, G. , Kornbluth, J. , 2009. Impaired NK cytolytic activity and enhanced tumor growth in NK lytic-associated molecule-deficient mice. J. Immunol.. 183, 6913–6921. [DOI] [PubMed] [Google Scholar]
- Hudspeth, K. , Silva-Santos, B. , Mavilio, D. , 2013. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front. Immunol.. 4, 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, H. , Hideshima, T. , Fulciniti, M. , Lutz, R.J. , Yasui, H. , Okawa, Y. , 2009. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res.. 15, 4028–4037. [DOI] [PubMed] [Google Scholar]
- Imai, C. , Iwamoto, S. , Campana, D. , 2005. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 106, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury, J.D. , Medeiros, L.J. , Rassidakis, G.Z. , McDonnell, T.J. , Abruzzo, L.V. , Lai, R. , 2003. Expression of MCL-1 in mantle cell lymphoma is associated with high-grade morphology, a high proliferative state, and p53 overexpression. J. Pathol.. 199, 90–97. [DOI] [PubMed] [Google Scholar]
- Long, E.O. , Kim, H.S. , Liu, D. , Peterson, M.E. , Rajagopalan, S. , 2013. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol.. 31, 227–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugini, L. , Cecchetti, S. , Huber, V. , Luciani, F. , Macchia, G. , Spadaro, F. , 2012. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol.. 189, 2833–2842. [DOI] [PubMed] [Google Scholar]
- Lutz, R.J. , Whiteman, K.R. , 2009. Antibody-maytansinoid conjugates for the treatment of myeloma. MAbs. 1, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki, G. , Hayes, G.M. , Naji, A. , Tyler, T. , Carosella, E.D. , Rouas-Freiss, N. , 2008. NK resistance of tumor cells from multiple myeloma and chronic lymphocytic leukemia patients: implication of HLA-G. Leukemia. 22, 998–1006. [DOI] [PubMed] [Google Scholar]
- Maruyama, I.N. , Rakow, T.L. , Maruyama, H.I. , 1995. cRACE: a simple method for identification of the 5' end of mRNAs. Nucleic Acid. Res.. 23, 3796–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel, J.M. , Pinilla-Ibarz, J. , Epling-Burnette, P.K. , 2012. Molecular action of lenalidomide in lymphocytes and hematologic malignancies. Adv. Hematol.. 2012, 513702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morett, A. , Bottino, C. , Vitale, M. , Pende, D. , Cantoni, C. , Mingari, M.C. , 2001. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol.. 19, 197–223. [DOI] [PubMed] [Google Scholar]
- Olson, J.A. , Zeiser, R. , Beilhack, A. , Goldman, J.J. , Negrin, R.S. , 2009. Tissue-specific homing and expansion of donor NK cells in allogeneic bone marrow transplantation. J. Immunol.. 183, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, C. , Ratanatharathorn, V. , Adams, P. , Braun, T. , Silver, S. , Ayash, L. , 2001. Allogeneic stem cell transplantation reduces disease progression compared to autologous transplantation in patients with multiple myeloma. Bone. Marrow Transpl.. 27, 801–807. [DOI] [PubMed] [Google Scholar]
- Rousseau, C. , Ferrer, L. , Supiot, S. , Bardiès, M. , Davodeau, F. , Faivre-Chauvet, A. , 2012. Dosimetry results suggest feasibility of radioimmunotherapy using anti-CD138 (B-B4) antibody in multiple myeloma patients. Tumour. Biol.. 33, 679–688. [DOI] [PubMed] [Google Scholar]
- Savan, R. , Chan, T. , Young, H.A. , 2010. Lentiviral gene transduction in human and mouse NK cell lines. Methods Mol. Biol.. 612, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönberg, K. , Hejazi, M. , Uhrberg, M. , 2012. Protocol for the clonal analysis of NK cell effector functions by multi-parameter flow cytometry. Methods Mol. Biol.. 903, 381–392. [DOI] [PubMed] [Google Scholar]
- Shimasaki, N. , Campana, D. , 2013. Natural killer cell reprogramming with chimeric immune receptors. Methods Mol. Biol.. 969, 203–220. [DOI] [PubMed] [Google Scholar]
- Sun, W.P. , Wang, F.M. , Xie, F. , Wang, G.Q. , Sun, J. , Yu, G.H. , 2007. A novel anti-human syndecan-1 (CD138) monoclonal antibody 4B3: characterization and application. Cell. Mol. Immunol.. 4, 209–214. [PubMed] [Google Scholar]
- Swift, B.E. , Williams, B.A. , Kosaka, Y. , Wang, X.H. , Medin, J.A. , Viswanathan, S. , 2012. Natural killer cell lines preferentially kill clonogenic multiple myeloma cells and decrease myeloma engraftment in a bioluminescent xenograft mouse model. Haematologica. 97, 1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam, Y.K. , Maki, G. , Miyagawa, B. , Hennemann, B. , Tonn, T. , Klingemann, H.G. , 1999. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum. Gene Ther.. 10, 1359–1373. [DOI] [PubMed] [Google Scholar]
- Tam, Y.K. , Miyagawa, B. , Ho, V.C. , Klingemann, H.G. , 1999. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J. Hematother.. 8, 281–290. [DOI] [PubMed] [Google Scholar]
- Tassev, D.V. , Cheng, M. , Cheung, N.K. , 2012. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther.. 19, 84–100. [DOI] [PubMed] [Google Scholar]
- Tonn, T. , Becker, S. , Esser, R. , Schwabe, D. , Seifried, E. , 2001. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematother. Stem null. Res.. 10, 535–544. [DOI] [PubMed] [Google Scholar]
- Uherek, C. , Tonn, T. , Uherek, B. , Becker, S. , Schnierle, B. , Klingemann, H.G. , 2002. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 100, 1265–1273. [PubMed] [Google Scholar]
- Vivier, E. , Raulet, D.H. , Moretta, A. , Caligiuri, M.A. , Zitvogel, L. , Lanier, L.L. , 2011. Innate or adaptive immunity? The example of natural killer cells. Science. 331, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijdenes, J. , Dore, J.M. , Clement, C. , Vermot-Desroches, C. , 2002. CD138. J. Biol. Regul. Homeostatic. Agents. 16, 152–155. [PubMed] [Google Scholar]
- Yan, Y. , Steinherz, P. , Klingemann, H.G. , Dennig, D. , Childs, B.H. , McGuirk, J. , 1998. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res.. 4, 2859–2868. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article.
Supplementary data