

Abstract
The human carcinoembryonic antigen (CEA) is a cell adhesion molecule involved in both homotypic and heterotypic interactions. The aberrant overexpression of CEA on adenocarcinoma cells correlates with their increased metastatic potential. Yet, the mechanism(s) by which its adhesive properties can lead to the implantation of circulating tumor cells and expansion of metastatic foci remains to be established. In this study, we demonstrate that the IgV‐like N terminal domain of CEA directly participates in the implantation of cancer cells through its homotypic and heterotypic binding properties. Specifically, we determined that the recombinant N terminal domain of CEA directly binds to fibronectin (Fn) with a dissociation constant in the nanomolar range (KD 16 ± 3 nM) and interacts with itself (KD 100 ± 17 nM) and more tightly to the IgC‐like A3 domain (KD 18 ± 3 nM). Disruption of these molecular associations through the addition of antibodies specific to the CEA N or A3B3 domains, or by adding soluble recombinant forms of the CEA N, A3 or A3B3 domains or a peptide corresponding to residues 108–115 of CEA resulted in the inhibition of CEA‐mediated intercellular aggregation and adherence events in vitro. Finally, pretreating CEA‐expressing murine colonic carcinoma cells (MC38.CEA) with rCEA N, A3 or A3B3 modules blocked their implantation and the establishment of tumor foci in vivo. Together, these results suggest a new mechanistic insight into how the CEA IgV‐like N domain participates in cellular events that can have a macroscopic impact in terms of cancer progression and metastasis.
Keywords: Metastasis, Tumor implantation, Carcinoembryonic antigen, Cellular adherence, Homotypic binding, Heterotypic association
Highlights
Deregulated CEA overexpression correlates with increased tumor metastasis.
The mechanism of CEA‐mediated engraftment of metastatic cells remains unknown.
We show that CEA participates in 2 classes of associations, via its N domain.
These associations promote cellular adherence to either Fn or immobilized CEA.
Inhibiting these interactions attenuates the engraftment of tumor cells in vivo.
1. Introduction
Metastatic cancers remain the primary cause of morbidity and mortality for cancer patients despite significant advances in the treatment of localized tumors (Psaila and Lyden, 2009). The prevention of metastasis combines early surgery (or radiation therapy in some cases) as well as systemic therapy given before or after surgery to target disseminated tumor cells that have detached from primary tumors to engraft at distal sites, and/or were not detected or accessible to surgical excision at the time of diagnosis (Psaila and Lyden, 2009; Klein, 2009). Tumor metastasis is a complex phenomenon where intercellular and cell‐matrix adhesion events involving tumor cells lead to the formation as well as expansion of metastatic foci (Orava et al., 2013; Psaila and Lyden, 2009; Klein, 2009; Fidler, 2003; Desgrosellier and Cheresh, 2010).
The carcinoembryonic antigen (CEA) is an important pro‐metastatic oncoprotein. First reported by Gold and Freedman in 1965, CEA is a 180‐kDa GPI anchored, glycoprotein that is aberrantly over‐expressed by epithelial cancers including cancers of the gastrointestinal tract, breast, lung, ovary and pancreas (Camacho‐Leal and Stanners, 2008; Samara et al., 2007; Benchimol et al., 1989). Structurally, CEA is an immunoglobulin‐like surface protein composed of 651 amino acids divided into seven distinct domains: an IgV‐like N domain, and six IgC‐like regions (A1, B1, A2, B2, A3 and B3) (Benchimol et al., 1989; Zhou et al., 1993; Taheri et al., 2000; Berinstein, 2002).
Originally described as an Ig‐like cell adhesion molecule, CEA expression plays a dual role in tissue remodeling as well as in host defense, under normal physiological conditions (Benchimol et al., 1989; Beauchemin and Arabzadeh, 2013; Kuespert et al., 2006; Hammarström, 1999; Ilantzis et al., 2002). However, the deregulated over‐expression of CEA by cancer cells promotes two forms of interactions: inhibitory interactions with other receptors on the cell surface that promote neogenesis and protection from anoikis, as well as interactions conducive to the implantation of circulating tumor cells (Zimmer and Thomas, 2001; Beauchemin and Arabzadeh, 2013; Li et al., 2010; Hostetter et al., 1990; Ordoñez et al., 2000; Samara et al., 2007). For instance, increased CEA expression on the surface of cancer cells leads to the de‐regulation of TGF‐β signaling, as a result of its direct interaction with TGF‐β receptor 1 (TGF‐β R1), thus promoting uncontrolled proliferation and invasion (Li et al., 2010). Additionally, CEA expression correlates with the inhibition of cell death induced by anoikis, the early inactivation of caspase‐9 and activation of the PI3‐K/Akt survival pathway as well as the inactivation of caspase‐8 (Camacho‐Leal and Stanners, 2008) presumably by directly binding TRAIL‐R2 (DR5; Ordoñez et al., 2000; Samara et al., 2007). Interestingly, both these interactions are mediated via the PELPK peptide motif (residues 108–112 found in the N domain of CEA) (Ordoñez et al., 2000; Samara et al., 2007).
A second class of interactions mediated by CEA favors cellular adherence events as a result of a combination of self‐associations (homotypic interactions) that promote intercellular aggregation and adhesion (Benchimol et al., 1989; Taheri et al., 2000; Samara et al., 2007; Zhou et al., 1993) as well as heterophilic interactions involving hnRNP M expressed by Kupffer cells, or α5β1 integrin which in turn leads to its association with fibronectin (Fn) (Thomas et al., 2011; Ordoñez et al., 2007; Camacho‐Leal et al., 2007).
Defining the exact mechanisms by which CEA mediates the engraftment of circulating tumor cells, and the subsequent expansion of metastatic tumor foci, represents a pre‐requisite for devising improved treatment strategies against metastatic cancers, aimed at blocking the adhesive properties of CEA. Specifically, we have shown that raising aptamers or mounting an antibody‐mediated immune response focused to the N domain of CEA block CEA‐dependent tumor implantation in vivo (Abdul‐Wahid et al., 2012; Orava et al., 2013). In this study, we tested the hypothesis that CEA directly participates in the implantation of cancer cells. We mapped the CEA domains responsible for its homotypic cellular adherence and its interaction with the ECM protein fibronectin (Fn). We report that the CEA IgV‐like N domain serves a key role in the formation of at least two classes of binding events leading to cellular engraftment and tumor foci formation. The first binding event involves the direct association of CEA with Fn, independently of the presence of human α5β1 integrin. The second binding event involves the formation of both cis‐ [N‐to‐N] and trans‐ [N‐to‐A3] homotypic complexes. Specific targeting of these interactions resulted in the blockage of tumor cell implantation both in vitro and in vivo.
2. Materials and methods
2.1. Antibodies, cells and culture conditions
CEA expression was detected using the mouse anti‐human CEA mAb Col1 (Life Technologies). Mouse anti‐GAPDH (Calbiochem; Billerica, MA) and rabbit anti‐α5 integrin (Cell Signaling Technology; Danvers, MA) were used to compare the endogenous expression levels of α5 integrin on MC38 and MC38.CEA cells. Rabbit anti‐CEA pAb H‐300 was purchased from Santa Cruz Biotechnology (Dallas, TX). Mouse anti‐CEA N and A3B3 domains antibodies were generated as previously reported (Abdul‐Wahid et al., 2012). Briefly, endotoxin contamination was removed from rCEA N or A3B3 domain preparations by passing solutions of the purified proteins through Detoxigel columns (Pierce, Thermo Scientific, Ontario, Canada). The detoxified N or A3B3 modules (100 μg) were then mixed with 100 μg poly I:C and injected (i.p.) into C57/Bl mice on days 1, 3 and 10. Animals were bled on day 15 and their sera collected, pooled and analyzed for the presence of CEA N‐ or A3B3‐specific antibodies.
Supplementary Table 1 lists the different cell lines used in this study, as well as the amount of CEA expressed by these cells. MC38 and MC38.CEA were kindly provided by Dr. J. Schlom (National Cancer Institute; Bethesda, Maryland). All cells were cultured at 37 °C in a humidified 5.0% CO2 atmosphere in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin (100 U/mL), and dihydrostreptomycin (100 μg/mL).
2.2. Generation of recombinant CEA modules
The rCEA modules generated for this study and their corresponding sequence numbering within CEA are listed in Figure 1. The full length tumor glycoform of CEA was a kind gift from Dr. J. Shively (City of Hope Irell & Manella Graduate School of Biological Sciences, Duarte, CA). The gene segment encoding CEA A1B1 domain was amplified using the CEA‐AB1 forward primer: (5′‐GAG CTG CCC AAG CCC CAT ATG TCC AGC AAC AAC TCC AAA CCC GTG GAG GAC AAG GAT GCT‐3′), and the CEA‐AB1 reverse primer (5″‐ATC CTC GGA TCC CTC GAG CTA ATG GTG ATG ATG ATG GTG ATG ATG TTT GGG TGG CTC TGC ATA GAC TGT GAT CGT CGT‐3″). The rCEA A2B2 domain was amplified using the CEA‐AB2 forward primer (5′‐AAC CCC GTG GAG CAT ATG GAT GCT GTA GCC‐3′) and the CEA‐AB2 reverse primer (5″‐TGT CCT CCA CGG GCT CGA GCT AAT GAT GAT GAT GAT GGT GAT GGT GCA GCT CCG CAG AGA‐3″). The rCEA A3 domain was amplified using the CEA‐A3 forward primer (5′‐TCCAACATC CAT ATG CAT CAT CAC CAT CAC CAT CAT CAT GAA AAC CTC TAT TTC CAA TCA GCC AGT GGC CAC AGC AGG ACT ACA GTC AAG‐3′) and the CEA‐A3 reverse primer (5″‐CGA GTG GCA GGA GAG GGA TCC CTC GAG CTA CGA AAG GTA AGA CGA GTC TGG GGG GGA AAT GAT GGG GGT GTC CGG CCC ATA‐3″). The above modules, in conjunction with rCEA N, FLAG‐tagged rCEA N and A3B3 domains were cloned between the NdeI and XhoI sites of pET30b (Novagen; Darmstadt, Germany), expressed in Escherichia coli BL21 (DE3) Star (Invitrogen; Oakville, Ontario) as poly histidine‐tagged proteins and purified as previously reported (Abdul‐Wahid et al., 2012). IgC‐like AB modules were refolded using the method of Michaux et al. (2008) where proteins were extensively dialyzed against a HEPES‐buffered (20 mM, pH7) solution supplemented with 5 mM β‐mercaptoethanol and 1 M 2‐methyl‐2,4‐pentanediol (MPD; Fisher Sci). Purified rCEA N, FLAG N, or A3 domain modules were concentrated by ultrafiltration and refolded by dilution in a buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl and 10 mM β‐mercaptoethanol.
Figure 1.
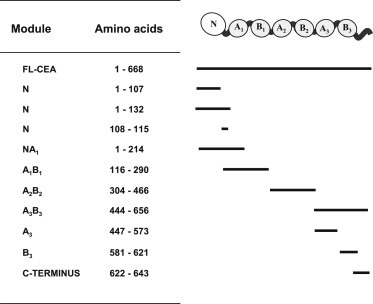
Recombinant and synthetic CEA modules used in this study.
Removal of the polyhistidine tag from the rCEA N or A3 domains was achieved using His‐tagged recombinant Tobacco Etch Virus (TEV) protease. The suspension containing digested as well as undigested and rTEV was mixed with ten volumes solubilization buffer (50 mM Tris (pH8), 8 M urea, 250 mM NaCl, and 10 mM β‐mercaptoethanol) and then subjected to affinity chromatography using Ni‐NTA columns. Untagged rCEA modules were collected in the flow through fraction and refolded as described above. The extent of cleavage and the purity of the final recombinant products were confirmed by SDS PAGE.
2.3. Peptide synthesis
Synthetic peptides corresponding to the IgC‐like B3 domain (CEA residues 581–621), the C‐terminal region of CEA (residues 622–643), the predicted Fn‐binding domain of CEA (residues 108–115; CEA N108–115), the Fn‐binding thrombospondin peptide (FnBP; GGWSHWS) and the human ribosomal stalk protein RPLP0 (P0; MGFGLFD) were assembled by solid phase synthesis on a PS3 Peptide Synthesizer (Protein Technologies Inc.; Tucson, AZ), using Wang resins and 9‐fluorenylmethoxycarbonyl (Fmoc) protected amino acids (Peptides International, Inc.; Louisville, KY). Fmoc‐protecting groups were removed in the presence of 5% piperazine/0.1 M HOBt in DMF while coupling reactions were activated with DIPEA (N, N‐diisopropylethylamine; Sigma–Aldrich), except for cysteine couplings where DIPEA was replaced with 2,4,6‐collidine (Sigma–Aldrich). Biotinylation of all synthetic peptides was performed directly on solid phase using a solution of Biotin/DIC (N,N′‐Diisopropylcarbodiimide)/HOBt in DMF for 3 h. Peptides were cleaved from their solid support using 82.5% TFA : 5% phenol : 5% H2O : 5% thioanisole : 2.5% EDT (Reagent K) for 2–4 h at RT and purified to homogeneity by RP‐HPLC (Waters; Milford, MA) on a C18 semi‐preparative (Phenomenex; Torrance, CA) column. Solvent system: S1, 5% acetonitrile +0.1% TFA; S2, 100% acetonitrile +0.1% TFA, linear gradient from 0% to 100% of S2 in 20 min, flow rate 7 mL/min. Peptide sequences were confirmed by MALDI‐TOF.
2.4. Yeast 2‐hybrid analysis of CEA homotypic interactions
Yeast 2‐hybrid analyses were performed as previously described (McCluskey et al., 2008). Briefly, the rCEA N‐domain was subcloned between the NdeI and BamHI sites of pGBKT7 (Clontech, Mountain View, CA), in frame with the GAL4 DNA‐binding domain, to create the bait vector. The prey modules were generated by subcloning the rCEA N, A1B1, A2B2, A3B3, or the human RPLP2 between the NdeI and XhoI sites of pGADT7(Clontech) in frame with the GAL4 activation domain. Bait and prey plasmids were transformed into Saccharomyces cerevisiae and bait‐prey proteins interactions detected by plating colony dilutions on selective growth media (McCluskey et al., 2008).
2.5. Enzyme linked immunosorbent assay‐based analysis of protein interactions
Protein interactions involving CEA domains were assessed using a modification of a previously published ELISA‐based binding assay (Abdul‐Wahid et al., 2012). Briefly, 96‐well flat‐bottomed Falcon microtiter plates (Becton–Dickinson Biosciences; Franklin Lakes, NJ) were coated with 1 μg of purified CEA modules per well in 100 μl of sodium carbonate (pH 9.5) for 24 h at 37 °C. Unbound proteins were removed using three washes with PBS‐0.05% Tween 20 (PBST), and wells were blocked with 200 μl of 1% BSA in PBS for 60 min at room temperature. Plates were then washed three times with PBST and incubated for 1 h at room temperature with the FLAG‐tagged rCEA N module (1 μg diluted in 100 μl of PBST) followed by three washes with 200 μl of PBST. Mapping of the A3B3 segment responsible for reciprocal homotypic binding was defined by competition ELISA examining the prevention of binding of FLAG‐tagged rCEA N and immobilized rCEA A3B3 modules by the addition of 1 μM of CEA A3, B3 or C‐terminal CEA modules. Bound FLAG‐tagged rCEA modules were detected using horseradish peroxidase (HRP) coupled anti‐FLAG M2 monoclonal antibody (mAb FLAG M2; Sigma–Aldrich) diluted to 1:5000 in PBST. 3,3′,5,5′‐tetramethylbenzidine (TMB; Sigma) was used as the HRP substrate and absorbance values were recorded at 450 nm.
For determining the binding of CEA to Fn, multi‐well plates were coated with 1 μg of Fn and incubated with His‐tagged rCEA N1–107 (kindly provided by Dr. S. Gray‐Owen; University of Toronto), rCEA N1–132, NA1, A1B1 or rhTNF‐α (which served as a control His‐tagged protein). The presence of bound His‐tagged proteins was detected using HRP‐coupled mAb His‐1 (1: 5000 dilution; Sigma–Aldrich).
Equilibrium dissociation constants for the CEA N domain interaction with itself, the A3 domain and with Fn were derived by ELISA as previously described (Sato et al., 2009; Smith et al., 2013; Jardim et al., 2000). Briefly, increasing concentrations of FLAG‐tagged rCEA N were added to multi‐well plates coated with 1 μg of either Fn, rCEA N or rCEA A3 domains. Plates were developed as described above and the obtained optical densities (450 nm) were used to derive the amount of bound protein from a standard curve constructed using concentrations of purified recombinant FLAG‐tagged rCEA N domain. The derivation of the amounts of bound FLAG‐tagged rCEA N from the recorded optical densities was done by applying a four parameter logistics equation: y = min + (max − min)/(1 + (x/EC50))ˆ(−Hill slope); where y represents the recorded optical density and x represents the amount of FLAG‐tagged rCEA N domain in each well. Binding curves of specifically‐bound protein ligand as a function of total concentration of FLAG‐tagged rCEA N domain and their related Scatchard plots were then constructed and used to derive the dissociation equilibrium constants for the interactions between FLAG‐tagged rCEA N and either rCEA N, A3 domains or Fn. The dissociation equilibrium constant values were calculated with PRISM software (version 5.01; Graph Pad Software for Science, San Diego, CA) using a nonlinear fit of the binding curve and analyses on the basis of a single site‐specific binding model. All binding experiments were repeated three times with each data set done in quadruplicates.
2.6. Pull down of CEA homotypic complexes
Ni‐NTA magnetic beads (Promega; Madison, Wisconsin) were saturated with 100 μg His‐tagged rCEA or control protein modules for 2 h at 37 °C. Unbound proteins were washed away and the coated beads were suspended in binding buffer (20 mM HEPES (pH7), 150 mM NaCl, 10 mM β‐mercaptoethanol) in the presence of untagged rCEA N‐domain (1 μM) and incubated for 2.5 h at room temperature with gentle shaking. The beads were then extensively washed with wash buffer (20 mM HEPES (pH7), 150 mM NaCl, 10 mM β‐mercaptoethanol, 1% Triton X‐100) to remove unbound rCEA N‐domain modules. The bound untagged CEA N‐domain modules were released from immobilized CEA N‐domain on Ni‐NTA beads by adding 8 M urea. The samples were resolved by SDS‐PAGE and the presence of released untagged N‐domain was visualized by Coomassie R‐250 staining.
For validating the direct interaction between Fn and the heptapeptide CEA N108–115, 5 μg of Fn were incubated with biotinylated heptapeptides (50 μg) with shaking for 2 h at room temperature. Streptavidin magnetic beads (Life Technologies; Oakville, Canada) were added to the biotin‐peptide/Fn mixtures, and the presence of peptide‐retrieved Fn was detected by western blotting, using rabbit anti‐human Fn pAb (1:1000; Sigma–Aldrich). The 7 amino acids Fn‐binding thrombospondin peptide (FnBP; GGWSHWS) was used as a positive control, whereas the human ribosomal stalk protein RPLP0 (P0; MGFGLFD) was used as an irrelevant peptide.
2.7. Disruption of CEA‐mediated intercellular aggregation
Disruption of CEA‐mediated intercellular aggregation by the addition of rCEA modules was assessed using a modification of the method described by Zhou et al. (1993). Briefly, 2.0 × 106 cells were suspended in the presence of 1 μM rCEA modules in a 2 mL volume of cell adhesion medium composed of DMEM supplemented with 1 mM EDTA and 1% FBS. The cell‐containing suspension was incubated a 37 °C with gentle shaking and the degree of disaggregation was determined by counting the number of single cells after 120 min. Experiments were performed four times and the percentage of single cells values reported represents the average (±SEM) of the ratio between single and total (single + aggregated) cells (Zhou et al., 1993).
2.8. Analysis of CEA‐dependent cell adhesion
The adherence of MC38.CEA cells to rCEA domain‐coated surfaces was measured in real‐time using an xCELLigence RTCA DP label‐free, impedance‐based cell sensing device (ACEA Biosciences; San Diego, CA). Cellular adhesion was monitored using MC38.CEA cells and parental CEA‐negative MC38 cells (2.5 × 104 cells per well) grown in complete medium as described above. Cell suspensions were dispensed into E‐plates where the bottom of each well harbored a gold film sensor. Wells were coated with 1 μg of a control protein (BSA), Fn, rCEA N, A3 or A3B3 modules. The adherence of cells to immobilized protein domains was monitored every minute over a period of 6 h. The inhibition of CEA‐dependent cellular adhesion by the addition of either rCEA modules or CEA‐specific pAbs was monitored by measuring the loss of cell adhesion. Briefly, MC38.CEA (2.5 × 104 cells) were pre‐treated with either rCEA modules (1 μM), anti‐CEA N‐domain or anti‐CEA A3B3 sera (1:100 final dilution) and then added to E‐plates coated with rCEA N domain (1 μg) N domain. The adhesion of MC38.CEA cells in the presence of control protein (BSA; 1 μM) or in the absence of either rCEA modules or sera served as controls. For the experiments examining the modulation of cellular adhesion to Fn‐coated surfaces, E‐plates were first coated with Fn (1 μg per well) and then incubated with 1 μM (in 100 μL) of either rCEA N, A3B3, BSA for 30 min at 37 °C, followed by the dispensing of 2.5 × 104 MC38.CEA cells. Changes in electrical impedance at the surface of E‐plates (reported as ΔCell Index units) were collected every minute for a period of 6 h and corrected for the effect of additives (i.e. surface coating and/or rCEA modules or pAbs).
2.9. Inhibition of MC38.CEA tumor implantation in vivo
Experiments aimed at interfering with the implantation of MC38.CEA cells in the peritoneal cavity of C57BL/6 mice were performed as previously described (Orava et al., 2013). Briefly, 2.0 × 105 MC38.CEA cells were pre‐treated with 1 μM of either rCEA N, A3 or A3B3 modules for 15 min on ice, washed to remove unbound protein, and subsequently injected in the peritoneal cavity of C57BL/6 mice. Non‐treated or control protein (BSA)‐treated MC38.CEA cells served as controls. After 21 days, mice were sacrificed and the number of peritoneal tumor nodules and their volumes were recorded following dissection, as previously described (Abdul‐Wahid et al., 2012). Specifically, the length and width of tumor nodules were measured using microcalipers. Tumor volumes were calculated using the modified formula where the volume of the tumor (mm3) equals [(x 2 × y)/2]; where x and y represent the transverse and longitudinal diameters of the tumor respectively. C57BL/6 mice (12–16 weeks‐old) were bred and kept under standard pathogen‐free conditions at the Sunnybrook Health Sciences Center Comparative Research Animal facility. All experiments were performed under the approval of the local animal welfare committee and in accordance with the rules and regulations of the Canadian Council for Animal Care.
2.10. Statistical and data analyses
Collected data sets were tested for normality, to confirm Gaussian distribution, using the Shapiro–Wilk normality test. Data sets were compared using either the Mann‐Whitney‐U‐test or the Student t‐test. Cellular adhesion kinetic data recorded using the xCelligence RTCA DP instrument were analyzed using the RTCA Data Analysis Software 1.0 (ACEA Biosciences). Analysis of statistical significance and plotting of graphs were all done using PRISM software (Graph Pad Software for Science). Statistical significance was acceptable when P ≤ 0.05.
3. Results
3.1. Mapping CEA domains involved in homotypic adhesion events
Dissecting the homotypic adhesion properties of CEA required the generation of its individual Ig‐like modules, as shown in Figure 1. The segment representing the CEA N domain (amino acids 1–132) was expressed as a bait module in a yeast 2‐hybrid assay to initially analyze the interaction between the N domain and all CEA Ig domains in order to map the interactive segments of CEA. We observed the growth of colonies co‐expressing the CEA N domain (bait modules) along with all the other CEA domains (as prey modules; Figure 2A). No growth of colonies was observed with yeast expressing either the CEA N domain (as bait) alone or in combination with recombinant human RPLP2 (which served as a negative control protein). This initial observation led us to analyze the relative binding of the CEA IgV‐like N domain to other CEA Ig‐like modules by ELISA. At a 1:1 molar ratio, binding was observed (Figure 2 panel B) between the FLAG‐tagged rCEA N domain and the untagged N domain itself. However, the ELISA signal was significantly greater for the N domain interacting with the recombinant CEA A3B3 domain. Importantly, the latter complex generated an ELISA signal comparable to that of the FLAG‐tagged N‐domain binding to the full length tumor glycoform of CEA (FL‐CEA; Figure 2B). These results are consistent with our previously reported observations (Abdul‐Wahid et al., 2012). In contrast, the binding of the FLAG‐tagged N domain to either rCEA A1B1 or A2B2 modules were found to be marginal, although statistically greater than the binding of the tagged N domain to our control protein (rhTNF‐α; Figure 2 panels B). These observations were confirmed by ELISA where the binding of His‐tagged rCEA modules was observed to an immobilized, non‐tagged, rCEA N domain module (Supplementary Figure 1).
Figure 2.
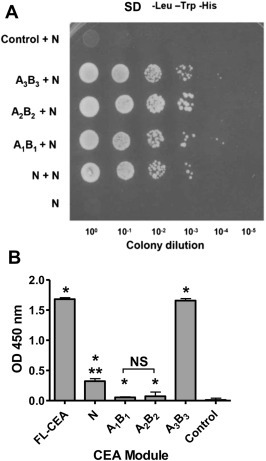
Mapping of the CEA segments responsible for homotypic interactions. A. Yeast‐2‐hybrid analysis of the interacting human CEA modules. A “bait” vector was constructed where the IgV‐like N domain of CEA was fused to the GAL4 DNA‐binding domain. The yeast strain AH109 was co‐transformed with this vector as well as with a vector expressing either the IgV‐like N‐, IgC‐like A1B1‐, A2B2‐, or A3B3‐domains fused to the GAL4 activation domain. The resulting yeast colonies were grown overnight and spotted (5 μl) as tenfold serial dilution onto SD medium lacking Trp, Leu, and His to select for interacting partners leading to colony growth. The lack of interaction between CEA N (as bait) and rhRPLP2 (as prey) served as a control. B. Confirmation of the homotypic interactions between the FLAG‐tagged N‐ and the Ig domains of CEA, or rhTNF‐α (control protein). Binding of FLAG‐rCEA N to either immobilized rCEA Ig domains, the full‐length tumor glycoform of CEA or to rhTNF‐α (negative control) was monitored by ELISA. The presence of bound FLAG‐tagged N domain was detected using an HRP‐coupled anti‐FLAG M2 (1:5000. NS: not statistically significant); * significant when compared to the control protein (P ≤ 0.05); ** significant when comparing the binding intensity of rCEA N to either A1B1 or A2B2. Statistical significance was established using a Student‐t‐test.
3.2. The A3 domain of CEA interacts directly with the N domain
The homotypic interaction between the N and the A3, B3 and C terminal regions of CEA was further defined by generating a recombinant A3 domain (residues 447–573) and synthesizing peptides corresponding to the B3 domain (residues 581–621) and the exposed C‐terminus (residues 622–643) of CEA. These segments were individually tested for their ability to inhibit the binding between the FLAG‐tagged N and the A3B3 module (Figure 3, panel A). Only the addition of the A3 domain (residues 447–573) was capable of significantly inhibiting the binding of the FLAG‐tagged N‐ to the immobilized rCEA A3B3 domain (Figure 3, panel A). Addition of either the B3 domain or the extracellular CEA C‐terminal peptide only weakly inhibited this interaction as was the case for rhTNF‐α, an irrelevant His‐tagged protein serving as a negative control (Figure 3 panel A). Pull‐down experiments of the untagged rCEA N domain with either His‐tagged rCEA N, A3B3 or the A3 domains further confirmed homotypic interactions between the N domain with itself (N‐to‐N) and with the A3 domain (N‐to‐A3) but not with an His‐tagged control protein (rhTNF‐α; Figure 3 panel B).
Figure 3.
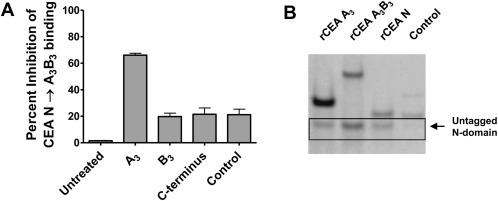
The CEA IgC‐like A3 domain is responsible for forming reciprocal homotypic complexes with the IgV‐like N domain. A. The interaction of the N domain of CEA with the A3B3 region was mapped to its A3 domain using a soluble recombinant CEA A3 module and synthetic peptides representing the B3 domain and the C terminus of CEA acting as inhibitors. Recombinant human rhTNF‐α served as a control protein. B. Pull‐down experiments using magnetic Ni‐NTA beads demonstrating specific interactions between untagged rCEA N domain and either His‐tagged rCEA A3B3 or A3 modules. His‐tagged rCEA N domain was used as a positive control, whereas His‐tagged rhTNF‐α served as a negative control protein and did not pull down untagged rCEA N domain.
3.3. Inhibition of CEA homotypic associations prevents cellular aggregation and adhesion
The relevance of N‐to‐N (cis) and N‐to‐A3 (trans) interactions towards homophilic cell adhesion was first investigated using a panel of CEA‐expressing cells. These cells were initially detached from their substratum and suspended in the presence of 1 μM of either rCEA N, A3B3 or A3 domains or an irrelevant His‐tagged control protein (rhTNF‐α), followed by the enumeration of the number of single vs. aggregated cells as a function of time.
The addition of rCEA modules to non CEA‐expressing cells (i.e. MC38 or HeLa cells) suspensions had no effect on the reversal of their intercellular aggregation (Figure 4). Inhibition of CEA‐mediated homotypic adhesion by the addition of the above rCEA modules resulted in a significant disruption of CEA‐dependent cellular aggregation (Figure 4). However, as most of these cell lines express other CEACAMs that also may interact with CEA, it is possible that the differences in aggregation might reflect inhibition of associations involving CEA and other CEACAMs.
Figure 4.
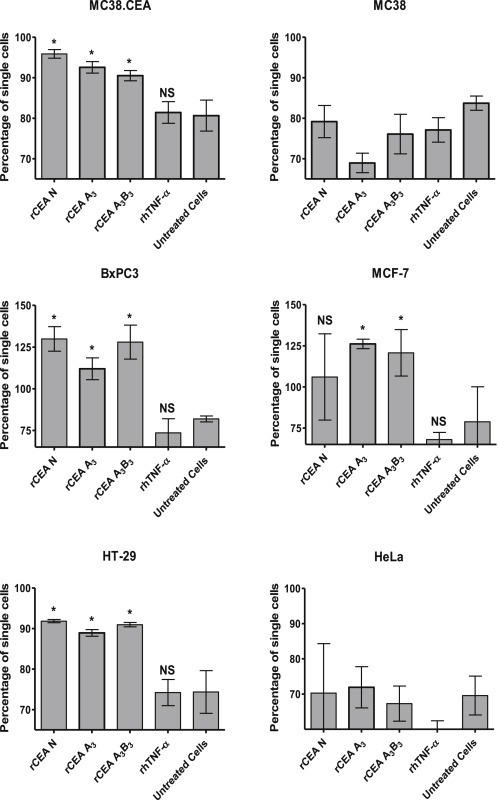
Effect of soluble rCEA modules on the time‐dependant aggregation of cells. CEA‐expressing (murine MC38.CEA, human BxPC3, human MCF‐7, human HT‐29) cells as well as CEA‐negative cells (murine MC38, human HeLa) were detached from their substratum and incubated in suspension with either the rCEA N, A3B3, or A3 modules or a control His‐tagged protein (rhTNF‐α) at a concentration of 1 μM protein per 106 cells per mL. Single cells were counted with a hemocytometer and reported as the percentage of single cells observed in the suspension after 2 h relative to the number of single cells present at time zero. Each histogram bar represents the average percentage of single cells (±SEM) calculated from four independent experiments. NS: not statistically significant; * significant when compared to untreated cells (P ≤ 0.05).
We subsequently sought to determine whether the specific disruption of CEA homotypic adhesion events could also influence cellular adherence to CEA‐coated surfaces. Accordingly, we monitored the adhesion of MC38.CEA cells to the surface of E‐plate wells pre‐coated with either rCEA N, A3B3 or A3 domains (Figure 5). Cell adhesion events were measured as changes in electrical impedance (in real time over a period of 6 h), relative to either BSA‐coated, or non‐coated wells, and is reported as ΔCell index values. MC38.CEA cells selectively adhered to E‐plate wells coated with rCEA N, A3B3 or A3 domains (Figure 5, panels A and B) while CEA− MC38 cells did not bind to these surfaces (data not shown). Pre‐treatment of MC38.CEA cells with 1 μM of either soluble rCEA N, A3B3 or A3 domains resulted in the specific disruption of their adherence to rCEA N‐coated surfaces (Figure 5, panels C and D), as did the addition of polyclonal antisera recognizing either the N or A3B3 domains (Figure 5E). In contrast, pre‐treatment of MC38.CEA cells with either BSA (a control protein) or pAb H‐300 (reactive with epitopes distributed in the first 300 amino acids of CEA) did not disrupt the adherence of MC38.CEA cells to rCEA N‐coated surfaces (Figure 5E). Together, these results indicate that CEA expression by tumor cells allowed them to aggregate and selectively adhere to CEA‐coated surfaces via the establishment of homophilic adhesion complexes involving the N domain.
Figure 5.
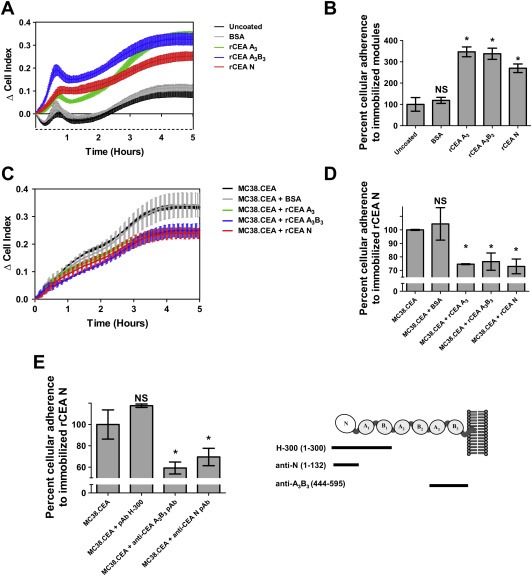
Disruption of CEA‐homotypic binding impedes the adherence of CEA‐expressing tumor cells to rCEA N domain‐coated surfaces. A. Selective adherence of MC38.CEA cells to gold sensor E‐plates coated with either 1 μg BSA (control protein), rCEA N, A3 or A3B3 modules. Cellular adhesion of MC38.CEA cells to CEA modules was directly measured as changes in electrical impedance recorded by a gold sensor located in each tissue culture well of E‐plate wells. Changes in impedance, reported as ΔCell Index values, were measured at 1 min interval over a period of 6 h. B. Bar graph representing the percentage of cells adhering to the coated E‐plate surfaces at the end of the adherence phase of MC38.CEA cells (6 h post cell addition) relative to uncoated wells, set at 100% after “(6h post cell addition)”. Panels C and D highlight the disruption of cellular adherence to CEA N domain‐coated E‐plates in the presence of (1 μM) rCEA N, A3B3, or A3 modules, but not BSA, to MC38.CEA cells prior to dispensing these cells into wells. NS: not statistically significant; * significant when compared to the control protein (P ≤ 0.05). Statistical significance was established using a Student‐t‐test. E. Addition of polyclonal antibodies recognizing epitopes within regions of the N and A3 B3 domains of CEA suggest that antibodies directed at epitopes within regions 1–132 of CEA N domain (pAb H‐300) and residues 444–595 of the A3 B3 domain (anti‐CEA A3B3 pAb) can specifically inhibit CEA‐mediated homophilic cellular adhesion. NS: not statistically significant; * significant when compared to untreated MC38.CEA cells (P ≤ 0.05). Analyses were performed using a Mann‐Whitney‐U‐test.
3.4. Direct association between CEA and fibronectin
The adherence of metastatic cancer cells at distal sites often involves interactions with extracellular matrix components such as fibronectin (Fn). Here, we observed that MC38.CEA cells (murine colorectal cancer cells stably expressing human CEA; Robbins et al., 1991) adhere more strongly to Fn‐coated surfaces than MC38 cells after 6 h (Figure 6A) suggesting that CEA may directly interact with Fn, in contrast to previous studies postulating an indirect interaction between CEA and Fn mediated through the association of CEA with α5β1 integrin (Camacho‐Leal et al., 2007, Figure 6 panels A and B).
Figure 6.

Preferential adherence of CEA‐expressing cancer cells to fibronectin is the result of a direct CEA N domain‐Fn interaction. CEA‐expressing (MC38.CEA) cells, but not the parental MC38 cells, preferentially adhere to immobilized Fn (Panels A and B). Cells (2.5 × 104) were dispensed to E‐plates wells pre‐coated with either 1 μg of Fn, BSA or in the absence of any immobilized protein. Cellular adhesion to Fn is reported as averaged ΔCell Index values (±SEM). C. Mapping the region of the CEA N domain responsible for Fn binding. Binding of His‐tagged rCEA segments spanning residues 1 to 290 of CEA to immobilized Fn was monitored by ELISA. The presence of bound His‐tagged proteins was detected using HRP‐coupled anti‐His monoclonal antibody (mAb His‐1; 1:5000). NS: not statistically significant; * significant when compared to control wells (P ≤ 0.05). Statistical analyses were performed using a Mann‐Whitney‐U‐test. D. Pull down of Fn by biotinylated CEA N108–115. Fn (5 μg) was incubated in the presence or absence of biotinylated heptapeptides (50 μg) and pulled down with magnetic Streptavidin beads. The presence of Fn was detected using anti‐Fn rabbit pAb (1:1000). The biotinylated 7‐mer peptides used were CEA N108‐115, FnBP, as well as the irrelevant P0 7‐mer. Presentation of soluble rCEA N domain significantly ameliorates the adherence of MC38.CEA cells to Fn‐coated surfaces (panels E and F). Blocking of cell surface CEA N domain through the addition of rCEA A3B3 (1 μM) results in a substantial reduction in adherence of MC38.CEA cells to Fn. NS: not statistically significant; * significant when compared to untreated cells (P ≤ 0.05). Statistically analyses were performed using a Student‐t‐test.
Using purified recombinant CEA domains and measuring their direct protein interactions by ELISA to immobilized fibronectin, we found that the N terminal segment of CEA directly bound fibronectin in a dose‐dependent manner, and was amenable to being out‐competed by the addition of soluble fibronectin (Figure 6C, Supplementary Figure 2A). Moreover, the amino acid sequence encompassing residues 108–115 of CEA (PELPKPSI) was found to be the critical region of CEA required for directly binding the CEA N domain to immobilized Fn (Figure 6C). The importance of this amino acid sequence in directly binding Fn was further confirmed when a synthetic biotinylated CEA N108‐115 peptide was used to pull‐down Fn (Figure 6D), as well as preventing the association between fibronectin and the CEA N domain (Supplementary Figure 2B).
Finally, adding rCEA N domain to E‐plate surfaces pre‐coated with Fn, before dispensing MC38.CEA cells, led to a small enhancement of cellular adherence by ∼20–25% (Figure 6E and F). In contrast, the addition of rCEA A3B3 reduced cellular adherence to Fn‐coated wells by ∼55% (Figure 6E and F) suggesting that the binding site of the N domain to A3 may be in close spatial proximity to the N domain region involved in its binding to Fn.
3.5. Derivation of equilibrium dissociation constants for CEA N‐to‐N, N‐to‐A3 and N‐to‐Fn interactions
The ability of the FLAG‐tagged rCEA N module to bind directly to immobilized Fn, rCEA N and A3 domains, in a dose dependent manner led to the derivation (2, 3, 6) of dissociation constants from ELISA‐based binding curves (Figure 7) using established protocols (Sato et al., 2009; Smith et al., 2013; Jardim et al., 2000). The calculated ratios of bound to free FLAG‐tagged rCEA N module with individual monomeric components revealed that N‐to‐N, N‐to‐A3 and N‐to‐Fn interactions to be of moderate to high affinity with K D values of 100 ± 17 nM, 18 ± 3 nM and 16 ± 3 nM for the binding of FLAG‐tagged rCEA N to immobilized rCEA N, rCEA A3 and Fn, respectively.
Figure 7.
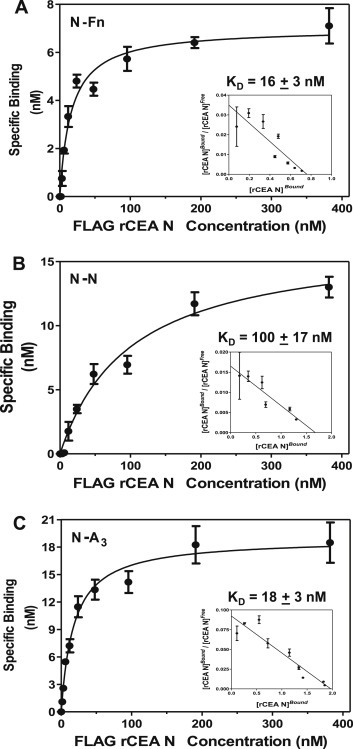
Derivation of dissociation constants for CEA‐dependent homotypic and heterotypic interactions. Wells were coated with either Fn (panel A) or untagged rCEA N (panel B) or rCEA A3 (panel C) domains. The presence of bound FLAG‐tagged rCEA N was quantified using HRP‐coupled anti‐FLAG M2 mAb (1: 5000). Each assay was performed three times with each dilution repeated in quadruplicate. The recorded average absorbance values at 450 nm were used to derive the concentration of bound FLAG‐tagged rCEA N using a standard curve initially constructed relating absorbance readings (at 450 nm) to known concentrations of FLAG‐tagged rCEA N domain. The resulting binding curves and dissociation constants were calculated using Graph Pad PRISM (version 5.01) software.
3.6. Disruption of CEA‐dependent associations impedes cellular attachment in vivo
The above observations suggested that the N domain of CEA mediates adhesion events favoring the binding of CEA‐expressing cancer cells to ECM components as well as cellular aggregation. Therefore, blocking N domain‐mediated associations may impede CEA‐dependent cell aggregation and implantation in vivo, resulting in either a lowering of the number of tumor nodules found and/or to smaller nodule volumes. MC38.CEA cells (2.0 × 105) were thus pre‐treated with 1 μM of either rCEA N, A3B3 or A3 domains, and injected in the peritoneal cavity of mice. Animals receiving non‐treated MC38.CEA cells or MC38.CEA cells pre‐treated with an irrelevant protein (BSA) were used as controls. As shown in Figure 8, the pre‐treatment of MC38.CEA cells with either rCEA N, A3B3 or A3 domains resulted in both a reduction in the number and size of the tumor nodules that developed in the peritoneal cavity of treated mice. These findings demonstrate the importance of interactions mediated by the CEA N domain on the implantation of tumor cells in vivo, and also suggests the possibility that blocking CEA N domain binding events may disrupt other heterotypic interactions (e.g. TGF‐β R1, hnRNP M and DR5).
Figure 8.
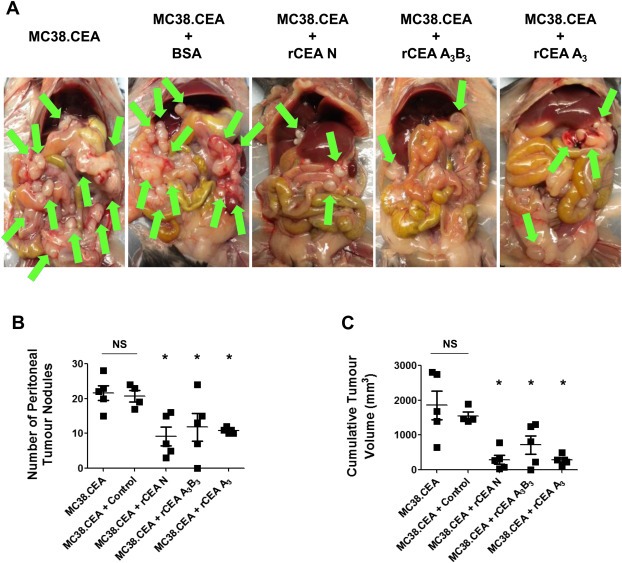
Blockage of CEA binding events disrupts the implantation of CEA‐expressing tumor cells in vivo. MC38.CEA cells (2.0 × 105) pre‐treated with either rCEA N, A3B3, or A3 modules (1 μM) were implanted (intraperitonally) into groups of C57BL/6 mice (n = 5). A. Photographs illustrate the presence of MC38.CEA tumor nodules (arrows) in the peritoneal cavity at Day 21 post‐implantation. The number of tumor nodules (panel B) and their respective cumulative volume (panel C) were assessed at Day 21 post‐tumor implantation. NS; not statistically significant when compared to untreated cells. Asterisk denotes statistical significance (P ≤ 0.05) when compared to control animals implanted with either MC38.CEA cells or MC38.CEA cells pre‐treated with control protein (BSA); Student‐t‐test.
4. Discussion
CEA is expressed on a variety of human adenocarcinomas (Berinstein, 2002). Since the aberrant overexpression of CEA by cancer cells is associated with cancer progression and tumor metastasis (Berinstein, 2002; Bast et al., 2001; Duffy, 2006), CEA has been used as a clinical biomarker for monitoring disease recurrence and the management of metastatic cancers (Berinstein, 2002; Bast et al., 2001; Duffy, 2006). Nonetheless, the connection between increasing levels of CEA in the serum and the increased likelihood for metastatic disease remain to be established.
In the present study, we define key molecular interactions underlying the binding properties of CEA, and in particular its N terminal Ig V domain, to itself as well as to fibronectin, an extracellular matrix element. We further demonstrate that these specific interactions are essential for the engraftment of disseminated CEA‐expressing tumor cells as well as the formation and expansion of tumor foci in vivo.
The biological functions of CEA revolve around its ability to form homotypic as well as heterotypic interactions (Taheri et al., 2000; Singer et al., 2010). The N domain of CEA can form both N‐to‐N (cis‐) and N‐to‐A3 (trans‐) domains homotypic associations (2, 3, 4, 5, this work; Zhou et al., 1993; Taheri et al., 2000). It has been established that the GFCC'C″ face of the N domain represents the interface defining the cis‐homotypic interaction with amino acids F29, S32, V39, Q44, I91, L95 and E99 being involved (Korotkova et al., 2008). Conversely, the CFG face of the CEA N domain has been hypothesized to be important for mediating trans‐homotypic interactions (Taheri et al., 2000).
Unlike other CEACAM family members (such as CEACAM‐1, ‐3, ‐6 or ‐8), CEA (CEACAM 5) favors trans‐homotypic domain binding (Figure 2) in that its binding can include IgC‐like domains and is not limited to making dimers involving opposing N domains (Zhou et al., 1993; Kuroki et al., 2001; Klaile et al., 2009). Although some evidence in the past suggested that the main reciprocal homophilic association occurs between the CEA N and A3B3 domains (Zhou et al., 1993), it was never shown that the CEA N domain interacted specifically with the A3 domain. In the present study, we purified recombinant domains of CEA and demonstrated using a combination of a yeast‐2‐hybrid screen, pull‐down experiments and ELISA‐based protein binding inhibition assays (Figures 2 and 3) that the CEA N domain bound to itself as well as all the CEA IgC‐like A3 domain.
The relevance of the observed N‐to‐N and N‐to‐A3 associations at a cellular level was subsequently proven using CEA‐mediated cellular adhesion/aggregation assays (Figures 4 and 5). Firstly, we demonstrated using a classical cell disaggregation assay (Zhou et al., 1995, 1990, 1993) that the addition of either the recombinant N or A3 domains of CEA were able to delay the kinetics of CEA‐mediated intercellular aggregation of cells expressing CEA on their surface (MC38.CEA, BxPC3, MCF‐7, HT‐29) as opposed to cells lacking CEA expression (MC38 and HeLa; Figure 4). In a second approach, the disruption of cellular adherence of MC38.CEA cells to CEA‐coated surfaces, was demonstrated by adding either soluble rCEA modules or polyclonal antisera specific to the recombinant N or A3 domains (Figure 5). The selective adherence of MC38.CEA cells to CEA‐coated surfaces as well as the blockage of cellular aggregation and adherence by the addition of either soluble rCEA modules or antibodies specific to the CEA N or A3B3 domains highlighted the importance of CEA in promoting cellular adherence, and possible engraftment in a manner that exceeds aggregation in suspension. Importantly, rCEA modules used in this study inhibited CEA‐dependent adherence and aggregation at a final concentration of 1 μM, a 2‐order of magnitude lower concentration than previously reported for added CEA chimeras (Zhou et al., 1993).
Our finding that the CEA N domain directly associates with fibronectin (Fn) was unexpected. It was previously thought that CEA‐expressing cancer cells preferentially adhered to Fn in an indirect manner via an association with α5β1 integrin (Ordoñez et al., 2007; Camacho‐Leal et al., 2007). Specifically, MC38.CEA cells, murine colorectal cancer cells transfected to stably express human CEA (Robbins et al., 1991), do not express the normal complement of human surface molecules that would normally synergize with CEA, including human α5β1 integrin. However, these cells express a murine homologue whose endogenous expression levels is not affected by the heterologous expression of CEA, since both MC38.CEA and the parental MC38 cells express comparable levels of murine α5 integrin (Supplementary Figure 3 panels A and B). Yet despite the presence of comparable levels of α5β1 integrin, CEA‐expressing MC38.CEA cells adhered more strongly to Fn‐coated surfaces as compared to CEA negative MC38 cells, suggesting that CEA directly interacts with Fn (Figure 6). Since previous studies have hinted at the existence of a role for the CEA N‐terminus in mediating an α5β1 integrin‐dependent adhesions to Fn, we used rCEA segments covering overlapping sequences of the CEA N‐terminus and mapped a region encompassing amino acid residues 108‐115 (PELPKPSI) as the Fn binding element of the CEA N domain (Figure 6, panels C, D); an interaction that occurs in the absence of human α5β1 integrin. Interestingly, these residues have been consistently implicated in heterotypic associations between CEA and a number of receptors (including TGF‐β R1, hnRNP M and DR5) linked to tumor metastasis (Zimmer and Thomas, 2013, 1990, 2010, 2000, 2007, 2001).
Importantly, the availability of purified, folded rCEA N and A3 domains allowed us to derive for the first time, dissociation constants (K D) for their monovalent interactions using ELISA‐based binding assays (Figure 7; Sato et al., 2009; Smith et al., 2013; Jardim et al., 2000). The calculated K D values were 16 ± 3 nM for the heterotypic N‐to‐Fn interaction; and 100 ± 17 nM, and 18 ± 3 nM for the homotypic CEA N‐to‐N and N‐to‐A3 domains interactions, respectively (Figure 7). These K D values are comparable to the dissociation constant (0.2 nM) reported for the interaction of integrin α8β1with nephronectin (Sato et al., 2009). Furthermore, the derived KD values suggest a significant affinity for each of these individual binding events [as interacting monomeric partners] that would be substantially magnified in terms of overall avidity within the context of a multivalent display of CEA molecules on cancer cells. This hypothesis was directly tested in vivo by assessing if recombinant, soluble rCEA N, A3 and A3B3 domains could block the engraftment of MC38.CEA murine cancer cells in the peritoneal cavity of mice. Indeed, the pre‐treatment of MC38.CEA cells with rCEA N, A3 and A3B3 modules, as a means of disrupting CEA N‐mediated interactions, led to the disruption of cellular implantation and subsequent expansion of nascent tumor foci in the peritoneal cavity of mice (Figure 8). This finding represents the first in vivo evidence directly linking the importance of this single N domain of CEA and its adhesive functions to the implantation of disseminated CEA‐expressing tumor cells.
In recent studies, we demonstrated that targeting the homotypic adhesion functions of the CEA N domain using DNA aptamers resulted in the reduction of the number and size of implanted MC38.CEA tumor cells in the peritoneal cavity of mice (Orava et al., 2013), whereas the adoptive transfer of B cells or passive protection with antisera from mice vaccinated with the CEA N domain, resulted in a complete protection against the development of peritoneal tumor nodules (Abdul‐Wahid et al., 2012). The better performance of the antisera in blocking tumor implantation over DNA aptamers may be related to the fact that DNA aptamers were shown to block CEA‐mediated interactions involving regions of the first 107 amino acids of CEA while the N domain epitopes recognized by the polyclonal sera spanned amino acid residues 1–132 of the CEA N domain and in particular, the PELPKPSI region of CEA that allows for a direct association with Fn (Abdul‐Wahid et al., 2012).
5. Conclusions
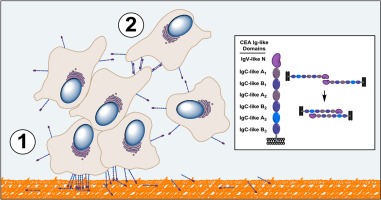
Together, these observations suggests that CEA N domain (residues 1–115) participates in at least two classes of binding events leading to cellular engraftment and tumor foci formation (as illustrated in Figure 9A). One binding event reflects the heterophilic interaction of membrane bound CEA to the extracellular matrix through one of its components, namely Fn (Figure 9B). Additional heterotypic interactions involving other ECM components and receptors (TGF‐β R1, hnRNP M and DR5) may also take place. A second class of binding events involves homophilic interactions linked to the N domain of CEA associating with itself and the A3 domain, resulting in cellular aggregation and proliferation leading to tumor foci formation (Figure 9B). This model provides a partial mechanistic rationale for the established fact that the expression of CEA on tumor cells correlates with the occurrence of metastases (Hostetter et al., 1990).
Figure 9.
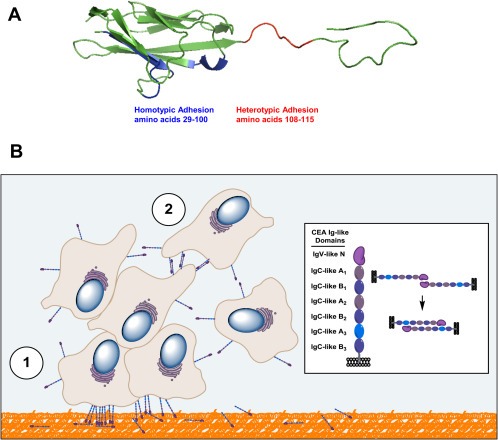
Proposed mechanistic model of CEA‐mediated hetero‐ and homotypic adhesion events leading to the engraftment and expansion of metastatic tumor foci. A. A ribbon structure of the CEA N domain highlighting the two areas necessary for the engraftment of CEA‐expressing tumor cells. Amino acid residues 29–100 (highlighted in blue) are present on the CFC′C″G of the CEA N domain and are necessary for homotypic interactions while amino acid residues 108–115 (highlighted in red) are required for the association of the CEA N domain with Fn. B. Representation of key binding events linking the CEA N domain to CEA‐expressing tumor cell implantation and aggregation. In this model, CEA‐expressing cells interact via their N domain with ECM proteins such as Fn (step 1, orange‐colored matrix), allowing such cells to engraft at a site distal to a primary tumor site. Surviving and proliferating CEA‐expressing cancer cells subsequently bind to each other through homotypic interactions involving their CEA N and A3 domains (step 2). Expanding CEA‐expressing cell populations lead to cellular aggregates through multivalent interactions leading to the occurrence of micrometastases.
Overall, the present study suggests that targeting the adhesive properties of the CEA N domain, either by vaccination (Abdul‐Wahid et al., 2012) or by using N domain‐specific antibodies or soluble N domain ligands (Orava et al., 2013), will inhibit the establishment of tumor foci leading to metastases. Such an intervention would be best applied as a preventive strategy such as a vaccination and be aimed at disease stages where tumor dissemination is minimal.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgments
The authors wish to express their gratitude to Dr. J. Schlom (NCI) for providing the MC38 and MC38.CEA cell lines, Dr. J. Shively for providing the full length tumour glycoform of CEA and Dr. S. Gray‐Owen for providing His‐tagged rCEA N1–107. The authors of this manuscript wish to also thank Dr. Melissa Brierley for help with the xCelligence RTCA, as well as Dr. Peter Lee and Ms. Michelle Martin for help with the handling of animals. This work was supported by the Canadian Breast Cancer Foundation.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.12.002.
Abdul-Wahid Aws, Huang Eric H.-B., Cydzik Marzena, Bolewska-Pedyczak Eleonora and Gariépy Jean, (2014), The carcinoembryonic antigen IgV‐like N domain plays a critical role in the implantation of metastatic tumor cells, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.12.002.
References
- Abdul-Wahid, A. , Huang, E.H. , Lu, H. , Flanagan, J. , Mallick, A.I. , Gariépy, J. , 2012. A focused immune response targeting the homotypic binding domain of the carcinoembryonic antigen blocks the establishment of tumor foci in vivo. Int. J. Cancer. 131, 2839–2851. [DOI] [PubMed] [Google Scholar]
- Bast, R.C. , Ravdin, P. , Hayes, D.F. , Bates, S. , Fritsche, H. , Jessup, J.M. , Kemeny, N. , Locker, G.Y. , Mennel, R.G. , Somerfield, M.R. , 2001. 2000 update of recommendations for the use of tumor markers in breast and colorectal cancer: clinical practice guidelines of the American society of clinical oncology. J. Clin. Oncol.. 19, 1865–1878. [DOI] [PubMed] [Google Scholar]
- Beauchemin, N. , Arabzadeh, A. , 2013. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev.. 32, 643–671. [DOI] [PubMed] [Google Scholar]
- Benchimol, S. , Fuks, A. , Jothy, S. , Beauchemin, N. , Shirota, K. , Stanners, C.P. , 1989. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell. 57, 327–334. [DOI] [PubMed] [Google Scholar]
- Berinstein, N.L. , 2002. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J. Clin. Oncol.. 20, 2197–2207. [DOI] [PubMed] [Google Scholar]
- Camacho-Leal, P. , Zhai, A.B. , Stanners, C.P. , 2007. A co-clustering model involving alpha5beta1 integrin for the biological effects of GPI-anchored human carcinoembryonic antigen (CEA). J. Cell Physiol.. 211, 791–802. [DOI] [PubMed] [Google Scholar]
- Camacho-Leal, P. , Stanners, C.P. , 2008. The human carcinoembryonic antigen (CEA) GPI anchor mediates anoikis inhibition by inactivation of the intrinsic death pathway. Oncogene. 27, 1545–1553. [DOI] [PubMed] [Google Scholar]
- Desgrosellier, J.S. , Cheresh, D.A. , 2010. Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer. 10, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy, M.J. , 2006. Serum tumor markers in breast cancer: are they of clinical value?. Clin. Chem.. 52, 345–351. [DOI] [PubMed] [Google Scholar]
- Fidler, I.J. , 2003. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 3, 453–458. [DOI] [PubMed] [Google Scholar]
- Gold, P. , Freedman, S.O. , 1965. Demonstration of tumor-specific antigens in human colonic carcinomata by immunological tolerance and absorption techniques. J. Exp. Med.. 121, 439–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström, S. , 1999. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol.. 9, 67–81. [DOI] [PubMed] [Google Scholar]
- Hostetter, R.B. , Augustus, L.B. , Mankarious, R. , Chi, K.F. , Fan, D. , Toth, C. , Thomas, P. , Jessup, J.M. , 1990. Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. J. Natl. Cancer Inst.. 82, 380–385. [DOI] [PubMed] [Google Scholar]
- Ilantzis, C. , DeMarte, L. , Screaton, R.A. , Stanners, C.P. , 2002. Deregulated expression of the human tumor marker CEA and CEA family member CEACAM6 disrupts tissue architecture and blocks colonocyte differentiation. Neoplasia. 4, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardim, A. , Liu, W. , Zheleznova, E. , Ullman, B. , 2000. Peroxisomal targeting signal-1 receptor protein PEX5 from Leishmania donovani. Molecular, biochemical, and immunocytochemical characterization. J. Biol. Chem.. 275, 13637–13644. [DOI] [PubMed] [Google Scholar]
- Kitsuki, H. , Katano, M. , Morisaki, T. , Torisu, M. , 1995. CEA-mediated homotypic aggregation of human colorectal carcinoma cells in a malignant effusion. Cancer Lett.. 88, 7–13. [DOI] [PubMed] [Google Scholar]
- Klaile, E. , Vorontsova, O. , Sigmundsson, K. , Müller, M.M. , Singer, B.B. , Ofverstedt, L.G. , Svensson, S. , Skoglund, U. , Obrink, B. , 2009. The CEACAM1 N-terminal Ig domain mediates cis- and trans-binding and is essential for allosteric rearrangements of CEACAM1 microclusters. J. Cell Biol.. 187, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, C.A. , 2009. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer. 9, 302–312. [DOI] [PubMed] [Google Scholar]
- Korotkova, N. , Yang, Y. , Le Trong, I. , Cota, E. , Demeler, B. , Marchant, J. , Thomas, W.E. , Stenkamp, R.E. , Moseley, S.L. , Matthews, S. , 2008. Binding of Dr adhesins of Escherichia coli to carcinoembryonic antigen triggers receptor dissociation. Mol. Microbiol.. 67, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuespert, K. , Pils, S. , Hauck, C.R. , 2006. CEACAMs: their role in physiology and pathophysiology. Curr. Opin. Cell Biol.. 18, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki, M. , Abe, H. , Imakiirei, T. , Liao, S. , Uchida, H. , Yamauchi, Y. , Oikawa, S. , Kuroki, M. , 2001. Identification and comparison of residues critical for cell-adhesion activities of two neutrophil CD66 antigens, CEACAM6 and CEACAM8. J. Leukoc. Biol.. 70, 543–550. [PubMed] [Google Scholar]
- Li, Y. , Cao, H. , Jiao, Z. , Pakala, S.B. , Sirigiri, D.N. , Li, W. , Kumar, R. , Mishra, L. , 2010. Carcinoembryonic antigen interacts with TGF-β receptor and inhibits TGF-β signaling in colorectal cancers. Cancer Res.. 70, 8159–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey, A.J. , Poon, G.M. , Bolewska-Pedyczak, E. , Srikumar, T. , Jeram, S.M. , Raught, B. , Gariépy, J. , 2008. The catalytic subunit of shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol.. 378, 375–386. [DOI] [PubMed] [Google Scholar]
- Michaux, C. , Pomroy, N.C. , Privé, G.G. , 2008. Refolding SDS-denatured proteins by the addition of amphipathic cosolvents. J. Mol. Biol.. 375, 1477–1488. [DOI] [PubMed] [Google Scholar]
- Orava, E.W. , Abdul-Wahid, A. , Huang, E.H. , Mallick, A.I. , Gariépy, J. , 2013. Blocking the attachment of cancer cells in vivo with DNA aptamers displaying anti-adhesive properties against the carcinoembryonic antigen. Mol. Oncol.. 7, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordoñez, C. , Screaton, R.A. , Ilantzis, C. , Stanners, C.P. , 2000. Human carcinoembryonic antigen functions as a general inhibitor of anoikis. Cancer Res.. 60, 3419–3424. [PubMed] [Google Scholar]
- Ordoñez, C. , Zhai, A.B. , Camacho-Leal, P. , Demarte, L. , Fan, M.M. , Stanners, C.P. , 2007. GPI-anchored CEA family glycoproteins CEA and CEACAM6 mediate their biological effects through enhanced integrin alpha5beta1-fibronectin interaction. J. Cell Physiol.. 210, 757–765. [DOI] [PubMed] [Google Scholar]
- Psaila, B. , Lyden, D. , 2009. The metastatic niche: adapting the foreign soil. Nat. Rev. Cancer. 9, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, P.F. , Kantor, J.A. , Salgaller, M. , Hand, P.H. , Fernsten, P.D. , Schlom, J. , 1991. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res.. 51, 3657–3662. [PubMed] [Google Scholar]
- Samara, R.N. , Laguinge, L.M. , Jessup, J.M. , 2007. Carcinoembryonic antigen inhibits anoikis in colorectal carcinoma cells by interfering with TRAIL-R2 (DR5) signaling. Cancer Res.. 67, 4774–4782. [DOI] [PubMed] [Google Scholar]
- Sato, Y. , Uemura, T. , Morimitsu, K. , Sato-Nishiuchi, R. , Manabe, R. , Takagi, J. , Yamada, M. , Sekiguchi, K. , 2009. Molecular basis of the recognition of nephronectin by integrin alpha8beta1. J. Biol. Chem.. 284, 14524–14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, B.B. , Scheffrahn, I. , Kammerer, R. , Suttorp, N. , Ergun, S. , Slevogt, H. , 2010. Deregulation of the CEACAM expression pattern causes undifferentiated cell growth in human lung adenocarcinoma cells. PLoS One. 5, 8747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, E. , Vekaria, R. , Brown, K.A. , Longstaff, C. , 2013. Kinetic regulation of the binding of prothrombin to phospholipid membranes. Mol Cell Biochem.. 382, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri, M. , Saragovi, U. , Fuks, A. , Makkerh, J. , Mort, J. , Stanners, C.P. , 2000. Self recognition in the Ig superfamily. Identification of precise subdomains in carcinoembryonic antigen required for intercellular adhesion. J. Biol. Chem.. 275, 26935–26943. [DOI] [PubMed] [Google Scholar]
- Thomas, P. , Forse, R.A. , Bajenova, O. , 2011. Carcinoembryonic antigen (CEA) and its receptor hnRNP M are mediators of metastasis and the inflammatory response in the liver. Clin. Exp. Metast.. 28, 923–932. [DOI] [PubMed] [Google Scholar]
- Zhou, H. , Fuks, A. , Stanners, C.P. , 1990. Specificity of intercellular adhesion mediated by various members of the immunoglobulin supergene family. Cell Growth Differ. 1, 209–215. [PubMed] [Google Scholar]
- Zhou, H. , Fuks, A. , Alcaraz, G. , Bolling, T.J. , Stanners, C.P. , 1993. Homophilic adhesion between Ig superfamily carcinoembryonic antigen molecules involves double reciprocal bonds. J. Cell Biol.. 122, 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, R. , Thomas, P. , 2001. Mutations in the carcinoembryonic antigen gene in colorectal cancer patients: implications on liver metastasis. Cancer Res.. 61, 2822–2826. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data