

Abstract
ERK 1/2 are found to be hyperactive in many cancers. Active ERK 1/2 (pERK 1/2) are known to protect cancer cells from undergoing death receptor‐mediated apoptosis, although the mechanism(s) behind this is poorly understood. Through in vitro kinase assays and mass‐spectrometry we demonstrate that pERK 1/2 can phosphorylate pro‐Caspase‐8 at S387. Also, in EGFR‐overexpressing Type I and II ovarian and breast cancer cell lines respectively, ERK 1/2 remain active only during the interphase. During this period, pERK 1/2 could inhibit Trail‐induced apoptosis, most effectively during the G1/S phase. By knocking‐down the endogenous pro‐Caspase‐8 using RNAi and replacing it with its non‐phosphorylatable counterpart (S387A), a significant increase in Caspase‐8 activity upon Trail stimulation was observed, even in the presence of pERK 1/2. Taken together, we propose that a combination of Trail and an inhibitor of ERK 1/2 activities could potentially enhance of Trail's effectiveness as an anti‐cancer agent in ERK 1/2 hyperactive cancer cells.
Keywords: Caspase‐8, Apoptosis, Cell‐cycle, pERK 1/2
Highlights
pERK 1/2 inhibit Caspase‐8 activation by phosphorylating it at its S387 residue.
pERK 1/2 protect both Type I and II cancer cells from Caspase‐8 derived apoptosis.
This phosphorylation occurs during the interphase, most prominently during the G1/S phase, of SKOV3 and MDA‐MB‐468 cells.
Replacing cellular Caspase‐8 with non‐phosphorylatable mutant enhances apoptosis.
Cdk1 and pERK 1/2 and possibly Cdk2 can protect cancer cells from apoptosis throughout the cell cycle.
1. Introduction
Apoptosis or programmed cell death is an essential tool for the body to eliminate excessive, damaged or harmful cells (Hengartner, 2000). Cells can undergo apoptosis through two distinct pathways. The extrinsic or death receptor‐mediated apoptotic pathway is triggered by the binding of the cell surface death receptors like FasR (CD95), Trail (DR4 and DR5) or TNF‐R with FasL, Trail or TNFα, respectively. This leads to the activation of pro‐Caspases‐8 and/or ‐10 into active Caspases‐8 and/or ‐10 (hereafter Caspases‐8 and ‐10) at the DISC (Death Inducing Signaling Complex). During the intrinsic or stress‐mediated apoptotic pathway, internal and external stresses lead to the release of cytochrome‐c from the mitochondria, which, together with Apaf‐1, activates pro‐Caspase‐9 into Caspase‐9.
These three caspases (pro‐Caspases‐8, ‐9 and ‐10) are together known as the initiator caspases since they can initiate the activation of the effector pro‐Caspases 3 and 7 into active Caspases‐3 and ‐7 (hereafter Caspases‐3 and ‐7), triggering the apoptotic cascade (Budihardjo et al., 1999; Hengartner, 2000; Kurokawa and Kornbluth, 2009; Lawen, 2003; Tran et al., 2001).
Cells in which Caspase‐8 alone is sufficient to activate pro‐Caspase‐3, in response to receptor‐mediated apoptotic stimuli, are termed as Type I cells. In Type II cells however, Caspase‐8 requires the assistance of the intrinsic apoptotic pathway for enhancing such an apoptotic stimuli and activating pro‐Caspase‐3. In the later cell types, Caspase‐8 has to first cleave and activate the BH3 Interacting Domain death agonist (BID) into truncated BID (tBID), which in turn activates the intrinsic apoptotic pathway (Holmstrom et al., 2000; Ozoren and El‐Deiry, 2002; Westphal and Kalthoff, 2003).
As much as it is important for the body to sustain a suitable level of apoptosis and maintain homeostasis, it is equally vital for the cancer cells to avoid apoptosis in order to proliferate irrespective of all the genetic mutations they accumulate over the course of their development (Hanahan and Weinberg, 2000, 2011). Cancer cells employ a number of different means to avoid apoptosis and preventing the activation of pro‐Caspase‐8 is one of them. The Extracellular signal Regulated Kinases 1/2 (ERK 1/2) have been implicated in adversely meddling with the extrinsic apoptotic pathway in response to cytotoxic agents in both Type I and Type II cancer cells, for a long time (Holmstrom et al., 1998, 2000, 2001).
ERK 1/2 are serine/threonine protein kinases and members of the Mitogen Activated Protein Kinase (MAPK) family. They act as transducers of extracellular stimuli as they are activated through the Ras/Raf/MEK/ERK cascade in response to growth and differentiation stimuli, ligands for G protein‐coupled receptors, cytokines and other mitogenic factors (Meloche and Pouyssegur, 2007; Pearson et al., 2001; Wada and Penninger, 2004). The active ERK 1/2 can induce, maintain, phosphorylate and regulate a battery of kinases like RSKs 1, 2 and 3; MNKs 1 and 2; transcription factors like c‐Jun, c‐Fos, ATF‐2, Elk1; hsp1; topoisomerase IIb, and Cyclin D1. By positively or negatively regulating numerous such substrates, pERK 1/2 can regulate gene expression, cell proliferation, apoptosis, differentiation, cell–matrix interactions and cell migration (Brunet et al., 1999; Meloche and Pouyssegur, 2007; Pucci et al., 2009; Roux and Blenis, 2004; Wada and Penninger, 2004; Yamamoto et al., 2006).
Details of the precise mechanism(s) behind the anti‐apoptotic roles of pERK 1/2 are lacking. In this communication, we have attempted to pin‐point the substrate(s) that are influenced by pERK 1/2 in order to regulate the extrinsic apoptotic pathway. For this purpose, EGFR receptor overexpressing human ovarian and breast cancer cell lines SKOV‐3 and MDA‐MB‐468 along with the colorectal carcinoma cell line HCT‐116 were chosen because of the presence of hyperactive ERK 1/2. Evidence provided here have led us to conclude that pERK 1/2 can regulate the extrinsic apoptotic pathway by phosphorylating pro‐Caspase‐8 at its S387 residue in a cell cycle specific manner.
2. Materials and methods
2.1. Cell culture, antibodies and chemicals
The ERK 1/2 overexpressing ovarian cancer cell line SKOV‐3 (HTB‐77) and breast cancer cell line MDA‐MB‐468 (HTB‐132) were purchased from ATCC and cultivated according to their guidelines. Peripheral Blood Lymphocytes (PBLs) were obtained from the blood of healthy donors as described previously (Matthess et al., 2010).
Antibodies were obtained from the following sources: ERK 1/2 (06‐182) from Millipore; BID (2002), pERK 1/2 (9101), PARP (9542), Caspase‐3 (9665), Caspase‐8 (9496), Caspase‐9 (9502), Bax (2772), BID (2002) and Cyclin B1 (4138) from Cell Signaling; Plk1 (sc‐17783), Cyclin E (sc‐198), Cyclin A (sc‐271645), GST (sc‐374171) from Santa Cruz Biotechnologies; pro‐Caspase‐8 (ALX‐804‐429) from Enzo Life Sciences; Flag (F1804) and β‐Actin (A5441) from Sigma; GFP (ab290) and XIAP (ab281151) from Abcam; secondary antibodies against Rabbit (NA934V) and Mouse (NXA931) from GE Healthcare. The polyclonal antibody against Caspase‐8 (pS387) was produced by Eurogentec by immunizing rabbits with the peptide EEQPYLEMDLSpSPQTR.
Reagents and kits were obtained from the following sources: The synthetic MEK 1/2 inhibitor PD98059 Millipore (513000); the purified kinases – ERK2 (P6080), CAMKII (P6060), CDK2 (P6025) were from NEB; CDK1 (0134‐0135‐1), PLK1 (0183‐0000‐1) from ProQinase; Aurora B (A31‐10G‐10) from SignalChem and Aurora A (BML‐SE406) from Enzo Life Sciences; Nocodazole (M1404) and Thymidine (T‐1895) from Sigma; RO3306 (217699) from Calbiochem; Trail (ALX‐201‐115) from Enzo Life Sciences; Cycloheximide (66‐81‐9) from Sigma–Aldrich; the PLA assay kit from Olink Biosciences; Protein G Sepharose Beads (17‐0618‐01) and Glutathione Sepharose A Beads (17‐0756‐01) from GE Healthcare; AnnexinV (556422) and 7AAD (559925) were from BD Biosciences. [γ‐32P] ATP (3000 Ci/mmol) from Amersham Pharmacia.
2.2. siRNAs and plasmids
The following siRNAs were either designed or purchased – siRNA‐pro‐Caspase‐8 (5′‐GAGCUAAAGUUAAAUAGGAUU‐3′) (Sigma), commercial siRNA‐ERK 1/2 (6560, Cell Signaling); and commercial siRNA‐ERK 1 (sc‐29307) and siRNA‐ERK2 (sc‐35335) from Santa Cruz Biotechnologies, siRNA‐BID (5′‐GAAGACAUCAUCCGGAAUA‐3′) (Sigma) (Wagner et al., 2004) scrambled siRNA (1027281, Qiagen). siRNA transfections were performed with Oligofectamine Reagent (12252‐011, Invitrogen) or Lipofectamine RNAiMax Transfection Reagent (13778‐075, Life Technologies), as described previously (Kappel et al., 2007; Spankuch‐Schmitt et al., 2002).
Pro‐Caspase‐8 WT, Caspase‐8 NT, p18 and p10 sub‐fragments were inserted into the pGex 5x‐3 vector (GE Healthcare) into the BamHI/EcoRI sites. Pro‐Caspase‐8 (WT) and its mutants – Pro‐Caspase‐8 S387A and Pro‐Caspase‐8 S387E were inserted into the 3xFlag‐tagged pcDNA3.1‐Hygro+ vector (Invitrogen) into the HindIII/XbaI sites. The resulting Flag‐tagged Caspase‐8 sequences were inserted into the pcDNA3.1‐V5 vector (Invitrogen). ERK1 and ERK2 were inserted into the GFP‐tagged pcDNA3.1‐Hygro+ vector (Invitrogen) into the BamHI/EcoRI (ERK1) and HindIII/XbaI (ERK2) sites. Sequences for the primers have been tabulated in Supplementary Table 1. Mutants were generated through site‐directed mutagenesis using the QuikChange protocol and Pfu Ultra II Fusion HS DNA polymerase (Stratagene).
2.3. Pull‐down and immunoprecipitation
Both the methods were performed as described previously (Raab et al., 2011).
2.4. In vitro kinase assay
Both radioactive (hot) and non‐radioactive (cold) in vitro kinase assays were performed in accordance with the methods described previously (Spankuch et al., 2004; Yuan et al., 2004).
2.5. Cell synchronization and cell cycle analysis
SKOV‐3 and MDA‐MB‐468 cells were first treated with Thymidine (2 mM) for 16 h, released into fresh medium for 10 h followed by treatment for 16 h with Thymidine (2 mM) (double thymidine‐treatment) or RO3306 (9 μM) (thymidine‐RO3306‐treatment) or Nocodazole (50 ng/ml) (thymidine‐Nocodazole‐treatment) to enrich the cells in G1/S, G2 and M phases, respectively. S phase cells were obtained by double thymidine treatment followed by the release of the resulting G1/S phase enriched cells into fresh medium for 2 h. Cell cycle distribution of the harvested cells were performed by Propidium Iodide (PI) staining, as has been mentioned earlier (Yuan et al., 2011), followed by their analysis through flow cytometry using a FACScan instrument (BD). FACS data were analyzed with the BD Cell Quest pro software (version 5.2.1, BD).
2.6. Apoptosis analysis
Apoptosis was induced by stimulating the cells with a combination of Trail (100 ng/ml) or FasL (100 ng/ml) or TNFα (20 ng/ml) in the presence of 10 μg/ml of Cycloheximide (CHX). Apoptosis was measured by staining the cells first with AnnexinV/7AAD according to the manufacturer's protocol followed by their analysis using a FACScan instrument (BD). All the resulting data was analyzed using the BD Cell Quest pro software (version 5.2.1, BD).
2.7. Proximity Ligation Assay
The Proximity Ligation Assay was performed as per the manufacturer's protocol. Briefly, it involves converting potential protein–protein interactions into DNA molecules by first targeting the two interacting proteins using specific antibodies against them, which has to be generated in two entirely different hosts. These primary antibodies are then targeted by PLA probes, each specific against the primary antibody host, conjugated with a short oligonucleotide sequence. These two oligonucleotides are then ligated using a ligase providing a template for a Rolling Circle Amplification (RCA). This template is formed only when the two proteins are located within 40 nm of each other, a distance considered to be close enough for favoring their potential interaction in vivo. The RCA is then amplified by a suitable polymerase and labeled with detectable probes which can then be visualized under an immunofluorescence microscope as a fluorescent dot (Weibrecht et al., 2010).
3. Results
3.1. pERK 1/2 can prevent death receptor‐mediated apoptosis
To confirm whether pERK 1/2 can indeed protect cancer cells from undergoing death receptor‐mediated apoptosis, we first treated the ovarian cancer cell line SKOV‐3 with the synthetic MEK 1/2 inhibitor PD98059 or with the vehicle control DMSO. These cells were then treated with Trail, FasL or TNFα, in order to activate the extrinsic apoptotic pathway, over different time‐periods. Immunoblotting of the cell lysates in all the three cases demonstrated that the treatment of the cells with PD98059 lead to an increase in pro‐Caspase‐8 activation indicating that pERK 1/2 may play a role in suppressing death receptor mediated apoptosis (Figure 1B–D). Also, Trail appeared to be more potent at inducing apoptosis than FasL or TNFα in this cell line.
Figure 1.

pERK 1/2 can prevent death receptor‐mediated apoptosis. (A) Schematic representation of the pro‐Caspase‐8 activation process. (B) Randomly growing EGFR‐overexpressing SKOV‐3 cells were pre‐treated for 1 h with 100 μM of PD98059 or an equal volume of the vehicle control DMSO. Apoptosis was induced in these cells by the death receptor activating agents Trail (100 ng/ml) or (B) FasL (100 ng/ml) or (C) TNFα (20 ng/ml) for 0, 2 and 3 h where the 0 h time point indicates the absence of any apoptosis induction. Cycloheximide (CHX) (10 μg/ml) was also used in each case to enhance the efficiency of these cytotoxic agents through its ability to inhibit the synthesis of anti‐apoptotic proteins like FLIP and XIAP. Lysates of the harvested cells were resolved by SDS‐PAGE and immunoblotted for pERK 1/2, ERK 1/2, pro‐Caspase‐8 (p55/53), and Caspase‐8 cleavage product (p18). The level of β‐Actin in the lysates was used as the loading control.
3.2. Pro‐Caspase‐8 is a substrate for pERK2 (p42 MAPK) in vitro
Since ERK 1/2 are kinases, we wanted to determine whether pERK 1/2 can regulate the extrinsic apoptotic pathway by directly phosphorylating the key component of the extrinsic apoptotic pathway – pro‐Caspase‐8. For this purpose, commercially available ERK2 along with a panel of other kinases like Aurora A and B; CDK1 and 2; and CAMKII were assessed for their ability to phosphorylate GST‐tagged full length pro‐Caspase‐8 protein (wild‐type) through an in vitro kinase assay in the presence of [γ‐32P] ATP. ERK2 as well as CDK1 and 2 were able to strongly phosphorylate the pro‐Caspase‐8 (Figure 2A).
Figure 2.

Pro‐Caspase‐8 is a substrate for pERK2 (p42 MAPK) in vitro. (A) GST‐tagged full length pro‐Caspase‐8 protein (GST‐pro‐Caspase‐8 WT) was incubated with commercially available Aurora A and B, CDK1 and 2, CAMKII, ERK2 kinases and [γ‐32P] ATP for 30 min at 30 °C, resolved by SDS‐PAGE and visualized by autoradiography. (B) GST‐tagged fusion proteins of pro‐Caspase‐8 (WT (wild type), NT (N‐terminus including the prodomain), p18 and p10) were subjected to phosphorylation by commercially available ERK2. Coomassie staining of the gel (lower panel) and its autoradiography (upper panel) were performed. (C) Mass‐spectrometric analysis of the p10 sub‐fragment of Caspase‐8 was performed in the presence or the absence of the ERK2 kinase. (D) GST‐pro‐Caspase‐8 WT or the mutant full length pro‐Caspase‐8 (GST‐pro‐Caspase‐8 S387A) proteins were subjected to in vitro kinase assay by the ERK2 kinase. Coomassie stained SDS‐PAGE gel (lower panel) and its corresponding autoradiograph (upper panel) are shown. (E) The same panel of commercially available kinases were used to phosphorylate the GST‐pro‐Caspase‐8 S387A protein through an in vitro kinase assay. (F) SKOV‐3 cells were transfected with the expression vectors for GFP‐ERK 1 or GFP‐ERK 2. The expressed proteins were immunoprecipitated using an anti‐GFP antibody. The immunoprecipitates and the lysates were analyzed for the levels of pERK 1/2 and GFP‐pERK 1 or GFP‐pERK 2 (upper panel). The immunoprecipitated kinases were assessed for their abilities to phosphorylate GST‐pro‐Caspase‐8 WT, GST‐pro‐Caspase‐8 S387A and GST‐Caspase‐8 p10 fusion proteins through an in vitro kinase assay (lower panel). (G) A polyclonal antibody was generated against a peptide including the phosphorylated S387 residue of pro‐Caspase‐8. GST‐pro‐Caspase‐8 WT and GST‐pro‐Caspase‐8 S387A were incubated with commercially available CDK1, PLK1, CAMKII and ERK2 kinases along with non‐radioactive ATP. Samples were analyzed for Caspase‐8 phosphorylation using the new antibody (Caspase‐8 pS387) and for the levels of pro‐Caspase‐8. (H) Increasing amounts of the GST‐pro‐Caspase‐8 WT protein were then incubated with equal amounts of the ERK2 kinase and ATP to perform a non‐radioactive kinase assay. The kinase or GST‐pro‐Caspase‐8 WT alone was used as negative controls. The combination of protein‐kinase was assessed for phospho‐Caspase‐8 (Caspase‐8 pS387), pro‐Caspase‐8 and pERK2.
To assess the site/domain at which ERK 1/2 phosphorylate pro‐Caspase‐8, we next performed an in vitro kinase assay involving the kinase and GST‐tagged fusion proteins of different pro‐Caspase‐8 sub‐fragments. It was observed that the major phosphorylation site for ERK2 on pro‐Caspase‐8 was located in its p10 sub‐fragment (Figure 2B).
The p10 sub‐fragment of Caspase‐8 was also subjected to Mass Spectrometric analysis in the presence or the absence of ERK2. The result revealed that the kinase phosphorylates the p10 sub‐fragment sequence EEQPYLEMDLSpSPQTR at the S387 residue (Figure 2C). To further confirm that S387 is indeed the site of ERK2‐mediated phosphorylation of pro‐Caspase‐8, a GST‐tagged full length pro‐Caspase‐8 mutant protein was generated (GST‐pro‐Caspase‐8 S387A) and subjected to an in vitro kinase assay in the presence of the kinase. A complete loss of phosphorylation signal was observed in case of the mutant protein (Figure 2D). In addition, a comparative in vitro kinase assay involving [γ‐32P] ATP was performed with GST‐pro‐Caspase‐8 S387A along with the panel of commercially available kinases. The phosphorylation signal for CDK2 reduced significantly while that for ERK2 and CDK1 were completely lost as compared to their respective phosphorylation signals in the presence of the GST‐pro‐Caspase‐8 WT protein (Figure 2A and E). This indicated that, while for the ERK2 and CDK1, S387 is the only phosphorylation site on pro‐Caspase‐8, it is a major but not the only phosphorylation site for CDK2.
These data indicated that at least in vitro, the ERK2 MAPK can phosphorylate pro‐Caspase‐8 at its S387 residue. But does ERK1 MAPK can also perform a similar function? Since, purified ERK1 kinase is not available commercially, we next transfected SKOV‐3 cells with expression vectors for GFP‐ERK 1 or GFP‐ERK 2 (Figure 2F, upper panel). These kinases were then immunoprecipitated and assessed for their ability to individually phosphorylate GST‐pro‐Caspase‐8 WT, S387A and p10 fusion proteins using an in vitro kinase assay. Both the kinases were able to phosphorylate pro‐Caspase‐8 at S387 in the WT and p10 fusion proteins while, the phospho signal disappeared completely when the S387 site was mutated (Figure 2F, lower panel). This confirmed that pro‐Caspase‐8 is phosphorylated at the same position (S387) by both ERK 1/2.
Based on these findings, we generated a polyclonal antibody against this phosphorylation site (Caspase‐8 pS387) by immunizing rabbits with the peptide YLEMDLSpSPQTRY. In order to assess the specificity of this antibody, GST‐pro‐Caspase‐8 WT or GST‐pro‐Caspase‐8 S387A were subjected to a non‐radioactive (cold) kinase assay in the presence of commercially available ERK2, CDK1, PLK1 and CAMKII. While the GST‐pro‐Caspase‐8 WT protein, phosphorylated at S387 by ERK2 and CDK1, was recognized by the new phospho‐specific antibody, it couldn't detect this signal in the GST‐pro‐Caspase‐8 S387A protein (Figure 2G). Furthermore, we also treated increasing amounts of the GST‐Caspase‐8 WT protein with equal amounts of the ERK2. Increasing levels of GST‐pro‐Caspase‐8 WT phosphorylation was detected by the antibody with corresponding increase in pro‐Caspase‐8 protein levels (Figure 2H). Also, as little as 50 ng of the phosphorylated Caspase‐8 was recognized by the new antibody while no phosphorylation signal was detected when the kinase or the substrate was present alone. Both these experiments (Figure 2G and H) thus confirm the sensitivity and the specificity of this Caspase‐8 pS387 antibody.
3.3. Pro‐Caspase‐8 can interact with pERK 1/2 in vivo
To determine whether these kinases interact with their substrate pro‐Caspase‐8 in vivo as well, we performed a Proximity Ligation Assay (PLA) in the SKOV‐3 cells. The number of florescent spots was counted under a fluorescent microscope and has been represented graphically. It was observed that the number of fluorescent spots greatly increased when the SKOV‐3 cells were treated with antibodies against both pERK 1/2 and pro‐Caspase‐8 followed by treatment with the PLA probes (Figure 3A). Each of these fluorescent spots represents the presence of both the kinases and pro‐Caspase‐8 at distances (40 nm) close enough to potentially interact with each other within a cell.
Figure 3.
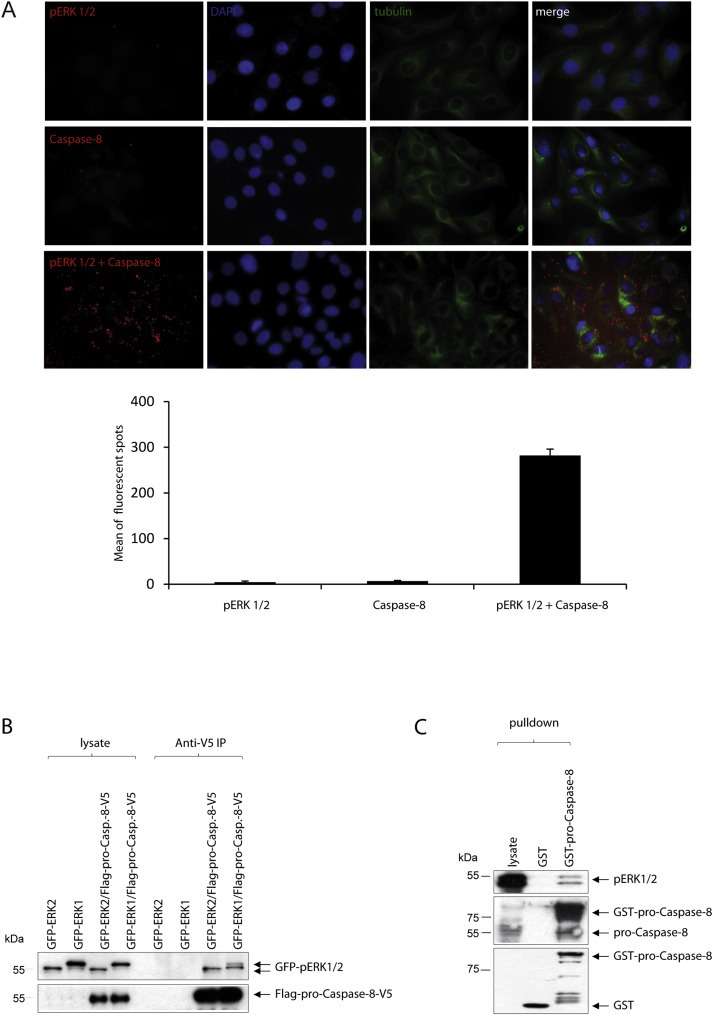
Pro‐Caspase‐8 can interact with pERK 1/2 in vivo. (A) Randomly growing SKOV‐3 cells were seeded on slides, fixed, permeablized, blocked and then labeled with the anti‐pERK 1/2 (rabbit) or anti‐pro‐Caspase‐8 (mouse) antibodies or both. The nuclear region of these cells was identified by staining their DNA with DAPI while their cellular cytoskeleton was shown by staining them with a GFP‐labeled anti‐tubulin antibody. The stained cells were subjected to an in situ Proximity Ligation Assay (PLA). Each of the fluorescent spots seen under an immunofluorescence microscope represented the close proximity, in the order of 40 nm, of two potentially interacting proteins within the cells. The number of fluorescent spots were counted in five different fields, each field having approximately 15–20 cells (anti‐pERK 1/2 or anti‐pro‐Caspase‐8 or both), and the mean number of spots per field was represented graphically. (B) Randomly growing HeLa cells were transfected with GFP‐ERK 1 or GFP‐ERK 2 or co‐transfected with the expression vectors for Flag‐ and V5‐tagged pro‐Caspase‐8. The pro‐Caspase‐8 was then immunoprecipitated using an anti‐V5 antibody. Immunoblots of the lysates and the immunoprecipitates were stained with anti‐Flag and anti‐GFP antibodies to assess for the presence of immunoprecipitated pro‐Caspase‐8 and for the expression or co‐immunoprecipitation of GFP‐ERK 1 or GFP‐ERK 2, respectively. (C) GST alone or GST‐pro‐Caspase‐8 was incubated with the lysates of randomly growing SKOV‐3 cells. pERK 1/2 that was pulled‐down by the GST‐pro‐Caspase‐8 was detected by immunoblotting.
To further evaluate whether pERK 1/2 can indeed bind to pro‐Caspase‐8 directly, SKOV‐3 cells were transfected with expression vectors for GFP‐ERK 1 or GFP‐ERK 2 along with expression vectors for Flag‐ and V5‐tagged pro‐Caspase‐8. When the immunoprecipitated Flag‐pro‐Caspase‐8‐V5 protein was then probed using western blot for pERK 1/2, it was observed that both of them could bind to the pro‐Caspase‐8 (Figure 3B).
We also performed a pull‐down assay to assess whether the GST‐pro‐Caspase‐8 protein can bind to endogenous ERK 1/2 or pERK 1/2. Immunoblot analysis demonstrated that the GST‐pro‐Caspase‐8 protein was able to pull‐down pERK 1/2 from the lysates of SKOV‐3 cells (Figure 3C). These experiments therefore indicate that pERK 1/2 and pro‐Caspase‐8 are interacting protein partners in vivo.
3.4. The cellular effects of pERK 1/2‐induced phosphorylation of pro‐Caspase‐8
Since the phosphorylation site S387 is located in close proximity to the Caspase‐8 cleavage site D384, we wanted to investigate whether pERK 1/2 can inhibit the extrinsic apoptotic pathway in cancer cells by phosphorylating pro‐Caspase‐8 at S387 and preventing its further cleavage, in vivo. To establish this, we selected two EGFR‐overexpressing cancer cell lines – SKOV‐3 and MDA‐MB‐468.
The ERK 1/2 activities in randomly growing SKOV‐3 cells were inhibited by PD98059 and then subjected to Trail‐induced apoptosis for different time periods. A marked increase in Caspase‐8 cleavage and a reduction in Caspase‐8 (pS387) levels were observed in the cell lysates when the ERK 1/2 activities were inhibited (Figure 4A). Caspase‐8 activity in these lysates was also measured using the Caspase‐8 Glo‐Assay and showed a similar time‐dependent increase in activity in the absence of pERK 1/2 (Figure 4B). The levels of overall apoptosis, in the presence or the absence of pERK 1/2, in these cells were evaluated by determining the incorporation of AnnexinV/7AAD in these cells through FACS analysis. It was observed that the percentage of overall apoptosis in the inhibitor‐treated cells increased significantly, almost doubling 2 h after Trail‐treatment till the end of the observation period (3 h), as compared to the DMSO‐treated controls (Figure 4C).
Figure 4.
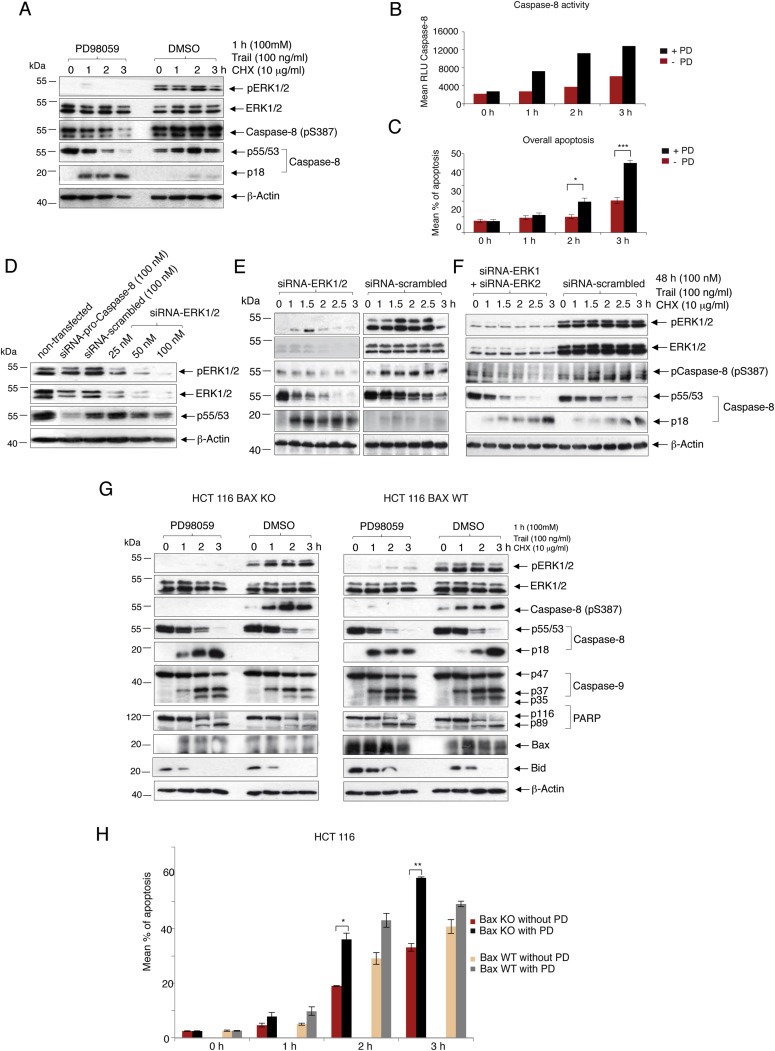
The cellular effects of pERK 1/2‐induced phosphorylation of pro‐Caspase‐8 in SKOV‐3 (Type I) and Bax KO‐ or Bax WT‐HCT 116 (Type II) cells. ERK 1/2 activation was inhibited in randomly growing SKOV‐3 cells by pre‐treatment with 100 μM PD98059 for 1 h. DMSO‐treated cells were used as negative control. Apoptosis was induced in these cells by treating with a combination of Trail (100 ng/ml) and CHX (100 μg/ml) for different time‐periods in the presence of PD98059 or DMSO. (A) Harvested cells were either lysed to assess for the presence of active (pERK 1/2) or whole ERK 1/2, pCaspase8 (S387), p55/53 and p18 through immunoblotting and (B) to measure the Caspase‐8 activity through a Caspase‐8 Glo‐Assay or (C) were used for measuring the overall apoptosis by AnnexinV/7AAD FACS analysis (mean ± SEM, n = 3). (D) SKOV‐3 cells were non‐transfected or transfected with 25, 50 and 100 nM of siRNA‐ERK 1/2 or 100 nM siRNA‐pro‐Caspase‐8 (positive control) or 100 nM of siRNA‐scrambled (negative control). 48 h after transfection, these cells were harvested, lysed and assessed for the presence of pERK 1/2, ERK 1/2 and p55/53. Protein loading was verified by detecting the amounts of β‐Actin in the lysates. (E) SKOV‐3 cells were transfected with 100 nM of either siRNA‐ERK 1/2 (Cell Signaling) or siRNA‐scrambled for 48 h. This was followed by initiation of apoptosis for different time‐periods using Trail and CHX. At the end of each time‐point, the cells were harvested and their lysates were immunoblotted to assess for the presence of pERK 1/2 and ERK 1/2, pCaspase8 (S387), p55/53, p18 and β‐Actin. (F) SKOV‐3 cells were transfected with a combination of 100 nM each of siRNA‐ERK 1 and siRNA‐ERK2 (Santa Cruz Biotechnologies) as well as an equal concentration of siRNA‐scrambled for 48 h. This was followed by initiation of apoptosis for different time‐periods using Trail and CHX. At the end of each time‐point, the cells were harvested and their lysates were immunoblotted to assess for the presence of pERK 1/2 and ERK 1/2, pCaspase8 (S387), p55/53, p18 and β‐Actin. Randomly growing Bax knock‐out (Bax KO)‐ or Bax wild‐type (Bax WT)‐HCT 116 cells were similarly subjected to Trail induced apoptosis for different time‐periods (G) Lysates of the harvested cells were used for detecting pERK 1/2, ERK 1/2, p‐Caspase‐8 (S387), pro‐Caspase‐8, p18, pro‐Caspase‐9 and its cleavage products (p37 and p35), Bax, PARP, BID and β‐Actin. (H) Harvested cells were also assessed for the levels of overall apoptosis (mean ± SEM, n = 3) (P‐value – * ≤0.05, ** ≤0.005, *** ≤0.0005).
Small molecule inhibitors like PD98059 are known to have off‐target specificity (Anastassiadis et al., 2011; Davis et al., 2011). To verify any possibility of the inhibitor enhancing Caspase‐8 induced apoptosis through any mechanism other than that involving the inhibition of pERK 1/2, the expressions of ERK 1/2 were inhibited with an siRNA‐ERK 1/2 (Cell Signaling). Once an optimum siRNA‐ERK 1/2 concentration was determined for the effective depletion for ERK 1/2 protein expression (100 nM) (Figure 4D), SKOV‐3 cells were treated with equal concentrations (100 nM) of the siRNA‐ERK 1/2 or the siRNA‐scrambled followed by Trail‐treatment over different durations. Immunoblots of the cell lysates showed a marked increase in Caspase‐8 cleavage and a reduction in Caspase‐8 (pS387) levels in the siRNA‐ERK 1/2‐treated cells as compared to the controls (Figure 4E). These observations were also validated in a similar experiment involving a combination of specific commercial siRNAs against ERK1 and ERK2 (Santa Cruz Biotechnologies). Immunoblotting the cell lysates once again showed an increase in Caspase‐8 cleavage and a reduction in Caspase‐8 (pS387) when ERK1 and 2 expressions were knocked down, as compared to the siRNA‐scrambled treated cells (Figure 4F). These two experiments (Figure 4E and F) provided further evidence that pro‐Caspase‐8 is phosphorylated at S387 and its processing depends on ERK 1/2 activities in vivo.
Similar to the SKOV‐3 cells, ERK 1/2 activation was also inhibited in the MDA‐MB‐468 cells using the small‐molecule inhibitor PD98059 followed by the induction of apoptosis using Trail (Supplementary Figure 1A). Western blot analysis of the cell lysates demonstrated a robust increase in Caspase‐8 cleavage associated with a reduction in the amounts of Caspase‐8 (pS387) in the inhibitor‐treated cells as compared to the controls (Supplementary Figure 1A). Caspase‐8 activity also increased significantly after Trail treatment in the absence of active ERK 1/2 (Supplementary Figure 1B). However, the difference in the Caspase‐3/7 activity between the inhibitor and the DMSO treated controls were surprisingly less pronounced (Supplementary Figure 1C). Similarly, the difference in the levels of overall apoptosis in the presence or the absence of the inhibitor was also very nominal (Supplementary Figure 1D). Taken together, these findings indicated that in the MDA‐MB‐468 cells, additional factors other than Caspase‐8 might be involved in driving the cells towards apoptosis in response to death receptor activation. We therefore decided to first establish the subtype (Type I or II) of these cells.
Towards this goal, we tried to silence the expression of the BID gene by RNAi in the MDA‐MB‐468 cells. siRNA‐scrambled transfected cells were used as negative controls. Apoptosis was induced in these cells by treating them with Trail for 2 h in the presence or the absence of pERK 1/2. For comparison, the SKOV‐3 cells were also subjected to similar treatment as the MDA‐MB‐468 cells. The MDA‐MB‐468 cells demonstrated the activation of the intrinsic apoptotic pathway in the presence of Trail, as measured by the levels of Caspase‐9, XIAP and Caspase‐3, while this was not the case with the SKOV‐3 cells (Supplementary Figure 2A and B). While the use of 100 nM of siRNA‐BID downregulated the BID gene expression effectively in the SKOV‐3 cells, substantial levels of the BID protein could still be detected in the MDA‐MB‐468 cells, which was sufficient to activate the intrinsic apoptotic pathway upon Trail stimulation. This indicates that while the SKOV‐3 cell line belong to the Type I subtype of cells, the MDA‐MB‐468 cells come under the Type II subtype.
Type II cancer cells frequently exhibit resistance to death receptor‐mediated apoptosis by inactivating the intrinsic apoptotic pathway (Matthess et al., 2010; Zhang and Fang, 2005). In order to mimic such conditions in the MDA‐MB‐468 cells, we decided to once again knock‐down the BID gene in these cells, pre‐treat them with or without PD98059 and then induce apoptosis with Trail over different time periods. Immunoblotting of the lysates showed an increase in the levels of Caspase‐8 cleavage corresponding with a decrease in the levels of Caspase‐8 (pS387) (Supplementary Figure 1E). This was also reflected upon measuring the Caspase‐8 activity (Supplementary Figure 1F). However, like their non‐transfected counterparts (Supplementary Figure 1C), there was no significant increase in Caspase‐3 activity upon PD98059 treatment in the siRNA‐BID transfected cells (Supplementary Figure 1G). It was also observed that while knocking down the BID protein levels did reduce the amount of overall apoptosis (Supplementary Figure 1H) as compared to the non‐transfected cells (Supplementary Figure 1D), it didn't lead to any significant difference in overall apoptosis in the presence or the absence of pERK 1/2. This once again reflects BID's ability to amplify even the slightest apoptotic signal derived from active Caspase‐8 and in turn successfully activate the intrinsic apoptotic pathway (Schungel et al., 2009), and our inability to completely knock‐down the BID gene in the MDA‐MB‐468 cells meant that we weren't able to effectively shut down the intrinsic apoptotic pathway in these cells. As a result, even in the DMSO treated cells, a small amount of active Caspase‐8 could cleave any residual BID which could in turn amplify this signal thereby overcoming the anti‐apoptotic functions of pERK 1/2.
We therefore induced apoptosis in the Type II colon cancer cell line HCT 116 (Ozoren and El‐Deiry, 2002) in which the pro‐apoptotic Bcl‐2 associated X protein (Bax) gene had been knocked out (Bax KO), rendering its intrinsic apoptotic pathway muted. Comparisons were made with HCT 116 cell line with normal Bax gene expression (Bax WT). Lysates of the cells showed that while BID was cleaved upon Trail treatment in both the Bax KO and Bax WT cells, there was comparatively less Caspase‐9 cleavage in the inhibitor treated Bax KO cells as compared to its DMSO treated counterparts. This was not the case in the Bax WT cells. A similar trend was seen with the PARP cleavage also (Figure 4G). However, there was a major decrease in the Caspase‐8 (p18) level in the DMSO treated Bax KO cells as compared to their Bax WT counterparts, after Trail treatment. It is possible that a fully functional intrinsic apoptotic pathway in the Bax WT cells leads to higher amounts of pro‐Caspase‐3 activation as compared to the Bax KO cells. Active Caspase‐3 has been reported to be able to cleave the p43/41 sub‐fragment of intermediately processed pro‐Caspase‐8 into Caspase‐8 (p18) (Ferreira et al., 2012). The Bax KO cells also showed a significant increase in the levels of overall apoptosis in the absence of pERK 1/2 after 2 and 3 h of Trail treatment as compared to the DMSO treated controls. This trend was not visible in the Bax WT cells (Figure 4H) indicating that inhibiting ERK 1/2 activities can sensitize even Type II cancer cells, with impaired intrinsic apoptotic pathway, to Trail induced apoptosis.
3.5. ERK 1/2 is inactive during mitosis in SKOV‐3 and MDA‐MB‐468 cell lines
The distribution of pERK 1/2 during the cell cycle and their roles in cancer cells have been debated for over two decades (Chambard et al., 2007; Harding et al., 2003; Meloche, 1995; Meloche and Pouyssegur, 2007; Yamamoto et al., 2006). However, to determine whether pERK 1/2 can equally protect the SKOV‐3 and MDA‐MB‐468 cell lines from Caspase‐8‐mediated apoptosis throughout their cell cycle, we wanted to assess their activation status during the different phases of the cell cycle in these cells.
The SKOV‐3 and MDA‐MB‐468 cells were enriched in G1/S, S, G2 and M phases. Lysates of these cells displayed that in case of both the cell lines, ERK 1/2 were almost completely inactivated during mitosis (Figure 5A and B). Apart from analyzing the levels of different cell cycle markers (Figure 5A and B), the distribution of these cells in the different phases of the cell cycle was also examined by PI‐FACS (Figure 5C and D).
Figure 5.

ERK 1/2 is inactive during mitosis in SKOV‐3 and MDA‐MB‐468 cell lines. SKOV‐3 and MDA‐MB‐468 cells were arrested in G1/S phase, G2 phase and M phase. Non‐synchronized (U/T) cells were used as negative control. Harvested cells were either lysed to assess for the levels of pERK 1/2 and ERK 1/2, the mitotic markers Cyclin B1 and PLK1, the G1/S and S phase marker Cyclin E and β‐Actin (A and B) or were directly fixed and treated with the fluorescent labelled DNA intercalating stain Propidium Iodide (PI) to determine their cell‐cycle distribution by measuring their DNA contents (C and D). (E) SKOV‐3 cells were arrested during the M phase using thymidine‐Nocodazole‐treatment. Mitotic cells were collected by shake‐off, washed twice with PBS and released into fresh medium. Cells were harvested every 3 h until 24 h, harvested, lysed and immunoblotted. The progress of these cells out of mitosis was followed by assessing for the mitotic markers Cyclin B1 and PLK1 and compared with the corresponding levels of pERK 1/2 and ERK 1/2, at each time‐point. Non‐synchronized (U/T) cells were used as the negative control.
Next, SKOV‐3 cells were first enriched in pro‐metaphase by thymidine‐Nocodazole‐treatment and then released into fresh medium over a period of 24 h. Cells were harvested at regular intervals of 3 h, lysed and immunoblotted. It was observed once again that ERK 1/2 remained inactive so long as the cells remained in mitosis. Immediately after the exit of these cells from mitosis, i.e. 12 h after their release from Nocodazole treatment, pERK 1/2 levels started to increase until the end of the observation period (Figure 5E).
Taken together, these results strongly suggest that at least in these EGFR‐overexpressing cell lines (SKOV‐3 and MDA‐MB‐468), the activities of ERK 1/2 are limited exclusively to the interphase of their respective cell cycles.
3.6. Caspase‐8 is more active during G1/S than G2 phase in the absence of pERK 1/2
Having demonstrated that ERK 1/2 are active during all but the mitotic phase in the SKOV‐3 and MDA‐MB‐468 cells, we asked whether their anti‐apoptotic properties are also similarly prevalent throughout the interphase in these cell lines. For this purpose, apoptosis was induced in SKOV‐3 cells enriched in the G1/S and G2 phases, in the presence or the absence of pERK 1/2. Comparisons were made with similarly treated randomly growing or M phase arrested SKOV‐3 cells.
The cell cycle distribution of the randomly growing, G1/S‐ and G2‐phase enriched cells was analyzed using PI‐FACS (Supplementary Figure 3A–C). Lysates of these cells were assessed for Caspase‐8 activity and presence of Caspase‐8 (pS387) through western blot (Figure 6A–D, upper panels). Overall apoptosis in these cells was also measured by AnnexinV/7AAD FACS analysis (Figure 6A–D, lower panels). It was observed that the inhibition of ERK 1/2 activation yielded the highest percentage of Caspase‐8 activity and also overall apoptosis in G1/S phase arrested cells as compared to the G2 phase arrested or randomly growing SKOV‐3 cells. These findings also corresponded with Caspase‐8 (pS387) levels which decreased the most in the G1/S phase cells in the presence of the inhibitor (Figure 6A–D, upper panels). Expectedly, the mitotic phase cells showed no apparent differences in the levels of Caspase‐8 (pS387), Caspase‐8 activity or overall apoptosis between the inhibitor (PD98059) treated or untreated samples. These findings show that pERK 1/2 have the strongest anti‐apoptotic role during the G1/S phase in SKOV‐3 cells.
Figure 6.

Caspase‐8 is more active during G1/S than G2 phase in the absence of pERK 1/2. Randomly growing SKOV‐3 cells (A) or those enriched in the G1/S (B), G2 (C) and M (D) phases were pre‐treated for 1 h with 100 μM PD98059 or an equal volume of DMSO as control and subjected to Trail induced apoptosis for different time‐periods. The 0 h time point represents the absence of any apoptosis induction. Shake‐off cells enriched in mitosis were released into fresh medium supplemented with 10 μg/ml of MG132 to prevent their exit from mitosis. Harvested cells from each time‐point in every phase were assessed for overall apoptosis or lysed for immunoblotting and stained with pERK 1/2, ERK 1/2, p55/53, p18, Caspase‐8 (pS387), Cyclin A, Cyclin B1 and β‐Actin. (mean ± SD, n = 3).
3.7. pERK 1/2 induced phosphorylation of pro‐Caspase‐8 inhibits its further activation in vivo
To investigate whether the phosphorylation of pro‐Caspase‐8 at S387 is actually responsible for the anti‐apoptotic properties of ERK 1/2, the endogenous pro‐Caspase‐8 in the SKOV‐3 cells was first silenced by an siRNA‐Caspase‐8 and then replaced with exogenous Flag‐tagged wild‐type (WT) or non‐phosphorylatable (S387A) or phospho‐mimicking (S387E) pro‐Caspase‐8 mutants. Non‐transfected (U/T) or siRNA‐scrambled transfected SKOV‐3 cells were used as the negative controls. Comparisons were also made with cells in which the endogenous pro‐Caspase‐8 had been knocked down but was not replaced with any exogenous Caspase‐8. All these cells were subjected to Trail‐induced apoptosis in the presence or the absence of pERK 1/2. SKOV‐3 cells in which the endogenous pro‐Caspase‐8 had been replaced with the Flag‐tagged pro‐Caspase‐8 (S387A) demonstrated the highest amount of Caspase‐8 (p18) as compared to the Flag‐tagged pro‐Caspase‐8 (WT) or the Flag‐tagged pro‐Caspase‐8 (S387E) transfected cells. Expectedly, the levels of Caspase‐8 (p18) were almost the same in the PD98059 or the DMSO treated cells (Figure 7A and B). It should be noted here that the SKOV‐3 cells were subjected to significant stresses as a result of the transfections of the different pro‐Caspase‐8. As a result, basal level of p18 was detected even in the absence of any Trail treatment (0 h), possibly as a consequence of stress‐induced activation of pro‐Caspase‐9 and subsequent pro‐Caspase‐3 activation, as has been mentioned earlier (Ferreira et al., 2012).
Figure 7.

pERK 1/2 induced phosphorylation of pro‐Caspase‐8 inhibits its further activation in vivo. Randomly growing SKOV‐3 cells were first transfected with 20 nM of siRNA‐Caspase‐8 targeting the untranslated region of pro‐Caspase‐8 or 20 nM of siRNA‐scrambled. These cells were either incubated for 24 h after transfection, the siRNA‐Caspase‐8 treated cells were re‐transfected with vectors encoding Flag‐tagged pro‐Caspase‐8 WT or the non‐phosphorylatable pro‐Caspase‐8 S387A or the phospho‐mimicking pro‐Caspase‐8 S387E and incubated for a further 24 h. Apoptosis was then induced in these cells with 100 ng/ml of Trail either in the presence (A) or the absence (B) of pERK 1/2. The 0 h time point represents the absence of any apoptosis induction. Lysates were analyzed for pERK 1/2, ERK 1/2, transfected Flag‐pro‐Caspase‐8, pro‐Caspase‐8 and its cleavage product (p18) and β‐Actin.
3.8. Inhibiting ERK 1/2 activation also sensitizes primary lymphocytes to Trail‐induced apoptosis
To determine whether pERK 1/2 can also inhibit pro‐Caspase‐8‐mediated apoptosis in primary cells, Peripheral Blood Lymphocytes (PBLs) were isolated from the blood of healthy donors and then stimulated with or without the cytokine Interleukin‐2 (IL‐2). These cells were then pre‐treated with either PD98059 or DMSO for 1 h and then subjected to Trail induced apoptosis over different time periods. Immunoblot of the cell lysates were analyzed for ERK 1/2 activities and also Caspase‐8 cleavage (Supplementary Figure 4). Similar to the cancer cell lines, an enhancement in Caspase‐8 cleavage was observed only in the absence of pERK 1/2. Interestingly, in the DMSO treated controls, no pERK 1/2 could be detected at the 3 h time point, which coincided with the appearance of the p18 sub‐fragment of Caspase‐8, thus hammering in the importance of pERK 1/2 in preventing pro‐Caspase‐8 activation, even in normal human cells.
4. Discussion
Normal human cells have to acquire novel capabilities through a series of genetic mutations in order to progressively develop into the more than 100 different known cancer types. Cancer cells are therefore susceptible to being targeted for apoptosis by the body's defensive mechanisms and avoiding this is essential for them to develop further (Hanahan and Weinberg, 2000, 2011). Owing to the enormous significance of pro‐Caspase‐8 in initiating apoptosis in both Type I and II cells, it is frequently subjected to inactivation in cancer cells (Fulda, 2009). Cellular kinases can often phosphorylate pro‐Caspase‐8 rendering it unable to undergo further processing even in the presence of death receptor activating agents. Over the years, different groups have been able to identify a handful of kinases that can carry out such phosphorylation. The p38 MAPK can phosphorylate pro‐Caspase‐8 at Ser364 (Alvarado‐Kristensson et al., 2004), the Src kinase at Tyr380 (Cursi et al., 2006), CDK1 at Ser387 (Matthess et al., 2010) and finally RSK2 at Thr263 (Peng et al., 2011). Active ERK 1/2 have also been reported to protect cancer cells from undergoing apoptosis in the presence of cytotoxic agents like Trail, FasL or TNFα and we intended to determine the mechanism(s) behind this anti‐apoptotic function of pERK 1/2 (Holmstrom et al., 1998, 2000, 2005, 2004, 2009, 2001, 2003).
The EGFR‐Ras‐Raf‐MEK‐ERK MAPK cascade is one of the most studied kinase pathways due to its wide ranging functions and the frequent deregulation of its members in cancer cells. One or more of the upstream kinases that activate ERK 1/2 are deregulated in a large variety of cancers (Fernandez‐Medarde and Santos, 2011; Lev et al., 2004; Lo, 2012; McCubrey et al., 2007; Pearson and Fabbro, 2004; Satyamoorthy et al., 2003; Steinmetz et al., 2004; Zhang et al., 2003; Zhou et al., 2010). As a direct consequence ERK 1/2 are found to be hyperactive in melanoma, ovarian, leukemia, breast, renal cell, prostate and other cancers as compared to their respective normal cell counterparts. Steinmetz et al. have also reported high levels of ERK 1/2 in the metastatic tumors of 75% ovarian cancer patients (Satyamoorthy et al., 2003; Steinmetz et al., 2004; Towatari et al., 1997). However, despite ERK 1/2's frequent hyperactivity in cancer cells and their roles in protecting cancer cells from undergoing apoptosis, the exact mechanisms behind their anti‐apoptotic functions remain elusive.
Our experiments with commercially available or immunoprecipitated ERK 1/2 demonstrate that both active ERK 1 and 2 can phosphorylate pro‐Caspase‐8 at S387 in vitro. Work done in the EGFR‐overexpressing SKOV‐3 (Type I) and MDA‐MB‐468 (Type II) cancer cell lines also show that this is a major site for the post‐translational modification of pro‐Caspase‐8 by pERK 1/2 in vivo, as rendering this phosphorylation site non‐phosphorylatable (S387A) in a replacement experiment, significantly enhances Trail‐induced apoptosis even in the presence of pERK 1/2 (Figure 7). It is interesting to note here that contrary to previously published report (Zhang and Fang, 2005), we did not observe any significant increase in the activities of ERK 1/2 upon Trail treatment, ruling this out as the reason behind pERK 1/2's anti‐apoptotic function at least in these cancer cell lines. It is possible though that these kinases are activated and regulated differently in different cancer cells.
In Type II cells, following Caspase‐8 induced activation, active BID (tBID) oligomerises the pro‐apoptotic Bax protein which in turn leads to the release of cytochrome‐c from the mitochondria culminating in the activation of pro‐Caspase‐9 (Eskes et al., 2000; Gillissen et al., 2013). Inhibiting ERK 1/2 activities resulted in a significant increase in overall apoptosis, upon Trail treatment, in the Bax KO cells as compared to the Bax WT cells (Figure 4). It is imperative to note here that the knock‐out of the Bax gene alone is not sufficient to silence the intrinsic apoptotic pathway completely. Bak, another pro‐apoptotic member of the Bcl‐2 family, has similar and overlapping function in activating the intrinsic apoptotic pathway as Bax (Fulda and Debatin, 2006; Zhang and Fang, 2005). However, this further emphasizes that inhibiting pERK 1/2 mediated phosphorylation of pro‐Caspase‐8 at its S387 can potentially sensitize even Type II cancer cells, with a muted intrinsic apoptotic pathway, to Trail‐induced apoptosis. It remains to be seen whether a double knockout (Bax/Bak KO) HCT‐116 cell line shows higher levels of overall apoptosis than its Bax WT and Bax KO counterparts. This is important since a blocked intrinsic apoptotic pathway is considered to cause poor prognosis in cancer patients while imparting resistance to Trail that activate the extrinsic apoptotic pathway or other chemotherapeutic agents that rely on inducing DNA damage to trigger apoptosis (Fulda et al., 2002; Zhang and Fang, 2005).
It has been variously claimed that ERK 1/2 remain active starting from the S phase, all the way till the end of mitosis and that they are absolutely essential in the G2‐M transition in HeLa and NIH 3T3 cells (Chambard et al., 2007) or get inactivated at the end of the G2 phase and have no roles to play in the mitotic entry of HeLa cells (Harding et al., 2003). Sustained levels of pERK 1/2 have also been reported to be essential for the G1‐S transition of a cell although they are not required for S phase entry and consequently are rapidly inactivated at the end of this transition (Meloche, 1995; Meloche and Pouyssegur, 2007). However, we have shown here that ERK 1/2 activities are limited to the interphase in the SKOV‐3 and the MDA‐MB‐468 cell lines (Figure 5).
Interestingly, although these two kinases remain active throughout the interphase in the SKOV‐3 cells, inhibiting their activation during the G1/S and the G2 phases didn't translate into equal enhancement in Trail‐induced apoptosis. In fact, inactivating them sensitized the SKOV‐3 cells to apoptosis the most during the G1/S phase (Figure 6). It is possible that during the G2 phase, there are other factors that also negatively influence the extrinsic apoptotic pathway and need to be further investigated. One example could be the kinase CDK2 which remains active during the late S and G2 phases (Hu et al., 2001). We have shown here through our work that CDK2 can also phosphorylate pro‐Caspase‐8 at its S387 residue in vitro (Figure 2A and E). It is possible that the reduced level of overall apoptosis, when pERK 1/2 is inhibited during the G2 phase or in randomly growing cells, compared to the G1/S phase, is due to synergism between pERK 1/2 and CDK2 to phosphorylate pro‐Caspase‐8, during the G2 phase. CDK2 has also been reported to be hyperactive in certain tumors and have been correlated with higher proliferative activity of the tumor cells (Dong et al., 2001). It is possible that in such tumors, CDK2 can phosphorylate pro‐Caspase‐8 at S387, even in the absence of pERK 1/2.
Our work with human primary blood lymphocytes demonstrate that the anti‐apoptotic functions of pERK 1/2 is not limited to only cancer cells but can occur even in normal cells with hyperactive ERK 1/2 (Supplementary Figure 4). It should be noted however, that Trail has been known to selectively target cancer cells and therefore are being evaluated in clinical trials (Bellail et al., 2009). It therefore, needs to be assessed whether the effectiveness of Trail, as an anti‐cancer therapeutic, can be enhanced by using it in combination with an inhibitor of ERK 1/2 synthesis or its activation. Since previous work done in our lab has shown that the key Cyclin Dependent Kinase, CDK1 can also phosphorylate pro‐Caspase‐8 at S387 during mitosis, thus inhibiting its further activation (Matthess et al., 2010), we propose here a model in which these three kinases, pERK 1/2 and CDK1/Cyclin B1, together can protect cancer cells from undergoing Caspase‐8 mediated apoptosis in response to cytotoxic agents like Trail, FasL or TNFα, throughout the entire duration of their cell cycle (Figure 8).
Figure 8.
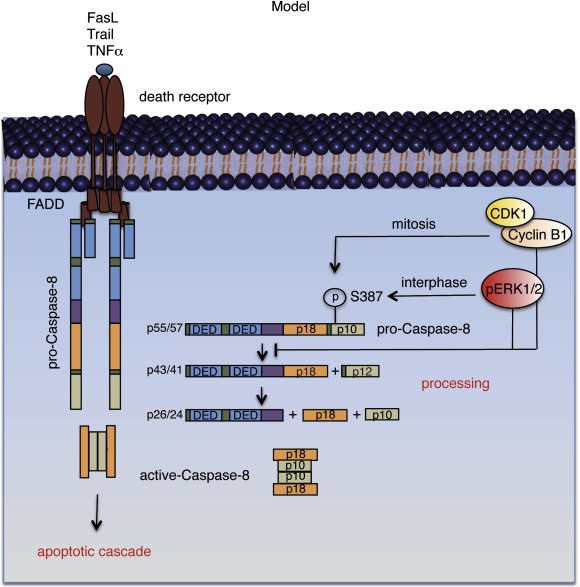
Proposed model for the inactivation of pro‐Caspase‐8 through phosphorylation at S387 by pERK 1/2 and CDK1/Cyclin B1 during the entire cell cycle in cancer cells. During the interphase of the cell cycle, ERK 1/2 remains in their active form, pERK 1/2. pERK 1/2 associates with pro‐Caspase‐8 and phosphorylates it at S387 preventing its further cleavage and the formation of the enzymatically active Caspase‐8. As a consequence, pERK 1/2 prevents pro‐Caspase‐8 from initiating the activation of downstream effector Caspases (pro‐Caspase‐3/7), in response to cytotoxic agents such as Trail, FasL and TNFα. While pERK 1/2 can protect cancer cells from undergoing death receptor pathway‐induced apoptosis during the interphase, CDK1/Cyclin B1 performs a similar function during mitosis also by phosphorylating pro‐Caspase‐8 at S387 and preventing its further activation, thereby covering the entire cell cycle.
5. Conclusion
We report that pERK 1/2 phosphorylate pro‐Caspase‐8 at its S387 residue during the interphase. In Type I cancer cells, replacing the endogenous pro‐Caspase‐8 with a non‐phosphorylatable pro‐Caspase‐8 mutant (S387A) enhances Trail induced apoptosis. Interestingly, inactivating ERK 1/2 activities could also significantly sensitize Type II cancer cells, with muted intrinsic apoptotic pathway, to Trail induced apoptosis. Combining a chemotherapeutic agent like Trail, which is known to selectively induce the apoptosis in cancer cells (Bellail et al., 2009), along with an inhibitor of ERK 1/2 activation, could therefore enhance Trail's efficiency against both Type I and II cancer cells.
Conflict of interest
The authors declare no potential conflict of interest.
Author contribution
Project Conception and design: Ranadip Mandal, Monika Raab and Klaus Strebhardt.
Experimental work and interpretation of data: Ranadip Mandal, Monika Raab and Yves Matthess.
Manuscript writing: Ranadip Mandal, Sven Becker, Rainald Knecht and Klaus Strebhardt.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
We thank Michael Karas and Stavroula Markoutsa for the MALDI‐TOF‐Mass Spectrometry analysis and Bert Vogelstein for providing us the Bax WT/KO HCT‐116 cells. This work was supported by grants from the German Cancer Consortium (DKTK), Deutsche Krebshilfe, the Carls and the BANSS Stiftung.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.11.003.
Mandal Ranadip, Raab Monika, Matthess Yves, Becker Sven, Knecht Rainald and Strebhardt Klaus, (2014), pERK 1/2 inhibit Caspase‐8 induced apoptosis in cancer cells by phosphorylating it in a cell cycle specific manner, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.11.003.
References
- Alvarado-Kristensson, M. , Melander, F. , Leandersson, K. , Ronnstrand, L. , Wernstedt, C. , Andersson, T. , 2004. p38-MAPK signals survival by phosphorylation of Caspase‐8 and caspase-3 in human neutrophils. J. Exp. Med.. 199, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis, T. , Deacon, S.W. , Devarajan, K. , Ma, H. , Peterson, J.R. , 2011. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol.. 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellail, A.C. , Qi, L. , Mulligan, P. , Chhabra, V. , Hao, C. , 2009. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev. Recent Clin. Trials. 4, 34–41. [DOI] [PubMed] [Google Scholar]
- Brunet, A. , Roux, D. , Lenormand, P. , Dowd, S. , Keyse, S. , Pouyssegur, J. , 1999. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J.. 18, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihardjo, I. , Oliver, H. , Lutter, M. , Luo, X. , Wang, X. , 1999. Biochemical pathways of caspase activation during apoptosis. Ann. Rev. Cell Dev. Biol.. 15, 269–290. [DOI] [PubMed] [Google Scholar]
- Chambard, J.C. , Lefloch, R. , Pouyssegur, J. , Lenormand, P. , 2007. ERK implication in cell cycle regulation. Biochim. Biophys. Acta. 1773, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Cursi, S. , Rufini, A. , Stagni, V. , Condo, I. , Matafora, V. , Bachi, A. , Bonifazi, A.P. , Coppola, L. , Superti-Furga, G. , Testi, R. , Barila, D. , 2006. Src kinase phosphorylates Caspase‐8 on Tyr380: a novel mechanism of apoptosis suppression. EMBO J.. 25, 1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, M.I. , Hunt, J.P. , Herrgard, S. , Ciceri, P. , Wodicka, L.M. , Pallares, G. , Hocker, M. , Treiber, D.K. , Zarrinkar, P.P. , 2011. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol.. 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Dong, Y. , Sui, L. , Tai, Y. , Sugimoto, K. , Tokuda, M. , 2001. The overexpression of cyclin-dependent kinase (CDK) 2 in laryngeal squamous cell carcinomas. Anticancer Res.. 21, 103–108. [PubMed] [Google Scholar]
- Eskes, R. , Desagher, S. , Antonsson, B. , Martinou, J.C. , 2000. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol.. 20, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Medarde, A. , Santos, E. , 2011. Ras in cancer and developmental diseases. Genes Cancer. 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, K.S. , Kreutz, C. , Macnelly, S. , Neubert, K. , Haber, A. , Bogyo, M. , Timmer, J. , Borner, C. , 2012. Caspase-3 feeds back on Caspase‐8, Bid and XIAP in type I Fas signaling in primary mouse hepatocytes. Apopt. Int. J. Program. Cell Death. 17, 503–515. [DOI] [PubMed] [Google Scholar]
- Fulda, S. , 2009. Caspase‐8 in cancer biology and therapy. Cancer Lett.. 281, 128–133. [DOI] [PubMed] [Google Scholar]
- Fulda, S. , Debatin, K.M. , 2006. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 25, 4798–4811. [DOI] [PubMed] [Google Scholar]
- Fulda, S. , Meyer, E. , Debatin, K.M. , 2002. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 21, 2283–2294. [DOI] [PubMed] [Google Scholar]
- Gillissen, B. , Richter, A. , Richter, A. , Overkamp, T. , Essmann, F. , Hemmati, P.G. , Preissner, R. , Belka, C. , Daniel, P.T. , 2013. Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent 'type I' mode. Cell Death Dis.. 4, e643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harding, A. , Giles, N. , Burgess, A. , Hancock, J.F. , Gabrielli, B.G. , 2003. Mechanism of mitosis-specific activation of MEK1. J. Biol. Chem.. 278, 16747–16754. [DOI] [PubMed] [Google Scholar]
- Hengartner, M.O. , 2000. The biochemistry of apoptosis. Nature. 407, 770–776. [DOI] [PubMed] [Google Scholar]
- Holmstrom, T.H. , Chow, S.C. , Elo, I. , Coffey, E.T. , Orrenius, S. , Sistonen, L. , Eriksson, J.E. , 1998. Suppression of Fas/APO-1-mediated apoptosis by mitogen-activated kinase signaling. J. Immunol.. 160, 2626–2636. [PubMed] [Google Scholar]
- Holmstrom, T.H. , Schmitz, I. , Soderstrom, T.S. , Poukkula, M. , Johnson, V.L. , Chow, S.C. , Krammer, P.H. , Eriksson, J.E. , 2000. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J.. 19, 5418–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B. , Mitra, J. , van den Heuvel, S. , Enders, G.H. , 2001. S and G2 phase roles for Cdk2 revealed by inducible expression of a dominant-negative mutant in human cells. Mol. Cell. Biol.. 21, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel, S. , Matthess, Y. , Kaufmann, M. , Strebhardt, K. , 2007. Silencing of mammalian genes by tetracycline-inducible shRNA expression. Nature Prot.. 2, 3257–3269. [DOI] [PubMed] [Google Scholar]
- Kurokawa, M. , Kornbluth, S. , 2009. Caspases and kinases in a death grip. Cell. 138, 838–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawen, A. , 2003. Apoptosis-an introduction. Bioessays. 25, 888–896. [DOI] [PubMed] [Google Scholar]
- Lee, M.W. , Bach, J.H. , Lee, H.J. , Lee, D.Y. , Joo, W.S. , Kim, Y.S. , Park, S.C. , Kim, K.Y. , Lee, W.B. , Kim, S.S. , 2005. The activation of ERK1/2 via a tyrosine kinase pathway attenuates trail-induced apoptosis in HeLa cells. Cancer Invest. 23, 586–592. [DOI] [PubMed] [Google Scholar]
- Lev, D.C. , Kim, L.S. , Melnikova, V. , Ruiz, M. , Ananthaswamy, H.N. , Price, J.E. , 2004. Dual blockade of EGFR and ERK1/2 phosphorylation potentiates growth inhibition of breast cancer cells. Br. J. Cancer. 91, 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Yan, S. , Zhou, T. , Terada, Y. , Erikson, R.L. , 2004. The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene. 23, 763–776. [DOI] [PubMed] [Google Scholar]
- Lo, R.S. , 2012. Receptor tyrosine kinases in cancer escape from BRAF inhibitors. Cell Res.. 22, 945–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthess, Y. , Raab, M. , Sanhaji, M. , Lavrik, I.N. , Strebhardt, K. , 2010. Cdk1/cyclin B1 controls Fas-mediated apoptosis by regulating Caspase‐8 activity. Mol. Cell. Biol.. 30, 5726–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey, J.A. , Steelman, L.S. , Chappell, W.H. , Abrams, S.L. , Wong, E.W. , Chang, F. , Lehmann, B. , Terrian, D.M. , Milella, M. , Tafuri, A. , Stivala, F. , Libra, M. , Basecke, J. , Evangelisti, C. , Martelli, A.M. , Franklin, R.A. , 2007. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta. 1773, 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche, S. , 1995. Cell cycle reentry of mammalian fibroblasts is accompanied by the sustained activation of p44mapk and p42mapk isoforms in the G1 phase and their inactivation at the G1/S transition. J. Cell Physiol.. 163, 577–588. [DOI] [PubMed] [Google Scholar]
- Meloche, S. , Pouyssegur, J. , 2007. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 26, 3227–3239. [DOI] [PubMed] [Google Scholar]
- Ozoren, N. , El-Deiry, W.S. , 2002. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia. 4, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, G. , Robinson, F. , Beers Gibson, T. , Xu, B.E. , Karandikar, M. , Berman, K. , Cobb, M.H. , 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev.. 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Pearson, M.A. , Fabbro, D. , 2004. Targeting protein kinases in cancer therapy: a success?. Expert Rev. Anticancer Ther.. 4, 1113–1124. [DOI] [PubMed] [Google Scholar]
- Peng, C. , Cho, Y.Y. , Zhu, F. , Zhang, J. , Wen, W. , Xu, Y. , Yao, K. , Ma, W.Y. , Bode, A.M. , Dong, Z. , 2011. Phosphorylation of Caspase‐8 (Thr-263) by ribosomal S6 kinase 2 (RSK2) mediates Caspase‐8 ubiquitination and stability. J. Biol. Chem.. 286, 6946–6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucci, B. , Indelicato, M. , Paradisi, V. , Reali, V. , Pellegrini, L. , Aventaggiato, M. , Karpinich, N.O. , Fini, M. , Russo, M.A. , Farber, J.L. , Tafani, M. , 2009. ERK-1 MAP kinase prevents TNF-induced apoptosis through bad phosphorylation and inhibition of Bax translocation in HeLa Cells. J. Cell Biochem.. 108, 1166–1174. [DOI] [PubMed] [Google Scholar]
- Raab, M. , Kappel, S. , Kramer, A. , Sanhaji, M. , Matthess, Y. , Kurunci-Csacsko, E. , Calzada-Wack, J. , Rathkolb, B. , Rozman, J. , Adler, T. , Busch, D.H. , Esposito, I. , Fuchs, H. , Gailus-Durner, V. , Klingenspor, M. , Wolf, E. , Sanger, N. , Prinz, F. , Angelis, M.H. , Seibler, J. , Yuan, J. , Bergmann, M. , Knecht, R. , Kreft, B. , Strebhardt, K. , 2011. Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nature Comm.. 2, 395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, P.P. , Blenis, J. , 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev.. 68, 320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamoorthy, K. , Li, G. , Gerrero, M.R. , Brose, M.S. , Volpe, P. , Weber, B.L. , Van Belle, P. , Elder, D.E. , Herlyn, M. , 2003. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res.. 63, 756–759. [PubMed] [Google Scholar]
- Schungel, S. , Buitrago-Molina, L.E. , Nalapareddy, P. , Lebofsky, M. , Manns, M.P. , Jaeschke, H. , Gross, A. , Vogel, A. , 2009. The strength of the Fas ligand signal determines whether hepatocytes act as type 1 or type 2 cells in murine livers. Hepatology. 50, 1558–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spankuch-Schmitt, B. , Bereiter-Hahn, J. , Kaufmann, M. , Strebhardt, K. , 2002. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J. Natl. Cancer Inst.. 94, 1863–1877. [DOI] [PubMed] [Google Scholar]
- Spankuch, B. , Matthess, Y. , Knecht, R. , Zimmer, B. , Kaufmann, M. , Strebhardt, K. , 2004. Cancer inhibition in nude mice after systemic application of U6 promoter-driven short hairpin RNAs against PLK1. J. Natl. Cancer Inst.. 96, 862–872. [DOI] [PubMed] [Google Scholar]
- Steinmetz, R. , Wagoner, H.A. , Zeng, P. , Hammond, J.R. , Hannon, T.S. , Meyers, J.L. , Pescovitz, O.H. , 2004. Mechanisms regulating the constitutive activation of the extracellular signal-regulated kinase (ERK) signaling pathway in ovarian cancer and the effect of ribonucleic acid interference for ERK1/2 on cancer cell proliferation. Mol. Endocrinol.. 18, 2570–2582. [DOI] [PubMed] [Google Scholar]
- Towatari, M. , Iida, H. , Tanimoto, M. , Iwata, H. , Hamaguchi, M. , Saito, H. , 1997. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia. 11, 479–484. [DOI] [PubMed] [Google Scholar]
- Tran, S.E. , Holmstrom, T.H. , Ahonen, M. , Kahari, V.M. , Eriksson, J.E. , 2001. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J. Biol. Chem.. 276, 16484–16490. [DOI] [PubMed] [Google Scholar]
- Wada, T. , Penninger, J.M. , 2004. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 23, 2838–2849. [DOI] [PubMed] [Google Scholar]
- Wagner, K.W. , Engels, I.H. , Deveraux, Q.L. , 2004. Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J. Biol. Chem.. 279, 35047–35052. [DOI] [PubMed] [Google Scholar]
- Weibrecht, I. , Leuchowius, K.J. , Clausson, C.M. , Conze, T. , Jarvius, M. , Howell, W.M. , Kamali-Moghaddam, M. , Soderberg, O. , 2010. Proximity ligation assays: a recent addition to the proteomics toolbox. Exp. Rev. Proteom.. 7, 401–409. [DOI] [PubMed] [Google Scholar]
- Westphal, S. , Kalthoff, H. , 2003. Apoptosis: targets in pancreatic cancer. Mol. Cancer. 2, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, T. , Ebisuya, M. , Ashida, F. , Okamoto, K. , Yonehara, S. , Nishida, E. , 2006. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr. Biol.. 16, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Yuan, J. , Sanhaji, M. , Kramer, A. , Reindl, W. , Hofmann, M. , Kreis, N.N. , Zimmer, B. , Berg, T. , Strebhardt, K. , 2011. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am. J. Pathol.. 179, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, J. , Yan, R. , Kramer, A. , Eckerdt, F. , Roller, M. , Kaufmann, M. , Strebhardt, K. , 2004. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene. 23, 5843–5852. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Fang, B. , 2005. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.. 12, 228–237. [DOI] [PubMed] [Google Scholar]
- Zhang, X.D. , Borrow, J.M. , Zhang, X.Y. , Nguyen, T. , Hersey, P. , 2003. Activation of ERK1/2 protects melanoma cells from TRAIL-induced apoptosis by inhibiting Smac/DIABLO release from mitochondria. Oncogene. 22, 2869–2881. [DOI] [PubMed] [Google Scholar]
- Zhou, L. , Tan, X. , Kamohara, H. , Wang, W. , Wang, B. , Liu, J. , Egami, H. , Baba, H. , Dai, X. , 2010. MEK1 and MEK2 isoforms regulate distinct functions in pancreatic cancer cells. Oncol. Rep.. 24, 251–255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data