

Abstract
In clinical practice, targeted therapies are usually administered together with chemotherapeutics. However, little is known whether conventional cytotoxic agents enhance the efficacy of targeted compounds, and whether a possible synergy would be dictated by drug‐sensitizing genetic alterations. To explore these issues, we leveraged the design of clinical studies in humans to conduct a multi‐arm trial in an ‘in‐cell’ format. Using the MET oncogene as a model target and a panel of genetically characterized cell lines as a reference population, we found that two different chemotherapeutic regimens – cisplatin and 5‐fluorouracil – exerted widespread cytotoxic activity that was not further enhanced by MET inhibition with a monovalent anti‐MET antibody. From a complementary perspective, targeted MET inhibition was successful in a selected complement of cells harboring MET genomic lesions. In this latter setting, addition of chemotherapy did not provide a therapeutic advantage. Mechanistically, chemotherapeutics did not influence the basal activity of MET in cells with normal MET genomic status nor did they contribute to neutralize MET signals in cells with MET amplification. These data suggest that tumors displaying MET aberrations achieve plateau responses by MET monotherapy and do not receive further benefit by addition of cytotoxic treatments.
Keywords: MET tyrosine kinase, Monoclonal antibodies, Chemotherapy
Highlights
MET targeted therapy is effective per se when tumors exhibit MET amplification.
MET targeted therapy outperforms standard cytotoxics in genetically defined patients.
MET targeted therapy is dispensable in not MET‐addicted tumors.
1. Introduction
The advent of rationally targeted therapeutics has resulted in a new model of ‘cancer precision medicine’ whereby drug treatment is matched to the presence or absence of defined molecular characteristics in the tumor of each patient (Yauch and Settleman, 2012). This approach is fostered by accumulating genomic data – which steadily highlight new potential druggable targets – and it is experimentally supported by preclinical platforms featuring extensive multidimensional annotations, including vast collections of cancer cell lines and patient‐derived xenografts (Barretina et al., 2012; Bertotti et al., 2011; Garnett et al., 2012; Sharma et al., 2010; Tentle et al., 2012). Notwithstanding the impressive efficacy of some agents in genetically enriched subpopulations, and because of sensible cautionary issues inherent in human experimentation, first‐line targeted monotherapy in ‘target‐positive’ patients remains daunting. In fact, it is currently common practice to combine targeted therapies with a chemotherapy backbone, in order to guarantee a ‘one‐fits‐for‐all’ basis likely to display some generic – but expected – activity. Inevitably, addition of chemotherapeutics confounds the value of molecular lesions in predicting sensitivity or resistance to targeted drugs and precludes lucid evaluation of whether response is attributable to selective blockade of the aberrant target, nonspecific cytotoxic activity of the chemotherapeutic compound(s), or both. Accordingly, it remains to be established whether chemotherapy can intensify the efficacy of targeted agents, or rational inactivation of the dominant oncoprotein in a genetically permissive context suffices to reach a ‘point of no return’ beyond which chemotherapy becomes superfluous.
These open issues well apply to the case of the MET receptor tyrosine kinase. It is now established that, at least in cellular and animal models, some cancer types rely on high‐grade amplification of the MET oncogene – and the ensuing hyper‐activation of the encoded kinase – for their growth and survival, and this dependence results in drastic impairment of cancer cell viability upon MET inactivation (‘oncogene addiction’: Bardelli et al., 2013; Bertotti et al., 2009; Comoglio et al., 2008; McDermott et al., 2007; Smolen et al., 2006). Such preclinical findings have been recently confirmed in patients with MET‐amplified esophago‐gastric adenocarcinomas, who, albeit very rare (2%), have received clinical benefit from therapy with MET small molecule inhibitors or antibodies (Catenacci et al., 2011; Lennerz et al., 2011). MET mainly activates anti‐apoptotic pathways. This suggests that tumors exhibiting constitutive MET signaling may be intrinsically poorly sensitive to chemotherapeutics, due to the incessant operativity of MET‐dependent survival signals, and that MET inhibition may potentiate the effects of chemotherapy in MET‐amplified tumors. However, this assumption has not been challenged experimentally.
Cancers exhibiting MET amplification account for 2–4% of the overall population (COSMIC database:http://www.sanger.ac.uk). In most solid tumors, MET displays normal gene copy number but its expression (and activity) can be transcriptionally induced by cues present in the tumor reactive stroma – such as inflammatory cytokines and pro‐angiogenic factors – and by exogenous stress stimuli such hypoxia or ionizing radiations (De Bacco et al., 2011; Pennacchietti et al., 2003; Trusolino et al., 2010). In this setting, MET upregulation provides pro‐survival and pro‐invasive advantages that exacerbate the tumor malignant phenotype (‘oncogene expedience’: Comoglio et al., 2008). At present, it is not known whether chemotherapeutics, similar to treatments such as radiotherapy (De Bacco et al., 2011), can induce MET overexpression. If this were the case, MET inhibition could sensitize cancer cells to chemotherapy in a wide spectrum of tumors, even in contexts in which MET is not genetically amplified.
Prompted by these quandaries, we decided to conduct an ‘in‐cell trial’ in which we comparatively assessed the effects of MET inhibition alone, chemotherapy alone, and a combination of chemotherapy and MET‐targeted therapy in a panel of cancer cell lines featuring normal or amplified levels of MET.
2. Material and methods
2.1. Cell lines and reagents
A549 (lung adenocarcinoma); NCI‐H1975, NCI‐H23, NCI‐H522, NCI‐H460 and HCC‐78 (non‐small cell lung cancer); NCI‐H2452, NCI‐H226 and NCI‐H28 (mesothelioma); NCI‐H441 and NCI‐H820 (lung papillary adenocarcinoma); NCI‐H322M (bronchio‐alveolar carcinoma); SW620, SW48 and DLD1 (colorectal adenocarcinoma); HT29 (rectosigmoid colon adenocarcinoma); MDA‐MB‐231 and MDA‐MB‐468 (breast adenocarcinoma); OVCAR8, OVCAR3, A2780 and SKOV3, (ovarian adenocarcinoma); U‐118 MG, SNB‐75 and SF‐268 (glioblastoma); CaKi (kidney carcinoma); HS746T (gastric adenocarcinoma) cell lines were purchased from ATCC (Rockville, MD); EBC‐1 (non‐small cell lung cancer) were acquired from HSRRB cell bank (Osaka, Japan); GTL16 (gastric adenocarcinoma) are a laboratory batch obtained from limiting dilutions of MKN45 (Giordano et al., 1988). All cells were kept in culture for less than 8 weeks and used between passage 2 and 20. Cells were grown in recommended media (Sigma Aldrich, St. Louis, MO) supplemented with 50 units/mL penicillin (Sigma Aldrich, St. Louis, MO), 50 mg/mL streptomycin (Sigma Aldrich, St. Louis, MO), 2 mM glutamine (Sigma Aldrich, St. Louis, MO) and 2% or 10% Foetal Bovine Serum (Lonza Sales Ltd, Basel, Switzerland) as indicated. Cells were maintained at 37 °C in 5% CO2 atmosphere. Cisplatin (Sigma Aldrich, St. Louis, MO), 5‐fluorouracil (Calbiochem, Merck Millipore, Darmstadt) and PHA‐665752 (Tocris Cookson Ltd, Bristol UK) were administered in 2% serum media and used at the indicated concentrations. MV‐DN30 was kindly provided by Dr. Petronzelli (Sigma Tau, Pomezia).
2.2. Immunoblots
Cells were lysed in EB buffer (10 mM Tris–HCl pH 7.4; 1% TRITON X‐100; 5 mM EDTA; 150 mM NaCl; 10% Glycerol; 50 mM NaF; 2 mM Na3VO4; 1 mM Phenylmethanesulfonyl fluoride and a cocktail of protease inhibitors) protein concentration was determined using a BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL). Equal amount of proteins were loaded on to SDS‐PAGE gels and transferred to nitrocellulose membrane supports (Hybond C+; Amersham, Buckinghamshire, UK). The membranes were decorated with the following antibodies: mouse monoclonal anti MET DL‐21 (Prat et al., 1991); goat polyclonal anti actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA); rabbit polyclonal anti p44/42 MAPK (ERK1/2) (Cell Signaling Technology, Danvers, MA, USA); rabbit polyclonal anti phospho p44/42 MAPK (ERK1/2) (Cell Signaling Technology, Danvers, MA, USA); rabbit polyclonal anti AKT (Cell Signaling Technology, Danvers, MA, USA); rabbit polyclonal anti phospho AKT (Ser473) (Cell Signaling Technology, Danvers, MA, USA); rabbit monoclonal anti phospho MET (Tyr1234/Y1235) (Cell Signaling Technology, Danvers, MA, USA); mouse monoclonal anti vinculin (Sigma, St Louis, MO, USA). Anti mouse, anti rabbit, anti goat and protA peroxidase conjugated secondary antibodies were from Amersham (Arlington Heights, VA). Signal detection was performed using ECL Western Blotting Substrate (Promega Corporation, Madison, Wisconsin, USA).
2.3. Cell viability assay
Cell viability assays were performed in 96 well plates and low serum conditions (2%). Proliferation rates were assessed using Cell Titre Glo Luminescence (Promega Corporation, Madison, Wisconsin, USA). All the experiments were performed at least two times in triplicates or quadruplicates as indicated (Benvenuti et al., 2011).
2.4. RNA extraction and quantitative real time PCR (Q PCR)
Total RNA was isolated using RNeasy Mini Kit (Quiagen, Gmbh, Hilden, Germany) according to manufacturer's instructions. One microgram of RNA was retro‐transcribed into cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to manufacturer's instructions. Q PCR was performed using Taq Man Gene Expression Assays for HGF, MET, UBC and GUSB (Applied Biosystems, Foster City, CA) with TaqMan PCR master mix (Applied Biosystems, Foster City, CA) and ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA). All samples were analyzed in triplicates.
2.5. Apoptosis assay
Treated cells were detached and incubated with FITC‐conjugated Annexin V (BD Bioscience). DAPI (Roche Applied Science) was used to identify dead cells. Samples were analyzed on CYAN flow cytometer (CyAn ADP, Beckman Coulter Life Sciences, Brea, CA, USA) equipped with 488 nm, 405 nm and 642 nm solid state lasers. Data were collected and processed using Summit 4.3 software (Beckman Coulter Life Sciences).
2.6. In vivo cell transplantation and therapy
All animal procedures were performed according to protocols approved by the Italian Ministry of Health and by the internal Ethical Committee for Animal Experimentation. EBC‐1 were injected subcutaneously (1.5 × 106 cells/mouse) into the posterior flanks of 6‐week‐old immunodeficient nu−/− female mice (Charles River Laboratories, Lecco, Italy). When tumors reached the size of approximately 50 mm3 mice were randomized and assigned to four arms (n = 6) and treated three times per week for 2 months as follow: placebo, MV‐DN30 (20 μg/mice), CDDP (24 μg/mice) or MV‐DN30 plus CDDP (20 μg/mice and 24 μg/mice respectively). Tumors were measured with a calliper and tumors' volume was calculated at the indicated days using the formula: 4/3π × (d/2)2 × (D/2), where d and D are the minor and the major tumor axis respectively. The general physical status of mice was monitored constantly. At the end of the experiments mice were euthanized and tumors extracted.
2.7. Statistical analysis
Experiments were performed in triplicates or quadruplicates as indicated. Standard deviation (SD) or standard error mean (SEM) of each result was reported. All statistical analyses were performed using Excel software (Microsoft Office 2010).
3. Results
3.1. ‘In‐cell trial’ design
Typically, preclinical analysis of anticancer compounds involves testing them on a small number of cancer cell lines that may be inadequate to capture the genetic heterogeneity of tumors on a population scale. Moreover, to the best of our knowledge, systematic comparative assessment of the activity of targeted drugs with or without standard chemotherapeutics has not been attempted so far. We therefore decided to gauge the sensitivity of a panel of 28 cancer cell lines, representing diverse tumor types (Supplementary Table 1) and expressing different levels of MET protein (Supplementary Figure 1), to four treatment modalities: (i) individual blockade of the MET oncoprotein by the monovalent DN30 monoclonal antibody (MV‐DN30), an engineered antibody that binds to MET with high affinity and promotes receptor shedding and down‐regulation (Pacchiana et al., 2010); (ii) cisplatin (cis‐diamminedichloroplatinum, CDDP), a DNA cross‐linking agent (Loehrer and Einhorn, 1984); (iii) 5‐fluorouracil (5FU) a thymidylate synthase inhibitor (Palumbo et al., 2013); and (iv) combination therapy with MV‐DN30 and either CDDP or 5FU. MV‐DN30 was administered at a fixed 125 nM concentration, a dosage that was found to achieve 50% growth inhibition (IC50) in an artificial system of BaF3 cells exhibiting interleukin‐3 independence as a consequence of MET ectopic overexpression (our unpublished observations). CDDP and 5FU were used at three increasing concentrations (0.1, 1.0 and 10 μM).
Figures 1 and 2 depict a series of waterfall plots showing the effect of the various therapeutic regimens on cancer cell growth after 48 h of compound administration, as measured by ATP cellular content (a proxy for cell numbers). For each treatment arm, responses are independently ranked by cell number compared with values of placebo‐treated cells. Cells featuring at least 20% reduction of viability upon treatment were scored as ‘responders’.
Figure 1.
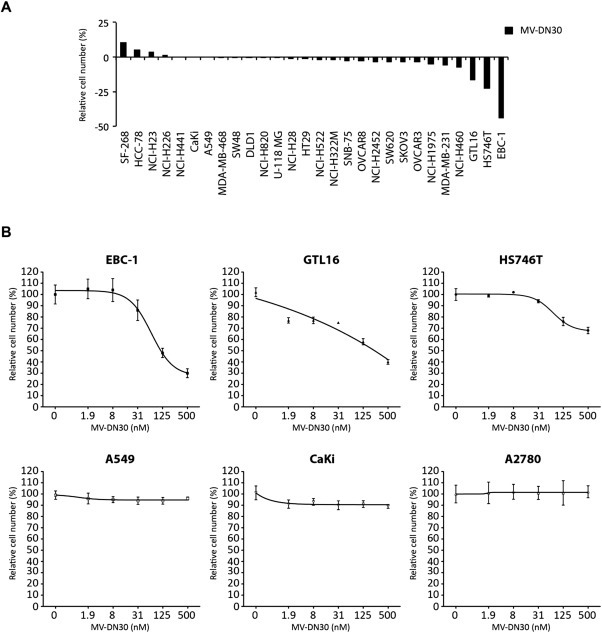
Effect of anti MET MV‐DN30 treatment on cancer cell growth. (A) Waterfall plot showing the effect of MV‐DN30 on cell growth in a panel of 28 human cancer‐derived cell lines. Each cell line was grown in the presence of MV‐DN30 (125 nM) or placebo as adherent cells. Cell growth was measured 48 h after treatment start and measured by ATP cellular content (a proxy for cell numbers). Growth rates are expressed as percentage over placebo‐treated cells. (B) Dose‐response curves performed using increasing concentrations of MV‐DN30 (1.9–500 nM) on MET addicted cells: EBC‐1, GTL16 and HS746T; and on control cells: A549 (expressing physiological levels of MET), CaKi (expressing constitutively active receptor without it being amplificated – not addicted) and A2780 (not expressing MET). Data are means ± SD of three independent experiments performed in triplicates (A) and quadruplicated (B). Error bars represent standard deviations.
Figure 2.
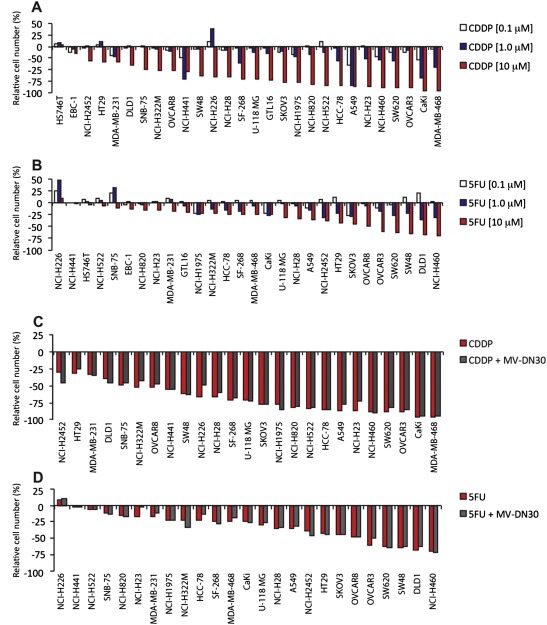
Effect of CDDP and 5FU treatment on growth of a panel of human cancer‐derived cell lines. Waterfall plots showing the effect of CDDP (A) and 5FU (B) on cancer cell growth. 28 human cancer‐derived cell lines were grown in the presence of increasing concentrations (0.1–1.0–10 μM) of either CDDP or 5FU as adherent cells. Cell growth was assessed 48 h after treatment start and measured by ATP cellular content. Growth rates are expressed as percentage over placebo‐treated cells. (C) Waterfall plot showing the effect of CDDP in combination with anti MET antibody on growth of not MET addicted cancer cells. Cells were incubated with: placebo; CDDP (10 μM) or MV‐DN30 plus CDDP (125 nM and 10 μM respectively). Growth rates are expressed as percentage over placebo‐treated cells. (D) Waterfall plot showing the effect of 5FU in combination with MV‐DN30 on growth of not addicted cancer cells. Cells were incubated with: placebo; 5FU (10 μM) or MV‐DN30 plus 5FU (125 nM and 10 μM respectively). Growth rates are expressed as percentage over placebo‐treated cells. Data are means ± SD of three independent experiments performed in triplicates.
3.2. Effects of anti‐MET monotherapy
Consistent with the notion that targeted therapies are efficacious only in genetically defined tumor subsets, the majority of the cell lines tested were largely refractory to MV‐DN30 monotherapy, with relative sensitivity displayed only by GTL16, HS746T and, to a higher extent, EBC‐1 (Figure 1A). Notably, these three responders all harbor high‐grade MET gene amplification, further attesting to increased MET gene copy number as a molecular predictor of sensitivity to MET inhibition (Bertotti et al., 2009; McDermott et al., 2007; Smolen et al., 2006).
Responsiveness to MV‐DN30 was more precisely evaluated in dose‐response assays with increasing antibody concentrations (1.9–500 nM) using MET‐amplified cell lines (EBC‐1, GTL16 and HS746T) as well cell lines with normal MET gene copy number: A549 (expressing normal level of MET), CaKi (overexpressing MET, without gene amplification) and A2780 (not expressing MET). In all MET‐amplified cell lines, MV‐DN30 reduced viability in a dose‐dependent manner and to various degrees, with EBC‐1 being the most sensitive to MET inactivation (Figure 1B). At variance, cells without MET amplification were completely resistant to MV‐DN30, even at high antibody concentrations (Figure 1B).
3.3. Effects of standard chemotherapy alone and in combination with MET inhibition
Different from targeted MET blockade, and in line with induction of pervasive cytotoxic effects, response to either CDDP or 5FU was more evenly distributed and on average more pronounced than that observed for MV‐DN30, with overall cellular toxicity increasing as a function of dosage (Figure 2A and B). We note, anecdotally, that EBC‐1 and HS746T cells proved to be particularly sensitive to MET inhibition, but were substantially refractory to either CDDP or 5FU (1, 2A, B). EBC‐1 cells also scored as ‘best responders’ in a large‐scale monotherapy screening with a MET small molecule inhibitor (McDermott et al., 2007). It is tempting to speculate that ‘MET addiction’ in EBC‐1 cells sustains constitutive survival pathways that may limit responsiveness to standard cytotoxic agents, an assumption that could have therapeutic relevance in patients with tumors exhibiting intense MET amplification.
For combinatorial treatments, we elected to use MV‐DN30 together with the higher concentration of either CDDP or 5FU (10 μM). In all lines, combination therapy was not superior to best agent monotherapy: addition of MV‐DN30 did not sensitize cells without MET amplification to chemotherapy (Figure 2C and D); similarly, addition of chemotherapy did not substantially improve response to MV‐DN30 in MET‐amplified cell lines, with no effect in HS746T and EBC‐1 and only slight additive activity in GTL16 (Figure 3A and B). This latter result was confirmed using the small molecule inhibitor PHA‐665752 as an alternative way to halt MET activity (Figure 3C and D).
Figure 3.
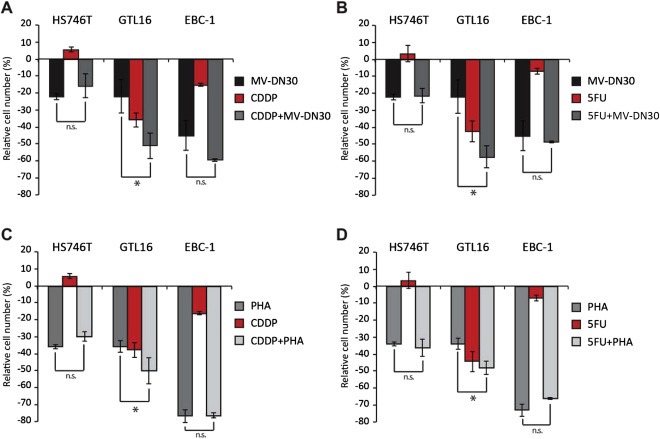
Effect of CDDP and 5FU treatment on growth of MET addicted cells. Effects of MV‐DN30 (125 nM), CDDP (10 μM) and 5FU (10 μM) as single agents or in combination (panels A and B) and of PHA‐665752 (100 nM), CDDP (10 μM) and 5FU (10 μM) as single agents or in combination (panels C and D) on growth of HS746T, GTL16 and EBC‐1 (MET addicted). Cell proliferation rates were determined 48 h after treatment start and measured by ATP cellular content. Data are means ± SD of three independent experiments performed in triplicates. Error bars represent standard deviations. *: P < 0.05 by Student's t test; n.s.: statistically not significant.
The evidence that MV‐DN30 and chemotherapy did not cooperate in cells expressing physiological levels of MET is in contrast with the observation that other cytotoxic insults, such as ionizing radiation, are in fact more detrimental when MET is inhibited, independent of MET genomic status (De Bacco et al., 2011). Mechanistically, ionizing radiation induces transcriptional upregulation and catalytic activation of MET; increased MET activity conveys anti‐apoptotic signals that prevent cell death induced by irradiation, and MET inhibition curtails such signals so that the efficacy of radiotherapy is enhanced. In coherence with the finding that MET inhibition did not exacerbate response to chemotherapy, time‐course experiments on A549 and NCI‐H441 revealed that treatment with CDDP or 5FU did not produce significant changes in MET expression, both at the mRNA and protein levels (Figure 4A and B), or even reduced MET protein levels, as observed for NCI‐H441 treated with CDDP (Figure 4A and B). Since growth arrest is usually accompanied by MET protein downregulation (Boccaccio et al., 1994), this decreased expression of MET is likely due to the more pronounced cytostatic activity of CDDP compared with 5FU (see Figure 2). As previously demonstrated, MET expression was upregulated by ionizing radiation (Figure 4B).
Figure 4.
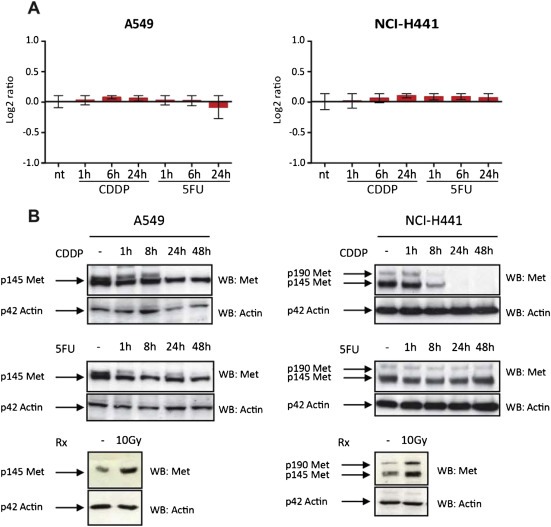
Effect of CDDP and 5FU treatment on MET transcription. (A) Time course experiment assessing MET mRNA level in A549 and NCI‐H441 cells treated with CDDP (10 μM) or 5FU (10 μM) in low serum conditions (2%). RNA was extracted 1, 6 or 24 h after treatment beginning and examined by Q PCR. Results were normalized on UBC and GUSB house keeping genes. Graphs show the fold change of treated cells versus not treated cells. Data are means ± SD of three independent experiments performed in triplicates. (B) Time course experiment assessing MET protein expression in A549 and NCI‐H441 cells treated with CDDP (10 μM) or 5FU (10 μM) in low serum conditions (2%). Cells were lysed 1, 8, 24 or 48 h after treatment beginning and proteins were separated on SDS PAGE gels (8%) as total extracts. MET protein level was checked probing the membranes with anti MET antibody. Actin was used as control for equal protein loading. – represent placebo treated cells. Irradiated cells (exposed to 10 Gy for 12 h) were used as controls of MET transcriptional upregulation.
Lack of synergy between MET inhibition and standard chemotherapy in MET‐amplified cells was also evident at the signaling level: treatment with MV‐DN30 effectively suppressed constitutive MET autophosphorylation and abrogated baseline phosphorylation of distal transducers such as Akt and MAPK. However, this overall signal neutralization was not further influenced by CDDP or 5FU (Figure 5A). This suggests that individual MET blockade in MET‐amplified tumors elicits maximal down‐modulation of downstream pathways that cannot be additively aggravated by conventional cytotoxics. Biologically, the rate of apoptosis induced by anti‐MET monotherapy, as assessed by annexin‐V staining, was only moderately increased by co‐treatment with CDDP or 5FU (Figure 5B). (We note again, ‘en passant’, that the apoptotic response of ‘MET addicted’ EBC‐1 was stronger following treatment with MV‐DN30 than following administration of chemotherapeutics; this is similar to that observed in the initial screen using viability assays as readouts).
Figure 5.
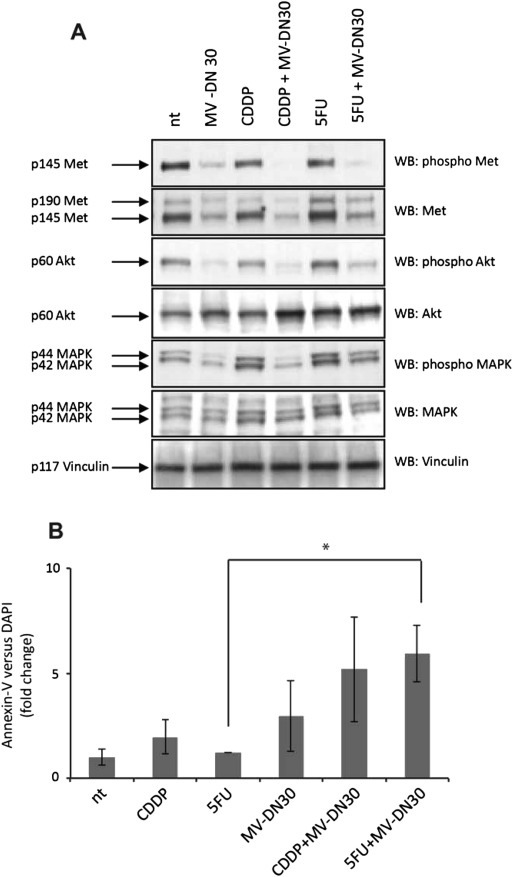
Analysis of MET signaling and apoptosis rates in response to MET inhibition alone or in combination with standard chemotherapy. (A) ECB‐1 cells were treated with MV‐DN30 (125 nM) alone or in combination with either CDDP (10 μM) or 5FU (10 μM) for 48 h. Cellular extracts were run on SDS PAGE gels and analyzed by western blotting using anti phospho MET and then reprobed with anti MET antibodies. MET signaling cascade was analysed probing the membranes with anti phospho MAPK and anti phospho AKT antibodies. Total MAPK and AKT levels were determined reprobing the same membranes. Vinculin was used as control for equal loading. (B) ECB‐1 cells were treated with MV‐DN30 (125 nM) alone or in combination with either CDDP (10 μM) or 5FU (10 μM) and apoptosis determined by flow cytometry analysis of Annexin V versus DAPI following 48 h of treatment. Data are means ± SD of three independent experiments. Error bars represent standard deviations. *: P < 0.05 by Student's t test.
Finally, we transferred the ‘in‐cell trial’ to an in vivo xenograft setting. Immunocompromised mice were injected with EBC‐1 cells and, after 15 days, randomized into four treatment arms: (i) placebo; (ii) MV‐DN30 alone; (iii) CDDP alone; and (iv) MV‐DN30 + CDDP. As expected, and consistent with the in vitro data, CDDP was poorly effective, whereas MV‐DN30 potently inhibited tumor growth. Again in accordance with the in vitro experiments, monotherapy with MV‐DN30 alone was as effective as the combination with CDDP, reinforcing the notion that the inhibitory effect of MET targeting in MET‐amplified cells was maximal and not further increased by concomitant chemotherapy (Figure 6). The same results were obtained using 5FU as chemotherapeutic drug (Supplementary Figure 2).
Figure 6.
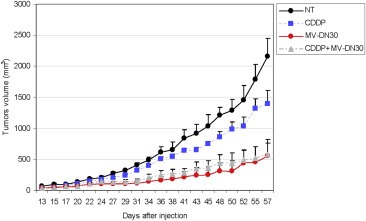
Effect of MV‐DN30 and CDDP as single agents and in combination on growth of tumor xenografts. EBC‐1 cells were injected subcutaneously on the right flanks of immunocompromised mice to induce the formation of experimental tumors. When tumors volume was approximately 50 mm3 (15 days post‐injection) mice were randomized into four arms (n = 6) and treated as follow: (i) placebo; (ii) MV‐DN30 (20 μg/mice); (iii) CDDP (24 μg/mice) and (iv) MV‐DN30 + CDDP (20 and 24 μg/mice respectively). Mice were treated three times per week for two months and tumors volume measured at the indicated days using a calliper. Values represent the mean tumor volume of each experimental group and error bars report standard errors.
4. Discussion
The first hint that targeted therapy in solid cancers can be more effective than chemotherapy – but only in patients with tumors bearing drug‐sensitizing mutations – emerged from studies in advanced non‐small cell lung cancer (NSCLC). In a randomized phase 3 trial comparing the EGFR small molecule inhibitor gefitinib versus a carboplatin‐paclitaxel doublet as first line treatment in advanced NSCLC, pre‐planned sub‐group analysis indicated that progression‐free survival was significantly longer for gefitinib than chemotherapy in patients with EGFR mutant tumors, and significantly longer for chemotherapy than gefitinib in patients with EGFR wild‐type tumors (Mok et al., 2009). This and other findings contributed to FDA approval of EGFR inhibitor monotherapy for the initial treatment of patients with EGFR mutation‐positive metastatic NSCLC. In this vein, again in advanced NSCLC, single‐agent inhibition of ALK by crizotinib has been recently shown to be superior to standard chemotherapy in ALK‐translocated cases (Shaw et al., 2013).
The notion that targeted therapy can outperform standard cytotoxics in genetically defined patient subpopulations does not imply that addition of chemotherapy is unproductive. At least in principle, one could argue that the effects of selective target inhibition can be potentiated by less specific but potentially more aggressive pro‐apoptotic insults, such as those triggered by chemotherapeutics. Although empirical, this line of thinking has informed clinical practice, and combination therapies with targeted drugs and conventional agents are now commonplace despite the risk of cumulative toxicity. Examples of FDA‐approved combinations include the anti‐HER2 monoclonal antibody trastuzumab and the mitotic inhibitor paclitaxel in the first‐line treatment of HER2‐amplified metastatic breast cancer (Slamon et al., 2001); trastuzumab and a cisplatin‐5FU doublet in HER2‐positive advanced gastric carcinoma (Bang et al., 2010), and the anti‐EGFR antibody cetuximab and cisplatin‐5FU in head and neck cancer (Vermorken et al., 2008). Although most of such combinations have received preclinical experimental validation in xenograft mouse models, their real impact in terms of therapeutic synergy awaits confirmation.
MET inhibitors are currently being tested in several investigational trials, but none of them has been certified for clinical use and conclusions about their efficacy remain immature (Gherardi et al., 2012). Schematically, studies with MET inhibitors are structured according to two major designs. On one side, recruitment is limited to ‘target‐positive’ cases – for instance, patients with MET‐amplified esophago‐gastric adenocarcinoma or MET‐mutant papillary renal carcinoma – and treatment involves targeted monotherapy, usually in patients who have failed prior cycles of conventional chemotherapy. On the other side, recruitment does not occur on a molecular basis and usually MET inhibitors are administered together with standard cytotoxics; trials of this kind are ongoing in NSCLC, colorectal tumors, and triple‐negative breast cancer.
Based on these premises we sought to dissect the relative contribution of anti‐MET targeted therapy versus standard chemotherapy to tumor growth inhibition and systematically coupled response annotation with the assessment of MET genomic status. To do this we embraced a reductionist approach in cancer cell lines, while trying to accommodate the informative merits of clinical trials. Hence, we selected a reference population – a panel of cancer cell lines of different tumor origin and with different genomic makeups, including some with MET gene amplification – and designed a multi‐arm study in which each cancer cell line was comparatively challenged with anti‐MET monotherapy, single‐agent chemotherapy, and combination therapy.
A first series of experiments demonstrated that chemotherapy was overall superior to specific MET blockade in cell lines with normal activity (and genetically wild‐type forms) of MET. This is not surprising, as lack of genetic lesions responsible for target hyperactivity usually predicts lack of efficacy of targeted intervention. The unexpected finding is that neutralization of MET, which commonly exerts cytoprotective activities in response to exogenous stress stimuli, did not drastically potentiate the effects of chemotherapy. The reason for this inadequacy is intrinsic to the mode of action of chemotherapeutic agents (at least those tested in this study). Both CDDP and 5FU did not induce transcriptional upregulation of MET, which is one stereotyped mechanism whereby cancer cells adapt to adverse events, including other cytotoxic therapeutic modalities such as ionizing radiation (De Bacco et al., 2011). The clinical relevance of this observation is straightforward: patients with tumors that are routinely treated with radiotherapy are likely to benefit from MET inhibition, irrespective of MET genetic status. At variance, MET inhibition is probably futile in patients with MET wild‐type tumors that receive standard chemotherapy. It cannot be excluded that, in the in vivo situation, chemotherapeutics could activate MET signaling in spite of their inability to boost MET expression. Indeed, the inflammatory phenotype that is commonly produced by standard chemotherapeutics might lead to increased local bioavailability of the MET ligand HGF in the tumor reactive stroma, with consequent paracrine activation of MET and emission of MET‐driven pro‐survival signals in cancer cells (Bhowmick et al., 2004). Our ‘in‐cell’ approach indirectly tackled this issue by performing viability assays in the presence of serum, which contains HGF, recapitulates in part the trophic composition of the tumor microenvironment, and sustains tonic activation of MET signaling. Our repeated observation that MET inhibition does not dramatically influence the outcome of chemotherapy, even in a context of basal MET activity, suggests that interception of MET activity is unlike to reinforce the effects of conventional cytotoxics also in the clinical setting.
Another point of interest of this investigation is that impairment of tumor growth in MET‐amplified cells appeared to be saturated by MET inhibition, with no additional activity of chemotherapy. Again, this is somehow unexpected because nullification of MET‐regulated survival signals is supposed to exacerbate cell death in the presence of pro‐apoptotic stimuli. It is likely that blockade of the dominant oncoprotein in a context of oncogene addiction is so catastrophic that any other insult becomes superfluous. Accordingly, MET obstruction resulted in massive abrogation of MET‐dependent downstream signals, with no further impact by addition of cytotoxic agents. When translated into the clinic, this finding supports the use of anti‐MET monotherapy in all cases in which a genetic basis for MET hyperactivity is evident, with salient consequences as far as toxicity issues and economic sustainability are concerned.
Cancer cell line‐based drug sensitivity screens are emerging as crucial tools to inform therapeutic decisions (Sharma et al., 2010; Trusolino and Bertotti, 2012). Their implementation is likely to become increasingly important as the pace of discovery of new investigational compounds is increasing and the rationale for drug combinations receives continuous support from mechanistic studies. Our work provides formal demonstration that MET targeted monotherapy is maximally effective per se whenever tumors exhibit the relevant genetic lesion and is dispensable – even when combined with chemotherapy – in genetically inappropriate contexts. These results add weight to the emerging paradigm that clinical trials need to rely on tumor molecular characterization as a prerequisite for patient stratification and should progressively dismiss post‐hoc, evidence‐based conclusions.
Authors' contributions
SB and PMC conceived and designed the experiments; SB, AA and LL performed the experiments; SB, LL and AG analyzed the data; LT and PMC wrote the paper.
Conflict of interest
The authors disclose no potential conflicts of interest.
Financial support
Paolo M. Comoglio: AIRC ‘IG’ n. 11852 and AIRC ‘Special Program Molecular Clinical Oncology 5xMille’ n. 9970; Livio Trusolino: AIRC ‘IG’ n. 10116.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
We thank Melisa Milan for passionate bench work and Dr. Elena Casanova for supervising the apoptosis experiments (FACS analysis); Dr. Francesca De Bacco for reagents and suggestions and Dr. Raffaella Albano for inestimable technical assistance. We thank Antonella Cignetto and Michela Bruno for secretarial help and Daniela Gramaglia for assistance in grant management.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.12.006.
Benvenuti Silvia, Gentile Alessandra, Lazzari Luca, Arnesano Addolorata, Trusolino Livio and Comoglio Paolo M., (2014), An ‘in‐cell trial’ to assess the efficacy of a monovalent anti‐MET antibody as monotherapy and in association with standard cytotoxics, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.12.006.
References
- Bang, Y.J. , Van Cutsem, E. , Feyereislova, A. , Chung, H.C. , Shen, L. , Sawaki, A. , Lordick, F. , Ohtsu, A. , Omuro, Y. , Satoh, T. , Aprile, G. , Kulikov, E. , Hill, J. , Lehle, M. , Ruschoff, J. , Kang, Y.K. , 2010. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 376, 687–697. [DOI] [PubMed] [Google Scholar]
- Bardelli, A. , Corso, S. , Bertotti, A. , Hobor, S. , Valtorta, E. , Siravegna, G. , Sartore-Bianchi, A. , Scala, E. , Cassingena, A. , Zecchin, D. , Apicella, M. , Migliardi, G. , Galimi, F. , Lauricella, C. , Zanon, C. , Perera, T. , Veronese, S. , Corti, G. , Amatu, A. , Gambacorta, M. , Diaz, L.A. , Sausen, M. , Velculescu, V.E. , Comoglio, P. , Trusolino, L. , Di Nicolantonio, F. , Giordano, S. , Siena, S. , 2013. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov.. 3, 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina, J. , Caponigro, G. , Stransky, N. , Venkatesan, K. , Margolin, A.A. , Kim, S. , Wilson, C.J. , Lehar, J. , Kryukov, G.V. , Sonkin, D. , Reddy, A. , Liu, M. , Murray, L. , Berger, M.F. , Monahan, J.E. , Morais, P. , Meltzer, J. , Korejwa, A. , Jane-Valbuena, J. , Mapa, F.A. , Thibault, J. , Bric-Furlong, E. , Raman, P. , Shipway, A. , Engels, I.H. , Cheng, J. , Yu, G.K. , Yu, J. , Aspesi, P. , de Silva, M. , Jagtap, K. , Jones, M.D. , Wang, L. , Hatton, C. , Palescandolo, E. , Gupta, S. , Mahan, S. , Sougnez, C. , Onofrio, R.C. , Liefeld, T. , MacConaill, L. , Winckler, W. , Reich, M. , Li, N. , Mesirov, J.P. , Gabriel, S.B. , Getz, G. , Ardlie, K. , Chan, V. , Myer, V.E. , Weber, B.L. , Porter, J. , Warmuth, M. , Finan, P. , Harris, J.L. , Meyerson, M. , Golub, T.R. , Morrissey, M.P. , Sellers, W.R. , Schlegel, R. , Garraway, L.A. , 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti, S. , Lazzari, L. , Arnesano, A. , Li Chiavi, G. , Gentile, A. , Comoglio, P.M. , 2011. Ron kinase transphosphorylation sustains MET oncogene addiction. Cancer Res.. 71, 1945–1955. [DOI] [PubMed] [Google Scholar]
- Bertotti, A. , Burbridge, M.F. , Gastaldi, S. , Galimi, F. , Torti, D. , Medico, E. , Giordano, S. , Corso, S. , Rolland-Valognes, G. , Lockhart, B.P. , Hickman, J.A. , Comoglio, P.M. , Trusolino, L. , 2009. Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci. Signal.. 2, 80 [DOI] [PubMed] [Google Scholar]
- Bertotti, A. , Migliardi, G. , Galimi, F. , Sassi, F. , Torti, D. , Isella, C. , Cora, D. , Di Nicolantonio, F. , Buscarino, M. , Petti, C. , Ribero, D. , Russolillo, N. , Muratore, A. , Massucco, P. , Pisacane, A. , Molinaro, L. , Valtorta, E. , Sartore-Bianchi, A. , Risio, M. , Capussotti, L. , Gambacorta, M. , Siena, S. , Medico, E. , Sapino, A. , Marsoni, S. , Comoglio, P.M. , Bardelli, A. , Trusolino, L. , 2011. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov.. 1, 508–523. [DOI] [PubMed] [Google Scholar]
- Bhowmick, N.A. , Neilson, E.G. , Moses, H.L. , 2004. Stromal fibroblasts in cancer initiation and progression. Nature. 432, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaccio, C. , Gaudino, G. , Gambarotta, G. , Galimi, F. , Comoglio, P.M. , 1994. Hepatocyte growth factor (HGF) receptor expression is inducible and is part of the delayed-early response to HGF. J. Biol. Chem.. 269, 12846–12851. [PubMed] [Google Scholar]
- Catenacci, D.V. , Henderson, L. , Xiao, S.Y. , Patel, P. , Yauch, R.L. , Hegde, P. , Zha, J. , Pandita, A. , Peterson, A. , Salgia, R. , 2011. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence. Cancer Discov.. 1, 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoglio, P.M. , Giordano, S. , Trusolino, L. , 2008. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug Discov.. 7, 504–516. [DOI] [PubMed] [Google Scholar]
- De Bacco, F. , Luraghi, P. , Medico, E. , Reato, G. , Girolami, F. , Perera, T. , Gabriele, P. , Comoglio, P.M. , Boccaccio, C. , 2011. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst.. 103, 645–661. [DOI] [PubMed] [Google Scholar]
- Garnett, M.J. , Edelman, E.J. , Heidorn, S.J. , Greenman, C.D. , Dastur, A. , Lau, K.W. , Greninger, P. , Thompson, I.R. , Luo, X. , Soares, J. , Liu, Q. , Iorio, F. , Surdez, D. , Chen, L. , Milano, R.J. , Bignell, G.R. , Tam, A.T. , Davies, H. , Stevenson, J.A. , Barthorpe, S. , Lutz, S.R. , Kogera, F. , Lawrence, K. , McLaren-Douglas, A. , Mitropoulos, X. , Mironenko, T. , Thi, H. , Richardson, L. , Zhou, W. , Jewitt, F. , Zhang, T. , O'Brien, P. , Boisvert, J.L. , Price, S. , Hur, W. , Yang, W. , Deng, X. , Butler, A. , Choi, H.G. , Chang, J.W. , Baselga, J. , Stamenkovic, I. , Engelman, J.A. , Sharma, S.V. , Delattre, O. , Saez-Rodrigue, J. , Gray, N.S. , Settleman, J. , Futreal, P.A. , Haber, D.A. , Stratton, M.R. , Ramaswamy, S. , McDermott, U. , Benes, C.H. , 2012. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardi, E. , Birchmeier, W. , Birchmeier, C. , Vande Woude, G. , 2012. Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer. 12, 89–103. [DOI] [PubMed] [Google Scholar]
- Giordano, S. , Di Renzo, M.F. , Ferracini, R. , Chiado-Piat, L. , Comoglio, P.M. , 1988. p145, a protein with associated tyrosine kinase activity in a human gastric carcinoma cell line. Mol. Cell. Biol.. 8, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennerz, J.K. , Kwak, E.L. , Ackerman, A. , Michael, M. , Fox, S.B. , Bergethon, K. , Lauwers, G.Y. , Christensen, J.G. , Wilner, K.D. , Haber, D.A. , Salgia, R. , Bang, Y.J. , Clark, J.W. , Solomon, B.J. , Iafrate, A.J. , 2011. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol.. 29, 4803–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehrer, P.J. , Einhorn, L.H. , 1984. Drugs five years later. Cisplatin. Ann. Intern. Med.. 100, 704–713. [DOI] [PubMed] [Google Scholar]
- McDermott, U. , Sharma, S.V. , Dowell, L. , Greninger, P. , Montagut, C. , Lamb, J. , Archibald, H. , Raudales, R. , Tam, A. , Lee, D. , Rothenberg, S.M. , Supko, J.G. , Sordella, R. , Ulkus, L.E. , Iafrate, A.J. , Maheswaran, S. , Njauw, C.N. , Tsao, H. , Drew, L. , Hanke, J.H. , Ma, X.J. , Erlander, M.G. , Gray, N.S. , Haber, D.A. , Settleman, J. , 2007. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. U. S. A.. 104, 19936–19941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok, T.S. , Wu, Y.L. , Thongprasert, S. , Yang, C.H. , Chu, D.T. , Saijo, N. , Sunpaweravong, P. , Han, B. , Margono, B. , Ichinose, Y. , Nishiwaki, Y. , Ohe, Y. , Yang, J.J. , Chewaskulyong, B. , Jiang, H. , Duffield, E.L. , Watkins, C.L. , Armour, A.A. , Fukuoka, M. , 2009. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med.. 361, 947–957. [DOI] [PubMed] [Google Scholar]
- Pacchiana, G. , Chiriaco, C. , Stella, M.C. , Petronzelli, F. , De Santis, R. , Galluzzo, M. , Carminati, P. , Comoglio, P.M. , Michieli, P. , Vigna, E. , 2010. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J. Biol. Chem.. 285, 36149–36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo, M.O. , Kavan, P. , Miller, W.H. , Panasci, L. , Assouline, S. , Johnson, N. , Cohen, V. , Patenaude, F. , Pollak, M. , Jagoe, R.T. , Batist, G. , 2013. Systemic cancer therapy: achievements and challenges that lie ahead. Front. Pharmacol.. 4, 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchietti, S. , Michieli, P. , Galluzzo, M. , Mazzone, M. , Giordano, S. , Comoglio, P.M. , 2003. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 3, 347–361. [DOI] [PubMed] [Google Scholar]
- Prat, M. , Crepaldi, T. , Gandino, L. , Giordano, S. , Longati, P. , Comoglio, P.M. , 1991. C-terminal truncated forms of Met, the hepatocyte growth factor receptor. Mol. Cell. Biol.. 11, 5954–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, S.V. , Haber, D.A. , Settleman, J. , 2010. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer. 10, 241–253. [DOI] [PubMed] [Google Scholar]
- Shaw, A.T. , Kim, D.W. , Nakagawa, K. , Seto, T. , Crino, L. , Ahn, M.J. , De Pas, T. , Besse, B. , Solomon, B.J. , Blackhall, F. , Wu, Y.L. , Thomas, M. , O'Byrne, K.J. , Moro-Sibilot, D. , Camidge, D.R. , Mok, T. , Hirsh, V. , Riely, G.J. , Iyer, S. , Tassell, V. , Polli, A. , Wilner, K.D. , Janne, P.A. , 2013. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med.. 368, 2385–2394. [DOI] [PubMed] [Google Scholar]
- Slamon, D.J. , Leyland-Jones, B. , Shak, S. , Fuchs, H. , Paton, V. , Bajamonde, A. , Fleming, T. , Eiermann, W. , Wolter, J. , Pegram, M. , Baselga, J. , Norton, L. , 2001. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med.. 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Smolen, G.A. , Sordella, R. , Muir, B. , Mohapatra, G. , Barmettler, A. , Archibald, H. , Kim, W.J. , Okimoto, R.A. , Bell, D.W. , Sgroi, D.C. , Christensen, J.G. , Settleman, J. , Haber, D.A. , 2006. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. U. S. A.. 103, 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentle, J.J. , Tan, A.C. , Weekes, C.D. , Jimeno, A. , Leong, S. , Pitts, T.M. , Arcaroli, J.J. , Messersmith, W.A. , Eckhardt, S.G. , 2012. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol.. 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusolino, L. , Bertotti, A. , 2012. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: six degrees of separation. Cancer Discov.. 2, 876–880. [DOI] [PubMed] [Google Scholar]
- Trusolino, L. , Bertotti, A. , Comoglio, P.M. , 2010. MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol.. 11, 834–848. [DOI] [PubMed] [Google Scholar]
- Vermorken, J.B. , Mesia, R. , Rivera, F. , Remenar, E. , Kawecki, A. , Rottey, S. , Erfan, J. , Zabolotnyy, D. , Kienzer, H.R. , Cupissol, D. , Peyrade, F. , Benasso, M. , Vynnychenko, I. , De Raucourt, D. , Bokemeyer, C. , Schueler, A. , Amellal, N. , Hitt, R. , 2008. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med.. 359, 1116–1127. [DOI] [PubMed] [Google Scholar]
- Yauch, R.L. , Settleman, J. , 2012. Recent advances in pathway-targeted cancer drug therapies emerging from cancer genome analysis. Curr. Opin. Genet. Dev.. 22, 45–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data