

Abstract
Hsp90 is an important driver of stabilization and activation of several oncogenic proteins in many key pathways in oncogenesis, including HER2. The present study demonstrated that synuclein gamma (SNCG) prevents the protein degradation and protects the function of HER2 in the condition when the function of Hsp90 is blocked. Disruption of Hsp90 resulted in a significant degradation of HER2 and the loss of activity. However, SNCG completely recovered Hsp90 disruption‐mediated losses of HER2 and the function. SNCG bound to HER2 in the presence and absence of Hsp90. Specifically, the C‐terminal (Gln106‐Asp127) of SNCG bound to the loop connecting αC helix and β4 sheet of the kinase domain of HER2. SNCG renders resistance to 17‐AAG‐induced tumor suppression in tumor xenograft. Crossing SNCG transgenic mice with HER2 mice stimulated HER2‐induced tumor growth and rendered resistance to Hsp90 disruption. The present study indicates that SNCG protects Hsp90 client protein of HER2, and renders resistance to Hsp90 disruption.
Keywords: Synuclein gamma, Chaperone, HER2 signaling, Breast cancer, Therapy resistance
Highlights
C‐terminal of SNCG bound to the loop connecting αC helix and β4 sheet of the kinase domain of HER2.
SNCG protects Hsp90 client protein of HER2.
SNCG renders resistance to Hsp90 disruption.
Abbreviations
- BCSG1
breast cancer specific gene 1
- ERα
estrogen receptor α
- Hsp
heat shock protein
- mTOR
mammalian target of rapamycin
- SNCA
synuclein α
- SNCB
synuclein β
- SNCG
synuclein γ
1. Introduction
Hsp90 is a ubiquitously expressed molecular chaperone that controls the folding, assembly, intracellular disposition, and proteolytic turnover of many proteins, most of which are involved in signal transduction processes (Neckers, 2007). Hsp90 keeps unstable proteins poised for activation until they are stabilized by conformational changes associated with signal transduction (Wandinger et al., 2008). So far, more than 200 client proteins are known to be regulated by Hsp90, including key mediators of signal transduction and aberrant oncogenic fusion proteins (Neckers, 2007; Wandinger et al., 2008; Bonvini et al., 2002; Nimmanapalli et al., 2001; Chiosis et al., 2001; Caldas‐Lopes et al., 2009). Loss of Hsp90 chaperone activity may result in misfolding of target proteins, ultimately leading to their ubiquitylation and proteasomal degradation (Wandinger et al., 2008). Because of these activities, Hsp90 is becoming an emerging therapeutic target for cancer. The clinical development of Hsp90 inhibitors, such as 17‐AAG, as cancer chemotherapeutics is progressing rapidly. Although 17‐AAG, which permits simultaneous inhibition of multiple Hsp90 regulated client oncoproteins, is a promising new anticancer agent, acquired resistance to 17‐AAG has been demonstrated in many different cancer cells (Workman et al., 2007; Gaspar et al., 2009; McCollum et al., 2006), indicating that other proteins or chaperones may also protect Hsp90 client proteins when the Hsp90 function is blocked.
We previously identified a breast cancer specific gene BCSG1, also known as synuclein γ (SNCG) (Ji et al., 1997). Synucleins are a family of small proteins consisting of 3 known members, synuclein α (SNCA), synuclein β (SNCB), and SNCG (Clayton and George, 1998). While synucleins are highly expressed in neuronal cells and have been specifically implicated in neurodegenerative diseases (Polymeropoulos et al., 1997; Spillantini et al., 1997), SNCG is not clearly involved in neurodegenerative diseases but is primarily involved in neoplastic diseases. So far, the abnormal expression of SNCG protein has been demonstrated in many different malignant diseases, including breast (Ji et al., 1997; Bruening et al., 2000; Wu et al., 2003; Guo et al., 2007), liver (Liu et al., 2005; Zhao et al., 2006), esophagus (Liu et al., 2005), colon (Liu et al., 2009, 2005, 2008, 2010), gastric (Liu et al., 2005), lung (Liu et al., 2005), prostate (Liu et al., 2005), pancreas (Li et al., 2004; Hibi et al., 2009), bladder (Iwaki et al., 2004), cervical cancers (Liu et al., 2005), ovarian cancer (Bruening et al., 2000; Czekierdowski et al., 2006), and glial tumors (Fung et al., 2003). In these studies, SNCG protein is abnormally expressed in a high percentage of tumor tissues but rarely expressed in tumor‐matched nonneoplastic adjacent tissues. The clinical relevance of SNCG expression on breast cancer prognosis was confirmed in clinical follow‐up studies (Wu et al., 2003; Guo et al., 2007). Patients with an SNCG‐positive tumor had a significantly shorter disease‐free survival and overall survival compared with the patients with no SNCG expression. SNCG is a new unfavorable prognostic marker for breast cancer progression and a potential target for breast cancer treatment. At the cellular level, SNCG increases metastasis (Jia et al., 1999) and hormone‐dependent tumor growth (Jiang et al., 2003, 2004), promotes genetic instability (Gupta et al., 2003; Inaba et al., 2005), and regulates estrogen receptor signaling (Jiang et al., 2004; Liu et al., 2007; Shi et al., 2010).
HER2‐positive tumors account for approximately 30% of all breast cancer and these tumors carry poor prognosis. The importance of HER2 in breast cancer led to the development of agents that aimed at reducing HER2 level or activity. Trastuzumab (Herceptin) and lapatinib are 2 agents that have gained FDA approval for treating HER2‐positive breast cancer. In spite of their impressive results, a significant fraction of patients still develop either primary or secondary resistance. Several possible mechanisms of resistance have been proposed and one of the most described mechanisms is the loss of PTEN activity and/or activation of PI3K/Akt/mTOR signaling pathway (Jones and Buzdar, 2009). Considering that HER2 is an Hsp90 client protein and requires interaction with Hsp90 and its chaperone to acquire proper protein function (Citri et al., 2004), the Hsp90 inhibitor such as 17‐AAG provides an alternative approach to target HER2 through dissociation of HER2 from the chaperone, which leads to HER2 degradation by a proteasome‐dependent manner (Xu et al., 2001). Our previous studies demonstrate that expression of SNCG was strongly correlated with lymph node involvement, metastasis, and HER2 status (Guo et al., 2007). In the present study, we evaluated the effect of SNCG on HER2 expression and function. The results indicate that SNCG specifically interacts with HER2 and protects HER2 stability and function under stressful conditions when the activity of Hsp90 is blocked.
2. Results
2.1. SNCG prevents Hsp90 disruption induced HER2 degradation
Clinical studies demonstrated that expression of SNCG is clinically associated with HER2 status (Guo et al., 2007). Since the stability and function of HER2 is regulated by Hsp90 and 17‐AAG acts primarily by promoting the degradation of HER2, we investigated whether expression of SNCG could prevent 17‐AAG‐induced HER2 degradation. Using previously established SNCG transfected MCF‐7 cell (Jiang et al., 2003, 2004), we demonstrated that while treatment of parental MCF‐7 and vector transfected MCF‐neo1 cells with 17‐AAG resulted in a significant decrease in HER2 expression, forced expression of SNCG completely prevented the induced HER2 degradation (Figure 1A). Treatment of the cells with Herceptin had no effect on HER2 expression levels. To investigate whether the reduction of HER2 was due to proteasomal degradation, we treated MCF‐7 cells with proteasome inhibitor MG132. The results of this treatment revealed that 17‐AAG‐induced HER2 degradation could be blocked by proteasome inhibitor (Figure 1B). These results indicate that dysfunction of Hsp90 by treatment with Hsp90 inhibitors destabilized the HER2 protein and removed the unstable HER2 by proteasomes.
Figure 1.
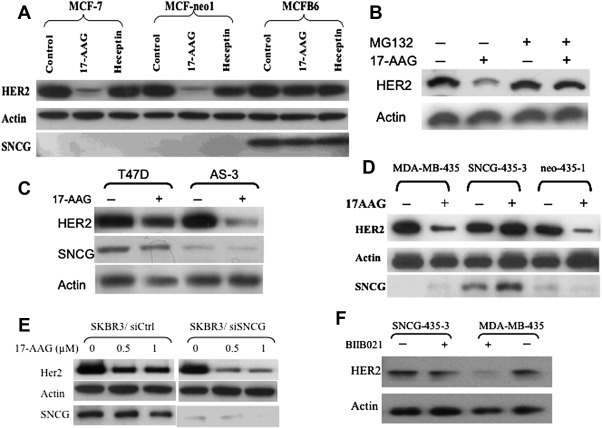
Prevention of HER2 degradation by SNCG. A. MCF‐7, MCF‐neo1, and MCFB6 cells were treated either with 17‐AAG (1 μM) or Herceptin (1 μM) for 12 h. HER2 and SNCG expression were analyzed by Western blot and normalized with actin. B. Reduction of HER2 was prevented by proteasome inhibitor treatment. MCF‐7 cells were pre‐treated with 20 μM of proteasome inhibitor MG 132 for 2 h followed by a treatment with 17‐AAG (1 μM) for 10 h. Cell lysates were analyzed for HER2 expression. C. T47D and its SNCG knockdown AS‐3 cells were treated with 17‐AAG (1 μM) for 12 h. HER2 and SNCG expression were analyzed by Western blot D. Parental MDA‐MB‐435, SNCG‐435‐3, and control neo‐435‐1 cells were treated with 1 μM 17‐AAG for 15‐h. E. Prevention of HER2 degradation in SKBR3 cells. Control siRNA (siCtrl) and SNCG siRNA infected cells treated with 17‐AAG (0.5 and 1 μM) for 12 h. HER2 and SNCG expression were analyzed by Western blot. F. Prevention of BIIB021‐induced HER2 degradation. Cells were treated with BIIB021 (80 nM, 24 h). HER2 expression was analyzed by Western blot.
The effect of SNCG expression on 17‐AAG‐induced HER2 degradation was further demonstrated by inhibiting endogenous SNCG expression in T47D cells using previously established stable SNCG knockdown T47D cell lines: AS‐3 cells (Jia et al., 1999). As demonstrated in Figure 1C, knockdown SNCG in T47D cells significantly increased sensitivity to 17‐AAG‐induced HER2 degradation. Since SNCG activates ERα transcriptional activity (Jiang et al., 2003, 2004) and ERα and HER2 cross activate each other, it is likely that the observed SNCG‐mediated HER2 protection in ER‐positive MCF‐7 and T47D cells is mediated indirectly by stimulation of ERα activity. To exclude this possibility, we first used ER‐negative and SNCG stably transfected MDA‐MB‐435 cells (Figure 1D). As we demonstrated in ER‐positive MCF‐7 cells, 17‐AAG treatment caused a significant reduction of HER2 levels in parental SNCG‐negative MDA‐MB‐435 cells and control vector transfected neo‐435‐1 cells. However, this 17‐AAG‐induced downregulation of HER2 was completely blocked by expression of SNCG in stably transfected SNCG‐435‐3 cells. Since there has been a controversy over the past several years about the true origin of the human MDA‐MB‐435 cell line, which might be derived from M14 melanoma cells (Chambers, 2009), next we used ER‐negative and SNCG‐positive SKBR3 breast cancer cell, which is considered as HER2 over‐expressed cell line. The effect of SNCG knockdown on HER2 expression in response to 17‐AAG treatment was determined in SKBR3 cells (Figure 1E). Treatment of SNCG knockdown cells with 17‐AAG significantly reduced HER2 expression levels. These data suggest that SNCG prevents HER2 degradation under stressful conditions, in which 17‐AAG blocks the chaperone function of Hsp90 and such protection of HER2 is ERα‐independent. We also tested the effect of SNCG on prevention of degradation of HER2 due to the Hsp90 disruption induced by different Hsp90 inhibitors. As shown in Figure 1F, treatment of MCF‐7 cells with BIIB021, a novel fully synthetic inhibitor that binds competitively with geldanamycin in the ATP‐binding pocket of Hsp90, resulted in a significant loss of HER2. Similar to what we observed for 17‐AAG, expression of SNCG prevented the BIIB021‐mediated loss of HER2.
2.2. SNCG prevents 17‐AAG‐induced downregulation of HER2 downstream effectors
Recent studies demonstrated manipulation of HER2 levels resulted in parallel changes in FoxM1 expression in breast cancer cells (Bektas et al., 2008; Francis et al., 2009). We next investigated whether 17‐AAG‐induced downregulation of HER2 can result in a concomitant decrease in FoxM1 expression and whether expression of SNCG can prevent the downregulation of FoxM1. In SNCG‐negative MDA‐MB‐435 cells, while downregulation of HER2 by blocking Hsp90 with 17‐AAG coincided with lower levels of FoxM1, enforced expression of SNCG in SNCG‐435‐3 cells prevented HER2 degradation and restored FoxM1 levels (Figure 2A). Similar results were also observed in the SNCK knockdown SKBR3 cells. These studies indicate that SNCG protects HER2 and its downstream effector FoxM1 in the absence of Hsp90 function.
Figure 2.
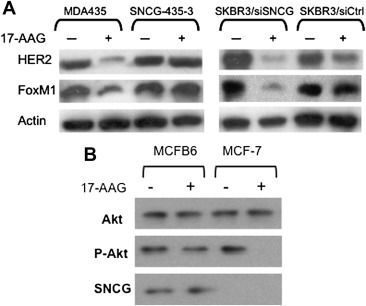
Protection of HER2 downstream effectors FoxM1 and Akt. A. MDA‐MB‐435, SNCG stably transfected SNCG‐435‐3, SKBR3, and SNCG knockdown SKBR3/siSNCG cells were treated with 17‐AAG (1 μM) for 12 h. HER2, FoxM1, and actin expression were analyzed by Western blot. B. Protection of Akt activity. MCF‐7 and SNCG stably transfected MCFB6 cells were treated with 1 μM 17‐AAG or 1 μM Herceptin for 12‐h. Total proteins were subjected to Western analysis of Akt, phosphorylated Akt, and SNCG.
We also investigated the effect of SNCG on the function and stability of another HER2 downstream effector Akt. As shown in Figure 2B, expression of SNCG did not activate Akt under normal conditions. However, under stressful conditions when 17‐AAG blocked the chaperoning function of Hsp90, the loss of Akt activity was completely protected by SNCG. 17‐AAG treatment only slightly decreased Akt expression, suggesting that the protein stability of Akt is not very sensitive to the inhibition of Hsp90 chaperoning function. Once activated Akt, however, vector transfected control MCF‐7 cells were highly sensitive to the inhibitory effect of 17‐AAG after 12 h of exposure; in contrast, SNCG over‐expressing MCFB6 cells were resistant to the inhibitory effect of this compound.
2.3. SNCG physically interacts with HER2
Because SNCG prevented 17‐AAG‐mediated degradation of HER2, we investigated if SNCG also physically interacts with HER2. We performed a GST pull‐down assay using the purified GST‐tagged SNCG protein to pull down HER2 (Figure 3A). The GST‐tagged SNCG was immobilized to GST beads and incubated with lysates of SKBR3 cells. The results of immunoblotting revealed that HER2 was specifically precipitated by immobilized GST‐SNCG, indicating that HER2 directly interacts with SNCG in vitro.
Figure 3.
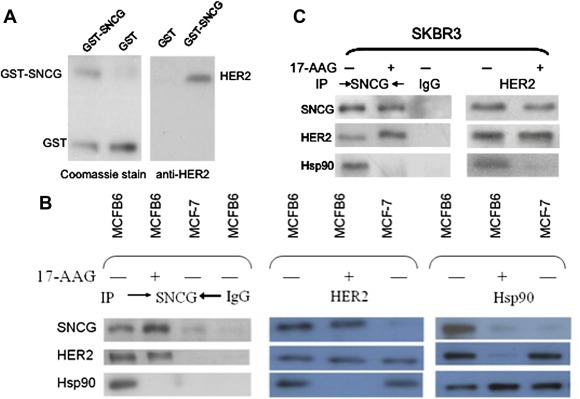
SNCG interacts with HER2. A. GST‐SNCG fusion protein was expressed in E. coli, purified, and stained with Coomassie Blue to demonstrate the expression of the fusion proteins (A, left panel). The SKBR3 cell extracts were subsequently incubated either with bead‐bound GST as a negative control or GST‐SNCG. After the beads were washed, proteins were subjected to Western blot for HER2 (A, right panel). B. Parental MCF‐7 and SNCG transfected MCFB6 cells were treated with or without 1 μM 17‐AAG for 15 h. Equal amount of protein was subjected to IP with antibodies against SNCG, HER2, and Hsp90 followed by Western blot for SNCG, HER2, and Hsp90. As a negative, IP with control IgG did not pull down SNCG, HER2, and Hsp90 in MCFB6 cells. C. SKBR3 cells were treated with or without 1 μM 17‐AAG for 15 h. Equal amount of protein was subjected to IP with antibodies against SNCG and HER2 followed by Western blot for SNCG, HER2, and Hsp90. As a negative, IP with control IgG did not pull down SNCG, HER2, and Hsp90 in SKBR3 cells.
Using SNCG transfected MCFB6 cells, we also determined if SNCG physically interacts with HER2 in cells in the presence and absence of 17‐AAG by immunoprecipitation (IP) assays. IP of SNCG in SNCG‐positive MCFB6 cells co‐precipitated HER2 and Hsp90 in the absence of 17‐AAG, indicating that SNCG participated in a chaperone complex with Hsp90 and HER2 in the absence of Hsp90 inhibitor. As negative controls, IP of SNCG in SNCG‐negative MCF‐7 cells and IP with control IgG in SNCG‐positive MCFB6 cells did not pull down HER2. Similarly, IP of HER2 co‐precipitated SNCG and Hsp90 and IP of Hsp90 co‐precipitated HER2 and SNCG in MCFB6 cells (Figure 3B). As expected, after cells were treated with 17‐AAG, Hsp90 dissociated from its client protein HER2. However, although SNCG dissociated from Hsp90, it still bound to HER2. Since SNCG still bound to HER2 even after Hsp90 was dissociated from its client protein, these data indicate that although SNCG participated in the chaperone complex with Hsp90, its function on HER2 was mediated by Hsp90‐independent pathways such as by direct binding and chaperoning HER2. We also investigated the in vivo interactions among endogenous SNCG, HER2 and Hsp90 in SKBR3 cells (Figure 3C). The same interaction pattern between endogenous SNCG and HER2 and Hsp90 was observed in SKBR3 cells, as we demonstrated in SNCG transfected MCF‐7 cells. When SKBR3 cells were cultured in the absence of 17‐AAG, endogenous SNCG was co‐precipitated with HER2 and Hsp90. However, after the treatment of 17‐AAG, Hsp90 dissociated from HER2 and SNCG, but SNCG still bound to Hsp90, indicating that the interaction between the endogenous SNCG, HER2, and Hsp90 proteins also occurs in the physiological situation.
2.4. Molecular modeling of interaction between SNCG and HER2
The kinase domain of HER2 holds an N‐terminal and C‐terminal lobes of which the region connecting the αC helix with β4 sheets in N‐terminal lobe was proven to interact with Hsp90 (Sidera et al., 2008; Citri et al., 2006). Disruption of this Hsp90‐HER2 interaction was shown to affect the stability of the HER2 (Citri et al., 2004). In order to evaluate the possible interactions between the HER2 kinase domain and SNCG, the structure of the HER2 kinase domain is modeled based on the coordinates of the active state conformation of ErbB‐1 kinase domain (PDB id: 2GS2) (Zhang et al., 2006); and the insilico interaction between the SNCG‐C terminal region (Manivel et al., 2011) and HER2 kinase domain was analyzed. The obtained interacting conformation (Figure 4A) reveals the specific orientation of SNCG‐C‐terminal region in which the residues Glu120‐Asp127 were placed in a position to make interactions with the residues Gly76‐Arg84 of the loop connecting the αC helix with β4 of HER2 kinase domain (Figure 4B). The orientation of SNCG was stabilized by the hydrophobic and hydrogen bonds between the residues of Gly76‐Arg84 of HER2 with the residues Glu120‐Asp127 of C‐terminal SNCG (Figure 4C), of which Gly78, Ser79 and Arg84 of HER2 involved in H‐bond between Glu120 and Asp127 of SNCG respectively. Meanwhile, a carbonyl–carbonyl interaction between Gly76 of HER2 and Glu120 of SNCG was also found to stabilize the interaction between the SNCG and HER2 kinase domain.
Figure 4.
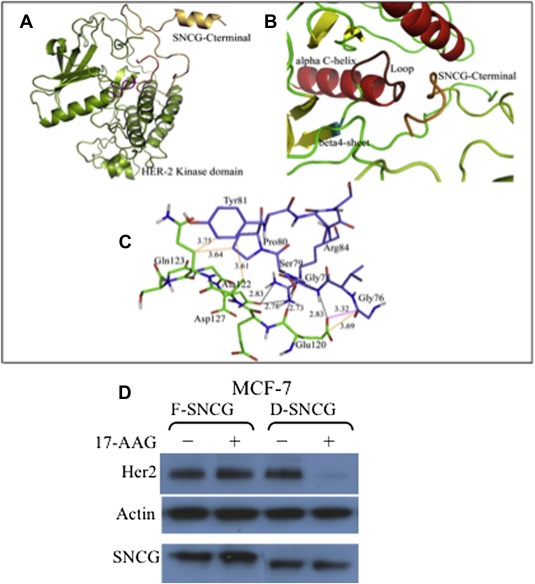
Structure analyses of the interaction between SNCG and HER2. A. Docked interacting conformation of HER2 (color code: lemon green) with the SNCG‐C terminal region (color code: orange). B. The interaction mode between the residues Glu120‐Asp127 of SNCG with the loop connecting the αC helix with β4 sheet in the N‐lobe of the predicted HER2 kinase domain. C. Residues involved in the specific HER2 (violet colored stick)‐SNCG (green colored stick) interaction. Hydrogen bonds: black dotted lines. Hydrophobic interactions: orange dotted lines. Carbonyl–carbonyl interactions: magenta dotted lines. D. MCF‐7 cells, engineered to express full length F‐SNCG and C‐terminal deleted D‐SNCG, were treated with 17‐AAG (1 μM, 15 h) and extracts were subjected to Western analysis of Her2, SNCG, and actin.
The molecular modeling study indicates that the C‐terminal aa 106–127 of SNCG interacts with its client proteins of HER2 and Akt. To confirm that the effects of SNCG on protection HER2 relays on its specific C‐terminal interaction with HER2, we constructed a deletion mutant of SNCG, which the C‐terminal region of 106–127 was deleted. While the wild type SNCG (F‐SNCG) was able to protect HER2 under the conditions that Hsp90 was disrupted, C‐terminal deleted SNCG (D‐SNCG) failed to protect HER2 under the same conditions (Figure 4D). These data are consistent with the molecular modeling analysis, which indicate that the C‐terminal aa 106–127 of SNCG binds to HER2 and protects HER2 from degradation.
2.5. SNCG renders resistance to 17‐AAG in xenograft model
To determine whether the protective effect of SNCG could be administered in vivo tumor xenograft model to a similar effect. We choose HER2 over‐expressing and SNCG‐negative MDA‐MB‐435 and its SNCG stably transfected SNCG‐435‐3 cell. We treated the mice with established MDA‐MB‐435 and SNCG‐435‐3 tumors with 17‐AAG. As shown in Figure 5A, treatment of MDA‐MB‐435 tumors resulted in a 62% tumor growth inhibition. However, SNCG‐435‐3 tumors were resistant to the treatment with only 25% tumor growth inhibition. A dose of 50 mg/kg 17‐AAG was sufficient to induce HER2 degradation in MDA‐MB‐435 tumors. In contrast, expression of SNCG restored HER2 in SNCG‐435‐3 tumors (Figure 5B).
Figure 5.
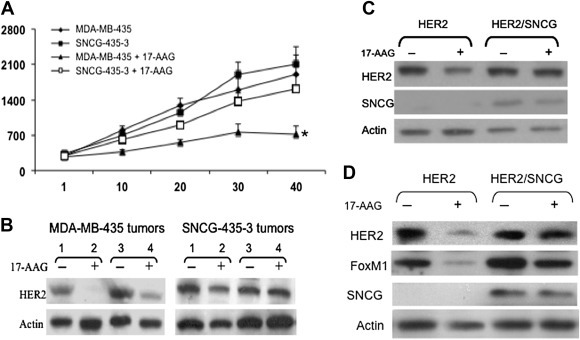
In vivo study of SNCG on protection of HRER2. A. Tumor growth in response to 17‐AAG treatment. Mice bearing established tumors were treated with either vehicle control or 17‐AAG (50 mg/kg, i.p., 3 times/week). All mice were sacrificed at day 40 following the first drug treatment. Statistical comparison for tumor size in 17‐AAG treated MDA‐MD‐435 mice relative to mice in other groups indicates *P < 0.01. B. Protection of HER2 expression. Tumors were harvested from two MDA‐MB‐435 and two SNCG‐435‐3 xenografts at day 40, 12 h following the last 17‐AAG treatment. Total protein was subjected to Western analyses of HER2, and actin. C. Protection of 17‐AAG‐induced HER2 degradation by SNCG in mammary organ culture. A pair (left and right) of inguinal mammary glands from a 14‐week virgin HER2 transgenic mouse and a HER2/SNCG bitransgenic mouse were cultured in the organ culture medium as described in Methods for 2 days followed by a 2‐day treatment with 1 μM 17‐AAG. The left mammary gland was used as control and the right gland was treated with 17‐AAG. Total protein was isolated and subjected to Western analysis. D. SNCG protects HER2 in HER2/SNCG bitransgenic mice. A 14‐month old HER2/SNCG bitransgenic mouse as well as an age‐matched HER2 transgenic mouse were treated with 17‐AAG (50 mg/kg, i.p., 3 times/week) for three weeks. Age‐matched control mice were treated with vehicle (5% Tween 80, 5% DMSO in PBS). At the end of 3‐week treatment, mice were sacrificed and the right inguinal gland was removed and subjected to Western analysis of HER2, FoxM1, SNCG, and actin.
2.6. Expression of SNCG in mammary gland protects HER2 and exacerbates HER2‐induced mammary tumor development
We developed a bitransgenic mice model that co‐express both HER2 and SNCG in the mammary epithelium by crossbreeding the previously established MMTV/SNCG transgenic mice (Liu et al., 2007) with MMTVneu HER2 transgenic mice. The effect of SNCG on HER2 was investigated in the mammary gland of SNCG/HER2 bitransgenic mice.
We first used ex vivo model involving a whole mouse mammary gland organ culture (MGOC) to study whether SNCG can prevent HER2 degradation due to the loss of Hsp90 chaperoning function. Treatment of mammary glands from HER2 transgenic mice in MGOC with 17‐AAG caused a significant reduction of HER2 expression. However, consistent with the in vitro effect in breast cancer cells, co‐expression of SNCG in HER2 transgenic mammary gland prevented 17‐AAG‐induced HER2 degradation, resulting in only a slight decrease in HER2 expression (Figure 5C). Next, we studied the effect of SNCG on the expression and function of HER2 on the transgenic mice treated with 17‐AAG. Treatment of HER2 transgenic mice with 17‐AAG resulted in a significant loss of HER2 and its downstream effector FoxM1. However, expression of SNCG in the SNCG/HER2 bitransgenic mice prevented the loss of HER2 and FoxM1 due to the disruption of Hsp90 functions (Figure 5D). These studies provide direct evidence supporting the chaperone role of SNCG on Hsp90 client proteins of HER2 sin a more relevant mammary gland.
Effect of SNCG on HER2‐induced mammary tumor was investigated in SNCG/HER2 bitransgenic mice. While expression of SNCG in mammary epithelium induces mammary hyperplasia, MMTV/SNCG transgenic mice fail to develop mammary tumors (Liu et al., 2007). Previous studies have shown that HER2 transgenic mice develop multifocal metastatic mammary tumors at ∼6 months of age (Siegel et al., 1999). Expression of SNCG in mammary glands significantly stimulated HER2‐induced mammary tumorigenesis (Table 1). The results reveal that bitransgenics show significantly reduced latency of palpable mammary tumor formation with 50% of the animals showing tumor formation at 145 days as opposed to 182 days for HER2 mice. Tumor growth was also significantly stimulated. At day 250, the average tumor size of bitransgenics was 1.45‐fold of that of HER2 mice. There was a slight increase in tumor incidence in bitransgenics, but it was not significant compared with HER2 mice. To determine whether 17‐AAG has an antitumor effect on transgenic mice and whether expression of SNCG can render resistance to 17‐AAG‐mediated antitumor effect on HER2 mice, we treated the tumor‐bearing mice with 17‐AAG for 1‐month period from day 220 to day 250. Tumors in HER2 mice receiving the treatment had a mean 65% reduction in volume when compared with tumors in mice that were treated with vehicle control (318 mm3 vs. 921 mm3). In contrast, SNCG/HER2 mice treated with 17‐AAG demonstrated a slight 22% tumor inhibition during the therapy (1045 mm3 vs. 1338 mm3). To exclude the possibility that the attenuated antitumor effect of 17‐AAG on SNCG/HER2 bitransgenic mice is due to the poor accessibility of larger tumors to the drug, in a separate experiment, we started the drug treatment at the earlier day for the bitransgenic mice in order to have a similar tumor size at the starting point. While treatment of HER2 mice with 17‐AAG resulted in a 59% tumor inhibition (from 821 mm3 to 337 mm3), expression of SNCG reduced tumor inhibition to 25% in the SNCG/HER2 bitransgenic mice (from 891 mm3 to 671 mm3). These data indicate that SNCG renders tumor resistance to Hsp90 disruption.
Table 1.
Expression of SNCG exacerbates HER2‐induced mammary tumor development and renders resistance to antitumor effect of 17‐AAG. The effect of SNCG on HER2‐mediated tumorigenesis was measured by tumor incidence (percentage of mice developing tumors at 250‐day period), tumor latency T50 (day with 50% of the animals showing tumor formation), and tumor size at the day 250. For treatment, mice bearing tumors were treated with either vehicle control or 17‐AAG (50 mg/kg, i.p., 3 times/week) for 1‐month period. There were 15 mice analyzed for each group. Tumor growth inhibition was calculated by comparison of treated vs. non‐treated mice. Statistical comparisons for both T50 and tumor size in SNCG/HER2 bitransgenic mice relative to HER2 transgenic mice indicate p < 0.01. Statistical comparison for tumor incidence in SNCG/HER2 mice relative to HER2 mice indicates p > 0.05. Statistical comparison for tumor size in 17‐AAG treated vs. non‐treated HER2 mice indicates P < 0.01. Statistical comparison for tumor size in 17‐AAG treated vs. non‐treated SNCG/HER2 mice indicates P > 0.05.
| Experimental group | Tumor incidence | T50 | Tumor size (mm3) |
|---|---|---|---|
| Genotypes | Tumor/total (%) | (Days) | Day 250 |
| SNCG | N/A | N/A | N/A |
| HER2 | 52 | 182 | 921 ± 198 |
| SNCG/HER2 | 58 | 145 | 1338 ± 281 |
| With Treatment of 17‐AAG at day 220–250 | |||
| HER2 | N/A | N/A | 318 ± 49 (65% ↓) |
| SNCG/HER2 | N/A | N/A | 1045 ± 136 (22% ↓) |
| Tumor size before 17‐AAG treatment (mm3) | Tumor size after 17‐AAG treatment (mm3) | ||
| HER2 | 821 ± 168 (day 230) | 337 ± 41 (59% ↓) | |
| SNCG/HER2 | 891 ± 229 (day 190) | 671 ± 78 (25% ↓) | |
3. Discussion
The present findings demonstrate a novel role for SNCG in the context of protection of stability and function of Hsp90 client protein HER2 under stressful conditions when the chaperone function of Hsp90 is blocked. In this article, we provide evidence that 1) SNCG prevents the degradation of HER2 under conditions when the chaperoning function of Hsp90 is blocked by 17‐AAG. The SNCG‐mediated protection was demonstrated in four different cell systems including overexpression of SNCG in SNCG‐negative MCF‐7 cells and MDA‐MB‐435 cells and knockdown endogenous SNCG expression in T47D and SKBR3 cells; 2) SNCG protects HER2 in tumor xenograft and in the mammary gland from HER2/SNCG bitransgenic mouse; 3) SNCG stimulates HER2‐induced mammary tumor growth and renders a resistance to 17‐AAG‐mediated antitumor effect in the HER2/SNCG bitransgenic mice; 4) SNCG binds to HER2 both in vitro in cell‐free system and in breast cancer cells; such interaction occurs even in the presence of 17‐AAG, in which Hsp90 dissociates from its client protein HER2; and 5) molecular modeling study indicates that SNCG binds to the loop connecting αC helix and β4 sheet of the kinase domain of HER2.
Although the biology and pathology of HER2 have been under extensive investigation, factors that regulate HER2 expression and function remain largely unknown. Over the past years, there has been increasing evidence that Hsp90 interacts with a great number of molecules including HER2 that are involved in the development and/or survival of cancer cells (Neckers, 2007; Wandinger et al., 2008; Bonvini et al., 2002; Nimmanapalli et al., 2001; Chiosis et al., 2001; Caldas‐Lopes et al., 2009). The role of Hsp90 in the regulation of HER2 has been attributed so far to stabilization of the receptor at the cell surface, via interaction with its cytoplasmic kinase domain, such that disruption of the HER2/Hsp90 association induced by 17‐AAG leads to proteasomal degradation of the receptor (Citri et al., 2004; Xu et al., 2001). However, resistance to Hsp90 inhibitors has been established and one of the mechanisms underlying resistance to Hsp90 disruption is the presence of other molecular chaperones that also regulate Hsp90 client proteins (Workman et al., 2007; Gaspar et al., 2009; McCollum et al., 2006). The chaperone‐like activity of SNCG has been demonstrated in the cell‐free system by assaying the aggregation of thermally denatured proteins; SNCG binding reduces the thermally induced degradation of alcohol dehydrogenase and insulin (Souza et al., 2000). Consistent with the previous report on chaperone‐like activity of SNCG, here we provided evidence that SNCG prevents the degradation and the loss of activity of HER2 when 17‐AAG blocks the chaperoning function of Hsp90. This protection on HER2 was demonstrated in breast cancer cells, tumor xenograft, and the mammary gland from HER2/SNCG bitransgenic mouse. These data suggest a critical role of SNCG in maintaining the stability and function of HER2 signaling pathway.
We previously demonstrated that SNCG participates in Hsp90‐based multi‐chaperone complex for ERα and stimulates ERα transcriptional activity (Jiang et al., 2004; Liu et al., 2007). In the present study, we also demonstrated that SNCG physically associates with HER2 and Hsp90. One of the critical questions that needs to be addressed is whether SNCG‐mediated effect on HER2 is manifested by Hsp90‐based chaperone complex or by its own chaperoning function. Using Hsp90 inhibitor 17‐AAG, our data demonstrated that treatment of cells with 17‐AAG resulted in a significant degradation of HER2. However, expression of SNCG completely recovered 17‐AAG‐mediated loss of HER2 expression. Furthermore, when cells were treated with 17‐AAG, while Hsp90 lost its ATPase function and dissociated from its client protein HER2, SNCG was still physically associated with and chaperoning HER2. These data suggest that SNCG, which can replace the chaperone function of Hsp90 on its client protein HER2, is an independent chaperone protein and its chaperoning function on HER2 is not dependent on Hsp90. Thus, SNCG and Hsp90 act cooperatively in regulating HER2 stability and function.
To study the mechanism through which SNCG protects various Hsp90 client proteins, we performed docking analysis of the interaction between SNCG and HER2. It is well established that disruption of interaction between Hsp90 and kinase domain of HER2, mediated by the loop connecting αC helix and β4 sheet of N‐lobe of kinase domain (Citri et al., 2006), results in the ubiquitin mediated degradation of HER2 (Xu et al., 2001). This region acts as the hinge controlling the movement of αC helix, which is also defined as the conserved chaperone‐binding region in some of the Hsp90 client kinase proteins including HER2 (Citri et al., 2006). Accordingly, the replacement of Hsp90 function by SNCG, which also acts as a chaperone, invoked the possibility of the interaction between SNCG and the chaperone‐binding region of HER2. Specifically, the C‐terminal tail region of SNCG, enriched with the highly charged residues, has been suggested to possess chaperone‐like activity and to mediate protein–protein interactions (Manivel et al., 2011). We provide evidence that the C‐terminal of SNCG binds directly to the loop connecting αC helix and β4 sheet of the kinase domain of HER2. We reason that this interaction stabilizes Hsp90 client proteins and maintains their function when the chaperone function of Hsp90 is disrupted.
17‐AAG seems to display significant Hsp90‐dependent antitumor activity. Although the exact mechanisms by which 17‐AAG kills cancer cells remain to be defined and may vary with cell types, studies suggest that abrogation of the survival pathways, such as HER2 signaling, may lead to apoptosis (Okawa et al., 2009). Using tumor xenograft model, we demonstrated that while SNCG expression renders resistance to 17‐AAG‐induced tumor suppression, compromising endogenous SNCG expression increased sensitivity to 17‐AAG‐induced cytotoxicity. Consistent with tumor xenograft model, our study demonstrated that SNCG antagonizes 17‐AAG‐mediated antitumor effect in SNCG/HER2 bitransgenic model. These data support the conclusion that protection of Hsp90 client proteins, such as HER2, is responsible for the antagonistic effect of SNCG against 17‐AAG. It should be pointed out that 17‐AAG binds to Hsp90 and promotes the degradation of various Hsp90 client proteins important in the proliferation and survival of malignant cells. Although the direct target of 17‐AAG is Hsp90, and HER2 is a Hsp90 client protein, multiple mechanisms may contribute to the SNCG‐mediated antitumor effect. It has been demonstrated that overexpression of SNCG leads to constitutive activation of ERK1/2 and inactivation of JNK in response to several chemotherapeutic agents (Hua et al., 2009). Nevertheless, we demonstrated that mammary specific expression of SNCG in HER2/SNCG bitransgenic mouse protects HER2, stimulates HER2‐mediated mammary tumorigenesis, and renders resistance to 17‐AAG‐mediated antitumor effect. We reason that the chaperoning function of SNCG on HER2 signaling and other Hsp90 regulated signaling pathways, such as PI3K/Akt/mTOR, may serve an underlying mechanism for promoting cancer progression and resistance to Hsp90 disruptors.
Unlike a typical chaperone Hsp90, which is essential for a range of indispensable functions in normal tissue, SNCG is not expressed in normal cells but aberrantly expressed in advanced malignant state through epigenetic control by demethylation of CpG sties within SNCG gene (Liu et al., 2005), suggesting that SNCG is a more tumor oriented chaperone. Targeting tumor specific chaperones, such as SNCG, represents a potential alternative to direct Hsp90 inhibition that may offer greater specificity. Notably, resistance to 17‐AAG has been frequently reported in cancer cells (Workman et al., 2007; Gaspar et al., 2009; McCollum et al., 2006). It is known that Hsp90 disruption induces unfolded protein response and leads to an ER stress (Davenport et al., 2007); and ER stress often causes a robust increase in the expression of molecular chaperones, which assist protein refolding (Szegezdi et al., 2006). This may represent a fundamental rationale for acquired resistance to Hsp90 disruption. Since SNCG is upregulated by ER stress (Hua et al., 2009), and we demonstrated here that SNCG protects HER2 when Hsp90 function is disrupted; targeting SNCG should be exploited to increase the clinical efficacy of Hsp90‐directed therapy. The study will potentially lead to a new molecular profile of the tumor for the optimal patient selection for Hsp90 disruption and a new strategy of combining SNCG targeting with Hsp90 disruption as a novel advantageous approach for treatment of cancer.
4. Materials and methods
4.1. Materials
Proteasome inhibitor MG132 and Hsp90 inhibitor 17‐AAG (Sigma) were dissolved in DMSO. Herceptin (Genentech) was dissolved in sterile water at 20 mg/ml. Antibodies used (Santa Cruz) were as follows: anti‐SNCG (goat polyclonal antibody E‐20, 1:300 dilution); anti‐Hsp90 (rabbit polyclonal antibody sc‐7947, 1:1000 dilution); anti‐HER2 (mouse monoclonal antibody sc‐71677); anti‐FoxM1 (rabbit polyclonal antibody sc‐13016); normal goat IgG (sc‐2028); normal rabbit IgG (sc‐2027) and anti‐actin (goat polyclonal antibody sc‐1615).
4.2. Cell lines, SNCG transfection, and knockdown
MCF‐neo1 (vector transfected MCF‐7 clone), and SNCG stably transfected MCFB6 (MCF‐7 clone) cells and SNCG‐435‐3 cells (MDA‐MB‐435 clone) were previously established and described (Jia et al., 1999; Jiang et al., 2003). Knockdown SNCG expression in T47D cells and establishment of two stably SNCG antisense transfected clones (AS‐3 and AS‐1) were previously selected and characterized (Jiang et al., 2003). To knockdown endogenous SNCG in SKBR3 cells, we used SNCG siRNA Lentiviral Particle Gene Silencers (42290‐v from Santa Cruz). SNCG siRNA consists of a pool of five target‐specific 19–25 nt siRNAs designed to knockdown SNCG gene expression. Subconfluent SKBR3 cells were infected with the lentiviral vectors according to manufacture's protocol.
4.3. In vitro pulldown assay using Glutathione‐S‐Transferase
This was performed as we previously described (Shi et al., 2010).
4.4. Preparation of recombinant SNCG protein (rSNCG)
Escherichia coli expressed rSNCG was prepared as we previously described (Guo et al., 2007).
4.5. Immunoprecipitation (IP)
Cells were cultured in 100 mm cell culture dishes in the stripped condition for 3 days. After the treatment with E2 or 17‐AAG, cells were lysed (1× PBS, 1% Triton X‐100, 10 mM sodium molybdate, 2 mg/ml aprotinin, 0.5 mg/ml leupeptin and 1 mM PMSF), disrupted by sonication, and centrifuged for 10 min at 10,000 g. Immunoprecipitation was carried out as we previously described Jiang et al., 2003, 2004, 2010
4.6. Mammary gland organ culture
As we previously described (Liu et al., 2007), a pair of inguinal whole mammary gland was removed from 14‐week virgin female MMTV/SNCG and HER2/SNCG transgenic mice and cultured in medium 199 containing 5% FCS, with medium changed every two days. The medium was supplemented with following components from Clonetics: bovine pituitary extract (52 μg/ml), insulin (5 μg/ml), EGF (10 ng/ml), and hydrocortisone (1 μg/ml). The glands were cultured in the organ culture for 2 days before addition of 1 μM 17‐AAG for additional 2 days. At day 4, the glands were subjected to protein extraction for Western analysis.
4.7. Tumor growth in athymic nude mice
A nude mouse tumorigenic assay was performed as we previously described (Jiang et al., 2004; Shi et al., 2010). Briefly, approximately 1 × 106 cells were injected into a 6‐week old female athymic nude mouse (Frederick Cancer Research and Development Center, Frederick, MD). Each animal received two injections, one on each side, in the mammary fat pads between the first and second nipples. Tumor size was determined at weekly intervals by three‐dimensional measurements (mm) using a caliper. For drug administration, 17‐AAG was dissolved in 5% Tween 80, 5% DMSO in PBS, and administrated at 50 mg/kg, i.p. 3 times/week as previously reported.
4.8. Generation of SNCG/HER2 bitransgenic mice and HER2‐induced mammary tumor
We previously established and characterized a mammary specific SNCG transgenic mouse model under the MMTV promoter (Liu et al., 2007). Transgenic males and females from the same family were mated to generate homozygous mice. We interbred homozygous MMTV/SNCG mice with homozygous MMTVneu mice (FVB/N‐Tg from Jackson Lab) to generated bitransgenic mice. Both MMTV/SNCG and MMTVneu mice were on the FVB background. Animals were genotyped using tail DNA (Shi et al., 2010). Mammary tumor formation was monitored in nulliparous mice by weekly physical palpation. Mice were terminated at 10 months and mammary tumors harvested from mice were subjected histologic analysis as we previously described (Jiang et al., 2004; Liu et al., 2007).
4.9. Molecular docking study
The coordinates of active state conformation of ErbB‐1 (PDB id: 2GS2) (Zhang et al., 2006) were used to model the structure of HER2 kinase domain by using MODELLER 9v7. The generated structure was optimized using the inbuilt options provided in the modeler tool. The insilico interaction study between the C‐terminal SNCG (Manivel et al., 2011) with the modeled HER2 kinase domain was carried out using the easy interface option of HADDOCK online server. The PyMol molecular visualization tool version 0.99 was used to analyze and to prepare the images and DimPlot was used to plot the interactions between SNCG and HER2.
4.10. Statistical analysis
Results were reported as the mean ± SD for typical experiments done in three replicate samples and compared by the Student's t test. Results were considered significantly different for p < 0.05. All experiments were done at least twice to ensure reproducibility of the results.
Acknowledgments
This study was supported in part by grants 81230054 and 81071819 from State Key Program of National Natural Science Foundation of China; a grant from National Key Technology R&D Program for the 12th Five‐year Plan of China (2013BAI01B06); and a grant from State Key Laboratory of Reproductive Medicine.
Shao Yongfeng, Wang Bingchan, Shi Dorothy, Miao Suyu, Manivel Panneerselvam, Krishna Ramadas, Chen Yiding, Eric Shi Y., (2014), Synuclein gamma protects HER2 and renders resistance to Hsp90 disruption, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.05.011.
The authors declare no conflict of interest.
References
- Bektas, N. , Haaf, A. , Veeck, J. , Wild, P.J. , Luscher-Firzlaff, J. , Hartmann, A. , Knuchel, R. , Dahl, E. , 2008. Tight correlation between expression of the Forkhead transcription factor FOXM1 and HER2 in human breast cancer. BMC cancer 8, 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvini, P. , Gastaldi, T. , Falini, B. , Rosolen, A. , 2002. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res. 62, 1559–1566. [PubMed] [Google Scholar]
- Bruening, W. , Giasson, B.I. , Klein-Szanto, A.J. , Lee, V.M. , Trojanowski, J.Q. , Godwin, A.K. , 2000. Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer 88, 2154–2163. [PubMed] [Google Scholar]
- Caldas-Lopes, E. , Cerchietti, L. , Ahn, J.H. , Clement, C.C. , Robles, A.I. , Rodina, A. , Moulick, K. , Taldone, T. , Gozman, A. , Guo, Y. , Wu, N. , de Stanchina, E. , 2009. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. U S A. 106, 8368–8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers, A.F. , 2009. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma?. Cancer Res. 69, 5292–5293. [DOI] [PubMed] [Google Scholar]
- Chiosis, G. , Timaul, M.N. , Lucas, B. , Munster, P.N. , Zheng, F.F. , Sepp-Lorenzino L,Rosen, N. , 2001. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol. 8, 289–299. [DOI] [PubMed] [Google Scholar]
- Citri, A. , Kochupurakkal, B.S. , Yarden, Y. , 2004. The achilles heel of ErbB-2/HER2: regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 3, 51–60. [PubMed] [Google Scholar]
- Citri, A. , Harari, D. , Shohat, G. , Ramakrishnan, P. , Gan, J. , Lavi, S. , Eisenstein, M. , Kimchi, A. , Wallach, D. , Pietrokovski, S. , Yarden, Y. , 2006. Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 281, 14361–14369. [DOI] [PubMed] [Google Scholar]
- Clayton, D.F. , George, J.M. , 1998. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254. [DOI] [PubMed] [Google Scholar]
- Czekierdowski, A. , Czekierdowska, S. , Wielgos, M. , Smolen, A. , Kaminski, P. , Kotarski, J. , 2006. The role of CpG islands hypomethylation and abnormal expression of neuronal protein synuclein-gamma (SNCG) in ovarian cancer. Neuro Endocrinol. Lett. 27, 381–386. [PubMed] [Google Scholar]
- Davenport, E.L. , Moore, H.E. , Dunlop, A.S. , Sharp, S.Y. , Workman, P. , Morgan, G.J. , Davies, F.E. , 2007. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 110, 2641–2649. [DOI] [PubMed] [Google Scholar]
- Francis, R.E. , Myatt, S.S. , Krol, J. , Hartman, J. , Peck, B. , McGovern, U.B. , Wang, J. , Guest, S.K. , Filipovic, A. , Gojis, O. , Palmieri, C. , Peston, D. , 2009. FoxM1 is a downstream target and marker of HER2 overexpression in breast cancer. Int. J. Oncol. 35, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung, K.M. , Rorke, L.B. , Giasson, B. , Lee, V.M. , Trojanowski, J.Q. , 2003. Expression of alpha-, beta-, and gamma-synuclein in glial tumors and medulloblastomas. Acta Neuropathol. 106, 167–175. [DOI] [PubMed] [Google Scholar]
- Gaspar, N. , Sharp, S.Y. , Pacey, S. , Jones, C. , Walton, M. , Vassal, G. , Eccles, S. , Pearson, A. , Workman, P. , 2009. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 69, 1966–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J. , Shou, C. , Meng, L. , Jiang, B. , Dong, B. , Yao, L. , Xie, L. , Zhang, J. , Chen, Y. , Budman, D.R. , Shi, Y.E. , 2007. Neuronal protein synuclein gamma predicts poor clinical outcome in breast cancer. Int. J. Cancer. 121, 1296–1305. [DOI] [PubMed] [Google Scholar]
- Gupta, A. , Inaba, S. , Wong, O.K. , Fang, G. , Liu, J. , 2003. Breast cancer-specific gene 1 interacts with the mitotic checkpoint kinase BubR1. Oncogene 22, 7593–7599. [DOI] [PubMed] [Google Scholar]
- Hibi, T. , Mori, T. , Fukuma, M. , Yamazaki, K. , Hashiguchi, A. , Yamada, T. , Tanabe, M. , Aiura, K. , Kawakami, T. , Ogiwara, A. , Kosuge, T. , Kitajima, M. , 2009. Synuclein-gamma is closely involved in perineural invasion and distant metastasis in mouse models and is a novel prognostic factor in pancreatic cancer. Clin. Cancer Res. 15, 2864–2871. [DOI] [PubMed] [Google Scholar]
- Hu, H. , Sun, L. , Guo, C. , Liu, Q. , Zhou, Z. , Peng, L. , Pan, J. , Yu, L. , Lou, J. , Yang, Z. , Zhao, P. , Ran, Y. , 2009. Tumor cell-microenvironment interaction models coupled with clinical validation reveal CCL2 and SNCG as two predictors of colorectal cancer hepatic metastasis. Clin. Cancer Res. 15, 5485–5493. [DOI] [PubMed] [Google Scholar]
- Hua, H. , Xu, L. , Wang, J. , Jing, J. , Luo, T. , Jiang, Y. , 2009. Up-regulation of gamma-synuclein contributes to cancer cell survival under endoplasmic reticulum stress. J. Pathol. 217, 507–515. [DOI] [PubMed] [Google Scholar]
- Inaba, S. , Li, C. , Shi, Y.E. , Song, D.Q. , Jiang, J.D. , Liu, J. , 2005. Synuclein gamma inhibits the mitotic checkpoint function and promotes chromosomal instability of breast cancer cells. Breast Cancer Res. Treat. 94, 25–35. [DOI] [PubMed] [Google Scholar]
- Iwaki, H. , Kageyama, S. , Isono, T. , Wakabayashi, Y. , Okada, Y. , Yoshimura, K. , Terai, A. , Arai, Y. , Iwamura, H. , Kawakita, M. , YoshiKi, T. , 2004. Diagnostic potential in bladder cancer of a panel of tumor markers (calreticulin, gamma -synuclein, and catechol-o-methyltransferase) identified by proteomic analysis. Cancer Sci. 95, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, H. , Liu, Y.E. , Jia, T. , Wang, M. , Liu, J. , Xiao, G. , Joseph, B.K. , Rosen, C. , Shi, Y.E. , 1997. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 57, 759–764. [PubMed] [Google Scholar]
- Jia, T. , Liu, Y.E. , Liu, J. , Shi, Y.E. , 1999. Stimulation of breast cancer invasion and metastasis by synuclein gamma. Cancer Res. 59, 742–747. [PubMed] [Google Scholar]
- Jiang, Y. , Liu, Y.E. , Lu, A. , Gupta, A. , Goldberg, I.D. , Liu, J. , Shi, Y.E. , 2003. Stimulation of estrogen receptor signaling by gamma synuclein. Cancer Res. 63, 3899–3903. [PubMed] [Google Scholar]
- Jiang, Y. , Liu, Y.E. , Goldberg, I.D. , Shi, Y.E. , 2004. Gamma synuclein, a novel heat-shock protein-associated chaperone, stimulates ligand-dependent estrogen receptor alpha signaling and mammary tumorigenesis. Cancer Res. 64, 4539–4546. [DOI] [PubMed] [Google Scholar]
- Jones, K.L. , Buzdar, A.U. , 2009. Evolving novel anti-HER2 strategies. Lancet Oncol. 10, 1179–1187. [DOI] [PubMed] [Google Scholar]
- Li, Z. , Sclabas, G.M. , Peng, B. , Hess, K.R. , Abbruzzese, J.L. , Evans, D.B. , Chiao, P.J. , 2004. Overexpression of synuclein-gamma in pancreatic adenocarcinoma. Cancer 101, 58–65. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Liu, W. , Wu, Y. , Zhou, Y. , Xue, R. , Luo, C. , Wang, L. , Zhao, W. , Jiang, J.D. , Liu, J. , 2005. Loss of epigenetic control of synuclein-gamma gene as a molecular indicator of metastasis in a wide range of human cancers. Cancer Res. 65, 7635–7643. [DOI] [PubMed] [Google Scholar]
- Liu, Y.E. , Pu, W. , Jiang, Y. , Shi, D. , Dackour, R. , Shi, Y.E. , 2007. Chaperoning of estrogen receptor and induction of mammary gland proliferation by neuronal protein synuclein gamma. Oncogene 26, 2115–2125. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Guo, J. , Qu, L. , Bing, D. , Meng, L. , Wu, J. , Shou, C. , 2008. Applications of novel monoclonal antibodies specific for synuclein-gamma in evaluating its levels in sera and cancer tissues from colorectal cancer patients. Cancer Lett. 269, 148–158. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Dong, B. , Lu, A. , Qu, L. , Xing, X. , Meng, L. , Wu, J. , Eric Shi, Y. , Shou, C. , 2010. Synuclein gamma predicts poor clinical outcome in colon cancer with normal levels of carcinoembryonic antigen. BMC cancer 10, 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manivel, P. , Muthukumaran, J. , Kannan, M. , Krishna, R. , 2011. Insight into residues involved in the structure and function of the breast cancer associated protein human gamma synuclein. J. Mol. Model. 17, 251–263. [DOI] [PubMed] [Google Scholar]
- McCollum, A.K. , Teneyck, C.J. , Sauer, B.M. , Toft, D.O. , Erlichman, C. , 2006. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 66, 10967–10975. [DOI] [PubMed] [Google Scholar]
- Neckers, L. , 2007. Heat shock protein 90: the cancer chaperone. J. Biosci. 32, 517–530. [DOI] [PubMed] [Google Scholar]
- Nimmanapalli, R. , O'Bryan, E. , Bhalla, K. , 2001. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 61, 1799–1804. [PubMed] [Google Scholar]
- Okawa, Y. , Hideshima, T. , Steed, P. , Vallet, S. , Hall, S. , Huang, K. , Rice, J. , Barabasz, A. , Foley, B. , Ikeda, H. , Raje, N. , Kiziltepe, T. , 2009. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood 113, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos, M.H. , Lavedan, C. , Leroy, E. , Ide, S.E. , Dehejia, A. , Dutra, A. , Pike, B. , Root, H. , Rubenstein, J. , Boyer, R. , Stenroos, E.S. , Chandrasekharappa, S. , 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Shi, Y.E. , Chen, Y. , Dackour, R. , Potters, L. , Wang, S. , Ding, Q. , Wang, Z. , Liu, Y.E. , 2010. Synuclein gamma stimulates membrane-initiated estrogen signaling by chaperoning estrogen receptor (ER)-alpha36, a variant of ER-alpha. Am. J. Pathol. 177, 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidera, K. , Gaitanou, M. , Stellas, D. , Matsas, R. , Patsavoudi, E. , 2008. A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J. Biol. Chem. 283, 2031–2041. [DOI] [PubMed] [Google Scholar]
- Siegel, P.M. , Ryan, E.D. , Cardiff, R.D. , Muller, W.J. , 1999. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 18, 2149–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza, J.M. , Giasson, B.I. , Lee, V.M. , Ischiropoulos, H. , 2000. Chaperone-like activity of synucleins. FEBS Lett. 474, 116–119. [DOI] [PubMed] [Google Scholar]
- Spillantini, M.G. , Schmidt, M.L. , Lee, V.M. , Trojanowski, J.Q. , Jakes, R. , Goedert, M. , 1997. Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Szegezdi, E. , Logue, S.E. , Gorman, A.M. , Samali, A. , 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandinger, S.K. , Richter, K. , Buchner, J. , 2008. The Hsp90 chaperone machinery. J. Biol. Chem. 283, 18473–18477. [DOI] [PubMed] [Google Scholar]
- Workman, P. , Burrows, F. , Neckers, L. , Rosen, N. , 2007. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 1113, 202–216. [DOI] [PubMed] [Google Scholar]
- Wu, K. , Weng, Z. , Tao, Q. , Lin, G. , Wu, X. , Qian, H. , Zhang, Y. , Ding, X. , Jiang, Y. , Shi, Y.E. , 2003. Stage-specific expression of breast cancer-specific gene gamma-synuclein. Cancer Epidemiol. Biomarkers Prev. 12, 920–925. [PubMed] [Google Scholar]
- Xu, W. , Mimnaugh, E. , Rosser, M.F. , Nicchitta, C. , Marcu, M. , Yarden, Y. , Neckers, L. , 2001. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J. Biol. Chem. 276, 3702–3708. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Gureasko, J. , Shen, K. , Cole, P.A. , Kuriyan, J. , 2006. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149. [DOI] [PubMed] [Google Scholar]
- Zhao, W. , Liu, H. , Liu, W. , Wu, Y. , Chen, W. , Jiang, B. , Zhou, Y. , Xue, R. , Luo, C. , Wang, L. , Jiang, J.D. , Liu, J. , 2006. Abnormal activation of the synuclein-gamma gene in hepatocellular carcinomas by epigenetic alteration. Int. J. Oncol. 28, 1081–1088. [PubMed] [Google Scholar]