

Abstract
A paradigm shift has occurred in the last decade from chemotherapy to targeted therapy for the management of many patients with advanced sarcoma. This work identifies a combination of targeted agents and doxorubicin that are effective against small cell sarcoma cell lines.
Three small cell sarcoma cell lines were studied: RD18 (rhabdomyosarcoma), A204 (undifferentiated sarcoma) and TC 71 (Ewing's sarcoma). Each cell line was exposed to increasing concentrations of vorinostat (HDAC inhibitor), 17‐DMAG (HSP90 inhibitor), abacavir (anti‐telomerase) or sorafenib (tyrosine kinase inhibitor) alone, combined with one another, or combined with doxorubicin. Cell viability, cell cycle analysis and apoptosis were assessed by MTS assay, propidium iodide‐Annexin V staining, and caspase 3/7 activity, respectively. The Chou and Talalay combination index (CI) was used to determine whether the effects were additive (CI = 1), synergistic (CI < 1) or antagonistic (CI > 1).
In monotherapy, targeted agents achieved 30–90% reductions in viability, with the exception of abacavir. Dual‐targeted combination therapies with vorinostat, sorafenib and 17‐DMAG demonstrated synergy. Abacavir was antagonistic with every other drug and was not further studied. Both vorinostat and 17‐DMAG synergized with doxorubicin, achieving 60% cell killing compared to 12% with doxorubicin alone. No synergy was observed for sorafenib with doxorubicin. The triple therapy vorinostat, 17‐DMAG and doxorubicin did not show synergy, but increased the subG1 population at 24H, from 30% to 70% compared to monotherapies with an increase in apoptosis.
This work provides evidence of synergy of combinations of vorinostat, 17‐DMAG and sorafenib in small cell sarcoma. In addition to doxorubicin, these combinations enhance doxorubicin cytotoxicity at therapeutically relevant concentrations.
Keywords: Small cell sarcoma, Targeted therapy, Cell line, Combination, Doxorubicin
Highlights
The optimal combination of targeted agents with doxorubicin in small cell sarcoma was screened.
Either vorinostat or 17DMAG synergized with doxorubicin.
Dual combination therapies of vorinostat, sorafenib and 17‐DMAG showed strong synergism.
Vorinostat, 17DMAG and doxorubicin combination achieved the highest cytotoxicity.
Abacavir was found antagonistic with each drug.
1. Introduction
Soft tissue sarcomas (STS) are a rare and heterogeneous group of tumors with poor prognosis. (Gatta et al., 2011; Rosenberg et al., 2012) Small cell sarcomas, such as rhabdomyosarcoma (RMS) and Ewing's sarcoma are chemosensitive tumors, but the relapse rate is high and median survival is poor in the metastatic setting.(Morioka et al., 2001) Doxorubicin is the standard of care in most sarcomas, but this regimen confers less than 10% survival at 5 years in metastatic sarcoma. (Blay et al., 2003; Dumont et al., 2013; Edmonson et al., 1993; Mouridsen et al., 1987; Pinedo et al., 1984; Van Glabbeke et al., 1999) The development of targeted therapies has changed the landscape of sarcoma research, bringing hope to patients and clinicians to achieve better response rates and survival. (Riedel, 2011; Smith and Riedel, 2011) Yet, many clinical trials of targeted therapies as single agents in sarcoma have failed to demonstrate efficacy. (Hawkins et al., 2013) As a result, targeted agents have occupied only specific niches and have not yet supplanted cytotoxic chemotherapies as standard of care.
Combining chemotherapies is a strategy that has long been used to take advantage of agents with different mechanisms of action and non‐overlapping toxicity profiles. To combine targeted agents in dual‐, or in poly‐therapy, may also delay or overcome resistance, and prevent relapse of patients treated with standard therapy. Targeted agents may allow reduced doses of standard cytotoxic chemotherapies, sparing patients toxicity or prolonged use. This may be particularly advantageous in relation to doxorubicin cardiotoxicity. Histone deacetylase (HDAC) inhibitors represent a new class of anti‐cancer drugs currently in development. Histone post‐translational modifications regulate accessibility of the DNA wrapped around histones to the nuclear transcription factors. In normal cells, histone acetylation leads to a more open chromatin conformation that is more favorable for transcription. HDAC is an enzyme responsible for removing acetyl groups from histones, thereby inhibiting DNA transcription. HDAC modifications have been identified in many tumors. (Taby and Issa, 2010) HDAC inhibitors induce differentiation, cell cycle arrest, apoptosis, and inhibition of tumor cell growth in preclinical assays. (Cain et al., 2013; Ma et al., 2009) Among HDAC inhibitors in clinical investigation, vorinostat, also known as suberoylanilide hydroxamic acid (SAHA), exerts antitumor activity on malignant cells that express a multi‐drug‐resistant phenotype. A synergistic action has been shown with ionizing radiation and inhibitors of kinases. Vorinostat has demonstrated promising results with in vivo models of prostate cancer and hematological malignancies, and is currently approved for cutaneous T cell lymphoma. (Wagner et al., 2010) Recently, several lines of evidence have suggested activity of vorinostat on sarcoma cells. Most of the models tested were osteosarcoma and Ewing's sarcoma. (Cain et al., 2013; Sonnemann et al., 2007; Wachtel and Schafer, 2010) However, in Ptch mutant mice with RMS, the combination of 5‐aza‐2'deoxycytidine, a hypomethylating agent, and valproic acid, one of the first HDAC inhibitor discovered, had anti‐tumor activity. (Ecke et al., 2009) Several phase I trials have been performed with HDAC inhibitors, including in children with refractory solid tumors.(Witt et al., 2012) They have also been tested in combination with sorafenib in a phase I trial (Dasari et al., 2013) and temsirolimus in a separate phase I trial (Coulter et al., 2013).
Heat shock proteins are chaperones responsible for protein folding and regulation of cell signaling pathways in normal cells. HSP90 is a member of the heat shock protein family, whose expression is increased when cells are exposed to stress such as elevated temperature, starvation or stress that causes protein denaturation or interferes with proper folding. (Smith et al., 1998; Smith and Riedel, 2011) Intracellular heat shock proteins are highly expressed in tumors and are essential to their survivalin the face of stressors such as hypoxia and chemotherapy. In particular, HSP90 is known to play a critical role in survival and growth of several types of cancer. Inhibition of HSP90 in RMS cell lines caused arrest of proliferation and migration and induced apoptosis in vitro, findings which which were confirmed in vivo in mice. (Lesko et al., 2007) In Ewing's sarcoma cell lines and mouse models, HSP90 inhibitors also demonstrated activity. (Ambati et al., 2014) As such, HSP90 inhibitors may be promising anti‐sarcoma agents, and rationale exists to combine with other targeted agents. The HSP90 antagonist 17‐DMAG is currently in clinical trials (Lesko et al., 2007; Martins et al., 2008; Terry et al., 2005; Wachtel and Schafer, 2010).
Sorafenib is a multi‐kinase inhibitor targeting vascular endothelial growth factor (VEGF) receptors 1 and 2, Flt‐3, c‐kit, Raf‐1, platelet‐derived growth factor receptors (PDGFR) alpha and beta, and fibroblast growth factor (FGFR) receptor 1.(Karaman et al., 2008; Wilhelm et al., 2006) These receptors may control pathways relevant to the pathogenesis of RMS, as suggested by the association of PDGFR alpha and beta expression and inferior outcome in RMS. (Armistead et al., 2007; Blandford et al., 2006) Moreover, RAF is activated by RAS and activating mutations of RAS are present in 35–50% of embryonal RMS. (Martinelli et al., 2009; Paulson et al., 2011) Finally, FGFR1 expression has been demonstrated in RMS, associated with epigenetic modification. (Goldstein et al., 2007)
The inhibition of these kinases results in anti‐tumor activity through the inhibition of cancer cell proliferation, angiogenesis and increase of apoptosis. (Wilhelm et al., 2004) Importantly, Sorafenib has demonstrated activity against synovial sarcoma in vitro, (Peng et al., 2009) and has been tested in a phase II study as a single agent on metastatic or recurrent sarcomas. (Pacey et al., 2011) The activity of sorafenib was notable against angiosarcoma and was minimal against other sarcomas, warranting further evaluation in these and other sarcoma subtypes, in combination with cytotoxic or kinase‐specific agents. (Vincenzi et al., 2013) Pazopanib as exemplified the use of TKI as single agents in STS (Van der Graaf et al., 2012).
Upregulation of telomerase protects tumor cells from aging and death. (Burger, 1999; Deng and Chang, 2007) An antiretroviral nucleoside analogue called abacavir (Ziagen) has potent antiretroviral activity and telomerase inhibitory activity in various cellular systems and animal studies. In solid tumors, particularly prostate cancer cell lines, abacavir caused a reduction of proliferation and senescence. (Carlini et al., 2010) It has not been tested on sarcoma cell lines so far.
Whereas the aforementioned drugs, Vorinostat, 17‐DMAG, Sorafenib, and Abacavir, have seemingly disparate targets, there is evidence that inhibition of multiple targets simultaneously may induce anti‐tumor effects by complementary mechanisms. For instance, in gastrointestinal stromal tumors (GIST), it has been observed that HSP90 is a target for vorinostat, which acetylates HSP90, dysregulating KIT protein folding and inducing apoptosis. (Muhlenberg et al., 2009) Therefore, early phase clinical trials are underway to test targeted therapy combination such as sorafenib and vorinostat in patients with sarcomas (Dasari et al., 2013).
Evidence‐based medicine is currently the best way to ensure quality and good practice but it may not be the best creative environment for basic or translational research. Indeed, science asks Nature to answer our questions by yes or no and may prevent creativity, exploring new paths, new drugs or other options. (Mallet, 2007) As opposed to this vision, we chose an empirical approach to conduct this work rather than a screening of sarcoma active proven agents.
This work aims to study targeted therapy combinations as a preclinical background to potential clinical trials. We sought to identify active dual‐ and triple‐combination therapies of targeted agents and to assemble them together and with standard chemotherapy doxorubicin in small cell sarcoma cell lines. The underlying question was to determine whether or not we could completely remove standard chemotherapy from cell treatment and still obtain acceptable efficacy on our model.
2. Material and method
2.1. Cell culture and reagents
TC71 human Ewing's sarcoma cells were cultured in RPMI 1640 with 10% fetal bovine serum, 2 mmol/L l‐glutamine. A204 human undifferentiated RMS cell line was cultured in McCoy's Medium with 10% fetal bovine serum, 2 mmol/L l‐glutamine. RD18 human RMS cell line was cultured in modified Dulbecco's Modified Eagle's Medium (DMEM) 12, supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum. All cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA). They were negative for Mycoplasma, as determined by UT–MD Anderson CCSG Characterized Cell Line Core and were maintained at 37 °C in a humidified incubator, with 5% CO2. Cell lines are described in Table 1. Vorinostat was provided by Merck & Co. Inc. (Whitehouse Station, NJ, USA), doxorubicin HCl was obtained from Ben Venue lab (Benford, OH), and 17‐DMAG was purchased from Invivogen (San Diego, CA). Sorafenib and Abacavir were purchased from the University of Texas–MD Anderson Cancer Center Pharmacy. The drugs were dissolved in dimethyl sulfoxide (DMSO) (Fisher Bioreagents) at 10 mmol/l and filtered through 0.22 micron filters, and aliquots were stored in −20 °C, protected from light.
Table 1.
Cell line description and P53 status.
| Cell line | Type | p53 | Fusion | References |
|---|---|---|---|---|
| A204 | Undifferentiated RMS | Wilde type | None | Xu J et al., Cancer Res. 2010 Aug 15 |
| RD18 | ERMS | Wilde type | None | Masuelli L et al., Front Biosci. 2012 Jan 1 |
| TC71 | Ewing | Mutant (637 C > T) | EWS‐FLI1 | Herrero‐Martín D et al., Br J Cancer. 2009 Jul 7; http://www‐p53.iarc.fr |
2.2. Viability evaluation
Cells were cultured in 100 mm dishes (Corning Life Sciences, Corning, NY), then seeded in 96‐well plates at a concentration of 5 × 10 cells per well, and incubated for 24 h, 48 h, 72 h with increasing concentrations of vorinostat, 17‐DMAG, abacavir and sorafenib, initially as monotherapy, then in triplicate combinations. The half‐maximal inhibitory concentration (IC50) of each targeted therapy as a single agent in each cell line was determined after 48 h of incubation. Based on these results, each agent was combined with doxorubicin. Each experiment was performed at least three times in each cell line, at the three different time points of incubation. The combination data were consistent throughout the cell lines and the most representative results were selected for the figures at 48 h of incubation. In some cases, doses were lowered to avoid excessive cell killing, which might preclude analysis of synergy. At the end of incubation, the reduction in cell viability compared with untreated cells was determined by MTS assay through the CellTiter 96 AQueous Non‐Radioactive Cell Proliferation Assay (Promega Corporation, Madison, WI), according to the manufacturer's instructions. Absorbance at 490 nm, reflecting the number of living cells in culture was measured using KC Junior software and microplate reader (Bio‐Tek Instruments, Winooski, VT). Relative cell viability (%) was calculated as the mean absorbance of replicate treatment‐wells minus the mean absorbance of replicate background wells, divided by the mean absorbance of replicate DMSO‐treated wells minus the mean absorbance of replicate background wells, multiplied by 100. Direct cell counting was used to pass and split the cells with a constant concentration of 30,000 cells/ml in 10 cm dishes and was done by counting nuclei (5–25 μm) using an automated Vi‐Cell Analyzer (Beckman Coulter) as described previously or Mozi Z cell counter (Orflo).
2.3. Cell cycles, apoptosis and caspase 3/7 activity assay
TC71 cells were treated with 0.15 uM of vorinostat, 5 uM of 17‐DMAG and 0.125 uM of doxorubicin using concomitant administration. Cell cycle assay was performed by propidium iodide (PI) staining (Roche, Indianapois, IN, USA) to identify the sub‐G1 population and analyzed by a FACS Canto II flow cytometer and FACS Diva 6.1 software (BD Biosciences, San Jose, CA), as previously described. (Reynoso et al., 2011) To assess apoptosis by flow cytometry, cells were stained with alexa fluor 488 annexin V and PI (Invitrogen, Carlsbad, CA USA), whereas caspase 3 and 7 activities were measured using the Apo‐One homogeneous Capase‐3/7 kit (Promega Inc., Madison, WI, USA) according to manufacturer's instructions.
2.4. Synergy analysis
The combination index (CI) method of Chou and Talalay was used to determine whether drug combinations were synergistic, additive, or antagonistic. (Chou, 2008, 2010, 1981, 2005) Briefly, the combination index was calculated based on the fraction of the cells affected (Fa) per dose, as measured in cell viability and apoptosis assays; CIs were generated using CalcuSyn software (Biosoft, Cambridge, UK). The combinations were considered synergistic if the CI was less than 1 with a significant synergism for a CI less than 0.8, additive for a CI equal to 1, or antagonistic for a CI superior to 1.
3. Results
3.1. Dual targeted therapies with vorinostat, sorafenib and 17‐DMAG
We first determined the inhibitory concentrations of targeted therapies as single agents in each cell line. The IC50 of abacavir was very high and not reached in RD18 cells. We based our combination data on the IC25 of each drug. We estimated the CI using Chou and Talalay method as reported in Table 2. Vorinostat, sorafenib and 17‐DMAG had a synergistic activity with one another, which was consistent across the different cell lines except in RD18 for the 17‐DMAG plus vorinostat combination. Vorinostat plus 17‐DMAG achieved the most synergy. Abacavir was found antagonistic with each drug and was not tested further.
Table 2.
IC50 of monotherapy and combinations and their CI – additive (CI = 1), synergistic (CI < 1) or antagonistic effect (CI > 1).
| Monotherapy | RD 18 | A204 | TC71 | |||
|---|---|---|---|---|---|---|
| IC50 (uM) | ||||||
| Sorafenib | 5 | 5 | 5 | |||
| 17‐DMAG | 10 | 10 | 3 | |||
| Abacavir | Not reached | 750 | 375 | |||
| Vorinostat | 3 | 1 | 2 | |||
| Combination | IC50 (uM) | CI | IC50 (uM) | CI | IC50 (uM) | CI |
| Sorafenib | 2.5 | 0.8 | 2.5 | 0.7 | 2.5 | 0.5 |
| 17‐DMAG | 5 | 10 | 5 | |||
| Sorafenib | 2.5 | 1.1 | 5 | 1.4 | 2.5 | 1.1 |
| Abacavir | 375 | 188 | 188 | |||
| Sorafenib | 5 | 0.7 | 5 | 0.9 | 2.5 | 0.8 |
| Vorinostat | 1.5 | 3 | 3 | |||
| 17‐DMAG | 10 | 1.1 | 5 | 1.2 | 5 | 1 |
| Abacavir | 188 | 750 | 188 | |||
| 17‐DMAG | 1.5 | 3 | 1.5 | 0.7 | 1.5 | 0.6 |
| Vorinostat | 0.75 | 3 | 0.75 | |||
| Abacavir | 750 | 1.9 | 750 | 3.7 | 188 | 1.4 |
| Vorinostat | 3 | 3 | 3 | |||
3.2. Targeted triple therapy
Drugs that were synergistic 2 by 2 were combined together. Cells were exposed to vorinostat, 17‐DMAG and sorafenib at the doses of 0.25, 0.5 and 1 uM for homogeneity. Cell viability assays showed that the most growth reduction was achieved by the triple therapy (Figure 1A and B). However, the action of the 3 drugs together was either additive or antagonistic compared to targeted dual therapies in all 3 cell lines (mean of CIs = 1.2).
Figure 1.
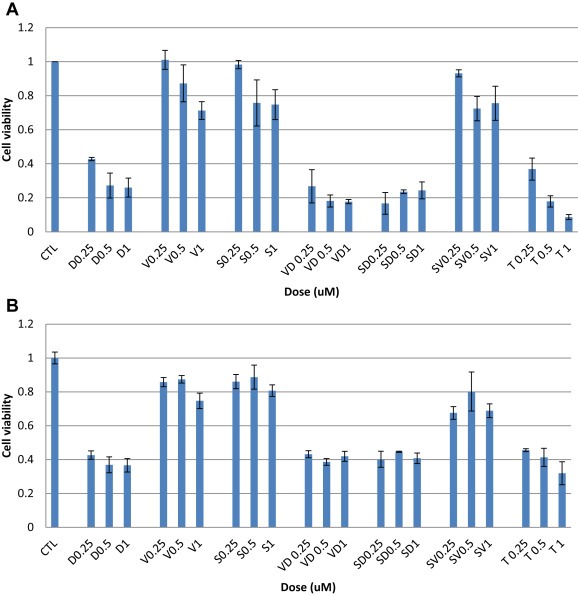
Cell viability of cell lines with triple therapy (A) TC71 and (B) RD18. S = sorafenib, D = 17‐DMAG, V = vorinostat, T = triple therapy.
3.3. Doxorubicin plus a targeted dual therapy
In order to identify an optimal combination of cytotoxic and targeted therapies, we first tested doxorubicin with each targeted agent: vorinostat, 17‐DMAG and sorafenib. (Figure 2). The combinations of doxorubicin and either 17‐DMAG or vorinostat achieved synergistic reductions of cell viability (Figure 2A–C; CI: 0.7–0.8), whereas almost no synergy was observed with doxorubicin and sorafenib. We further tested combination therapies consisting of doxorubicin with vorinostat plus 17‐DMAG (Figure 3A) or sorafenib plus 17‐DMAG (Figure 3B). A modest reduction in cell viability was observed with the combination of doxorubicin plus vorinostat or 17‐DMAG, while no additional effect was observed with doxorubicin and the dual‐targeted therapy of vorinostat plus 17‐DMAG. Similarly, no additional benefit was observed for the combination of doxorubicin, sorafenib, and 17‐DMAG (Figure 3A and B). Both CIs were above 1 for these triple combinations, suggesting an additive effect.
Figure 2.
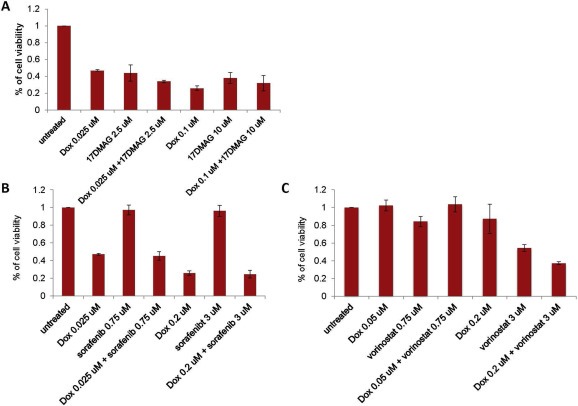
Cell viability of doxorubicin plus either (A) 17‐DMAG (RD18), (B) sorafenib (RD18) or (C) vorinostat (TC71).
Figure 3.
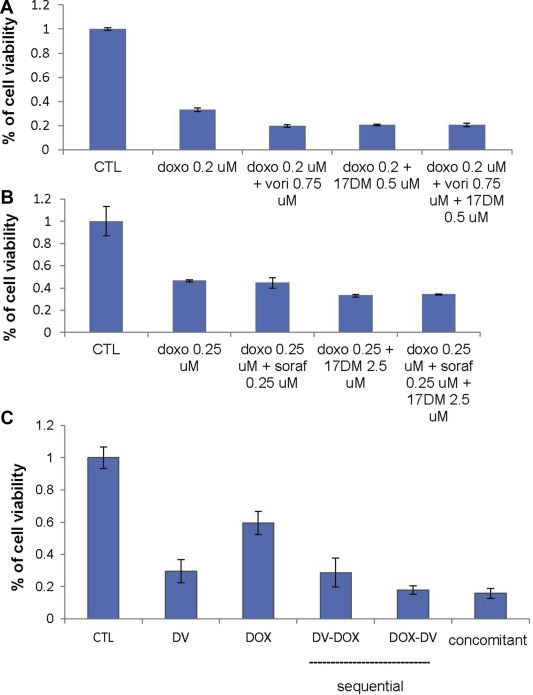
Cell viability of doxorubicin plus 17‐DMAG and (A) vorinostat (TC71) or (B) sorafenib (RD18) and (C) drug sequence effect viability through sequential or concomitant administration of doxorubicin plus vorinostat‐17‐DMAG (RD18).
3.4. Drug sequence effect on viability
To understand the mechanism of action and the interactions between targeted therapies and standard cytotoxic chemotherapy agents like doxorubicin, we studied further the triple combination consisting of doxorubicin, vorinostat and 17‐DMAG using different treatment sequences. We tested cell viability to assess the optimal timing of each therapeutic class. Our results show that initial treatment with dual targeted therapy is more active than initial chemotherapy. Furthermore, serial treatment with multiple agents was not superior to concomitant treatment (Figure 3C).
3.5. Apoptosis induction by doxorubicin plus vorinostat‐17‐DMAG therapy
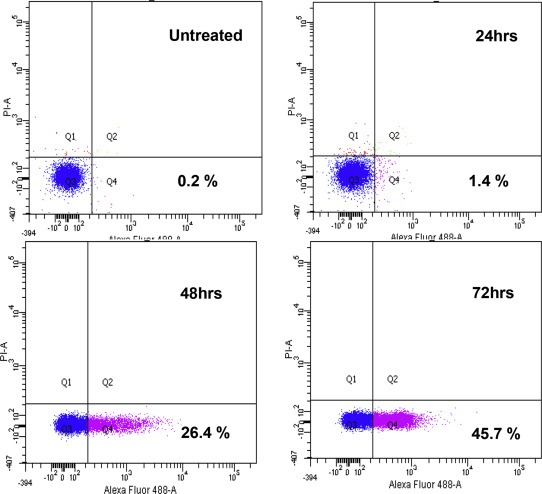
The triple therapy of vorinostat, 17‐DMAG and doxorubicin did not demonstrate synergism (CI:1.2) with regard to reductions in cell viability, but cell cycle analysis showed that this triple therapy transiently increased the subG1 population at 24H, from 30% in monotherapy to 70% (Figure 4A and B). The mechanism of action was further investigated and showed an increase in caspase 3 dependent apoptosis with the triple combination (Fig. 5) and early caspase‐independent apoptosis (11–36%) (Figure 6).
Figure 4.
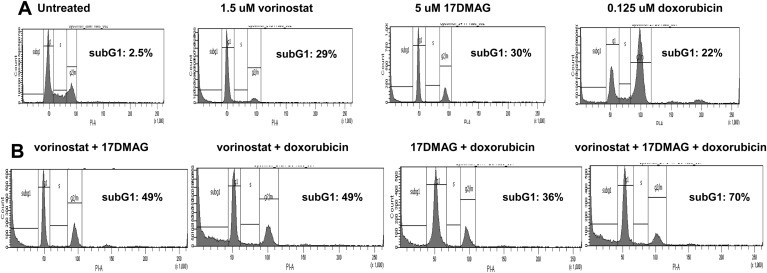
Cell cycle distribution of TC71 cells treated with vorinostat, doxorubicin and 17‐DMAG as (A) monotherapy and (B) in combination.
Figure 5.
Measure of apoptosis by Caspase 3/7 activity of doxorubicin, vorinostat and 17‐DMAG combination in TC71 cell line.
Figure 6.
Apoptosis measured with Alexa Fluor 488 Annexin V and PI staining of doxorubicin, vorinostat and 17‐DMAG combination in TC71 cell line.
4. Discussion
While standard treatment of small cell sarcomas still involves cytotoxic chemotherapy agents, targeted therapies represent a new paradigm to enrich the therapeutic palette of this disease. This work provides preclinical data on synergism of dual targeted therapy combinations, including vorinostat, 17‐DMAG and sorafenib, in small cell sarcoma cell lines. Targeted triple therapy not including doxorubicin does not achieve synergism, but causes a modest reduction in cell viability. In combination with doxorubicin, dual‐targeted therapy combination enhances the cytotoxicity of doxorubicin at therapeutically‐relevant concentrations. The selected combination of doxorubicin plus the dual‐targeted therapies vorinostat and 17‐DMAG achieved synergistic induction of caspase‐mediated and caspase‐independent apoptosis. According to our data, the most effective combinations included the HDAC inhibitor vorinostat, suggesting that the molecular mechanism underlying the synergy observed in our study involves acetylation of key proteins, such as histones, housekeeping proteins, p53 or proteins implicated in the downstream signaling pathways of the other targeted agents. Our results do not support the use of targeted‐only polytherapy, as the triple combination of targeted agents did not achieve synergy.
In the era of personalized medicine, targeted therapies may represent a more precise and relevant approach. However, a better understanding of the biology tumors, particularly sarcomas, is needed. Many sarcomas have specific chromosomal translocations such as PAX3 or PAX7‐FOXO1 in alveolar RMS (Sorensen et al., 2002), loss of heterozygosity in chromosome 11 (Scrable et al., 1989), or FLT1‐EWS1 in Ewing's sarcoma. (Bonin et al., 1993; Davis et al., 1995; Scrable et al., 1989; Turc‐Carel et al., 1988) These translocations might sensitize cells to DNA disrupting agents such as trabectedin. (Le Cesne et al., 2012) The key tumor suppressor gene p53 might also be important in tumor response to targeted agents. The majority of Ewing's sarcomas are wild type for p53 (Hamelin et al., 1994; Komuro et al., 1993), and studies have demonstrated that the chimeric protein EWS‐FLI1 silences p53 activity (Ban et al., 2008; Li et al., 2010). Recent data suggest that the molecular mechanism of p53 abrogation by EWS‐FLI1 involves a decrease of acetylation of p53 and increase of mdm2‐mediated p53 degradation (Li et al., 2012). In view of this, it is possible that use of HDAC inhibitors may re‐acetylate p53 and results in an increase of p53 stability and activity. In our study, the TC71 cell line, which achieved the greatest viability reductions in combination therapies, is mutated for P53 but was sensitive to vorinostat. In contrast to Ewing's sarcoma, P53 is mutated in the majority of RMS (Felix et al., 1992). In vitro, the wild‐type P53 A204 cells were sensitive to different agents in monotherapy, but the combinations failed to achieve synergy compared to other cell lines. Finally, RD18 was mutated for P53 and was sensitive to monotherapies, as well as double‐ and triple‐combinations.
In vitro studies such as ours present inherent limitations. First, the cell lines may not represent the real spectrum of small cell sarcoma, here exemplified by p53 status. However, cell lines offer the possibility to explore agents and combinations that would not have been possible using a mouse model. It allowed us to explore even inactive combinations or drugs in monotherapy, following an empirical approach. In fact, an agent such as abacavir, whose mechanism of action (anti‐telomerase) would theoretically have made a good candidate for early phase clinical trial, was shown to have no effect in monotherapy. However, an activity might have been seen in combination with other agents. (Elmore and Holt, 2007) Secondly, the doses used for the combinations had to be determined through a step by step process to avoid over cell killing. This explains the fact that the doses for dual and triple therapies were very low with none activity or higher viability artifacts observed in the monotherapies shown as controls for comparison purpose.
Finally, scientific progress throughout history has often been due to revolutions, random findings that complete changes of paradigm rather than a step‐to‐step progression of knowledge. (Kuhn, 1972) The analogy of trench warfare versus a significant battle has also been evoked in oncology clinical research. (Stewart and Kurzrock, 2009) The empirical approach presents the advantage of considering other pathways and therapeutic classes and is open to creativity, rather than expecting an answer to what is thought to be the next step.
Many challenges remain with regards to clinical trial design for two, three or more targeted agents alone or combined with standard chemotherapy. Preclinical studies such as ours offer some background to test targeted multiple therapies together with doxorubicin in small cell sarcoma.
Acknowledgments
This work was supported by the Amschwand Sarcoma Cancer Foundation and the ARC foundation for Cancer Research.
Dumont S.N., Yang D., Dumont A.G., Reynoso D., Blay J-Y., Trent J.C., (2014), Targeted polytherapy in small cell sarcoma and its association with doxorubicin, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.05.016.
References
- Ambati, S.R. , Lopes, E.C. , Kosugi, K. , Mony, U. , Zehir, A. , Shah, S.K. , Taldone, T. , Moreira, A.L. , Meyers, P.A. , Chiosis, G. , Moore, M.A. , 2014. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 8, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armistead, P.M. , Salganick, J. , Roh, J.S. , Steinert, D.M. , Patel, S. , Munsell, M. , El-Naggar, A.K. , Benjamin, R.S. , Zhang, W. , Trent, J.C. , 2007. Expression of receptor tyrosine kinases and apoptotic molecules in rhabdomyosarcoma: correlation with overall survival in 105 patients. Cancer. 110, 2293–2303. [DOI] [PubMed] [Google Scholar]
- Ban, J. , Bennani-Baiti, I.M. , Kauer, M. , Schaefer, K.L. , Poremba, C. , Jug, G. , Schwentner, R. , Smrzka, O. , Muehlbacher, K. , Aryee, D.N. , Kovar, H. , 2008. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing's sarcoma. Cancer Res. 68, 7100–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandford, M.C. , Barr, F.G. , Lynch, J.C. , Randall, R.L. , Qualman, S.J. , Keller, C. , 2006. Rhabdomyosarcomas utilize developmental, myogenic growth factors for disease advantage: a report from the children's oncology group. Pediatr. Blood Cancer. 46, 329–338. [DOI] [PubMed] [Google Scholar]
- Blay, J.Y. , Van Glabbeke, M. , Verweij, J. , Van Oosterom, A.T. , Le Cesne, A. , Oosterhuis, J.W. , Judson, I. , Nielsen, O.S. , 2003. Advanced soft-tissue sarcoma: a disease that is potentially curable for a subset of patients treated with chemotherapy. Eur. J. Cancer. 39, 64–69. [DOI] [PubMed] [Google Scholar]
- Bonin, G. , Scamps, C. , Turc-Carel, C. , Lipinski, M. , 1993. Chimeric EWS-FLI1 transcript in a Ewing cell line with a complex t(11;22;14) translocation. Cancer Res. 53, 3655–3657. [PubMed] [Google Scholar]
- Burger, A.M. , 1999. Telomerase in cancer diagnosis and therapy: a clinical perspective. BioDrugs Clinical Immunother. Biopharm. Gene Ther. 12, 413–422. [DOI] [PubMed] [Google Scholar]
- Cain, J.E. , McCaw, A. , Jayasekara, W.S. , Rossello, F.J. , Marini, K.D. , Irving, A.T. , Kansara, M. , Thomas, D.M. , Ashley, D.M. , Watkins, D.N. , 2013. Sustained low-dose treatment with the histone deacetylase inhibitor LBH589 induces terminal differentiation of osteosarcoma cells. Sarcoma. 2013, 608964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlini, F. , Ridolfi, B. , Molinari, A. , Parisi, C. , Bozzuto, G. , Toccacieli, L. , Formisano, G. , De Orsi, D. , Paradisi, S. , Grober, O.M. , Ravo, M. , Weisz, A. , Arcieri, R. , Vella, S. , Gaudi, S. , 2010. The reverse transcription inhibitor abacavir shows anticancer activity in prostate cancer cell lines. PLoS One. 5, e14221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.C. , 2008. Preclinical versus clinical drug combination studies. Leuk. Lymp. 49, 2059–2080. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , Talalay, P. , 1981. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur. J. Biochem. FEBS. 115, 207–216. [DOI] [PubMed] [Google Scholar]
- Coulter, D.W. , Walko, C. , Patel, J. , Moats-Staats, B.M. , McFadden, A. , Smith, S.V. , Khan, W.A. , Bridges, A.S. , Deal, A.M. , Oesterheld, J. , Davis, I.J. , Blatt, J. , 2013. Valproic acid reduces the tolerability of temsirolimus in children and adolescents with solid tumors. Antican. Drugs. 24, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari, A. , Gore, L. , Messersmith, W.A. , Diab, S. , Jimeno, A. , Weekes, C.D. , Lewis, K.D. , Drabkin, H.A. , Flaig, T.W. , Camidge, D.R. , 2013. A phase I study of sorafenib and vorinostat in patients with advanced solid tumors with expanded cohorts in renal cell carcinoma and non-small cell lung cancer. Invest New Drugs. 31, 115–125. [DOI] [PubMed] [Google Scholar]
- Davis, R.J. , Bennicelli, J.L. , Macina, R.A. , Nycum, L.M. , Biegel, J.A. , Barr, F.G. , 1995. Structural characterization of the FKHR gene and its rearrangement in alveolar rhabdomyosarcoma. Hum. Mol. Genetics. 4, 2355–2362. [DOI] [PubMed] [Google Scholar]
- Deng, Y. , Chang, S. , 2007. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab Invest. 87, 1071–1076. [DOI] [PubMed] [Google Scholar]
- Dumont, S.N. , Araujo, D.M. , Munsell, M.F. , Salganick, J.A. , Dumont, A.G. , Raymond, K.A. , Linassier, C. , Patel, S. , Benjamin, R.S. , Trent, J.C. , 2013. Management and outcome of 239 adolescent and adult rhabdomyosarcoma patients. Cancer med. 2, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecke, I. , Petry, F. , Rosenberger, A. , Tauber, S. , Monkemeyer, S. , Hess, I. , Dullin, C. , Kimmina, S. , Pirngruber, J. , Johnsen, S.A. , Uhmann, A. , Nitzki, F. , Wojnowski, L. , Schulz-Schaeffer, W. , Witt, O. , Hahn, H. , 2009. Antitumor effects of a combined 5-aza-2'deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res. 69, 887–895. [DOI] [PubMed] [Google Scholar]
- Edmonson, J.H. , Ryan, L.M. , Blum, R.H. , Brooks, J.S. , Shiraki, M. , Frytak, S. , Parkinson, D.R. , 1993. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J. Clin. Oncol. 11, 1269–1275. [DOI] [PubMed] [Google Scholar]
- Elmore, L.W. , Holt, S.E. , 2007. Telomerase inhibition as an adjuvant anticancer therapy: it is more than just a waiting game. Exp. Opinion Therap. Targets. 11, 427–430. [DOI] [PubMed] [Google Scholar]
- Felix, C.A. , Kappel, C.C. , Mitsudomi, T. , Nau, M.M. , Tsokos, M. , Crouch, G.D. , Nisen, P.D. , Winick, N.J. , Helman, L.J. , 1992. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res. 52, 2243–2247. [PubMed] [Google Scholar]
- Gatta, G. , Van der Zwan, J.M. , Casali, P.G. , Siesling, S. , Dei Tos, A.P. , Kunkler, I. , Otter, R. , Licitra, L. , Mallone, S. , Tavilla, A. , Trama, A. , Capocaccia, R. , 2011. Rare cancers are not so rare: the rare cancer burden in Europe. Eur. J. Cancer Oxf. Engl. 1990). 47, 2493–2511. [DOI] [PubMed] [Google Scholar]
- Goldstein, M. , Meller, I. , Orr-Urtreger, A. , 2007. FGFR1 over-expression in primary rhabdomyosarcoma tumors is associated with hypomethylation of a 5' CpG island and abnormal expression of the AKT1, NOG, and BMP4 genes. Genes Chrom. Cancer. 46, 1028–1038. [DOI] [PubMed] [Google Scholar]
- Hamelin, R. , Zucman, J. , Melot, T. , Delattre, O. , Thomas, G. , 1994. p53 mutations in human tumors with chimeric EWS/FLI-1 genes. Int. J. Cancer J. Int. du Cancer. 57, 336–340. [DOI] [PubMed] [Google Scholar]
- Hawkins, D.S. , Spunt, S.L. , Skapek, S.X. , 2013. Children's Oncology Group's 2013 blueprint for research: soft tissue sarcomas. Pediatr. Blood Cancer. 60, 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman, M.W. , Herrgard, S. , Treiber, D.K. , Gallant, P. , Atteridge, C.E. , Campbell, B.T. , Chan, K.W. , Ciceri, P. , Davis, M.I. , Edeen, P.T. , Faraoni, R. , Floyd, M. , Hunt, J.P. , Lockhart, D.J. , Milanov, Z.V. , Morrison, M.J. , Pallares, G. , Patel, H.K. , Pritchard, S. , Wodicka, L.M. , Zarrinkar, P.P. , 2008. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Komuro, H. , Hayashi, Y. , Kawamura, M. , Hayashi, K. , Kaneko, Y. , Kamoshita, S. , Hanada, R. , Yamamoto, K. , Hongo, T. , Yamada, M. , 1993. Mutations of the p53 gene are involved in Ewing's sarcomas but not in neuroblastomas. Cancer Res. 53, 5284–5288. [PubMed] [Google Scholar]
- Kuhn, T.S. , 1972. La structure des révolutions scientifiques trad. [de la 2e édition américaine]. Flammarion, Paris [Google Scholar]
- Le Cesne, A. , Cresta, S. , Maki, R.G. , Blay, J.Y. , Verweij, J. , Poveda, A. , Casali, P.G. , Balana, C. , Schoffski, P. , Grosso, F. , Lardelli, P. , Nieto, A. , Alfaro, V. , Demetri, G.D. , 2012. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur. J. Cancer. 48, 3036–3044. [DOI] [PubMed] [Google Scholar]
- Lesko, E. , Gozdzik, J. , Kijowski, J. , Jenner, B. , Wiecha, O. , Majka, M. , 2007. HSP90 antagonist, geldanamycin, inhibits proliferation, induces apoptosis and blocks migration of rhabdomyosarcoma cells in vitro and seeding into bone marrow in vivo. Antican. Drugs. 18, 1173–1181. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Li, X. , Fan, G. , Fukushi, J. , Matsumoto, Y. , Iwamoto, Y. , Zhu, Y. , 2012. Impairment of p53 acetylation by EWS-Fli1 chimeric protein in Ewing family tumors. Cancer Lett. 320, 14–22. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Tanaka, K. , Fan, X. , Nakatani, F. , Li, X. , Nakamura, T. , Takasaki, M. , Yamamoto, S. , Iwamoto, Y. , 2010. Inhibition of the transcriptional function of p53 by EWS-Fli1 chimeric protein in Ewing Family Tumors. Cancer Lett. 294, 57–65. [DOI] [PubMed] [Google Scholar]
- Ma, X. , Ezzeldin, H.H. , Diasio, R.B. , 2009. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 69, 1911–1934. [DOI] [PubMed] [Google Scholar]
- Mallet, D. , 2007. La médecine entre science et Existence Collection Espace Éthique Vuilbert; Paris: 246 [Google Scholar]
- Martinelli, S. , McDowell, H.P. , Vigne, S.D. , Kokai, G. , Uccini, S. , Tartaglia, M. , Dominici, C. , 2009. RAS signaling dysregulation in human embryonal Rhabdomyosarcoma. Genes Chrom. Cancer. 48, 975–982. [DOI] [PubMed] [Google Scholar]
- Martins, A.S. , Ordonez, J.L. , Garcia-Sanchez, A. , Herrero, D. , Sevillano, V. , Osuna, D. , Mackintosh, C. , Caballero, G. , Otero, A.P. , Poremba, C. , Madoz-Gurpide, J. , de Alava, E. , 2008. A pivotal role for heat shock protein 90 in Ewing sarcoma resistance to anti-insulin-like growth factor 1 receptor treatment: in vitro and in vivo study. Cancer Res. 68, 6260–6270. [DOI] [PubMed] [Google Scholar]
- Morioka, H. , Yabe, H. , Morii, T. , Yamada, R. , Kato, S. , Yuasa, S. , Yano, T. , 2001. In vitro chemosensitivity of human soft tissue sarcoma. Anticancer Res. 21, 4147–4151. [PubMed] [Google Scholar]
- Mouridsen, H.T. , Bastholt, L. , Somers, R. , Santoro, A. , Bramwell, V. , Mulder, J.H. , Van Oosterom, A.T. , Buesa, J. , Pinedo, H.M. , Thomas, D. , 1987. Adriamycin versus epirubicin in advanced soft tissue sarcomas. A randomized phase II/phase III study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer Clin. Oncol. 23, 1477–1483. [DOI] [PubMed] [Google Scholar]
- Muhlenberg, T. , Zhang, Y. , Wagner, A.J. , Grabellus, F. , Bradner, J. , Taeger, G. , Lang, H. , Taguchi, T. , Schuler, M. , Fletcher, J.A. , Bauer, S. , 2009. Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res. 69, 6941–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey, S. , Ratain, M.J. , Flaherty, K.T. , Kaye, S.B. , Cupit, L. , Rowinsky, E.K. , Xia, C. , O'Dwyer, P.J. , Judson, I.R. , 2011. Efficacy and safety of sorafenib in a subset of patients with advanced soft tissue sarcoma from a Phase II randomized discontinuation trial. Invest. New Drugs. 29, 481–488. [DOI] [PubMed] [Google Scholar]
- Paulson, V. , Chandler, G. , Rakheja, D. , Galindo, R.L. , Wilson, K. , Amatruda, J.F. , Cameron, S. , 2011. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromos. Cancer. 50, 397–408. [DOI] [PubMed] [Google Scholar]
- Peng, C.L. , Guo, W. , Ji, T. , Ren, T. , Yang, Y. , Li, D.S. , Qu, H.Y. , Li, X. , Tang, S. , Yan, T.Q. , Tang, X.D. , 2009. Sorafenib induces growth inhibition and apoptosis in human synovial sarcoma cells via inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol. Ther. 8, [DOI] [PubMed] [Google Scholar]
- Pinedo, H.M. , Bramwell, V.H. , Mouridsen, H.T. , Somers, R. , Vendrik, C.P. , Santoro, A. , Buesa, J. , Wagener, T. , Van Oosterom, A.T. , Van Unnik, J.A. , 1984. Cyvadic in advanced soft tissue sarcoma: a randomized study comparing two schedules. A study of the EORTC Soft Tissue and Bone Sarcoma Group. Cancer. 53, 1825–1832. [DOI] [PubMed] [Google Scholar]
- Reynolds, C.P. , Maurer, B.J. , 2005. Evaluating response to antineoplastic drug combinations in tissue culture models. Methods Mol. Med. 110, 173–183. [DOI] [PubMed] [Google Scholar]
- Reynoso, D. , Nolden, L.K. , Yang, D. , Dumont, S.N. , Conley, A.P. , Dumont, A.G. , Zhou, K. , Duensing, A. , Trent, J.C. , 2011. Synergistic induction of apoptosis by the Bcl-2 inhibitor ABT-737 and imatinib mesylate in gastrointestinal stromal tumor cells. Mol. Oncol. 5, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel, R.F. , 2011. Targeted agents for sarcoma: is individualized therapy possible in such a diverse tumor type?. Semin. Oncol. 38, (Suppl 3) S30–S42. [DOI] [PubMed] [Google Scholar]
- Rosenberg, A.R. , Skapek, S.X. , Hawkins, D.S. , 2012. The inconvenience of convenience cohorts: rhabdomyosarcoma and the PAX-FOXO1 biomarker. Cancer Epidemiol. Biomarkers Prev.: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 21, 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrable, H. , Witte, D. , Shimada, H. , Seemayer, T. , Sheng, W.W. , Soukup, S. , Koufos, A. , Houghton, P. , Lampkin, B. , Cavenee, W. , 1989. Molecular differential pathology of rhabdomyosarcoma. Genes Chrom. Cancer. 1, 23–35. [DOI] [PubMed] [Google Scholar]
- Smith, D.F. , Whitesell, L. , Katsanis, E. , 1998. Molecular chaperones: biology and prospects for pharmacological intervention. Pharmacol. Rev. 50, 493–514. [PubMed] [Google Scholar]
- Smith, J.L. , Riedel, R.F. , 2011. Emerging therapeutic targets for soft tissue sarcoma. Curr. Oncol. Rep. 13, 350–358. [DOI] [PubMed] [Google Scholar]
- Sonnemann, J. , Dreyer, L. , Hartwig, M. , Palani, C.D. , Hong le, T.T. , Klier, U. , Broker, B. , Volker, U. , Beck, J.F. , 2007. Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing's sarcoma cells. J. Cancer Res. Clin. Oncol. 133, 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, P.H. , Lynch, J.C. , Qualman, S.J. , Tirabosco, R. , Lim, J.F. , Maurer, H.M. , Bridge, J.A. , Crist, W.M. , Triche, T.J. , Barr, F.G. , 2002. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J. Clin. Oncol. 20, 2672–2679. [DOI] [PubMed] [Google Scholar]
- Stewart, D.J. , Kurzrock, R. , 2009. Cancer: the road to Amiens. J. Clin. Oncol. 27, 328–333. [DOI] [PubMed] [Google Scholar]
- Taby, R. , Issa, J.P. , 2010. Cancer epigenetics. CA Cancer J. Clin. 60, 376–392. [DOI] [PubMed] [Google Scholar]
- Terry, J. , Lubieniecka, J.M. , Kwan, W. , Liu, S. , Nielsen, T.O. , 2005. Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin prevents synovial sarcoma proliferation via apoptosis in in vitro models. Clin. Cancer Res. 11, 5631–5638. [DOI] [PubMed] [Google Scholar]
- Turc-Carel, C. , Aurias, A. , Mugneret, F. , Lizard, S. , Sidaner, I. , Volk, C. , Thiery, J.P. , Olschwang, S. , Philip, I. , Berger, M.P. , 1988. Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22) (q24;q12). Cancer Genet. Cytogen. 32, 229–238. [DOI] [PubMed] [Google Scholar]
- Van der Graaf, W.T. , Blay, J.Y. , Chawla, S.P. , Kim, D.W. , Bui-Nguyen, B. , Casali, P.G. , Schoffski, P. , Aglietta, M. , Staddon, A.P. , Beppu, Y. , Le Cesne, A. , Gelderblom, H. , Judson, I.R. , Araki, N. , Ouali, M. , Marreaud, S. , Hodge, R. , Dewji, M.R. , Coens, C. , Demetri, G.D. , Fletcher, C.D. , Dei Tos, A.P. , Hohenberger, P. , 2012. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 379, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Van Glabbeke, M. , Van Oosterom, A.T. , Oosterhuis, J.W. , Mouridsen, H. , Crowther, D. , Somers, R. , Verweij, J. , Santoro, A. , Buesa, J. , Tursz, T. , 1999. Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens–a European Organization for research and treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J. Clin. Oncol. 17, 150–157. [DOI] [PubMed] [Google Scholar]
- Vincenzi, B. , Silletta, M. , Schiavon, G. , Frezza, A.M. , Del Vescovo, R. , Zobel, B.B. , Santini, D. , Dei Tos, A.P. , Tonini, G. , 2013. Sorafenib and dacarbazine in soft tissue sarcoma: a single institution experience. Expert Opin. Invest. Drugs. 22, 1–7. [DOI] [PubMed] [Google Scholar]
- Wachtel, M. , Schafer, B.W. , 2010. Targets for cancer therapy in childhood sarcomas. Cancer Treat. Rev. 36, 318–327. [DOI] [PubMed] [Google Scholar]
- Wagner, J.M. , Hackanson, B. , Lubbert, M. , Jung, M. , 2010. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenetics. 1, 117–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm, S. , Carter, C. , Lynch, M. , Lowinger, T. , Dumas, J. , Smith, R.A. , Schwartz, B. , Simantov, R. , Kelley, S. , 2006. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835–844. [DOI] [PubMed] [Google Scholar]
- Wilhelm, S.M. , Carter, C. , Tang, L. , Wilkie, D. , McNabola, A. , Rong, H. , Chen, C. , Zhang, X. , Vincent, P. , McHugh, M. , Cao, Y. , Shujath, J. , Gawlak, S. , Eveleigh, D. , Rowley, B. , Liu, L. , Adnane, L. , Lynch, M. , Auclair, D. , Taylor, I. , Gedrich, R. , Voznesensky, A. , Riedl, B. , Post, L.E. , Bollag, G. , Trail, P.A. , 2004. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109. [DOI] [PubMed] [Google Scholar]
- Witt, O. , Milde, T. , Deubzer, H.E. , Oehme, I. , Witt, R. , Kulozik, A. , Eisenmenger, A. , Abel, U. , Karapanagiotou-Schenkel, I. , 2012. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klin Padiatr. 224, 398–403. [DOI] [PubMed] [Google Scholar]