

Abstract
Background
Given our preclinical data showing synergy between dovitinib and paclitaxel in preclinical models we conducted this phase I trial aiming to define the recommended phase II‐dose (RP2D) on the basis of toxicity and pharmacodynamic criteria while searching for genetic variants that could sensitize patients to the regimen under study.
Patients and methods
A 3+3 escalation schedule was adopted. Seriated FGF23 and dovitinib and paclitaxel pharmacokinetic profiles were determined along a single‐agent dovitinib “priming‐phase” followed by a dovitinib + paclitaxel combination phase. RECIST 1.1 criteria and NCI CTCAE V.4.0 were used. In fresh pre‐treatment tumor biopsy samples, FGFR1, 2 and 3 amplifications were revealed by FISH probes; 32 missense variants were genotyped in tumors and peripheral blood mononuclear cells with Taqman genotyping assays (FGFR1‐3 and RET). Constructs encoding for wild‐type and variant genes associated with clinical benefit were transfected into HEK‐293 cells for preclinical experiments checking constitutive activation and dovitinib sensitivity of the variants.
Results
twelve patients were recruited in three dose‐levels. At level 1B (200 mg dovitinib 5‐days‐on/2‐days‐off plus 60 mg/m 2‐week of paclitaxel) more than 50% FGF23 upregulation was observed and no dose‐limiting‐toxicities (DLTs) occurred. The most frequent toxicities were asthenia, neutropenia, nausea/vomiting and transaminitis. Two patients with progressive disease prior to trial inclusion achieved prolonged disease stabilization. Both had the germline variant G2071A in the RET gene, which led to constitutive activation of the protein product and Y‐905 phosphorylation, both in transfectants and in patients with the alteration. This variant was sensitive to dovitinib; in addition both patients experienced progression upon medication withdrawal.
Conclusions
Level 1B was the RP2D as it provided adequate pharmacodynamic exposure to dovitinib. The G2071A germline variant act as a genetic modifier that renders different tumors sensitive to dovitinib.
Keywords: Phase I, Dovitinib, Antiangiogenic, Clinical trial, RET genetic variant, Sensitizing genetic variant
Highlights
We conducted a phase‐I trial of the combination of dovitinib plus paclitaxel.
The dose escalation was guided by pharmacodynamic and toxicity criteria.
We tested amplifications and mutations at the genes encoding for dovitinib targets.
Only one variant, RET‐G2071A, was found in two patients.
G2071A encoded a constitutively active, dovitinib‐sensitive RET, that rendered the patients sensitive.
1. Introduction
Multiple small‐molecule antiangiogenics with multi‐kinase inhibitor activities have undergone clinical development. Novel agents of this class seem to be at least as effective as sunitinib or sorafenib, but show a better tolerability profile (Motzer et al., 2013, 2014, 2011). Some of these novel agents inhibit oncogenic‐addiction driving kinases on top of pro‐angiogenic kinases. Dovitinib is one of these agents, with activity against VEGFR1‐3, PDGFRA/B, FGFR1‐3, KIT, RET and FTL3 at <50 nM (Andre et al., 2013; Angevin et al., 2013; Lee et al., 2005). Dovitinib is administered with a schedule of 5‐days‐on/2‐days‐off at a dose of 500 mg daily. Combining an agent of this type with chemotherapy is appealing because in monotherapy small‐molecule antiangiogenics are active against renal or liver cancer, although they show little or no activity against the most frequent epithelial malignancies. We demonstrated synergistic effects of combining chemotherapy plus dovitinib in preclinical models of pancreatic cancer (Hernandez‐Agudo et al., 2013). In several pilot experiments with patient‐derived xenograft models we observed additive or synergistic effects in breast, pancreas, and lung cancer models combining dovitinib with paclitaxel or gemcitabine, but not with adriamycin (Hernandez‐Agudo et al., 2013, and unpublished data). The wide clinical applications of weekly paclitaxel led us to choose it as the partner for dovitinib in this trial.
Late‐phase trials combining similar agents with chemotherapy based on the early‐phase‐defined RP2Ds according to toxicity criteria suggest that long‐term administration these RP2Ds might be non‐tolerable (Bergh et al., 2012; Paz‐Ares et al., 2012; Reck et al., 2010). Thus we incorporated pharmacodynamic measurements to guide the determination of the RP2D in case the patients showed adequate drug exposure at non‐toxic doses. Finally, because of the promiscuous activity of dovitinib against several oncogenic kinases, we determined several potential sensitizing genetic alterations that could narrow‐down the potential patient populations more likely to benefit from dovitinib‐based regimens in the future.
2. Patients and methods
2.1. Patient eligibility
Patients were eligible for this phase I trial if they had a histologically documented advanced solid malignancy for which no standard therapy existed. Other eligibility criteria included: signed informed consent; evaluable disease according to RECIST 1.1 criteria (Eisenhauer et al., 2009); recovery to any toxicity to less than grade 2 according to the NCI CTCAE V4.0 and adequate organ function, defined as follows (must comply all of them): ECOG 0‐2, life expectancy higher than 3 months, adequate hematologic function (absolute neutrophil count higher than 1500/mm3; platelet count higher than 75,000/mm3; hemoglobin higher than 8 g/dl); serum creatinine below 1.5×upper limit of normal; adequate liver function (serum bilirubine below 1.5×upper limit of normal, GOT and GPT below 2.5×upper limit of normal in presence or absence of liver metastasis); potassium between 3 and 5.5 mmol/L and sodium between 130 and 150 mmol/L.
Exclusion criteria included: LVEF <50%, long QTc or other significant heart conditions; active significant endocrine conditions including diabetes; and concurrent treatment with CYP inducers or QTc‐prolonging drugs.
This clinical trial was conducted in accordance with Good Clinical Practice guidelines and was approved by the review board and local ethics committees at each participating center.
2.2. Study design, dose escalation, treatment regimen and procedures
This was a phase I, open‐label dose‐escalation study. A classic 3+3 design was adopted. The treatment schedule is depicted in Figure 1.
Figure 1.
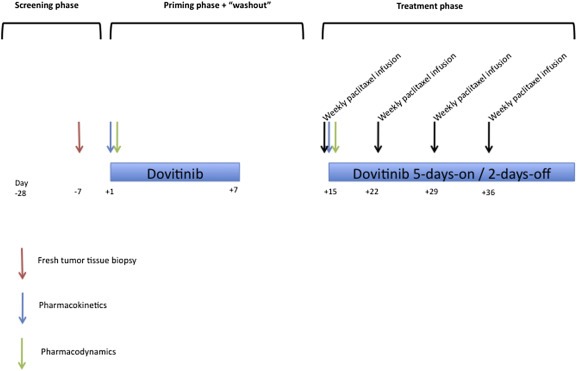
Treatment schedule. The screening included conventional assessments plus an image‐guided tumor biopsy (within 7 days before to the first dovitinib dose) and LVEF determination. Dovitinib was then administered orally for a week (“priming phase”); a pharmacokinetic profile was obtained on an inpatient basis on day 1. Blood samples for pharmacodynamic determinations were obtained at 0 and +24 h. After a 1 week “wash‐out” period, the treatment phase started with concurrent treatment of dovitinib + paclitaxel. A second pharmacokinetic profile (so that the paclitaxel‐induced modifications in dovitinib pharmacokinetics could be studied) including as well paclitaxel determinations, in order to compare the pharmacokinetics in combination with dovitinib with historical data of weekly paclitaxel (Fennelly et al., 1997), and two additional samples for pharmacodynamic determinations were obtained. The patients were discharged after 24 h and the treatment was continued with dovitinib administered orally on a schedule of 5‐days‐on/2‐days‐off in combination with weekly paclitaxel, in 28‐days cycles. Patients were visited and evaluated for toxicity weekly beginning with the first dose of dovitinib and until the end of the first cycle, bi‐weekly during the second and third cycles, and subsequently on a monthly basis. RECIST evaluations were performed every 2 cycles.
The starting dose (level 1) was set at 80 mg/m2 of i.v. weekly paclitaxel plus 200 mg/day of dovitinib (5/2). The dose‐limiting toxicity (DLT) period assessment was from the first administration of dovitinib to the end of the first cycle (42 days). However, no dose escalation was scheduled until all patients from one level had completed 2 cycles of treatment (70 days).
The primary objectives of the study were to assess the toxicity and safety of the combination, and to determine the RP2D (based on toxicity and pharmacodynamic data) and DLTs. The primary endpoints were frequency and severity of adverse events and incidence of DLTs, and plasma up‐regulation of FGF23 plasma levels, a biomarker indicative of inhibition of FGFR1 activity (Andre et al., 2013; Kim et al., 2011). A DLT was defined as any of the following: grade ≥3 febrile neutropenia; grade ≥4 neutropenia >7 days, or grade ≥4 thrombocytopenia or anemia; or any non‐hematologic event more severe than non‐tolerable grade 2 that required reduction or delay beyond 1 week. The secondary objectives were to evaluate the potential pharmacokinetic interactions between the two agents and to preliminary evaluate the efficacy of the combination depending on genetic alterations of the targets of dovitinib. The secondary endpoints were disease response, progression‐free survival and determination of mutations/amplifications of FGFR1‐2‐3 and RET. Dovitinib inhibits FGFR1‐2‐3, RET, and other targets (VEGFR1‐3, PDGFRA/B, CKIT and FTL3). These 4 targets were chosen because of two reasons: first, FTL3 and PDGFRB, and PDGFRB and CKIT, are mostly known to be mutated and related with sensitivity to other inhibitors in hematologic malignancies (Heinrich et al., 2008a) and GISTs (Heinrich et al., 2008b). The target population to be included in the CNIO‐BR‐002 trial was non‐hematologic tumors, and the likelihood of including GIST patients due to competitive trials in this malignancy, very low. Second, VEGFR1‐3 mutations and SNPs have been widely studied as potential biomarkers for antiangiogenics, mostly with negative results with the exception of some SNPs (Garcia‐Donas et al., 2011). However, recent findings from several TCGH studies, pointed towards FGFR1‐2‐3 and RET as potential novel, yet unexplored, oncogenic addiction drivers. All the genetic variants tested were assayed both in tumor tissue and peripheral blood mononuclear samples in order to determine their germline or somatic origin.
2.3. Pharmacokinetic sampling; pharmacodynamic and genetic determinations and correlative studies with wild‐type and mutant‐RET variants
Fasted dovitinib alone (day 1 of the priming phase) or in combination with 1‐h infusion of paclitaxel (day 1 of the combination phase) pharmacokinetics were determined by using a validated liquid chromatography with tandem mass spectrometry method with a lower limit of quantification (LLOQ) of 1.0 ng/mL. Plasma paclitaxel concentrations were determined by using a validated high‐performance liquid chromatography method with a LLOQ of 5.0 ng/mL. Noncompartmental pharmacokinetic analysis was conducted using the Phoenix software (Pharsight Inc., Mountain View, CA) to determine key pharmacokinetic parameters including Area Under the concentration‐time Curve (AUC) and the maximum plasma concentration of dovitinib (Cmax).
Free FGF23 was determined by using the FGF23‐ELISA Kit from Millipore, following manufacturer's instructions.
Tumor biopsy samples were obtained within 7 days before to treatment start and snap‐frozen in liquid nitrogen in OCT‐blocks. Amplifications in the FGFR1/2 and FGFR3 loci were determined with probes from Kreatech and Zytolight, respectively, by counting the signal over >200 interphasic nuclei and setting a cut‐off threshold of 2.
The 32 missense substitutions in FGFR1, 2, 3 and RET genes, gathered in Table 1, were determined with Taqman SNP genotyping assays (Life Technologies), using the primers‐probes sequences offered in Supplementary Table 1. They were collected from COSMIC (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) and dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) databases, and were selected based on their occurrence rate. Of note, these rates correspond to the knowledge about these genes available back in 2011, when the study was designed and the Taqman SNP genotyping assays prepared. The known variants in 2011 are depicted in Supplementary Table 2. Those variants with the highest frequency were selected, after excluding synonymous‐coding variants.
Table 1.
Genetic variants determined both in the tumor biopsies and peripheral blood mononuclear cells.
| Gene | Transcript mutation | Protein mutation | rsID | COSMIC ID | MAF | dbSNP (mut) | COSMIC (mut) | Clinical significance |
|---|---|---|---|---|---|---|---|---|
| FGFR1 | c.374C > T | p.S125L | rs121913473 | COSM601 | NA | 1 | 1 | NA |
| c.754C > A | p.P252T | rs121913472 | COSM12834 | NA | 1 | 1 | NA | |
| c.755C > G | p.P252R | rs121909627 | NA | NA | 1 | NA | pathogenic | |
| FGFR2 | c.755C > G | p.S252W | rs79184941 | COSM36903 | NA | 3 | 48 | pathogenic |
| c.1647T > A | p.N549K | rs121913476 | COSM36912 | NA | 1 | 20 | NA | |
| c.1144T > C | p.C382R | rs121913474 | COSM36906 | NA | 1 | 9 | NA | |
| c.758C > G | p.P253R | rs77543610 | COSM49170 | NA | 2 | 7 | pathogenic | |
| c.870G > C | p.W290C | rs121918499 | COSM41286 | NA | 1 | 3 | pathogenic | |
| c.929A > G | p.K310R | rs121913475 | COSM36901 | NA | 1 | 2 | NA | |
| c.1124A > G | p.Y375C | rs121913478 | COSM36904 | NA | 2 | 8 | pathogenic | |
| FGFR3 | c.746C > G | p.S249C | rs121913483 | COSM715 | NA | 1 | 1243 | pathogenic |
| c.1118A > G | p.Y373C | rs121913485 | COSM718 | NA | 1 | 407 | pathogenic | |
| c.742C > T | p.R248C | rs121913482 | COSM714 | NA | 1 | 240 | pathogenic | |
| c.1108G > T | p.G370C | rs121913479 | COSM716 | NA | 1 | 114 | pathogenic | |
| c.1111A > T | p.S371C | rs121913484 | COSM17461 | NA | 1 | 56 | pathogenic | |
| c.2089G > T | p.G697C | rs121913480 | COSM24802 | NA | 1 | 44 | NA | |
| c.1948A > C | p.K650Q | rs78311289 | COSM726 | NA | 2 | 5 | pathogenic | |
| c.1948A > G | p.K650E | rs78311289 | COSM719 | NA | 0 | 46 | pathogenic | |
| c.1949A > C | p.K650T | rs78311289 | COSM731 | NA | 0 | 5 | pathogenic | |
| c.1949A > T | p.K650M | rs78311289 | COSM720 | NA | 0 | 36 | pathogenic | |
| c.1950G > T | p.K650N | rs78311289 | COSM1428730 | NA | 5 | 1 | pathogenic | |
| c.1172C > A | p.A391E | rs28931615 | COSM721 | NA | 3 | 32 | pathogenic | |
| c.749C > G | p.P250R | rs4647924 | NA | NA | 4 | NA | pathogenic | |
| c.1138G > A | p.G380R | rs28931614 | COSM24842 | NA | 2 | 12 | pathogenic | |
| c.1620C > G | p.N540K | rs28933068 | NA | NA | 3 | NA | pathogenic | |
| RET | c.2753T > C | p.M918T | rs74799832 | COSM965 | NA | 3 | 290 | pathogenic |
| c.1900T > C | p.C634R | rs75076352 | COSM966 | NA | 5 | 12 | pathogenic | |
| c.1901G > A | p.C634Y | rs75996173 | COSM974 | NA | 7 | 7 | pathogenic | |
| c.1902C > G | p.C634W | rs77709286 | COSM975 | NA | 3 | 5 | pathogenic | |
| c.2304G > C | p.E768D | rs78014899 | COSM21338 | NA | 2 | 4 | pathogenic | |
| c.2071G > A | p.G691S | rs1799939 | COSM1666596 | 0.155 | 22 | 1 | Benign | |
| c.2944C > T | p.R982C | rs17158558 | NA | 0.017 | 14 | 0 | other |
All variants were tested both in tumor and germline material.
NA: Non available.
MAF: Minor allele frequency.
dbSNP (mut) and COSMIC (mut) columns report on the number of reported samples with the variant (may/2014).
Thirty micrograms of total protein from tumor areas with >90% epithelial content were mixed with Laemmly buffer, denatured and separated by 7.5 SDS‐PAGE and transferred onto nitrocellulose membranes. Membranes were incubated with antibodies for RET and phospho‐tyrosine 905‐RET (both from Cell Signaling Technology, #3223 and #3221 respectively) and solved with a conventional chemoluminescence detection system.
Regarding the experiments with wild type and mutant RET variants, the methods were as follows:
The pWZL‐Neo‐RET from Addgene (20614) was used to express the RET mutant containing the A substitution in codon 691 (AGT). The RET wild type construct was generated by using the QuikChange® II Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol. Briefly, PCR reaction was carried out with 10 ng of template vector, 125 ng of each primer, 200 uM dNTPs, and 2.5 U pfue Turbo DNA polymerase using the following cycling conditions:1 min at 95 °C, 18 cycles of 50 s at 95 °C, 50 s at 60 °C and 13 min at 68 °C, followed by 7 min at 68 °C. After PCR reaction plasmid were amplified in XL10‐Gold ultracompetent cells (Strategene) and substitution was confirmed by DNA sequencing.
Primers containing the A–G substitution in codon 691 (AGT to GGT) were synthesized by Sigma as follows: forward (5′‐CAGCTACTCCTCTTCCGGTGCC CGCCGGCCCTCGC‐3′) and reverse (5′‐GCGAGGGCCGGCGGGCACCGGA AGAGGAGTAGCTG‐3′).
HEK‐293 cell line used for this study was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Sigma). HEK‐293 cells were plated at a density of 0.5 × 106 cells/p100‐mm‐diameter plates overnight in DMEM‐10% FBS. Transfection was performed after 24 h with 5 ug of the indicated plasmids conjugated to 0.5 uL Lipofectamine 2000 (Invitrogen) in Opti‐MEM® reduced‐serum medium (Gibco). Antibiotic selection with Neoymycin was started 48 h after transfection, and maintained until clones were observed (4 weeks approximately). More than 10 clones were picked and successfully expanded both for wild‐type and variant RET stable transfectants. Three different clones per genotype were routinely tested for experimental purposes; representative data are shown.
Stable clones (wild type or variants) were exposed for 2 h to 250 nM of dovitinib or vehicle. The cells were washed with PBS and scraped into a tube containing washing buffer (150 mM NaCl, 50 mM HEPES, protease and phosphatase Inhibitor Cocktail (Thermo Scientific, Rockford, IL), vortexed and centrifuged at 12,000 rcf at 4 °C, 2 min. The supernatants were discarded. Pellets were resuspended in lysis buffer (Cell Signaling Technology, Beverly, MA), homogenized, sonicated for 15 min at 4 °C, centrifuged and supernatant collected. Protein concentration was measured for each sample by with a BSA protein kit (Pierce, Rockford, IL) and spectrophotometry. Equal amounts of total protein (30 μg) were mixed with Laemmli buffer, denatured, and separated by 7.5% SDS‐PAGE and then transferred on nitrocellulose membranas (Bio‐Rad Laboratories, Mississauga, Ontario, Canada) following the manufacturer's instructions. Non‐specific binding sites were blocked for 1 h with Tris buffered saline (TBS))‐Tween 1%–5% of Bovine Serum Albumin (BSA) (Sigma–Aldrich) at RT. Membranes were incubated with antibodies for RET (C31B4clone) (1:1000; Cell Signaling Technology, Beverly, MA) and phospho‐RET (1:1000; Cell Signaling Technology, Beverly, MA). Tubulin was detected using a monoclonal antibody against γ‐tubulin (1:5000; Sigma–Aldrich, Oakville, Ontario, Canada). After overning incubation at 4 °C, blots were rinsed with Tris buffered saline (TBS)‐Tween 1% and then incubated with goat anti‐rabbit horseradish peroxidase‐linked immunoglobulin G (IgG) or goat anti‐mouse horseradish peroxidase‐linked IgG for 1 h at RT. After rinsing, immunoreactive bands were visualized with an enhanced chemiluminescence detection system (ECL, Amersham International, Cardiff, UK).
2.4. Statistics
The safety analysis population comprised all patients who received one or more doses of study drugs. Because of the number of patients enrolled in the trial, the analyses were performed with descriptive statistics.
3. Results
Between April 18, 2012 and April 15, 2013, 12 patients were registered. Patient characteristics are summarized in Table 2.
Table 2.
Baseline demographics and patient characteristicsa.
| Characteristic | Value |
|---|---|
| Age (median; range) | 62.5 (37.9–73.4) |
| Sex (male/female) | 7 (64%)/4 (36%) |
| ECOG (0/1) | 3 (27%)/8 (73%) |
| Baseline FEV1 (median; range) | 69.2 (55–75) |
| Tumor type | |
| ‐ Lung | 1 (9%) |
| ‐ Breast | 1 (9%) |
| ‐ Pancreas | 1 (9%) |
| ‐ Uterine carcinosarcoma | 1 (9%) |
| ‐ Adenoid cystic carcinoma | 1 (9%) |
| ‐ Epithelioid hemangioendothelioma | 1 (9%) |
| ‐ Colorectal | 5 (46%) |
| ‐ Prior number of systemic regimens for metastatic disease (median; range) | 4 (1–9) |
| ‐ Patients with prior antiangiogenic treatment | 7 (64%) |
One patient withdrew consent prior to receiving the first treatment dose. Eleven patients are evaluable for toxicity and efficacy.
3.1. Dose escalation, DLTs, adverse events and drug exposure
One patient enrolled in level 1 (with colorectal cancer metastatic to brain and lungs) experienced G4 neutropenia plus G2 non‐tolerable mucositis on day +50. The patient was admitted to the hospital. On day 6 the patient presented respiratory insufficiency; the chest X‐ray showed diffuse interstitial infiltrate and the patient died 24 h later. Definitive classification of this event [(nosocomial infection secondary to the neutropenia vs disease progression (lymphangitic carcinomatosis)] was not possible. The other two patients did not experience severe toxicity. Because of the possibly‐related G5 event, although outside the DLT‐period, the dose‐level was expanded. The fourth patient experienced a DLT (G3 asthenia + diarrhea plus G4 GGT elevation). The protocol was stopped for safety reasons and amended with new dose levels.
Recruitment re‐started at level 1B (60 mg/m2 of weekly paclitaxel plus 200 mg/day of dovitinib 5/2); no DLTs were registered (3 patients). The first two patients enrolled at level 2B (60 mg/m2 of weekly paclitaxel plus 300 mg/day of dovitinib) did not experience DLTs; the third, experienced G3 asthenia; therefore a fourth patient was enrolled. However, the first patient at level was found dead at his home, 10 weeks after starting treatment and right before disease re‐evaluation (no autopsy was performed). At that point, the sponsor applied to the Spanish Drug Agency for early study closure on the grounds of safety reasons and study termination was mandated. All the patients still receiving medication stopped participation in the trial despite lack of significant toxicity.
Twenty‐seven priming‐phases and 27 cycles were administered. The toxic events per level and number of cycles are depicted in Table 3. No meaningful toxicities occurred during the priming phase. The median relative dose intensities were 97% for dovitinib and 76% for paclitaxel. At dose level 1B, the relative dose intensity was >97% for both drugs.
Table 3.
Toxicity per cycle and level occurring in at least 10% of the cycles (excluding alopecia).
| Level, toxicity type | Grade 1–2 (%) | Grade 3 (%) | Grade 4 (%) | Grade 5a (%) |
|---|---|---|---|---|
| Level 1 (5 cycles administered) | ||||
| Diarrhea | 40% | 20% | – | – |
| Mucositis | 40% | 20% | – | – |
| Asthenia | 80% | 20% | – | – |
| Lymphopenia | – | 40% | – | – |
| Neutropenia | 40% | 20% | 40% | – |
| Thrombopenia | 20% | 20% | – | – |
| Hypophosphatemia | 40% | 40% | – | – |
| Nausea/vomiting | 60% | – | – | – |
| ALT elevation | 20% | – | – | – |
| AST elevation | 60% | – | – | – |
| GGT elevation | 40% | – | 20% | – |
| Hypoalbuminemia | 80% | |||
| Hypomagnesemia | 40% | – | – | – |
| Cough | 20% | – | – | – |
| Level 1B (14 cycles administered) | ||||
| Diarrhea | 50% | – | – | – |
| Mucositis | 7% | – | – | – |
| Asthenia | 86% | – | – | – |
| Lymphopenia | 28% | – | – | – |
| Neutropenia | 21% | – | – | – |
| Thrombopenia | 14% | – | – | – |
| Hypophosphatemia | 14% | – | – | – |
| Nausea/vomiting | 65% | – | – | – |
| ALT elevation | 21% | – | – | – |
| AST elevation | – | – | – | – |
| GGT elevation | – | – | 57%b | – |
| Hypoalbuminemia | – | – | 14% | – |
| Hypomagnesemia | 14% | – | – | – |
| Peripheral neuropathy | 57% | – | – | – |
| Cough | 36% | – | – | – |
| Hypoproteinemia | 36% | – | – | – |
| Onycolysis | 36% | – | – | – |
| Peripheral edema | 36% | – | – | – |
| Rash | 29% | – | – | – |
| Taste alterations | 21% | – | – | – |
| Level 2B (8 cycles administered) | ||||
| Diarrhea | 62% | – | – | – |
| Mucositis | – | – | – | – |
| Asthenia | 75% | 12% | – | – |
| Lymphopenia | 37% | 12% | – | – |
| Neutropenia | 12% | – | – | – |
| Hypophosphatemia | 25% | – | – | – |
| Nausea/vomiting | 25% | – | – | – |
| FA elevation | 75% | 25% | – | – |
| ALT elevation | 50% | – | – | – |
| AST elevation | 62% | – | – | – |
| GGT elevation | 75% | 12% | 12% | – |
| Hypoalbuminemia | 12% | 12% | – | – |
| Peripheral neuropathy | 25% | – | – | – |
| Cough | 12% | – | – | – |
| Hypoproteinemia | 37% | – | – | – |
| Onycolysis | 12% | – | – | – |
| Peripheral edema | 25% | – | – | – |
| Rash | 25% | – | – | – |
| Taste alterations | 50% | – | – | – |
| Phlebitis | – | 25% | – | – |
| Lipase elevation | 12% | – | – | – |
The two grade 5 events cannot be related or ruled out definitively to be disease progression or grade 5 infection (patient in level 1), or disease progression or grade 5 bleeding (patient in level 2B).
One patient presented elevated GGT for 8 cycles and since the beginning; the patient entered protocol with a waiver form.
3.2. Pharmacokinetics and pharmacodynamics
Dovitinib pharmacokinetic parameters in the absence and in the presence of paclitaxel are provided in Figure 2. Detailed parameters for both agents are included in Table 4. An increase in dovitinib pharmacokinetic parameters can be observed. The paclitaxel exposure is similar to that reported in the literature for single‐agent weekly paclitaxel, suggesting adequate exposure to the cytotoxic agent (Fennelly et al., 1997; Ready et al., 2007). The FGF23 plasma concentration time course per dose level followed the kinetics depicted in Figure 2B. Increases in FGF23 plasma concentration of 50%–100% have been correlated with reduced pERK in tumors and clinical activity in previous trials, despite different schedules (Andre et al., 2013; Kim et al., 2011), suggesting adequate average pharmacodynamic effect at all tested levels.
Figure 2.
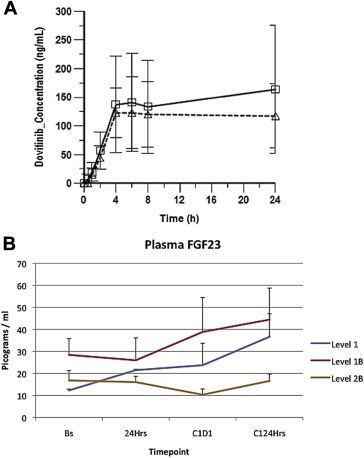
Pharmacokinetics and pharmacodynamics. (A) Dovitinib plasma concentration‐time profiles in the presence (solid‐line with squars) and absence (dotted‐line with triangles) of paclitaxel. (B) Average plasmatic FGF23 levels per timepoint (baseline, 24 h after the first dovitinib dose, before the first dovitinib dose of the combination phase and 24 h later) and dose levels. Error bars: standard error of the mean.
Table 4.
Dovitinib pharmacokinetic parameters in the presence and absence of paclitaxel and paclitaxel pharmacokinetic parameters.
| Absence of paclitaxel | Presence of paclitaxel | |
|---|---|---|
| Dovitinib PK: | ||
| Cmax (ng/mL) | 135 ± 52 | 186 ± 94 |
| AUC (h.ng/mL) | 2590 ± 1137 | 3167 ± 1875 |
| Paclitaxel PK: | ||
| Cmax (ng/mL) | NA | 556 ± 271 |
| AUC (h.ng/mL) | NA | 3441 ± 235 |
3.3. Selective activity over G2071A RET‐activating germline genetic variant
No patient tested positive for either amplification of FGFR1, 2 or 3, or 31 of the 32 genetic variants in the FGFR genes or RET‐genes. Two patients with epithelioid hemangioendothelioma and adenoid cystic carcinoma, respectively, were found to harbor a G‐to‐A coding variant in the 2071 position of the RET gene (exon 11), leading to a glycine to serine change in position 691 (Figure 3A). The variant was of germline origin, as we confirmed the G‐to‐A change in peripheral‐blood mononuclear cells in both patients. This variant is predicted to generate two novel serine phosphorylation sites in the RET protein, and to possibly enhance its kinase activity (Lantieri et al., 2013). This variant is associated with earlier onset in sporadic medullary thyroid carcinoma (Robledo et al., 2003), it is aggregated in patients with sporadic medullary thyroid carcinoma (Elisei et al., 2004) and it is frequently found in desmoplastic melanoma (Narita et al., 2009). In vitro studies have shown that this variant is associated with increased oncogenic signaling and cell replication/invasion (Sawai et al., 2005). Although meta‐analyses do not suggest its role as a hereditary cancer gene, these reports suggest that it functions as a genetic modifier or even a low‐penetrance gene.
Figure 3.
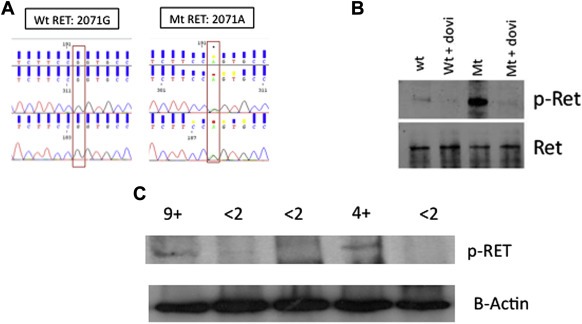
Activating RET variant sensitive to dovitinib. (A) Electropherogram showing the wild‐type sequence (G, left panel) and the variant sequence (A, right panel) in the 2071 position. (B) HEK‐293 cells were transfected with a wild‐type or a mutant‐RET‐encoding plasmid and blotted for anti‐905‐Y‐RET, in the presence and in the absence of dovitinib. It can be appreciated how the baseline levels of RET phosphorylation are highly increased in the mutant variant, despite similar total RET levels, whereas it is completely sensitive to dovitinib exposure. Virtually no residual phosphorylation of RET remains after incubation with 250 nM of dovitinib (2 h). (C) Western blot of anti‐905‐Y‐RET performed with lysates from tumor biopsy sample (only 5 biopsy samples yielded sufficient tissue after performing the FISH and mutational analysis). The two patients showing therapeutic benefit are those with the RET genetic variant, showing baseline increased phosphorylation. The numbers above the bands indicate how long (in months) the disease was controlled.
We transfected HEK293 cells with plasmids encoding wild‐type and G2071A variants and tested the phosphorylation levels of 905‐Y‐RET, the site that translates “active” conformational state. We found that the G2071A variant shows >10‐fold higher phosphorylation rates in this tyrosine site, whereas in resting conditions, the phosphorylation of the wild‐type variant is negligible (Figure 3B). Interestingly, this constitutively active variant was sensitive to dovitinib (Figure 3B). HEK293 cells did not express endogenous RET levels, what avoids potential confounding factors (Figure S1A); the effects of dovitinib in non‐transfected or empty‐vector‐transfected HEK293 are provided in Figure S1B.
The two patients with the genetic variant had constitutive 905‐Y‐RET phosphorylation (Figure 3C). Both patients were experiencing disease progression that was documented by two consecutive CT scans performed within 3 months before enrollment. Once they received trial medication, both achieved disease control. One had disease control for more than 9 months and discontinued the trial because of cumulative toxicity deemed related to paclitaxel. The other patient discontinued the trial because of the sponsor's decision to close the trial; this patient was without toxicity or progression at 4 months after registration. Both patients experienced disease progression after medication withdrawal. Thus, the progressive disease was controlled by the medication in the patients harboring the dovitinib‐sensitive, RET‐hyperactivated variant, and this control was lost upon medication withdrawal, suggesting a cause–effect relationship between dovitinib exposure and disease control in these two cases.
3.4. Disease control in the remainder patients
At the time of trial report, 6 (55%) patients had died; five of them died because of disease progression (one during treatment and the remainder after discontinuing the trial). There were no tumor responses. Four patients were not evaluable. One of these four was not evaluable because of early trial closure, one because of toxicity and two because of exitus before to the first scheduled evaluation.
Out of the 7 evaluable patients, 5 (71%) presented disease progression as best response. The other two patients were the patients with the active RET‐variant. Thus, no disease control was achieved in absence of this variant. The median overall survival and progression‐free survival (including the patients with the variant) were 6 months (95% C.I.: 2.7–9.7) and 2.7 months (95% C.I.: 2.2–3.4), respectively. Best response, PFS and OS at the individual level are provided in Supplementary Table 3.
4. Discussion
Dovitinib is a multikinase inhibitor agent with activity against angiogenic and tumor stromal regulators and several oncogenic kinases.
Agents of this class usually face two challenges for adequate development in solid malignancies: first, besides the case of liver and kidney cancer, they require combination with chemotherapy for being effective in most epithelial cancers. However, these combinations are often toxic in the long term. Long‐term administration at the single‐agent RP2D is substantially toxic, as recently reported in the randomized phase III trial of renal cell carcinoma patients with one‐third of the patients requiring dose reductions and approximately half of the patients requiring dose interruptions (Motzer et al., 2013, 2014). Patients usually are administered medications for longer periods in phase III than in phase I trials; this is a common problem with multi‐kinase inhibitors. Long‐term toxicity is an even more severe issue in chemotherapy plus multi‐kinase inhibitor trials (Bergh et al., 2012; Paz‐Ares et al., 2012; Reck et al., 2010). However, patients might be adequately exposed to both drugs with doses below the maximum tolerated dose, what can be determined by pharmacodynamic parameters. Thus, we designed this trial with a pharmacodynamic endpoint, because it was possible that the patients were adequately exposed to dovitinib at doses less than the maximum‐tolerated doses. The second challenge is that targeted agents are unlike to exert their activity in unselected patient populations; thus, it is appealing to find easy‐to‐determine markers that narrow‐down patients subgroups where activity is more likely to occur – for this reason, we assessed the status of genetic variants of dovitinib targets with high probability of functional implications (missense variants with possible function enhancer properties).
The examination of the PK data (Figure 2, Table 4) shows an almost 40% increase in the dovitinib concentration at 24 h, >40% increase in the Cmax and almost 25% increase in the AUC, when administered in combination with paclitaxel compared with in monotherapy. However, the small number of patients led to large standard errors that complicate the data interpretation. The paclitaxel exposure in this study (Table 4) was within the ranged of exposure observed in the literature (Fennelly et al., 1997; Ready et al., 2007). Among the side effects that determined DLTs probably only transaminitis can be unequivocally attributed to dovitinib. At this dose level, previous studies have not shown high incidence of transaminitis, but those studies administered dovitinib in monotherapy. Potentially, plasmatic accumulation of dovitinib attributable to the combination could account for this. Regarding neutropenia, the effects of dovitinib on FTL3, a receptor involved in normal myeloid maturation, might enhance the otherwise mild myelosuppressor effects of the administered paclitaxel doses. Interestingly, we showed adequate pharmacodynamic modulation in the three dose levels (Figure 2B), with similar fold‐increments of FGF23 as those reported in the literature (Andre et al., 2013; Kim et al., 2011). The pharmacodynamic effects plus the pharmacokinetic data suggesting accumulation of dovitinib makes us believe that the exposure to the targeted agent at level 1B is sufficient. Together with the lack of toxicity at this level, we recommend it as the RP2D. Although the early trial closure because of sponsor decision did not allow us to, it would have been interesting to further explore this dose level by including more patients. However, this is currently a limitation of our study because of the actions taken.
A second point of importance in the era of targeted therapies is finding biomarkers that can narrow‐down potential patients populations more likely to benefit from a given drug. Germline genetic variations, although associated with less penetrant phenotypes, are easier and cheaper to determine. Germline and somatic RET variants have been implicated mostly in endocrine malignancies. Here we report for the first time the potential role of a germline variant in RET as a potential biomarker of activity for a targeted therapy. This variant caused an aminoacid change in position 691 that was hypothesized to lead to increased RET‐kinase activity (Lantieri et al., 2013). Here, we show that such is actually the case, as evidenced by the striking increase in constitutive tyrosine phosphorylation in position 905 (Figure 3B). This variant has not been associated with MEN syndromes or with hereditary endocrine cancers, although the gain‐of‐function effects in the protein are evident in the literature in preclinical and epidemiologic studies (Elisei et al., 2004; Narita et al., 2009; Robledo et al., 2003; Sawai et al., 2005). The gain of function is probably insufficient to transform, but it may act as an enhancer. Although not a bonafide oncogene, the minor‐allele‐frequency of this variant is between 15 and 20%; conversely, oncogenic variants are usually highly sensitizing to targeted therapies but their incidence is usually low. Like oncogenic variants, this gain‐of‐function variant is sensitive to dovitinib according to our preclinical data (Figure 3B). Regarding the clinical data, 2/11 patients enrolled in this trial had this germline genetic variant present. Interestingly, although RET has not been implicated yet in the biology of epithelioid hemangioendothelioma or adenoid cystic carcinoma, we show how both cases had increased RET activation (Figure 3C), what could render these patients sensitive to dovitinib. Both patients had progressive disease before entering the trial and were the only two patients showing signs of activity, suggesting that this is a sensitizing polymorphism. A retrospective study involving archival samples from a recently completed randomized trial with dovitinib is currently ongoing, and should clarify this point, in order to justify, or not, prospective interventional studies in G2071A genetically‐enriched patient cohorts.
Funding
This work was supported by the Fondo de Investigación Sanitaria (Ministry of Health, Spain), grant numbers FIS PI‐2010/0288 and FIS PI‐2013/00430. MQF is a recipient of an AECC Scientific Foundation “Beca de Retorno 2010” grant. Unrestricted donations from Rosae Foundation and AVON España S.A.U. Novartis Spain provided funding support.
The disclosed funding bodies and agencies did not have any direct role in the conduction or design of the study.
Disclosure
The authors have declared no conflicts of interest. Eugene Tan is employed by Novartis. He performed the pharmacokinetic assays and analysis in this study.
Author contribution
-
‐
Conception, design, and study supervision: Miguel Quintela‐Fandino, Ramon Colomer.
-
‐
Trial coordination: Miguel Quintela‐Fandino, Ramon Salazar.
-
‐
Patient recruitment and treatment: Miguel Quintela‐Fandino, Ramon Salazar, Marta Gil, Antonio Gonzalez‐Martin, Raul Marquez, Raquel Bratos, Juan Guerra.
-
‐
Administrative support and trial logistics: Antonio Lopez.
-
‐
Correlative studies design and interpretation: Miguel Quintela‐Fandino, Maria Jose Bueno.
-
‐
Sample handling, processing and storage, database maintenance: Jesus Sanchez, Tamara Mondejar.
-
‐
Genetic analysis: Luis Lombardia.
-
‐
Pharmacokinetics: Eugene Tan.
-
‐
Data collection: all.
-
‐
Manuscript writing and approval: all.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Figure S1 (A) Naïve, untransfected HEK293 cells do not show meaningful native levels of RET protein. HEK293 lysates are offered together with WT‐RET transfected HEK293, in order to compare the baseline levels of total and phosphorylated (Y905) RET. (B) Untransfected and empty‐vector transfected (stable clones) were exposed to 250 nM dovitinib or vehicle for 2 h. As no endogenous RET protein was observed (and thus no p‐RET), we provide B‐tubulin as housekeeping.
{kind=link}
Acknowledgments
We thank Novartis Spain for the provision of sufficient dovitinib strengths to conduct the clinical trial.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.07.005.
Quintela-Fandino Miguel, Bueno Maria J., Lombardia Luis, Gil Marta, Gonzalez-Martin Antonio, Marquez Raul, Bratos Raquel, Guerra Juan, Tan Eugene, Lopez Antonio, Colomer Ramon, Salazar Ramon, (2014), Selective activity over a constitutively active RET‐variant of the oral multikinase inhibitor dovitinib: Results of the CNIO‐BR002 phase I‐trial, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.07.005.
References
- Andre, F. , Bachelot, T. , Campone, M. , Dalenc, F. , Perez-Garcia, J.M. , Hurvitz, S.A. , Turner, N. , Rugo, H. , Smith, J.W. , Deudon, S. , Shi, M. , Zhang, Y. , Kay, A. , Porta, D.G. , Yovine, A. , Baselga, J. , 2013. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin. Cancer Res. 19, 3693–3702. [DOI] [PubMed] [Google Scholar]
- Angevin, E. , Lopez-Martin, J.A. , Lin, C.C. , Gschwend, J.E. , Harzstark, A. , Castellano, D. , Soria, J.C. , Sen, P. , Chang, J. , Shi, M. , Kay, A. , Escudier, B. , 2013. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin. Cancer Res. 19, 1257–1268. [DOI] [PubMed] [Google Scholar]
- Bergh, J. , Bondarenko, I.M. , Lichinitser, M.R. , Liljegren, A. , Greil, R. , Voytko, N.L. , Makhson, A.N. , Cortes, J. , Lortholary, A. , Bischoff, J. , Chan, A. , Delaloge, S. , Huang, X. , Kern, K.A. , Giorgetti, C. , 2012. First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: results of a prospective, randomized phase III study. J. Clin. Oncol. 30, 921–929. [DOI] [PubMed] [Google Scholar]
- Eisenhauer, E.A. , Therasse, P. , Bogaerts, J. , Schwartz, L.H. , Sargent, D. , Ford, R. , Dancey, J. , Arbuck, S. , Gwyther, S. , Mooney, M. , Rubinstein, L. , Shankar, L. , Dodd, L. , Kaplan, R. , Lacombe, D. , Verweij, J. , 2009. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Elisei, R. , Cosci, B. , Romei, C. , Bottici, V. , Sculli, M. , Lari, R. , Barale, R. , Pacini, F. , Pinchera, A. , 2004. RET exon 11 (G691S) polymorphism is significantly more frequent in sporadic medullary thyroid carcinoma than in the general population. J. Clin. Endocrinol. Metab. 89, 3579–3584. [DOI] [PubMed] [Google Scholar]
- Fennelly, D. , Aghajanian, C. , Shapiro, F. , O'Flaherty, C. , McKenzie, M. , O'Connor, C. , Tong, W. , Norton, L. , Spriggs, D. , 1997. Phase I and pharmacologic study of paclitaxel administered weekly in patients with relapsed ovarian cancer. J. Clin. Oncol. 15, 187–192. [DOI] [PubMed] [Google Scholar]
- Garcia-Donas, J. , Esteban, E. , Leandro-Garcia, L.J. , Castellano, D.E. , del Alba, A.G. , Climent, M.A. , Arranz, J.A. , Gallardo, E. , Puente, J. , Bellmunt, J. , Mellado, B. , Martinez, E. , Moreno, F. , Font, A. , Robledo, M. , Rodriguez-Antona, C. , 2011. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 12, 1143–1150. [DOI] [PubMed] [Google Scholar]
- Heinrich, M.C. , Joensuu, H. , Demetri, G.D. , Corless, C.L. , Apperley, J. , Fletcher, J.A. , Soulieres, D. , Dirnhofer, S. , Harlow, A. , Town, A. , McKinley, A. , Supple, S.G. , Seymour, J. , Di Scala, L. , van Oosterom, A. , Herrmann, R. , Nikolova, Z. , McArthur, A.G. , 2008. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin. Cancer Res. 14, 2717–2725. [DOI] [PubMed] [Google Scholar]
- Heinrich, M.C. , Maki, R.G. , Corless, C.L. , Antonescu, C.R. , Harlow, A. , Griffith, D. , Town, A. , McKinley, A. , Ou, W.B. , Fletcher, J.A. , Fletcher, C.D. , Huang, X. , Cohen, D.P. , Baum, C.M. , Demetri, G.D. , 2008. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 26, 5352–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Agudo, E. , Soto, M. , Mondejar, T. , Sanchez, J. , Mulero, F. , Desco, M. , Quintela-Fandino, M. , 2013. 18F-misonidazole positron-emission tomography (FMISO-PET) as an early biomarker of vascular normalization in response to antiangiogenic therapy. Eur. J. Cancer 49, S155 [Google Scholar]
- Kim, K.B. , Chesney, J. , Robinson, D. , Gardner, H. , Shi, M.M. , Kirkwood, J.M. , 2011. Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin. Cancer Res. 17, 7451–7461. [DOI] [PubMed] [Google Scholar]
- Lantieri, F. , Caroli, F. , Ceccherini, I. , Griseri, P. , 2013. The involvement of the RET variant G691S in medullary thyroid carcinoma enlightened by a meta-analysis study. Int. J. Cancer 132, 2808–2819. [DOI] [PubMed] [Google Scholar]
- Lee, S.H. , Lopes de Menezes, D. , Vora, J. , Harris, A. , Ye, H. , Nordahl, L. , Garrett, E. , Samara, E. , Aukerman, S.L. , Gelb, A.B. , Heise, C. , 2005. In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin. Cancer Res. 11, 3633–3641. [DOI] [PubMed] [Google Scholar]
- Motzer, R.J. , Hutson, T.E. , Cella, D. , Reeves, J. , Hawkins, R. , Guo, J. , Nathan, P. , Staehler, M. , de Souza, P. , Merchan, J.R. , Boleti, E. , Fife, K. , Jin, J. , Jones, R. , Uemura, H. , De Giorgi, U. , Harmenberg, U. , Wang, J. , Sternberg, C.N. , Deen, K. , McCann, L. , Hackshaw, M.D. , Crescenzo, R. , Pandite, L.N. , Choueiri, T.K. , 2013. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 369, 722–731. [DOI] [PubMed] [Google Scholar]
- Motzer, R.J. , Porta, C. , Vogelzang, N.J. , Sternberg, C.N. , Szczylik, C. , Zolnierek, J. , Kollmannsberger, C. , Rha, S.Y. , Bjarnason, G.A. , Melichar, B. , De Giorgi, U. , Grunwald, V. , Davis, I.D. , Lee, J.L. , Esteban, E. , Urbanowitz, G. , Cai, C. , Squires, M. , Marker, M. , Shi, M.M. , Escudier, B. , 2014. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol. 15, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita, N. , Tanemura, A. , Murali, R. , Scolyer, R.A. , Huang, S. , Arigami, T. , Yanagita, S. , Chong, K.K. , Thompson, J.F. , Morton, D.L. , Hoon, D.S. , 2009. Functional RET G691S polymorphism in cutaneous malignant melanoma. Oncogene 28, 3058–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares, L.G. , Biesma, B. , Heigener, D. , von Pawel, J. , Eisen, T. , Bennouna, J. , Zhang, L. , Liao, M. , Sun, Y. , Gans, S. , Syrigos, K. , Le Marie, E. , Gottfried, M. , Vansteenkiste, J. , Alberola, V. , Strauss, U.P. , Montegriffo, E. , Ong, T.J. , Santoro, A. , 2012. Phase III, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 30, 3084–3092. [DOI] [PubMed] [Google Scholar]
- Ready, N.E. , Lipton, A. , Zhu, Y. , Statkevich, P. , Frank, E. , Curtis, D. , Bukowski, R.M. , 2007. Phase I study of the farnesyltransferase inhibitor lonafarnib with weekly paclitaxel in patients with solid tumors. Clin. Cancer Res. 13, 576–583. [DOI] [PubMed] [Google Scholar]
- Reck, M. , Frickhofen, N. , Cedres, S. , Gatzemeier, U. , Heigener, D. , Fuhr, H.G. , Thall, A. , Lanzalone, S. , Stephenson, P. , Ruiz-Garcia, A. , Chao, R. , Felip, E. , 2010. Sunitinib in combination with gemcitabine plus cisplatin for advanced non-small cell lung cancer: a phase I dose-escalation study. Lung Cancer 70, 180–187. [DOI] [PubMed] [Google Scholar]
- Rini, B.I. , Escudier, B. , Tomczak, P. , Kaprin, A. , Szczylik, C. , Hutson, T.E. , Michaelson, M.D. , Gorbunova, V.A. , Gore, M.E. , Rusakov, I.G. , Negrier, S. , Ou, Y.C. , Castellano, D. , Lim, H.Y. , Uemura, H. , Tarazi, J. , Cella, D. , Chen, C. , Rosbrook, B. , Kim, S. , Motzer, R.J. , 2011. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 378, 1931–1939. [DOI] [PubMed] [Google Scholar]
- Robledo, M. , Gil, L. , Pollan, M. , Cebrian, A. , Ruiz, S. , Azanedo, M. , Benitez, J. , Menarguez, J. , Rojas, J.M. , 2003. Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res. 63, 1814–1817. [PubMed] [Google Scholar]
- Sawai, H. , Okada, Y. , Kazanjian, K. , Kim, J. , Hasan, S. , Hines, O.J. , Reber, H.A. , Hoon, D.S. , Eibl, G. , 2005. The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Cancer Res. 65, 11536–11544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Figure S1 (A) Naïve, untransfected HEK293 cells do not show meaningful native levels of RET protein. HEK293 lysates are offered together with WT‐RET transfected HEK293, in order to compare the baseline levels of total and phosphorylated (Y905) RET. (B) Untransfected and empty‐vector transfected (stable clones) were exposed to 250 nM dovitinib or vehicle for 2 h. As no endogenous RET protein was observed (and thus no p‐RET), we provide B‐tubulin as housekeeping.