

Abstract
A number of immunotherapies, in particular immune checkpoint targeting antibodies and adoptive T‐cell therapies, are starting to transform the treatment of advanced cancers. The likelihood to respond to these immunotherapies differs strongly across tumor types, with response rates for checkpoint targeting being the highest in advanced melanoma, renal cell cancer and non‐small cell lung cancer. However, also non‐responsiveness is observed, indicating the presence of intrinsic resistance or naturally acquired resistance. In addition, a subgroup of patients that do initially respond to immunotherapy will later recur, thereby also pointing towards a role of therapy‐induced acquired resistance.
Here, we review our current understanding of both intrinsic and acquired resistance mechanisms in cancer immunotherapy, and discuss potential strategies to overcome them.
Keywords: Cancer immunotherapy, Immune resistance, T‐cell response
Highlights
Intrinsic and acquired immune resistance determine the efficiency of cancer immunotherapy.
An effective immune response requires T‐cell activation, homing and effector function.
The development of immune resistance can be evaluated on the basis on these three requirements.
A personalized approach to assess immune resistance is likely to increase the efficacy of immunotherapy.
1. Introduction
For many tumor types, including melanoma, renal cell cancer, colon cancer, ovarian cancer, and some subtypes of breast cancer, the presence of lymphocytic infiltrates within the tumor is highly correlated with improved outcome (Alexe et al., 2007; Clemente et al., 1996; Erdag et al., 2012; Galon et al., 2006; Hwang et al., 2012; Mahmoud et al., 2011; Nakano et al., 2001; Zhang et al., 2003). These infiltrates mostly consist of CD4+ and CD8+ T‐cells, and especially for melanoma it has been well established that part of these T‐cells recognize tumor‐associated antigens (Coulie et al., 1994; Kawakami et al., 1994). The fact that these cells can have direct tumoricidal potential is well illustrated by the clinical effects of adoptive transfer of ex vivo expanded tumor‐infiltrating lymphocytes (TIL) in metastatic melanoma patients. In several small clinical trials, response rates varying from 40% to 70% have been observed in highly selected metastatic melanoma patients (Dudley et al., 2010, 2008). In a more recent intent‐to‐treat analysis in a TIL trial for melanoma, a response rate of 30% has been reported (Besser et al., 2013). Within these studies, the absolute numbers of CD8+ T‐cells infused is strongly correlated with response to treatment, suggesting an important role for MHC class I restricted, cytotoxic T‐lymphocyte (CTL) mediated tumor killing (Besser et al., 2013; Radvanyi et al., 2012). Direct evidence in support of such a role has been obtained through the administration of TIL products enriched for CD8+ T‐cells, which showed a response rate comparable to that seen with unselected TIL products (Dudley et al., 2013). In addition to tumor‐reactive CD8+ T‐cells, it is clear that TIL products can also contain CD4+ T‐cell populations that are tumor‐reactive, and there is evidence for an anti‐tumoral effect of such tumor‐reactive CD4+ populations in melanoma and cholangiocarcinoma (Hunder et al., 2008; Tran et al., 2014).
A second, much more widely used, group of immunotherapeutic strategies that target the same cellular compartment focuses on the administration of antibodies that bind to immune checkpoint molecules, thereby (re)activating an endogenous tumor‐specific T‐cell immune response. Administration of ipilimumab, an antibody that binds the inhibitory receptor cytotoxic T‐lymphocyte antigen 4 (CTLA‐4) on T‐cells, has shown a four month increase in median overall survival in phase III trials, leading to FDA and EMA approval (Hodi et al., 2010; Robert et al., 2011). An analysis of a large cohort of melanoma patients treated following this registration shows a long‐term survival in 20–25% of treated metastatic melanoma patients (Prieto et al., 2012), a number that compares favorably to the 8–10% seen previously in patients treated with chemotherapy. More recently, objective response rates up to 50% have been reported in phase I/II trials testing antibodies that target another checkpoint molecule, programmed cell death protein 1 (PD‐1) or its ligand (PD‐L1). Importantly, clinical responses upon PD‐1 – PD‐L1 targeting have been observed in malignancies other than melanoma, such as renal cell carcinoma (RCC) and non‐small cell lung cancer (NSCLC) (Brahmer et al., 2012; Hamid et al., 2013; Topalian et al., 2012).
These encouraging clinical results have rightfully put immunotherapy at the forefront of oncological practice. Nevertheless, it is important to note that a substantial number of patients still derive no or only limited benefit for reasons largely unknown, sometimes at the cost of severe toxicities. The disparity in response rates observed between different immunotherapeutic treatment modalities, but also across tumor types strongly suggests a role for immune resistance. Further evidence for such resistance comes from patients treated with immunotherapy who experience an initial decrease in overall tumor burden but eventually succumb to disease recurrence. In the following sections we describe the relevance of different classes of immunotherapy resistance in oncology and contrast this with therapy resistance seen with targeted therapies. Furthermore, we describe the strategies that may be taken to obtain a better understanding of immunotherapy resistance, and how this knowledge can be used clinically.
2. Requirements for an optimal anti‐tumor T‐cell response
To understand at which levels resistance to T cell‐based cancer immunotherapy may occur, it is important to first describe the key elements that are required for a successful T‐cell response that leads to cancer regression. To do so, we subdivide this process into three discrete steps (Figure 1).
Figure 1.
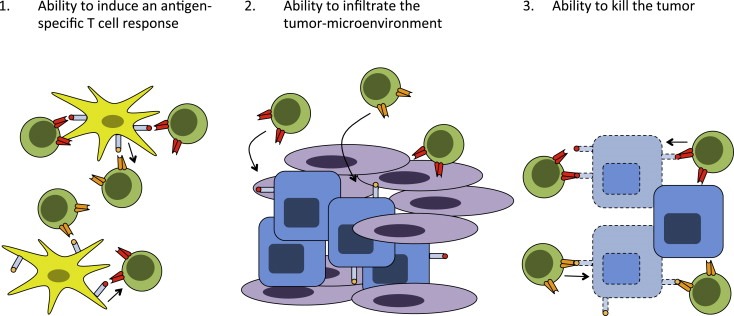
Key elements for an effective anti‐tumor T‐cell response. The development of an effective anti‐tumor T‐cell response follows three distinct steps: 1.) Priming and activation of naïve antigen‐specific T‐cells; 2.) Migration and infiltration of activated T‐cells through the vasculature and tumor‐surrounding stroma; 3.) Recognition of cognate peptide in the context of MHC and release of cytolytic granules to mediate tumor cell killing.
First, T‐cells need to be properly activated by professional antigen‐presenting cells (APCs) in peripheral lymphoid organs. For this to occur, two things are required: A). Dendritic cells (DCs) need to display tumor antigens (derived from apoptotic or necrotic tumor cells) in the context of MHC class I or II for which an antigen‐specific T‐cell repertoire is present. B). These DCs need to have received maturation signals that instruct the development of an effector T‐cell response, rather than T‐cell anergy or the expansion of regulatory T (Treg) cells.
Second, following priming in peripheral lymphoid organs, the activated T‐cells need to home to the tumor, extravasate through the endothelium and infiltrate via the surrounding stromal tissue into the tumor before they can bind to their target. This both requires certain phenotypic characteristics, such as expression of chemokine receptors, on the T‐cells and the expression of cell adhesion molecules/chemokines by the vascular endothelium for cells to pass the endothelial barrier and invade the tumor (Harlin et al., 2009). T‐cells that have been inefficiently activated, because of lack of co‐stimulatory molecule expression on APCs, or as a result of ineffective priming, can become anergic. By the same token, also when T‐cell priming is efficient, but the tumor lacks the inflammatory signals to attract these cells, the tumor‐specific immune response will be of little value.
Third, the T‐cell receptors on the infiltrating T‐cells need to contact peptide MHC complexes on the tumor cell surface, in the case of CD8+ cells, to release lytic granules in the immune synapse thereby mediating tumor destruction. Furthermore, the environment that the T‐cells encounter needs to permit such cytolytic activity. Negative feedback loops that regulate T‐cell activity at effector sites are abundant and are essential to prevent run‐away immune responses, but can also inhibit T‐cell mediated tumor regression.
Having described the requirements for an optimal anti‐tumor immune response, we can now make a subdivision of cancer immune resistance into three distinct classes (Figure 2A) that are further detailed below.
Figure 2.
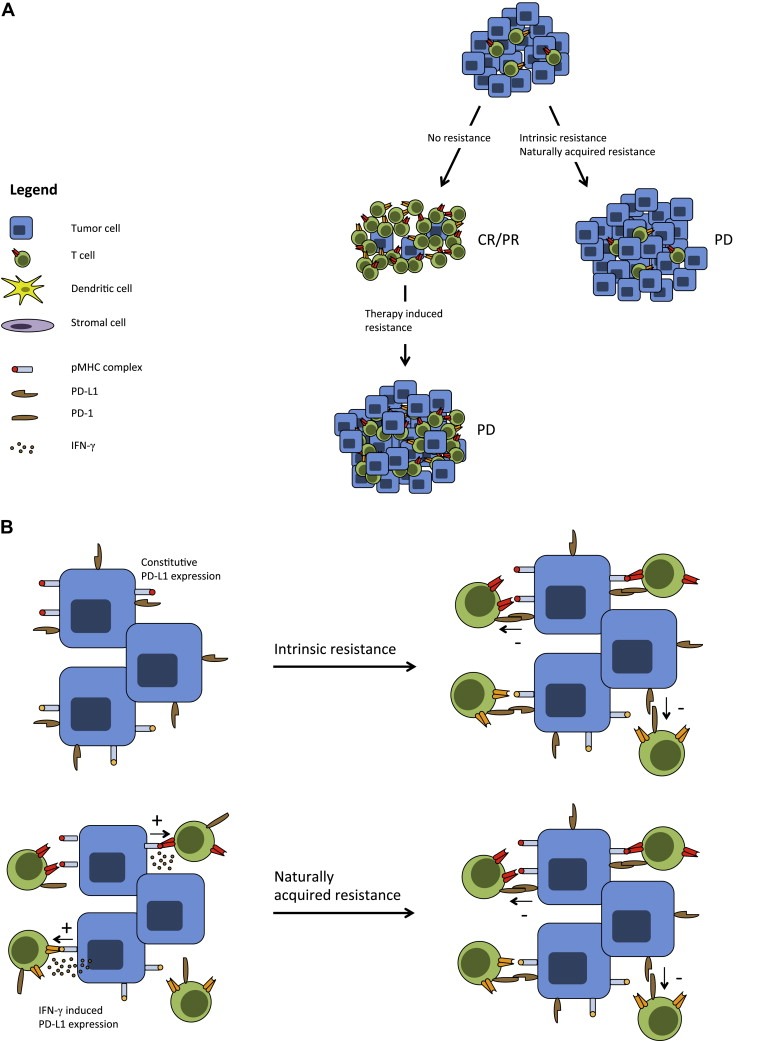
Categories of immune resistance. A. Several categories of immune resistance can be distinguished in either treatment‐naïve or treatment experienced cancer patients. In the first group, tumor‐infiltrating T‐cells are either absent or scanty indicative of intrinsic resistance or naturally acquired resistance. These patients are unlikely to respond to immune modulatory treatments. In the second group of patients, the degree of immune infiltration is sufficient to establish tumor regression upon immunotherapy initiation. However, due to several potentially overlapping mechanisms the tumor becomes resistant to this immune pressure and therapy‐induced resistance ensues. B. Mechanisms of intrinsic and naturally acquired resistance are exemplified by PD‐L1 expression and subsequent effector function inhibition of antigen‐specific T‐cells. Upper panel shows tumor cells that constitutively express PD‐L1 as a result of genetic alterations related to the oncogenic process. Lower panel shows induced expression of PD‐L1 mediated by IFN‐γ producing T‐cells.
3. Intrinsic resistance
First, there are non‐responding patients that lack anti‐tumor immune activity and that also fail to elicit a T‐cell response that has substantial tumoricidal potential upon immunotherapy, indicating intrinsic resistance. This form of resistance can be the result of a failing anti‐tumor immune response either locally or systemically. A first class of systemic immune failure is observed in patients that are unable to elicit a potent immune response to a large variety of truly foreign antigens, such as viruses. This is for example observed in severely immune‐compromised HIV patients or transplantation patients who carry an increased risk of virally induced neoplasias (Butel, 2000). Also elderly people may lose the capacity to mount a sufficiently strong systemic immune response upon foreign antigen exposure, possibly caused by a decrease in the diversity of the total T‐cell pool (Akbar and Fletcher, 2005; Khan et al., 2004; Messaoudi et al., 2004). This effect is demonstrated in elderly patients who suffer from the reactivation of silent viruses, such as VZV that causes shingles, or infection with Merkel cell polyomavirus that can cause Merkel cell carcinoma (Feng et al., 2008). Although speculative, it is conceivable that such patient subgroups are less capable of mounting an anti‐tumor immune response that is efficient enough to eradicate cancer cells. A second class of systemic intrinsic resistance is formed by tumors that express few antigens that can be seen as foreign by the immune system. Tumor antigens can be subdivided in distinct antigen classes that together form the antigenic landscape of a particular tumor. Specifically, many of the antigens that are (over‐) expressed by tumors are also expressed on healthy tissues. For at least some of these ‘self antigens’, the avidity of the available T‐cell repertoire will be low because of T‐cell tolerance (Kvistborg et al., 2013). As a second class of antigens, human tumors can express epitopes that are truly foreign to the immune system, either derived from viral proteins or from mutant epitopes formed as a consequence of mutations. Recent evidence suggests that recognition of such neo‐antigens may be of particular importance for tumor control (Brown et al., 2014; Champiat et al., 2014; Heemskerk et al., 2013; Robbins et al., 2013; van Rooij et al., 2013). Consequently, non‐viral tumors with a low mutational load, such as pediatric and many of the liquid tumors, may be more likely to evade immune detection than tumors with a high mutational load, such as melanoma or smoking‐related non‐small cell lung cancer (NSCLC) (Alexandrov et al., 2013; Vogelstein et al., 2013).
Local intrinsic immune resistance may manifest itself in several ways. In some patients tumors may completely lack lymphocytic infiltrates. Assuming that in at least some of these patients, a systemic tumor‐specific T‐cell response was induced (data are presently lacking on this), this would signify the presence of a non‐inflammatory tumor microenvironment that hampers infiltration of immune cells that would otherwise be able to recognize the tumor (Gajewski et al., 2010; Taube et al., 2012).
Assuming that tumor‐specific T‐cells are properly activated and capable of homing to the tumor, the tumor microenvironment can pose the last barrier for T‐cells to exert their effector functions thereby giving rise to intrinsic resistance. It has been described that expression of PD‐L1, which is the main ligand for the T‐cell inhibitory molecule PD‐1, can be induced upon loss of the tumor‐suppressor gene PTEN and activation of the PI3K pathway in glioblastoma cell lines (Parsa et al., 2007) (Figure 2B). Additionally, the secretion of inhibitory molecules such as TGF‐β, IL‐10 and IDO can have a direct negative effect on T‐cell function in the microenvironment (Braun et al., 2005; Geissmann et al., 1999; Pickup et al., 2013; Steinbrink et al., 1999), but also indirectly via the recruitment of tolerogenic immature DCs, myeloid derived‐suppressor cells (MDSCs) or (inducible) regulatory CD4+ T‐cells (Gabrilovich et al., 2012; Lutz and Schuler, 2002; Strauss et al., 2007; Vukmanovic‐Stejic et al., 2006). It is important to point out though that the presence of a T‐cell infiltrate within a progressing tumor does not necessarily imply local inhibition of T‐cell function as the mechanism of intrinsic resistance. Specifically, for most cancer types where T‐cell infiltration is apparent we do not presently know to what extent this T‐cell infiltrate consists of tumor‐specific T‐cells or of bystander cells, and only in the former case, local inhibition needs to be considered as a barrier to immune control.
Intrinsic resistance is not unique to immunotherapy but can also be observed in patients treated with targeted therapies. A well‐described example of this is the different sensitivities of tumors that carry the BRAF V600E mutations to drugs such as vemurafenib that bind the mutant BRAF protein. Specifically, whereas the majority of melanoma patients with a BRAF V600E mutation show a rapid (albeit often transient, see below) tumor regression upon treatment with BRAF inhibitors, patients with BRAF V600E colorectal cancer are unresponsive to these drugs. Recent work has demonstrated that this intrinsic resistance is due to EGFR expression in the BRAF mutant colorectal tumors, and that sensitivity can be imposed by concomitant EGFR inhibition (Prahallad et al., 2012; Sun et al., 2014). While obtained in an entirely different therapeutic field, these data illustrate that intrinsic resistance can occur as a coincidental side effect of the oncogenic process, and can be overcome upon a better understanding of this process.
4. Naturally acquired resistance
Naturally acquired resistance is special in that it is unique to immunotherapy. This form of resistance is defined as a reduced sensitivity that is not induced by cancer immunotherapy but that develops as a consequence of naturally occurring immune pressure. In this group of patients, there will generally be signs of an ongoing immune response in peripheral blood or tumor tissue, but they will fail to derive benefit from immune modulatory treatment.
In the case of naturally acquired resistance, there is presently little evidence for altered T‐cell activation or homing. Rather, this form of resistance may mostly manifest itself as mechanisms that interfere with T‐cell activity within the tumor microenvironment. Multiple inhibitory feedback mechanisms can play a role here, including the expression of a variety of (potentially overlapping) checkpoint molecules that dampen the immune response, such as LAG‐3, TIM‐3 and BTLA (Pardoll, 2012). As an example, when tumor‐infiltrating effector T‐cells start to produce IFN‐γ upon binding of cognate antigen, this will induce PD‐L1 expression on the tumor cell surface, which serves to limit further T‐cell effector function by engaging the immune checkpoint molecule PD‐1 (Taube et al., 2012) (Figure 2B).
In addition, a naturally occurring immune response may select for tumor cell subpopulations with loss of MHC class I expression, or other defects in the antigen processing machinery, thereby cloaking the tumor cell from the immune system (del Campo et al., 2014; Khong et al., 2004; Restifo et al., 1996). A similar immune evasive effect may be achieved through selection of tumor subclones present within heterogeneous tumors lacking one or multiple antigens that are subject to strong Darwinian selection, a process called immune‐editing (Dunn et al., 2002; Khong and Restifo, 2002; Matsushita et al., 2012). Strong evidence for immune‐editing has been obtained in mouse model systems. However, other murine studies suggest that antigen loss may be less of an issue in cases in which the release of IFN‐γ and TNF‐α by CTLs leads to the destruction of tumor stroma (Zhang et al., 2008). Human data on this topic are at present lacking but may conceivably be obtained with the recently developed abilities to describe T‐cell responses against (mutant) antigens within individual patients.
5. Therapy‐induced resistance
A third class of resistance is observed when patients that initially respond to immunotherapy relapse, which we define as therapy‐induced resistance. This type of resistance is well known in patients treated with classical cytotoxic agents or with targeted agents, such as BRAF inhibitors (Chapman et al., 2011). Natural resistance upon treatment with such targeted therapies, where virtually all patients eventually relapse, can be due to selection of resistant tumor clones already present at low numbers at the start of treatment, or of newly mutated resistant clones. This stands in stark contrast with immunotherapy‐treated patients where durable complete responses are often already observed after a single‐modality treatment. Although immunotherapy‐induced clinical responses can last up to years, a subgroup of patients experiences only temporary disease regression (Di Giacomo et al., 2013; Prieto et al., 2012; Rosenberg et al., 2011). The general mechanisms of therapy‐induced resistance will be very similar to those mentioned previously in the setting of naturally acquired resistance: When a properly activated T‐cell pool with homing capacity is present, an equilibrium between effector T‐cells and the tumor is reached locally, which at some point in time tips the balance in favor of renewed tumor growth.
6. Strategies to study resistance mechanisms
To increase our understanding of immunotherapy resistance, we suggest to analyze this process on the basis of the three different nodes that are involved in an effective anti‐tumor immune response.
First, a diverse T‐cell pool is required that can respond to a wide variety of tumor‐associated antigens. The currently used immunotherapeutic strategies that exploit the activity of the endogenous T‐cell compartment appear predominantly effective in tumors with median to high mutational loads, consistent with a role of neo‐antigen recognition in tumor control. While the occurrence of neo‐antigen reactive T‐cells appears to be a common trait in human melanoma (Robbins et al., 2013; van Rooij et al., 2013), more direct evidence for their role in tumor control is still lacking. Longitudinal immune monitoring of neo‐antigen‐specific T‐cells in a setting of cancer immunotherapy, using polychromatic flow cytometry or mass cytometry (Bendall et al., 2011) should be of value here. In a similar manner, assessment of immune competence (i.e. the ability to elicit a polyfunctional T‐cell response) on a per patient basis could help guide eligibility for immunotherapeutic intervention.
Second, to study the homing capacity of endogenously activated T‐cells in the context of immune escape, we need to address to what extent the tumor microenvironment is capable of triggering T‐cell infiltration. To study this, pre‐therapy tumor biopsies can be taken from patients included in immunotherapy trials and predictive gene‐expression signatures established that correlate with ongoing or subsequent T‐cell infiltration or with clinical benefit (Ji et al., 2012).
The third and final step that needs to be analyzed is the ability of T‐cells to release their effector functions at the site where it is needed. Several feedback mechanisms are at play here. Importantly though a hierarchy has not yet been determined, and such a hierarchy 1). Is likely to differ between tumor types; 2). Is within tumor types likely to differ depending on the specific genetic alterations; 3). May for a given tumor conceivably even vary depending on the site of metastasis. Recent work has emphasized the role of PD‐L1 expression as an important regulator of local T‐cell effector function. Using PD‐L1 expression as a biomarker grouped responding patients in an anti‐PD‐1 phase I clinical study, although absence of expression did not exclude a response to therapy (Weber et al., 2013). These data underscore the value of biomarker discovery not only for the early phases of the endogenous immune response (e.g. local inflammation) but also for the later effector phase. Notably though, patients with colorectal cancer only infrequently show responses to PD‐1 blockade, even though these tumors have high mutational loads and T‐cell infiltrates within these tumors has been shown to form a prognostic factor superior to the standard TNM classification (Galon et al., 2014; Pages et al., 2009). These data are consistent with the hypothesis that within these tumors, another inhibitory pathway could be dominant.
7. Strategies to overcome resistance
The efficacy of many immunotherapeutic strategies is dependent on the strength of the endogenous T‐cell response, including the level of tolerance towards the antigens recognized. Therefore, patients with an impaired capacity to mount immune responses, or who carry tumors that express few strong T‐cell antigens may gain most clinical benefit from strategies that create the missing tumor‐reactive T‐cell pool. This may be achieved by the adoptive transfer of T‐cells genetically modified to express an exogenous CAR or TCR capable of effective target killing (Morgan et al., 2006; Robbins et al., 2011) (Grupp et al., 2013; Kalos et al., 2011; Porter et al., 2011). In patients with a weak T‐cell response against tumor antigens, low‐frequency tumor‐specific T‐cell populations may be enriched from PBMNC or, perhaps preferable, from TIL, in order to steer reactivity towards predefined tumor‐associated epitopes and thereby augment the anti‐tumor response. Alternatively, expression of co‐stimulatory and co‐inhibitory markers in fresh tumor digest has been shown to define the tumor‐reactive T‐cell subset in melanoma lesions, offering a potential means to create TIL products that are enriched for tumor reactivity without the need for prior knowledge on antigen‐specificity (Gros et al., 2014; Ye et al., 2014).
Patients that have the capacity to mount a systemic T‐cell response but where tumors do not permit the infiltration of immune cells or prevent the initiation of a local endogenous immune response, are unlikely to benefit from treatment regimens that rely on such local immune responses. Such patients might benefit more from pre‐conditioning regimens that promote an immune supportive tumor microenvironment by providing ‘danger signals’ and the establishment of an inflammatory signature (Gajewski, 2012). This may conceivably be achieved through the induction of immunogenic cell death either by chemotherapy, radiotherapy or even local injection of Toll‐like receptor (TLR) agonists (Kroemer et al., 2013). For cancer types with a relatively low mutational load, such as ovarian, breast or pancreatic cancer, the use of DNA damaging agents could lead to an increase in the mutation frequency and as such broaden (albeit in a non‐clonal manner) the epitope landscape (Segal et al., 2008; Zhang et al., 2014).
Patients that do exhibit an endogenous anti‐tumor immune response in all the relative compartments discussed before are eligible for at least several immunotherapeutic interventions such as ipilimumab (anti‐CTLA‐4) and nivolumab (anti‐PD‐1). As with many classical cytotoxic agents, combination therapy could be of importance here in preventing escape from immune pressure. In this context, the nodes in the tumor immune interaction that are targeted should be as little overlapping as possible and preferably complementary in nature. The combination of ipilimumab (thought to be involved in the early priming phase of T‐cell activation) and nivolumab (thought to be involved in the later effector phase of T‐cell activation) has already shown higher response rates than either treatment modality alone (Wolchok et al., 2013). Presently, little is known about optimal timing or sequencing of available therapies but there is increasing evidence that patients failing one type of immunotherapy can respond to another, indicating independently operating resistance mechanisms than can be targeted accordingly. As an example, patients that have not benefitted from ipilimumab treatment still can have a meaningful objective response to anti‐PD‐1 treatment (Hamid et al., 2013) and vice versa (Weber et al., 2013). Patients failing ipilimumab treatment can likewise develop a durable complete remission upon TIL therapy (Besser et al., 2013).
8. Conclusion
In cancer immunotherapy, future rational treatment decision‐making should be based on the specific node that is affected in the tumor immune system interaction: 1.) Are antigen‐specific T‐cells efficiently activated in the treatment‐naïve host? 2.) Is there infiltration of those T‐cells into the tumor? and 3.) Are the tumor‐infiltrating T‐cells able to exert their function at that site? To achieve this, efforts should be focused on the discovery and implementation of simple biomarkers at each stage in the immune response that can predict whether a patient is likely to respond to a specific type of immunotherapy or not. Considering the variation in response rates of immunotherapy within one tumor entity and between tumor types, immunotherapy finds itself at the point where a patient‐specific approach is required in order to achieve durable tumor control in a larger group of patients.
Acknowledgments
This work was supported by Dutch Cancer Society grant KWF 2012‐5463 to T.N.M.S., and by a SU2C‐CRI Cancer Immunology Translational Cancer Research Grant to T.N.M.S.
Kelderman Sander , Schumacher Ton N.M. and Haanen John B.A.G. , ( 2014. ), Acquired and intrinsic resistance in cancer immunotherapy , Molecular Oncology , 8 , doi: 10.1016/j.molonc.2014.07.011.
References
- Akbar, A.N. , Fletcher, J.M. , 2005. Memory T cell homeostasis and senescence during aging. Curr. Opin. Immunol.. 17, 480–485. [DOI] [PubMed] [Google Scholar]
- Alexandrov, L.B. , 2013. Signatures of mutational processes in human cancer. Nature. 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexe, G. , 2007. High expression of lymphocyte-associated genes in node-negative HER2+ breast cancers correlates with lower recurrence rates. Cancer Res.. 67, 10669–10676. [DOI] [PubMed] [Google Scholar]
- Bendall, S.C. , 2011. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser, M.J. , 2013. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res.. 19, 4792–4800. [DOI] [PubMed] [Google Scholar]
- Brahmer, J.R. , 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med.. 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, D. , 2005. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 106, 2375–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S.D. , 2014. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res.. 24, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butel, J.S. , 2000. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 21, 405–426. [DOI] [PubMed] [Google Scholar]
- Champiat, S. , 2014. Exomics and immunogenics: bridging mutational load and immune checkpoints efficacy. Oncoimmunology. 3, e27817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, P.B. , 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med.. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente, C.G. , 1996. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 77, 1303–1310. [DOI] [PubMed] [Google Scholar]
- Coulie, P.G. , 1994. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med.. 180, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Campo, A.B. , 2014. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int. J. Cancer. 134, 102–113. [DOI] [PubMed] [Google Scholar]
- Di Giacomo, A.M. , 2013. Long-term survival and immunological parameters in metastatic melanoma patients who responded to ipilimumab 10 mg/kg within an expanded access programme. Cancer Immunol. Immunother.. 62, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, M.E. , 2010. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin. Cancer Res.. 16, 6122–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, M.E. , 2013. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J. Clin. Oncol.. 31, 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, M.E. , 2008. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol.. 26, 5233–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, G.P. , 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol.. 3, 991–998. [DOI] [PubMed] [Google Scholar]
- Erdag, G. , 2012. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res.. 72, 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, H. , 2008. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 319, 1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich, D.I. , 2012. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol.. 12, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski, T.F. , 2012. Cancer immunotherapy. Mol. Oncol.. 6, 242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski, T.F. , 2010. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J.. 16, 399–403. [DOI] [PubMed] [Google Scholar]
- Galon, J. , 2006. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- Galon, J. , 2014. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol.. 232, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann, F. , 1999. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J. Immunol.. 162, 4567–4575. [PubMed] [Google Scholar]
- Gros, A. , 2014. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 124, 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, S.A. , 2013. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med.. 368, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid, O. , 2013. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med.. 369, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlin, H. , 2009. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res.. 69, 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk, B. , 2013. The cancer antigenome. EMBO J.. 32, 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi, F.S. , 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med.. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunder, N.N. , 2008. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med.. 358, 2698–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, W.T. , 2012. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol. Oncol.. 124, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, R.R. , 2012. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother.. 61, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos, M. , 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med.. 3, 95ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami, Y. , 1994. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. U S A. 91, 6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, N. , 2004. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J. Immunol.. 173, 7481–7489. [DOI] [PubMed] [Google Scholar]
- Khong, H.T. , Restifo, N.P. , 2002. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol.. 3, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong, H.T. , 2004. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J. Immunother.. 27, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer, G. , 2013. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol.. 31, 51–72. [DOI] [PubMed] [Google Scholar]
- Kvistborg, P. , 2013. Human cancer regression antigens. Curr. Opin. Immunol.. 25, 284–290. [DOI] [PubMed] [Google Scholar]
- Lutz, M.B. , Schuler, G. , 2002. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity?. Trends Immunol.. 23, 445–449. [DOI] [PubMed] [Google Scholar]
- Mahmoud, S.M. , 2011. An evaluation of the clinical significance of FOXP3+ infiltrating cells in human breast cancer. Breast Cancer Res. Treat.. 127, 99–108. [DOI] [PubMed] [Google Scholar]
- Matsushita, H. , 2012. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 482, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi, I. , 2004. Age-related CD8 T cell clonal expansions constrict CD8 T cell repertoire and have the potential to impair immune defense. J. Exp. Med.. 200, 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, R.A. , 2006. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 314, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, O. , 2001. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res.. 61, 5132–5136. [PubMed] [Google Scholar]
- Pages, F. , 2009. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol.. 27, 5944–5951. [DOI] [PubMed] [Google Scholar]
- Pardoll, D.M. , 2012. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa, A.T. , 2007. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med.. 13, 84–88. [DOI] [PubMed] [Google Scholar]
- Pickup, M. , 2013. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer. 13, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, D.L. , 2011. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med.. 365, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad, A. , 2012. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Prieto, P.A. , 2012. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin. Cancer Res.. 18, 2039–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radvanyi, L.G. , 2012. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res.. 18, 6758–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo, N.P. , 1996. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst.. 88, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, P.F. , 2013. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med.. 19, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, P.F. , 2011. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol.. 29, 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert, C. , 2011. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med.. 364, 2517–2526. [DOI] [PubMed] [Google Scholar]
- Rosenberg, S.A. , 2011. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res.. 17, 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal, N.H. , 2008. Epitope landscape in breast and colorectal cancer. Cancer Res.. 68, 889–892. [DOI] [PubMed] [Google Scholar]
- Steinbrink, K. , 1999. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood. 93, 1634–1642. [PubMed] [Google Scholar]
- Strauss, L. , 2007. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin. Cancer Res.. 13, 4345–4354. [DOI] [PubMed] [Google Scholar]
- Sun, C. , 2014. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 508, 118–122. [DOI] [PubMed] [Google Scholar]
- Taube, J.M. , 2012. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl Med.. 4, 127ra137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, S.L. , 2012. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med.. 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E. , 2014. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 344, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij, N. , 2013. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol.. 31, e439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B. , 2013. Cancer genome landscapes. Science. 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukmanovic-Stejic, M. , 2006. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J. Clin. Invest.. 116, 2423–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, J.S. , 2013. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol.. 31, 4311–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok, J.D. , 2013. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med.. 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Q. , 2014. CD137 accurately identifies and enriches for naturally-occurring tumor-reactive T cells in tumor. Clin. Cancer Res.. 20, 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , 2008. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J. Clin. Invest.. 118, 1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G.N. , 2014. TP53 K351N mutation-associated platinum resistance after neoadjuvant chemotherapy in patients with advanced ovarian cancer. Gynecol. Oncol.. 132, 752–757. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , 2003. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med.. 348, 203–213. [DOI] [PubMed] [Google Scholar]