

Abstract
A major limitation of targeted anticancer therapies is intrinsic or acquired resistance. This review emphasizes similarities in the mechanisms of resistance to endocrine therapies in breast cancer and those seen with the new generation of targeted cancer therapeutics. Resistance to single‐agent cancer therapeutics is frequently the result of reactivation of the signaling pathway, indicating that a major limitation of targeted agents lies in their inability to fully block the cancer‐relevant signaling pathway. The development of mechanism‐based combinations of targeted therapies together with non‐invasive molecular disease monitoring is a logical way forward to delay and ultimately overcome drug resistance development.
Keywords: Anticancer therapy, Drug resistance, Targeted therapy, Pathway reactivation, Endocrine therapy, Drug combinations
Highlights
Resistance to cancer (targeted) therapies remains a major problem in the clinic.
We highlight similarities between resistance mechanisms to different targeted therapies.
Resistance to targeted monotherapy is frequently driven by pathway reactivation.
Rational combinations of targeted therapies need to be developed.
Non‐invasive molecular disease monitoring shows promise to guide these combinations.
Abbreviations
- aCGH
array comparative genomic hybridization
- ALK
anaplastic lymphoma kinase
- BCAR1
breast cancer anti-estrogen resistance 1
- BCR-ABL
breakpoint cluster region protein – Abelson murine leukemia viral oncogene homolog 1
- CML
chronic myeloid leukemia
- RC
colorectal cancer
- EGFR
epidermal growth factor receptor
- EMT
epithelial-to-mesenchymal transition
- ERα
estrogen receptor alpha
- ESR1
estrogen receptor 1
- FISH
fluorescent in situ hybridization
- GBM
glioblastoma multiforme
- GEMMs
genetically engineered mouse models
- IGF1R
insulin-like growth factor 1 receptor
- IHC
immunohistochemistry
- MAPK
mitogen-activated protein kinase
- MLPA
multiplex ligation-dependent probe amplification
- NCOA3
nuclear receptor coactivatior 3
- NSCLC
non-small cell lung cancer
- ORF
open-reading frame
- pCR
pathological complete response
- PELP1
proline, glutamate and leucine rich protein 1
- PDGFRB
beta-type platelet-derived growth factor receptor
- PDX
patient-derived xenograft
- PIK3CA
phosphatidylinositol 3-kinase catalytic subunit
- PKA
protein kinase A
- qPCR
quantitative polymerase chain reaction
- ROS1
c-ros oncogene 1
- RTK
receptor tyrosine kinases
- SERM
selective estrogen receptor modulator
- shRNA
short-hairpin RNA
- SRC
v-src avian sarcoma viral oncogene homolog
- TGFβ
transforming growth factor beta
- TNBC
triple-negative breast cancer
- TKI
tyrosine kinase inhibitor
1. Introduction
Resistance to therapy remains a major challenge in oncology. Resistance comes in two flavors: (1) early intrinsic resistance (also known as innate or de novo resistance) or fast adaptive tumor responses, and (2) late acquired resistance, resulting from clonal evolution of resistant variants. Anticancer drug resistance has been studied since the 1960s (Brockman, 1963), but has gained momentum after the introduction of targeted cancer therapeutics and several technological advances such as RNA interference (Brummelkamp et al., 2002) and next generation DNA/RNA sequencing. Selective targeting of activated pathways has proven to be effective, but the observed responses are usually partial and not durable when using single agent therapies. This translates clinically in prolonged progression‐free survival, but similar overall survival compared to standard of care. Examples where prolonged progression‐free survival has been achieved without giving rise to improved overall survival are crizotinib (ALK‐TKI) in advanced ALK‐positive Non‐Small Cell Lung Cancer (NSCLC) and gefitinib (EGFR‐TKI) in EGFR‐mutated NSCLC (Maemondo et al., 2010; Shaw et al., 2013). An exception is the case of the BRAFV600E‐specific inhibitor vemurafenib in BRAF V600E‐mutated metastatic melanoma. Patients with metastatic melanoma have a median survival of 6–10 months and activating BRAF mutation was associated with shortened survival in patients with metastatic disease (Long et al., 2011). A phase 3 trial with vemurafenib in BRAF V600E‐mutated metastatic melanoma showed significant improvement in both progression‐free survival and overall survival with vemurafenib compared to chemotherapy in an early interim analysis of overall survival (Chapman et al., 2011). Although the median duration of follow‐up in this study was too short to draw strong conclusions, long follow‐up data of a phase 2 trial with vemurafenib in the same clinical setting confirmed these early results and showed increase in median overall survival to approximately 16 months (Sosman et al., 2012).
Here we review the recent insights into mechanisms of resistance to targeted therapies. We focus on the reactivation of signaling pathways as a recurrent pattern of resistance development to single‐agent targeted therapies. We first discuss the resistance mechanisms to endocrine therapy in breast cancer, the first targeted therapy introduced in the clinic. We will use this as an example to highlight that the mechanisms of resistance to endocrine therapy that have been identified in breast cancer are seen all over again with the new pathways‐targeted therapies in other cancers. Finally, we argue that synthetic lethal combinations of targeted therapies together with non‐invasive molecular disease monitoring are a promising way forward to fight drug resistance.
2. Endocrine resistance in breast cancer
The synthesis of competitive inhibitors of the binding of the hormone estrogen to its receptor (ERα) in the 1970s led to the development of the first targeted cancer drug: tamoxifen. Tamoxifen is a triphenylethylene derivative classified as a selective estrogen receptor modulator (SERM). It impairs the mitogenic function of ERα in breast cancer by competing with estrogen for binding to the receptor. The binding of tamoxifen to the ERα changes the receptor conformation, which is distinct from the conformational change that is induced by estrogen binding. This conformation change prevents the formation of the ERα complex with its essential transcriptional co‐activators and thereby inhibits ERα‐mediated transcription (Shiau et al., 1998). A second class of endocrine drugs that target estrogen synthesis has been developed subsequently: the aromatase inhibitors. Aromatase is the enzyme responsible for the estrogen synthesis from androgenic substrates (extra‐ovarian synthesis) (Smith and Dowsett, 2003). Aromatase inhibitors cannot inhibit the estradiol production in the ovaries themselves and are therefore not active in premenopausal patients without ovarian suppression. Consequently, tamoxifen is typically given to premenopausal patients, whereas aromatase inhibitors are given to postmenopausal patients, although postmenopausal sequential treatment of tamoxifen and aromatase inhibitors is often prescribed as well.
Almost 70% of breast cancers are classified as ERα‐positive by IHC, and endocrine therapies targeting estrogen action (anti‐estrogens and aromatase inhibitors) are only effective in ERα‐positive breast cancers. Expression of ERα protein is strongly predictive of response to endocrine therapies (Davies et al., 2011). However, approximately one third of ERα‐positive early breast cancers do not respond to endocrine therapy (intrinsic resistance) or relapse after an initial response (acquired resistance) (EBCTCG, 2005). The proportion of breast cancer patients with advanced or metastatic disease that relapses during or after endocrine therapy is even higher. It is important to note that patients who develop resistance to one kind of endocrine treatment can still respond to another type (Wang et al., 2009; Yoo et al., 2011). The various mechanisms underlying resistance to endocrine therapy that have been proposed and studied are outlined below. They can be classified in three main categories: (1) alterations of the drug target (i.e. ESR1/ERα), (2) alterations in downstream and upstream effectors of ERα signaling, and (3) bypass mechanisms (Table 1).
Table 1.
Similarity in the mechanisms of resistance to endocrine therapies in breast cancer and to targeted therapies in other cancers.
| Drug resistance mechanism | Endocrine therapies breast cancer | Targeted therapies in other cancers | |
|---|---|---|---|
| Example | Example | Drug (cancer type) | |
| Alterations of the drug target | |||
| Mutation | ESR1 mutation/translocationa | ABL T315I mutationoEGFR T790M mutationpALK F1147L mutationqROS1 mutationr | Imatinib (CML)EGFR‐TKIs (NSCLC)Crizotinib (NSCLC)Crizotinib (NSCLC) |
| EGFR ectodomain mutations | Cetuximab (CRC) | ||
| Amplification | ESR1 amplificationb | BCR‐ABL amplificationtBRAF amplificationuEGFR amplificationv | Imatinib (CML)BRAF‐inhibitor (melanoma)EGFR‐TKI (NSCLC) |
| Receptor downregulation | Loss of ERα expressionc | Loss of mutant EGFR expressionw | EGFR‐TKI (GBM) |
| Splice variant | ERα splice variantsd | BRAF splice variantx | BRAF‐inhibitor (melanoma) |
| Pathway activation | |||
| By upstream components | HER family activationePI3K‐AKT‐mTOR activationf | RTK activationy | BRAF‐inhibitor (melanoma, CRC, thyroid); MEK inhibitor (TNBC); AKT inhibitor |
| RAS pathway activationg | RAS mutationsz | BRAF‐inhibitor (melanoma) | |
| BCAR1 overexpressionh | |||
| PKAi | |||
| By downstream components | AIB1 (SRC3) overexpressionj | KRAS mutation and amplificationaa | Cetuximab (CRC) |
| MYC activationk | PTEN loss and PIK3CA mutationab | Trastuzumab (breast cancer) | |
| MEK1/2 mutationsac | BRAF‐/MEK‐inhibitor (melanoma) | ||
| Bypass mechanism | |||
| Notch activationl | PKA activationad | BRAF‐MEK‐inhibitor (melanoma) | |
| Metabolic activity of CYP2D6m | COT expressionae | BRAF‐inhibitor (melanoma) | |
| Loss of CDK10 expressionn | MET amplificationaf | EGFR‐TKI (NSCLC); cetuximab (CRC) | |
| PI3K pathway activationag | BRAF‐inhibitor (melanoma) | ||
| HGF secretionah | BRAF‐inhibitor (melanoma) | ||
| EMT ai | Multiple TKIs | ||
(Cancer Genome Atlas Network, 2012; Jeselsohn et al., 2014; Li et al., 2013; Robinson et al., 2013; Toy et al., 2013).
(Albertson, 2012; Ooi et al., 2012).
(Encarnacion et al., 1993; Gutierrez et al., 2005; Johnston et al., 1995; Kuukasjarvi et al., 1996).
(Groenendijk et al., 2013; Herynk and Fuqua, 2004; Shi et al., 2009).
(Arpino et al., 2004; De Laurentiis et al., 2005; Gutierrez et al., 2005; Osborne et al., 2003).
(Miller et al., 2011a; Yamnik and Holz, 2010).
(McGlynn et al., 2009; Yamnik and Holz, 2010).
(van der Flier et al., 2000).
(Holm et al., 2009; Michalides et al., 2004).
(Osborne et al., 2003).
(Miller et al., 2011b).
(Magnani et al., 2013; Rizzo et al., 2008).
(Goetz et al., 2005; Hoskins et al., 2009; Singh et al., 2011).
(Iorns et al., 2008).
(Gorre et al., 2001; Michor et al., 2005).
(Kobayashi et al., 2005; Sequist et al., 2011).
(Choi et al., 2010; Katayama et al., 2012; Sasaki et al., 2010).
(Awad et al., 2013).
(Montagut et al., 2012).
(Gorre et al., 2001).
(Shi et al., 2012).
(Sequist et al., 2011).
(Nathanson et al., 2014).
(Poulikakos et al., 2011; Shi et al., 2014).
(Chandarlapaty et al., 2011; Corcoran et al., 2012; Duncan et al., 2012; Girotti et al., 2013; Montero‐Conde et al., 2013; Nazarian et al., 2010; Prahallad et al., 2012; Sun et al., 2014a,b; Villanueva et al., 2010).
(Nazarian et al., 2010; Shi et al., 2014; Trunzer et al., 2013).
(Diaz et al., 2012; Misale et al., 2012; Valtorta et al., 2013).
(Berns et al., 2007; Majewski et al., 2014; Nagata et al., 2004).
(Emery et al., 2009; Trunzer et al., 2013; Van Allen et al., 2014; Wagle et al., 2011).
(Johannessen et al., 2013).
(Johannessen et al., 2010).
(Bardelli et al., 2013; Engelman et al., 2007; Turke et al., 2010; Sequist et al., 2011).
(Shi et al., 2014).
(Straussman et al., 2012).
(Brunen et al., 2013; Byers et al., 2013; Fuchs et al., 2008; Huang et al., 2012; Salt et al., 2014, Thomson et al., 2005; Yauch et al., 2005).
2.1. Alterations of ESR1 and its encoded protein ERα
Patients with the highest ERα protein expression benefit slightly more from tamoxifen compared to patients with low receptor expression, but the latter group still have substantial benefit (Davies et al., 2011). However, response to tamoxifen is rare in ERα‐negative breast cancer. A portion of ERα‐positive tumors becomes independent of estrogen signaling after which they loose ERα expression and, hence, are tamoxifen resistant. Gutierrez et al. studied the ERα expression in paired clinical breast cancer samples from before the start of tamoxifen treatment and after tumor progression (Gutierrez et al., 2005). They found loss of ERα expression in 17% of ERα‐positive tumors at the time of tumor progression. This was in line with earlier reports showing that ERα loss occurs in 15–30% of the tumors at the time of recurrence (Encarnacion et al., 1993; Johnston et al., 1995; Kuukasjarvi et al., 1996). Loss of ERα was associated with tamoxifen resistance (Johnston et al., 1995) and can be used as a predictor of poor response to subsequent endocrine therapy (Kuukasjarvi et al., 1996).
Mutations in ESR1, the gene coding for ERα, were proposed as yet another possible mechanism of endocrine therapy resistance. However, ESR1 mutations were only found in a very low percentage of primary breast cancers, if at all present (Cancer Genome Atlas Network, 2012). In a recent report by Li et al., ESR1 ligand‐binding domain mutations were identified in patient‐derived xenografts (PDXs) derived from primary or metastatic breast cancer patients who were resistant to hormonal treatment (Li et al., 2013). This report was followed by three independent studies showing that activating ESR1 mutations are relatively frequent in advanced ERα‐positive hormone‐resistant breast cancers (Jeselsohn et al., 2014; Robinson et al., 2013; Toy et al., 2013). The mutation frequency was somewhat variable in the different cohorts they analyzed (range: 11–55%), with an average frequency of 17.3% in 167 cases. The authors also show that these ESR1 mutations were not present at detectable levels in the primary tumors, indicating that the specific mutations are either clonally selected or arise de novo under the pressure of endocrine therapy with tamoxifen or aromatase inhibitors. In addition, the study by Jeselsohn et al. showed absence of these mutations in any stage of ERα‐negative disease (Jeselsohn et al., 2014). The mutations are clustered in the ligand‐binding domain of ERα, similar to what was reported by Li et al. for ESR1 mutations in the PDXs. Mutations in this domain result in ligand‐independent constitutive activity. On the other hand, Toy et al. reported no significant difference in sensitivity to aromatase inhibitors for ESR1 wild‐type or mutant patients, but the number of patients was small. The three studies together demonstrate that mutant ERα can still bind anti‐estrogens like tamoxifen, although higher doses are required. Further studies are needed to fully address the association with ESR1 mutations and clinical outcome.
The study by Li et al. on breast cancer patients‐derived xenografts reported an ESR1/YAP1 gene rearrangement that replaces the ligand‐binding and AF2 domains of ESR1 (Li et al., 2013). This translocation induces estradiol‐independent growth and makes cells insensitive to the selective ER down‐regulator fulvestrant. Similar ESR1 translocations that preserve the DNA‐binding and AF1 domains of ESR1 were detected by the authors in the TCGA breast cancer RNA‐seq data (Li et al., 2013). The frequency of these in‐frame ESR1 translocations might be very low, but can still be of clinical significance considering the high number of breast cancer patients.
Several different ERα variant mRNAs have been described in human breast cancer. Almost all of these naturally occurring variants are mRNA splicing variants, in which one or more exons are absent from the ERα mRNA. In most ERα splicing variants, except for variants lacking exon 3 or 4, translation runs out of frame after the site of splicing, leading to a truncated protein (Herynk and Fuqua, 2004). One identified variant, the ERα36, is transcribed from an alternative promoter in the first intron of the ESR1 gene (Shi et al., 2009). ERα36 mediates in vitro membrane‐initiated effects of estrogen signaling, and tamoxifen treatment fails to block the ERα36 mediated activation of the MAPK pathway. Shi et al. also found that breast cancer patients with high ERα36 expression are less likely to benefit from tamoxifen treatment. Two ERα splice variants, the ERΔ3 and the ERΔ7 variant, were described as dominant negative receptor forms in the presence of wild‐type ERα (Herynk and Fuqua, 2004). The expression of the ERΔ7 is associated with a Basal subtype of breast cancers expressing ERα mRNA (Groenendijk et al., 2013).
Gene amplification is defined as a focal high‐level copy number increase of a region of genomic DNA. Gene amplification usually goes hand in hand with gene overexpression. Overexpression of ERα in cell lines caused broad anti‐estrogen resistance in vitro (Tolhurst et al., 2011). Holst et al. reported in 2007, using FISH on tissue microarrays, that ESR1 was amplified in 21% of breast cancers and that these amplifications were present in breast cancers with high ERα expression. Multiple groups have since tried to reproduce these findings, reporting a considerable difference in amplification frequencies when using in situ hybridization compared to biochemical methods (aCGH, MLPA, qPCR) (as summarized in Albertson, 2012). It was later found that the large clustered FISH signals, interpreted as ESR1 amplification, are sensitive to RNase treatment (Ooi et al., 2012). This indicated that the FISH probe was detecting accumulation of ESR1 mRNA transcripts in the nucleus of cells expressing high levels of ERα, providing an explanation for the controversy in ESR1 amplification frequencies between different studies. Although this finding of RNase sensitivity of ESR1 FISH signals has yet to be reproduced, it seems likely that the amplification frequency will be around 5% of breast cancers. Despite this small percentage, there might still be a (modest) role of ESR1 amplification in endocrine therapy resistance.
2.2. Alterations in upstream and downstream effectors of ERα signaling
Both preclinical and clinical studies suggest that crosstalk between ERα and growth factor and stress kinase signaling pathways is involved in endocrine therapy resistance (as reviewed in Musgrove and Sutherland, 2009; Osborne and Schiff, 2011). Among those, the crosstalk between ERα and the HER family is best studied. The ERBB/HER family consists of 4 structurally related membrane receptors: HER1 (EGFR), HER2 (ERBB2/NEU), HER3, and HER4. The tyrosine kinase HER2 is amplified in 10–20% of ERα‐positive breast cancers. It has unique receptor features compared to the other receptors of the family: it has no specific ligand, but forms homodimers with itself and heterodimers with other ERBB receptors. Tamoxifen activates HER2 via the membrane functions of ERα, and this activated HER2 phosphorylates both ERα and AIB1 (SRC3). There is clinical evidence that tamoxifen is less effective in ERα‐positive breast cancer overexpressing HER2 (De Laurentiis et al., 2005; Osborne et al., 2003). Furthermore, a small percentage of tumors originally negative for HER2 showed HER2 amplification or overexpression after tumor progression with tamoxifen (Gutierrez et al., 2005). Although HER1 (EGFR) signaling downstream is not identical to HER2 signaling, there is significant overlap. In a study by Arpino et al. it was shown that patients with EGFR overexpression were less likely to respond to tamoxifen and had shorter time to treatment failure (Arpino et al., 2004). Aromatase inhibitors may be different, because ligand deprivation shuts off all nuclear and membrane ERα activity, disallowing crosstalk of ERα with other pathways.
Increasing evidence supports an interaction between the mTOR pathway and ERα signaling, and hyperactivation of the PI3K‐AKT‐mTOR pathway promotes anti‐estrogen resistance (Miller et al., 2011a). The mTOR‐substrate S6 kinase 1 can phosphorylate the AF1 domain of ERα, which is responsible for ligand‐independent receptor activation (Yamnik and Holz, 2010). The clinical benefit of inhibiting mTOR in combination with endocrine therapy has already been demonstrated. The mTOR inhibitor everolimus combined with an aromatase inhibitor improved progression‐free survival compared with aromatase inhibitor alone in postmenopausal ERα‐positive advanced breast cancer (BOLERO‐2 trial) (Baselga et al., 2012b). Furthermore, the TAMRAD trial reported that tamoxifen plus everolimus increased clinical benefit, time‐to‐progression and overall survival compared with tamoxifen alone in postmenopausal women with aromatase‐inhibitor‐resistant metastatic breast cancer (Bachelot et al., 2012).
Expression and activation of the RAS/RAF1/MAPK pathway is associated with poor outcome on tamoxifen (McGlynn et al., 2009). MAPK activation can phosphorylate serine‐118 and serine‐167 of ERα, resulting in ligand‐independent activation of the receptor and transcription of estrogen‐regulated genes and proliferation (Yamnik and Holz, 2010).
Phosphorylation of ERα at serine‐305 by protein kinase A (PKA) has been associated with tamoxifen resistance (Michalides et al., 2004). This phosphorylation induces a conformational change in the receptor, which prevents the induction to the inactive state by tamoxifen. Patients whose tumors express serine‐305 phosphorylated ERα (Holm et al., 2009) or patients with downregulation of a negative regulator of PKA (Michalides et al., 2004) do not benefit from tamoxifen. Phosphorylation of ERα at other sites (serine‐118 and serine‐167) is associated with anti‐estrogen sensitivity as well, although different effects are reported for pre‐ versus postmenopausal women (Beelen et al., 2012).
The function of ERα is regulated by interactions with other transcription factors and through post‐translational modification of the receptor itself or its interactors. Overexpression and increased phosphorylation (hence activity) of ERα co‐activators leads to constitutive ERα transcriptional output. The activity of nuclear receptor coactivator 3 (NCOA3, also known as AIB1 or SRC3) is associated with reduced responsiveness to tamoxifen in patients (Osborne et al., 2003). The ERα coactivator PELP1 localizes to the cytoplasm in many breast cancers, where it functions as scaffold for the interaction of ERα with SRC that subsequently recruits other cofactors (Gururaj et al., 2006). Modeling cytoplasmic PELP1 localization in vitro makes tumor cells hypersensitive to estrogen but resistant to tamoxifen. The SRC substrate BCAR1 (also known as CAS) can induce tamoxifen resistance in vitro upon overexpression and breast cancers overexpressing BCAR1 are less responsive to tamoxifen (van der Flier et al., 2000).
There is extensive evidence from in vitro studies that cell cycle regulators play a role in endocrine therapy resistance. However, clinical evidence supporting this notion is scarce, especially for the prognosis‐independent predictive value of cell cycle regulators. Cyclin D1 and MYC are the best studied, although studies for the role of cyclin D1 have yielded contradictory results (Beelen et al., 2012). MYC is amplified in around 15% of breast cancers; MYC protein overexpression and MYC activation signature were predictive for the response to endocrine therapy in a large cohort of ERα‐positive breast cancers (Miller et al., 2011b).
2.3. Bypass mechanisms
Reprogramming of the chromatin landscape through epigenetic modifications or changes in genome accessibility may play a central role in endocrine therapy resistance. Magnani et al. showed significant differences in the transcriptional programs of endocrine therapy‐resistant and ‐responsive breast cancer cell lines (Magnani et al., 2013). The histone H3 lysine 36 trimethylation (H3K36me3) mark is found in actively transcribed gene bodies. Only around 12% of H3K36me3 sites were shared between endocrine therapy‐resistant and ‐responsive cells (Magnani et al., 2013). Furthermore, the authors showed that most of the open chromatin sites are cell type‐specific and only a minority is shared between resistant and responsive cells. The genome‐wide reprogramming of the chromatin landscape disengage the resistant cells from classical ERα signaling and promotes Notch signaling (Magnani et al., 2013; Poulikakos et al., 2011). Crosstalk between the Notch pathway and ERα signaling in breast cancer has been reported previously (Rizzo et al., 2008). Pharmacological targeting of Notch signaling can be of interest for endocrine therapy resistant breast cancers (Haughian et al., 2012).
Tamoxifen is converted in the liver into its active metabolites endoxifen and 4‐hydroxytamoxifen and this conversion is catalyzed by the polymorphic enzyme P450 2D6 (CYP2D6) (Hoskins et al., 2009). CYP2D6 is located on chromosome 22q13.1, and polymorphisms in this gene can significantly affect enzymatic activity. Approximately 6–10% of Caucasian women carry less active or inactive alleles of this gene and are consequently less responsive to tamoxifen (Goetz et al., 2005; Hoskins et al., 2009; Singh et al., 2011). In addition, the metabolic activity of CYP2D6 can be lowered by certain commonly prescribed medications, like for example certain selective serotonin or noradrenaline reuptake inhibitors (Hemeryck and Belpaire, 2002).
Finally, Iorns et al. showed both in vitro and in patient series that loss of CDK10 expression resulted in resistance to tamoxifen (Iorns et al., 2008). Loss of CDK10 activates RAF1 (CRAF) and the downstream MEK‐ERK signaling cascade, resulting in cyclin D1 expression and subsequent independence of estrogen signaling.
3. Drug resistance to more recent targeted therapies
The recent DNA and RNA sequencing projects in several cancer types by The Cancer Genomic Atlas (cancergenome.nih.gov) and other consortia have revealed recurrent somatic alterations in several genes that are considered to be drivers of oncogenesis. They activate crucial oncoproteins such as RAS, BRAF, EGFR, PIK3CA, ALK, BCR‐ABL and many others. These driver alterations can give rise to a tumor dependency on a particular signaling pathway or module. These dependencies, called ‘oncogene addiction’, can result from activation of genes that stimulate a pathway or inactivation of genes that inhibit a signaling pathway. Tumor dependencies can also be the result of other factors, like hormone dependency or lineage dependency. The addiction of certain cancer types to specific signaling pathways or modules in those pathways creates an Achilles heel for tumor maintenance that can be exploited therapeutically (Weinstein, 2002). However, examples of pure ‘oncogene addiction’ from a single oncogenic event are rare. Most cancers contain many mutations, making such tumors potentially less dependent on a single oncogenic event. Another complication is that signaling pathways are interconnected and these interactions are often dynamic (Lemmon and Schlessinger, 2010). A third factor limiting the success of targeted single‐agent therapy is the heterogeneity of tumors, especially in the advanced or metastatic setting. Similar to resistance mechanisms for endocrine therapy, the mechanisms underlying resistance to other targeted therapies can be classified in three main categories (Table 1): (1) alterations of the drug target (Figure 1(a)), (2) alterations in upstream and downstream effectors resulting in pathway reactivation (Figure 1(b) and (c)) and (3) bypass mechanisms (Figure 1(d)). In that sense, the recently‐identified mechanisms of resistance to targeted cancer drugs represent a clear case of “déjà vu all over again” (a quote from the legendary American baseball player Yogi Berra (Berra, 1998)).
Figure 1.
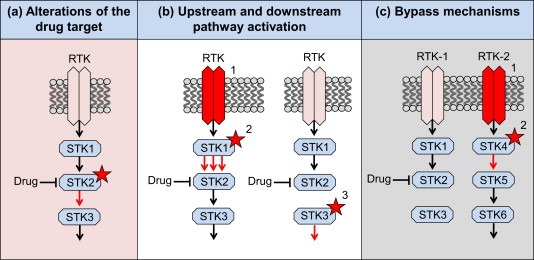
Schematic representation of three recurrent mechanisms of resistance to targeted therapies. RTK: receptor tyrosine kinase. STK: serine/threonine kinase. (a) Alterations of the drug target, e.g. mutation, amplification or splice variants. (b) Pathway reactivation through (1) RTK activation or (2) mutation or amplification of an upstream component or (3) by mutation or amplification of a downstream component. (c) Bypass by parallel pathway activation through (1) activation of a second RTK (by itself or through feedback regulation) or (2) mutation of a parallel STK.
3.1. Alterations of the drug target
One of the mechanisms of resistance is mutation of the target kinase domain, which reduces the ability of the drug to inhibit the kinase, a so‐called ‘gatekeeper’ mutation. Mutations in the ABL kinase domain (T315 residue) confer resistance to the ABL‐inhibitor imatinib in chronic myeloid leukemia (CML) patients with the Philadelphia‐chromosome translocation (Gorre et al., 2001). These mutations were shown to be pre‐existing in subpopulations of tumor cells positively selected by the treatment (Michor et al., 2005). Another well‐known example is the EGFR T790M mutation, which abrogates the inhibitory activity of EGFR‐TKIs and is present in half of the patients that develop resistance to EGFR‐TKIs (Kobayashi et al., 2005; Sequist et al., 2011). A subset of these patients also developed EGFR amplification, where the T790M allele is selectively amplified (Sequist et al., 2011). Analogous to the T790M mutation in EGFR are mutations in the kinase domain of ALK that are found at the time of resistance to crizotinib in tumors with ALK‐fusions (Choi et al., 2010; Katayama et al., 2012; Sasaki et al., 2010).
Resistance by mutating the drug target was also reported for ROS1. Chromosomal translocations that create fusion proteins involving the tyrosine kinase domains of ALK or ROS1 were identified in lung adenocarcinoma, and are of particular interest because of the availability of an active inhibitor of these kinases: the multi‐targeted TKI crizotinib. Crizotinib has shown efficacy in the treatment of lung cancers harboring ROS1 translocations (Davies et al., 2012; Ou Si et al., 2013). Resistance to crizotinib developed in a patient with a CD74‐ROS1 translocated metastatic lung adenocarcinoma. This resistance was caused by a mutation in the ROS1 kinase domain that made the kinase insensitive to crizotinib (Awad et al., 2013).
Some colorectal cancers with KRAS or BRAF mutations are addicted to the MAPK pathway. Because MEK1/2 are the only substrates of RAF kinases, their inhibition is explored as a treatment strategy to block the MAPK signaling in KRAS and BRAF mutated tumors. However, tumors can bypass this blockade by amplifying the signal that goes through this pathway, for example by amplification of the driving oncogene. In vitro, it was shown that KRAS or BRAF mutated colorectal cancer cell lines made resistant to the MEK‐inhibitor had a marked upregulation of their respective driving oncogene due to amplification (Corcoran et al., 2010; Little et al., 2011). Amplification of mutant BRAF was also demonstrated in melanoma patients treated with BRAF‐inhibitor (Shi et al., 2012). Similarly, resistance to imatinib was associated with progressive BCR‐ABL gene amplification (Gorre et al., 2001).
Alternative splicing of BRAF has been implicated in drug resistance to vemurafenib in BRAF‐mutated melanoma. In vitro, in a subset of cells resistant to vemurafenib, a BRAF V600E‐splicing variant was identified. The splice variant encodes a version of BRAF lacking the RAS‐binding domain and is resistant to the BRAF‐inhibitor. This splicing variant for BRAF V600E was also found by PCR analysis in 6 of the 19 tumors derived from melanoma patients having developed vemurafenib resistance (Poulikakos et al., 2011). Mutations in NRAS were identified in 4 out of 19 progression samples and were mutually exclusive with BRAF‐splicing variants. BRAF‐splicing variants were not detected in two samples derived from patients with de novo resistance or in melanoma samples that were never treated with vemurafenib. However, it remains questionable whether these splice variants are in fact acquired during the treatment or were already present in a small subpopulation that was too small for the detection. In a different study analyzing tumor samples from patients whose melanomas developed acquired resistance to either vemurafenib or dabrafenib monotherapy, alternative BRAF splicing was detected in 6 out of 48 (13%) tumors after progression (Shi et al., 2014).
Another type of drug target modification is the finding of an acquired EGFR ectodomain mutation (S492R) that prevents cetuximab binding and confers resistance to cetuximab. This mutation was identified in two out of ten patients with disease progression after cetuximab treatment (Montagut et al., 2012).
The selective sensitivity of tumor cells to targeted drugs relies on the expression of and dependency on the oncogenic target. An example is the sensitivity of tumor cells that express mutant EGFR to EGFR‐TKIs. As we discussed for endocrine therapy, the downregulation of the oncogenic target is one way for tumor cells to acquire resistance. Single‐cell analyses of glioblastoma multiforme (GBM) PDX models and clinical samples from GBM patients treated with EGFR‐TKIs have demonstrated that tumor cells reversibly up‐ or downregulate mutant EGFR expression to reach an optimal state for tumor growth (Nathanson et al., 2014). Resistance to EGFR‐TKIs occurred by elimination of mutant EGFR from extrachromosomal DNA. The withdrawal of the drug was followed by the re‐emergence of EGFR mutations on extrachromosomal DNA. The re‐emergence of EGFR mutations suggests that these cells regain sensitivity to EGFR‐TKIs, as was indeed shown in the paper. The benefit from intermittent dosing of TKIs instead of continuous dosing is supported by the clinical observation that patients can regain sensitivity to a drug after a so‐called ‘drug holiday’ (Yano et al., 2005). Recently, it was reported that discontinuous dosing of vemurafenib in a PDX model of BRAF V600E‐mutant melanoma delayed the development of drug resistance (Das Thakur et al., 2013). The reversible drug resistance suggests that epigenetic regulatory mechanisms play a role in acquiring this reversible‐resistance state. Data supporting the existence of such an epigenetic mechanism came from an shRNA screen performed in our laboratory to address the question whether changes in expression of chromatin regulators could confer resistance to BRAF‐inhibitors (Sun et al., 2014b). We found that suppression of sex determining region Y‐box 10 (SOX10) in melanoma caused activation of TGFβ signaling, which led to transcriptional upregulation of EGFR and PDGFRB, causing drug resistance. In a heterogenous population of melanoma cells having varying levels of SOX10 suppression, cells with low SOX10 and consequently high EGFR expression were rapidly enriched in the presence of the BRAF‐inhibitor, but this was reversed when the drug treatment was discontinued. These data highlight that conditions that confer selective advantage during drug exposure can be a liability in the absence of drug (Sun et al., 2014b).
3.2. Signaling pathway reactivation mediated by alterations in upstream or downstream signaling
Activation of receptor tyrosine kinases (RTKs) represents a major mechanism for upstream signaling activation. RTKs are high‐affinity cell surface receptors for many growth factors, cytokines and hormones. In humans, a total of 58 receptor tyrosine kinase proteins have been identified that can be divided in 20 subclasses or subfamilies (Lemmon and Schlessinger, 2010). All RTKs have a similar molecular architecture, with ligand‐binding domains in the extracellular region, a single transmembrane helix, and a cytoplasmic region that contains the protein tyrosine kinase domain plus additional regulatory regions. The mechanisms of RTK activation and regulation have been excellently reviewed by Lemmon and Schlessinger (2010). The RTKs are important drug targets, although resistance will eventually develop against every inhibitor of a single RTK. It became clear that all pathways previously thought to be linear are in fact highly interconnected into a complex and dynamic signaling network, with RTKs functioning as key regulatory nodes. However, knowing the components of a signaling pathway or network is not sufficient, due to the presence of positive and negative feedback mechanisms. In recent years, the upregulation of RTKs by feedback mechanisms, following the inhibition of selective kinases has received much attention. We discuss several examples where RTKs play a role in acquired resistance to targeted therapies in the following paragraph.
As discussed, the clinical activity of the BRAFV600E‐specific inhibitor in BRAF‐mutated melanoma is well‐established. In sharp contrast to this is the observation that inhibition of BRAF in BRAF V600E‐mutant colorectal cancer (CRC) did not result in clinical benefit. It was found that this lack of response to BRAF inhibition in colon cancer is caused by the feedback activation of EGFR upon BRAF inhibition, suggesting a drug combination strategy to make colon cancer cells BRAF‐inhibitor responsive (Corcoran et al., 2012; Prahallad et al., 2012). RTK upregulation also plays a role in the acquired resistance of melanomas to BRAFV600E‐inhibition (Girotti et al., 2013; Nazarian et al., 2010; Sun et al., 2014b; Villanueva et al., 2010). Induction of PDGFRβ emerged as a dominant feature of acquired resistance in a subset of melanoma cell lines, patient‐derived biopsies and short‐term cultures (Nazarian et al., 2010). Furthermore, EGFR or SRC family kinase signaling activation was observed after resistance to vemurafenib in melanoma patients (Girotti et al., 2013; Sun et al., 2014b). The third malignancy with a high frequency of BRAF‐mutations is thyroid cancer. Thyroid cancers, like colorectal cancers, are also relatively resistant to BRAF‐inhibitors. It was reported that the majority of BRAF‐mutant thyroid cancer cell lines are insensitive due to a feedback mechanism that induces HER3 transcription (Montero‐Conde et al., 2013). HER3 activation was part of a generalized hyperactivation of RTKs, but HER3 together with HER2 rapidly reactivates the MAPK signaling.
Another node in the MAPK pathway, MEK, has been explored as a target in triple‐negative breast cancer (TNBC). The inhibition of MEK with a specific inhibitor in TNBC cell lines and in genetically engineered mouse models (GEMMs) resulted in rapid reprogramming of the kinome through induced expression and activation of multiple RTKs that bypassed the initial ERK inhibition (Duncan et al., 2012). The inhibition of MEK caused a rapid loss of ERK activity, resulting in rapid MYC degradation, which in turn induced expression and activation of several RTKs. Apart from inhibition of the MAPK pathway, inhibition of the PI3K‐AKT pathway is also associated with activated RTK signaling. Inhibition of AKT induces a conserved set of RTKs, including HER3, IGF1R and insulin receptor (Chandarlapaty et al., 2011). This activation is partially due to mTORC1 inhibition (O'Reilly et al., 2006) and partially due to a FOXO‐dependent activation of receptor expression.
Using a kinome‐centered synthetic lethality screen, we found in our laboratory that suppression of HER3 sensitizes KRAS mutant lung and colon cancer cells to MEK inhibitors (Sun et al., 2014a). It was found that MEK inhibition results in MYC‐dependent transcriptional upregulation of HER3, which is responsible for intrinsic drug resistance. Drugs targeting both EGFR and HER2, each capable of forming heterodimers with HER3, can reverse unresponsiveness to MEK inhibition by decreasing inhibitory phosphorylation of the pro‐apoptotic BCL2‐associated proteins BAD and BIM (Sun et al., 2014a).
The now almost classic example of a reactivated pathway by second mutation resulting from downstream pathway activation is the emergence of KRAS alterations in KRAS wild‐type colorectal cancers treated with the EGFR monoclonal antibody cetuximab or panitumumab. Several studies have identified somatic mutations in KRAS, a downstream effector of EGFR, as a biomarker of intrinsic resistance to anti‐EGFR therapy in CRC (Amado et al., 2008; Karapetis et al., 2008). It was recently shown that a proportion of CRC patients with an initial response to anti‐EGFR therapy, developed alterations (e.g. mutation or focal amplification) in the KRAS gene at the time of progression (Diaz et al., 2012; Misale et al., 2012; Valtorta et al., 2013). Importantly, these mutations can be detected non‐invasively in the blood of patients, several months before radiographic detection of disease progression. Mathematical modeling indicated that the mutations were present in expanded subclones before the start of anti‐EGFR therapy (Diaz et al., 2012) although new mutations can also arise (Misale et al., 2012). Downstream secondary mutations were also found as a resistance mechanism to MEK and BRAF inhibition. Mutations in MEK1 or MEK2 were found in tumor samples after disease progression (Emery et al., 2009; Trunzer et al., 2013; Van Allen et al., 2014; Wagle et al., 2011).
The anti‐HER2 monoclonal antibody trastuzumab, known in the clinic as herceptin®, is used in breast cancers overexpressing the RTK HER2. HER2 is overexpressed by either focal amplification or, in a small percentage, by mutation (Bose et al., 2013). Using RNA interference as a screening tool for drug resistance, it was found that loss of PTEN conferred resistance to trastuzumab and that activation of the PI3K pathway is predictive for response to trastuzumab in clinical samples (Berns et al., 2007; Nagata et al., 2004). There is also clinical evidence that women with HER2‐positive breast cancer who also carry activating mutations in PIK3CA are less likely to benefit from neo‐adjuvant HER2‐targeted therapies than those without a PIK3CA mutation (Majewski et al., 2014).
It has been shown that tumor regressions with vemurafenib treatment in BRAF‐mutant metastatic melanoma patients are dependent on the MAPK pathway blockade (Bollag et al., 2010). An inhibition of the pathway below a certain threshold was insufficient for tumor regression. Modulating the signaling output of the inhibited pathway around the threshold required for tumor inhibition can have major consequences for tumor response. In spite of the high response rates of BRAF‐mutant melanomas to vemurafenib and the improvement in overall survival, all patients eventually develop disease progression. Based on studies investigating resistance mechanisms to vemurafenib in BRAF V600E‐mutated melanoma, the mitogen‐activated protein kinase (MAPK) pathway is frequently reactivated in relapsed tumors (Shi et al., 2014). Melanomas can acquire resistance to the BRAFV600E‐inhibitor by expressing high levels of activated RAS resulting from NRAS or KRAS mutations. High levels of activated RAS upstream of RAF lead to subsequent MAPK pathway reactivation (Nazarian et al., 2010; Shi et al., 2014; Trunzer et al., 2013). Another example of this phenomenon is the identification of COT, also known as MAP3K8, as a MAPK pathway agonist. This was found through expression of all kinase and kinase‐related open reading frames (ORFs) in cells that are sensitive to the BRAF‐inhibitor (Johannessen et al., 2010). COT activates ERK through a MEK‐dependent mechanism that does not require RAF signaling. Indeed, high COT expression is associated with acquired resistance in patients that relapsed following treatment with MEK or RAF inhibitors (Johannessen et al., 2010).
3.3. Bypass mechanisms
Pathway reactivation is a common and recurrent mechanism of resistance to RTK‐inhibitors, MAPK pathway inhibitors and PI3K‐AKT pathway inhibitors. However, activation of parallel pathways to bypass pathway inhibition also contributes to intrinsic and acquired resistance. Whole‐exome sequencing identified upregulation of PI3K‐AKT signaling as a second core resistance mechanism to BRAF inhibition (Shi et al., 2014). Mutations in the PI3K‐PTEN‐AKT pathway were present in 22% of the cases with acquired resistance to BRAF‐inhibition, in most cases overlapping with alterations in the MAPK pathway.
Engelman et al. have shown that amplification of MET occurred in 22% of lung cancer samples from patients that developed resistance to gefitinib or erlotinib (Engelman et al., 2007). Furthermore, it was shown that subpopulations of cells with MET amplification were present prior to drug exposure (Turke et al., 2010). Although the frequency might be lower than the initially reported 22% (Sequist et al., 2011), amplification of MET is driving HER3‐dependent activation of the PI3K‐AKT pathway. This acquired activation of the PI3K‐AKT pathway during EGFR‐TKI is in other cases caused by PIK3CA mutation (Sequist et al., 2011). Takezawa et al. showed in vitro and in vivo the importance of HER2 in mediating the sensitivity of EGFR‐mutant tumor cells to EGFR‐TKIs (Takezawa et al., 2012). HER2 was amplified in 12% of tumors with acquired resistance and mutually exclusive with T790M mutation (Takezawa et al., 2012).
Amplification of the MET receptor is also associated with acquired resistance in CRC patients treated with anti‐EGFR monoclonal antibodies that do not develop KRAS mutations (Bardelli et al., 2013). The prevalence of MET amplification was only 1% in unselected metastatic CRC samples, but increased to 12.5% in cetuximab‐resistant, genetically pre‐selected xenopatients, indicating that MET amplification characterizes a significant fraction of cetuximab‐resistant cases that are wild‐type for KRAS, BRAF, NRAS, PIK3CA and HER2. In an unselected cohort, the clinical validation of MET amplification as a biomarker of resistance to EGFR therapy in metastatic CRC requires large (retrospective) studies.
By expressing 15,906 human ORFs to identify genes whose upregulation conferred resistance to MAPK‐pathway inhibition, it was found that, beside genes activating ERK signaling, cAMP and cAMP‐dependent protein kinase (PKA) activation could confer resistance to MAPK‐pathway inhibition in melanoma cells (Johannessen et al., 2013). PKA, once activated, can phosphorylate several substrates and modulate the activity of several transcription factors. Phosphorylation of two transcription factors downstream of cAMP and PKA, CREB and ATF1, was measured in tumor biopsies before or during treatment and after relapse with the BRAF‐inhibitor or the BRAF‐MEK‐inhibitor combination. Levels of CREB and ATF1 phosphorylation were suppressed after treatment with the BRAF‐inhibitor or the BRAF‐MEK‐inhibitor combination. In contrast, the levels of CREB and ATF1 phosphorylation in patients with a relapse were equal to the levels of phosphorylation in the pre‐treatment cohort.
There are many studies on the role of tumor microenvironment in tumor growth and metastasis, but only a few on a possible role in drug resistance. Nearly all studies on drug resistance have focused on cell‐autonomous mechanisms of drug resistance, disregarding the effects of the tumor microenvironment. Technical challenges to properly model the microenvironment in vitro and in vivo is a major contributor of this publication bias. A co‐culture model of tumor cells with stromal cells showed that the effect of targeted cancer drugs on tumor cells is greatly diminished when the tumor cells are cultured in the presence of stromal cells (Straussman et al., 2012). This stroma‐mediated resistance mechanism was more striking for targeted therapies compared with cytotoxic chemotherapy. They further zoomed in on the role of stroma‐mediated BRAF‐inhibitor resistance and found that stromal secretion of hepatocyte growth factor (HGF) resulted in activation of MET receptor and thereby reactivation of the MAKP and PI3K‐AKT pathway, causing BRAF‐inhibitor resistance (Straussman et al., 2012).
The evidence that TGFβ‐signaling plays a role in resistance against targeted and conventional cancer therapies further supports the importance of indirect mechanisms of drug resistance. TGFβ‐signaling is an important player in the epithelial‐to‐mesenchymal transition (EMT), but also regulates the immune response of the extracellular matrix and activates direct or indirect multiple signaling pathways (Brunen et al., 2013). Activation of TGFβ‐signaling was found sufficient to induce TKI resistance in multiple cancer types (Huang et al., 2012). Although the mechanism is still poorly understood, the process of EMT and the EMT‐phenotype both have been linked several times to drug sensitivity and drug resistance (Byers et al., 2013; Fuchs et al., 2008; Salt et al., 2014; Thomson et al., 2005; Yauch et al., 2005).
4. What we have learned from drug resistance and how to deal with it
An understanding of the recurrent patterns of resistance, as discussed in this review, is necessary to design strategies to overcome intrinsic resistance and delay acquired resistance to targeted cancer drugs. To address this, significant efforts are now underway to (1) develop better drugs or more effective drug combinations to fully suppress the oncogenic signaling pathways and (2) development of non‐invasive assays to monitor drug response and the emergence of drug resistance (Figure 2).
Figure 2.
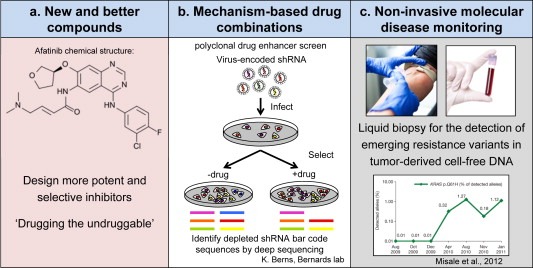
Strategies to overcome intrinsic resistance and delay acquired resistance. (a) The design of more selective and potent inhibitors, also for what is currently ‘undruggable’. (b) The development of more effective drug combinations, e.g. by investigating mechanisms of drug resistance in the clinic or by in vitro and in vivo synthetic lethality screens. (c) Repeated non‐invasive detection of emerging resistance variants, for example in tumor‐derived cell‐free DNA (cfDNA), to dynamically adapt the combination strategy.
4.1. Developing better drugs and effective drug combinations
The continuous effort to design more selective and potent inhibitors will definitely result in increased response rates to these inhibitors (Figure 2(a)). An example is the development of inhibitors that can inhibit the ‘gatekeeper’ mutations that are responsible for a large part of the resistance to some TKIs. Patients with EGFR‐mutant NSCLC who have acquired resistance through the T790M mutation (‘gatekeeper’ mutation) have few treatment options. Second‐generation EGFR inhibitors have been developed that also inhibit T790M‐mutant EGFR. One of these second‐generation EGFR inhibitors, afatinib, was FDA‐approved last year as first‐line treatment for EGFR‐mutated NSCLC. However, in contrast to previous in vitro and in vivo studies with second‐generation EGFR TKIs, Kim et al. reported the T790M mutation as a resistance mechanism for afatinib as well (Kim et al., 2012). This might be related to the nondiscriminatory action of this compound toward wild‐type and mutant EGFR. A mutant‐selective irreversible inhibitor might be more appropriate to overcome this problem. However, it should be noted that the emergence of the T790M mutation might take longer with second‐generation inhibitors compared to first‐generation EGFR‐TKIs, like erlotinib and gefitinib. Fortunately, new drugs have been designed that inhibit T790M‐mutant EGFR. Several of these inhibitors have been described (Zhou et al., 2009), and one of them is currently being evaluated in phase 1 and 2 clinical trials in EGFR‐mutant NSCLC after showing promising results in in vitro and in vivo validations (Walter et al., 2013). An important note to add here is that the greater potency of targeted therapies may result in increased heterogeneity of resistance mechanisms to these therapies.
The L1196M ‘gatekeeper’ mutation in ALK, conferring resistance to the ALK‐inhibitor crizotinib, can be inhibited by a newly designed potent, selective, and orally available ALK‐inhibitor (Sakamoto et al., 2011). Similarly, a compound was designed that targets the T315I mutation in ABL (O'Hare et al., 2009). Based on results of the phase 2 trial, this drug named ponatinib was FDA‐approved in 2012 for patients with resistant, BCR‐ABL positive CML. However, on 9 October 2013, the FDA issued a partial clinical hold on new trial enrollment for ponatinib due to an increased number of blood clots observed in patients taking the drug in the phase 3 trial (EPIC trial). This trial was discontinued shortly thereafter.
What we have learned from the identified resistance mechanisms? An outstanding lesson is that drug resistance to targeted single‐agent therapies is frequently driven by pathway reactivation, suggesting that a major limitation of targeted agents lies in their inability to fully block the pathway of interest. We therefore need to identify and target points of coalescence: nodes onto which multiple independent resistance mechanisms and tumor dependencies may arise. Furthermore, this points to the need for more potent and mechanism‐based combination therapies (Figure 2(b)). A recent example is the design of clinical trials with the dual combination of BRAF‐inhibitors and EGFR inhibition, or a triple combination with adding PI3K‐ or MEK‐inhibition in BRAF‐mutant colorectal cancer (ClinicalTrials.gov identifiers NCT01719380, NCT01750918 and NCT01791309).
Another example is the combination of the BRAF‐inhibitor with a MEK‐inhibitor in patients with metastatic BRAF‐mutant melanoma, after the identification of MAPK pathway reactivation as a frequent resistance mechanism to monotherapy with the BRAF‐inhibitor. Complete inhibition of the MAPK pathway could potentially be achieved by combining the BRAF‐inhibitor with a MEK (MAP2K)‐inhibitor. The clinical trial with the combination of the BRAFV600E‐inhibitor dabrafenib and the MEK‐inhibitor trametinib reported an improved median progression‐free survival of the combination by almost 4 months compared to dabrafenib monotherapy (Flaherty et al., 2012). Toxicity of combinations with targeted therapies has always been regarded as the limiting factor for this approach. However, the study by Flaherty et al. showed that dabrafenib and trametinib could be safely combined at full monotherapy doses (Flaherty et al., 2012). In fact, cutaneous squamous‐cell carcinoma was seen in only 7% of patients receiving the combination, in comparison to 19% of patients receiving BRAF monotherapy. The development of new generation targeted therapies with alternative dosing strategies to overcome toxicities will allow previously toxic combinations to be developed.
Although the progression‐free survival of patients with BRAF‐mutant melanoma increased by almost 4 months using the combination the BRAF‐inhibitor with the MEK‐inhibitor, resistance still developed in most patients. In a follow‐up report of this trial, the authors investigated the resistance mechanisms for 5 patients that experienced clinical benefit before developing progressive disease (Wagle et al., 2014). Despite the dual inhibition of the MAPK pathway, they surprisingly identified in 3 out of 5 cases a resistance mechanism that engages MAPK‐effectors: in one patient an acquired MEK2‐mutation was found, in another patient the previously reported BRAF‐splice variant and the third patient a focal BRAF amplification. The prevalence of MAPK pathway alterations in these resistant tumors underlines the reliance of BRAF‐mutant melanomas on MEK/ERK signaling and the incomplete inhibition of the pathway despite using two drugs hitting this pathway. More potent MEK inhibition by increasing the dose of the MEK‐inhibitor or, in the future, ERK‐inhibitors, may provide a solution for overcoming resistance to the combination.
Several combinations with endocrine therapy in breast cancer are being tested. As discussed, the combination of the mTOR inhibitor everolimus with tamoxifen or an aromatase inhibitor showed significant clinical benefit for advanced or metastatic ERα‐positive breast cancer. Furthermore, dual targeting of HER2‐positive tumors with trastuzumab (HER2 monoclonal antibody) and lapatinib (EGFR/HER2‐TKI) was undertaken because of the synergistic interaction between the two compounds in preclinical models (Scaltriti et al., 2009; Xia et al., 2005). In the NeoALTTO study, the use of lapatinib, trastuzumab, and their combination was assessed as neo‐adjuvant therapy for women with HER2‐positive early breast cancer (Baselga et al., 2012a). The pathological complete response (pCR) rate was significantly higher in the group treated with the combination than in the group treated with trastuzumab alone (difference 21.1%, 9.1–34.2, p = 0.0001). Furthermore, the combination of pertuzumab (an anti‐HER2 humanized monoclonal antibody that inhibits receptor dimerization) plus trastuzumab plus docetaxel in patients with HER2‐positive metastatic breast cancer significantly prolonged progression‐free survival and overall survival as compared to placebo plus trastuzumab plus docetaxel (Baselga et al., 2012c; Swain et al., 2013).
Finally, combination of the CDK4/6 inhibitor palbociclib with the aromatase inhibitor letrozole demonstrated clinical benefit in advanced ERα‐positive breast cancer. The combination improved progression‐free survival in a phase 2 trial from 7.5 months for patients treated with letrozole monotherapy to 26.1 months for those in the combination arm and was well tolerated (Finn et al., 2012). These positive results are currently validated in a large phase 3 clinical trial (ClinicalTrials.gov identifier NCT01740427).
4.2. Future developments
Although effective drug combinations are indispensable to deal upfront with resistance, the extensive tumor heterogeneity in some tumors is likely to limit the durability of responses to these combinations. In fact, as mentioned above, even resistance to combination therapies was already observed in the clinic. Mathematical modeling showed that the presence of a single mutation conferring cross‐resistance to each of the two drugs will not lead to sustained improvement for the majority of patients (Bozic et al., 2013). Sequential non‐invasive detection of emerging resistance variants will be essential to dynamically adapt the combination strategy (Figure 2(c)). A potential source to identify these variants is circulating, cell‐free DNA (cfDNA) in the blood, reviewed in (Crowley et al., 2013). This was used to identify the emergence of KRAS mutations or MET amplifications during treatment of CRC patients with anti‐EGFR therapy (Bardelli et al., 2013; Diaz et al., 2012; Misale et al., 2012). Currently, the sensitivity of these assays is around 0.1–1% variant frequencies, which currently limits the utility to finding hotspot mutations in predefined genes. Future improvements in sequencing accuracy will undoubtedly increase the sensitivity of cfDNA analysis. With the continuous drop in sequencing cost, it may soon become feasible to monitor apparently healthy individuals for the presence of occult cancer. When this becomes reality, stage IV metastatic disease may become rare, with associated increase in cancer survival.
Until now, clinicians could often only wait until drug resistance develops and then try, often without solid scientific rationale a second line therapy. These sequential therapies generally form a perfect recipe for certain treatment failure. However, we are now entering an era in which we should be able to anticipate the next move of the cancer cell, due to increasing understanding of the resistance mechanisms, and develop rational combination therapies. Moreover, even if we fail to predict the next move of the cancer, we will be able to monitor its progression from early stages on using non‐invasive diagnostic approaches. By analogy, to win a game of chess, it is imperative to predict the next likely move of the opponent. We are now entering a new era of cancer therapy in which we will keep getting better in predicting and understanding the next move of the cancer to evade therapy. As a result, our chances of winning should increase proportionally. However, as Yogi Berra said: “It is tough to make predictions, especially about the future”.
Financial support
The work in our laboratory is supported by grants from the European Research Council (grant no. 250043) (ERC), the EU FP7 Programs COLTHERES (grant no. 259015) and RATHER (grant no. 258967), the Netherlands Organization for Scientific Research (NWO) and the Dutch Cancer Society (KWF).
Groenendijk Floris H., Bernards René, (2014), Drug resistance to targeted therapies: Déjà vu all over again, Molecular Oncolog. 8, doi: 10.1016/j.molonc.2014.05.004.
References
- Albertson, D.G. , 2012. ESR1 amplification in breast cancer: controversy resolved?. J. Pathol. 227, 1–3. [DOI] [PubMed] [Google Scholar]
- Amado, R.G. , Wolf, M. , Peeters, M. , Van Cutsem, E. , Siena, S. , Freeman, D.J. , Juan, T. , Sikorski, R. , Suggs, S. , Radinsky, R. , Patterson, S.D. , Chang, D.D. , 2008. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 1626–1634. [DOI] [PubMed] [Google Scholar]
- Arpino, G. , Green, S.J. , Allred, D.C. , Lew, D. , Martino, S. , Osborne, C.K. , Elledge, R.M. , 2004. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin. Cancer Res. 10, 5670–5676. [DOI] [PubMed] [Google Scholar]
- Awad, M.M. , Katayama, R. , McTigue, M. , Liu, W. , Deng, Y.L. , Brooun, A. , Friboulet, L. , Huang, D. , Falk, M.D. , Timofeevski, S. , Wilner, K.D. , Lockerman, E.L. , Khan, T.M. , Mahmood, S. , Gainor, J.F. , Digumarthy, S.R. , Stone, J.R. , Mino-Kenudson, M. , Christensen, J.G. , Iafrate, A.J. , Engelman, J.A. , Shaw, A.T. , 2013. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N. Engl. J. Med. 368, 2395–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelot, T. , Bourgier, C. , Cropet, C. , Ray-Coquard, I. , Ferrero, J.M. , Freyer, G. , Abadie-Lacourtoisie, S. , Eymard, J.C. , Debled, M. , Spaeth, D. , Legouffe, E. , Allouache, D. , El Kouri, C. , Pujade-Lauraine, E. , 2012. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J. Clin. Oncol. 30, 2718–2724. [DOI] [PubMed] [Google Scholar]
- Bardelli, A. , Corso, S. , Bertotti, A. , Hobor, S. , Valtorta, E. , Siravegna, G. , Sartore-Bianchi, A. , Scala, E. , Cassingena, A. , Zecchin, D. , Apicella, M. , Migliardi, G. , Galimi, F. , Lauricella, C. , Zanon, C. , Perera, T. , Veronese, S. , Corti, G. , Amatu, A. , Gambacorta, M. , Diaz, L.A. , Sausen, M. , Velculescu, V.E. , Comoglio, P. , Trusolino, L. , Di Nicolantonio, F. , Giordano, S. , Siena, S. , 2013. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 3, 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga, J. , Bradbury, I. , Eidtmann, H. , Di Cosimo, S. , de Azambuja, E. , Aura, C. , Gomez, H. , Dinh, P. , Fauria, K. , Van Dooren, V. , Aktan, G. , Goldhirsch, A. , Chang, T.W. , Horvath, Z. , Coccia-Portugal, M. , Domont, J. , Tseng, L.M. , Kunz, G. , Sohn, J.H. , Semiglazov, V. , Lerzo, G. , Palacova, M. , Probachai, V. , Pusztai, L. , Untch, M. , Gelber, R.D. , Piccart-Gebhart, M. , Neo, A.S.T. , 2012. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lance. 379, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga, J. , Campone, M. , Piccart, M. , Burris, H.A. , Rugo, H.S. , Sahmoud, T. , Noguchi, S. , Gnant, M. , Pritchard, K.I. , Lebrun, F. , Beck, J.T. , Ito, Y. , Yardley, D. , Deleu, I. , Perez, A. , Bachelot, T. , Vittori, L. , Xu, Z. , Mukhopadhyay, P. , Lebwohl, D. , Hortobagyi, G.N. , 2012. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 366, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga, J. , Cortes, J. , Kim, S.B. , Im, S.A. , Hegg, R. , Im, Y.H. , Roman, L. , Pedrini, J.L. , Pienkowski, T. , Knott, A. , Clark, E. , Benyunes, M.C. , Ross, G. , Swain, S.M. , 2012. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 366, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelen, K. , Zwart, W. , Linn, S.C. , 2012. Can predictive biomarkers in breast cancer guide adjuvant endocrine therapy?. Nat. Rev. Clin. Oncol. 9, 529–541. [DOI] [PubMed] [Google Scholar]
- Berns, K. , Horlings, H.M. , Hennessy, B.T. , Madiredjo, M. , Hijmans, E.M. , Beelen, K. , Linn, S.C. , Gonzalez-Angulo, A.M. , Stemke-Hale, K. , Hauptmann, M. , Beijersbergen, R.L. , Mills, G.B. , van de Vijver, M.J. , Bernards, R. , 2007. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cel. 12, 395–402. [DOI] [PubMed] [Google Scholar]
- Berns, K. , Bernards, R. , 2013. Understanding resistance to targeted cancer drugs through loss of function genetic screens. Drug Resist Updat. 15, 268–275. [DOI] [PubMed] [Google Scholar]
- Berra, Y. , 1998. The Yogi Book: “I Really Didn't Say Everything I Said!” Workman Publishing Company, Inc; [Google Scholar]
- Bollag, G. , Hirth, P. , Tsai, J. , Zhang, J. , Ibrahim, P.N. , Cho, H. , Spevak, W. , Zhang, C. , Zhang, Y. , Habets, G. , Burton, E.A. , Wong, B. , Tsang, G. , West, B.L. , Powell, B. , Shellooe, R. , Marimuthu, A. , Nguyen, H. , Zhang, K.Y. , Artis, D.R. , Schlessinger, J. , Su, F. , Higgins, B. , Iyer, R. , D'Andrea, K. , Koehler, A. , Stumm, M. , Lin, P.S. , Lee, R.J. , Grippo, J. , Puzanov, I. , Kim, K.B. , Ribas, A. , McArthur, G.A. , Sosman, J.A. , Chapman, P.B. , Flaherty, K.T. , Xu, X. , Nathanson, K.L. , Nolop, K. , 2010. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Natur. 467, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, R. , Kavuri, S.M. , Searleman, A.C. , Shen, W. , Shen, D. , Koboldt, D.C. , Monsey, J. , Goel, N. , Aronson, A.B. , Li, S. , Ma, C.X. , Ding, L. , Mardis, E.R. , Ellis, M.J. , 2013. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 3, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozic, I. , Reiter, J.G. , Allen, B. , Antal, T. , Chatterjee, K. , Shah, P. , Moon, Y.S. , Yaqubie, A. , Kelly, N. , Le, D.T. , Lipson, E.J. , Chapman, P.B. , Diaz, L.A. , Vogelstein, B. , Nowak, M.A. , 2013. Evolutionary dynamics of cancer in response to targeted combination therapy. Elif. 2, e00747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman, R.W. , 1963. Mechanisms of resistance to anticancer agents. Adv. Cancer Res. 7, 129–234. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T.R. , Bernards, R. , Agami, R. , 2002. A system for stable expression of short interfering RNAs in mammalian cells. Scienc. 296, 550–553. [DOI] [PubMed] [Google Scholar]
- Brunen, D. , Willems, S.M. , Kellner, U. , Midgley, R. , Simon, I. , Bernards, R. , 2013. TGF-beta: an emerging player in drug resistance. Cell Cycl. 12, 2960–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers, L.A. , Diao, L. , Wang, J. , Saintigny, P. , Girard, L. , Peyton, M. , Shen, L. , Fan, Y. , Giri, U. , Tumula, P.K. , Nilsson, M.B. , Gudikote, J. , Tran, H. , Cardnell, R.J. , Bearss, D.J. , Warner, S.L. , Foulks, J.M. , Kanner, S.B. , Gandhi, V. , Krett, N. , Rosen, S.T. , Kim, E.S. , Herbst, R.S. , Blumenschein, G.R. , Lee, J.J. , Lippman, S.M. , Ang, K.K. , Mills, G.B. , Hong, W.K. , Weinstein, J.N. , Wistuba, Coombes, K.R. , Minna, J.D. , Heymach, J.V. , 2013. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network, 2012. Comprehensive molecular portraits of human breast tumours. Natur. 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty, S. , Sawai, A. , Scaltriti, M. , Rodrik-Outmezguine, V. , Grbovic-Huezo, O. , Serra, V. , Majumder, P.K. , Baselga, J. , Rosen, N. , 2011. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cel. 19, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, P.B. , Hauschild, A. , Robert, C. , Haanen, J.B. , Ascierto, P. , Larkin, J. , Dummer, R. , Garbe, C. , Testori, A. , Maio, M. , Hogg, D. , Lorigan, P. , Lebbe, C. , Jouary, T. , Schadendorf, D. , Ribas, A. , O'Day, S.J. , Sosman, J.A. , Kirkwood, J.M. , Eggermont, A.M. , Dreno, B. , Nolop, K. , Li, J. , Nelson, B. , Hou, J. , Lee, R.J. , Flaherty, K.T. , McArthur, G.A. , 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y.L. , Soda, M. , Yamashita, Y. , Ueno, T. , Takashima, J. , Nakajima, T. , Yatabe, Y. , Takeuchi, K. , Hamada, T. , Haruta, H. , Ishikawa, Y. , Kimura, H. , Mitsudomi, T. , Tanio, Y. , Mano, H. , 2010. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 363, 1734–1739. [DOI] [PubMed] [Google Scholar]
- Corcoran, R.B. , Dias-Santagata, D. , Bergethon, K. , Iafrate, A.J. , Settleman, J. , Engelman, J.A. , 2010. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci. Signal. 3, ra84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran, R.B. , Ebi, H. , Turke, A.B. , Coffee, E.M. , Nishino, M. , Cogdill, A.P. , Brown, R.D. , Della Pelle, P. , Dias-Santagata, D. , Hung, K.E. , Flaherty, K.T. , Piris, A. , Wargo, J.A. , Settleman, J. , Mino-Kenudson, M. , Engelman, J.A. , 2012. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley, E. , Di Nicolantonio, F. , Loupakis, F. , Bardelli, A. , 2013. Liquid biopsy: monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 10, 472–484. [DOI] [PubMed] [Google Scholar]
- Das Thakur, M. , Salangsang, F. , Landman, A.S. , Sellers, W.R. , Pryer, N.K. , Levesque, M.P. , Dummer, R. , McMahon, M. , Stuart, D.D. , 2013. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Natur. 494, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, C. , Godwin, J. , Gray, R. , Clarke, M. , Cutter, D. , Darby, S. , McGale, P. , Pan, H.C. , Taylor, C. , Wang, Y.C. , Dowsett, M. , Ingle, J. , Peto, R. , 2011. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lance. 378, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, K.D. , Le, A.T. , Theodoro, M.F. , Skokan, M.C. , Aisner, D.L. , Berge, E.M. , Terracciano, L.M. , Cappuzzo, F. , Incarbone, M. , Roncalli, M. , Alloisio, M. , Santoro, A. , Camidge, D.R. , Varella-Garcia, M. , Doebele, R.C. , 2012. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 18, 4570–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Laurentiis, M. , Arpino, G. , Massarelli, E. , Ruggiero, A. , Carlomagno, C. , Ciardiello, F. , Tortora, G. , D'Agostino, D. , Caputo, F. , Cancello, G. , Montagna, E. , Malorni, L. , Zinno, L. , Lauria, R. , Bianco, A.R. , De Placido, S. , 2005. A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clin. Cancer Res. 11, 4741–4748. [DOI] [PubMed] [Google Scholar]
- Diaz, L.A. , Williams, R.T. , Wu, J. , Kinde, I. , Hecht, J.R. , Berlin, J. , Allen, B. , Bozic, I. , Reiter, J.G. , Nowak, M.A. , Kinzler, K.W. , Oliner, K.S. , Vogelstein, B. , 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Natur. 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, J.S. , Whittle, M.C. , Nakamura, K. , Abell, A.N. , Midland, A.A. , Zawistowski, J.S. , Johnson, N.L. , Granger, D.A. , Jordan, N.V. , Darr, D.B. , Usary, J. , Kuan, P.F. , Smalley, D.M. , Major, B. , He, X. , Hoadley, K.A. , Zhou, B. , Sharpless, N.E. , Perou, C.M. , Kim, W.Y. , Gomez, S.M. , Chen, X. , Jin, J. , Frye, S.V. , Earp, H.S. , Graves, L.M. , Johnson, G.L. , 2012. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cel. 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EBCTCG, 2005. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lance. 365, 1687–1717. [DOI] [PubMed] [Google Scholar]
- Emery, C.M. , Vijayendran, K.G. , Zipser, M.C. , Sawyer, A.M. , Niu, L. , Kim, J.J. , Hatton, C. , Chopra, R. , Oberholzer, P.A. , Karpova, M.B. , MacConaill, L.E. , Zhang, J. , Gray, N.S. , Sellers, W.R. , Dummer, R. , Garraway, L.A. , 2009. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 20411–20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encarnacion, C.A. , Ciocca, D.R. , McGuire, W.L. , Clark, G.M. , Fuqua, S.A. , Osborne, C.K. , 1993. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res. Treat. 26, 237–246. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Janne, P.A. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Scienc. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Finn, R.S. , Crown, J.P. , Lang, I. , Boer, K. , Bondarenko, I.M. , Kulyk, S.O. , Ettl, J. , Patel, R. , Pinter, T. , Schmidt, M. , Shparyk, Y. , Thummala, A.R. , Voytko, N.L. , Breazna, A. , Kim, S.T. , Randolph, S. , Slamon, D.J. , 2012. Results of a randomized phase 2 study of PD 0332991, a cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2− advanced breast cancer (BC). Cancer Res. 72, (24 Suppl.) Abstract nr S1–S6 [Google Scholar]
- Flaherty, K.T. , Infante, J.R. , Daud, A. , Gonzalez, R. , Kefford, R.F. , Sosman, J. , Hamid, O. , Schuchter, L. , Cebon, J. , Ibrahim, N. , Kudchadkar, R. , Burris, H.A. , Falchook, G. , Algazi, A. , Lewis, K. , Long, G.V. , Puzanov, I. , Lebowitz, P. , Singh, A. , Little, S. , Sun, P. , Allred, A. , Ouellet, D. , Kim, K.B. , Patel, K. , Weber, J. , 2012. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, B.C. , Fujii, T. , Dorfman, J.D. , Goodwin, J.M. , Zhu, A.X. , Lanuti, M. , Tanabe, K.K. , 2008. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 68, 2391–2399. [DOI] [PubMed] [Google Scholar]
- Girotti, M.R. , Pedersen, M. , Sanchez-Laorden, B. , Viros, A. , Turajlic, S. , Niculescu-Duvaz, D. , Zambon, A. , Sinclair, J. , Hayes, A. , Gore, M. , Lorigan, P. , Springer, C. , Larkin, J. , Jorgensen, C. , Marais, R. , 2013. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 3, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz, M.P. , Rae, J.M. , Suman, V.J. , Safgren, S.L. , Ames, M.M. , Visscher, D.W. , Reynolds, C. , Couch, F.J. , Lingle, W.L. , Flockhart, D.A. , Desta, Z. , Perez, E.A. , Ingle, J.N. , 2005. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J. Clin. Oncol. 23, 9312–9318. [DOI] [PubMed] [Google Scholar]
- Gorre, M.E. , Mohammed, M. , Ellwood, K. , Hsu, N. , Paquette, R. , Rao, P.N. , Sawyers, C.L. , 2001. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Scienc. 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Groenendijk, F.H. , Zwart, W. , Floore, A. , Akbari, S. , Bernards, R. , 2013. Estrogen receptor splice variants as a potential source of false-positive estrogen receptor status in breast cancer diagnostics. Breast Cancer Res. Treat. 140, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururaj, A.E. , Rayala, S.K. , Vadlamudi, R.K. , Kumar, R. , 2006. Novel mechanisms of resistance to endocrine therapy: genomic and nongenomic considerations. Clin. Cancer Res. 12, 1001s–1007s. [DOI] [PubMed] [Google Scholar]
- Gutierrez, M.C. , Detre, S. , Johnston, S. , Mohsin, S.K. , Shou, J. , Allred, D.C. , Schiff, R. , Osborne, C.K. , Dowsett, M. , 2005. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 23, 2469–2476. [DOI] [PubMed] [Google Scholar]
- Haughian, J.M. , Pinto, M.P. , Harrell, J.C. , Bliesner, B.S. , Joensuu, K.M. , Dye, W.W. , Sartorius, C.A. , Tan, A.C. , Heikkila, P. , Perou, C.M. , Horwitz, K.B. , 2012. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc. Natl. Acad. Sci. U.S.A. 109, 2742–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemeryck, A. , Belpaire, F.M. , 2002. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr. Drug Metab. 3, 13–37. [DOI] [PubMed] [Google Scholar]
- Herynk, M.H. , Fuqua, S.A. , 2004. Estrogen receptor mutations in human disease. Endocr. Rev. 25, 869–898. [DOI] [PubMed] [Google Scholar]
- Holm, C. , Kok, M. , Michalides, R. , Fles, R. , Koornstra, R.H. , Wesseling, J. , Hauptmann, M. , Neefjes, J. , Peterse, J.L. , Stal, O. , Landberg, G. , Linn, S.C. , 2009. Phosphorylation of the oestrogen receptor alpha at serine 305 and prediction of tamoxifen resistance in breast cancer. J. Pathol. 217, 372–379. [DOI] [PubMed] [Google Scholar]
- Hoskins, J.M. , Carey, L.A. , McLeod, H.L. , 2009. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat. Rev. Cance. 9, 576–586. [DOI] [PubMed] [Google Scholar]
- Huang, S. , Holzel, M. , Knijnenburg, T. , Schlicker, A. , Roepman, P. , McDermott, U. , Garnett, M. , Grernrum, W. , Sun, C. , Prahallad, A. , Groenendijk, F.H. , Mittempergher, L. , Nijkamp, W. , Neefjes, J. , Salazar, R. , Ten Dijke, P. , Uramoto, H. , Tanaka, F. , Beijersbergen, R.L. , Wessels, L.F. , Bernards, R. , 2012. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cel. 151, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorns, E. , Turner, N.C. , Elliott, R. , Syed, N. , Garrone, O. , Gasco, M. , Tutt, A.N. , Crook, T. , Lord, C.J. , Ashworth, A. , 2008. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cel. 13, 91–104. [DOI] [PubMed] [Google Scholar]
- Jeselsohn, R. , Yelensky, R. , Buchwalter, G. , Frampton, G. , Meric-Bernstam, F. , Gonzalez-Angulo, A.M. , Ferrer-Lozano, J. , Perez-Fidalgo, J.A. , Cristofanilli, M. , Gomez, H. , Arteaga, C.L. , Giltnane, J. , Balko, J.M. , Cronin, M.T. , Jarosz, M. , Sun, J. , Hawryluk, M. , Lipson, D. , Otto, G. , Ross, J.S. , Dvir, A. , Soussan-Gutman, L. , Wolf, I. , Rubinek, T. , Gilmore, L. , Schnitt, S. , Come, S.E. , Pusztai, L. , Stephens, P. , Brown, M. , Miller, V.A. , 2014. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 20, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen, C.M. , Boehm, J.S. , Kim, S.Y. , Thomas, S.R. , Wardwell, L. , Johnson, L.A. , Emery, C.M. , Stransky, N. , Cogdill, A.P. , Barretina, J. , Caponigro, G. , Hieronymus, H. , Murray, R.R. , Salehi-Ashtiani, K. , Hill, D.E. , Vidal, M. , Zhao, J.J. , Yang, X. , Alkan, O. , Kim, S. , Harris, J.L. , Wilson, C.J. , Myer, V.E. , Finan, P.M. , Root, D.E. , Roberts, T.M. , Golub, T. , Flaherty, K.T. , Dummer, R. , Weber, B.L. , Sellers, W.R. , Schlegel, R. , Wargo, J.A. , Hahn, W.C. , Garraway, L.A. , 2010. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Natur. 468, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen, C.M. , Johnson, L.A. , Piccioni, F. , Townes, A. , Frederick, D.T. , Donahue, M.K. , Narayan, R. , Flaherty, K.T. , Wargo, J.A. , Root, D.E. , Garraway, L.A. , 2013. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Natur. 504, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, S.R. , Saccani-Jotti, G. , Smith, I.E. , Salter, J. , Newby, J. , Coppen, M. , Ebbs, S.R. , Dowsett, M. , 1995. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 55, 3331–3338. [PubMed] [Google Scholar]
- Karapetis, C.S. , Khambata-Ford, S. , Jonker, D.J. , O'Callaghan, C.J. , Tu, D. , Tebbutt, N.C. , Simes, R.J. , Chalchal, H. , Shapiro, J.D. , Robitaille, S. , Price, T.J. , Shepherd, L. , Au, H.J. , Langer, C. , Moore, M.J. , Zalcberg, J.R. , 2008. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765. [DOI] [PubMed] [Google Scholar]
- Katayama, R. , Shaw, A.T. , Khan, T.M. , Mino-Kenudson, M. , Solomon, B.J. , Halmos, B. , Jessop, N.A. , Wain, J.C. , Yeo, A.T. , Benes, C. , Drew, L. , Saeh, J.C. , Crosby, K. , Sequist, L.V. , Iafrate, A.J. , Engelman, J.A. , 2012. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 4, 120ra117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Ko, J. , Cui, Z. , Abolhoda, A. , Ahn, J.S. , Ou, S.H. , Ahn, M.J. , Park, K. , 2012. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol. Cancer Ther. 11, 784–791. [DOI] [PubMed] [Google Scholar]
- Kobayashi, S. , Boggon, T.J. , Dayaram, T. , Janne, P.A. , Kocher, O. , Meyerson, M. , Johnson, B.E. , Eck, M.J. , Tenen, D.G. , Halmos, B. , 2005. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792. [DOI] [PubMed] [Google Scholar]
- Kuukasjarvi, T. , Kononen, J. , Helin, H. , Holli, K. , Isola, J. , 1996. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. 14, 2584–2589. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A. , Schlessinger, J. , 2010. Cell signaling by receptor tyrosine kinases. Cel. 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Shen, D. , Shao, J. , Crowder, R. , Liu, W. , Prat, A. , He, X. , Liu, S. , Hoog, J. , Lu, C. , Ding, L. , Griffith, O.L. , Miller, C. , Larson, D. , Fulton, R.S. , Harrison, M. , Mooney, T. , McMichael, J.F. , Luo, J. , Tao, Y. , Goncalves, R. , Schlosberg, C. , Hiken, J.F. , Saied, L. , Sanchez, C. , Giuntoli, T. , Bumb, C. , Cooper, C. , Kitchens, R.T. , Lin, A. , Phommaly, C. , Davies, S.R. , Zhang, J. , Kavuri, M.S. , McEachern, D. , Dong, Y.Y. , Ma, C. , Pluard, T. , Naughton, M. , Bose, R. , Suresh, R. , McDowell, R. , Michel, L. , Aft, R. , Gillanders, W. , DeSchryver, K. , Wilson, R.K. , Wang, S. , Mills, G.B. , Gonzalez-Angulo, A. , Edwards, J.R. , Maher, C. , Perou, C.M. , Mardis, E.R. , Ellis, M.J. , 2013. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, A.S. , Balmanno, K. , Sale, M.J. , Newman, S. , Dry, J.R. , Hampson, M. , Edwards, P.A. , Smith, P.D. , Cook, S.J. , 2011. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci. Signal. 4, ra17 [DOI] [PubMed] [Google Scholar]
- Long, G.V. , Menzies, A.M. , Nagrial, A.M. , Haydu, L.E. , Hamilton, A.L. , Mann, G.J. , Hughes, T.M. , Thompson, J.F. , Scolyer, R.A. , Kefford, R.F. , 2011. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 29, 1239–1246. [DOI] [PubMed] [Google Scholar]
- Maemondo, M. , Inoue, A. , Kobayashi, K. , Sugawara, S. , Oizumi, S. , Isobe, H. , Gemma, A. , Harada, M. , Yoshizawa, H. , Kinoshita, I. , Fujita, Y. , Okinaga, S. , Hirano, H. , Yoshimori, K. , Harada, T. , Ogura, T. , Ando, M. , Miyazawa, H. , Tanaka, T. , Saijo, Y. , Hagiwara, K. , Morita, S. , Nukiwa, T. , North-East Japan Study, G2010. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388. [DOI] [PubMed] [Google Scholar]
- Magnani, L. , Stoeck, A. , Zhang, X. , Lanczky, A. , Mirabella, A.C. , Wang, T.L. , Gyorffy, B. , Lupien, M. , 2013. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 110, E1490–E1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski, I.J., Nuciforo, P., Mittempergher, L., Bosma, A.J., Eidtmann, H., Holmes, E., Sotiriou, C., Fumagalli, D., Jimenez, J., Aura, C., Prudkin, L., Delgado, M.C.D., de la Pena, L., Loi, S., GSK TBDef, de Azambuja, E., Harbeck, N., Piccart-Gebhart, M., Bernards, R., Baselga, J., 2014. PIK3CA mutations are associated with resistance to neoadjuvant HER2-targeted therapies in breast cancer, submitted for publication.
- McGlynn, L.M. , Kirkegaard, T. , Edwards, J. , Tovey, S. , Cameron, D. , Twelves, C. , Bartlett, J.M. , Cooke, T.G. , 2009. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin. Cancer Res. 15, 1487–1495. [DOI] [PubMed] [Google Scholar]
- Michalides, R. , Griekspoor, A. , Balkenende, A. , Verwoerd, D. , Janssen, L. , Jalink, K. , Floore, A. , Velds, A. , van't Veer, L. , Neefjes, J. , 2004. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cel. 5, 597–605. [DOI] [PubMed] [Google Scholar]
- Michor, F. , Hughes, T.P. , Iwasa, Y. , Branford, S. , Shah, N.P. , Sawyers, C.L. , Nowak, M.A. , 2005. Dynamics of chronic myeloid leukaemia. Natur. 435, 1267–1270. [DOI] [PubMed] [Google Scholar]
- Miller, T.W. , Balko, J.M. , Arteaga, C.L. , 2011. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J. Clin. Oncol. 29, 4452–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, T.W. , Balko, J.M. , Ghazoui, Z. , Dunbier, A. , Anderson, H. , Dowsett, M. , Gonzalez-Angulo, A.M. , Mills, G.B. , Miller, W.R. , Wu, H. , Shyr, Y. , Arteaga, C.L. , 2011. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin. Cancer Res. 17, 2024–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale, S. , Yaeger, R. , Hobor, S. , Scala, E. , Janakiraman, M. , Liska, D. , Valtorta, E. , Schiavo, R. , Buscarino, M. , Siravegna, G. , Bencardino, K. , Cercek, A. , Chen, C.T. , Veronese, S. , Zanon, C. , Sartore-Bianchi, A. , Gambacorta, M. , Gallicchio, M. , Vakiani, E. , Boscaro, V. , Medico, E. , Weiser, M. , Siena, S. , Di Nicolantonio, F. , Solit, D. , Bardelli, A. , 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Natur. 486, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagut, C. , Dalmases, A. , Bellosillo, B. , Crespo, M. , Pairet, S. , Iglesias, M. , Salido, M. , Gallen, M. , Marsters, S. , Tsai, S.P. , Minoche, A. , Seshagiri, S. , Serrano, S. , Himmelbauer, H. , Bellmunt, J. , Rovira, A. , Settleman, J. , Bosch, F. , Albanell, J. , 2012. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 18, 221–223. [DOI] [PubMed] [Google Scholar]
- Montero-Conde, C. , Ruiz-Llorente, S. , Dominguez, J.M. , Knauf, J.A. , Viale, A. , Sherman, E.J. , Ryder, M. , Ghossein, R.A. , Rosen, N. , Fagin, J.A. , 2013. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 3, 520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove, E.A. , Sutherland, R.L. , 2009. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cance. 9, 631–643. [DOI] [PubMed] [Google Scholar]
- Nagata, Y. , Lan, K.H. , Zhou, X. , Tan, M. , Esteva, F.J. , Sahin, A.A. , Klos, K.S. , Li, P. , Monia, B.P. , Nguyen, N.T. , Hortobagyi, G.N. , Hung, M.C. , Yu, D. , 2004. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cel. 6, 117–127. [DOI] [PubMed] [Google Scholar]
- Nathanson, D.A. , Gini, B. , Mottahedeh, J. , Visnyei, K. , Koga, T. , Gomez, G. , Eskin, A. , Hwang, K. , Wang, J. , Masui, K. , Paucar, A. , Yang, H. , Ohashi, M. , Zhu, S. , Wykosky, J. , Reed, R. , Nelson, S.F. , Cloughesy, T.F. , James, C.D. , Rao, P.N. , Kornblum, H.I. , Heath, J.R. , Cavenee, W.K. , Furnari, F.B. , Mischel, P.S. , 2014. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Scienc. 343, 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian, R. , Shi, H. , Wang, Q. , Kong, X. , Koya, R.C. , Lee, H. , Chen, Z. , Lee, M.K. , Attar, N. , Sazegar, H. , Chodon, T. , Nelson, S.F. , McArthur, G. , Sosman, J.A. , Ribas, A. , Lo, R.S. , 2010. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Natur. 468, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hare, T. , Shakespeare, W.C. , Zhu, X. , Eide, C.A. , Rivera, V.M. , Wang, F. , Adrian, L.T. , Zhou, T. , Huang, W.S. , Xu, Q. , Metcalf, C.A. , Tyner, J.W. , Loriaux, M.M. , Corbin, A.S. , Wardwell, S. , Ning, Y. , Keats, J.A. , Wang, Y. , Sundaramoorthi, R. , Thomas, M. , Zhou, D. , Snodgrass, J. , Commodore, L. , Sawyer, T.K. , Dalgarno, D.C. , Deininger, M.W. , Druker, B.J. , Clackson, T. , 2009. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cel. 16, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, K.E. , Rojo, F. , She, Q.B. , Solit, D. , Mills, G.B. , Smith, D. , Lane, H. , Hofmann, F. , Hicklin, D.J. , Ludwig, D.L. , Baselga, J. , Rosen, N. , 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi, A. , Inokuchi, M. , Harada, S. , Inazawa, J. , Tajiri, R. , Kitamura, S.S. , Ikeda, H. , Kawashima, H. , Dobashi, Y. , 2012. Gene amplification of ESR1 in breast cancers – fact or fiction? A fluorescence in situ hybridization and multiplex ligation-dependent probe amplification study. J. Pathol. 227, 8–16. [DOI] [PubMed] [Google Scholar]
- Osborne, C.K. , Bardou, V. , Hopp, T.A. , Chamness, G.C. , Hilsenbeck, S.G. , Fuqua, S.A. , Wong, J. , Allred, D.C. , Clark, G.M. , Schiff, R. , 2003. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 95, 353–361. [DOI] [PubMed] [Google Scholar]
- Osborne, C.K. , Schiff, R. , 2011. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Si, B.Y. , Camidge, D.R. , Riely, G.J. , Salgia, R. , Shapiro, G. , Solomon, B.J. , Engelman, J.A. , Kwak, E.L. , Clark, J.W. , Tye, L. , Wilner, K.D. , Stephenson, P. , Varella-Garcia, M. , Bergethon, K. , Iafrate, A.J. , Shaw, A.T. , 2013. Efficacy and safety of crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer (NSCLC). J. Clin. Oncol. 13, (Suppl.) abstr 8032 [Google Scholar]
- Poulikakos, P.I. , Persaud, Y. , Janakiraman, M. , Kong, X. , Ng, C. , Moriceau, G. , Shi, H. , Atefi, M. , Titz, B. , Gabay, M.T. , Salton, M. , Dahlman, K.B. , Tadi, M. , Wargo, J.A. , Flaherty, K.T. , Kelley, M.C. , Misteli, T. , Chapman, P.B. , Sosman, J.A. , Graeber, T.G. , Ribas, A. , Lo, R.S. , Rosen, N. , Solit, D.B. , 2011. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Natur. 480, 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad, A. , Sun, C. , Huang, S. , Di Nicolantonio, F. , Salazar, R. , Zecchin, D. , Beijersbergen, R.L. , Bardelli, A. , Bernards, R. , 2012. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Natur. 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Rizzo, P. , Miao, H. , D'Souza, G. , Osipo, C. , Song, L.L. , Yun, J. , Zhao, H. , Mascarenhas, J. , Wyatt, D. , Antico, G. , Hao, L. , Yao, K. , Rajan, P. , Hicks, C. , Siziopikou, K. , Selvaggi, S. , Bashir, A. , Bhandari, D. , Marchese, A. , Lendahl, U. , Qin, J.Z. , Tonetti, D.A. , Albain, K. , Nickoloff, B.J. , Miele, L. , 2008. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 68, 5226–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, D.R. , Wu, Y.M. , Vats, P. , Su, F. , Lonigro, R.J. , Cao, X. , Kalyana-Sundaram, S. , Wang, R. , Ning, Y. , Hodges, L. , Gursky, A. , Siddiqui, J. , Tomlins, S.A. , Roychowdhury, S. , Pienta, K.J. , Kim, S.Y. , Roberts, J.S. , Rae, J.M. , Van Poznak, C.H. , Hayes, D.F. , Chugh, R. , Kunju, L.P. , Talpaz, M. , Schott, A.F. , Chinnaiyan, A.M. , 2013. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 45, 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, H. , Tsukaguchi, T. , Hiroshima, S. , Kodama, T. , Kobayashi, T. , Fukami, T.A. , Oikawa, N. , Tsukuda, T. , Ishii, N. , Aoki, Y. , 2011. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cel. 19, 679–690. [DOI] [PubMed] [Google Scholar]
- Salt, M.B. , Bandyopadhyay, S. , McCormick, F. , 2014. Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov. 4, 186–199. [DOI] [PubMed] [Google Scholar]
- Sasaki, T. , Okuda, K. , Zheng, W. , Butrynski, J. , Capelletti, M. , Wang, L. , Gray, N.S. , Wilner, K. , Christensen, J.G. , Demetri, G. , Shapiro, G.I. , Rodig, S.J. , Eck, M.J. , Janne, P.A. , 2010. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 70, 10038–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti, M. , Verma, C. , Guzman, M. , Jimenez, J. , Parra, J.L. , Pedersen, K. , Smith, D.J. , Landolfi, S. , Ramon y Cajal, S. , Arribas, J. , Baselga, J. , 2009. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogen. 28, 803–814. [DOI] [PubMed] [Google Scholar]
- Sequist, L.V. , Waltman, B.A. , Dias-Santagata, D. , Digumarthy, S. , Turke, A.B. , Fidias, P. , Bergethon, K. , Shaw, A.T. , Gettinger, S. , Cosper, A.K. , Akhavanfard, S. , Heist, R.S. , Temel, J. , Christensen, J.G. , Wain, J.C. , Lynch, T.J. , Vernovsky, K. , Mark, E.J. , Lanuti, M. , Iafrate, A.J. , Mino-Kenudson, M. , Engelman, J.A. , 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, A.T. , Kim, D.W. , Nakagawa, K. , Seto, T. , Crino, L. , Ahn, M.J. , De Pas, T. , Besse, B. , Solomon, B.J. , Blackhall, F. , Wu, Y.L. , Thomas, M. , O'Byrne, K.J. , Moro-Sibilot, D. , Camidge, D.R. , Mok, T. , Hirsh, V. , Riely, G.J. , Iyer, S. , Tassell, V. , Polli, A. , Wilner, K.D. , Janne, P.A. , 2013. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 368, 2385–2394. [DOI] [PubMed] [Google Scholar]
- Shi, H. , Hugo, W. , Kong, X. , Hong, A. , Koya, R.C. , Moriceau, G. , Chodon, T. , Guo, R. , Johnson, D.B. , Dahlman, K.B. , Kelley, M.C. , Kefford, R.F. , Chmielowski, B. , Glaspy, J.A. , Sosman, J.A. , van Baren, N. , Long, G.V. , Ribas, A. , Lo, R.S. , 2014. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H. , Moriceau, G. , Kong, X. , Lee, M.K. , Lee, H. , Koya, R.C. , Ng, C. , Chodon, T. , Scolyer, R.A. , Dahlman, K.B. , Sosman, J.A. , Kefford, R.F. , Long, G.V. , Nelson, S.F. , Ribas, A. , Lo, R.S. , 2012. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 3, 724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, L. , Dong, B. , Li, Z. , Lu, Y. , Ouyang, T. , Li, J. , Wang, T. , Fan, Z. , Fan, T. , Lin, B. , Wang, Z. , Xie, Y. , 2009. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J. Clin. Oncol. 27, 3423–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau, A.K. , Barstad, D. , Loria, P.M. , Cheng, L. , Kushner, P.J. , Agard, D.A. , Greene, G.L. , 1998. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cel. 95, 927–937. [DOI] [PubMed] [Google Scholar]
- Singh, M.S. , Francis, P.A. , Michael, M. , 2011. Tamoxifen, cytochrome P450 genes and breast cancer clinical outcomes. Breas. 20, 111–118. [DOI] [PubMed] [Google Scholar]
- Smith, I.E. , Dowsett, M. , 2003. Aromatase inhibitors in breast cancer. N. Engl. J. Med. 348, 2431–2442. [DOI] [PubMed] [Google Scholar]
- Sosman, J.A. , Kim, K.B. , Schuchter, L. , Gonzalez, R. , Pavlick, A.C. , Weber, J.S. , McArthur, G.A. , Hutson, T.E. , Moschos, S.J. , Flaherty, K.T. , Hersey, P. , Kefford, R. , Lawrence, D. , Puzanov, I. , Lewis, K.D. , Amaravadi, R.K. , Chmielowski, B. , Lawrence, H.J. , Shyr, Y. , Ye, F. , Li, J. , Nolop, K.B. , Lee, R.J. , Joe, A.K. , Ribas, A. , 2012. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 366, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman, R. , Morikawa, T. , Shee, K. , Barzily-Rokni, M. , Qian, Z.R. , Du, J. , Davis, A. , Mongare, M.M. , Gould, J. , Frederick, D.T. , Cooper, Z.A. , Chapman, P.B. , Solit, D.B. , Ribas, A. , Lo, R.S. , Flaherty, K.T. , Ogino, S. , Wargo, J.A. , Golub, T.R. , 2012. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Natur. 487, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C. , Hobor, S. , Bertotti, A. , Zecchin, D. , Huang, S. , Galimi, F. , Cottino, F. , Prahallad, A. , Grernrum, W. , Tzani, A. , Schlicker, A. , Wessels, L.F. , Smit, E.F. , Thunnissen, E. , Halonen, P. , Lieftink, C. , Beijersbergen, R.L. , Di Nicolantonio, F. , Bardelli, A. , Trusolino, L. , Bernards, R. , 2014. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 7, 86–93. [DOI] [PubMed] [Google Scholar]
- Sun, C. , Wang, L. , Huang, S. , Heynen, G.J. , Prahallad, A. , Robert, C. , Haanen, J. , Blank, C. , Wesseling, J. , Willems, S.M. , Zecchin, D. , Hobor, S. , Bajpe, P.K. , Lieftink, C. , Mateus, C. , Vagner, S. , Grernrum, W. , Hofland, I. , Schlicker, A. , Wessels, L.F. , Beijersbergen, R.L. , Bardelli, A. , Di Nicolantonio, F. , Eggermont, A.M. , Bernards, R. , 2014. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Natur. 508, 118–122. [DOI] [PubMed] [Google Scholar]
- Swain, S.M. , Kim, S.B. , Cortes, J. , Ro, J. , Semiglazov, V. , Campone, M. , Ciruelos, E. , Ferrero, J.M. , Schneeweiss, A. , Knott, A. , Clark, E. , Ross, G. , Benyunes, M.C. , Baselga, J. , 2013. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 14, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa, K. , Pirazzoli, V. , Arcila, M.E. , Nebhan, C.A. , Song, X. , de Stanchina, E. , Ohashi, K. , Janjigian, Y.Y. , Spitzler, P.J. , Melnick, M.A. , Riely, G.J. , Kris, M.G. , Miller, V.A. , Ladanyi, M. , Politi, K. , Pao, W. , 2012. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, S. , Buck, E. , Petti, F. , Griffin, G. , Brown, E. , Ramnarine, N. , Iwata, K.K. , Gibson, N. , Haley, J.D. , 2005. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 65, 9455–9462. [DOI] [PubMed] [Google Scholar]
- Tolhurst, R.S. , Thomas, R.S. , Kyle, F.J. , Patel, H. , Periyasamy, M. , Photiou, A. , Thiruchelvam, P.T. , Lai, C.F. , Al-Sabbagh, M. , Fisher, R.A. , Barry, S. , Crnogorac-Jurcevic, T. , Martin, L.A. , Dowsett, M. , Charles Coombes, R. , Kamalati, T. , Ali, S. , Buluwela, L. , 2011. Transient over-expression of estrogen receptor-alpha in breast cancer cells promotes cell survival and estrogen-independent growth. Breast Cancer Res. Treat. 128, 357–368. [DOI] [PubMed] [Google Scholar]
- Toy, W. , Shen, Y. , Won, H. , Green, B. , Sakr, R.A. , Will, M. , Li, Z. , Gala, K. , Fanning, S. , King, T.A. , Hudis, C. , Chen, D. , Taran, T. , Hortobagyi, G. , Greene, G. , Berger, M. , Baselga, J. , Chandarlapaty, S. , 2013. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 45, 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trunzer, K. , Pavlick, A.C. , Schuchter, L. , Gonzalez, R. , McArthur, G.A. , Hutson, T.E. , Moschos, S.J. , Flaherty, K.T. , Kim, K.B. , Weber, J.S. , Hersey, P. , Long, G.V. , Lawrence, D. , Ott, P.A. , Amaravadi, R.K. , Lewis, K.D. , Puzanov, I. , Lo, R.S. , Koehler, A. , Kockx, M. , Spleiss, O. , Schell-Steven, A. , Gilbert, H.N. , Cockey, L. , Bollag, G. , Lee, R.J. , Joe, A.K. , Sosman, J.A. , Ribas, A. , 2013. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 31, 1767–1774. [DOI] [PubMed] [Google Scholar]
- Turke, A.B. , Zejnullahu, K. , Wu, Y.L. , Song, Y. , Dias-Santagata, D. , Lifshits, E. , Toschi, L. , Rogers, A. , Mok, T. , Sequist, L. , Lindeman, N.I. , Murphy, C. , Akhavanfard, S. , Yeap, B.Y. , Xiao, Y. , Capelletti, M. , Iafrate, A.J. , Lee, C. , Christensen, J.G. , Engelman, J.A. , Janne, P.A. , 2010. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cel. 17, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtorta, E. , Misale, S. , Sartore-Bianchi, A. , Nagtegaal, I.D. , Paraf, F. , Lauricella, C. , Dimartino, V. , Hobor, S. , Jacobs, B. , Ercolani, C. , Lamba, S. , Scala, E. , Veronese, S. , Laurent-Puig, P. , Siena, S. , Tejpar, S. , Mottolese, M. , Punt, C.J. , Gambacorta, M. , Bardelli, A. , Di Nicolantonio, F. , 2013. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cance. 133, 1259–1265. [DOI] [PubMed] [Google Scholar]
- Van Allen, E.M. , Wagle, N. , Sucker, A. , Treacy, D.J. , Johannessen, C.M. , Goetz, E.M. , Place, C.S. , Taylor-Weiner, A. , Whittaker, S. , Kryukov, G.V. , Hodis, E. , Rosenberg, M. , McKenna, A. , Cibulskis, K. , Farlow, D. , Zimmer, L. , Hillen, U. , Gutzmer, R. , Goldinger, S.M. , Ugurel, S. , Gogas, H.J. , Egberts, F. , Berking, C. , Trefzer, U. , Loquai, C. , Weide, B. , Hassel, J.C. , Gabriel, S.B. , Carter, S.L. , Getz, G. , Garraway, L.A. , Schadendorf, D. , 2014. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 4, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier, S. , Brinkman, A. , Look, M.P. , Kok, E.M. , Meijer-van Gelder, M.E. , Klijn, J.G. , Dorssers, L.C. , Foekens, J.A. , 2000. Bcar1/p130Cas protein and primary breast cancer: prognosis and response to tamoxifen treatment. J. Natl. Cancer Inst. 92, 120–127. [DOI] [PubMed] [Google Scholar]
- Villanueva, J. , Vultur, A. , Lee, J.T. , Somasundaram, R. , Fukunaga-Kalabis, M. , Cipolla, A.K. , Wubbenhorst, B. , Xu, X. , Gimotty, P.A. , Kee, D. , Santiago-Walker, A.E. , Letrero, R. , D'Andrea, K. , Pushparajan, A. , Hayden, J.E. , Brown, K.D. , Laquerre, S. , McArthur, G.A. , Sosman, J.A. , Nathanson, K.L. , Herlyn, M. , 2010. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cel. 18, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle, N. , Emery, C. , Berger, M.F. , Davis, M.J. , Sawyer, A. , Pochanard, P. , Kehoe, S.M. , Johannessen, C.M. , Macconaill, L.E. , Hahn, W.C. , Meyerson, M. , Garraway, L.A. , 2011. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 29, 3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle, N. , Van Allen, E.M. , Treacy, D.J. , Frederick, D.T. , Cooper, Z.A. , Taylor-Weiner, A. , Rosenberg, M. , Goetz, E.M. , Sullivan, R.J. , Farlow, D.N. , Friedrich, D.C. , Anderka, K. , Perrin, D. , Johannessen, C.M. , McKenna, A. , Cibulskis, K. , Kryukov, G. , Hodis, E. , Lawrence, D.P. , Fisher, S. , Getz, G. , Gabriel, S.B. , Carter, S.L. , Flaherty, K.T. , Wargo, J.A. , Garraway, L.A. , 2014. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 4, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, A.O. , Sjin, R.T. , Haringsma, H.J. , Ohashi, K. , Sun, J. , Lee, K. , Dubrovskiy, A. , Labenski, M. , Zhu, Z. , Wang, Z. , Sheets, M. , St Martin, T. , Karp, R. , van Kalken, D. , Chaturvedi, P. , Niu, D. , Nacht, M. , Petter, R.C. , Westlin, W. , Lin, K. , Jaw-Tsai, S. , Raponi, M. , Van Dyke, T. , Etter, J. , Weaver, Z. , Pao, W. , Singh, J. , Simmons, A.D. , Harding, T.C. , Allen, A. , 2013. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 3, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Jain, S. , Coombes, C.R. , Palmieri, C. , 2009. Fulvestrant in advanced breast cancer following tamoxifen and aromatase inhibition: a single center experience. Breast J. 15, 247–253. [DOI] [PubMed] [Google Scholar]
- Weinstein, I.B. , 2002. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Scienc. 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Xia, W. , Gerard, C.M. , Liu, L. , Baudson, N.M. , Ory, T.L. , Spector, N.L. , 2005. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogen. 24, 6213–6221. [DOI] [PubMed] [Google Scholar]
- Yamnik, R.L. , Holz, M.K. , 2010. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Lett. 584, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, S. , Nakataki, E. , Ohtsuka, S. , Inayama, M. , Tomimoto, H. , Edakuni, N. , Kakiuchi, S. , Nishikubo, N. , Muguruma, H. , Sone, S. , 2005. Retreatment of lung adenocarcinoma patients with gefitinib who had experienced favorable results from their initial treatment with this selective epidermal growth factor receptor inhibitor: a report of three cases. Oncol. Res. 15, 107–111. [PubMed] [Google Scholar]
- Yauch, R.L. , Januario, T. , Eberhard, D.A. , Cavet, G. , Zhu, W. , Fu, L. , Pham, T.Q. , Soriano, R. , Stinson, J. , Seshagiri, S. , Modrusan, Z. , Lin, C.Y. , O'Neill, V. , Amler, L.C. , 2005. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 11, 8686–8698. [DOI] [PubMed] [Google Scholar]
- Yoo, C. , Kim, S.B. , Ahn, J.H. , Jung, K.H. , Ahn, Y. , Gong, G. , Kim, H.H. , Kim, H.J. , Son, B.H. , Ahn, S.H. , 2011. Efficacy of fulvestrant in heavily pretreated postmenopausal women with advanced breast cancer: a preliminary report. J. Breast Cance. 14, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, W. , Ercan, D. , Chen, L. , Yun, C.H. , Li, D. , Capelletti, M. , Cortot, A.B. , Chirieac, L. , Iacob, R.E. , Padera, R. , Engen, J.R. , Wong, K.K. , Eck, M.J. , Gray, N.S. , Janne, P.A. , 2009. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Natur. 462, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]