

Abstract
In early breast cancer, local relapses represent a determinant and not simply an indicator of risk for distant relapse and death. Notably, 90% of local recurrences occur at or close to the same quadrant of the primary cancer. Relevance of PI3K/mTOR/p70S6K signaling in breast tumorigenesis is very well documented. However, the pathway/s involved in the process of breast cancer local relapse are not well understood. The ribosomal protein p70S6K has been implicated in breast cancer cell response to post‐surgical inflammation, supporting the hypothesis that it may be crucial also for breast cancer recurrence. Here, we show that p70S6K activity is required for the survival of breast cancer cells challenged in “hostile” microenvironments. We found that impairment of p70S6K activity in breast cancer cells strongly decreased their tumor take rate in nude mice. In line with this observation, if cells were challenged to grow in anchorage independence or in clonogenic assay, growth of colonies was strongly dependent on an intact p70S6K signaling. This in vitro finding was particularly evident when breast cancer cells were grown in the presence of wound fluids harvested following surgery from breast cancer patients, suggesting that the stimuli present in the post‐surgical setting at least partially relied on activity of p70S6K to stimulate breast cancer relapse. From a mechanistic point of view, our results indicated that p70S6K signaling was able to activate Gli1 and up‐regulate the anti‐apoptotic protein Bcl2, thereby activating a survival response in breast cancer cells challenged in hostile settings. Our work highlights a previously poorly recognized function of p70S6K in preserving breast cancer cell survival, which could eventually be responsible for local relapse and opens the way to the design of new and more specific therapies aiming to restrain the deleterious effects of wound response.
Keywords: Breast cancer, p70S6K, mTOR, Xenograft, Survival, Proliferation
Highlights
p70S6K is activated in breast cancer cells exposed to post‐surgical wound fluids.
p70S6K activity is necessary to prevent cell death in “hostile” environments.
Inhibition of p70S6K1 leads to different outcomes respect to inhibition of mTOR.
The p70S6K/Gli1/Bcl2 signaling axis is necessary to elicit a survival response.
p70S6K represents a promising target to prevent breast cancer local recurrence.
1. Introduction
Following breast conserving surgery, local relapse is a common event that not only represents a disturbing factor but may also worsen the prognosis of early breast cancer patients (Benson et al., 2009; Huston and Simmons, 2005). Several studies have shown that locoregional recurrence is a determinant and not simply an indicator of risk of distant relapse and death (Benson et al., 2009). The importance of restraining local recurrence in breast cancer patients was recently overviewed, conclusively showing that treatments improving local control have definite effects on long‐term survival (Clarke et al., 2005).
Notably, 90% of local recurrences occur at or close to the same quadrant of the primary cancer, supporting the idea that local disease develops from re‐growth of residual cancer cells, left behind in the peritumoral tissue after surgery (Benson et al., 2009). It was hypothesized that the perturbation of the microenvironment by primary tumor removal and the subsequent wound healing process may result in stimulation of postsurgical residual disease (Baker et al., 1989, 2001, 2007).
This hypothesis was supported by our (Belletti et al., 2008b) and others' (Tagliabue et al., 2003) in vitro studies that demonstrated the stimulatory effect of post‐surgical drainage fluids (hereafter referred to as wound fluids, WF) on breast cancer cells proliferation and invasion.
The PI3K/mTOR/p70S6K axis is known to regulate a signal transduction cascade that promotes cell growth, survival and metabolism. It is aberrantly activated in many types of cancer, playing a major role in breast cancer cell proliferation and anti‐cancer drug resistance (Ghayad and Cohen, 2010). The 70‐kDa ribosomal protein S6 kinase (p70S6K, hereafter p70S6K) is a serine/threonine kinase (Fenton and Gout, 2011, 1997, 2010, 2010, 1997), downstream target of mTOR, a master regulator of cell growth and proliferation that integrates signals from multiple inputs (Hay and Sonenberg, 2004). p70S6K plays important roles in cell growth, proliferation and differentiation by regulating ribosome biogenesis, cell cycle progression and metabolism (Düvel et al., 2010; Kawasome et al., 1998; Shin et al., 2011).
Many data suggest an involvement of p70S6K in breast cancer. At genomic level, 17q23 amplification was observed in approximately 10% of all primary breast cancer cases causing increased copy number of the p70S6K gene, RPS6KB1, elevated p70S6K expression and proliferative advantage in breast cancer cell lines (Couch et al., 1999; Monni et al., 2001). Our previous work uncovered that p70S6K is activated in breast cancer cell in response to post‐surgical drainage fluids, supporting the hypothesis that it may be crucial also for breast cancer recurrence (Belletti et al., 2008b). We recently addressed this issue and demonstrated that p70S6K activity is robustly induced by surgery also in human patients and that its inhibition strongly impaired breast cancer local relapse in a mouse model of breast cancer (Segatto et al., 2013).
Here, we characterize the role of p70S6K during breast cancer growth and investigate the mechanism whereby p70S6K may foster tumor initiation and drive breast cancer local relapse.
2. Materials and methods
2.1. Study approval
All animal experiments were reviewed and approved by the CRO Institutional Animal Care and Use Committee and were conducted according to that committee's guidelines.
Wound Fluids (WF) were collected at CRO of Aviano, Italy. Specific informed consent was obtained from all patients. Scientific use of biological material was approved by Ethics Committee of the CRO of Aviano, Italy.
2.2. Cell culture and development of stable cell lines
MDA MB 231 (basal, ER−, PR−, HER2−), MDA MB 453 (luminal, ER−, PR−, HER2−), MCF‐7 (luminal, ER+, PR+, HER2−), BT‐474 (luminal, ER−, PR−, HER2+), BT‐549 (basal, ER−, PR−, HER2−), HBL‐100 (basal, ER−, PR−, HER2−) and SK‐BR‐3 (luminal, ER−, PR−, HER2+) mammary carcinoma cell lines (Neve et al., 2006) were obtained from ATCC (LGC Standards) and grown in Dulbecco modified Eagle medium (DMEM, Lonza) supplemented with 10% fetal bovine serum (FBS, SIGMA). Stable cell clones were obtained by retroviral transduction with a vector carrying Puromycin resistance (murine stem cell virus retroviral vectors, MSCV; Clontech), following the manufacturer's instructions, subcloned to encode for constitutively active mutant HA‐tagged p70S6K‐FER (carrying the substitutions F5A‐E389‐R3A) (Schalm and Blenis, 2002) or for kinase inactive mutant HA‐p70S6K‐KR (carrying the substitution K100R) (Schalm and Blenis, 2002). The expression vectors have been kindly provided by John Blenis (Harvard Medical School, Boston). p70S6K‐silenced mammary carcinoma cells were generated by lentiviral transduction of pLKO vectors encoding for human shRNAs of the MISSiON system (pLKO lentiviral vector, SIGMA). Briefly, 293FT cells were transfected with pLP1, pLP2, pLP/VSV‐G (Invitrogen recombinant lentivirus producing system) plus pLKOshRNA (sh1_TCRN0000194766, sh2_TCRN0000003159, sh3_TCRN0000003162) by standard calcium phosphate protocol. After 48 and 72 h, conditional medium containing lentiviral particles was harvested and used to transduce target cells. Cell clones and pools were selected in complete medium supplemented with 1.5 μg/ml puromycin.
All cell lines were authenticated by BMR Genomics srl Padova, Italia, on January 2012 according to Cell ID™ System (Promega) protocol and using Genemapper ID Ver 3.2.1, to identify DNA STR profiles.
2.3. Preparation of protein lysates, immunoprecipitation and immunoblotting analysis
MDA MB 231, MDA MB 453, MCF‐7, BT‐474, BT‐549, HBL‐100 and SK‐BR‐3 mammary carcinoma cell lines were serum starved in DMEM containing 0.1% bovine serum albumin (BSA, SIGMA) and then stimulated with 10% FBS or 5% WF for the indicated time points. Where indicated, cells were also pre‐treated for 30 min with the following inhibitors: PF‐4708671 (p70S6K1 inhibitor, 10 μM, SIGMA), Temsirolimus (Rapamycin analogue, 10, 20, 100 nM, Wyeth), LY‐294002 (PI3K reversible inhibitor, 20 μM, SIGMA), U0126 (MEK1/2 inhibitor, 10 μM, Calbiochem), Wortmannin (PI3K covalent inhibitor, 100 nM, Calbiochem) and Staurosporine (kinase inhibitor, 5 μM, Calbiochem).
To extract total proteins cells were scraped on ice using cold NP40 lysis buffer (0.5% NP40; 50 mM HEPES pH 7; 250 mM NaCl; 5 mM EDTA; 0.5 mM EGTA, pH 8) plus a protease inhibitor cocktail (Complete™, Roche) and supplemented with 1 mM Na3VO4 (SIGMA), 10 mM NaF (SIGMA) and 1 mM DTT (SIGMA).
To extract total proteins from tumor specimens, the same procedure was used, except that tissue disruption was first achieved by using the TissueLyser II (Qiagen).
Immunoprecipitation (IP) experiment was performed using 0.5 mg of total lysate in HNTG buffer (20 mM HEPES, 150 mM NaCl, 10% Glycerol, 0.1% Triton X‐100, protease inhibitor cocktail, 1 mM Na3VO4, 10 mM NaF and 1 mM DTT) with the specific primary antibodies (HA agarose‐conjugated, #A2095 SIGMA; GLI1, #2534 Cell Signaling), gently rocking overnight at 4 °C. When primary antibodies were not agarose conjugated, protein A or protein G Sepharose 4 Fast Flow (Amersham Biosciences) was added for the last 2 h of incubation. IPs were then washed six times in HNTG buffer and resuspended in 3× Laemmli Sample Buffer (5× Laemmli buffer composition: 50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 0.05% bromophenol blue and 125 mM beta‐mercaptoethanol).
For immunoblotting analysis, proteins were separated in 4–20% SDS‐PAGE (Criterion Precast Gel, Biorad) and transferred to nitrocellulose membranes (GE Healthcare). Membranes were blocked with 5% dried milk in TBS‐0.1% Tween20 or in Odyssey Blocking Buffer (Licor, Biosciences) and incubated at 4 °C overnight with primary antibodies. Then, membranes were incubated 1 h at RT with IR‐conjugated (Alexa Fluor 680, Invitrogen or IRDye 800, Rockland) secondary antibodies for infrared detection (Odyssey Infrared Detection System, Licor).
Primary antibodies AKT (sc‐1618), ERK1 (sc‐94), p70S6K1 (sc‐8418), vinculin (sc‐7694) were purchased from Santa Cruz; pT202/204 ERK1/2 (#9101), pS473 AKT (#4060), pT389 p70S6K1 (#9234), S6 (#2217), pS235/236 S6 (#4858), pS240/244 S6 (#5364), pT32 FOXO3a (#9464), GLI1 (#2534) were purchased from Cell Signaling; HA (#PRB101C) was purchased from Covance; Bcl2 (#OP60) was purchased from Calbiochem. pS84 GLI1 mouse monoclonal antibody was a kind gift of Dr Mien‐Chie Hung, MD Anderson Cancer Center, USA.
2.4. Wound fluid collection
Drainage wound fluids (WF) were collected over the 24 h after surgery from unselected patients undergone breast‐conserving surgery, as described previously (Belletti et al., 2008b). The assays were then performed using pools of all fluids.
2.5. Histological analysis and immunohistochemistry
Mouse samples were fixed in formalin (overnight at 4 °C) and processed for standard paraffin embedding. Histological sections (5 μm thick) were made from the paraffin blocks, deparaffinated with xylene, and stained with hematoxylin and eosin.
For immunohistochemistry analysis, routine deparaffinization of all sections mounted on positive charge slides was carried out according to standard procedures, followed by rehydration through serial ethanol treatments. Slides were immersed in citrate buffer [0.01 M sodium citrate (pH 6.0)] and heated in a microwave oven at 600 W (three times for 5 min each) to enhance antigen retrieval. Endogenous peroxidase was blocked with 0.3% hydrogen peroxide in methanol for 30 min. Sections were immunostained for 1 h at room temperature with anti‐Bcl2 antibody (#OP60, Calbiochem). The primary antibody was omitted and replaced with preimmune serum in the negative control. Sections were reacted with biotinylated anti‐mouse antibody and streptavidin–biotin–peroxidase (Histostain‐SP Kit, Zymed Laboratories, San Francisco, CA). Diaminobenzidine was used as a chromogene substrate. Finally, sections were washed in distilled water and weakly counterstained with Harry's modified hematoxylin.
2.6. Immunofluorescence analysis
For immunofluorescence staining, cells seeded on coverslips were fixed in PBS 4% paraformaledehyde (PFA) at room temperature (RT), permeabilized in PBS 0.2% Triton X‐100 and blocked in PBS‐1% BSA. Incubation with primary antibodies was performed overnight at 4 °C in PBS‐1% BSA, then samples were washed in PBS and incubated with secondary antibodies (Alexa Fluor 488‐, 546‐ or 633‐conjugated anti‐mouse or anti‐rabbit, Life Technologies) for 1 h at RT. Primary antibodies: pS84 GLI1 mouse monoclonal antibody was a kind gift of Dr Mien‐Chie Hung, MD Anderson, USA; pS473 AKT was purchased from Cell Signaling (cat #4060). Antibody incubation was followed by nuclear staining with 5 μg/ml Propidium Iodide (PI) in PBS for 20 min at RT. Coverslips were mounted in Mowiol 488 (Calbiochem‐Novabiochem) containing 2.5% (w/v) 1,4‐diazabicyclo (2,2,2)octane (DABCO, Sigma). For immunofluorescence performed on tumors, fresh mouse tumor specimens were included in OCT and stored at −80 °C. Histological sections (5 μm thick) were made from the OCT blocks and fixed 20′ at RT in 4% PFA before blocking in 10% normal goat serum. Sections were immunoprobed at 4 °C O.N. with the following primary antibodies: anti‐Bcl2 (#OP60, Calbiochem), anti‐GLI1 (#2534 Cell Signaling) or anti‐pS84 GLI1 (kind gift of Dr Mien‐Chie Hung, MD Anderson, USA). The primary antibody was omitted and replaced with preimmune serum for the negative control. Sections were then probed with Alexa Fluor 488‐, 546‐ or 633‐conjugated secondary anti‐mouse (Life Technologies) or anti‐rabbit for 1 h at RT, followed by nuclear staining with PI or TO‐PRO‐3 Iodide (Life Technologies) for 20′ at RT. Stained cells and sections were then observed using a confocal laser‐scanning microscope (TSP2 Leica) interfaced with a Leica DMIRE2 fluorescent microscope. Collected images were then processed and fluorescence quantified using the Volocity 6.0.1® computer program (Perkin–Elmer).
2.7. Proliferation assays and clonogenic assay
For growth curve, 5–9 × 104 cells/well (depending on the cell type) were seeded in 6‐well plates in complete medium or in serum free medium supplemented with 3% WF (SFM‐3% WF), in triplicate. Where indicated, PF‐4708671 (p70S6K1 inhibitor, SIGMA) or Temsirolimus (Rapamycin analogue, Wyeth) were added in the medium at the concentration indicated. Fresh medium, with or without inhibitors, was added every other day. At the indicated times, cells were detached by trypsin‐EDTA and counted by Trypan Blue exclusion test.
To evaluate the anchorage‐independent cell growth, MDA MB 453 cells (1.5 × 104) were re‐suspended in 2 ml top agar medium (DMEM‐10% FBS or SFM‐5% WF, 0.4% Low Melting Agarose, SIGMA) and quickly overlaid on a previously gelified 0.6% bottom agar medium (DMEM‐10% FBS, 0.6% Low Melting Agarose, SIGMA). The experiments were performed in six‐well tissue culture plates, in triplicate. Fresh medium was added to the wells twice a week as a feeder layer. After three weeks, the number of colonies was counted in 10 randomly chosen fields, at 10× magnification.
For colony assay, MDA MB 231 untreated or pre‐treated for 48 h with PF‐4708671 (p70S6K1 inhibitor, 10 μM, SIGMA) or Temsirolimus (Rapamycin analogue, 100 nM, Wyeth), used alone or in combination, were trypsinized, counted and seeded at density 500, 1000, 2000 or 20,000 cells/100 mm dish, as indicated, and incubated in complete growth medium in the presence of the inhibitors or left untreated. Two weeks later plates were stained with crystal violet and colonies were counted. In a subset of experiments, MDA MB 231 at density 20,000 cells/100 mm dish were seeded on coverslips to perform immunofluorescence analysis or were collected and lysed to perform WB analysis.
2.8. Motility assays
Motility was studied using chemotaxis and standard Matrigel invasion assay, performed essentially as previously described (Baldassarre et al., 2005, 2008, 2008, 2010).
Briefly, chemotaxis was performed by labeling cells with DiI fluorescent vital dye (Molecular Probes) for 20 min at 37 °C and plating 1 × 105 cells in the upper chamber of transwell‐like inserts, carrying a fluorescence‐shielding porous polyethylene terephthalate membrane with 8 μm pores (HTS Fluoroblok, BD) and then incubated at 37 °C for the indicated time. The lower chamber was filled with complete medium or serum free medium supplemented with 5% WF, as indicated. Migrating cells were evaluated by reading at different time points with a Spectrafluor (Tecan) the lower and the upper sides of the membrane. The experiments were performed at least 3 times in duplicate.
For invasion experiments, HTS Fluoroblok transwells were coated overnight at 4 °C with a layer of Matrigel (6 μg, Cultrex BME) diluted in DMEM 0.1% BSA. Cells were then processed as described above and after an overnight incubation percent of invading cells was calculated.
Where indicated, PF‐4708671 and Temsirolimus were mixed with the cells at the time of seeding, at the final concentration of 10 μM or 100 nM, respectively.
For evasion assay, 7.5 × 103 cells were included in Matrigel (Cultrex, BME) drops at the final concentration of 6 mg/ml (12 μl of matrix volume per drops). Matrigel was diluted in DMEM 0.1% BSA. The drops, sufficiently spaced from one another, were dispensed in cell culture dishes and maintained for 1 h at 37 °C upside down to let jellify. Then, the dishes were turned up and the drops incubated in complete medium or serum free medium supplemented with 5% WF. The evasion ability was evaluated 6 days after inclusion by measuring the distance covered by crystal violet‐stained cells exited from the drops (5 drops/cell lines/experiment). Images were collected using a stereo microscope Leica M205FA.
2.9. In vivo experiments
Primary tumors were established by injection of 2 × 106 MDA MB 231 control or derived cell clones bilaterally in the fat pads of the thoracic mammary glands of female athymic nude mice (Harlan, 6–8 weeks old). Growth of primary tumors was monitored by measuring tumor length (L) and width (W), and calculating tumor volume based on the formula L × W 2/2.
In the long‐term treatment experiment, after the appearance of palpable primary tumors generated from MDA MB 231 control cells, animals were intraperitoneally treated with PF‐4708671 (25 mg/kg, i.e. 600 μg/mouse) or Temsirolimus (12.5 mg/kg, i.e. 300 μg/mouse) twice a week, for three weeks. During treatment, growth of primary tumors was monitored and measured. Mice were sacrificed at the endpoint of the experiment.
To analyze the tumor take rate, mice were injected with 2 × 104 or 2 × 105 or 7.5 × 105 or 2 × 106 MDA MB 231 control or modified cells resuspended in 50 μl Matrigel/PBS (1:1). Growth of primary tumors was monitored up to 7 weeks. As an alternative approach, mice were injected with 1 × 105, 2 × 105 or 4 × 105 MDAMB231 control‐ or p70KR expressing‐cells, without Matrigel, in 100 μl of PBS. Growth of primary tumors was monitored up to 10 weeks.
To evaluate the effect of the three‐days schedule of treatment on tumor cells in vivo, primary tumors were established by injection of 2 × 106 MDA MB 231 control cells, bilaterally in thoracic MFP. When primary tumors reached a volume of 50–100 mm3, mice were intraperitoneally treated with PF‐4708671 (3 mice/group, 1200 μg/mouse) or with Temsirolimus (3 mice/group, 300 μg or 600 μg/mouse) or vehicle (3 mice/group, PBS) daily, for three consecutive days. Mice were sacrificed one day after the last treatment. Tumors were collected and stored for subsequent analyses.
2.10. Kinexus antibody MicroArray
Kinexus antibody microarray analysis was performed on MDA MB 231, pre‐treated with PF‐4708671 10 μM or with Temsirolimus 100 nM and stimulated with WF for 20 min. The KinexTM antibody microarray service utilizes antibody microarrays to track the differential binding of dye‐labeled proteins in lysates prepared from cells and tissues. Full KinexTM Service uses the KAM‐850 chip with two samples analyzed in duplicate with over 850 antibodies. Over 510 pan‐specific antibodies used in the chip provide for the detection of 189 protein kinases, 31 protein phosphatases, 142 regulatory subunits and other cell signaling proteins that regulate cell proliferation, stress and apoptosis.
2.11. Statistical analysis
Data were examined using the two‐tailed Student t test or unpaired two‐tailed Mann–Whitney U test. Differences were considered significant at p < 0.05. The computer software PRISM (version 4, GraphPad, Inc.) was used to make graphs and all statistical analyses.
3. Results
3.1. Wound fluids efficiently activate p70S6K in breast cancer cell lines
Our previous studies demonstrated that WF are very rich in cytokines and growth factors and very efficiently stimulated breast cancer cell proliferation, motility and invasion (Belletti et al., 2008b). Using a large panel of breast cancer cell lines, we confirmed that in breast cancer cells activation of p70S6K signaling is a common event following exposure to WF (Figure 1). The analysis of the p70S6K target phospho‐S6 showed that, with few exceptions, 5% WF stimulated p70S6K signaling at even higher extent than 10% serum.
Figure 1.
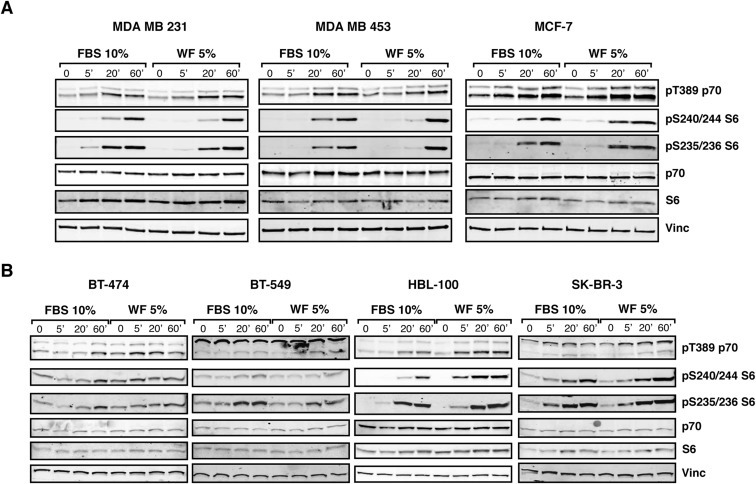
In breast cancer cell lines, p70S6K is efficiently activated following stimulation with wound fluids. A. Western blot analysis of MDA MB 231, MDA MB 453 and MCF‐7 cell lines serum starved and then stimulated for the indicated times with 10% serum (FBS) or 5% wound fluids (WF). B. Same as in (A), but using BT‐474, BT‐549, HBL‐100 and SK‐BR‐3 cell lines, as indicated. Vinculin expression was used as loading control.
We then wondered whether this strong activation was a mere downstream signaling event or played an active role in the response of breast cancer cells to the surgery‐induced inflammation. We thus set up different cellular models in order to evaluate the role of p70S6K. To test which breast cancer model would better fit to our studies, we interrogated the TCGA dataset for p70S6K expression and activity in 403 cancer specimens analyzed by reverse‐phase protein array, RPPA (Cancer Genome Atlas Network, 2012). The analysis of the TCGA dataset showed that levels of p70S6K were similar among basal‐like and luminal A breast cancer and slightly higher in luminal B ones (Supplementary Figure S1A, left panel). However, p70S6K activation, measured as S6 phosphorylation both at Ser235/236 and Ser240/244, was significantly higher in basal‐like breast cancer respect to the luminal subtypes (Supplementary Figure S1A, middle and right panels). Then we interrogated the same dataset sorting human breast cancer specimens for their receptor status. Results from this analysis showed that activation of p70S6K signaling was consistently increased in receptor negative specimens, particularly in the triple negative ones (TNBC) (Supplementary Figure S1B). Based on these notions, we decided to raise p70S6K activity in receptor positive breast cancer cells displaying a mildly aggressive behavior (luminal, MCF‐7) and to lower it in triple negative breast cancer cells displaying a highly aggressive behavior (basal‐like, MDA MB 231 and luminal, MDA MB 453) (Supplementary Figure S2). To this aim, cells were transfected with p70S6K mutants encoding for constitutively active p70S6K (HA‐p70FER, carrying the substitutions F5A‐E389‐R3A) (Supplementary Figure S2A and B) or for kinase inactive p70S6K (HA‐p70KR, carrying the substitution K100R) (Schalm and Blenis, 2002) (Supplementary Figure S2A and C) or transduced with lentiviral particles encoding for specific anti human p70S6K sh‐RNA (Supplementary Figure S2D and E). MCF‐7 expressing the constitutively active form of p70S6K (p70FER) displayed increased phosphorylation of the downstream target S6, both in the presence or absence of WF (Supplementary Figure S2B). Time course analysis of MDA MB 231 and MDA MB 453 cells expressing the kinase inactive mutant (p70KR) or silenced for p70S6K and stimulated with WF, resulted in decreased p70S6K downstream signaling (Supplementary Figure S2 and not shown). Moreover, we used a specific inhibitor of p70S6K1 activity, PF‐4708671 (hereafter PF) (Pearce et al., 2010a; Segatto et al., 2013), as well as the clinically approved mTOR inhibitor, rapamycin analogue Temsirolimus (hereafter Tems). Treatment with either inhibitor in the presence of serum or WF equally resulted in efficient dampening of p70S6K specific activity (Supplementary Figure S3A–D), while other pathways remained unaltered (Supplementary Figure S3E). Treatment of breast cancer cells with other signaling pathway inhibitors indicated that downregulation of p70S6K activity observed with PF and Tems was specific and not a general consequence of signaling shut down (Supplementary Figure S3E).
3.2. Impact of p70S6K activity on primary tumor growth
To evaluate whether the impairment of p70S6K signaling would impact on breast tumorigenesis, we used an orthotopic xenograft model of breast cancer and injected MDA MB 231 control‐ or p70KR‐cells in mouse mammary fat pads (MFP), using Matrigel to support their initial survival. As expected, impaired p70S6K activity strongly decreased primary breast tumor growth (Figure 2A and B). Next, we injected decreasing numbers of MDA MB 231 control‐ or p70KR‐cells and waited until tumors reached similar volumes before excising the masses (Figure 2C and D). p70KR‐cells grew slower and reached the same tumor mass approximately one week later respect to controls (Figure 2C and D). Then, in order to challenge their ability to initiate tumor growth without any environmental support and possibly discern among the ability of p70S6K signaling to impact on proliferation or survival of breast cancer cells in vivo, we injected cells in MFP without Matrigel. It is interesting to note how in this setting, an intact p70S6K signaling was extremely critical for tumor initiation (67% vs 0%, in control‐ and p70KR‐cells, at low cell number, 1 × 105, n = 12) (Figure 2E). More careful analysis of tumors arising from 2 × 105 and 4 × 105 injected cells suggested that impairment of p70S6K signaling significantly increased the tumor latency (average of 9 vs 26 days, Figure 2F) but, once tumors appeared, their growth rate was very similar (Figure 2G). This observation pointed out that, in the process of tumor initiation, p70S6K signaling played a major role in survival rather than in proliferation of breast cancer cells.
Figure 2.
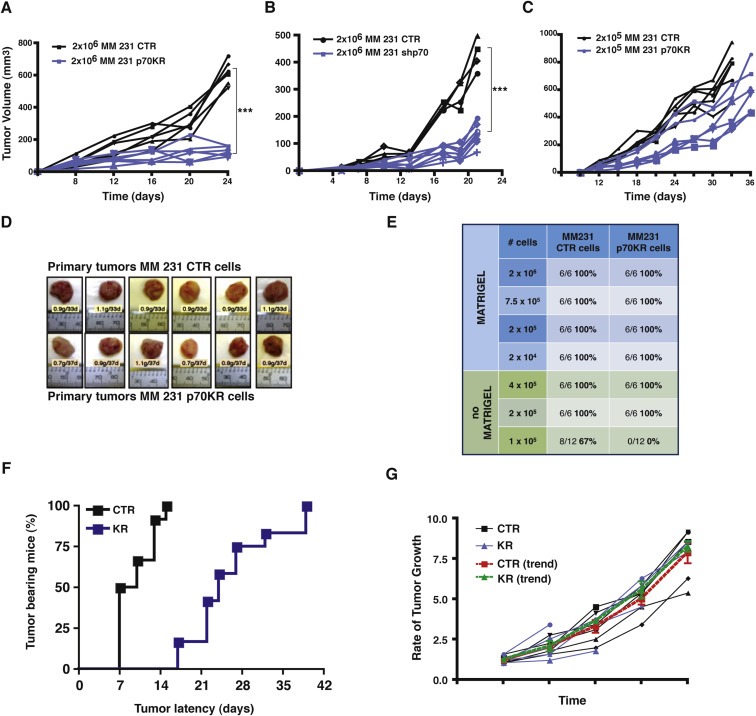
Primary tumor growth is altered by impairing p70S6K activity. A. Growth curves, expressed as tumor volume (mm3), of primary tumors derived from injection of MDA MB 231 cells (2 × 106) stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR), in thoracic mammary fat pads of nude mice (2 FP/mouse) in 50 μl Matrigel/PBS (1:1). Mice were sacrificed after 3 weeks. Statistical significance was calculated using the Student's t‐test, pooling together values of the last measurement of controls vs p70KR. Three asterisks (***) indicate a p value ≤ 0.005. B. Same as in (A) but using MDA MB 231 cells silenced for p70S6K (shp70) in place of the MDA MB 231 cells expressing the p70KR mutant. C. Growth curves, expressed as tumor volume (mm3), of primary tumors derived from injection of MDA MB 231 cells (2 × 105) stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR), in the nude mouse mammary fat pads (2 MFP/mouse) in 50 μl Matrigel/PBS (1:1). Mice were sacrificed when tumor volume was >600 mm3. After 7 weeks, all mice were sacrificed. D. Pictures of the primary tumor masses excised from mice described in (C), injected with MDA MB 231 control cells (upper images) or with p70KR expressing cells (lower images). E. Tumor take‐rate assessed by injection of the indicated numbers of MDA MB 231 cells, stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR), in the presence of absence of Matrigel. F. Graph reports the time dependent appearance of primary tumors derived from injection of MDA MB 231 cells (2 × 105 and 4 × 105) stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR), in the nude mouse mammary fat pads (2 MFP/mouse) without Matrigel. G. Graph reports the rate of tumor growth, independently from the time of appearance, in mice described in (F). Values are expressed as ratio of the tumor volume over the value of 20 mm3, considered as cut off. The red and the green lines represent the trend of growth of the MDA MB 231 CTR and p70KR, respectively.
Primary tumors from both control‐ and p70KR‐cells displayed very similar pathological features (Figure 3A, left panels). However, while tumors arisen from control‐cells invaded the surrounding tissue, the local lymph‐hematological district and the axillary/brachial lymphnodes at considerable extent, tumors from p70KR‐cells did the same at much lower extent (Figure 3A–C).
Figure 3.
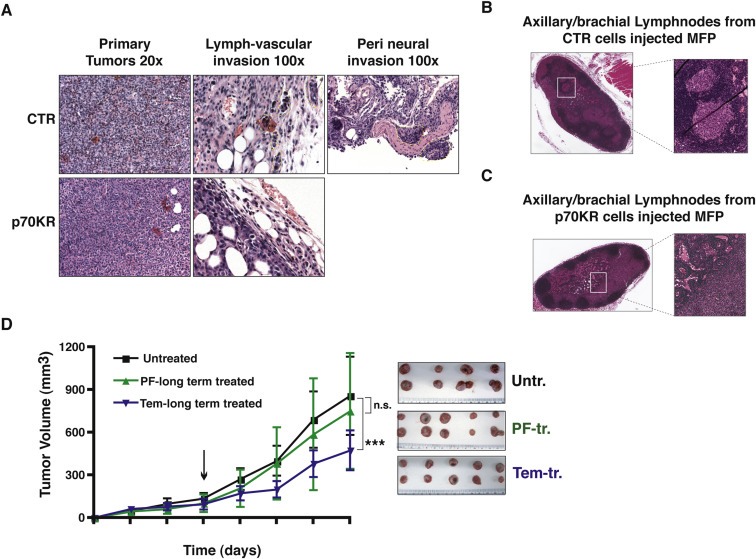
Invasion of surrounding tissue and local lymph‐hematological district was blocked by impairing p70S6K activity. A. Representative images of primary tumor histology (20× objective), stained by hematoxylin and eosin. Presence of lymph‐vascular and perineural invasion by control cells is evidenced by yellow dashed line (100× objective). B and C. Representative images of axillary/brachial lymphnodes, stained by hematoxylin and eosin. Presence of lymphnode metastasis in mice injected with control cells (B), and not with p70KR cells (C), is evidenced. D. Growth curves, expressed as tumor volume (mm3), of primary tumors derived from injection of MDAMB231 control cells (2 × 106), in the thoracic mammary fat pads of nude mice (2 MFP/mouse). Mice were intraperitoneally injected with PF‐4708671 (25 mg/kg, i.e. 600 μg/mouse) or Temsirolimus (12.5 mg/kg, i.e. 300 μg/mouse) or vehicle (untreated), twice a week for three weeks. Data represent the mean (±S.D.) of 10 tumors/treatment. Picture on the right shows the primary tumors, excised at the end of the treatment. Statistical significance was calculated using the Student's t‐test, pooling together values of the last measurement of controls vs p70KR. Three asterisks (***) indicate a p value ≤ 0.005. Difference in tumor volume between Untreated and PF‐treated tumors was not significant (n.s.).
Next, to establish the role of p70S6K in a preclinical model, we used an alternative approach, mimicking a neo‐adjuvant setting. We bilaterally injected MDA MB 231 control cells in mouse MFP and, from appearance of a palpable mass, we treated mice with PF, Tems or vehicle twice a week, for three weeks. No particular sufferance or toxicity was observed during treatment, either with PF or Tems. In line with the established role of mTOR in cell growth, Tems treated mice displayed a considerable decrease in their tumor growth throughout the course of treatment (Figure 3D). Treatment with PF did not elicit a significant decrease in the growth of established primary tumors, indicating that specific p70S6K1 activity is not essential during this stage of tumorigenesis (Figure 3D), in agreement with what observed using p70KR expressing cells (Figure 2G).
3.3. p70S6K activity positively contributes to proliferation and invasion of breast cancer cell lines
Our data in the mouse model indicated that inhibition of p70S6K specifically impairs the ability of breast cancer cells to survive, especially when challenged in more stringent conditions. To better understand the mechanism underlying our in vivo findings and the effects of alteration of p70S6K activity in breast cancer cells we analyzed their proliferative and migratory behavior in vitro. In particular, we focused our experiments on the cell response to WF and to culture conditions that could challenge their ability to survive (hereafter referred as hostile microenvironment).
In line with the well established role of p70S6K in cell proliferation (Fingar and Blenis, 2004; Fenton and Gout, 2011; Maruani et al., 2012), growth curve assays of MCF‐7 cells over expressing the constitutively active form of p70S6K revealed a considerable increase of their proliferative potential (Figure 4A). Conversely, when p70S6K activity was altered by either silencing or expressing the p70KR mutant, moderate but consistent decrease of cell proliferation was always observed (Figure 4B and C and Supplementary Figure S4A and B). Consistently, growth of the parental cells in the continuous presence of PF or Tem led to a similar reduction of proliferation rate (Supplementary Figure S4D). 5% WF used as source of growth stimuli in place of 10% FBS, very efficiently stimulated breast cancer cell proliferation, nonetheless dampening p70S6K signaling was sufficient to obtain effective proliferation arrest (Figure 4B and D and Supplementary Figure S4C). More importantly, using anchorage independent growth assays in soft agar, in the presence of either FBS or WF, we verified that impairment of p70S6K activity by expression of p70KR significantly decreased the cell ability to survive and grow (Figure 4E, CTR vs KR, in FBS p = 0.002). In particular, when WF was used as source of survival and growth stimuli the difference was extremely significant (CTR vs KR, in WF p < 0.0001). This again suggested that, under this stringent condition, the necessity of a robust p70S6K signaling was of particular relevance for the survival of the isolated cells.
Figure 4.
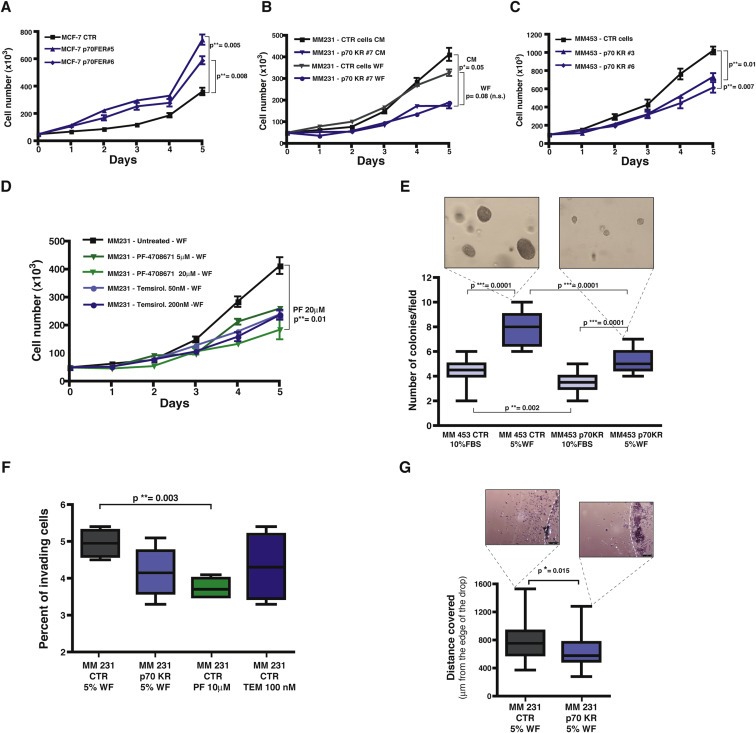
Growth and motility of breast cancer cell lines is altered by impairing p70S6K activity. A. Growth curve analysis of MCF‐7 cell line stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a constitutively active form of p70S6K (FER). Two independent cell clones have been evaluated. Cells (50 × 103/well) have been seeded in complete medium (10% FBS) on day 0, and then counted by Trypan Blue exclusion test, every day for 5 days. Data represents the mean (±S.D.) of two independent experiments performed in triplicate. B. Growth curve analysis of MDA MB 231 cell line stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR#7). Cells (50 × 103/well) have been seeded on day 0, in complete medium (10% FBS, CM) or in the presence of 3% wound fluids (WF), as indicated, and then counted by Trypan Blue exclusion test, every day for 5 days. Data represents the mean (±S.D.) of two independent experiments performed in triplicate. C. Growth curve analysis of MDA MB 453 cell line stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for the kinase inactive mutant of p70S6K (KR). Two independent cell clones have been evaluated. Cells (90 × 103/well) have been seeded in complete medium (10% FBS) on day 0, and then counted by Trypan Blue exclusion test, every day for 5 days. Data represents the mean (±S.D.) of two independent experiments performed in triplicate. D. Growth curve analysis of MDA MB 231 cell line in the presence of the indicated inhibitors. Cells (50 × 103/well) have been seeded in medium containing 3% wound fluids (WF) on day 0, in the presence of PF‐4708671 (5 μM or 20 μM) or Temsirolimus (50 nM or 200 nM) or vehicle (untreated) and then counted by Trypan Blue exclusion test, every day for 5 days. Fresh medium containing the inhibitor was replaced on day 3. Data represents the mean (±S.D.) of two independent experiments performed in triplicate. E. Anchorage independent growth of MDA MB 453 cells stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR). Cells (15 × 103/well) were included in soft agar in presence of complete medium (10% FBS) or 5% wound fluids (WF), for 3 weeks. Medium with serum or wound fluid was replaced twice a week. Graph reports the count of colony number/field evaluated in each condition, using a 10× objective of a contrast phase microscope. At least 20 fields/condition were counted. Pictures show the size of the colonies of MDA MB 453 CTR and p70KR, in the WF culture condition. F. Matrigel invasion assay of MDA MB 231 cells, stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR) or treated with the indicated inhibitor. Cells (1 × 105/transwell) have been seeded on top of a transwell chamber, pre‐coated with 6 μg Matrigel, in duplicate, in the presence of 5% wound fluids (WF) plus PF‐4708671 (10 μM) or Temsirolimus (100 nM), where indicated. The percent of invading cells after 12 h is shown and represents the mean (±S.D.) of two independent experiments performed in duplicate. G. Matrigel evasion assay of MDA MB 231 cells stably transduced with a retroviral empty vector (CTR) or with a retroviral vector encoding for a kinase inactive mutant of p70S6K (KR). Cells (7.5 × 103/12 μl drop) were included in Matrigel in the presence of 5% wound fluids (WF) for 6 days, then fixed and analyzed for the distance covered from the edge of the drop. In all panels, statistical significance was calculated using the Student's t‐test. A p value ≤ 0.05 was considered significant.
Then, given the decreased invasive profile showed by p70KR‐expressing cells in vivo (Figure 3A–C), we looked at the possibility that alteration of p70S6K activity could affect the migratory behavior of breast cancer cells. p70 silenced‐, p70KR expressing‐ and PF or Tems inhibited‐cells all displayed only a modest, although reliable, decrease in their migration rate, when tested in transwell assays (Supplementary Figure S5A–D). The reversal approach, i.e. the overexpression of the constitutively active form of p70, also gave rise to moderate but reproducible increase of the migratory ability (Supplementary Figure S5E). Then, we challenged cells in three‐dimensional (3D)‐invasion or evasion assays, in order to more accurately mimic the in vivo microenvironment. In line with what observed in vivo, invasion of a 3D‐matrix highlighted more significant differences between control cells and cells with impaired p70 signaling, particularly when p70S6K1, and not mTOR, was attacked (Figure 4F, CTR vs PF‐treated p = 0.003; Figure 4G, CTR vs KR p = 0.015).
Altogether, these results support the notion that p70S6K activity positively contributes to proliferation and invasion programs of breast cancer epithelial cells. More importantly, in line with the tumor take rate in vivo, our in vitro data pointed to a specific role played by p70S6K in preserving breast cancer cells survival when challenged in anchorage independent growth.
3.4. p70S6K activity controls survival of breast cancer cells
Results obtained so far together with our previous observation that p70S6K was necessary for breast cancer local recurrence (Segatto et al., 2013) suggested that an intact p70S6K signaling is important for cell survival more than for cell growth. Moreover, our recent work using a mouse model of breast cancer highlighted that the inhibition of mTOR did not lead to the same clinical outcomes of p70S6K1 inhibition in terms of local relapse. A shortage in the negative feedback loop mediated by mTOR leading to increased AKT activation was detected only in Tems‐treated cells, both in the in vitro and in vivo models (Segatto et al., 2013).
To get more molecular insights into this mechanism that was likely to subtend to the decreased survival observed in p70S6K‐impaired cells in hostile environments (2, 4), we performed a phosphoproteomic array comparing the levels of activation and/or expression of more than 500 proteins in Tems‐treated respect to PF‐treated cells. Normalized data filtered for a threshold of 15% of error range confirmed the hyper‐activation of AKT previously observed (Segatto et al., 2013) and highlighted the contextual up‐regulation of the pro‐survival protein Bcl2 (Supplementary Table S1). Western Blot analysis of cells exposed to increasing doses of Tems uncovered a dose‐dependent AKT activation, coupled with phosphorylation of its downstream target FOXO3A and up‐regulation of Bcl2 (Figure 5A).
Figure 5.
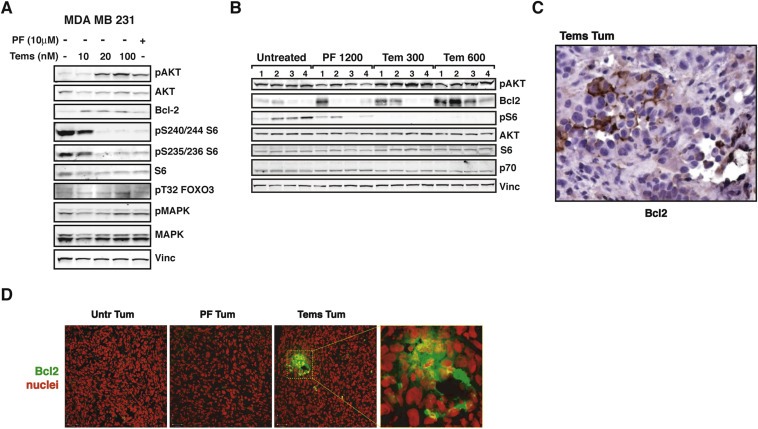
p70S6K1 signaling results in upregulation of Bcl2 in breast cancer cells. A. Western blot analysis of MDA MB 231 cells treated with the indicated inhibitor (PF‐4708671 10 μM or Temsirolimus 10, 20 and 100 nM) or left untreated. Vinculin expression was used as loading control. B. Western blot analysis of primary tumor lysates derived from injection of MDA MB 231 control cells (2 × 106), in nude mouse thoracic mammary fat pads. When tumors reached a volume of 50–100 mm3, mice were intraperitoneally injected with PF‐4708671 (50 mg/kg, i.e. 1200 μg/mouse) or Temsirolimus (12.5 mg/kg, i.e. 300 μg/mouse or 25 mg/kg, i.e. 600 μg/mouse) or vehicle (Untreated) for three consecutive days and then sacrificed. C. Immunohistochemistry analysis of Bcl2 on tumor section from a Tems‐treated mouse of the experiment described in (B). Nor untreated tumors nor those from PF‐treated mice displayed positivity for Bcl2 (magnification 200×). D. Immunofluorescence analysis on tumor sections from experiment described in (B), acquired by confocal microscopy. The panels show the merge of the immunostaining for Bcl2 (AF‐488, pseudocolored in green) with the nuclear staining with propidium iodide (pseudocolored in red). Bars correspond to 35 μm. The panel at the right shows higher magnification of the dashed area.
These findings prompted us to verify whether these molecular events occurred in vivo as well. We bilaterally injected mouse MFP with MDA MB 231 cells and, when palpable masses appeared, we treated tumors for three consecutive days with vehicle, PF or Tems. Then, mice were sacrificed and tumors analyzed. Also in this context, AKT hyper‐activation coupled with Bcl2 up‐regulation were consistently detected in Tems‐treated breast tumors, especially when drug dose was augmented (Figure 5B). Notably, staining of Bcl2, by either IHC (Figure 5C) or IF (Figure 5D), highlighted a specific and discrete pattern of expression inside the tumor, with agglomerations of Bcl2‐positive cells immersed in a Bcl2‐negative mass. This featured pattern could also explain why Bcl2 expression, although overall higher in Tem‐treated tumors respect to PF‐treated ones, was variable among the single tumors (Figure 5B). This observation is also supported by our analysis of protein lysates extracted from paired primary‐ vs relapsed‐tumors (untreated). Although a consistent increase in Bcl2 expression level was always observed in specimens from recurrent disease respect to its paired primary tumor, the absolute level of Bcl2 expression was very variable (data not shown).
This finding supported the notion that, under prolonged Tems treatment, isolated cells activated a survival response that is avoided when only p70S6K1 is specifically inhibited.
Next, we asked what was the mechanism by which p70S6K could regulate Bcl2. It has been recently demonstrated that p70S6K1 specifically phosphorylates Gli1 on S84 and activates it, promoting its nuclear translocation (Wang et al., 2012). Gli1 is a transcription factor mediator of the Hedgehog signaling (Ng and Curran, 2011), known for its crosstalk with p70S6K signaling and for its pro‐survival activity (Wang et al., 2012; Gruber Filbin et al., 2013). Since our experiments highlighted that Bcl2 was critically upregulated in surviving cells and Bcl2 is a transcriptional target of Gli1 (Bigelow et al., 2004), we have evaluated whether Gli1 pathway was implicated. First, we performed IF on the same tumors analyzed above for Bcl2 and stained tumor cells for Gli1 and for pSer84‐Gli1 (Figure 6A). Gli1 and even more pSer84‐Gli1 were well expressed in untreated tumors and in Tems‐treated ones, while their expression was dramatically lower in specimens from PF‐treated masses. These results were confirmed by immunoprecipitating (IP) Gli1 from the same tumors (Figure 6B).
Figure 6.
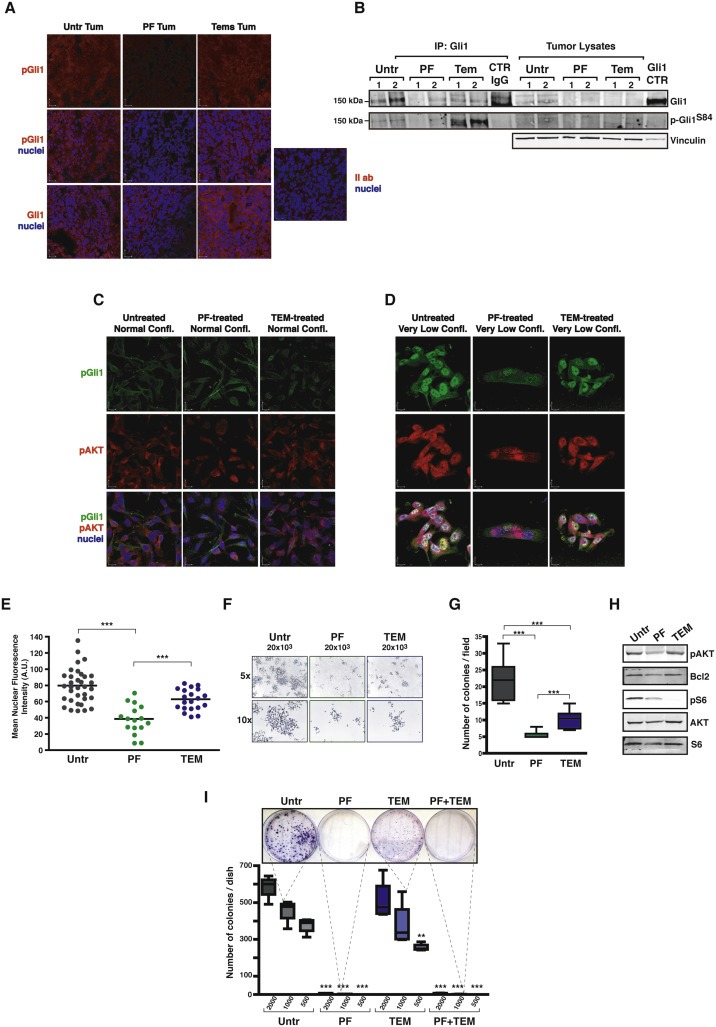
Pro‐survival response of breast cancer cells relies on the p70S6K1/Gli1 axis. A. Immunofluorescence analysis of tumor sections from experiment described in Figure 5B, acquired by confocal microscopy. Panels show the immunostaining for pSer84Gli1 (AF‐546, pseudocolored in red) alone or merged with the nuclear staining with TO‐PRO‐3 Iodide (pseudocolored in blue) or for Gli1 (AF‐546, pseudocolored in red) merged with the nuclear staining with TO‐PRO‐3 Iodide (pseudocolored in blue). Panel on the right represent staining omitting only the primary antibody. Bars correspond to 22 μm. B. Western Blot analysis of tumor protein lysates from experiment described in Figure 5B, immunoprecipitated for Gli1 and blotted for Gli1 and pS84‐Gli1, as indicated. Molecular weight marker of 150 kDa is indicated on the left. Vinculin was used as loading control of lysates. Gli1 CTR indicates a lysate of TOV112D cells, positive control for Gli1 expression. C. Immunofluorescence analysis of MDA MB 231 cells seeded at normal confluence conditions and treated as indicated, acquired by confocal microscopy. Panels show the immunostaining for pSer84Gli1 (AF‐488, pseudocolored in green) or for pAKT (AF‐633, pseudocolored in red) or merged with the nuclear staining with Propidium Iodide (pseudocolored in blue). Bars correspond to 15 μm. D. Same as in (C) except that MDA MB 231 cells were seeded at very low confluence. Bars correspond to 11 μm. E. Graph reports the mean nuclear fluorescence intensity, expressed as arbitrary units (A.U.) of the cells in (D). Values have been calculated measuring at least 16 cells/treatment and using the Volocity software (Perkin Elmer). F. Pictures refer to MDA MB 231 cells plated at very low density (20 × 103 cell/100 mm dish) in the presence of the indicated treatments or left untreated. Both 5× and 10× objectives have been used to capture images. G. Graph reports the number of colonies/field from the experiment described in (F). H. Western blot analysis of protein lysates from cells of the experiment described in (F). I. Colony assay of MDA MB 231 cells plated at ultra‐low density (2 × 103, 1 × 103 or 500 cell/100 mm dish, as indicated) and treated as indicated. Colonies were stained with crystal violet, then photographed (picture above) and counted (graph below). Data represent the counts of two different experiments, performed in duplicate. In all panels, statistical significance was calculated using the Student's t‐test. Two asterisks (**) indicate a p value ≤ 0.01. Three asterisks (***) indicate a p value ≤ 0.005.
These results suggested that the p70S6K1/Gli1 axis could be responsible for the survival observed in breast cancer cells challenged by hostile microenvironment. To prove this hypothesis, we plated cells at normal confluence (70%) or very sparse (single isolated cells), treated them with PF, Tem or vehicle and after 72 h analyzed cells by IF. pGli1, as well as pAKT, were expressed at very low level in cells plated at normal confluence, with only slight difference among the treatments (Figure 6C). However, when cell were plated at very low confluence, expression of pGli1 and pAKT greatly augmented in untreated and in Tem‐treated cells, while it remained very low in PF‐treated ones (Figure 6D). Importantly, pGli1 mainly located in the nuclei of expressing cells, strongly indicating that, under these culture conditions, Gli1 was active (Figure 6D and E). It is interesting to note that PF treatment not only affected pGli and pAKT expression, but also strongly impacted on the survival and subsequent growth of the isolated colonies. As shown in Figure 6F and G, colony size and number were extremely diminished by both Tem and PF treatment, but particularly by the latter one. Proteins extracted from these cells further confirmed that PF‐treated cells consistently displayed diminished Bcl2 expression, thus corroborating our previous observation (Figure 6H). Combined treatment with both inhibitors (PF + Tem) in the presence of WF stimulation recapitulated what observed after PF‐single treatment, both at protein and mRNA level (Supplementary Figure S6A and B), suggesting that specific inhibition of p70SK1 with PF could not be replaced by the inhibition of mTOR. These data are in accord with our previous findings showing how mTOR inhibition was not able to prevent tumor recurrences as PF did (Segatto et al., 2013). Finally, to evaluate whether this mechanism had important functional consequences, we plated a colony assay at very low cell concentration (2 × 103, 1 × 103, 500 cells/100 mm dish). MDA MB 231 cells grew very well also in these stringent culture conditions (Figure 6I). However, inhibition of p70S6K1 led to a striking failure of their survival and to cell death (Figure 6I), resembling what observed in vivo in the take rate of p70KR MDA MB 231 cells at 1 × 105 cells (0% of take rate, Figure 2E).
4. Discussion
The results of our study clearly show that p70S6K signaling is strongly involved in the response of breast cancer cells to post‐surgical inflammatory stimuli. Breast cancer is often a multifocal and multicentric disease, but often these tumor foci are dormant and referred to as "occult cancer" (Bissell and Hines, 2011). Following surgery, clinically undetected clusters of neoplastic cells may remain, either locally or at distant sites that may eventually develop into clinically detectable recurrence. As long as the microenvironment surrounding these tumors provided tumor‐suppressive signals these masses will not progress. For these neoplastic clusters to grow into frank cancer a subversion of tissue homeostasis must occur and signals from the microenvironment need to arrive and awake breast cancer cells (Bissell and Hines, 2011).
Surgery itself does undoubtedly represent one of such subverting factors. It is already known that surgery may modify the growth kinetics of breast cancer micro metastasis (Demicheli et al., 2001). In support of this hypothesis, a recent clinical study comparing radical mastectomy with breast conserving surgery reported that women undergone radical mastectomy without radiotherapy have a significant increased risk of loco‐regional recurrence compared to those treated with breast conserving surgery. The authors suggest that after extensive mastectomy an excessive wound response might stimulate the secretion of as‐yet uncharacterized growth factor(s) that precipitates loco‐regional recurrence (Abdulkarim et al., 2011). This assumption indirectly confirms our (Belletti et al., 2008b; Segatto et al., 2013) and others' (Tagliabue et al., 2003) observations and points to the wound healing process as a driving force for generation of local recurrences.
p70SK protein overexpression in breast cancer has been already associated with increased risk of locoregional recurrence (van der Hage et al., 2004) and with aggressive disease and poor prognosis (Akar et al., 2010), but the mechanisms associated with the role of p70S6K in these processes are not well understood. Our recent work directly correlated the activity of p70S6K with occurrence of breast cancer local relapse (Segatto et al., 2013) and here a mechanistic link is finally provided. Our data show that p70S6K activity in breast cancer cells is required for triggering a survival response via AKT and Bcl2. When breast cancer cells are grown in “hostile” culture conditions (soft agar, Figure 4E; polyHEMA, Segatto et al., 2013; breast microenvironment without Matrigel, Figure 2E) a robust p70S6K signaling is required to overcome apoptosis and then eventually grow. Our in vivo experiments indicate that p70S6K activity strongly contributed to growth of primary breast tumor (Figure 2A). However, more careful analysis indicated that impairment of p70S6K signaling increased the tumor latency (average of 9 vs 26 days, Figure 2F) but, once tumors appeared, their growth rate was very similar (Figure 2G). This observation pointed out that, in the process of tumor initiation, p70S6K signaling plays a major role in survival rather than in proliferation of breast cancer cells. Many data clearly support this conclusion and indicate that these two functions of p70S6K, one in proliferation and one in survival, have different weight depending on the cellular context. When cells are challenged to survive in “hostile” conditions, such as when isolated cells are immersed in stringent environments (soft agar, Figure 4E; take rate at low cell numbers, Figure 2E; colony assay, Figure 6G, G and I) p70S6K role in survival prevails (and precedes) the role in proliferation. On the other hand, when cells are analyzed in “normal” culture conditions (growth curves, Supplementary Fig. S4) or injected in vivo at high number (Figure 2A and B) or with the support of matrigel (Figure 2E), its role in proliferation prevails and is easily observable.
From a molecular point of view, we have identified the crosstalk between p70S6K signaling and the Hedgehog‐Gli1 pathway, as crucial to activate the survival response needed by breast cancer cells to escape apoptosis in critical contexts (Figure 6), as recently suggested by others in other cancers (Wang et al., 2012; Gruber Filbin et al., 2013).
Taken together, our data implicate that p70S6K activity has a major impact on the early steps of tumor growth while only a minor impact in the control of growth of the bulk mass. This observation is suggestive of potential similar effects of p70S6K signaling in all contexts where breast cancer cells face microenvironmental challenges, such as local and, possibly, distant recurrence, as suggested by others (Akar et al., 2010).
Importantly, our results suggest that p70S6K may represent a promising predictive marker for locoregional recurrence; it may help in guiding choice of optimal local therapy and be targeted for efficient peri‐tumoral "sterilization" of microenvironment following surgery. This is of particular importance since it is now well established that local therapy does not only have an impact on locoregional control but also on patients' survival (Clarke et al., 2005).
Current standard treatment for early breast cancer patients is breast‐conserving surgery followed by external beam radiotherapy. Radiotherapy produces a reduction of about two thirds in local recurrence, largely irrespective of the type of patient or treatment (Clarke et al., 2005). Radiotherapy and chemotherapy are spaced out to allow the recovery of normal tissues between treatments. However, surviving cancer cells could proliferate during the intervals, leading to treatment failure. Our in vitro and in vivo data suggest that interfering with p70S6K activity in this time frame could represent a reliable strategy to overcome breast cancer cells repopulation and efficiently block locoregional recurrence.
Drug development for cancer therapy has relied largely on proliferation as an endpoint. However, targeting the crosstalk between cancer cells and components of the microenvironment is likely to provide much more profound clinical benefits. In light of our results, we propose that targeting p70S6K may dampen the dialog occurring between the inflammatory stroma following surgery and the breast cancer cells at a stage that may result crucial for the fate of the tumor as well as the patient.
Author contribution
IS, SB, MS, LF and JA performed the laboratory work and analyzed the data. SM and MM provided patients' wound fluids. AV performed pathological review and immunohistochemical staining of the mouse tissues. IS, GB and BB designed the experiments. BB conceived the study. BB and GB coordinated the study. BB wrote the manuscript. IS, SB, SM, AC, AV, GB and BB critically discussed the data and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Italian Association for Cancer Research (AIRC) to BB (IG 10459), from Regione Friuli Venezia Giulia to GB, from CRO Intramural Grant to GB and SM, from the “Friuli Exchange Program” to SB and to Outgoing AIRC/Marie Curie International Fellowship in Cancer Research to SB.
Conflicts of interest
The authors declare that they have no competing interests.
Supporting information
The following is the Supplementary data related to this article:
Supplementary data
Acknowledgments
We are grateful to the patients who participated to this study by donating post‐surgery wound fluids and to the clinical teams who facilitated sample and data acquisition. We thank Sara D'Andrea and Sara Benevol for expert technical assistance and all members of the S.C.I.C.C. lab for helpful scientific discussion.
Supplementary material 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.02.006.
Segatto Ilenia, Berton Stefania, Sonego Maura, Massarut Samuele, Fabris Linda, Armenia Joshua, Mileto Mario, Colombatti Alfonso, Vecchione Andrea, Baldassarre Gustavo and Belletti Barbara, (2014), p70S6 kinase mediates breast cancer cell survival in response to surgical wound fluid stimulation, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.02.006.
References
- Abdulkarim, B.S. , Cuartero, J. , Hanson, J. , Deschênes, J. , Lesniak, D. , Sabri, S. , 2011. Increased risk of locoregional recurrence for women with T1-2N0 triple-negative breast cancer treated with modified radical mastectomy without adjuvant radiation therapy compared with breast-conserving therapy. J. Clin. Oncol.. 29, 2852–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar, U. , Ozpolat, B. , Mehta, K. , Lopez-Berestein, G. , Zhang, D. , Ueno, N.T. , Hortobagyi, G.N. , Arun, B. , 2010. Targeting p70S6K prevented lung metastasis in a breast cancer xenograft model. Mol. Cancer Ther.. 9, 1180–1187. [DOI] [PubMed] [Google Scholar]
- Baker, D.G. , Masterson, T.M. , Pace, R. , Constable, W.C. , Wanebo, H. , 1989. The influence of the surgical wound on local tumor recurrence. Surgery. 106, 525–532. [PubMed] [Google Scholar]
- Baldassarre, G. , Belletti, B. , Nicoloso, M.S. , Schiappacassi, M. , Vecchione, A. , Spessotto, P. , Morrione, A. , Canzonieri, V. , Colombatti, A. , 2005. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 7, 51–63. [DOI] [PubMed] [Google Scholar]
- Belletti, B. , Nicoloso, M.S. , Schiappacassi, M. , Berton, S. , Lovat, F. , Wolf, K. , Canzonieri, V. , D'Andrea, S. , Zucchetto, A. , Friedl, P. , Colombatti, A. , Baldassarre, G. , 2008. Stathmin activity influences sarcoma cell shape, motility, and metastatic potential. Mol. Biol. Cell. 19, 2003–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belletti, B. , Vaidya, J.S. , D'Andrea, S. , Entschladen, F. , Roncadin, M. , Lovat, F. , Berton, S. , Perin, T. , Candiani, E. , Reccanello, S. , Veronesi, A. , Canzonieri, V. , Trovò, M.G. , Zaenker, K.S. , Colombatti, A. , Baldassarre, G. , Massarut, S. , 2008. Targeted intraoperative radiotherapy impairs the stimulation of breast cancer cell proliferation and invasion caused by surgical wounding. Clin. Cancer Res.. 14, 1325–1332. [DOI] [PubMed] [Google Scholar]
- Belletti, B. , Pellizzari, I. , Berton, S. , Fabris, L. , Wolf, K. , Lovat, F. , Schiappacassi, M. , D'Andrea, S. , Nicoloso, M.S. , Lovisa, S. , Sonego, M. , Defilippi, P. , Vecchione, A. , Colombatti, A. , Friedl, P. , Baldassarre, G. , 2010. p27kip1 controls cell morphology and motility by regulating microtubule-dependent lipid raft recycling. Mol. Cell. Biol.. 30, 2229–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, J.R. , Jatoi, I. , Keisch, M. , Esteva, F.J. , Makris, A. , Jordan, V.C. , 2009. Early breast cancer. Lancet. 373, 1463–1479. [DOI] [PubMed] [Google Scholar]
- Bigelow, R.L. , Chari, N.S. , Unden, A.B. , Spurgers, K.B. , Lee, S. , Roop, D.R. , Toftgard, R. , McDonnell, T.J. , 2004. Transcriptional regulation of bcl-2 mediated by the sonic Hedgehog signaling pathway through gli-1. J. Biol. Chem.. 279, 1197–1205. [DOI] [PubMed] [Google Scholar]
- Bissell, M.J. , Hines, W.C. , 2011. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med.. 17, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network, 2012. Comprehensive molecular portraits of human breast tumours. Nature. 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, M. , Collins, R. , Darby, S. , Davies, C. , Elphinstone, P. , Evans, E. , Godwin, J. , Gray, R. , Hicks, C. , James, S. , MacKinnon, E. , McGale, P. , McHugh, T. , Peto, R. , Taylor, C. , Wang, Y. , Early Breast Cancer Trialists' Collaborative Group (EBCTCG)2005. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15 year survival: an overview of the randomised trials. Lancet. 366, 2087–2106. [DOI] [PubMed] [Google Scholar]
- Couch, F.J. , Wang, X.Y. , Wu, G.J. , Qian, J. , Jenkins, R.B. , James, C.D. , 1999. Localization of PS6K to chromosomal region 17q23 and determination of its amplification in breast cancer. Cancer Res.. 59, 1408–1411. [PubMed] [Google Scholar]
- Demicheli, R. , Valagussa, P. , Bonadonna, G. , 2001. Does surgery modify growth kinetics of breast cancer micrometastases?. Br. J. Cancer. 85, 490–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demicheli, R. , Retsky, M.W. , Hrushesky, W.J. , Baum, M. , 2007. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat. Clin. Pract. Oncol.. 12, 699–710. [DOI] [PubMed] [Google Scholar]
- Düvel, K. , Yecies, J.L. , Menon, S. , Raman, P. , Lipovsky, A.I. , Souza, A.L. , Triantafellow, E. , Ma, Q. , Gorski, R. , Cleaver, S. , Vander Heiden, M.G. , MacKeigan, J.P. , Finan, P.M. , Clish, C.B. , Murphy, L.O. , Manning, B.D. , 2010. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell. 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton, T.,R. , Gout, I.T. , 2011. Functions and regulation of the 70 kDa ribosomal S6 kinase. Int. J. Biochem. Cell Biol.. 43, 47–59. [DOI] [PubMed] [Google Scholar]
- Fingar, D.C. , Blenis, J. , 2004. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 23, 3151–3171. [DOI] [PubMed] [Google Scholar]
- Ghayad, S.E. , Cohen, P.A. , 2010. Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent Pat. Anticancer Drug Discov.. 5, 29–57. [DOI] [PubMed] [Google Scholar]
- Gruber Filbin, M. , Dabral, S.K. , Pazyra-Murphy, M.F. , Ramkissoon, S. , Kung, A.L. , Pak, E. , Chung, J. , Theisen, M.A. , Sun, Y. , Franchetti, Y. , Sun, Y. , Shulman, D.S. , Redjal, N. , Tabak, B. , Beroukhim, R. , Wang, Q. , Zhao, J. , Dorsch, M. , Buonamici, S. , Ligon, K.L. , Kelleher, J.F. , Segal, R.A. , 2013. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nat. Med.. 19, 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay, N. , Sonenberg, N. , 2004. Upstream and downstream of mTOR. Genes Dev.. 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- Huston, T.L. , Simmons, R.M. , 2005. Locally recurrent breast cancer after conservation therapy. Am. J. Surg.. 189, 229–235. [DOI] [PubMed] [Google Scholar]
- Jefferies, H.B. , Fumagalli, S. , Dennis, P.B. , Reinhard, C. , Pearson, R.B. , Thomas, G. , 1997. Rapamycin suppresses 5′ TOP mRNA translation through inhibition of p70S6K. EMBO J.. 16, 3693–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasome, H. , Papst, P. , Webb, S. , Keller, G.M. , Johnson, G.L. , Gelfand, E.W. , Terada, N. , 1998. Targeted disruption of p70(s6k) defines its role in protein synthesis and rapamycin sensitivity. Proc. Natl. Acad. Sci. U. S. A.. 95, 5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruani, D.M. , Spiegel, T.N. , Harris, E.N. , Shachter, A.S. , Unger, H.A. , Herrero-Gonz·lez, S. , Holz, M.K. , 2012. Estrogenic regulation of S6K1 expression creates a positive regulatory loop in control of breast cancer cell proliferation. Oncogene. 31, 5073–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monni, O. , Barlund, M. , Mousses, S. , Kononen, J. , Sauter, G. , Heiskanen, M. , Paavola, P. , Avela, K. , Chen, Y. , Bittner, M.L. , Kallioniemi, A. , 2001. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc. Natl. Acad. Sci. U. S. A.. 98, 5711–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve, R.M. , Chin, K. , Fridlyand, J. , Yeh, J. , Baehner, F.L. , Fevr, T. , Clark, L. , Bayani, N. , Coppe, J.P. , Tong, F. , Speed, T. , Spellman, P.T. , DeVries, S. , Lapuk, A. , Wang, N.J. , Kuo, W.L. , Stilwell, J.L. , Pinkel, D. , Albertson, D.G. , Waldman, F.M. , McCormick, F. , Dickson, R.B. , Johnson, M.D. , Lippman, M. , Ethier, S. , Gazdar, A. , Gray, J.W. , 2006. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, J.M. , Curran, T. , 2011. The Hedgehog's tale: developing strategies for targeting cancer. Nat. Rev. Cancer. 11, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce, L.R. , Alton, G.R. , Richter, D.T. , Kath, J.C. , Lingardo, L. , Chapman, J. , Hwang, C. , Alessi, D.R. , 2010. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J.. 431, 245–255. [DOI] [PubMed] [Google Scholar]
- Pearce, L.R. , Komander, D. , Alessi, D.R. , 2010. The nuts and the bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol.. 11, 9–22. [DOI] [PubMed] [Google Scholar]
- Schalm, S.S. , Blenis, J. , 2002. Identification of a conserved motif required for mTOR signaling. Curr. Biol.. 12, 632–639. [DOI] [PubMed] [Google Scholar]
- Segatto, I. , Berton, S. , Sonego, M. , Massarut, S. , D'Andrea, S. , Perin, T. , Fabris, L. , Armenia, J. , Rampioni, G. , Lovisa, S. , Schiappacassi, M. , Colombatti, A. , Bristow, R.G. , Vecchione, A. , Baldassarre, G. , Belletti, B. , 2013. Inhibition of breast cancer local relapse by targeting p70S6 kinase activity. J. Mol. Cell Biol.. 6, 428–431. [DOI] [PubMed] [Google Scholar]
- Shin, S. , Wolgamott, L. , Yu, Y. , Blenis, J. , Yoon, S.O. , 2011. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc. Natl. Acad. Sci. U. S. A.. 108, E1204–E1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabue, E. , Agresti, R. , Carcangiu, M.L. , Ghirelli, C. , Morelli, D. , Campiglio, M. , Martel, M. , Giovanazzi, R. , Greco, M. , Balsari, A. , Ménard, S. , 2003. Role of HER2 in wound-induced breast carcinoma proliferation. Lancet. 362, 527–533. [DOI] [PubMed] [Google Scholar]
- van der Hage, J.A. , van den Broek, L.J. , Legrand, C. , Clahsen, P.C. , Bosch, C.J. , Robanus-Maandag, E.C. , van de Velde, C.J. , van de Vijver, M.J. , 2004. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br. J. Cancer. 90, 1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Manteuffel, S.R. , Dennis, P.B. , Pullen, N. , Gingras, A.C. , Sonenberg, N. , Thomas, G. , 1997. The insulin-induced signaling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70S6K. Mol. Cell. Biol.. 17, 5426–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Ding, Q. , Yen, C.J. , Xia, W. , Izzo, J.G. , Lang, J.Y. , Li, C.W. , Hsu, J.L. , Miller, S.A. , Wang, X. , Lee, D.F. , Hsu, J.M. , Huo, L. , Labaff, A.M. , Liu, D. , Huang, T.H. , Lai, C.C. , Tsai, F.J. , Chang, W.C. , Chen, C.H. , Wu, T.T. , Buttar, N.S. , Wang, K.K. , Wu, Y. , Wang, H. , Ajani, J. , Hung, M.C. , 2012. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 21, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the Supplementary data related to this article:
Supplementary data