

Abstract
Chronic inflammation is one of the main causes of cancer, yet the molecular mechanism underlying this effect is not fully understood. In this study, we identified FAT10 as a potential target gene of STAT3, the expression of which is synergistically induced by NFκB co‐stimulation. STAT3 binding stabilizes NFκB on the FAT10 promoter and leads to maximum induction of FAT10 gene expression. Increased FAT10 represses the transcriptional activity of the tumor suppressor p53, a protein that accelerates the protein degradation of FAT10. This FAT10‐p53 double‐negative regulation is critical in the control of tumorigenesis, as overexpressed FAT10 facilitates the tumor progression in the solid tumor model. In conclusion, transcriptional synergy between STAT3 and NFκB functions to put weight on FAT10 in the mutually inhibitory FAT10‐p53 regulatory loop and thus favors tumorigenesis under inflammatory conditions.
Keywords: STAT3, FAT10, p53, Inflammation, Tumorigenesis
Highlights
FAT10, a novel target gene of STAT3, links inflammation and tumorigenesis.
STAT3 and NFκB synergistically act for maximum induction of FAT10 expression.
FAT10 suppresses transcriptional activity of p53 and accelerates tumorigenesis.
Double negative regulatory loop present between tumor suppressor p53 and FAT10.
Abbreviations
- TNF-α
Tumor necrosis factor-α
- IL-1β
Interleukin-1β
- STAT3
Signal transducer and activator of transcription 3
- JAK2
Janus kinase 2
- RITA
Reactivation of p53 and induction of tumor cell apoptosis
- CHX
Cycloheximide
- LMP2
Low molecular mass polypeptide 2
- RLU
Relative luciferase units
1. Introduction
Chronic inflammation is one of main risk factors for cancer (Coussens and Werb, 2002; Trinchieri, 2012). It contributes many aspects of carcinogenesis, including cellular proliferation, survival, angiogenesis, and metastasis. Tissue injury or microbial infection at local sites activates immune cells to produce inflammatory cytokines, including tumor necrosis factor‐α (TNF‐α), Interleukin‐1β (IL‐1β), and IL‐6. These inflammatory cytokines produced during acute inflammation aid the healing and regeneration of injured tissue, but sustained production of these cytokines triggers the accumulation of DNA damage and abnormal tissue healing (Grivennikov et al., 2010; Lu et al., 2006). Therefore, inflammatory conditions are generated to protect the body against infection, but provide a niche for cancer development by promoting proliferation or protecting malignant cells from cell death.
In addition to inflammatory cytokines, the transcription factors that mediate the expression of inflammatory effectors are also strongly implicated in carcinogenesis (Li et al., 2011a). During inflammation, secreted cytokines, such as IL‐6, IL‐10, or IL‐17, transmit an intracellular signal through STAT3 and activate the expression of genes that function in cell cycle progression, survival, and stress responses (Aggarwal et al., 2006; Trinchieri, 2012). The tumorigenic effect of STAT3 was initially proposed from the effects of constitutively active STAT3 in nude mice and in human cancers (Bromberg et al., 1999; Xu et al., 2009). An aberrantly active form of STAT3 is commonly found in various human cancer cell lines and primary tumors, including those of the prostate, breast, head, and neck. Tumors with higher STAT3 expression tend to be more aggressive (Mathews et al., 2010). On the other hand, NFκB initiates the transcriptional activation of inflammatory genes, pro‐survival genes, and proliferation‐promoting genes (DiDonato et al., 2012; Grivennikov and Karin, 2010b). Interestingly, STAT3 and NFκB are specifically activated by different groups of inflammatory cytokines, yet co‐regulate similar groups of anti‐apoptotic, pro‐angiogenic, and proliferative genes, with diverse regulatory mechanisms (Grivennikov and Karin, 2010b; Yu et al., 2009). For example, activated STAT3 and NFκB independently bind to cognate DNA elements in the Bcl2/VEGF promoter through direct DNA‐protein interactions, and synergistically enhance target gene expression (Lee et al., 2011; Yu et al., 2009). Activated STAT3 also increases the nuclear retention of NFκB through p300‐mediated acetylation of RelA/p65 (Lee et al., 2009). Depending on the relative amount or activity of each transcription factor under a given condition, the expression profiles of inflammatory mediators or oncogenes alter significantly, which determines the physiological status of the responding cells.
Due to their regulatory roles in the progression of inflammation and carcinogenesis, it is important to identify the physiological target genes of STAT3 and NFκB that contribute to inflammation‐mediated carcinogenesis. Using a combined approach of microarray analysis and computational programs that scan for STAT3 target genes, we have identified potential candidate genes that are overexpressed in both cancer and inflammatory conditions. FAT10 is an 18 kDa protein that is overexpressed in various cancers, including liver, colon, gastrointestinal, and gynecological tumors (Lee et al., 2003). FAT10 is primarily induced by inflammatory cytokines, TNF‐α, but the presence of secondary cytokines, such as IFN‐γ, can increase FAT10 expression further (Lukasiak et al., 2008). In this paper, we demonstrate that IL‐6‐activated STAT3 functions to synergistically enhance NFκB‐mediated FAT10 expression, thus facilitating FAT10‐mediated p53 inhibition, and therefore shifting the balance to favor inflammation‐mediated tumorigenesis.
2. Materials and methods
2.1. Plasmids and siRNA
A full‐length form of human FAT10 and p53 cDNA were cloned into the pFLAG‐CMV1 (Sigma–Aldrich, Saint Louis, MO). For non‐conjugating mutant form of FAT10, the last two amino acids of FAT10 were changed from GG to AA (Raasi et al., 2001). For the p53 non‐functional mutant, a point mutation (CGC[Arg] → CAC[His]) at 175 residues was introduced using site‐directed mutagenesis (Ory et al., 1994). To generate pFAT102.0kb‐luc reporter plasmid, genomic DNA that spans −2.0 kb∼+209 bp region of FAT10 promoters was PCR amplified from the genomic DNA of HepG2 and subcloned into the pGL3‐basic vector (Promega, Madison, WI). Deletion mutants of pFAT102.0kb‐luc were generated as listed in Table S1. STAT3 shRNAs were cloned into pSilencer 2.1 U6‐neo vector (Ambion, Austin, TX). An artificial p53RE‐containing reporter plasmid (p53RE‐luc) was from Dr. Jene Choi (Song et al., 2010). SiRNAs used were synthesized by GenePharma (Shanghai, China) (Table S2).
2.2. In silico screening
Publically available microarrays were obtained from the NCBI Gene Expression Omnibus (GEO) database. For datasets from inflammation in the colon, lung, and liver, a total of 113 arrays from GSE4183, GSE9452, GSE10616, GSE8581, GSE1871, GDS1239 and GSE6764 were used. For the cancer dataset, a total of 153 arrays from the colon (GSE4183 and GDS2609), lung (GSE6044), and liver (GSE6764 and GSE14323) were used. Each dataset was normalized by GC‐RMA for each GSE series and then by a quantile normalization method (Wu et al., 2004). A total of 130 genes with more than two fold induction, compared to control, in both inflammation and cancer datasets were selected. To identify genes that have a similar expression pattern to STAT3, we constructed another expression data matrix by combining 566 Affymetrix CEL files related to inflammation in diverse tissues, which was first normalized by GC‐RMA for each GSE series and then by a quantile normalization method. Given the expression data matrix X, we evaluated a permutation P‐value that is given by
where (similarity between the expression pattern of gene i and STAT3), y i=x i − x STAT3, and is the permuted similarity by randomly permuting the columns of x i. The permutation P‐value was then corrected by the Benjamini–Hochberg FDR controlling procedure (Benjamini and Hochberg, 1995). Further analysis for the STAT binding within candidate gene promoter regions was performed using STAT‐Finder program (Oh et al., 2009).
2.3. Cell culture and transfection
All cells except HepG2 and TC‐1 were maintained in DMEM with 10% fetal bovine serum (FBS)(Hyclone, Logan, UT). HepG2 and TC‐1 were cultured in MEM and RPMI1640 with 10% FBS, respectively. Plasmids were transfected by an Effectene transfection reagent (Qiagene, Hilden, Germany), following the manufacturer's instructions. For cytokine stimulation, rhIL‐6 (10 ng/ml) plus IL‐6sR (10 ng/ml), TNF‐α (10 ng/ml), IL‐1β (10 ng/ml), or IFN‐γ (10 ng/ml) were treated as indicated (all from R&D Systems, Minneapolis, MN).
2.4. Chromatin immunoprecipitation (ChIP)
Cells were fixed in 1% formaldehyde for 20 min and were lysed in sonication buffer (50 mM HEPES [pH 7.9], 140 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate, 0.1% SDS with protease inhibitors). DNA was fragmented using a Vibra Cell Sonicator with a microtip (VC 130 PB; Sonics, Newton, CT), with an average size of 200bp. For immunoprecipitation, STAT3 or p65 antibodies (Santa Cruz Technology) and Protein G Plus/Protein A‐Agarose beads (Calbiochem, Merck KGaA, Darmstadt, Germany) were used. Stepwise washing steps were performed twice with TSE‐1 (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl [pH 8.1]), TSE‐2 (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 500 mM NaCl, 20 mM Tris–HCl [pH 8.1]), Buffer‐3 (0.25 M LiCl, 1% NP‐40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl [pH 8.1]) and TE buffer. After reversal of the cross‐linking in 65 °C for 9 h and DNA precipitation, enriched DNA was analyzed by quantitative PCR. Primer information is provided in Table S3. STAT3 and NFκB specific binding were normalized with input control.
2.5. RNA isolation and analysis
Total RNA was extracted from cell lines and mouse solid tumors using RNAiso reagent (Takara Bio, Shiga, Japan). For cDNA synthesis from RNA, 1 μg of total RNA was reverse‐transcribed with oligo‐dT primer using ImProm‐II Reverse Transcription System (Promega). Quantitative real‐time PCR was performed with StepOne plus (Applied Biosystems) using SYBR‐Premix Ex Taq (Takara). Primer information is provided in Table S4.
2.6. Reporter assay
HepG2, Huh7, HCT116, or HEK293 cells in 24‐well plates were transfected with p53RE‐luc, p21WAF1/CIP1‐luc, or various FAT102.0kb‐luc plasmids (300 ng) together with pRL‐βActin (30 ng) for normalization of transfection efficiency. FLAG‐p53 and/or FLAG‐FAT10 were co‐transfected in some experiments. Dual luciferase assay was performed according to the manufacturer's instructions (Promega).
2.7. Immunoblot analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris–HCl [pH7.5], 150 mM NaCl, 1% NP‐40, 0.5% Na‐deoxycholate, 0.1% SDS with protease inhibitors). Total cell lysates (40 μg per sample) were used for electrophoresis in SDS‐polyacrylamide gels. After Protein transfer from SDS‐PAGE to nitrocellulose membrane (0.45 μm pore size, Whatman International Ltd., Kent, UK) and skim milk blocking, antibodies against the FLAG epitope (Sigma), STAT3 (Transduction Laboratories, Lexington, KY), p‐STAT1 and 3 (Cell Signaling Technology, Danvers, MA), STAT1 and 3 (Santa Cruz Technology), p53 (Cell Signaling Technology), cleaved caspase‐3 (Cell Signaling Technology) and FAT10 (ENZO Life Science Int Inc, Plymoth Meeting, PA) were used. Immunoreactive signals were detected by enhanced chemiluminescence with horseradish peroxidase (Pierce Biotechnology, ThermoFisher Scientific, Waltham, MA). Protein band intensity was quantified by using the ImageJ program (Schneider et al., 2012).
2.8. MTT assay
TC‐1 cells in 24‐well plates were incubated with 0.5 mg/ml Thiazolyl Blue Tetrazolium Bromide (MTT, Sigma) dissolved in PBS for 3hr in 37 °C. After removal the MTT solution, 150 μl of isopropanol : DMSO (9:1) solution was used for incubation for 15 min in 37 °C. MTT value was measured by ELISA in 560 nm and 690 nm (reference).
2.9. Apoptosis assay
HCT116 cells were resuspended in 1× Binding buffer (10 mM Hepes [pH 7.4], 140 mM NaCl, 2.5 mM CaCl2) and were stained with FITC‐conjugated anti‐Annexin V antibody (BD biosciences, Franklin Lakes, NJ) and PI (Sigma–Aldrich) for 20 min at room temperature. Apoptotic cells were then analyzed by Flow cytometry (FACSCalibur, Becton Dickinson, San Jose, CA).
2.10. Mouse experiments
To induce acute inflammation, 100 μg of LPS (Escherichia coli strain 0111:B4, Difco, Detroit, MI) was injected into C57BL/6J mice (8 weeks of age) intraperitoneally in 100 μl PBS. 0, 3 and 6 h after LPS injection, mice were sacrificed by cervical dislocation and the livers were harvested for RNA extraction. For the solid tumor model, 1 × 106 TC‐1 cells (mouse lung epithelial cell line) were subcutaneously injected into the C57BL/6J mice (5 weeks of age) in 100 μl PBS (Seo et al., 2011). Tumor size was measured on days 5, 10, 15 and 20 after injection. After 20 days, mice were sacrificed and tumor mass was measured. For the FAT10 over‐expressing TC‐1 cell line, a puromycin‐resistant gene cassette from the pKO Select Puro V810 vector (Lexicon Genetics Inc, The Woodlands, TX) was subcloned into the pFLAG‐FAT10 vector, then transfected into the TC‐1 cells. Puromycin (5 μg/ml) was used to stably select FAT10‐overexpressing TC‐1 cells. Approval of the study protocol was obtained from the POSTECH Institutional Animal Care and Use Committee. All animal experiments were carried out according to the principles of the NIH Guide for the Care and Use of Laboratory Animals.
2.11. Statistical analysis
All data were presented as mean ± S.E.M and were representative of at least two‐independent experiments with triplication. Statistical differences were calculated using unpaired, two‐tailed student's t‐test with Prism 5 software (GraphPad Software inc., La Jolla, CA). In general, p values less than 0.05 were considered significant.
3. Results
3.1. In silico selection of FAT10 as a potential target gene of STAT3
To identify potential regulatory genes that link inflammation and carcinogenesis, we searched STAT3 target genes that are highly induced during both inflammation and cancer (Figure 1A). For this purpose, we specifically used datasets derived from the colon, liver, and lung, where inflammation exhibited a strong clinical correlation with tumor progression. Since it has previously been reported that STAT3 expressions positively correlate with their target gene expressions (Hutchins et al., 2012; Lassmann et al., 2007; Narimatsu et al., 2001), expression signatures of those genes that were significantly induced in both chronic inflammation and cancer were compared to that of STAT3 (Figure 1B). Among the genes that show a similar expression profile to STAT3, a total of seven genes that exceeded a cut‐off P‐value of less than 10−2 were examined for the presence of STAT binding sites in their promoter regions (Figure 1C). Among them, FAT10 was the most likely target gene of STAT3, as it contains a total of three STAT binding sites in its proximal and distal promoter regions. Expression of FAT10 was also dramatically induced by lipopolysaccharide (LPS)‐initiated inflammatory condition in the mouse liver (Figure 1D). Upon stimulation by inflammatory cytokine IL‐6, about two‐fold induction of FAT10 gene expression was routinely observed after 6 h of stimulation. This induction was significantly reduced by AG490 treatment, an inhibitor of JAK2 tyrosine kinase that phosphorylates STAT3 as well as STAT1 (Gorina et al., 2005) (Figure 1E). Based on these data, we selected FAT10 for further analysis.
Figure 1.
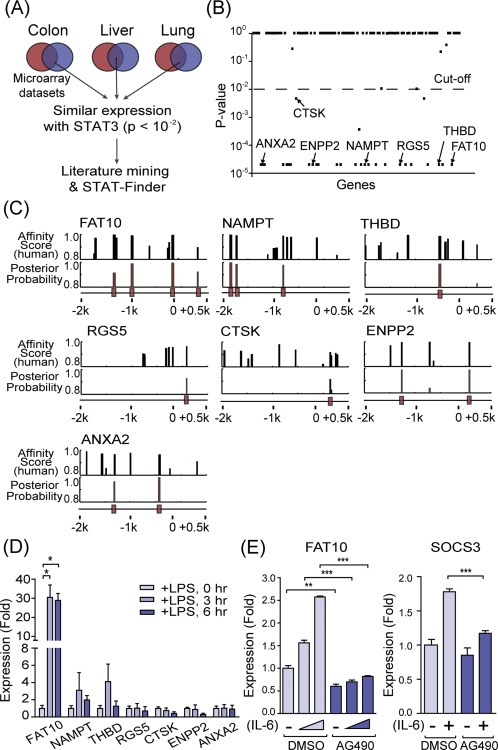
In silico screening of STAT3 target genes induced under conditions of inflammation and cancer. (A) Schematic diagram of the in silico screening. Microarray datasets from chronic inflammation (black circles) and cancers (gray circles) in colon, liver, and lung tissues were used. Overlapping genes were analyzed for expressions and STAT binding sites in their promoter regions. (B) Correlated expression of 112 genes (dot) with STAT3. Genes with P‐values less than 0.01 were selected for further analysis. (C) Prediction of STAT binding sites in the promoter region (from −2.0 kb to +0.5 kb) using STAT‐Finder program. The black bars at the bottom indicate the predicted STAT binding sites which is conserved in the six mammalian species, a posterior probability, higher than 0.8. (D) Expression of candidate genes during acute inflammation were analyzed in the liver of mice injected (i.p.) with 100 μg of LPS. qRT‐PCR analysis normalized with β‐actin. (E) Left, FAT10 expression was analyzed with qRT‐PCR after IL‐6 (10 ng/ml, 0, 1, and 3 h) stimulation in HepG2 cells, with pretreatment with DMSO or AG490 (100 μM, 12hr). Right, SOCS3 expression after 1hr of IL‐6 (10 ng/ml) stimulation. *P < 0.05, **P < 0.01 and ***P < 0.001 by Student's t‐test.
3.2. FAT10 was synergistically induced by co‐treatment of pro‐inflammatory cytokines IL‐6 and TNF‐α
It has previously been reported that FAT10 expression is strongly induced by pro‐inflammatory cytokines TNF‐α or IFN‐γ (Lukasiak et al., 2008). In contrast to these cytokines, IL‐6 stimulation exhibited slight FAT10 induction (Figure 2A). However, when cells were co‐treated with IL‐6 and TNF‐α, FAT10 expression was synergistically increased (Figure 2B). IFN‐γ was not induced in this culture condition (Fig. S1). Time‐course analysis of FAT10 mRNA expression demonstrated that FAT10 reached a maximum steady induction at 12–15 h after TNF‐α treatment (Figure 2C). Under IL‐6 and TNF‐α co‐stimulation, a maximum stable induction was observed at a similar time point after stimulation, but the fold induction was significantly increased. Similarly, a synergistic effect was detected when FAT10 proteins were measured. Although FAT10 protein was barely detectable at basal or IL‐6 alone stimulated cells, a slightly increased FAT10 protein by TNF‐α stimulation was significantly enhanced by IL‐6 co‐stimulation (Figure 2D). To test the effect of the constitutive STAT3 for sustaining the FAT10 expression, we then analyzed FAT10 expression at longer time points after TNF‐α stimulation. As shown in Figure 2E, transient NFκB activation by TNF‐α stimulation was sufficient to enhance and sustain the FAT10 production. This result suggests that IL‐6‐mediated STAT3 activation helps to strengthen the TNF‐α‐mediated NFκB transcriptional induction of FAT10.
Figure 2.
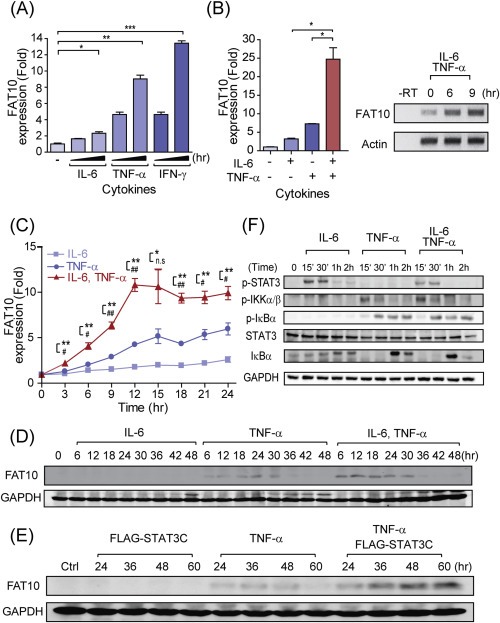
Synergistic induction of FAT10 by combination treatment of pro‐inflammatory cytokines, IL‐6 and TNF‐α. (A) HepG2 cells were treated with IL‐6, TNF‐α, or IFN‐γ (10 ng/ml each) for 0, 3, and 6 h, then FAT10 expression was analyzed with qRT‐PCR. (B) Left, Quantitative RT‐PCR of FAT10 expression with IL‐6, and/or TNF‐α stimulation (10 ng/ml each) for 6 h in HepG2 cells. Right, RT‐PCR of FAT10 expression with IL‐6 and TNF‐α stimulation in HepG2 cells. (C) Kinetics of FAT10 mRNA induction with IL‐6, and/or TNF‐α stimulation (10 ng/ml each) in HepG2 cells. *P‐value between TNF‐α and IL‐6, #P‐value between IL‐6 plus TNF‐α and IL‐6 (D) Kinetics of FAT10 protein induction with IL‐6, and/or TNF‐α stimulation (10 ng/ml each) in HepG2 cells. (E) Cells were transfected with FLAG‐STAT3C for 12hr, before TNF‐α (10 ng/ml) stimulation. Endogenous FAT10 and GAPDH were detected by immunoblot analysis. (F) Comparison of the STAT3 and NFκB activation after IL‐6 and/or TNF‐α stimulation (10 ng/ml each) by immunoblot analysis. For NFκB activation, phosphorylations of IKKα/β and IκBα proteins as well as degradation of IκBα were examined. Tyr705 phosphorylation in STAT3 was used for STAT3 activation. n.s, not significant, *,#P < 0.05 and **,##P < 0.01, and ***P < 0.001 by Student's t‐test.
To investigate whether this synergistic effect by co‐stimulation of IL‐6 and TNF‐α was caused by the enhanced activation of NFκB, phosphorylation of IKKα/β and IκBα were examined in the whole cellular lysates (Figure 2F) (Oeckinghaus et al., 2011). IL‐6 normally induces the phosphorylation of STAT3 after 15–30 min of stimulation. Neither the phosphorylation of IKKα/β and IκBα nor the degradation of IκBα was detected after IL‐6 stimulation. In contrast, both the IKKα/β and IκBα phosphorylation and the degradation of IκBα were similarly observed after treatment with TNF‐α alone and along with IL‐6, at 15 min, 30 min, and 1 h of stimulation, respectively. The rapid induction of IκBα protein after degradation indicates the presence of positive IκBα regulation by TNF‐α, as previously reported (Brown et al., 1993; Yamamoto et al., 2003). These results suggest that the transcriptional synergy between IL‐6 and TNF‐α in FAT10 induction occurs after the activation step of transcription factors.
3.3. Transcriptional synergy between IL‐6 and TNF‐α depends on the distal STAT binding site in the FAT10 promoter region
To investigate the transcriptional synergy between IL‐6 and TNF‐α in the FAT10 induction, A 2.2 kb genomic DNA fragment spanning the promoter and 5′ UTR of human FAT10 was used. This fragment (pFAT102.0kb‐luc) was sufficient to exhibit the transcriptional synergy between IL‐6 and TNF‐α (Figure 3A). In the FAT10 promoter, there are three STAT binding sites, together with at least five NFκB binding sites with affinity scores exceeding the cut‐off value (>0.9) (Figure 3A). Among them, the STAT binding site (STAT1) located in the distal promoter region was flanked by four NFκB binding sites (NFκB1‐4), while another STAT binding site (STAT3), near the transcription start site (TSS), partly overlapped with the NFκB binding site (NFκB5). To identify DNA element in the promoter region that is responsible for the synergistic FAT10 induction, we generated a series of deletion constructs and evaluated their contribution to gene expression in the presence of IL‐6 and/or TNF‐α stimulation. When the distal STAT‐NFκB binding cluster was deleted (pFAT10‐1.0kb‐luc or pFAT10‐2.0kb del STAT1‐ NFκB 1‐4‐luc), both individual IL‐6‐ or TNF‐α‐induced induction of FAT10 expression, as well as the synergistic induction by IL‐6 and TNF‐α co‐treatment, were completely blocked (Figure 3B). In contrast, a reporter construct (pFAT10‐2.0kb del STAT3‐NFκB5‐luc) that lacks the proximal STAT‐NFκB cluster exhibited normal TNF‐α‐induced promoter activity as well as the synergistic induction by IL‐6 co‐treatment (Figure 3B). These results suggest that the STAT‐NFκB cluster in the distal region, but not in the proximal region, is important in the regulation of TNF‐α‐mediated FAT10 induction, as well as in IL‐6‐mediated synergistic induction.
Figure 3.
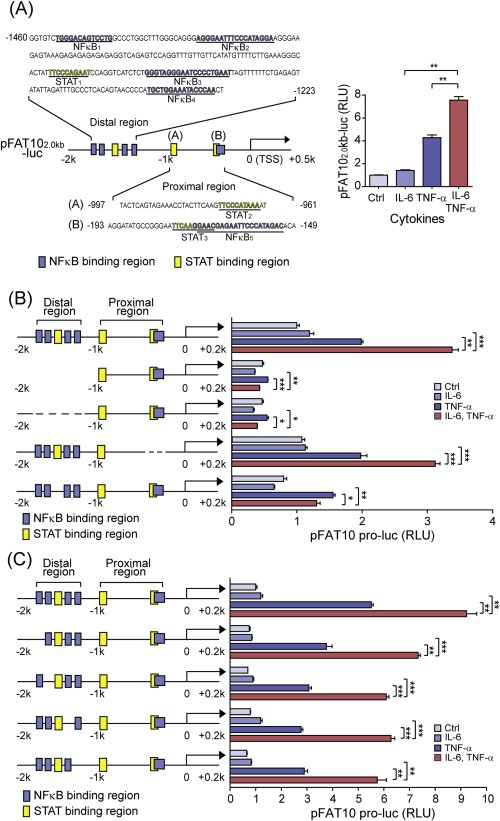
Transcriptional synergy depends on the distal STAT binding site in the FAT10 promoter. (A) Left, Diagram of the pFAT102.0kb‐luc construct, the 2.2 kb genomic DNA fragment of the FAT10, with predicted STAT and NFκB binding sites. Right, FAT10 promoter activation with pFAT102.0kb‐luc was measured after IL‐6 and/or TNF‐α stimulation (10 ng/ml each) for 6 h. (B, C) HepG2 cells were transfected with various reporter constructs, as indicated, and the synergistic FAT10 promoter activation was measured after IL‐6 and/or TNF‐α stimulation for 6 h. *P < 0.05, **P < 0.01, and ***P < 0.001 by Student's t‐test.
To confirm the role of distal STAT‐NFκB cluster in the IL‐6‐mediated synergistic induction of FAT10, another construct (pFAT10‐2.0kb del STAT1‐luc) that lacks the distal STAT binding site was generated, and its effect on FAT10 promoter activity was examined after stimulation (Figure 3B). This deletion construct shows comparable levels of TNF‐α‐induced promoter activity as the pFAT102.0kb‐luc plasmid. However, IL‐6‐mediated synergistic induction of TNF‐α‐dependent FAT10 promoter activity was completely impaired. In contrast, individual deletion of the NFκB binding sites in the distal STAT‐NFκB cluster did not affect either TNF‐α‐induced FAT10 promoter activation or the synergy between TNF‐α and IL‐6 (Figure 3C). However, NFκB binding in the distal STAT‐NFκB cluster was critical to mediate TNF‐α‐dependent FAT10 promoter activation, as total alteration of distal NFκB binding sites blocks FAT10 induction completely (Fig. S2). These results indicate that the STAT3 binding in the FAT10 promoter region is essential to mediate transcriptional synergy between TNF‐α and IL‐6.
3.4. Stable recruitment of p65 to the distal STAT‐NFκB cluster depends on STAT3 binding in the distal STAT binding site in the FAT10 promoter region
To identify the transcription factors responsible for controlling the transcriptional synergy of FAT10 induction, both STAT3 and NFκB (p65) binding to the proximal and distal STAT‐NFκB cluster of the FAT10 promoter were investigated using ChIP assay. Under IL‐6 stimulation, activated STAT3 was mainly recruited to the distal STAT‐NFκB cluster but not to the proximal STAT‐ NFκB cluster, and its binding was further increased by co‐treatment with TNF‐α (Figure 4A). Similarly, TNF‐α‐activated NFκB binding in the FAT10 promoter was mainly observed in the distal STAT‐NFκB cluster (Figure 4B). As observed for STAT3 binding, NFκB binding to the distal STAT‐NFκB cluster was further increased by co‐stimulation with IL‐6, suggesting molecular crosstalk between STAT3 and NFκB in the promoter region of FAT10, which explains the transcriptional synergy between TNF‐α and IL‐6. To confirm this transcriptional synergy, we silenced STAT3 expression and measured NFκB binding in the promoter region of FAT10 under IL‐6‐ and/or TNF‐α−stimulated conditions (Figure 4C and D). When STAT3 was silenced, both TNF‐α‐induced NFκB binding and the IL‐6‐mediated synergistic bindings of NFκB in the distal STAT‐NFκB cluster were significantly diminished. Silencing of STAT3 was independently confirmed by measuring its recruitment to the distal STAT‐NFκB cluster (D2) in FAT10 promoter, and to the promoter of SOCS3, a known STAT3 target gene (Figure 4E). These data suggest that activated STAT3 plays critical roles in the transcriptional induction of FAT10. STAT3 binding in the distal STAT‐NFκB cluster might facilitate the recruitment of NFκB to the adjacent DNA element, thereby enabling the formation of a stable transcriptional complex on the promoter of the FAT10 gene to activate synergistic gene induction.
Figure 4.
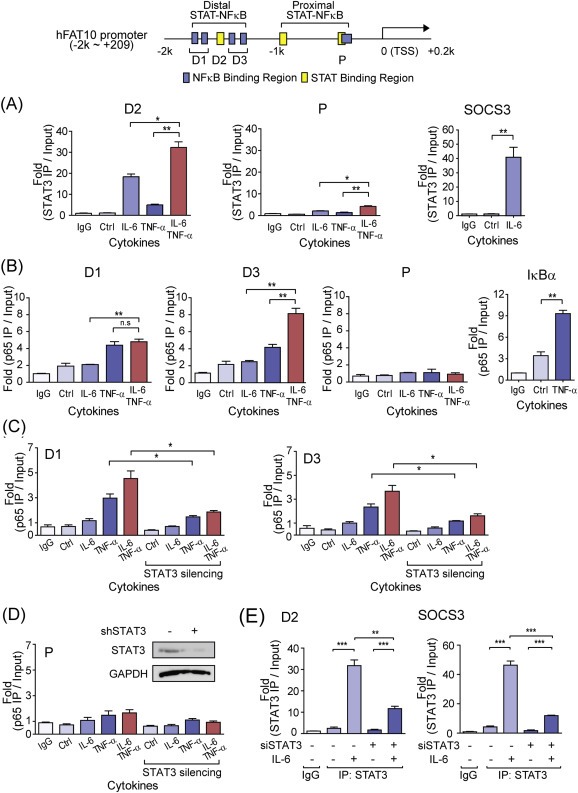
Stable recruitment of p65 to the distal STAT‐NFκB cluster in the FAT10 promoter and transcriptional synergy induced by IL‐6 and TNF‐α. Top, Diagram of the human FAT10 locus with PCR primers (D1, D2, D3, and P) used in this study. Relative binding of STAT3 or p65 to the FAT10 promoter was determined by ChIP analysis in the HepG2 cells, stimulated with IL‐6 and/or TNF‐α for 30 min (A) STAT3 binding to the distal (D2) and proximal (P) region of hFAT10 promoter. SOCS3 was used as a positive control for STAT3 binding. (B) p65 binding to the distal (D1, D3) and proximal (P) region of FAT10 promoter. IκBα were used as a positive control for NFκB binding. (C, D) Relative binding of p65 to the distal (C) or proximal (D) region of FAT10 promoter in the STAT3 silenced HepG2 cells. Cells were stimulated with IL‐6 and/or TNF‐α for 30 min (E) STAT3 binding to the distal (D2) region of FAT10 promoter after STAT3 silencing. All samples were normalized to intergenic input control. n.s, not significant, *P < 0.05 and **P < 0.01 by t‐test.
3.5. STAT3 and STAT1 compensate each other for FAT10 expression
Since IL‐6 activates both STAT1 and STAT3 (Dawn et al., 2004), we next examined the contribution of STAT1 in the transcriptional induction of FAT10 genes. Upon IL‐6 stimulation, STAT1 binding to the distal STAT‐NFκB cluster but not to the proximal STAT‐NFκB cluster was observed, but its binding was relatively weaker than STAT3 binding (Figure 5A). Furthermore, unlike STAT3, synergistic recruitment of STAT1 to the distal STAT‐NFκB cluster after co‐stimulation with TNF‐α was not detected. However, STAT1 binding to the promoter region of FAT10 was significantly enhanced when STAT3 was silenced, indicating that STAT1 and STAT3 compete for promoter binding, and STAT1 substitutes STAT3 when STAT3 is limited (Figure 5B). In support of this finding, FAT10 expression was hardly affected by individual knockdown of STAT1 or STAT3, and was efficiently repressed only when both proteins were silenced (Figure 5C). It is noteworthy to mention that the endogenous STAT3 level was increased when STAT1 was silenced, and the endogenous STAT1 was increased when STAT3 was silenced. Although it has previously been reported that STAT1 and STAT3 compete for the opposite function in the tumorigenesis, it seems STAT1 and STAT3 compensate for FAT10 expression in our system (Huang et al., 2002; Souissi et al., 2012; Xu et al., 2009).
Figure 5.
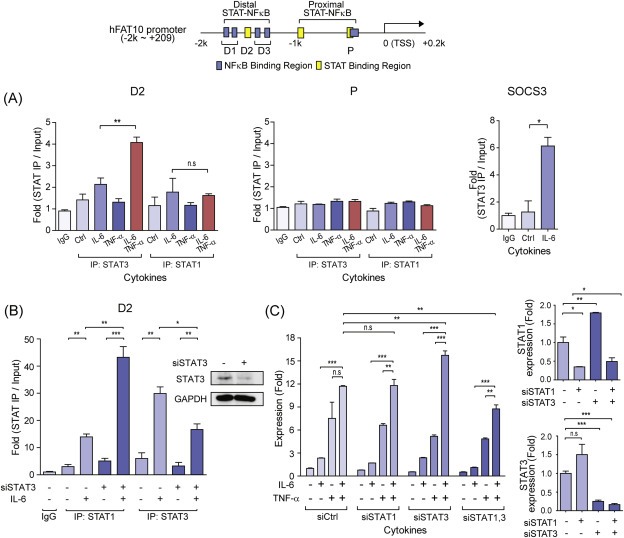
STAT3 and STAT1 compensate for FAT10 expression. Top, Diagram of the human FAT10 locus with PCR primer regions (D1, D2, D3, and P) used in this study. Relative binding of STAT1 or STAT3 to the FAT10 promoter was determined by ChIP analysis in the HepG2 cells, stimulated with IL‐6 and/or TNF‐α (10 ng/ml each) for 30 min (A) ChIP analysis of distal (D2) and proximal (P) STAT binding region of FAT10 promoter with STAT1 or STAT3 after stimulation with indicated cytokines in HepG2 cells. SOCS3 was used as positive control for STAT3 binding. (B) ChIP analysis of distal STAT binding region (D2) of FAT10 promoter with STAT1 or STAT3 after STAT3 silencing, stimulated with IL‐6 (10 ng/ml) for 30 min. STAT3 silencing was confirmed by western blotting by STAT3 antibody. All ChIP samples were normalized to intergenic input control. (C) Left, quantitative RT‐PCR of FAT10 expression after IL‐6 and/or TNF‐α stimulation (10 ng/ml each) for 3 h in the STAT1 and/or STAT3 silenced HepG2 cells. Right, relative STAT1 and STAT3 expressions after STAT1 and/or STAT3 silencing were shown. n.s, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001 by Student's t‐test.
3.6. Overexpressed FAT10 suppresses transcriptional activity of p53 and accelerates tumorigenesis
FAT10 controls diverse cellular processes, such as apoptosis, cell cycle, and immune responses, by covalently modifying its target proteins (Ren et al., 2011). Recently, the tumor suppressor p53 has been identified as a target substrate of FAT10, and its transcriptional activity is increased by FAT10 conjugation (Li et al., 2011b). However, under the inflammatory conditions that induce FAT10 expression, we observed that transcriptional activity of p53 was decreased, measured by the luciferase activity of an artificial p53‐reporter construct (Figure 6A), or of the natural promoter of p21WAF1/CIP1, a target gene of p53 (Figure 6B). To investigate whether FAT10 directly alters transcriptional activity of p53, exogenous FAT10 was transiently expressed in Huh‐7, HCT116, HEK293 or MEF cells, and the transcriptional activity of p53 was measured without further stimulation (Figure 6C,D and E). Since it is already known that p53 represses endogenous FAT10 expression (Zhang et al., 2006), we used Flag‐tagged FAT10, whose expression is controlled under a CMV promoter, and is independent to p53. In every cell tested, transiently expressed Flag‐FAT10 reduced transcriptional activity of exogenous Flag‐p53 or endogenous p53, without significant alterations on p53 protein levels. When FAT10 was silenced, p53 transcriptional activity was increased, at both basal and cytokine stimulated conditions (Figure 6F). In support of this finding, FAT10 silencing significantly increased the cellular apoptosis (Figure 6G), at both basal and p53 stimulated conditions. To specifically activate transcriptional activity of p53, we used RITA (reactivation of p53 and induction of tumor cell apoptosis) (Issaeva et al., 2004).
Figure 6.
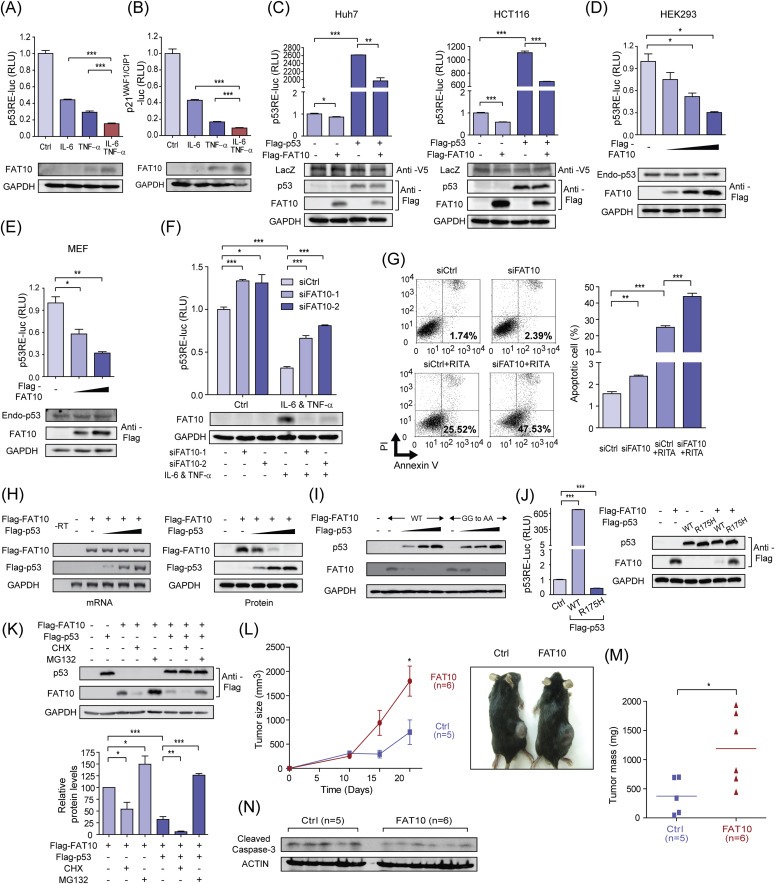
Overexpressed FAT10 suppresses transcriptional activity of p53 and accelerates tumorigenesis. (A, B) HepG2 cells were transfected with reporter construct of artificial p53‐responsive element (A, p53RE‐luc), or p53‐responsible natural promoter (B, p21WAF1/CIP1‐luc), and luciferase activity was determined 12 h after IL‐6 and/or TNF‐α stimulation (10 ng/ml each). Bottom panels show endogenous FAT10 expression at each condition. (C) Huh7 and HCT116 cells were transiently transfected with expression plasmids of FLAG‐p53 or FLAG‐FAT10, and transcriptional activity of p53 was measured by reporter construct with an artificial p53‐responsive element (p53RE‐luc). V5‐LacZ plasmid was used as a transfection control. (D, E) HEK293 (D) or MEF (E) cells were transiently transfected with increasing amounts of FLAG‐FAT10 plasmids, and transcriptional activity of p53 was measured. Endogenous p53 was measured by western blot analysis. (F) HepG2 cells were transfected with control or FAT10‐specific siRNAs, and transcriptional activity of p53 was measured (top). Endogenous FAT10 expression was determined (bottom). (G) HCT116 cells were transfected with control or FAT10‐specific siRNAs and treated with 30 μM RITA for 12 h to induce apoptosis. Flow cytometry analysis was then performed with annexin V and PI double‐staining. Left, one of the three independent experiments is shown. Right, the graph shows the mean +/− standard deviations of apoptotic cells. (H) Exogenously over‐expressed FAT10 mRNA and protein were measured by RT‐PCR (Left) and western blot analysis (Right), respectively, with increased FLAG‐p53 overexpression in the HCT116 p53‐null cells. RT‐PCR primers were designed to specifically detect mRNAs of Flag‐FAT10 or Flag‐p53. (I) WT or conjugation‐mutant forms of FAT10 (GG to AA) were similarly degraded by p53 overexpression, in a dose‐dependent manner in the HCT116 p53‐null cells. (J) Left, Transcriptional activity of p53 (WT) or p53 (R175H) mutant was determined by luciferase assay with p53RE‐luc in HCT116 p53‐null cells. Right, FAT10 degradation by p53 (WT) or p53 (R175H) mutant was tested in HCT116 p53‐null cells. (K) Top, Proteasome‐dependent degradation of FAT10 by exogenous p53 was shown in the HCT116 p53‐null cells. Cells were treated with CHX (50 μg/ml) or MG132 (20 μM) for 3 h, and expression of exogenous p53 and FAT10 proteins was detected. Bottom, band intensity was determined from three‐independent experiments by imageJ programs. (L) Control‐ or FAT10‐overexpressing TC‐1 cells were injected subcutaneously and growth of the solid tumor was measured at day 5, 10, 15, 20. Ctrl (n = 5), FAT10 (n = 6). (M) Tumor mass was measured after scarification at day 20. Dashed lines indicate the median value. (N) Cleaved caspase‐3 was detected in the sacrificed mouse tumors. *P < 0.05, **P < 0.01 and ***P < 0.001 by Student's t‐test.
In this set of experiments, we routinely observed that protein levels of exogenous Flag‐FAT10 were significantly lowered in the p53‐overexpressing cells (Figure 6C). To confirm this phenomenon, cells were transfected with increasing amounts of Flag‐p53, and both mRNA and protein levels of Flag‐FAT10 were measured (Figure 6H). With increasing Flag‐p53 expression, Flag‐FAT10 protein rapidly disappeared, while transfected Flag‐FAT10 mRNA was not significantly affected. This result indicates that degradation of FAT10 protein might be regulated in a p53‐dependent manner. For this p53‐mediated FAT10 degradation, conjugation activity of FAT10 was not required, as the non‐conjugating form of the FAT10 mutant (FAT10AA) was similarly affected by p53 (Figure 6I). Fattylation was intact, as FAT10 forms high molecular weight conjugates and E1 and E2 ligases responsible for fattylation of target proteins, USE1 and UBA6, were expressed (Fig. S3). Also, p53‐mediated FAT10 degradation requires the transcriptional activity of p53, as the transcriptionally inactive p53 (R175H) mutant failed to degrade FAT10 protein (Figure 6J). Treatment with MG132, a 26S proteasome inhibitor, abolished the inhibitory effect of overexpressed p53 on FAT10 protein, indicating that the proteasome‐dependent degradation of FAT10 protein was mediated by p53 protein (Figure 6K). These results all together suggest that p53 induces the expression of a cellular factor that controls the protein stability of FAT10.
To investigate the involvement of FAT10 in the tumorigenesis, we finally generated a FAT10‐overexpressing TC‐1 mouse lung cancer cell, and tested its effect in the solid tumor mouse model. FAT10 over‐expression itself did not significantly affect cell proliferation, but repressed p53 transcriptional activity in TC‐1 cells, as observed in other cells (Fig. S4). Empty vectors and FAT10‐overexpressing TC‐1 cells were then subcutaneously administered to C57BL/6J mice, and tumor growth was examined over two weeks. In FAT10‐overexpressing TC‐1 cells, more facilitated tumor growth relative to the empty vector‐transfected control cancer cells was observed (Figure 6L and M). Consequently, less cleavage of caspase‐3 were observed in the tumors derived from the FAT10‐overexpressing TC‐1 injected mice (Figure 6N). Taken together, these data indicates that the maintenance of the balanced levels of FAT10 and p53 is important for fine‐tuned regulation of p53, and overexpressed FAT10 functions to repress the transcriptional activity of p53, thereby augmenting tumor growth and tumorigenesis.
4. Discussion
In this study, we demonstrated that transcriptional synergy between STAT3 and NFκB functions to boost FAT10 expression, a gene that negatively balances cellular levels of p53 and thereby controls the p53‐mediated tumorigenesis (Figure 7). We and others showed that FAT10 and p53 are mutually inhibitory, with multiple layers of negative regulatory loops functioning at various levels of transcription and protein degradation. Under inflammatory conditions that STAT3 is active, or in a cellular condition that constitutively active STAT3 is present, transient NFκB activation mediated by inflammatory cytokines might lead to massive FAT10 production, which is sufficient to repress p53 activity and thereby favors tumorigenesis.
Figure 7.
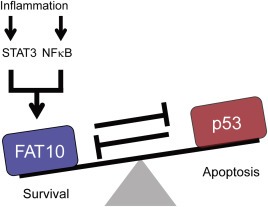
Proposed model of STAT3 and its target gene FAT10 in the regulation of carcinogenesis. At steady‐state, tumor suppressor p53 is counter‐balanced by FAT10. Under inflammatory situation, transcriptional synergy between STAT3 and NFκB boosts FAT10 production, which overcomes the inhibition by p53 and inhibits p53.
Due to the close link between chronic inflammation and the progression of cancer, the molecular mechanisms of inflammation that favor carcinogenesis have been actively searched (Aggarwal et al., 2006; Kundu and Surh, 2012; Kuraishy et al., 2011). Among them, the over‐activation of STAT3 and NFκB plays a critical role in the transition between inflammation and carcinogenesis, as multiple lines of evidences support this possibility. First, constitutive forms of both STAT3 and NFκB are frequently observed in human cancers (Grivennikov and Karin, 2010a; Yu et al., 2009). Second, there are multiple levels of crosstalk between STAT3 and NFκB, which strengthens signaling activity derived from them. For example, independent binding of STAT3 and NFκB to their cognate DNA elements synergistically enhances target gene expression, as reported in Bcl‐2 or VEGF (Lee et al., 2011; Yu et al., 2009). Unphosphorylated STAT3 physically interacts with NFκB through direct protein–protein association, and aids transcription of NFκB target genes, such as RANTES (Yang et al., 2007). In addition, STAT3‐mediated signaling activity is required to maintain constitutive NFκB level, which up‐regulates anti‐apoptotic genes and oncogenes, thus helping the initiation and maintenance of cancer (Lee et al., 2009).
It has previously been suggested that STAT3 and NFκB functions as a p53 repressor, since the blockage of STAT3 or NFκB activity up‐regulates p53 and p53‐mediated apoptosis in human cancers (Niu et al., 2005; Tergaonkar et al., 2002). However, the molecular mechanism that explains this phenomenon has not been adequately proposed. Here, we propose that FAT10 might be a molecular link that explains the inhibitory effect of STAT3 and NFκB on p53 activity. However, further study is required in order to figure out the detailed inhibitory mechanisms of FAT10 on p53.
FAT10 is a diubiquitin‐like protein, and is reported to be over‐expressed in the various human cancers, like liver, colon, and gastrointestinal tumors. These tumors are also reported with constitutively activated form of STAT3 (Ji et al., 2009; Lee et al., 2003; Xu et al., 2009). FAT10 is known to induce the low molecular mass polypeptide 2 (LMP2), an immunoproteasome subunit, which increases IκBα degradation, and results in enhanced NFκB activation (Gong et al., 2010). Furthermore, FAT10 silencing increased apoptosis induced by TNF‐α, indicating that FAT10 works in anti‐apoptotic processes (Ren et al., 2011). In this report, we also reported that FAT10 plays essential role in the maintenance of the normal range of p53 activity.
Although there is a report indicating that FAT10 conjugation increases p53 activity in some conditions (Li et al., 2011b), we have demonstrated that FAT10 and p53 are mutually inhibitory. Upon transient stimulation with inflammatory signals, FAT10 is temporarily induced but soon returns to its basal level due to its p53‐FAT10 double negative loop. A similar mode of regulation is also reported in the IKKβ‐dependent NFκB signaling pathways driven by inflammatory signals. This double negative regulation makes NFκB signaling temporary, to allow effective control of the acute phase response during inflammation, but to prevent sustained NFκB signaling that might lead to carcinogenesis (Gudkov et al., 2011).
Using an in silico screening strategy, we aimed to identify novel target genes of STAT3 that might link inflammation and tumorigenesis. Towards this end, we initially selected a group of genes that are co‐expressed in both inflammation and cancer conditions. Then, we used two criteria to select STAT3 target genes; first, we compared their gene expression signature with that of STAT3, and second, we searched their promoter sequences for STAT3 binding sites. This approach was quite effective, but due to signaling redundancy of JAK2 kinase and similarity in the DNA binding sequences, our in silico screening approach could not distinguish between STAT1 and STAT3 effectively. Nonetheless, transcriptional synergy of FAT10 induced by IL‐6 and TNF‐α co‐stimulation was limited to only STAT3, and not STAT1, even though IL‐6 activates both STAT1 and STAT3. When STAT3 expression is blocked, STAT1 occupies the STAT binding region of the FAT10 promoter and controls its expression. This result is somewhat surprising, since STAT1 and STAT3 are reported to antagonizing each other (Hu and Ivashkiv, 2009; Souissi et al., 2012; Wang et al., 2011). Since STAT1 generally promotes apoptosis via induction of the pro‐apoptotic gene, such as Bax, and STAT3 induces anti‐apoptotic genes, such as Bcl‐xL and Bcl‐2 (Liu et al., 2010; Wang et al., 2012), STAT1 and STAT3 have been proposed to compete for tumorigenesis. However, our finding provides a substitute role of STAT1 and STAT3 in the regulation of FAT10, which balances with the tumor suppressor p53 for proper control of inflammation and tumorigenesis. Therefore more extensive research has to be performed to fully understand the composite contribution of STAT family members in the regulation of inflammation and cancer progression.
5. Competing interests
The authors have declared that no competing interests exist.
Supporting information
The following is/are the supplementary data related to this article:
Fig. S1. IFN‐γ was not induced by IL‐6 and TNF‐α co‐treatment. HepG2 cells were stimulated with IL‐6 and TNF‐α (10 ng/ml each) for 6 h and IFN‐γ induction was examined by RT‐PCR. As a positive control, PMA (100 ng/ml)/Ionomycin (1 ng/ml) treated Jurkat cells were used.
Fig. S2. FAT10 promoter assay with distal NFκB deletion mutant. HepG2 cells were transfected with four NFκB binding sites deletion mutant in distal region, as indicated, and the synergistic FAT10 promoter activation was measured after IL‐6 and/or TNF‐α stimulation for 6 h. n.s, not significant, **P < 0.01, and ***P < 0.001 by Student's t‐test.
Fig. S3. Conjugate formation of FAT10. (A) UBA6 and USE1 (E1 and E2 enzyme for FAT10 conjugation) expression was analyzed with RT‐PCR in HepG2, HCT116 (p53‐null), and HEK293 cells. (B) HEK293FT cells were transfected with FAT10 (GG) or conjugation‐mutant forms of FAT10 (AA) for 48 h, then MG132 (20 μM) was treated for 6 h. After cell lysis and IP with anti‐FLAG, immunoblot analysis was performed with anti‐FLAG.
Fig. S4. Control‐ or FAT10‐overexpressing TC‐1 in the solid tumor model. (A, B) Cell proliferation of control‐ or FAT10‐overexpressing TC‐1 cells were examined by cell counting (A) and MTT assay (B). (C) Transcriptional activity of p53 was compared between control‐ or FAT10‐overexpressing TC‐1 cells. (D) Diagram of primer sets used for detecting endogenous or exogenous FAT10. (E, F) Flag‐FAT10 over‐expression was confirmed in the control‐ or FAT10‐overexpressing TC‐1 stable cells (E) and in the sacrificed mice tumors (F). (G) Flag‐FAT10 over‐expression was examined in control‐ or FAT10‐overexpressing TC‐1 stable cells after DMSO or MG132 (10 μM, 12hr) by western blotting. **P < 0.01 by Student's t‐test.
{kind=link}
Supplementary data
Acknowledgments
We thank Dr. Young Min Oh for technical advice on in silico screening and Nguyen Thi Hong Nhung for technical support. This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF‐2012R1A2A2A01007525, NRF‐2012R1A4A1028200).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.01.007
Choi Yongwook, Kim Jong Kyoung and Yoo Joo-Yeon, (2014), NFκB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.01.007.
References
- Aggarwal, B.B. , Shishodia, S. , Sandur, S.K. , Pandey, M.K. , Sethi, G. , 2006. Inflammation and cancer: how hot is the link?. Biochem. Pharmacol.. 72, 1605–1621. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , Hochberg, Y. , 1995. Controlling the false discovery rate – a practical and powerful approach to multiple testing. J. Roy Stat. Soc. B Met.. 57, 289–300. [Google Scholar]
- Bromberg, J.F. , Wrzeszczynska, M.H. , Devgan, G. , Zhao, Y. , Pestell, R.G. , Albanese, C. , Darnell, J.E. , 1999. Stat3 as an oncogene. Cell. 98, 295–303. [DOI] [PubMed] [Google Scholar]
- Brown, K. , Park, S. , Kanno, T. , Franzoso, G. , Siebenlist, U. , 1993. Mutual regulation of the transcriptional activator NF-kappa B and its inhibitor, I kappa B-alpha. Proc. Natl. Acad. Sci. U S A. 90, 2532–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens, L.M. , Werb, Z. , 2002. Inflammation and cancer. Nature. 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawn, B. , Xuan, Y.T. , Guo, Y. , Rezazadeh, A. , Stein, A.B. , Hunt, G. , Wu, W.J. , Tan, W. , Bolli, R. , 2004. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc. Res.. 64, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato, J.A. , Mercurio, F. , Karin, M. , 2012. NF-kappaB and the link between inflammation and cancer. Immunol. Rev.. 246, 379–400. [DOI] [PubMed] [Google Scholar]
- Gong, P. , Canaan, A. , Wang, B. , Leventhal, J. , Snyder, A. , Nair, V. , Cohen, C.D. , Kretzler, M. , D'Agati, V. , Weissman, S. , Ross, M.J. , 2010. The ubiquitin-like protein FAT10 mediates NF-kappaB activation. J. Am. Soc. Nephrol.. 21, 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina, R. , Petegnief, V. , Chamorro, A. , Planas, A.M. , 2005. AG490 prevents cell death after exposure of rat astrocytes to hydrogen peroxide or proinflammatory cytokines: involvement of the Jak2/STAT pathway. J. Neurochem.. 92, 505–518. [DOI] [PubMed] [Google Scholar]
- Grivennikov, S.I. , Greten, F.R. , Karin, M. , 2010. Immunity, inflammation, and cancer. Cell. 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov, S.I. , Karin, M. , 2010. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev.. 21, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov, S.I. , Karin, M. , 2010. Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev.. 20, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov, A.V. , Gurova, K.V. , Komarova, E.A. , 2011. Inflammation and p53: a tale of two stresses. Genes Cancer. 2, 503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Ivashkiv, L.B. , 2009. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 31, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. , Bucana, C.D. , Van Arsdall, M. , Fidler, I.J. , 2002. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 21, 2504–2512. [DOI] [PubMed] [Google Scholar]
- Hutchins, A.P. , Poulain, S. , Miranda-Saavedra, D. , 2012. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 119, e110–119. [DOI] [PubMed] [Google Scholar]
- Issaeva, N. , Bozko, P. , Enge, M. , Protopopova, M. , Verhoef, L.G. , Masucci, M. , Pramanik, A. , Selivanova, G. , 2004. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med.. 10, 1321–1328. [DOI] [PubMed] [Google Scholar]
- Ji, F. , Jin, X. , Jiao, C.H. , Xu, Q.W. , Wang, Z.W. , Chen, Y.L. , 2009. FAT10 level in human gastric cancer and its relation with mutant p53 level, lymph node metastasis and TNM staging. World J. Gastroenterol.. 15, 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu, J.K. , Surh, Y.J. , 2012. Emerging avenues linking inflammation and cancer. Free Radic. Biol. Med.. 52, 2013–2037. [DOI] [PubMed] [Google Scholar]
- Kuraishy, A. , Karin, M. , Grivennikov, S.I. , 2011. Tumor promotion via injury- and death-induced inflammation. Immunity. 35, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann, S. , Schuster, I. , Walch, A. , Gobel, H. , Jutting, U. , Makowiec, F. , Hopt, U. , Werner, M. , 2007. STAT3 mRNA and protein expression in colorectal cancer: effects on STAT3-inducible targets linked to cell survival and proliferation. J. Clin. Pathol.. 60, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C.G. , Ren, J. , Cheong, I.S. , Ban, K.H. , Ooi, L.L. , Yong Tan, S. , Kan, A. , Nuchprayoon, I. , Jin, R. , Lee, K.H. , Choti, M. , Lee, L.A. , 2003. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene. 22, 2592–2603. [DOI] [PubMed] [Google Scholar]
- Lee, H. , Deng, J. , Xin, H. , Liu, Y. , Pardoll, D. , Yu, H. , 2011. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-kappaB in tumors. Cancer Res.. 71, 3772–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. , Herrmann, A. , Deng, J.H. , Kujawski, M. , Niu, G. , Li, Z. , Forman, S. , Jove, R. , Pardoll, D.M. , Yu, H. , 2009. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 15, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Grivennikov, S.I. , Karin, M. , 2011. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell. 19, 429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T. , Santockyte, R. , Yu, S. , Shen, R.F. , Tekle, E. , Lee, C.G. , Yang, D.C. , Chock, P.B. , 2011. FAT10 modifies p53 and upregulates its transcriptional activity. Arch. Biochem. Biophys.. 509, 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Li, P.K. , Li, C. , Lin, J. , 2010. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J. Biol. Chem.. 285, 27429–27439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H. , Ouyang, W. , Huang, C. , 2006. Inflammation, a key event in cancer development. Mol. Cancer Res.. 4, 221–233. [DOI] [PubMed] [Google Scholar]
- Lukasiak, S. , Schiller, C. , Oehlschlaeger, P. , Schmidtke, G. , Krause, P. , Legler, D.F. , Autschbach, F. , Schirmacher, P. , Breuhahn, K. , Groettrup, M. , 2008. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene. 27, 6068–6074. [DOI] [PubMed] [Google Scholar]
- Mathews, L.A. , Hurt, E.M. , Zhang, X. , Farrar, W.L. , 2010. Epigenetic regulation of CpG promoter methylation in invasive prostate cancer cells. Mol. Cancer. 9, 267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narimatsu, M. , Maeda, H. , Itoh, S. , Atsumi, T. , Ohtani, T. , Nishida, K. , Itoh, M. , Kamimura, D. , Park, S.J. , Mizuno, K. , Miyazaki, J. , Hibi, M. , Ishihara, K. , Nakajima, K. , Hirano, T. , 2001. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol. Cell Biol.. 21, 6615–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, G. , Wright, K.L. , Ma, Y. , Wright, G.M. , Huang, M. , Irby, R. , Briggs, J. , Karras, J. , Cress, W.D. , Pardoll, D. , Jove, R. , Chen, J. , Yu, H. , 2005. Role of Stat3 in regulating p53 expression and function. Mol. Cell Biol.. 25, 7432–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus, A. , Hayden, M.S. , Ghosh, S. , 2011. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol.. 12, 695–708. [DOI] [PubMed] [Google Scholar]
- Oh, Y.M. , Kim, J.K. , Choi, Y. , Choi, S. , Yoo, J.Y. , 2009. Prediction and experimental validation of novel STAT3 target genes in human cancer cells. PloS One. 4, e6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory, K. , Legros, Y. , Auguin, C. , Soussi, T. , 1994. Analysis of the most representative tumour-derived p53 mutants reveals that changes in protein conformation are not correlated with loss of transactivation or inhibition of cell proliferation. EMBO J.. 13, 3496–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasi, S. , Schmidtke, G. , Groettrup, M. , 2001. The ubiquitin-like protein FAT10 forms covalent conjugates and induces apoptosis. J. Biol. Chem.. 276, 35334–35343. [DOI] [PubMed] [Google Scholar]
- Ren, J. , Wang, Y. , Gao, Y. , Mehta, S.B. , Lee, C.G. , 2011. FAT10 mediates the effect of TNF-alpha in inducing chromosomal instability. J. Cell Sci.. 124, 3665–3675. [DOI] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. , Eliceiri, K.W. , 2012. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, S.H. , Kim, K.S. , Park, S.H. , Suh, Y.S. , Kim, S.J. , Jeun, S.S. , Sung, Y.C. , 2011. The effects of mesenchymal stem cells injected via different routes on modified IL-12-mediated antitumor activity. Gene Ther.. 18, 488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J.Y. , Han, H.S. , Sabapathy, K. , Lee, B.M. , Yu, E. , Choi, J. , 2010. Expression of a homeostatic regulator, Wip1 (wild-type p53-induced phosphatase), is temporally induced by c-Jun and p53 in response to UV irradiation. J. Biol. Chem.. 285, 9067–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souissi, I. , Ladam, P. , Cognet, J.A. , Le Coquil, S. , Varin-Blank, N. , Baran-Marszak, F. , Metelev, V. , Fagard, R. , 2012. A STAT3-inhibitory hairpin decoy oligodeoxynucleotide discriminates between STAT1 and STAT3 and induces death in a human colon carcinoma cell line. Mol. Cancer. 11, 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tergaonkar, V. , Pando, M. , Vafa, O. , Wahl, G. , Verma, I. , 2002. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 1, 493–503. [DOI] [PubMed] [Google Scholar]
- Trinchieri, G. , 2012. Cancer and inflammation: an old intuition with rapidly evolving new concepts (*). Annu. Rev. Immunol.. 30, 677–706. [DOI] [PubMed] [Google Scholar]
- Wang, W.B. , Levy, D.E. , Lee, C.K. , 2011. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol.. 187, 2578–2585. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Liu, Q. , Ihsan, A. , Huang, L. , Dai, M. , Hao, H. , Cheng, G. , Liu, Z. , Wang, Y. , Yuan, Z. , 2012. JAK/STAT pathway plays a critical role in the proinflammatory gene expression and apoptosis of RAW264.7 cells induced by trichothecenes as DON and T-2 toxin. Toxicol. Sci. An Official J. Soc. Toxicol.. 127, 412–424. [DOI] [PubMed] [Google Scholar]
- Wu, Z.J. , Irizarry, R.A. , Gentleman, R. , Martinez-Murillo, F. , Spencer, F. , 2004. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc.. 99, 909–917. [Google Scholar]
- Xu, X. , Kasembeli, M.M. , Jiang, X. , Tweardy, B.J. , Tweardy, D.J. , 2009. Chemical probes that competitively and selectively inhibit Stat3 activation. PloS One. 4, e4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, Y. , Verma, U.N. , Prajapati, S. , Kwak, Y.T. , Gaynor, R.B. , 2003. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 423, 655–659. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Liao, X. , Agarwal, M.K. , Barnes, L. , Auron, P.E. , Stark, G.R. , 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev.. 21, 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Pardoll, D. , Jove, R. , 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D.W. , Jeang, K.T. , Lee, C.G. , 2006. p53 negatively regulates the expression of FAT10, a gene upregulated in various cancers. Oncogene. 25, 2318–2327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is/are the supplementary data related to this article:
Fig. S1. IFN‐γ was not induced by IL‐6 and TNF‐α co‐treatment. HepG2 cells were stimulated with IL‐6 and TNF‐α (10 ng/ml each) for 6 h and IFN‐γ induction was examined by RT‐PCR. As a positive control, PMA (100 ng/ml)/Ionomycin (1 ng/ml) treated Jurkat cells were used.
Fig. S2. FAT10 promoter assay with distal NFκB deletion mutant. HepG2 cells were transfected with four NFκB binding sites deletion mutant in distal region, as indicated, and the synergistic FAT10 promoter activation was measured after IL‐6 and/or TNF‐α stimulation for 6 h. n.s, not significant, **P < 0.01, and ***P < 0.001 by Student's t‐test.
Fig. S3. Conjugate formation of FAT10. (A) UBA6 and USE1 (E1 and E2 enzyme for FAT10 conjugation) expression was analyzed with RT‐PCR in HepG2, HCT116 (p53‐null), and HEK293 cells. (B) HEK293FT cells were transfected with FAT10 (GG) or conjugation‐mutant forms of FAT10 (AA) for 48 h, then MG132 (20 μM) was treated for 6 h. After cell lysis and IP with anti‐FLAG, immunoblot analysis was performed with anti‐FLAG.
Fig. S4. Control‐ or FAT10‐overexpressing TC‐1 in the solid tumor model. (A, B) Cell proliferation of control‐ or FAT10‐overexpressing TC‐1 cells were examined by cell counting (A) and MTT assay (B). (C) Transcriptional activity of p53 was compared between control‐ or FAT10‐overexpressing TC‐1 cells. (D) Diagram of primer sets used for detecting endogenous or exogenous FAT10. (E, F) Flag‐FAT10 over‐expression was confirmed in the control‐ or FAT10‐overexpressing TC‐1 stable cells (E) and in the sacrificed mice tumors (F). (G) Flag‐FAT10 over‐expression was examined in control‐ or FAT10‐overexpressing TC‐1 stable cells after DMSO or MG132 (10 μM, 12hr) by western blotting. **P < 0.01 by Student's t‐test.
Supplementary data