

Abstract
Targeted inhibition of MET/RON signaling by tyrosine kinase inhibitor BMS‐777607 for cancer treatment is currently under clinical trials. We have previously shown that BMS‐777607 induces chemoresistance in vitro by causing polyploidy, which hampers therapeutic efficacy. Here, we studied polyploidy‐associated senescence induced by BMS‐777607 in breast cancer cells and its prevention by mTOR inhibitor AZD8055, leading to increased chemosensitivity. In breast cancer T‐47D and ZR‐75‐1 cells, BMS‐777607 induced phenotypic changes including enlarged cellular size, flattened morphology, increased DNA content, and activity of senescence‐associated β‐galactosidase. These changes were accompanied by increased p21/WAF1 expression and decreased Retinoblastoma Ser780 phosphorylation, indicating that BMS‐777607 induces not only polyploidy but also senescence. The appearance of senescence was associated with polyploidy in which β‐galactosidase is exclusively expressed in polyploid cells. Survivin expression was increased in polyploid/senescent cells as analyzed by Western blotting. Increased survivin accumulated both in the nucleus and cytoplasm and dissociated with condensed DNA and mitotic spindle at the metaphase. Abnormal accumulation of survivin also rendered polyploid/senescent cells insensitive to cytotoxic activities of YM155, a DNA damaging agent with a suppressive effect on survivin gene transcription. AZD8055, a specific mTOR inhibitor, effectively prevented BMS‐777607‐induced polyploidy and senescence and restored survivin expression and its nuclear localization to normal levels. Although a synergism was not observed, BMS‐777607 plus AZD8055 increased cancer cell sensitivity toward different cytotoxic chemotherapeutics. In conclusion, BMS‐777607‐induced chemoresistance is associated with cell polyploidy and senescence. Inhibition of mTOR signaling by AZD8055 prevents BMS‐777607‐induced polyploidy/senescence and increases breast cancer cell chemosensitivity.
Keywords: Breast cancer, Receptor tyrosine kinase, Small molecule inhibitor, Senescence, Polyploidy, mTOR signaling, Chemosensitivity
Abbreviations
- AXL
Greek word anexelekto
- BCR
breakpoint cluster region
- BRAF
B-Rapidly Accelerated Fibrosarcoma
- DAPI
4′,6′-diamidino-2-phenylindole
- EMT
epithelial to mesenchymal transition
- EGFR
epithelial growth factor receptor
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FLT-3
Fms-like tyrosine kinase 3
- IGF-1R
insulin-like growth factor receptor-1
- mAb
monoclonal antibody
- MER
monocytes, epithelial and reproductive tissue
- MET
mesenchymal to epithelial transition
- mTOR
mammalian target of rapamycin
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- WAF1
wild-type p53-activated fragment 1
- PI3K
Phosphatidylinositide 3-kinase
- Rb
retinoblastoma
- RON
recepteur d'origine nantais
- RTK
receptor tyrosine kinase
- SABG
senescence-associated β-galactosidase
- siRNA
small interfering RNA
- TKI
tyrosine kinase inhibitor
- TYRO-3
tyrosine protein kinase receptor-3
1. Introduction
Receptor tyrosine kinases (RTK)2 MET and RON belong to a unique RTK subfamily implicated in epithelial tumorigenesis and malignancy (Gherardi et al., 2012, 2013, 2013). In breast cancer, overexpression of MET and RON is a pathogenic factor that facilitates the tumorigenic progression and provides the prognostic value for patient survival (Beviglia et al., 1997; Lee et al., 2005; Knight et al., 2013; Kretschmann et al., 2010). Moreover, increased MET and RON expression has been validated as a drug target for breast cancer therapy (Blumenschein et al., 2012, 2013, 2013). Various TKIs such as foretinib, MGCD265, and MK8033 and therapeutic monoclonal antibodies including onartuzumab and IMC‐RON8 (also known as Narnatumab) have been developed (Choueiri et al., 2013; Eder et al., 2010; Belalcazar et al., 2012; Northrup et al., 2013; Merchant et al., 2013; Zou et al., 2013). Currently, these TKIs and therapeutic antibodies are under clinical trials for advanced cancers that harbor aberrant MET and/or RON signaling. Preliminary results from these clinical studies indicate potential for tumor growth control and prolonged patient survival.
The major challenge faced in targeted cancer therapy is the development of acquired resistance to specific TKIs or therapeutic antibodies (Shien et al., 2013; Wheeler et al., 2010). Although clinical trials are still underway, acquired resistance to MET and RON dual TKIs has been reported in preclinical studies (Diamond et al., 2013; McDermott et al., 2010; Cepero et al., 2010; Qi et al., 2011). Currently, three major mechanisms of acquired resistance have been identified in kinase‐targeted cancer therapy (Engelman and Jänne, 2008; Garraway and Jänne, 2012). Genetic alterations including point mutation, gene amplification and alternative mRNA splicing are the forms of acquired resistance occurring in target proteins such as MET, EGFR, BCR‐ABL, and p16BRAF (Engelman and Jänne, 2008; Garraway and Jänne, 2012). In MET, a point mutation in the kinase activation loop (Y1230) destabilizes the autoinhibitory conformation of the protein leading to the hyper‐activation status (Qi et al., 2011). Activation of an alternative signaling pathway(s) also increases survival of cancer cells under targeted conditions (Engelman and Jänne, 2008; Garraway and Jänne, 2012). In lung cancers, acquired resistance caused by gefitinib‐induced inhibition of EGFR is featured by activation of the MET pathway with increased PI3K‐AKT signaling (Engelman et al., 2007). In targeting IGF1R for the treatment of childhood sarcoma, increased RON expression and signaling have emerged as a compensatory mechanism for the survival of tumor cells (Potratz et al., 2010). In addition, activation of the EGFR pathway or induction of KRAS gene amplification has been found to mediate acquired resistance to MET/RON targeted TKIs (Cepero et al., 2010). Another mechanism involved in acquired resistance is the change of cellular phenotype such as EMT (Kim et al., 2013; Ahmed et al., 2010). Cancer cells with EMT are highly resistant to TKI‐targeted therapy due to their acquisition of certain stem/progenitor cell characteristics, which are known as hallmark featured for drug resistance (Kim et al., 2013; Ahmed et al., 2010). Thus, understanding the mechanisms underlying acquired resistance by cancer cells is urgently needed.
BMS‐777607 is a synthetic TKI selective for the MET superfamily (Schroeder et al., 2009). BMS‐777607 primarily targets RON (IC50: 1.8 nM), MET (IC50: 3.9 nM), TYRO‐3 (IC50: 4.3), and AXL (1.1 nM) (Schroeder et al., 2009). At relatively high concentrations, BMS‐777607 also inhibits other targets such as MER (IC50: 14.0 nM), FLT‐3 (IC50: 16 nM), and aurora kinase B (AuKB, IC50: 78 nM) (Schroeder et al., 2009). Thus, BMS‐777607 is best viewed as a multi‐kinase inhibitor. Preclinical studies have shown that BMS‐777607 in vitro inhibits MET and RON signaling and suppresses various tumorigenic activities including cell growth and migration (Schroeder et al., 2009; Dai and Siemann, 2010; Sharma et al., 2013). Studies from tumor xenograft models also confirm that BMS‐777607 effectively inhibits tumor growth in a dose‐dependent manner (Schroeder et al., 2009). However, BMS‐777607 treatment also causes cancer cell chemoresistance manifested by the off‐target effect (Sharma et al., 2013). We have previously shown that treatment of breast, colon, and pancreatic cancer cells in vitro with BMS‐777607 induces extensive polyploidy. This effect is caused by inhibition of AuKB, resulting in cell cycle arrest at pro‐metaphase and failure to undergo cytokinesis (Sharma et al., 2013). Polyploid cells are long‐lived and acquire resistance to cytotoxic chemotherapeutics (Sharma et al., 2013; Davis et al., 2008). Thus, BMS‐777607‐induced phenotypic change owing to its off‐target effect opens a pathogenic avenue leading to acquired chemoresistance. In other words, the off‐target effect could constitute a mechanism of acquired resistance in targeted cancer therapy.
The present study seeks to find a pharmacological means to prevent BMS‐777607‐induced chemoresistance and to increase the therapeutic efficacy of BMS‐777607 against cancer cells. Currently, BMS‐777607 is under clinical phase I trials for treatment of advanced cancers (Clinical trials IDs: NCT01721148). Considering its negative impact on cellular phenotype, which may affect therapeutic efficacy, we have tried to determine cellular signaling proteins or pathways that act as the effector molecule in BMS‐777067‐induced chemoresistance. Moreover, we are interested in using pharmacological approaches to prevent or attenuate BMS‐777607‐induced resistance and to sensitize cancer cells to cytotoxic chemotherapeutics. We believe that results from this study should increase understanding of the therapeutic mechanism of BMS‐777607 and to improve its efficacy in kinase‐targeted cancer treatment.
2. Materials and methods
2.1. Cell lines and reagents
Breast cancer T‐47D and ZR‐75‐1 cells were from American Type Cell Culture (Manassas, VA). Mouse mAb Zt/g4 and rabbit polyclonal IgG antibody R5029 specific to human RON were used as previously described (Wang et al., 2007; Yao et al., 2011). Mouse or rabbit IgG antibodies specific to p53, p21/WAF1, survivin, α‐tubulin, Rb, phospho‐Rb at Ser780 residue, mTOR, phospho‐mTOR, p70/850S6K, phorspho‐p70/85S6K, and other signaling proteins were from Cell Signaling (Danvers, MA). BMS‐777607, AZD8055, rapamycin, and YM155 were from Selleck Chemicals (Houston, TX). Doxorubicin, cisplatin, and paclitaxel were from Fisher Scientific (Hanover Park, IL).
2.2. Assay for senescence‐associated β‐galactosidase (SABG) activity
T‐47D and ZR‐75‐1 cells (12,000 cells per well in a 24‐well plate in triplicate) in RPMI‐1640 with 5% FBS were treated with various amounts of BMS‐777607, YM155, AZD8055, or their different combinations for various time periods. SAGB activities from control and experimental cells were detected using a Senescence Cells Histochemical Staining Kit (Cat#: CS0030, Sigma–Aldrich, Inc., Saint Louis, MO). Images were photographed at magnification of ×200 using Olympus BK71 microscope equipped with a DSU confocal/fluorescent apparatus.
2.3. Transfection of siRNA to knockdown survivin expression
Survivin‐specific siRNA and control scramble RNA was from Cell Signaling (Danvers MA). T‐47D and ZR‐75‐1 cells were transfected with 100 nM siRNA or scramble RNA according to the manufacture's instruction. After incubation for 24 h, cells were treated with or without 5 μM BMS‐777607 for additional 72 h followed by Western blotting to determine levels of survivin. Transfected cells also were observed for morphological changes to determine polyploidy and analyzed by flow cytometer to study cell cycle change.
2.4. Western blot analysis
The method was performed as previously described (Wang et al., 1994; Yao et al., 2006). Cellular proteins (50 μg per sample) from cell lysate were separated in an 8% or 12% SDS‐PAGE under reduced conditions. Signaling proteins including p53, p21/WAF1, survivin, p70/85S6K, and others at the regular or phosphorylated status were detected using specific antibodies corresponding to individual proteins. The reactions were visualized using enhanced chemiluminescent reagents and recorded. Membranes also were reprobed with β‐actin antibodies to ensure equal sample loading.
2.5. Immunofluorescent detection of cellular proteins
The method was performed as previously described (Sharma et al., 2013). Briefly, cells at 1 × 105 cells per well in a 6‐well plate were cultured in RPMI‐1640 with 5% FBS and treated with various amounts of BMS‐777607, AZD8055, or their combinations for various time periods. Detection of survivin and α‐tubulin were performed by fixing cells with 4% paraformaldehyde solution and permeabilizing with 0.1% triton ×100, followed by incubation with individual specific antibodies. The detecting antibodies were goat anti‐rabbit IgG coupled with FITC or rhodamine. Normal mouse IgG was used as the control. Nuclear DNAs were stained with DAPI. Cellular immunofluorescence was observed under the Olympus BK71 microscope equipped with DUS/fluorescent apparatus as previously described (Sharma et al., 2013).
2.6. Assays for cell cycle, viability and DNA content
The effect of BMS‐777607, YM155, AZD8055, or chemotherapeutics on cell viability was determined by the MTS assay as previously described (Padhye et al., 2011). For measuring cellular DNA contents, cells were treated with 5 μM BMS‐777607, 25 nM AZD8055, or their combinations for 72 h, labeled with propidium iodide, and analyzed by an Accuri Flow Cytometer as previously described (Sharma et al., 2013).
2.7. Statistical analysis
GraphPad Prism 6 software was used for statistical analysis. Results are shown as mean ± SD. The data between control and experimental groups were compared using Student t test. Statistical differences at p < 0.05 were considered significant.
3. Results
3.1. Induction of cellular senescence by BMS‐777607 in breast cancer cells
Previous studies have shown that BMS‐777607 causes extensive polyploidy in breast cancer cells (Sharma et al., 2013). In this study we determined whether BMS‐777607 also induces cellular senescence in relation to polyploidy. T47‐D and ZR‐75‐1 cells were treated with various doses of BMS‐777607 for 72 h. SABG activity was detected in both T‐47D and ZR‐75‐1 cells in a dose‐dependent manner (Figure 1A). The percentages of SABG‐positive cells were associated with increased doses of BMS‐777607. The kinetic effect of BMS‐777607 on induction of senescence is shown in Figure 1B. The number of SABG‐positive cells was increased in a progressive, time‐dependent manner in both cell lines tested. At 72 h, more than 45% of T‐47D and 50% of ZR‐75‐1 cells, respectively, were positive for SABG activity (Figure 1B). The percentages were further increased up to more than 70% of T‐47D and ZR‐75‐1 cells after cells were treated for 120 h. Thus, BMS‐777607 induces not only polyploidy but also senescence in breast cancer cells.
Figure 1.
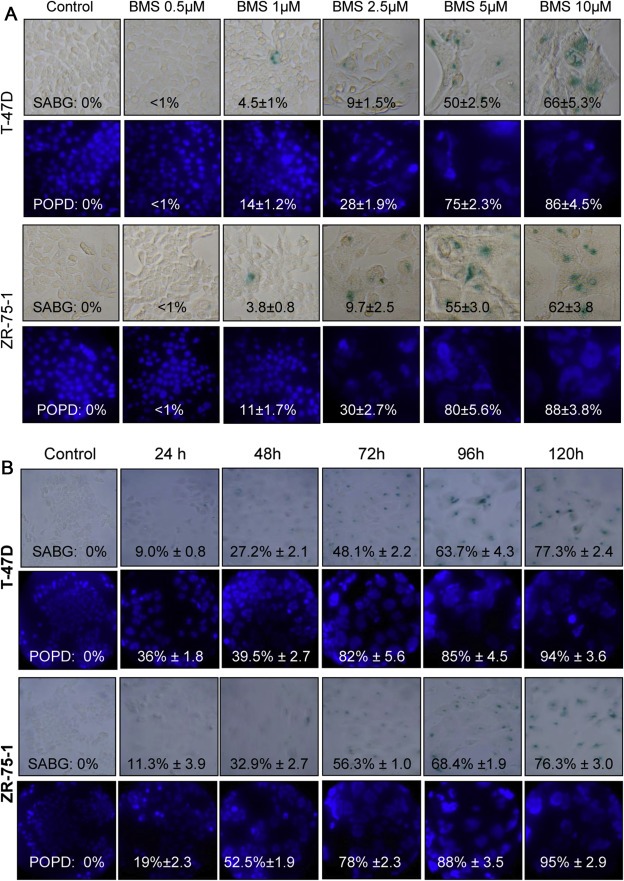
Induction of cellular senescence in breast cancer cells by BMS‐777607. T‐47D and ZR‐75‐1 cells (12,000 cells per well in triplicate in a 24‐well tissue culture plate) were incubated at 37 °C in RPMI‐1640 with 5% FBS and treated with different amounts of BMS‐777607 for 72 h (A) or with 5 μM BMS‐777607 up to 120 h to observe the kinetic effect (B). SBAG was detected using the senescence detection assay. Nuclear DNA was stained with DAPI followed by photography. The percentages of SAGB or polyploid cells were determined by counting 300 cells in three randomly selected areas as previously described (29). Results shown here are from one of three experiments with similar results.
The dynamics between SABG activity and polyploidy formation were compared. A progressive increase in the number of polyploid cells was observed in a time‐dependent manner in both cell lines tested (Supplementary Figure 1). Specifically, at 72 h after treatment with 5 μM BMS‐777607, more than 70% of T‐47D and ZR‐75‐1 cells underwent polyploidy. The percentages of SABG positive cells also were progressively increased. Compared with percentages of SABG‐positive cells, the number of polyploid cells were relatively higher than that of SABG‐positive cells in both cell lines tested (Supplementary Figure 1).
To determine the relationship between cells undergoing senescence and/or polyploidy, we analyzed 400 cells from both T‐47D and ZR‐75‐1 cells 72 h after BMS‐777607 treatment. All SABG‐positive cells (100%) showed polyploid features (Figure 1). However, not all polyploid cells stained positive for SABG activity. As shown in Figure 1, about 20% of T‐47D and ZR‐75‐1 polyploid cells were SABG negative, respectively. Moreover, in cells showing no polyploidy, SABG were all negative. Similar results also were observed in cells treated with BMS‐777607 for 120 h. These results indicate that SABG was exclusively expressed in polyploid cells. Thus, BMS‐777607‐induced senescence is associated with polyploidy and we referred these cells as polyploid/senescent cells.
3.2. Abnormal accumulation and localization of survivin in BMS‐777607‐induced senescent/polyploid cells
To determine signaling proteins relevant to polyploidy and senescence after BMS‐777607 treatment, we analyzed tumor suppressor protein p53 (Crescenzi et al., 2013), p21/WAF1 (Romanov et al., 2012), survivin (Wang et al., 2011), and Rb (Henley and Dick, 2012; Dick and Rubin, 2013) (Figure 2A). Levels of p53 were not changed as shown by Western blot analysis, although T47‐D cells express a mutant and ZR‐75‐1 cells a wild‐type p53 protein (Crescenzi et al., 2013). Expression of p21/WAF1 in both T‐47D and ZR‐75‐1 cells increased in a progressive and time‐dependent manner regardless the p53 status. Increased survivin expression was observed in a time‐dependent manner in BMS‐777607‐treated cells, which also was not associated with the p53 status in both cell lines tested. In addition, we observed dynamic expression and phosphorylation of Rb upon BMS‐777607 treatment. It appears that BMS‐777607 exerts a complex, two phase effect on Rb. Rb protein expression and its phosphorylation at Ser780 residue in T‐47D cells increased within 48 h and then reduced to the control levels but diminished in ZR‐75‐1 cells. Again, decreased Rb expression does not appear to be correlated with p53 status. Thus, results in Figure 2A demonstrate that expression of p21/WAF1 and survivin is up‐regulated in BMS‐777607‐induced polyploid/senescent cells regardless the status of p53. The overall effect of BMS‐777607 on Rb is down‐regulation of protein expression and phosphorylation.
Figure 2.
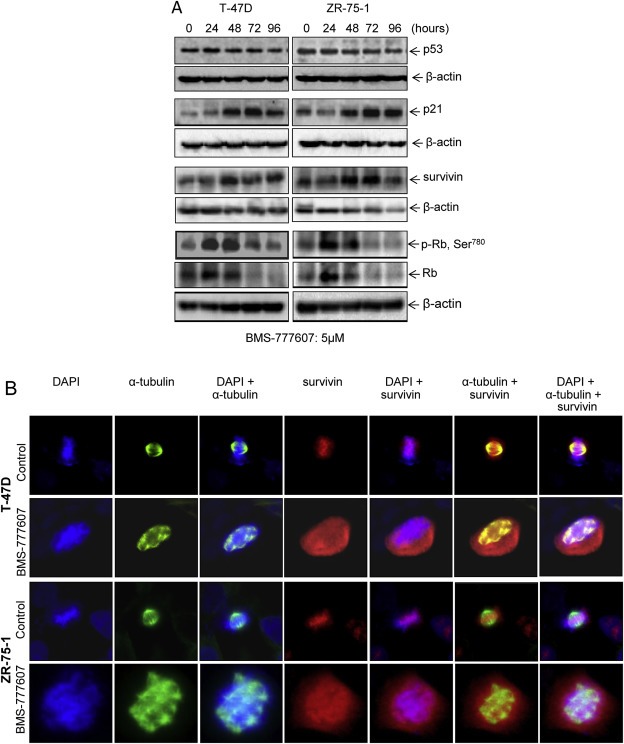
Altered expressions of p21/WAF1, survivin, and retinoblastoma protein in BMS‐777607‐induced polyploid/senescent breast cancer cells. (A) BMS‐777607 increases p21/WAF1 and survivin expression but down‐regulates Rb expression. T‐47D and ZR‐75‐1 cells (2 × 106 cells in 60 mm diameter culture dish) were treated with 5 μM BMS‐777607 for different time intervals. Cellular proteins (50 μg per sample) from cell lysates were subjected to Western blot analysis using individual antibodies specific to p53, p21/WAF1, survivin, regular and phospho‐Rb. B‐actin was used as the loading control. (B) Abnormal accumulation of survivin and its disassociation with condensed DNA and mitotic spindle. Both T‐47D and ZR‐75‐1 cells were treated with 5 μM BMS‐777607 for 72 h followed by immunofluorescent analysis using antibodies specific to survivin and α‐tubulin. Cells were also stained with DAPI for nuclear DNA. Images shown here are from one of two experiments with similar results.
We further studied survivin by immunofluorescent analysis in light of its expression pattern and function in cell survival, cell cycle, and drug resistance (Kelly et al., 2011; Berezov et al., 2012). In control cells at metaphase, survivin is predominantly localized in the nucleus, associated with condensed DNA, and involved in the assembly of a bipolar mitotic spindle (Figure 2B). However, large amounts of survivin accumulated both in the nucleus and cytoplasm of BMS‐777607‐induced polyploid/senescent cells with disorganized multi‐polar spindles. Survivin was disassociated with condensed polyploid DNA and was not involved in the assembly of mitotic spindle (Figure 2B). These results suggest that abnormal accumulation of survivin both in the nucleus and cytoplasm and its dissociation with condensed DNA and mitotic spindle are pathogenic features of BMS‐777607‐induced polyploid/senescent cells.
3.3. Association of abnormal accumulation of survivin with increased survival of polyploid/senescent cells
To study the role of survivin in more detail, we used specific siRNA to silence survivin expression in T‐47D and ZR‐75‐1 cells (Supplementary Figure 2). Knockdown of survivin expression did not affect cell morphology in both cell lines tested (Figure 3A). BMS‐777607‐induced polyploidy was not affected by scramble RNA. Surprisingly, levels of BMS‐777607‐induced polyploidy were not affected after transfection with specific siRNA. We further analyzed cell cycles by flow cytometer in cells transfected with survivin siRNA (Figure 3B). In control T‐47D and ZR‐75‐1 cells, certain cells (<5%) appeared as polyploidy with 8N DNA content. Knockdown of survivin expression only moderately affects cell cycles with decreased 4N DNA content. The profiles of BMS‐777607‐induced polyploidy were not significantly affected by specific siRNA. Thus, knockdown of survivin appears to play no role in BMS‐777607‐induced polyploidy.
Figure 3.
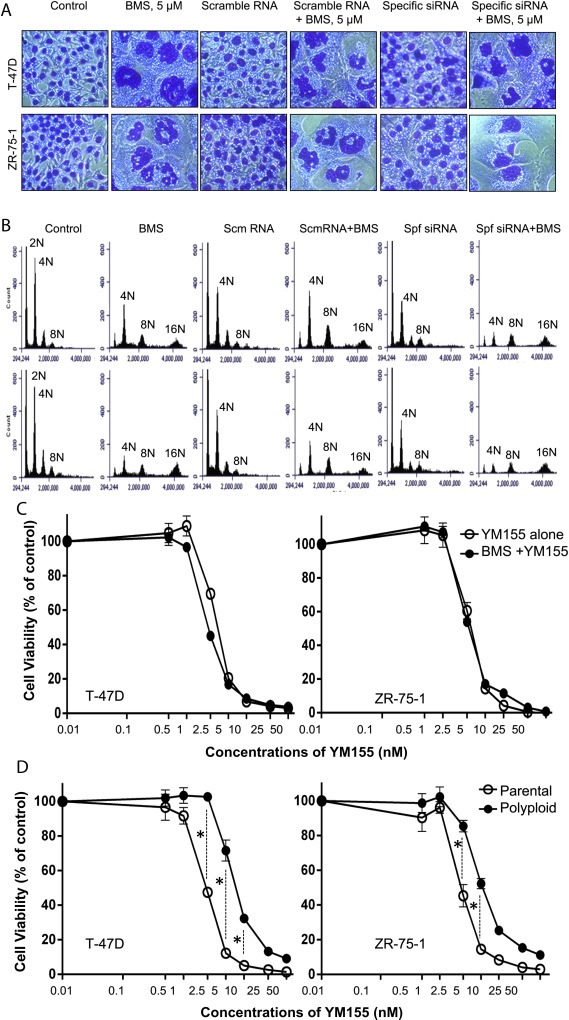
Abnormal accumulation of survivin correlates with increased survival of polyploid/senescent cells: T‐47D and ZR‐75‐1 cells (2 × 105 cells per well in duplicate in a 60 mm diameter culture plate) in RPMI‐1640 with 5% FBS were treated with survivin‐specific‐ siRNA or control scramble RNA for 24 h followed by treatment with BMS‐777607, YM155, and their different combinations for 72 h. (A) Cellular polyploidy was determined by morphological analysis with the appearance of the irregular shaped nuclear DNA as described in Materials and methods. (B) Cell cycle analysis was performed by Accuri Flow Cytometer after labeling cells with propidium iodide. (C) Sensitivities of parental T‐47D and ZR‐75‐1 cells to YM155 and YM155 plus BMS‐777607 were determined by treatment of cells (8000 cells per well in triplicate in a 96‐well plate) with different amounts of YM155 with or without 5 μM BMS‐777607 for 72 h. Cell viability was measured by the MTS assay. (D) Sensitivity of polyploid/senescent cells to YM155 was determined first by treatment of cells with 5 μM BMS‐777607 for 72 h. Polyploid/senescent cells were collected, re‐cultured in a 96‐well plate (8000 cells per well in triplicate), and then treated with different amounts of YM155 for an additional 72 h. Parental cells without pre‐BMS‐777607 treatment were used as the control. Cell viability was measured by the MTS assay. Data shown here are from one of three experiments with similar results. P, parental cells; P/S, polyploid/senescent cells; *, statistical significance (p < 0.05).
We then selected compound YM155 to treat polyploid/senescent cells. YM155 was originally identified as a selective transcription inhibitor of survivin (Nakahara et al., 2011). However, YM155 actually acts primarily as a DNA damaging agent with a broad transcription‐repressive activity including inhibition of survivin gene transcription (Glaros et al., 2012). Treatment of cells with YM155 alone dramatically reduced cell viability (Supplementary Figure 3A and B). In combination with BMS‐777607, YM155 at various doses had no effect on senescence and polyploidy induced by 5 μM BMS‐777607. In cells treated with BMS‐777607 plus 5 nM YM155, more than 50% and 70% of remaining viable T‐47D and ZR‐75‐1 cells showed senescence and polyploidy, respectively (Supplementary Figure 3). Thus, results from Figure 3A, B and Supplementary Figure 3 demonstrate that knockdown or inhibition of survivin expression does not affect BMS‐777607‐induced senescence and polyploidy. Abnormal accumulation of survivin is a feature in BMS‐777607‐induced polyploidy and senescence.
Finally, we determined if abnormal accumulation of survivin in polyploid/senescent cells is associated with increased cell resistance to cytotoxic agents. Again, we used YM155 that is highly cytotoxic to T‐47D and ZR‐75‐1 cells. Less than 10% of viable cells remained 72 h after 25 nM YM155 was applied (Figure 3C). Also, BMS‐777607 had no synergistic effect with YM155 on cell viability. However, when polyploid/senescent cells were used in compared to parental cells, a statistical difference in response to YM155 was observed (Figure 3D). At doses ranging from 2.5 nM, 5 nM, and 10 nM, the percentages of cell viability from polyploid/senescent cells from both cell lines were significantly higher than those of control cells. The calculated IC50 values increased from 2.2 nM to 2.3 nM for parental T‐47D and ZR‐75‐1 cells to 7.1 nM and 5.4 nM for polyploid/senescent T‐47D and ZR‐75‐1 cells, respectively. The calculated IC50 ratios were 3.1 for polyploid to parental T‐47D cells and 2.3 for polyploid to parental ZR‐75‐1 cells. Thus, abnormal accumulation of survivin is associated with increased resistance to YM155‐induced cytotoxicity. Nevertheless, the percentages of cell viability between parental and polyploid/senescent cells reached similar levels when YM155 was used at higher concentrations.
3.4. Prevention of BMS‐777607‐induced senescence, polyploidy, and survivin accumulation by mTOR inhibitor AZD8055
To find chemical inhibitors that regulate BMS‐777607‐induced senescence and polyploidy, several chemical inhibitors were screened and AZD8055, a specific mTOR inhibitor (Chresta et al., 2010), was found to have a preventive effect on BMS‐777607‐induced senescence (Figure 4A). In both T‐47D and ZR‐75‐1 cells, BMS‐777607‐induced SABG activity was almost completely prevented by 25 nM AZD8055, which showed no effect on cell viability. Interestingly, an increase in BMS‐777607 concentration up to 10 μM overcomes the preventive effect of AZD8055, indicating that a delicate balance between the doses of the two inhibitors is critically important to obtain optimal results. Moreover, BMS‐777607‐induced polyploidy was prevented by AZD8055 as evident by DAPI staining of nuclear DNAs (Figures 4A and B). Morphologically, the enlarged T47‐D and ZR‐75‐1 cells were restored to their original epithelial appearance in the presence of AZD8055 (Figures 4A and B). Similar results also were found when rapamycin was used (Supplementary Figure 4). Cell cycle analysis further confirmed that AZD8055 prevents BMS‐777607‐induced 8N chromosomes in both T‐47D and ZR‐75‐1 cells after cells were treated for 3 days (Figure 4C) or up to 7 days (Supplementary Figure 5). Colonogenic studies revealed that AZD8055 at 25 nM moderately affected cells growth after a 7 day incubation but its synergistic effect with BMS‐777607 was not observed (Supplementary Figure 6). Thus, AZD8055‐mediated inhibition of mTOR activity affected cell growth, which was able to prevent BMS‐777607‐induced polyploidy and senescence in breast cancer cells.
Figure 4.
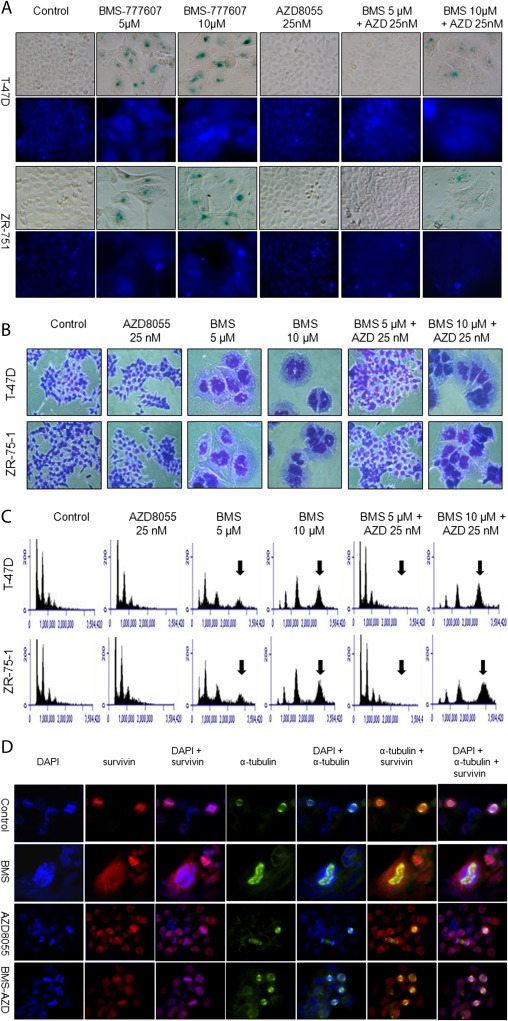
Preventive effect of AZD8055 on BMS‐777607‐induced senescence, polyploidy, and survivin expression in breast cancer cells. T‐47D and ZR‐75‐1 cells were cultured as described in Figure 3A and then treated for 72 h with BMS‐777607, AZD8055, and their different combinations. (A) Senescence was determined by detecting SABG and polyploidy was documented by staining nuclear DNA with DAPI. (B) The morphological appearance of polyploid cells also was determined by H&E staining and then photographed. (C) DNA contents were measured by the propidium iodide staining method followed by follow cytometric analysis. Arrows indicate 8N DNA contents. (D) Expression and localization of survivin and α‐tubulin were determined by immunofluorescent analysis using specific antibodies, followed by FITC or rhodamine‐labeled second antibodies as previously described (Sharma et al., 2013). Cells at metaphase were analyzed for expression and localization of survivin, α‐ tubulin, and their association with condensed DNA. Images were made at magnification of ×400. Data shown here are from one of three experiments with similar results.
We further studied the effect of AZD8055 on BMS‐777607‐mediated survivin expression using immunofluorescent methods. Consistent with results in Figure 2, abnormal accumulation of survivin induced by BMS‐777607 was observed in polyploid/senescent T‐47D cells, which was associated with disorganized multi‐polar‐like mitotic spindle assembly (Figure 4D). AZD8055 at 25 nM alone had no effect on induction of polyploidy. The level and pattern of survivin expression appeared to be normal in comparison to those of control cells. However, in cells treated with BMS‐777607 plus AZD8055, polyploidy was prevented. Significantly, the levels and patterns of survivin expression appeared to be restored to levels comparable to those of control cells. Moreover, survivin in association with condensed DNA and the bipolar mitotic spindle re‐appeared in cells treated with both agents (Figure 4D). Similar results also were observed in ZR‐75‐1 cells (Supplementary Figure 7). Significantly, we observed similar results in T‐47D cells treated with another mTOR inhibitor rapamycin (Supplementary Figure 4B). Thus, results in Figure 4D, together with those in Supplementary Figures 4–7, demonstrate that inhibition of mTOR signaling by AZD8055 prevented the abnormal accumulation of survivin in BMS‐777607‐treated cells and restored normal survivin patterns in association with nuclear DNA and mitotic spindle.
3.5. Effect of AZD8055 on expression and phosphorylation of p70/85S6K, Rb, and P21/WAF1 by BMS‐777607‐treated breast cancer cells
The data in Figure 4 suggest the importance of mTOR activity in BMS‐777607 for senescence and polyploidy. To confirm this, we first determined whether mTOR signaling is active in breast cancer cells. Results from Western blotting revealed constitutive phosphorylation of mTOR and its substrate p70/85S6K in both T‐47D and ZR‐75‐1 cells (Figure 5A). As expected, AZD8055, BMS‐777607 alone or in combination with BMS‐777607 did not affect mTOR phosphorylation but did significantly inhibit Thr421/Ser424 phosphorylation of mTOR substrate p70/85S6K. These results indicate that mTOR signaling is constitutively active in T‐47D and ZR‐75‐1 cells. Inhibition of the mTOR pathway by AZD8055 is associated with prevention of BMS‐777607‐induced senescence and polyploidy.
Figure 5.

Effect of BMS‐777607, AZD8055, and their combinations on expression and phosphorylation of mTOR, p70/85S6K, Rb, and p21/WAF1 in breast cancer cells. T‐47D and ZR‐75‐1 cells (3 × 106 cells per dish in DMEM with 5%FBS) were treated for 24 h with 5 μM BMS‐777607, 25 nM AZD8055, or their combination. Cellular proteins (50 μg per sample) from cell lysates were separated in an 8% or 12% SDS‐PAGE under reduced conditions. (A) The phosphorylation status of mTOR and its substrate p70/85S6K. Western blot analysis was performed using individual antibodies specific to phospho‐mTOR, mTOR, phospho‐p70/85S6K, and p70/85S6K, respectively. Levels of β‐actin were used as the loading control. (B) Expression and phosphorylation of Rb and p21/WAF1. Rb phosphorylation was determined using antibodies specific to phospho‐Ser780 residue in the Rb protein. Levels of the Rb and p21/WAF1 protein were detected by individual antibodies specific to Rb and p21/WAF1. Levels of β‐actin were used as the loading control.
An interesting observation was a change of Rb expression and phosphorylation after addition of BMS‐777607, AZD8055, or a combination of both (Figure 5B). Rb phosphorylation at Ser780 residue is known to be critical in regulating cellular senescence and polyploidy (46). In both T‐47D and ZR‐75‐1 cells, BMS‐777607 appeared to decrease Rb expression and Ser780 phosphorylation after a 72 h treatment. AZD8055 increased Rb phosphorylation in both cell lines tested. However, the effect of AZD8055 on Rb protein expression varied between T‐47D and ZR‐75‐1 cells. Increased Rb protein expression was observed in ZR‐75‐1 but not in T‐47D cells. The combinational treatment restored Rb phosphorylation and protein expression to control levels. In addition, we found that BMS‐777607 but not AZD8055 strongly induced p21/WAF1 expression. Also, increased p21/WAF1 expression was not affected by addition of AZD8055. These results indicate that Rb expression and phosphorylation status were affected by treatment with BMS‐777607, AZD8055 and their combinations. These changes could be associated with development or prevention of cellular senescence and polyploidy.
3.6. Prevention by AZD8055 of BMS‐777067‐induced polyploidy and senescence sensitizes breast cancer cells to chemotherapeutics
We tested whether such synergism might exist in breast cancer cells as well. Cells were treated with various amounts of AZD8066 with or without 5 μM BMS‐777607 for 72 h. Only a slight decrease in cell viability was observed in both cell lines tested (Supplementary Figure 8). In both cell lines tested, the IC50 values from cells treated with both compounds did not differ significantly from cells treated with a single compound. Thus, no synergism exists between BMS‐777607 and AZD8055.
Next, we determined whether BMS‐777607 plus AZD8055 increases sensitivity of breast cancer cells to chemoagents. We treated T‐47D and ZR‐75‐1 cells with or without BMS‐777607, AZD8055, or their combinations for 72 h. Cells were then treated with cisplatin, paclitaxel, and doxorubicin at their IC50 doses (Sharma et al., 2013). The IC50 doses were used to indicate cell sensitivity to chemoagents. As shown in Table 1, the percentages of cell viability from cisplatin, paclitaxel, and doxorubicin was slightly reduced in both T‐47D and ZR‐75‐1 cells treated with 25 nM AZD8055 in comparison to those from control cells treated with single chemoagents. This indicates that AZD8055 pretreatment does not exert significant synergistic effect with the three chemotherapeutics tested. In contrast, BMS‐777607 treatment resulted in increased chemoresistance in both T‐47D and ZR‐75‐1 cells in response to the three chemoagents. The percentages of cell viability were, respectively, 80.1%, 95.9%, and 95.9% for cisplatin, paclitaxel, and doxorubicin in T‐47D cells and at 89.8%, 88.4%, and 102.2% for cisplatin, paclitaxel, and doxorubicin in ZR‐75‐1 cells, respectively. However, treatment of T‐47D and ZR‐75‐1 cells with BMS‐777607 plus AZD8055 restored cellular sensitivity to cytotoxic activities of cisplatin, paclitaxel, and doxorubicin, respectively. The percentages of cell viability were, respectively, reduced to 43.4%, 53.0%, and 50.8% for T4‐7D cells and 49.2%, 51.5%, 44.2% for ZR‐75‐1 cells after treatment with cisplatin, paclitaxel, and doxorubicin. Significantly, reduced viability was comparable to the IC50 levels observed from control cells.
Table 1.
Effect of BMS‐777607 plus AZD8055 on sensitizing breast cancer cells to cytotoxic chemotherapeutics.
| Cell lines | %Cell viability after chemoagent treatment | |||||
|---|---|---|---|---|---|---|
| Cisplatin | Sensitivity index | Paclitaxel | Sensitivity index | Doxorubicin | Sensitivity index | |
| T‐47D cells pretreated | ||||||
| Control | 50.5 ± 3.9 | 1.0 | 50.9 ± 2.6 | 1.0 | 59.8 ± 2.6 | 1.0 |
| AZD8055 | 44.7 ± 2.3 | 0.89 | 51.7 ± 1.2 | 1.02 | 59.0 ± 4.7 | 0.99 |
| BMS777607 | 80.1 ± 3.2 | 1.59 | 95.9 ± 18.5 | 1.88 | 95.9 ± 18.5 | 1.60 |
| AZD + BMS | 43.4 ± 1.6 | 0.86 | 53.0 ± 1.5 | 1.04 | 50.8 ± 10.8 | 0.85 |
| ZR‐75‐1 cells pretreated | ||||||
| Control | 47.7 ± 4.3 | 1.0 | 47.6 ± 3.9 | 1.0 | 48.1 ± 16.8 | 1.0 |
| AZD8055 | 40.9 ± 1.4 | 0.86 | 46.4 ± 1.6 | 0.97 | 42.2 ± 7.6 | 0.88 |
| BMS777607 | 89.8 ± 5.0 | 1.88 | 88.4 ± 5.6 | 1.86 | 102.2 ± 12.5 | 2.14 |
| AZD + BMS | 49.2 ± 4.0 | 1.03 | 51.5 ± 3.2 | 1.08 | 44.2 ± 12.4 | 0.92 |
Cells cultured in RPMI‐1640 with 5% FBS and treated with or without 25 nM AZD8055, 5 μM BMS‐777607, and their combinations for 72 h. The remaining viable cells were collected and placed in 8000 cells per well in a 96‐well plate in triplicate and treated for additional 72 h with 3.7 μM cisplatin, 4 nM paclitaxel, and 300 nM doxorubicin for T‐47D cells and with 1.6 μM cisplatin, 6 nM paclitaxel, and 450 nM doxorubicin for ZR‐75‐1 cells, respectively. The amounts of individual chemoagents were selected based on calculated IC50 doses according to our previous studies (29). Cell viability was measured by the MTS assay. Viabilities from control, AZD8055, BMS‐777607, or BMS777607 + AZD8055 treated cells were set as 100%. Sensitivity index from cells treated with single chemoagents was set as 1.0.
4. Discussion
This report is our ongoing effort to determine the therapeutic potential of BMS777607 in breast cancer cells. In this study, we discovered that BMS‐777607 not only induces polyploidy, but also causes senescence, both of which contribute to chemoresistance. In polyploid/senescent cells, abnormal accumulation of survivin and its disassociation with condensed DNA and mitotic spindle were observed. Survivin is known to regulate cell survival and mitosis (Berezov et al., 2012). In our studies, abnormal accumulation of survivin is associated with polyploid/senescent cells insensitive to cytotoxic activity of YM155, a DNA damaging agent with suppressive effect on survivin gene transcription (Glaros et al., 2012; Yamauchi et al., 2012). We further observed that mTOR signaling is participated in BMS‐777607‐induced cellular polyploidy and senescence. By blocking mTOR signaling, AZD8055 prevents BMS‐777607‐induced polyploidy and senescence. Significantly, AZD8055 blocks the effect of BMS‐777607 on abnormal survivin accumulation and facilitates survivin in association with condensed DNA and mitotic spindles in the metaphase. Furthermore, we demonstrated that BMS‐777607 in combination with AZD8055 sensitizes breast cancer cells to cytotoxic activities of different chemotherapeutics. Thus, therapeutic activity of BMS‐777607 is associated with polyploidy and senescence, which render cancer cells insensitive to chemotherapy. Inhibition of mTOR signaling by AZD8055 prevents BMS‐777607 induced polyploidy and senescence. The coordination of BMS‐777607 with AZD8055 provides a pharmacological means to overcome BMS‐777607‐induced polyploidy/senescence and to increase chemosensitivity of breast cancer cells.
Senescence is often observed in cancer cells exposed to chemotherapy agents (Pare et al., 2013). Functionally, senescence is featured by growth arrest, resistance to apoptosis, and altered gene expression (Pare et al., 2013). Senescent cells typically display a flattened, enlarged morphology and most prominently express senescence‐associated galactosidase activities in their lysosomes (Pare et al., 2013). We found that BMS‐777607 induces not only polyploidy but also senescence (Figures 1 and 4). T‐47D and ZR‐75‐1 cells exhibit several features of BMS‐777607‐induced senescence. First, a relatively high concentration of up to 5 μM BMS‐777607 and a prolonged treatment for more than 48 h are required to induce senescence in a significant portion of cells. This suggests that the effect of BMS‐777607 is not mediated by direct inhibition of RON/MET signaling. Instead, it is caused mainly by its off‐target effect on other cellular proteins such as aurora kinase B (Schroeder et al., 2009). Second, SABG activities are exclusively detected in polyploid cells but not in cells showing regular morphology and DNA content. However, not all polyploidy cells show SABG activity. These observations suggest that the process of senescence requires additional signaling event(s). Third, BMS‐777607‐induced polyploidy/senescence is accompanied by abnormal accumulation of survivin, increased p21/WAF1 expression, and decreased Rb phosphorylation at Ser780 residue and its protein expression. These biochemical changes typify cellular senescence (Pare et al., 2013). At present, we do not know the underlying mechanisms for the development of senescence. Inhibition of aurora kinase B by BMS‐777607 appears to have an impact on cell cycle, which facilitates activation of the cellular senescence progress. Nevertheless, our discovery that BMS‐777607 induces not only polyploidy but also senescence provides mechanistic insight into the therapeutic profiles of BMS‐777607 against breast cancer cells.
The role of survivin in cell survival and chemoresistance has been widely reported (Wang et al., 2011; Kelly et al., 2011; Berezov et al., 2012). Survivin also acts as an integral component of the chromosome passenger protein complex and participates in chromosome segregation during mitosis (Campisi, 2013). We found that in breast cancer cells at metaphase, survivin is localized in the nucleus and associated with chromosomes aligned at the equatorial plate with α‐tubulin showing a bipolar spindle pattern. However, in BMS‐777607‐treated polyploid/senescent cells, survivin is abnormally accumulated both in the nucleus and cytoplasm and disassociated with condensed DNA and mitotic spindle. Such abnormalities could have a disruptive effect on the assembly of the chromosome passenger protein complex leading to impaired cell mitosis. Nevertheless, we observed that knockdown of survivin by specific siRNA had no effect on BMS‐777607‐induced polyploidy and senescence. Currently, the mechanisms underlying abnormal accumulation of survivin in BMS‐777607‐treated cells is unknown. However, mTOR inhibitor AZD8055 prevents abnormal accumulation and cytoplasmic localization of survivin in BMS‐777607‐treated cells. In this sense, mTOR signaling seems to be required for abnormal accumulation of survivin. Considering these facts, we reason that survivin expression, localization, and its association with chromosomes are abnormally altered upon BMS‐777607 treatment. Such alterations could have a pathogenic impact on BMS‐777607‐induced chemoresistance.
We tried to correlate abnormal accumulation of survivin with cellular sensitivity toward DNA damaging agent YM155 (Glaros et al., 2012), previously used as a specific transcription inhibitor of survivin (Nakahara et al., 2011). Consistent with a previous report (Pare et al., 2013), both T‐47D and ZR‐75‐1 cells are highly sensitive to YM155‐induced cytotoxicity. However, at a dose that is sufficient to block the survivin gene transcription, YM155 does not prevent BMS‐777607‐induced senescence and polyploidy in both breast cell lines tested. Moreover, we observed that polyploid/senescent cells with abnormal accumulation of survivin have developed resistance to YM155‐induced cytotoxic activity. These data suggest that the amount of survivin accumulated in polyploid/senescent cells is associated with increased cell survival. While it appears that survivin does not play a role in the development of polyploidy and senescence, it could be critical in preventing chromosome segregation and in impairing cytokinesis (Campisi, 2013). It is likely that abnormal accumulation of survivin protects polyploid/senescent breast cancer cells from the cytotoxic effect of chemoagents and facilitates the development of chemoresistance.
The finding that inhibition of mTOR signaling prevents BMS‐777607‐induced polyploidy and senescence is significant in terms of sensitizing breast cancer cells to chemotherapeutics. The mTOR pathway regulates cell cycle, survival, senescence, and transcription (Cornu et al., 2013). In breast cancer T‐47D and ZR‐75‐1 cells, mTOR and its downstream signaling molecules such as p70/85S6K are constitutively activated as evident by their phosphorylation. AZD8055 inhibits p70/85S6K phosphorylation leading to inhibition of mTOR signaling. As shown in the cell cycle analysis and the colonogenic assay (Supplementary Figures 5 and 6), AZD8055 is able to prevent cells from entering cell cycle (probably in the stage of DNA synthesis) by affecting DNA replication and thus impairs BMS‐777607‐induced polyploidy. Considering the preventive effect of AZD8055 on polyploidy and senescence, the data from combination treatment strongly suggest that mTOR signaling is critical in BMS‐777607‐mediated polyploidy and senescence.
The effects of BMS‐777607, AZD8055 and their combination on p21/WAF1 and Rb phosphorylation and protein expression are complex (Figures 2 and 5). From our observation, the overall effect of BMS‐777607 is increased p21/WAF1 expression and decreased Rb phosphorylation at Ser780 residue, both of which are biochemical features of senescent cells (Campisi, 2013). Specifically, in BMS‐777607‐treated cells, Rb expression and its Ser780 phosphorylation first increased within 48 h and then diminished after 72 h upon addition of BMS‐777607. This dual effect indicates a dynamic change in the Rb expression and activation status during the progression of cells toward polyploidy and senescence. On the other hand, AZD8055 alone not only causes inactivation of Rb by increasing its phosphorylation at Ser780 residue, but also reduces p21/WAF1 expression, which are the opposite effects of BMS‐777607. Nevertheless, the outcome of BMS‐777607 in combination with AZD8055 is the restoration of Rb expression and phosphorylation to control levels and increased p21/WAF1 expression. We notice that BMS‐777607‐increased p21/WAF1 expression occurs in a p53‐independent manner (Figure 2A). The p53 status also is not discriminated in AuKB‐mediated polyploidy since both T‐47D (harboring a mutant p53) and ZR‐75‐1 (expressing a wild‐type p53) showed extensive polyploidy after BMS‐777607 treatment. It is intriguing that inhibition of mTOR by AZD8055 fails to prevent the increased p21/WAF1 expression despite its ability to overcome the occurrence of polyploid/senescent phenotype. Thus, the complicated signaling interaction after addition of AZD8055 to BMS‐777607‐treated cells is responsible for the inability of cells to develop polyploidy and senescence.
To increase cancer cell drug responsiveness, we first sought to determine the possibility of a synergism between BMS‐777607 and AZD8055 in reduction of cell viability. However, the combination treatment did not exert any synergistic effect on breast cancer cells. We then addressed whether pretreatment with BMS‐777607 plus AZD8055 would increases sensitivity of breast cancer cells in response to chemotherapeutics. The results obtained from these experiments indicate that this is the case (Table 1). We observed increased sensitivity of T‐47D and ZR‐75‐1 cells against three individual chemotherapeutics, as judged by IC50 values, compared to those of cells treated with BMS‐777607 alone. In all cases, the effect of AZD8055 on prevention of BMS‐777607‐induced polyploidy and senescence appears to be necessary for optimal efficacy. We reason that in order to achieve the maximal therapeutic effect of BMS‐777607 on breast cancer cells through targeting RON and/or MET, it is critically important to prevent its off‐target effects such as polyploidy and senescence. Inclusion of mTOR inhibitor is a pharmacological approach to achieve this goal. As demonstrated in this study, breast cancers cells pretreated with BMS‐777607 plus AZD8055 display increased chemosensitivity in response to different chemotherapeutics.
The use of AZD8055 or BMS‐777607 in combination with chemoagents for cancer treatment requires clinical validation. Both AZD8055 and BMS‐777607 is currently under clinical trials. For BMS‐777607, it is likely that the combination therapy achieves the better therapeutic efficacy than the single drug application. However, due to differences in targeting signaling proteins, the clinical indications of AZD8055 or BMS‐777607 will be different. Currently, we have no evidence against the use of BMS‐7777607 for cancer treatment due to its off‐target effect. The outcome of clinical trials will validate its usefulness. Nevertheless, our study provides insight into the therapeutic mechanism of BMS‐777607 on cancer cells. Such studies should help to improve the therapeutic efficacy of BMS‐777607 and to minimize its off‐target effect.
5. Conclusions
The current study demonstrates that therapeutic activity of BMS‐777607 is hampered by polyploidy and senescence, which render cancer cells insensitive to chemotherapy. A working model is proposed to describe the complex cooperation between BMS‐777607 and AZD8055 in regulating cell phenotype (Supplementary Figure 9). Abnormal accumulation of survivin and its dissociation with condensed DNA and mitotic spindle are pathogenic features in BMS‐777607‐induced polyploid/senescent cells. Suppression of mTOR signaling by AZD8055 prevents BMS‐777607‐induced polyploidy and senescence. The coordination of BMS‐777607 with AZD8055 provides a pharmacological means to overcome BMS‐777607‐induced polyploidy/senescence and to increase chemosensitivity of breast cancer cells.
Conflict of interest
The authors declare no conflict of interest regarding this article.
Supporting information
The following is the supplementary material related to this article:
Supplementary data
Acknowledgments
We greatly appreciate the assistance of Ms. Susan Denney (Texas Tech University Health Sciences Center School of Pharmacy, Amarillo, TX, USA) in editing the manuscript. This work was supported in part by NIH grant R01 CA91980, funds from the Amarillo Area Foundation and from subproject #2011ZZ01 from State Key Laboratory for Diagnosis & Treatment of Infectious Diseases in First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, P.R. China (M.H. Wang). R. Zhang was supported by NIH grants R01 CA112029 and CA121211 and J. Zhou was supported by Natural Science Foundation of China (81161120537, 30930080, 91229125, 81001231, and 81370078).
Supplementary material 1.
1.1.
Supplementary material related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.12.014.
Sharma Sharad, Yao Hang-Ping, Zhou Yong-Qing, Zhou Jianwei, Zhang Ruiwen and Wang Ming-Hai, (2014), Prevention of BMS‐777607‐induced polyploidy/senescence by mTOR inhibitor AZD8055 sensitizes breast cancer cells to cytotoxic chemotherapeutics, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.12.014.
Contributor Information
Sharad Sharma, Email: sharad.sharama@ttuhsc.edu.
Hang-Ping Yao, Email: yaohangping@zju.edu.cn.
Yong-Qing Zhou, Email: zhouyq56@hotmail.com.
Jianwei Zhou, Email: jwzhou@njmu.edu.cn.
Ruiwen Zhang, Email: Ruiwen.zhang@ttuhsc.edu.
Ming-Hai Wang, Email: minghai.wang@ttuhsc.edu.
References
- Ahmed, N. , Abubaker, K. , Findlay, J. , Quinn, M. , 2010. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets. 10, 268–278. [DOI] [PubMed] [Google Scholar]
- Belalcazar, A. , Azaña, D. , Perez, C.A. , Raez, L.E. , Santos, E.S. , 2012. Targeting the Met pathway in lung cancer. Expert Rev. Anticancer Ther.. 12, 519–528. [DOI] [PubMed] [Google Scholar]
- Berezov, A. , Cai, Z. , Freudenberg, J.A. , Zhang, H. , Cheng, X. , Thompson, T. , 2012. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene. 31, 1938–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beviglia, L. , Matsumoto, K. , Lin, C.S. , Ziober, B.L. , Kramer, R.H. , 1997. Expression of the c-Met/HGF receptor in human breast carcinoma: correlation with tumor progression. Int. J. Cancer. 74, 301–309. [DOI] [PubMed] [Google Scholar]
- Blumenschein, G.R. , Mills, G.B. , Gonzalez-Angulo, A.M. , 2012. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol.. 30, 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi, J. , 2013. Aging, cellular senescence, and cancer. Annu. Rev. Physiol.. 75, 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepero, V. , Sierra, J.R. , Corso, S. , Ghiso, E. , Casorzo, L. , Perera, T. , Comoglio, P.M. , Giordano, S. , 2010. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res.. 70, 7580–7590. [DOI] [PubMed] [Google Scholar]
- Choueiri, T.K. , Vaishampayan, U. , Rosenberg, J.E. , Logan, T.F. , Harzstark, A.L. , Bukowski, R.M. , Rini, B.I. , Srinivas, S. , Stein, M.N. , Adams, L.M. , Ottesen, L.H. , Laubscher, K.H. , Sherman, L. , McDermott, D.F. , Haas, N.B. , Flaherty, K.T. , Ross, R. , Eisenberg, P. , Meltzer, P.S. , Merino, M.J. , Bottaro, D.P. , Linehan, W.M. , Srinivasan, R. , 2013. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol.. 31, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chresta, C.M. , Davies, B.R. , Hickson, I. , Harding, T. , Cosulich, S. , Critchlow, S.E. , Vincent, J.P. , Ellston, R. , Jones, D. , Sini, P. , James, D. , Howard, Z. , Dudley, P. , Hughes, G. , Smith, L. , Maguire, S. , Hummersone, M. , Malagu, K. , Menear, K. , Jenkins, R. , Jacobsen, M. , Smith, G.C. , Guichard, S. , Pass, M. , 2010. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res.. 70, 288–298. [DOI] [PubMed] [Google Scholar]
- Cornu, M. , Albert, V. , Hall, M.N. , 2013. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev.. 23, 53–62. [DOI] [PubMed] [Google Scholar]
- Crescenzi, E. , Raia, Z. , Pacifico, F. , Mellone, S. , Moscato, F. , Palumbo, G. , Leonardi, A. , 2013. Down-regulation of wild-type p53-induced phosphatase 1 (Wip1) plays a critical role in regulating several p53-dependent functions in premature senescent tumor cells. J. Biol. Chem.. 288, 16212–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Siemann, D.W. , 2010. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol. Cancer Ther.. 9, 1554–1561. [DOI] [PubMed] [Google Scholar]
- Davis, E. , Teng, H. , Bilican, B. , Parker, M.I. , Liu, B. , Carriera, S. , 2008. Ectopic Tbx2 expression results in polyploidy and cisplatin resistance. Oncogene. 27, 976–984. [DOI] [PubMed] [Google Scholar]
- Diamond, J.R. , Salgia, R. , Varella-Garcia, M. , Kanteti, R. , LoRusso, P.M. , Clark, J.W. , 2013. Initial clinical sensitivity and acquired resistance to MET inhibition in MET-mutated papillary renal cell carcinoma. J. Clin. Oncol.. 31, e254–e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick, F.A. , Rubin, S.M. , 2013. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol.. 14, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder, J.P. , Shapiro, G.I. , Appleman, L.J. , Zhu, A.X. , Miles, D. , Keer, H. , 2010. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin. Cancer Res.. 16, 3507–3516. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Jänne, P.A. , 2008. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res.. 14, 2895–2899. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Jänne, P.A. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Garraway, L.A. , Jänne, P.A. , 2012. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov.. 2, 214–226. [DOI] [PubMed] [Google Scholar]
- Gherardi, E. , Birchmeier, W. , Birchmeier, C. , Vande Woude, G. , 2012. Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer. 12, 89–103. [DOI] [PubMed] [Google Scholar]
- Glaros, T.G. , Stockwin, L.H. , Mullendore, M.E. , Smith, B. , Morrison, B.L. , Newton, D.L. , 2012. The “survivin suppressants” NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother. Pharmacol.. 70, 207–212. [DOI] [PubMed] [Google Scholar]
- Henley, S.A. , Dick, F.A. , 2012. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div.. 7, 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, R.J. , Lopez-Chavez, A. , Citrin, D. , Janik, J.E. , Morris, J.C. , 2011. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol. Cancer. 10, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.P. , Han, S.W. , Song, S.H. , Jeong, E.G. , Lee, M.Y. , Hwang, D. , Im, S.A. , Bang, Y.J. , Kim, T.Y. , 2013 Jul 22. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene. 10.1038/onc.2013.285 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Knight, J.F. , Lesurf, R. , Zhao, H. , Pinnaduwage, D. , Davis, R.R. , Saleh, S.M. , Zuo, D. , Naujokas, M.A. , Chughtai, N. , Herschkowitz, J.I. , Prat, A. , Mulligan, A.M. , Muller, W.J. , Cardiff, R.D. , Gregg, J.P. , Andrulis, I.L. , Hallett, M.T. , Park, M. , 2013. Met synergizes with p53 loss to induce mammary tumors that possess features of claudin-low breast cancer. Proc. Natl. Acad. Sci. U.S.A.. 110, E1301–E1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmann, K.L. , Eyob, H. , Buys, S.S. , Welm, A.L. , 2010. The macrophage stimulating protein/Ron pathway as a potential therapeutic target to impede multiple mechanisms involved in breast cancer progression. Curr. Drug Targets. 11, 1157–1168. [DOI] [PubMed] [Google Scholar]
- Lee, W.Y. , Chen, H.H. , Chow, N.H. , Su, W.C. , Lin, P.W. , Guo, H.R. , 2005. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin. Cancer Res.. 11, 2222–2228. [DOI] [PubMed] [Google Scholar]
- McDermott, U. , Pusapati, R.V. , Christensen, J.G. , Gray, N.S. , Settleman, J. , 2010. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res.. 70, 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant, M. , Ma, X. , Maun, H.R. , Zheng, Z. , Peng, J. , Romero, M. , Huang, A. , Yang, N.Y. , Nishimura, M. , Greve, J. , Santell, L. , Zhang, Y.W. , Su, Y. , Kaufman, D.W. , Billeci, K.L. , Mai, E. , Moffat, B. , Lim, A. , Duenas, E.T. , Phillips, H.S. , Xiang, H. , Young, J.C. , Vande Woude, G.F. , Dennis, M.S. , Reilly, D.E. , Schwall, R.H. , Starovasnik, M.A. , Lazarus, R.A. , Yansura, D.G. , 2013. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. U.S.A.. 110, E2987–E2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara, T. , Kita, A. , Yamanaka, K. , Mori, M. , Amino, N. , Takeuchi, M. , Tominaga, F. , Kinoyama, I. , Matsuhisa, A. , Kudou, M. , Sasamata, M. , 2011. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci.. 102, 614–621. [DOI] [PubMed] [Google Scholar]
- Northrup, A.B. , Katcher, M.H. , Altman, M.D. , Chenard, M. , Daniels, M.H. , Deshmukh, S.V. , Falcone, D. , Guerin, D.J. , Hatch, H. , Li, C. , Lu, W. , Lutterbach, B. , Allison, T.J. , Patel, S.B. , Reilly, J.F. , Reutershan, M. , Rickert, K.W. , Rosenstein, C. , Soisson, S.M. , Szewczak, A.A. , Walker, D. , Wilson, K. , Young, J.R. , Pan, B.S. , Dinsmore, C.J. , 2013. Discovery of 1-[3-(1-Methyl-1H-pyrazol-4-yl)-5-oxo-5H-benzo [4,5]cyclohepta[1,2-b]pyridin-7-yl]-N-(pyridin-2-ylmethyl)methanesul-fonamide (MK-8033): a Specific c-Met/Ron dual kinase inhibitor with preferential affinity for the activated state of c-Met. J. Med. Chem.. 56, 2294–2310. [DOI] [PubMed] [Google Scholar]
- Padhye, S.S. , Guin, S. , Yao, H.P. , Zhou, Y.Q. , Zhang, R. , Wang, M.H. , 2011. Sustained expression of the RON receptor tyrosine kinase by pancreatic cancer stem cells as a potential targeting moiety for antibody-directed chemotherapeutics. Mol. Pharm.. 8, 2310–2319. [DOI] [PubMed] [Google Scholar]
- Pare, R. , Yang, T. , Shin, J.S. , Lee, C.S. , 2013. The significance of the senescence pathway in breast cancer progression. J. Clin. Pathol.. 66, 491–495. [DOI] [PubMed] [Google Scholar]
- Potratz, J.C. , Saunders, D.N. , Wai, D.H. , Ng, T.L. , McKinney, S.E. , Carboni, J.M. , Gottardis, M.M. , Triche, T.J. , Jürgens, H. , Pollak, M.N. , Aparicio, S.A. , Sorensen, P.H. , 2010. Synthetic lethality screens reveal RPS6 and MST1R as 6modifiers of insulin-like growth factor-1 receptor inhibitor activity in childhood sarcomas. Cancer Res.. 70, 8770–8781. [DOI] [PubMed] [Google Scholar]
- Qi, J. , McTigue, M.A. , Rogers, A. , Lifshits, E. , Christensen, J.G. , Jänne, P.A. , 2011. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res.. 71, 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov, V.S. , Pospelov, V.A. , Pospelova, T.V. , 2012. Cyclin-dependent kinase inhibitor p21(Waf1): contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc.). 77, 575–584. [DOI] [PubMed] [Google Scholar]
- Schroeder, G.M. , An, Y. , Cai, Z.W. , Chen, X.T. , Clark, C. , Cornelius, L.A. , Dai, J. , Gullo-Brown, J. , Gupta, A. , Henley, B. , Hunt, J.T. , Jeyaseelan, R. , Kamath, A. , Kim, K. , Lippy, J. , Lombardo, L.J. , Manne, V. , Oppenheimer, S. , Sack, J.S. , Schmidt, R.J. , Shen, G. , Stefanski, K. , Tokarski, J.S. , Trainor, G.L. , Wautlet, B.S. , Wei, D. , Williams, D.K. , Zhang, Y. , Zhang, Y. , Fargnoli, J. , Borzilleri, R.M. , 2009. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2 dihy-dropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem.. 52, 1251–1258. [DOI] [PubMed] [Google Scholar]
- Sharma, S. , Zeng, Z.J. , Zhuang, C.M. , Zhou, Y.Q. , Yao, H.P. , Hu, X. , 2013. Small-molecule inhibitor BMS-777607 induces breast cancer cell polyploidy with increased resistance to cytotoxic chemotherapy agents. Mol. Cancer Ther.. 12, 725–736. [DOI] [PubMed] [Google Scholar]
- Shien, K. , Toyooka, S. , Yamamoto, H. , Soh, J. , Jida, M. , Thu, K.L. , 2013. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res.. 73, 3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M.H. , Lee, W. , Luo, Y.L. , Weis, M.T. , Yao, H.P. , 2007. Altered expression of the RON receptor tyrosine kinase in various epithelial cancers and its contribution to tumorigenic phenotypes in thyroid cancer cells. J. Pathol.. 213, 402–411. [DOI] [PubMed] [Google Scholar]
- Wang, M.H. , Ronsin, C. , Gesnel, M.C. , Coupey, L. , Skeel, A. , Leonard, E.J. , Breathnach, R. , 1994. Identification of the ron gene product as the receptor for the human macrophage stimulating protein. Science. 266, 117–119. [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Wu, P.C. , Roberson, R.S. , Luk, B.V. , Ivanova, I. , Chu, E. , Wu, D.Y. , 2011. Survivin and escaping in therapy-induced cellular senescence. Int. J. Cancer. 128, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, D.L. , Dunn, E.F. , Harari, P.M. , 2010. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol.. 7, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi, T. , Nakamura, N. , Hiramoto, M. , Yuri, M. , Yokota, H. , Naitou, M. , Takeuchi, M. , Yamanaka, K. , Kita, A. , Nakahara, T. , Kinoyama, I. , Matsuhisa, A. , Kaneko, N. , Koutoku, H. , Sasamata, M. , Kobori, M. , Katou, M. , Tawara, S. , Kawabata, S. , Furuichi, K. , 2012. Sepantronium bromide (YM155) induces disruption of the ILF3/p54(nrb) complex, which is required for survivin expression. Biochem. Biophys. Res. Commun.. 425, 711–716. [DOI] [PubMed] [Google Scholar]
- Yao, H.P. , Luo, Y.L. , Feng, L. , Cheng, L.F. , Lu, Y. , Li, W. , Wang, M.H. , 2006. Agonistic monoclonal antibodies potentiate tumorigenic and invasive activities of splicing variant of the RON receptor tyrosine kinase. Cancer Biol. Ther.. 5, 1179–1186. [DOI] [PubMed] [Google Scholar]
- Yao, H.P. , Zhou, Y.Q. , Ma, Q. , Guin, S. , Padhye, S.S. , Zhang, R.W. , Wang, M.H. , 2011. The monoclonal antibody Zt/f2 targeting RON receptor tyrosine kinase as potential therapeutics against tumor growth-mediated by colon cancer cells. Mol. Cancer. 10, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, H.P. , Zhuang, C.M. , Zhou, Y.Q. , Zeng, J.Y. , Zhang, R. , Wang, M.H. , 2013. Oncogenic variant RON160 expression in breast cancer and its potential as a therapeutic target by small molecule tyrosine kinase inhibitor. Curr. Cancer Drug Targets. 13, 686–697. [DOI] [PubMed] [Google Scholar]
- Yao, H.P. , Zhou, Y.Q. , Zhang, R. , Wang, M.H. , 2013. MSP-RON signaling in cancer: pathogenesis and therapeutic potential. Nat. Rev. Cancer. 13, 466–481. [DOI] [PubMed] [Google Scholar]
- Zou, Y. , Howell, G.M. , Humphrey, L.E. , Wang, J. , Brattain, M.G. , 2013. Ron knockdown and ron monoclonal antibody IMC-RON8 sensitize pancreatic cancer to Histone Deacetylase inhibitors (HDACi). PLoS One. 8, e69092–e69100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary material related to this article:
Supplementary data