

Abstract
Activated forms of the platelet derived growth factor receptor alpha (PDGFRα) have been described in various tumors, including FIP1L1‐PDGFRα in patients with myeloproliferative diseases associated with hypereosinophilia and the PDGFRαD842V mutant in gastrointestinal stromal tumors and inflammatory fibroid polyps.
To gain a better insight into the signal transduction mechanisms of PDGFRα oncogenes, we mutated twelve potentially phosphorylated tyrosine residues of FIP1L1‐PDGFRα and identified three mutations that affected cell proliferation. In particular, mutation of tyrosine 720 in FIP1L1‐PDGFRα or PDGFRαD842V inhibited cell growth and blocked ERK signaling in Ba/F3 cells. This mutation also decreased myeloproliferation in transplanted mice and the proliferation of human CD34+ hematopoietic progenitors transduced with FIP1L1‐PDGFRα. We showed that the non‐receptor protein tyrosine phosphatase SHP2 bound directly to tyrosine 720 of FIP1L1‐PDGFRα. SHP2 knock‐down decreased proliferation of Ba/F3 cells transformed with FIP1L1‐PDGFRα and PDGFRαD842V and affected ERK signaling, but not STAT5 phosphorylation. Remarkably, SHP2 was not essential for cell proliferation and ERK phosphorylation induced by the wild‐type PDGF receptor in response to ligand stimulation, suggesting a shift in the function of SHP2 downstream of oncogenic receptors.
In conclusion, our results indicate that SHP2 is required for cell transformation and ERK activation by mutant PDGF receptors.
Keywords: PDGFRA, PTPN11, SHP2, STAT5, Chronic eosinophilic leukemia
Abbreviations
- ERK
extracellular signal-regulated kinase
- FBS
fetal bovine serum
- FLT3
Fms-like tyrosine kinase 3
- FPα
FIP1L1-PDGFRα
- GIST
gastrointestinal stromal tumors
- MAPK
mitogen-activated protein kinases
- PDGF
platelet-derived growth factor
- PI3K
phosphatidylinositol-3-kinase
- PLCγ
phospholipase C gamma
- PKB
protein kinase B
- PTPN11
protein tyrosine phosphatase, non-receptor type 11
- SCF
stem cell factor
- ShRNA
short hairpin RNA
- SH2
SRC homology 2
- SHP2
SH2 domain-containing phosphatase 2
- SRE
serum response element
- STAT
signal transducer and activator of transcription
1. Introduction
The fusion of the FIP1L1 gene with PDGFRA is generated by a cryptic deletion on chromosome 4q12 and is responsible for the development of myeloid neoplasms associated with hypereosinophilia, a disease that is also referred to as chronic eosinophilic leukemia (Vardiman et al., 2009).
FIP1L1 (Factor interacting with Pap1‐like 1) is the homologue of a yeast gene, FIP required for mRNA polyadenylation (Ezeokonkwo et al.). PDGFRA encodes the platelet‐derived growth factor receptor α chain (PDGFRα), which belongs to the receptor‐tyrosine kinase family (Andrae et al., 2008; Toffalini and Demoulin, 2010). All breakpoints identified to date in PDGFRA are located within exon 12, which encodes the juxtamembrane domain, an inhibitory sequence located between the transmembrane and the kinase domains (Cools et al., 2003a). A partial deletion of this domain is sufficient to constitutively activate the tyrosine kinase activity of PDGFRα (Stover et al., 2006). Most patients respond well to the tyrosine kinase inhibitor imatinib mesylate (Glivec), which blocks PDGF receptors as well as ABL and c‐KIT (Gleich et al., 2002; Metzgeroth et al., 2008). Nevertheless, some patients acquire imatinib‐resistant mutations, such as T674I or D842V (Lierman et al., 2009).
Expression of FIP1L1‐PDGFRα (FPα) in the Ba/F3 hematopoietic cell line and in CD34+ human hematopoietic progenitors promotes cytokine‐independent cell growth (Buitenhuis et al., 2007; Cools et al., 2003a; Montano‐Almendras et al., 2012). In Ba/F3 cells, the FIP1L1 part can be replaced by a simple tag, suggesting that it is dispensable for FPα activation (Stover et al., 2006). By contrast, deletion of the FIP1L1 part decreased the impact of the oncoprotein in human hematopoietic progenitors (Buitenhuis et al., 2007). We observed that FPα escapes the normal degradation of activated receptors, leading to the accumulation of the oncoprotein and an enhanced transformation potential (Toffalini et al., 2009).
In addition to fusion genes, point mutations in PDGFRA were identified in various cancers, including gastrointestinal stromal tumor (GIST), glioma, FPα‐negative hypereosinophilic syndrome and inflammatory fibroid polyps (Elling et al., 2011; Heinrich et al., 2003; Huss et al., 2012; Velghe et al., 2013). The most common activating mutation is D842V, which is located in the activation loop of PDGFRα (Dewaele et al., 2008). It is present in 8% of all patients with GIST and is resistant to imatinib (Corless et al., 2005; Dewaele et al., 2008; Elling et al., 2011). Recently, this mutation was reported in a few patients diagnosed with multiple myeloma (Mulligan et al., 2013).
Signal transduction by wild‐type PDGFRα has been extensively studied (Heldin et al., 1998). The activated kinase domain phosphorylates at least ten tyrosine residues within the cytosolic part of the receptor. These phosphorylated tyrosines act as docking sites for the Src homology 2 (SH2) domains of multiple signaling mediators, including SRC kinases, the SHP2 phosphatase, the signal transducers and activators of transcription (STAT), phospholipase Cγ, phosphatidylinositol‐3 kinase (PI3K) and adaptor proteins such as GRB2, SHC and NCK (Heldin et al., 1998). Much redundancy has been found among phosphorylated tyrosines and signaling molecules as these pathways regulate broadly overlapping sets of genes, which promote cell survival and proliferation (Fambrough et al., 1999).
SHP2, encoded by the PTPN11 gene, is a ubiquitously expressed non‐receptor protein tyrosine phosphatase, which contains two N‐terminal SH2 domains and a C‐terminal protein tyrosine phosphatase domain. Germline PTPN11 mutations were reported in Noonan and LEOPARD syndromes, whereas somatic mutations occur in several neoplasms, such as juvenile myelomonocytic leukemia (Chan et al., 2008). The full activation of SHP2 requires the binding of the two SH2 domains to a doubly phosphorylated peptide (Heldin et al., 1998; Pluskey et al., 1995). In this respect, tyrosine residues 720 and 754 in PDGFRα have been described to bind SHP2 and could have a role in SHP2 activation (Bazenet et al., 1996; Rupp et al., 1994). A second possible activation mechanism implicates the association between the SH2 domains and one or two phosphorylated tyrosines located in the C‐terminal tail of SHP2 (Lu et al., 2001; Neel et al., 2003). SHP2 regulates many signaling pathways such as JAK/STAT, PI3K/PKB and RAS/mitogen‐activated protein kinases (MAPK). Besides its catalytic role, SHP2 also plays an adaptor role by recruiting signaling molecules such as STAT, GAB1/2 and GRB2, which is an essential component of the MAPK pathway (Kallin et al., 2004; Liu and Qu, 2011; Neel et al., 2003). SHP2 controls the activation of the RAS/MAPK pathway by PDGF at least in some cell types (Araki et al., 2003; Bennett et al., 1994; Ronnstrand et al., 1999; Zhang et al., 2004). Two reports also suggested that SHP2 is required for chemotaxis but not for proliferation induced by PDGF (Bazenet et al., 1996; Ronnstrand et al., 1999).
While signaling by wild‐type PDGF receptors has been intensively studied for almost three decades, surprisingly little is known about the pathways required for cell transformation by oncogenic PDGF receptor mutants. In the present study, we identified tyrosine 720 as a critical site for SHP2 recruitment by FPα, activation of ERK and transformation of hematopoietic cells. SHP2 was similarly important for the D842V mutant but not for the wild‐type receptor.
2. Material and methods
2.1. Antibodies, inhibitors and constructs
Anti‐PDGFRα (951), anti‐phosphotyrosine (PY99) and anti‐STAT5 (C‐17) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz). Anti‐phospho‐PLCγ1 (Tyr783), anti‐phospho‐ERK1/2 (Thr202/Tyr204), anti‐phospho‐SHP2 (Tyr542), anti‐SHP2 and anti‐PLCγ antibodies were purchased from Cell Signaling. The anti‐ERK2 (EET) rabbit polyclonal antiserum was previously described (Leevers and Marshall, 1992). Anti‐phospho‐STAT5 (Tyr694) antibodies were purchased from Cell Signaling and Signalway Antibody (SAB). A mouse monoclonal antibody against β‐actin (clone AC‐15) was purchased from Sigma. Imatinib was purchased from LC laboratories (Woburn, MA, USA). PDGF‐BB, stem cell factor (SCF) and FLT3 ligand (FLT3L) were obtained from PeproTech.
The RNAi Consortium lentiviral mouse PTPN11/SHP2 shRNA was obtained from Thermo scientific. Three constructs (TRCN0000029875, TRCN0000029877 and TRCN0000029878) were used for this study. The negative pLKO.1‐puro shScramble control was purchased from Addgene (#1864). pLKO.1‐puro Turbo GFP was purchased from Sigma (#SHC003). The human FIP1L1‐PDGFRA fusion cDNA was described earlier (Cools et al., 2003a) and comprised the first 923 nucleotides of FIP1L1 (Ensembl Transcript ID ENST00000337488) and the last 1573 nucleotides of PDGFRA (Ensembl Transcript ID ENST00000257290). FIP1L1‐PDGFRα in the retroviral pMSCV‐GFP vector was previously described (Cools et al., 2003a) and subcloned in the AgeI site of the lentiviral pTM895‐GFP vector, which was a kind gift from Pr. T. Michiels (Brussels, Belgium). Wild‐type PDGFRA cloned in pEF‐MYC‐CYTO (Invitrogen) was described previously (Velghe et al., 2013). Point mutations were created by site directed mutagenesis using the QuickChange™ XL‐II kit (Stratagene) according to manufacturer instructions. All constructs were checked by sequencing.
2.2. Transfection, infection, and thymidine incorporation assay
The human embryonic kidney (HEK)‐293T cells and IL‐3‐dependent Ba/F3 murine hematopoietic cell line were maintained in Dulbecco's Modified Eagle's Medium (DMEM, Gibco) with 10% fetal bovine serum (FBS), 50 U/ml penicillin and 50 μg/ml streptomycin (Gibco). Retroviral particles were produced by HEK‐293T cells transfected by the calcium phosphate method as previously described (Toffalini et al., 2009). Two days after transfection, Ba/F3 cells were infected once, as described (Toffalini et al., 2010), and sorted after 24 h by flow cytometry according to GFP expression. Lentiviral particles were produced by HEK‐293T cells. Cells were seeded in 10 cm plates. After one day, plasmid DNA (18 μg) was co‐transfected with the packaging plasmids pCMV‐dr8.2 dvpr (10 μg), pCMV‐VSV‐G (6 μg) and pRSV‐Rev (6 μg). The DNA was diluted to a final volume of 675 μl with water and mixed with 750 μl of BBS buffer (50 mM N,N‐bis‐(2‐hydroxyethyl)‐2‐aminoethane‐sulfonic acid, pH 7, 280 mM NaCl, 1.5 mM Na2HPO4) and 75 μl CaCl2 2.5 M. The solution was incubated for 20 min at room temperature. Chloroquine (25 μM) was added to HEK‐293T cells for 20 min at 37 °C. Precipitates (1.5 ml) were added to each plate. Four hours after transfection, cells were washed and incubated in medium during 48 h. Supernatants were used to infect Ba/F3 cells twice as described (Medves et al., 2010). Cells were sorted by flow cytometry after 24 h according to GFP expression. Ba/F3 cells expressing shRNA or GFP were created by lentiviral infection as described above. Cells were selected with 2 μg/ml puromycin and IL‐3. Puromycin was removed 24 h before experiments.
Human pEF‐MYC‐CYTO PDGFRA (wild‐type, D842V, Y720F or D842V/Y720F) was introduced in Ba/F3 cells by electroporation (200 V, 75 Ω, 1300 μF) with 50 μg of DNA. Cells were selected in the presence of G418 (3 mg/ml) and sorted by flow cytometry as described (Velghe et al., 2013).
Cell proliferation was measured by [3H]‐thymidine incorporation assays as described (Toffalini et al., 2009).
2.3. Luciferase assays
HEK‐293T cells were seeded in 12‐well plates (300 000 cells/well). After one day, cells were co‐transfected by calcium phosphate method with a luciferase construct controlled by serum response elements (pSRE‐luc, 0.125 μg), the pDRIVEchEF1‐RU5 vector (pEF1‐β‐galactosidase, 0.15 μg, Invitrogen) as internal control and the wild‐type human pcDNA3‐SHP2 or different mutant forms of SHP2. The mutants pcDNA3‐SHP2 C463S and Y546/584F were created using the QuikChange™ XL‐II mutagenesis protocol (Stratagene). After 24 h, cells were lysed and the luciferase activity was monitored using a GLOMAX® instrument (Turner Biosystems). The data are represented as the average ratio between the luciferase and the β‐galactosidase activity and normalized to condition with the empty vectors.
2.4. Binding assays
One mg of peptide [CDESTRSYVILSFEN, CDESTRSpYVILSFEN or CDESTRSpYGGGSFEN] was diluted in 100 μl of DMSO and mixed with 400 μl of coupling buffer (50 mM Tris, 5 mM EDTA‐Na, pH 8.5), incubated with 500 μl sulfolink coupling resin (#20401, Thermo scientific) during 15 min at 4 °C with constant shaking and then for 30 min at room temperature. Residual reactive sites were blocked with one volume (500 μl) of l‐cysteine (50 mM) during 15 min at 4 °C with constant shaking and 30 min at room temperature. Beads were washed, suspended in 1 ml of PBS and mixed with 200 ng of recombinant human active SHP2 (#1894‐SH‐100, R&D SYSTEMS) diluted in binding buffer (50 mM Tris, 150 mM NaCl, 1 mg/ml BSA, 0.5% Triton, 2 mM DTT, pH 7.5). Beads were incubated for 2 h at 4 °C with constant shaking, washed once with PBS and three times with buffer (50 mM Tris, 150 mM NaCl, 0.5% Triton, pH 7.5). Bound proteins were analyzed by Western blot.
2.5. Human CD34+ cell isolation, infection and culture
CD34+ cells were purified from umbilical cord blood after informed consent of the mother (ethical committee approval #B403201213787), cultured and transduced with lentiviral particles as described (Medves et al., 2011; Montano‐Almendras et al., 2012). Cells were sorted by flow cytometry according to GFP expression.
2.6. Retrovirus production, bone marrow cell transduction and transplantation
This experiment was performed essentially as previously described (Cools et al., 2003b). Briefly, retroviral particles used to infect primary murine bone marrow were generated by transient co‐transfection of HEK‐293T cells with a retroviral pMSCV construct (pMSCV‐GFP, ‐FPα or ‐FPα Y720F) with a packaging vector (pIK6.1 MCV.Ecopac). Virus supernatants were collected and tested as previously described (Schwaller et al., 1998). BALB/c mice were purchased from Charles River. Twelve‐week old BALB/c male donor mice were sacrificed by cervical dislocation. Bone marrow was flushed from femurs and tibias. After red blood cells lysis, hematopoietic stem cells and progenitors were enriched by removing lineage‐positive cells according to manufacturer instructions (#19756, StemCell Technologies). Cells were cultured overnight with murine IL‐3 (10 ng/ml, PeproTech), IL‐6 (10 ng/ml, PeproTech) and Stem Cell Factor (50 ng/ml, PeproTech) in RPMI 1640 (Invitrogen) with 20% of FBS, penicillin and streptomycin (transplant medium) for 24 h. One million cells were seeded in untreated 6‐well plates (#734‐0948, BD Falcon™) with 1 ml of viral supernatant and 2 ml of transplant medium containing 8 mg/ml Polybrene® and centrifuged for 90 min at 2500 rpm. Cells were incubated in transplant medium for 24 h. The day after, the percentage of GFP‐positive cells was measured by flow cytometry. One million cells (300 μl) were injected into the tail vein of sublethally irradiated (5 Gy) 6–8 week old female recipient mice. Mice were housed in individually ventilated cages. Peripheral blood was analyzed by differential cell counts (Animal Blood Cell counter, SCIL). Alternatively, blood was treated with red blood cell lysis buffer and analyzed by flow cytometry for GFP expression.
2.7. Flow cytometry
Intracellular staining and flow cytometry were performed as described (Toffalini et al., 2010). For signaling experiment, cells were washed to remove IL‐3 and starved for 4 h. As a positive control, some cells were restimulated with IL‐3 for 20 min after starvation (data not shown). Cells were treated with imatinib during starvation, as a control. The cells were incubated with the anti‐phospho‐STAT5 (Tyr694) or the anti‐phospho‐ERK1/2 (Thr202/Tyr204) antibodies conjugated to Alexa‐Fluor 647 (BD Transduction Laboratories). Results were expressed as percentage of positive cells.
2.8. Immunoprecipitation and western blot
5 × 106 Ba/F3 cells expressing FPα, wild‐type PDGRα or mutants were lysed in Triton X‐100 buffer as described (Toffalini et al., 2009). In immunoprecipitation experiments, cell lysates were incubated overnight with the anti‐SHP2 antibody (1 μg) and antibody complexes were collected using protein‐A/G‐beads as described (Toffalini et al., 2009). Western blot analysis was performed as previously described (Toffalini et al., 2009).
2.9. Statistics
All experiments were repeated at least three times with identical results, unless otherwise stated. In most figures, the average of multiple replicate experiments is shown with standard error of the mean (S.E.M.). Individual representative experiments are shown with standard deviation (S.D.). Statistical analysis was performed using Student t‐test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
3. Results
3.1. Tyrosine residue 720 is required for proliferation induced by FPα
To better characterize the signaling pathways activated by FPα, we mutated twelve potentially phosphorylated tyrosine residues into phenylalanine (Figure 1A). Ten different tyrosines have been shown to be phosphorylated in wild‐type PDGFRα (Heldin et al., 1998). We will refer to these tyrosines using their location in the wild‐type PDGFRα amino‐acid sequence. These residues were mutated either individually or two by two. For instance, the two binding sites for the PI3K subunit p85 (Y731 and Y742), for SHP2 (Y720 and Y754) or phospholipase Cγ (Y988 and Y1018) were mutated together. In addition, two tyrosines within the FIP1L1 part of FPα, located at position 110 and 113 (Figure 1A), were also reported to be phosphorylated in a phosphoproteomics study of the EOL‐1 eosinophilic leukemia cell line and were included in the present study (Goss et al., 2006).
Figure 1.
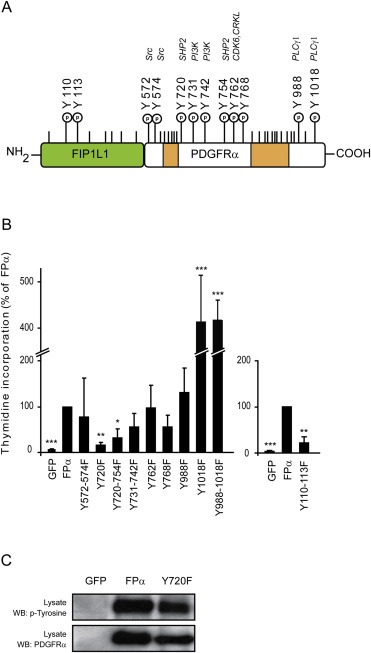
Identification of the FPα tyrosines residues required for cell growth. A. Schematic representation of the tyrosine residues present in FPα. The N‐terminal FIP1L1 portion of the fusion is represented in green. The split kinase domain of PDGFRα is indicated by orange boxes. Phosphotyrosines (P) in the PDGFRα portion of the fusion are numbered according to the wild‐type PDGFRα sequence. Some sites have been identified by a phosphoproteomics study in the FPα+ cell line Eol‐1 (Goss et al., 2006). The kinase domain was not drawn to scale. B. Ba/F3 cells were transduced with retroviral (left panel) or lentiviral plasmid (right panel) expressing GFP alone, FPα or the indicated FPα mutants in the presence of IL‐3. GFP‐expressing cells were sorted by flow cytometry. Sorted cells were washed to remove IL‐3, seeded and cell proliferation was assessed by measuring the [3H]‐thymidine incorporation after 24 h. The proliferation of cells transduced with FPα was used as a reference (100%). The average of three independent experiments is shown with S.E.M. All cell lines proliferated similarly in the presence of IL‐3 (data not shown). C. Tyrosine phosphorylation and FPα expression were assessed by western blot on total cell lysates of transduced Ba/F3 cells cultured without IL‐3 for 4 h.
The FPα mutants were introduced in the IL‐3‐dependent Ba/F3 cell line using a retroviral vector, as described (Cools et al., 2003, 2010, 2009). Homogenous cell populations were sorted by flow cytometry based on green fluorescent protein expression (GFP), which was driven by an internal ribosomal entry site (IRES) located after the FPα sequence. We performed [3H]‐thymidine incorporation assays as a read‐out of proliferation (Toffalini et al., 2010). In the absence of IL‐3, cells expressing the Y720F or the Y110/113F construct showed a significantly decreased proliferation. A similar effect was observed with the double mutant Y720/754F. Interestingly, proliferation of FPα mutated at Y1018 alone or in combination with Y988 was enhanced, suggesting that this mutation may disrupt a negative signaling pathway. The proliferation of Ba/F3 expressing the other mutants did not significantly differ from cells expressing FPα (Figure 1B). All cell lines grew similarly in the presence of IL‐3 (data not shown).
In the present study, we focused on the Y720F mutation, which produced the most dramatic effect. First, we tested whether this mutation affected the overall phosphorylation of FPα on tyrosines by western blot. Figure 1C shows that FPα auto‐phosphorylation was not affected, indicating that this mutation had no impact on the FPα tyrosine kinase activity. Altogether, these results suggested that phosphorylated Y720 may recruit signaling proteins that are required for FPα‐induced proliferation.
3.2. Mutation of residue Y720 impairs both myeloproliferation in mice and proliferation of human hematopoietic progenitors in vitro
The strong impact of a single tyrosine mutation in FPα transformation was surprising because previous studies on wild‐type PDGF receptors and the ETV6‐PDGFRβ fusion pointed to a high level of redundancy between individual phosphorylated tyrosine residues (Tallquist et al., 2003; Tomasson et al., 2000). To rule out an artifact of the Ba/F3 model, we tested the ability of the FPα Y720F mutant to transform mouse hematopoietic cells in vivo. To this end, we used the previously described bone marrow transplantation mouse leukemia model. BALB/c mice were transplanted with lineage‐negative cells bearing either non mutated FPα or the Y720F mutant. Previous reports showed that FPα induces a myeloproliferative disorder in this model, with increased proliferation of myeloid cells in the peripheral blood and splenomegaly (Stover et al., 2006). As a control, we confirmed that the expression levels of the mutants were similar in transduced cells before transplantation (Figure 2A). Three weeks after transplantation, the proliferation of the FPα and FPα Y720F myeloid cells in the blood was analyzed by flow cytometry based on GFP expression. The percentage of white blood cells expressing GFP was significantly different between the two conditions, indicating that the Y720F mutation severely reduced the FPα‐driven disease (Figure 2B). In agreement with this observation, the spleen weight was reduced in the Y720F mice compared with FPα mice, further illustrating the reduced oncogenic activity of Y720F (Figure 2C). Together, these results suggest that the Y720F mutation limits the development of a FPα‐driven myeloproliferative phenotype in mice.
Figure 2.
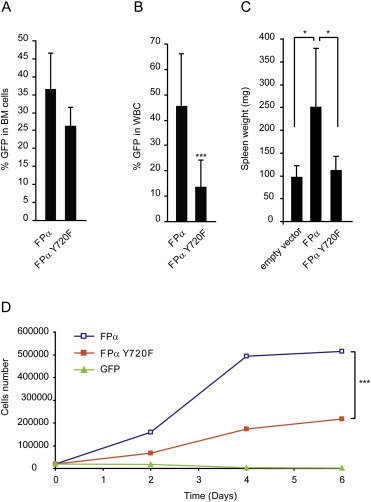
Importance of residue Y720 in vivo and in human CD34+ progenitors. A. GFP expression was measured by flow cytometry analysis in mouse hematopoietic stem cells and primitive progenitors. The cells were isolated from bone marrow, depleted in lineage‐positive cells and transduced with GFP, FPα or FPα Y720F retroviral particles. The average GFP fluorescence value (ten mice per group) is shown with S.D. B. 106 cells were transplanted in BALB/c mice. Three weeks after transplantation, GFP expression was measured by flow cytometry in white blood cells (WBC). The average value (ten mice per group) is shown with S.D. C. Mice were sacrificed after 3 weeks and spleens were weighted (n = 5). A unilateral Student t‐test was performed. D. Proliferation test was performed in CD34+ cells isolated from human umbilical cord blood, transduced with GFP (green triangles), FPα (open blue squares) or FPα‐Y720F (orange squares) and seeded at 3.104 cells/well in presence of recombinant human SCF and FLT3L both at 25 ng/ml. Viable cells were counted in the presence of Trypan blue. One representative experiment out of four is shown.
We have previously demonstrated that FPα stimulates the proliferation of human CD34+ stem and progenitor cells isolated from umbilical cord blood (Montano‐Almendras et al., 2012). Figure 2D shows that CD34+ cells transduced with a lentivirus expressing FPα proliferated to a higher extent compared with cells expressing FPα Y720F in liquid cultures. After six days, the number of cells transduced with the mutant was significantly decreased compared with FPα cells.
These experiments confirmed the importance of residue Y720 in vivo and in primary human hematopoietic cells.
3.3. Mutation of residue Y720 disrupts FPα signaling pathways
Previous studies have shown that the transcription factor STAT5 and the kinases ERK1/2 are important signaling mediators of FPα (Buitenhuis et al., 2007; Cools et al., 2003a). We first tested the activation of these mediators by western blot using phospho‐specific antibodies directed against key phosphorylated sites that are predictive of the activity of these two factors. We observed that STAT5 phosphorylation in Ba/F3 cells expressing the Y720F mutant decreased considerably compared with the FPα condition (Figure 3A). As a control, we treated cells with imatinib, a selective inhibitor of PDGFR, which inhibited STAT5 phosphorylation as expected. This result was confirmed by flow cytometry after intracellular staining with phospho‐specific antibodies (Figure 3B). We next analyzed the ERK1/2 pathway, which was also activated by FPα in an imatinib‐dependent manner (Figure 3A). The mutation of Y720 abolished phosphorylation of ERK1/2 by FPα. The same results were obtained by flow cytometry after intracellular staining (Figure 3C). Thus, a decreased activation of STAT5 and ERK1/2 by this mutant could explain its decreased ability to stimulate proliferation.
Figure 3.
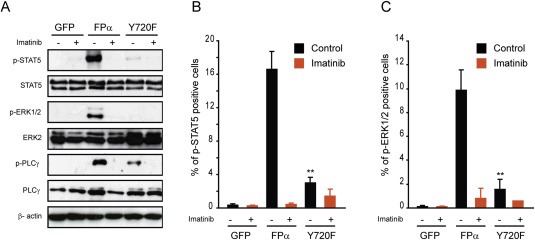
Mutation of residue Y720 affects signaling induced by FPα. A. Western blot analysis of Ba/F3 cells transduced with lentiviral plasmid encoding GFP, FPα or FPα Y720F. Cells were washed and cultured without IL‐3 for 4 h before being treated or not with imatinib (500 nM), as indicated. Total cell lysates were analyzed by western blot with the indicated antibodies. B–C. Flow cytometry analysis of transduced Ba/F3 stained with fluorescent anti‐phospho‐STAT5 (left panel) or anti‐phospho‐ERK1/2 (right panel) antibodies. 106 cells were permeabilized, washed and incubated without IL‐3 before applying the staining. As a negative control, cells were treated with imatinib (500 nM). The percentage of positive cells compared with background staining is indicated. The average of two independent experiments is shown with S.E.M.
We next evaluated the phosphorylation of PLCγ in Ba/F3 cells expressing FPα (Figure 3A). In line with recent results showing that PLCγ is activated by PDGFRβ fusion proteins (Medves et al., 2010), FPα was also found capable of including a strong PLCγ phosphorylation. The extent of the phosphorylation was decreased in the case of the Y720F mutant although not completely suppressed, indicating that this mutation does not disrupt unselectively all signaling pathways activated by FPα. Altogether, these findings point towards an important role of residue Y720 in FPα signaling.
3.4. FPα Y720 binds to SHP2
Previous reports suggested that the protein tyrosine phosphatase SHP2 interacts with tyrosine 720 in wild‐type PDGFRα, even though a direct binding has not been formally demonstrated (Bazenet et al., 1996; Heldin et al., 1998). In addition, SHP2 is a known regulator of the ERK pathway (Araki et al., 2003). To evaluate the interaction between FPα and SHP2, we performed co‐immunoprecipitation experiments. SHP2 was able to co‐immunoprecipitate with FPα but not with the Y720F mutant in Ba/F3 cells (Figure 4A). Moreover, we showed that the immunoprecipitated SHP2 can interact with the phosphorylated form of FPα but not FPα Y720F (Figure 4B) even though they were similarly phosphorylated in Ba/F3 cells (Figure 1C). This experiment suggested that SHP2 is recruited by tyrosine 720 of FPα, in line with the published results on wild‐type PDGFRα. Accordingly, mutation of Y720 also abolished the phosphorylation of SHP2 induced by FPα (Figure 4B). To establish whether SHP2 binds directly to FPα Y720, we tested the ability of a recombinant SHP2 protein to bind a phosphorylated peptide of 15 residues corresponding to the sequence surrounding Y720. Figure 4C shows that SHP2 could directly bind the phospho‐Y720 peptide and that this binding required Y720 phosphorylation. As SHP2 was shown to interact with hydrophobic residues located close to phosphorylated tyrosines, we substituted the val‐ile‐leu sequence that is adjacent to tyr720 by three glycine residues (Martinelli et al., 2008; Sweeney et al., 2005). This mutation prevented the interaction of recombinant SHP2 with the phosphorylated peptide, indicating that the recruitment is sequence‐specific. Based on these experiments, we concluded that SHP2 is able to bind directly to phosphorylated Y720 and the adjacent hydrophobic residues of FPα.
Figure 4.
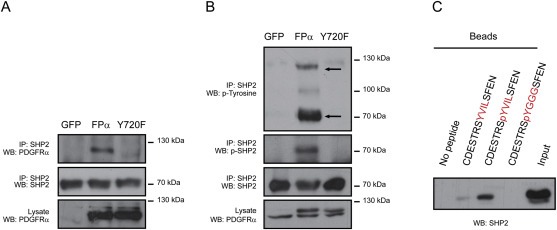
SHP2 binds to phosphorylated tyrosine 720. A‐B. SHP2 co‐immunoprecipitation with FPα. Ba/F3 cells were transduced with lentivirus expressing GFP alone, FPα or FPα Y720F and cultured without IL‐3 for 4 h. SHP2 was immunoprecipitated from cell lysates and analyzed by western blot with anti‐SHP2, anti‐PDGFRα, anti‐phospho‐Y542‐SHP2 or anti‐phosphotyrosine (PY99) antibodies. The upper and lower arrows on panel B correspond respectively to the molecular weight of FPα and SHP2. C. Interaction of SHP2 protein with a phosphopeptide corresponding to the Y720 phosphorylation site of PDGFRα. Purified recombinant SHP2 protein was incubated with beads coupled to the phospho‐peptide and interaction was detected by western blot analysis after extensive washing of the beads. As a control, the experiment was performed with a peptide containing the non‐phosphorylated tyrosine and a mutated phospho‐peptide. Input represents 200 ng of recombinant SHP2 protein.
3.5. SHP2 is required for ERK1/2 activation and proliferation by FPα
To further investigate the role of SHP2 in cell transformation by FPα, we used a lentiviral vector expressing shRNA that targets SHP2 (referred to as shSHP2), a control shRNA consisting of a scramble sequence (shScramble) or GFP. Ba/F3 cells were transduced with viral particles and selected in the presence of puromycin.
We analyzed SHP2 expression in puromycin‐resistant Ba/F3 cells by western blot. Figure 5A shows that three different shRNA suppressed efficiently the expression of SHP2. Accordingly, SHP2 phosphorylation was not detectable in cells expressing shSHP2. We next measured the proliferation of the cell lines expressing the different types of shRNA. The results show that SHP2 knockdown in Ba/F3 cells expressing FPα significantly reduced thymidine incorporation compared with control shRNA or GFP (Figure 5B).
Figure 5.
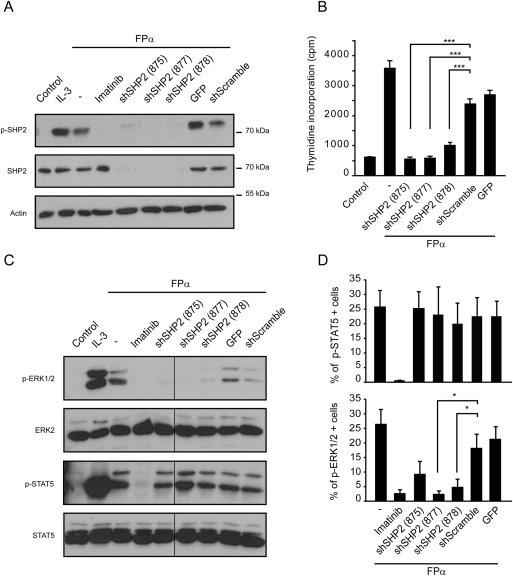
SHP2 is required for ERK1/2 activation and proliferation induced by FPα. A. Ba/F3 cells expressing FPα were transduced with three different shSHP2 (875, 877, 878), a control shScramble, or a turbo‐GFP vector. Cells were selected in the presence of puromycin and IL‐3. Cells were washed, cultured without IL‐3 and treated or not with imatinib (1 μM) for 4 h, as indicated. Total cell lysates were analyzed by western blot with the indicated antibodies. As a positive control of SHP2 phosphorylation, Ba/F3 cells were re‐stimulated with IL‐3 for 15 min after starvation. B. [3H]‐thymidine incorporation assay was performed in transduced Ba/F3 cells washed to remove IL‐3, seeded as described in materials and methods. Cell proliferation was assessed after 24 h. One representative experiment is shown. C. Western blot analysis of transduced Ba/F3 cells treated as in A. Total cell lysates were analyzed with the indicated antibodies. The vertical line indicates that lanes were cropped from the image of a single western blot membrane. D. Flow cytometry analysis of transduced Ba/F3 cells stained with fluorescent anti‐phospho‐STAT5 (upper panel) or anti‐phospho‐ERK1/2 (lower panel) antibodies. 5.105 cells were permeabilized, washed and incubated without IL‐3 before applying the staining. As a negative control, cells were treated with imatinib (1 μM). The average of three independent experiments is shown with S.E.M.
We next examined the effect of SHP2 downregulation on STAT5 and ERK1/2 activation. Expression of shSHP2 in FPα cells significantly decreased ERK1/2 phosphorylation but had no effect on STAT5 activation (Figure 5C). These results were confirmed by flow cytometry after intracellular staining (Figure 5D). Taken together, these experiments showed that SHP2 is required for proliferation and ERK1/2 activation by FPα but not for STAT5 activation.
3.6. The catalytic activity of SHP2 is needed for ERK1/2 activation by FPα
MAPK regulation by SHP2 depends on its catalytic activity and on its adaptor role although the relative importance of these two molecular functions has been much debated (Dance et al., 2008; Neel et al., 2003).
To tackle this issue, we used a luciferase reporter assay in which FPα stimulates the activity of a promoter driven by serum‐response elements (SRE) in transiently transfected HEK‐293T cells. This promoter is highly sensitive to MAPK activation. In line with results obtained in Ba/F3 cells, we also observed a decrease in luciferase activity when cells expressed the Y720F mutant compared with FPα (Figure 6A). The importance of SHP2 phosphatase activity was determined using a catalytically inactive dominant negative SHP2 mutant (C463S) (Walter et al., 1999). In 293T cells transfected with FPα, the expression of SHP2 C463S induced a significant decrease in MAPK activation compared with wild‐type SHP2, confirming the role of this phosphatase. To test the implication of the adaptor role of SHP2, we mutated the two key tyrosine residues that act as docking sites for Grb2, namely Y546 and Y584 (Dance et al., 2008). The impact of the SHP2 Y546/584F mutation was tested in the luciferase assay described above. This mutant did not affect MAPK activation by FPα (Figure 6B). As a control, cells were co‐transfected with a constitutively activated form of mouse M‐Ras Q71K (Demoulin et al., 2000; Louahed et al., 1999) in presence or not of the mutant SHP2. As expected, MAPK pathway was activated in a SHP2‐independent manner by activated RAS (Figure 6C, D). These results confirmed that the catalytic domain of SHP2 promotes ERK activation by FPα.
Figure 6.
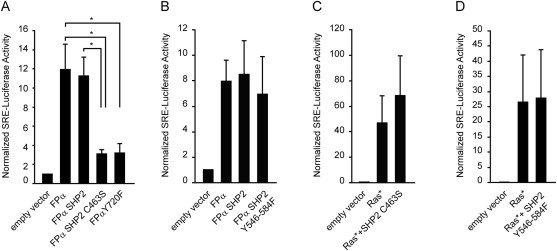
The catalytic activity of SHP2 is required for EKR1/2 activation. 293T cells were co‐transfected with FPα and SHP2, SHP2 C463S (A) or SHP2 Y546F/Y584F (B) and a luciferase construct cloned downstream of a SRE‐driven promoter. As a control, the co‐transfection of a constitutive active form of M‐Ras with SHP2 mutants in 293T cells was performed (C and D). The average of four independent experiments is shown with S.E.M.
3.7. Implication of SHP2 downstream PDGFRαD842V and wild‐type receptor
We next analyzed whether SHP2 was needed for cell transformation by other oncogenic mutants of PDGFRα. The most common PDGFRα mutation, D842V, was used for further experiments and combined with Y720F (Corless et al., 2005). Mutants were stably transfected in Ba/F3 cells by electroporation (Velghe et al., 2013). After sorting Ba/F3 cells expressing PDGFRαD842V or PDGFRαD842V/Y720F by flow cytometry, western blot demonstrated equivalent expression and phosphorylation between the two conditions in cells treated or not with IL‐3 (Figure 7A). The expression of the mutants was also validated by flow cytometry analysis and RT‐QPCR (data not shown). We previously reported the activation of STAT5 and ERK1/2 in Ba/F3 cells transduced with the D842V mutant (Velghe et al., 2013). We next compared the ability of D842V and D842V/Y720F to activate these signaling pathways (Figure 7A). In line with results obtained for FPα, mutation of Y720F strongly decreased SHP2 and ERK1/2 phosphorylation by the D842V mutant in absence of IL‐3. Interestingly, Y720F did not change the phosphorylation of STAT5 by D842V, unlike FPα. Finally, we measured the proliferation in cells cultured in the absence of cytokines or with PDGF. The results showed a significant decrease of thymidine incorporation in cells expressing D842V/Y720F, confirming the importance of Y720 (Figure 7B). This experiment also showed that PDGF treatment did not compensate for the loss of the Y720 residue. In control experiments, the cells transduced with the mutants or the empty vectors proliferated to the same extent in the presence of IL‐3 (data not shown).
Figure 7.
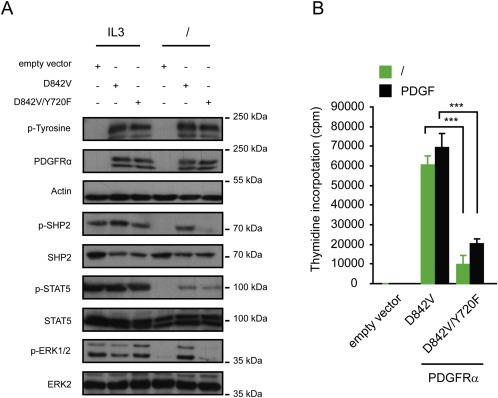
Mutation of Y720 did not affect overall PDGFRαD842V phosphorylation but affects signaling induced by PDGFRαD842V. A. Western blot analysis of Ba/F3 cells electroporated with plasmid pEF‐MYC‐CYTO empty vector, PDGFRαD842V or PDGFRαD842V/Y720F. Cells expressing PDGFR mutant were washed and cultured with or without IL‐3 for 4 h. Total cell lysates were analyzed by western blot with the indicated antibodies. The upper band represents the mature glycosylated membrane form of the receptor and the lower band correspond to the immature hypoglycosylated form which is located in the reticulum, as described (Heinrich et al., 2003). As a positive control of phosphorylation, Ba/F3 cells electroporated with the empty vector were treated without IL‐3 for 3 h 45 min and then with IL‐3 for 15 min. As a negative control of phosphorylation, Ba/F3 cells expressing empty vector were treated without IL‐3 for 4 h. Anti‐ actine, SHP2, STAT5 and ERK antibodies were used as control of protein loading. B. [3H]‐thymidine incorporation assay was performed in transduced Ba/F3 cells washed and cultured without IL‐3 or with PDGF‐BB (25 ng/ml). Cell proliferation was assessed after 72 h. As a negative control, Ba/F3 cells were transduced with the empty vector. One representative experiment is shown.
We next evaluated the requirement for SHP2 downstream PDGFRαD842V using shRNA (Figure 8A). SHP2 knockdown impaired ERK1/2 phosphorylation and cell proliferation induced by D842V, but did not affect STAT5 phosphorylation (Figure 8A and B). This indicated that SHP2 plays a similar role downstream D842V and FPα. These results contrasted with previous studies showing that Y720 and SHP2 are not essential for cell proliferation induced by wild‐type PDGFR receptors (Bazenet et al., 1996; Ronnstrand et al., 1999). Since these results had been obtained in a different model system, we introduced wild‐type PDGFRα in Ba/F3 cells and tested the impact of SHP2‐specific shRNA. Although SHP2 was phosphorylated upon PDGF‐stimulation (Figure 8A, upper right panel), proliferation of Ba/F3‐PDGFRα cells in response to PDGF was only modestly affected by shSHP2 (Figure 8C). When thymidine incorporation level of the control condition (i.e. in the absence of cytokine) was subtracted, shSHP2 had no impact on the mitogenic effect of PDGF. In agreement with these results, SHP2 deficiency did not affect the phosphorylation of ERK1/2 and STAT5 upon stimulation of the wild‐type receptor by PDGF (Figure 8A).
Figure 8.
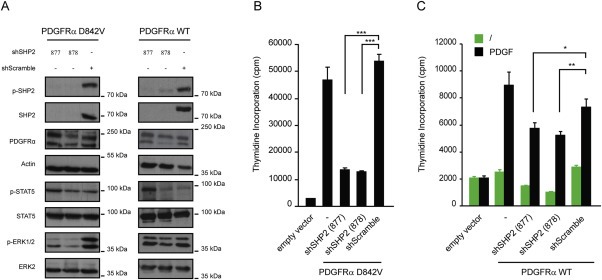
Role of SHP2 downstream of PDGFRαD842V and wild‐type PDGFRα. A. Western blot analysis of Ba/F3 cells expressing pEF‐MYC‐CYTO PDGFRαD842V or wild‐type (WT) PDGFRα transduced with two different shSHP2 (877, 878) or a control shScramble. Cells were washed and cultured without IL‐3 for 4 h. Cells expressing PDGFRα WT are stimulated with PDGF‐BB (25 ng/ml). Total cell lysates were analyzed by western blot with the indicated antibodies. B. [3H]‐thymidine incorporation assay was performed in transduced Ba/F3 cells washed and cultured without IL‐3. Cell proliferation was assessed after 24 h. As a positive control, Ba/F3 cells transduced with PDGFRαD842V were treated without IL‐3 for 24 h. Ba/F3 PDGFRαD842V shScramble cells were used as reference. One representative experiment is shown. C. [3H]‐thymidine incorporation assay was performed in transduced Ba/F3 cells washed and cultured without IL‐3 or with PDGF‐BB (25 ng/ml). Cell proliferation was assessed after 24 h. As a positive control, Ba/F3 cells transduced with PDGFRα WT were treated without PDGF‐BB for 24 h. As a negative control, Ba/F3 cells transduced with the empty vector were treated without cytokines for 24 h. Ba/F3 PDGFRα WT shScramble cells were used as reference. One representative experiment is shown.
In conclusion, using shRNA, we demonstrated that SHP2 is essential for the stimulation of cell proliferation and ERK1/2 activation by the FPα and D842V oncogenes but not by wild‐type PDGFRα.
4. Discussion
Our results demonstrate that SHP2 is required for cell proliferation and ERK1/2 activation downstream of FPα and D842V but not downstream of wild‐type PDGF receptors. This was shown by mutating a specific SHP2 binding site and by knocking‐down SHP2 with shRNA. Luciferase assay also confirmed the crucial importance of SHP2 for ERK1/2 activation. Wild‐type and mutant PDGFRα similarly induced the phosphorylation of SHP2 and ERK1/2, but only mutant oncogenic receptors required SHP2 for ERK activation and proliferation. This was consistent with previously published results which demonstrated that phosphorylation of tyrosine 720 in wild‐type PDGFRα is required for binding of SHP2 but not for activation of Ras and proliferation (Bazenet et al., 1996).
Our results show that wild‐type PDGFRα activate ERK more efficiently in a SHP2‐independent manner. Different mechanisms downstream wild‐type PDGFRα ‐ but not the oncogenes ‐ could co‐exist to activate the MAPK pathway, in such a way that ERK1/2 activation is still possible via GRB2 despite the depletion of SHP2 in cells expressing wild‐type PDGFRα.
Similar switches in signaling were reported for KIT and FLT3 (Toffalini and Demoulin, 2010). For instance, SRC activation is required to stimulate proliferation by wild‐type KIT but not by the constitutive mutant KITD816V (Sun et al., 2009). It is also well illustrated for FLT3: the internal tandem duplications (FLT3‐ITD, found in acute myeloid leukemia), induces SRC and STAT5 activation, unlike the wild‐type receptor (Leischner et al., 2012).
A functional role for SHP2 in oncogenic FPα and PDGFRαD842V‐induced cell transformation has not been previously demonstrated. Others studies suggested the implication of SHP2 in leukemogenesis induced by others mutants of the same receptor family such as KIT and FLT3 (Mali et al., 2012; Nabinger et al., 2013).
In our model, we also measured the activity of STAT5. SHP2 knockdown did not decrease the phosphorylation of STAT5 in Ba/F3 cells expressing FPα, D842V or wild‐type PDGFRα. By contrast, a role of SHP2 in STAT5 activation has been suggested in different studies (Xu and Qu, 2008). SHP2 was reported to directly dephosphorylate STAT5 (Chen et al., 2004, 2003) or indirectly enhance STAT5 activation (Ali et al., 2003). However, this does not seem to apply to PDGF receptor signaling. So far no direct binding sites for STAT5 have been reported in PDGFRα, by contrast to PDGFRβ (Valgeirsdottir et al., 1998). In our experiments, tyrosine 720 was involved in STAT5 activation by FPα, possibly by recruiting STAT5 directly or via an adaptor protein. However, we were unable to detect a co‐immunoprecipitation of STAT5 with FPα (data not shown). Unlike FPα, D842V activated STAT5 independently of residue Y720. This may be related to the different subcellular localizations of the two oncogenes (Toffalini and Demoulin, 2010). Another tyrosine may also be phosphorylated specifically in D842V and acts as alternative docking site for STAT5. The mechanism by which STAT5 is activated by mutant and wild‐type PDGFRα requires further investigations.
Our experiment with phosphopeptides shows that SHP2 is capable of binding directly to Y720 in vitro. Moreover, amino acids surrounding Y720 in FPα (RSpYVIL) matches the consensus site for SHP2 SH2 domain binding, except at position −2, where a hydrophobic residue is more frequently found (Martinelli et al., 2008; Sweeney et al., 2005). Our data are in agreement with the study of Bazenet et al., who also suggested a direct interaction using an alternative method (Bazenet et al., 1996). In conclusion, our results point to a direct binding of SHP2 to Y720. Nevertheless, we can not rule out the existence of more complex recruitment mechanisms in vivo. Finally, Y754 in the PDGFRα has been also described to bind SHP2 (Rupp et al., 1994). However, the phosphorylation of Y754 was only reported in the heterodimeric PDGFRα/β complex. If Y754 is phosphorylated in FPα, which remains to be established, it is clearly not sufficient to recruit SHP2, based on our co‐imunoprecipitation experiments. In addition, the double mutant FPα Y720/754F presented the same proliferation defect as the single mutant Y720F. Nevertheless, Y754 may act as a minor binding site for the second SH2 domain of SHP2, stabilizing the protein complex.
Interestingly, our results show that residues Y110/113, which have been reported to be phosphorylated in a previous study (Goss et al., 2006), are also involved in cell proliferation. In line with our results, Buitenhuis and colleagues showed that this domain of FPα has a role in human hematopoietic cell proliferation (Buitenhuis et al., 2007). It remains to be determined whether Y110/113 act as classical docking sites for signaling proteins or have a completely different function. Moreover, we observed that mutation of residue Y1018 led to a significant increase in cell proliferation, suggesting that a negative signaling pathway may be associated with this residue. In addition to PLCγ, one report suggested that this site recruits the E3 ubiquitin ligase Cbl (Reddi et al., 2007). While it is well established that Cbl promotes the degradation of wild‐type PDGF receptors, its role in FPα signaling is not yet clear (Miyake et al., 1999; Reddi et al., 2007; Toffalini et al., 2009).
This study demonstrates the important role of SHP2 for cell transformation by FPα and PDGFRαD842V. The SHP2‐MAPK pathway may therefore represent a potential therapeutic target, particularly for patients with the D842V mutation, which is resistance to most tyrosine kinase inhibitors (Corless et al., 2005).
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank the members of the MEXP unit for their constant support, Dominique Latinne, Hélène Schoemans and the cord blood bank teams of UCL and UZ Leuven, Vincent Stroobant for chemical peptide production, Nicolas Dauguet for cell sorting and Dr D. Sarbassov for Addgene plasmid 1864. This project was supported by grants from the Salus Sanguinis foundation and from “Actions de Recherches Consertées” (Communauté Française de Belgique, Belgium). L.A.N. was the recipient of a grant from FRS‐FNRS – Télévie.
Noël Laura A., Arts Florence A., Montano-Almendras Carmen P., Cox Luk, Gielen Olga, Toffalini Federica, Marbehant Catherine Y., Cools Jan and Demoulin Jean-Baptiste, (2014), The tyrosine phosphatase SHP2 is required for cell transformation by the receptor tyrosine kinase mutants FIP1L1‐PDGFRα and PDGFRα D842V, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.02.003.
Contributor Information
Laura A. Noël, Email: laura.noel@uclouvain.be
Florence A. Arts, Email: florence.arts@uclouvain.be
Carmen P. Montano-Almendras, Email: carmen.patricia.montano@gmail.com
Luk Cox, Email: Luk.Cox@cme.vib-kuleuven.be.
Olga Gielen, Email: Olga.Gielen@cme.vib-kuleuven.be.
Federica Toffalini, Email: federica.toffalini@gmail.com.
Catherine Y. Marbehant, Email: catherine.marbehant@gmail.com
Jan Cools, Email: jan.cools@cme.vib-kuleuven.be.
Jean-Baptiste Demoulin, Email: jb.demoulin@uclouvain.be.
References
- Ali, S. , Nouhi, Z. , Chughtai, N. , 2003. SHP-2 regulates SOCS-1-mediated Janus kinase-2 ubiquitination/degradation downstream of the prolactin receptor. J. Biol. Chem.. 278, 52021–52031. [DOI] [PubMed] [Google Scholar]
- Andrae, J. , Gallini, R. , Betsholtz, C. , 2008. Role of platelet-derived growth factors in physiology and medicine. Genes Dev.. 22, 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, T. , Nawa, H. , Neel, B.G. , 2003. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J. Biol. Chem.. 278, 41677–41684. [DOI] [PubMed] [Google Scholar]
- Bazenet, C.E. , Gelderloos, J.A. , Kazlauskas, A. , 1996. Phosphorylation of tyrosine 720 in the platelet-derived growth factor alpha receptor is required for binding of Grb2 and SHP-2 but not for activation of Ras or cell proliferation. Mol. Cell Biol. 16, 6926–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, A.M. , Tang, T.L. , Sugimoto, S. , Walsh, C.T. , Neel, B.G. , 1994. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc. Natl. Acad. Sci. U S A. 91, 7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitenhuis, M. , Verhagen, L.P. , Cools, J. , Coffer, P.J. , 2007. Molecular mechanisms underlying FIP1L1-PDGFRA-mediated myeloproliferation. Cancer Res.. 67, 3759–3766. [DOI] [PubMed] [Google Scholar]
- Chan, G. , Kalaitzidis, D. , Neel, B.G. , 2008. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev.. 27, 179–192. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Yu, W.M. , Bunting, K.D. , Qu, C.K. , 2004. A negative role of SHP-2 tyrosine phosphatase in growth factor-dependent hematopoietic cell survival. Oncogene. 23, 3659–3669. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Wen, R. , Yang, S. , Schuman, J. , Zhang, E.E. , Yi, T. , Feng, G.S. , Wang, D. , 2003. Identification of Shp-2 as a Stat5A phosphatase. J. Biol. Chem.. 278, 16520–16527. [DOI] [PubMed] [Google Scholar]
- Cools, J. , DeAngelo, D.J. , Gotlib, J. , Stover, E.H. , Legare, R.D. , Cortes, J. , Kutok, J. , Clark, J. , Galinsky, I. , Griffin, J.D. , Cross, N.C. , Tefferi, A. , Malone, J. , Alam, R. , Schrier, S.L. , Schmid, J. , Rose, M. , Vandenberghe, P. , Verhoef, G. , Boogaerts, M. , Wlodarska, I. , Kantarjian, H. , Marynen, P. , Coutre, S.E. , Stone, R. , Gilliland, D.G. , 2003. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med.. 348, 1201–1214. [DOI] [PubMed] [Google Scholar]
- Cools, J. , Stover, E.H. , Boulton, C.L. , Gotlib, J. , Legare, R.D. , Amaral, S.M. , Curley, D.P. , Duclos, N. , Rowan, R. , Kutok, J.L. , Lee, B.H. , Williams, I.R. , Coutre, S.E. , Stone, R.M. , DeAngelo, D.J. , Marynen, P. , Manley, P.W. , Meyer, T. , Fabbro, D. , Neuberg, D. , Weisberg, E. , Griffin, J.D. , Gilliland, D.G. , 2003. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRalpha-induced myeloproliferative disease. Cancer Cell. 3, 459–469. [DOI] [PubMed] [Google Scholar]
- Corless, C.L. , Schroeder, A. , Griffith, D. , Town, A. , McGreevey, L. , Harrell, P. , Shiraga, S. , Bainbridge, T. , Morich, J. , Heinrich, M.C. , 2005. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol.. 23, 5357–5364. [DOI] [PubMed] [Google Scholar]
- Dance, M. , Montagner, A. , Salles, J.P. , Yart, A. , Raynal, P. , 2008. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 20, 453–459. [DOI] [PubMed] [Google Scholar]
- Demoulin, J.B. , Uyttenhove, C. , Lejeune, D. , Mui, A. , Groner, B. , Renauld, J.C. , 2000. STAT5 activation is required for interleukin-9-dependent growth and transformation of lymphoid cells. Cancer Res.. 60, 3971–3977. [PubMed] [Google Scholar]
- Dewaele, B. , Wasag, B. , Cools, J. , Sciot, R. , Prenen, H. , Vandenberghe, P. , Wozniak, A. , Schoffski, P. , Marynen, P. , Debiec-Rychter, M. , 2008. Activity of dasatinib, a dual SRC/ABL kinase inhibitor, and IPI-504, a heat shock protein 90 inhibitor, against gastrointestinal stromal tumor-associated PDGFRAD842V mutation. Clin. Cancer Res.. 14, 5749–5758. [DOI] [PubMed] [Google Scholar]
- Elling, C. , Erben, P. , Walz, C. , Frickenhaus, M. , Schemionek, M. , Stehling, M. , Serve, H. , Cross, N.C. , Hochhaus, A. , Hofmann, W.K. , Berdel, W.E. , Muller-Tidow, C. , Reiter, A. , Koschmieder, S. , 2011. Novel imatinib-sensitive PDGFRA-activating point mutations in hypereosinophilic syndrome induce growth factor independence and leukemia-like disease. Blood. 117, 2935–2943. [DOI] [PubMed] [Google Scholar]
- Ezeokonkwo, C., Zhelkovsky, A., Lee, R., Bohm, A., Moore, C.L., A flexible linker region in Fip1 is needed for efficient mRNA polyadenylation. Rna 17, 652–664. [DOI] [PMC free article] [PubMed]
- Fambrough, D. , McClure, K. , Kazlauskas, A. , Lander, E.S. , 1999. Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell. 97, 727–741. [DOI] [PubMed] [Google Scholar]
- Gleich, G.J. , Leiferman, K.M. , Pardanani, A. , Tefferi, A. , Butterfield, J.H. , 2002. Treatment of hypereosinophilic syndrome with imatinib mesilate. Lancet. 359, 1577–1578. [DOI] [PubMed] [Google Scholar]
- Goss, V.L. , Lee, K.A. , Moritz, A. , Nardone, J. , Spek, E.J. , MacNeill, J. , Rush, J. , Comb, M.J. , Polakiewicz, R.D. , 2006. A common phosphotyrosine signature for the Bcr–Abl kinase. Blood. 107, 4888–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich, M.C. , Corless, C.L. , Duensing, A. , McGreevey, L. , Chen, C.J. , Joseph, N. , Singer, S. , Griffith, D.J. , Haley, A. , Town, A. , Demetri, G.D. , Fletcher, C.D. , Fletcher, J.A. , 2003. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 299, 708–710. [DOI] [PubMed] [Google Scholar]
- Heldin, C.H. , Östman, A. , Rönnstrand, L. , 1998. Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta. 1378, F79–F113. [DOI] [PubMed] [Google Scholar]
- Huss, S. , Wardelmann, E. , Goltz, D. , Binot, E. , Hartmann, W. , Merkelbach-Bruse, S. , Buttner, R. , Schildhaus, H.U. , 2012. Activating PDGFRA mutations in inflammatory fibroid polyps occur in exons 12, 14 and 18 and are associated with tumour localization. Histopathology. 61, 59–68. [DOI] [PubMed] [Google Scholar]
- Kallin, A. , Demoulin, J.B. , Nishida, K. , Hirano, T. , Ronnstrand, L. , Heldin, C.H. , 2004. Gab1 contributes to cytoskeletal reorganization and chemotaxis in response to platelet-derived growth factor. J. Biol. Chem.. 279, 17897–17904. [DOI] [PubMed] [Google Scholar]
- Leevers, S.J. , Marshall, C.J. , 1992. Activation of extracellular signal-regulated kinase, ERK2, by p21ras oncoprotein. Embo J.. 11, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leischner, H. , Albers, C. , Grundler, R. , Razumovskaya, E. , Spiekermann, K. , Bohlander, S. , Ronnstrand, L. , Gotze, K. , Peschel, C. , Duyster, J. , 2012. SRC is a signaling mediator in FLT3-ITD- but not in FLT3-TKD-positive AML. Blood. 119, 4026–4033. [DOI] [PubMed] [Google Scholar]
- Lierman, E. , Michaux, L. , Beullens, E. , Pierre, P. , Marynen, P. , Cools, J. , Vandenberghe, P. , 2009. FIP1L1-PDGFRalpha D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRalpha T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 23, 845–851. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Qu, C.K. , 2011. Protein tyrosine phosphatase SHP-2 (PTPN11) in Hematopoiesis and leukemogenesis. J. Signal Transduction. 2011, 195239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louahed, J. , Grasso, L. , De Smet, C. , Van Roost, E. , Wildmann, C. , Nicolaides, N.C. , Levitt, R.C. , Renauld, J.C. , 1999. Interleukin-9-induced expression of M-Ras/R-Ras3 oncogene in T-helper clones. Blood. 94, 1701–1710. [PubMed] [Google Scholar]
- Lu, W. , Gong, D. , Bar-Sagi, D. , Cole, P.A. , 2001. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol. Cell. 8, 759–769. [DOI] [PubMed] [Google Scholar]
- Mali, R.S. , Ma, P. , Zeng, L.F. , Martin, H. , Ramdas, B. , He, Y. , Sims, E. , Nabinger, S. , Ghosh, J. , Sharma, N. , Munugalavadla, V. , Chatterjee, A. , Li, S. , Sandusky, G. , Craig, A.W. , Bunting, K.D. , Feng, G.S. , Chan, R.J. , Zhang, Z.Y. , Kapur, R. , 2012. Role of SHP2 phosphatase in KIT-induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood. 120, 2669–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli, S. , Torreri, P. , Tinti, M. , Stella, L. , Bocchinfuso, G. , Flex, E. , Grottesi, A. , Ceccarini, M. , Palleschi, A. , Cesareni, G. , Castagnoli, L. , Petrucci, T.C. , Gelb, B.D. , Tartaglia, M. , 2008. Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes. Hum. Mol. Genet.. 17, 2018–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medves, S. , Duhoux, F.P. , Ferrant, A. , Toffalini, F. , Ameye, G. , Libouton, J.M. , Poirel, H.A. , Demoulin, J.B. , 2010. KANK1, a candidate tumor suppressor gene, is fused to PDGFRB in an imatinib-responsive myeloid neoplasm with severe thrombocythemia. Leukemia. 24, 1052–1055. [DOI] [PubMed] [Google Scholar]
- Medves, S. , Noel, L.A. , Montano-Almendras, C.P. , Albu, R.I. , Schoemans, H. , Constantinescu, S.N. , Demoulin, J.B. , 2011. Multiple oligomerization domains of KANK1-PDGFRB are required for JAK2-independent hematopoietic cell proliferation and signaling via STAT5 and ERK. Haematologica. 96, 1406–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzgeroth, G. , Walz, C. , Erben, P. , Popp, H. , Schmitt-Graeff, A. , Haferlach, C. , Fabarius, A. , Schnittger, S. , Grimwade, D. , Cross, N.C. , Hehlmann, R. , Hochhaus, A. , Reiter, A. , 2008. Safety and efficacy of imatinib in chronic eosinophilic leukaemia and hypereosinophilic syndrome: a phase-II study. Br. J. Haematol.. 143, 707–715. [DOI] [PubMed] [Google Scholar]
- Miyake, S. , Mullane-Robinson, K.P. , Lill, N.L. , Douillard, P. , Band, H. , 1999. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. A critical role for Cbl tyrosine kinase-binding domain. J. Biol. Chem.. 274, 16619–16628. [DOI] [PubMed] [Google Scholar]
- Montano-Almendras, C.P. , Essaghir, A. , Schoemans, H. , Varis, I. , Noel, L.A. , Velghe, A.I. , Latinne, D. , Knoops, L. , Demoulin, J.B. , 2012. ETV6-PDGFRB and FIP1L1-PDGFRA stimulate human hematopoietic progenitor proliferation and differentiation into eosinophils: role of nuclear factor [kappa]B. Haematologica. 97, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan, G. , Lichter, D.I. , Di Bacco, A. , Blakemore, S.J. , Berger, A. , Koenig, E. , Bernard, H. , Trepicchio, W. , Li, B. , Neuwirth, R. , Chattopadhyay, N. , Bolen, J.B. , Dorner, A.J. , van de Velde, H. , Ricci, D. , Jagannath, S. , Berenson, J.R. , Richardson, P.G. , Stadtmauer, E.A. , Orlowski, R.Z. , Lonial, S. , Anderson, K.C. , Sonneveld, P. , San Miguel, J.F. , Esseltine, D.L. , Schu, M. , 2013. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood. 123, 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabinger, S.C. , Li, X.J. , Ramdas, B. , He, Y. , Zhang, X. , Zeng, L. , Richine, B. , Bowling, J.D. , Fukuda, S. , Goenka, S. , Liu, Z. , Feng, G.S. , Yu, M. , Sandusky, G.E. , Boswell, H.S. , Zhang, Z.Y. , Kapur, R. , Chan, R.J. , 2013. The protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyperproliferation and malignant disease in vivo. Leukemia. 27, 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel, B.G. , Gu, H. , Pao, L. , 2003. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci.. 28, 284–293. [DOI] [PubMed] [Google Scholar]
- Pluskey, S. , Wandless, T.J. , Walsh, C.T. , Shoelson, S.E. , 1995. Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J. Biol. Chem.. 270, 2897–2900. [DOI] [PubMed] [Google Scholar]
- Reddi, A.L. , Ying, G. , Duan, L. , Chen, G. , Dimri, M. , Douillard, P. , Druker, B.J. , Naramura, M. , Band, V. , Band, H. , 2007. Binding of Cbl to a phospholipase Cgamma1-docking site on platelet-derived growth factor receptor beta provides a dual mechanism of negative regulation. J. Biol. Chem.. 282, 29336–29347. [DOI] [PubMed] [Google Scholar]
- Ronnstrand, L. , Arvidsson, A.K. , Kallin, A. , Rorsman, C. , Hellman, U. , Engstrom, U. , Wernstedt, C. , Heldin, C.H. , 1999. SHP-2 binds to Tyr763 and Tyr1009 in the PDGF beta-receptor and mediates PDGF-induced activation of the Ras/MAP kinase pathway and chemotaxis. Oncogene. 18, 3696–3702. [DOI] [PubMed] [Google Scholar]
- Rupp, E. , Siegbahn, A. , Ronnstrand, L. , Wernstedt, C. , Claesson-Welsh, L. , Heldin, C.H. , 1994. A unique autophosphorylation site in the platelet-derived growth factor alpha receptor from a heterodimeric receptor complex. Eur. J. Biochem./FEBS. 225, 29–41. [DOI] [PubMed] [Google Scholar]
- Schwaller, J. , Frantsve, J. , Aster, J. , Williams, I.R. , Tomasson, M.H. , Ross, T.S. , Peeters, P. , Van Rompaey, L. , Van Etten, R.A. , Ilaria, R. , Marynen, P. , Gilliland, D.G. , 1998. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. Embo J.. 17, 5321–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover, E.H. , Chen, J. , Folens, C. , Lee, B.H. , Mentens, N. , Marynen, P. , Williams, I.R. , Gilliland, D.G. , Cools, J. , 2006. Activation of FIP1L1-PDGFRalpha requires disruption of the juxtamembrane domain of PDGFRalpha and is FIP1L1-independent. Proc. Natl. Acad. Sci. U S A. 103, 8078–8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, J. , Pedersen, M. , Ronnstrand, L. , 2009. The D816V mutation of c-Kit circumvents a requirement for Src family kinases in c-Kit signal transduction. J. Biol. Chem.. 284, 11039–11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, M.C. , Wavreille, A.S. , Park, J. , Butchar, J.P. , Tridandapani, S. , Pei, D. , 2005. Decoding protein-protein interactions through combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry. 44, 14932–14947. [DOI] [PubMed] [Google Scholar]
- Tallquist, M.D. , French, W.J. , Soriano, P. , 2003. Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol.. 1, E52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffalini, F. , Demoulin, J.B. , 2010. New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood. 116, 2429–2437. [DOI] [PubMed] [Google Scholar]
- Toffalini, F. , Hellberg, C. , Demoulin, J.B. , 2010. Critical role of the platelet-derived growth factor receptor (PDGFR)-beta transmembrane domain in the TEL-PDGFRbeta cytosolic oncoprotein. J. Biol. Chem.. 285, 12268–12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffalini, F. , Kallin, A. , Vandenberghe, P. , Pierre, P. , Michaux, L. , Cools, J. , Demoulin, J.B. , 2009. The fusion proteins TEL-PDGFRbeta and FIP1L1-PDGFRalpha escape ubiquitination and degradation. Haematologica. 94, 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasson, M.H. , Sternberg, D.W. , Williams, I.R. , Carroll, M. , Cain, D. , Aster, J.C. , Ilaria, R.L. , Van Etten, R.A. , Gilliland, D.G. , 2000. Fatal myeloproliferation, induced in mice by TEL/PDGFbetaR expression, depends on PDGFbetaR tyrosines 579/581. J. Clin. Invest.. 105, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valgeirsdottir, S. , Paukku, K. , Silvennoinen, O. , Heldin, C.H. , Claesson-Welsh, L. , 1998. Activation of Stat5 by platelet-derived growth factor (PDGF) is dependent on phosphorylation sites in PDGF beta-receptor juxtamembrane and kinase insert domains. Oncogene. 16, 505–515. [DOI] [PubMed] [Google Scholar]
- Vardiman, J.W. , Thiele, J. , Arber, D.A. , Brunning, R.D. , Borowitz, M.J. , Porwit, A. , Harris, N.L. , Le Beau, M.M. , Hellstrom-Lindberg, E. , Tefferi, A. , Bloomfield, C.D. , 2009. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 114, 937–951. [DOI] [PubMed] [Google Scholar]
- Velghe, A.I. , Van Cauwenberghe, S. , Polyansky, A.A. , Chand, D. , Montano-Almendras, C.P. , Charni, S. , Hallberg, B. , Essaghir, A. , Demoulin, J.B. , 2013. PDGFRA alterations in cancer: characterization of a gain-of-function V536E transmembrane mutant as well as loss-of-function and passenger mutations. Oncogene. 10.1038/onc.2013.218 [DOI] [PubMed] [Google Scholar]
- Walter, A.O. , Peng, Z.Y. , Cartwright, C.A. , 1999. The Shp-2 tyrosine phosphatase activates the Src tyrosine kinase by a non-enzymatic mechanism. Oncogene. 18, 1911–1920. [DOI] [PubMed] [Google Scholar]
- Xu, D. , Qu, C.K. , 2008. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. A J. Virtual Libr.. 13, 4925–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S.Q. , Yang, W. , Kontaridis, M.I. , Bivona, T.G. , Wen, G. , Araki, T. , Luo, J. , Thompson, J.A. , Schraven, B.L. , Philips, M.R. , Neel, B.G. , 2004. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol. Cell. 13, 341–355. [DOI] [PubMed] [Google Scholar]